
Ginsenoside Rb1 induces autophagic lipid degradation via miR-128 targeting TFEB
Zhuoqun
Meng†
a,
Jianing
Lu†
ac,
Guangcai
Ge†
a,
Guang
Wang
d,
Ran
Zhang
a,
Yuhan
Li
a,
Shuang
Guan
 *ab and
Jing
Lu
*ab
*ab and
Jing
Lu
*ab
aCollege of Food Science and Engineering, Jilin University, Changchun, Jilin 130062, People's Republic of China. E-mail: gshuang1973@126.com; lu_jing@jlu.edu.cn; Tel: +86-0431-87836351 Tel: +86-0431-87836376
bKey Laboratory of Zoonosis, Ministry of Education College of Veterinary Medicine, Jilin University, Changchun, Jilin 130062, People's Republic of China
cHealth Technology College, Jilin Sport University, Changchun, Jilin 130062, People's Republic of China
dOffice of Laboratory and Equipment Management, Jilin University, Changchun, Jilin 130000, People's Republic of China
First published on 9th December 2022
Abstract
In recent years, the effect of lipid metabolism on health has attracted more and more attention. Ginseng is a traditional Chinese herbal medicine in China and is widely used as food in Asia. Ginsenoside Rb1 (Gs-Rb1) is the most abundant ingredient in ginsenoside, which has a variety of biological activities. In this study, we found that Gs-Rb1 can reduce lipid accumulation in mice and HepG2 cells induced by a high-fat diet (HFD) and palmitic acid (PA). At the same time, we also found that Gs-Rb1 could stimulate the autophagic flux of HFD-fed mice and PA-treated HepG2 cells, and it is further verified by adding the autophagy activator rapamycin (Rapa) and autophagy inhibitor chloroquine (CQ). Furthermore, we found that Gs-Rb1 promoted the nucleus translocation of the transcription factor EB (TFEB) and the target role of miR-128, thus stimulating autophagic flux. Therefore, our results showed that Gs-Rb1 enhanced the transcription of TFEB and its downstream lysosome-related genes by inhibiting miR-128, improved the degradation ability of lysosomes to autophagosomes, and then promoted autophagic lipid degradation.
1 Introduction
Hepatocyte lipid metabolism is always in a dynamic equilibrium state. When this steady state is broken due to diet, drugs or other induction factors, it will lead to lipid metabolism disorder and abnormal lipid accumulation. Furthermore, it may induce liver cirrhosis, liver cancer and other diseases, which seriously endanger human health. The main lipid components in hepatocytes are triglycerides (TGs) and cholesterol esters (CEs), which exist in the form of lipid droplets (LDs). Recent studies have found that autophagy can also degrade LDs.1,2 This process is called “lipophagy” and has been found in a variety of cells such as hepatocytes.3,4Autophagy is a stable self-degradation mechanism in cells, which can degrade aging and dead organelles, misfolded proteins and other molecules. It can promote the reuse of intracellular substances and provide energy for cell survival. As a special type of autophagy, lipophagy plays an important role in maintaining intracellular lipid balance.5 When cellular autophagy is initiated, the LC3 protein on the membrane of the autophagosome appears on the surface of LDs. LC3-II interacts with the LD envelope protein to wrap the LDs in the autophagosome and then fuses with the lysosome to form the autolysosome. LDs are degraded into free fatty acid (FFA) in the autolysosome. Enhanced autophagy reduces lipid accumulation in liver cells and improves lipid metabolism disorder, which is an important mechanism for improving lipid metabolism disorder.6 Martinez-Lopez et al. found that in addition to the classical way of degrading LDs by cytosolic lipase, LDs are also isolated in autophagosomes, and regulate the membrane curvature through autophagy-related proteins.7 As LD components were fused with lysosomes, they were decomposed by lysosomal enzymes, resulting in impaired autophagosome fusion and transport leading to lipid accumulation and steatosis in the liver.7 In addition to promoting lipid degradation, activation of autophagy also inhibits lipid biosynthesis. Huang et al. found that VOdipic-Cl significantly inhibited LD formation by activating autophagy, alleviated the increase of TG levels in the serum and liver of high fat diet (HFD)-fed mice, and reduced the accumulation of liver lipids.8 A small amount of lipid accumulation can stimulate autophagy and enhance the degradation of LDs, while excessive lipid accumulation can inhibit autophagy.8 HFD could enhance autophagy on the one hand, and damage autophagy, on the other hand, leading to lipid accumulation in the later stage. Therefore, the role and mechanism of autophagy in hepatocyte lipid metabolism have become a new focus and direction.
TFEB (transcription factor EB) is a master regulator of lysosomal biogenesis and autophagy.9–11 When autophagy is triggered or when lysosomes are under stress conditions, TFEB proteins translocate from the cytoplasm to the nucleus, thereby inducing the expression of hundreds of autophagy and lysosome-related genes.10,11 As one of the regulatory factors of TFEB, microRNAs (miRNAs) have attracted much attention in recent years. miRNAs are a class of noncoding RNAs with a length of approximately 20–24 nucleotides that control the expression level of their target genes.12 They play a role in the differentiation of many tissues and organs and participate in the pathogenesis of many human diseases.12 Some researchers have proven that TFEB is an effective target of miR-128 through luciferase activity experiments.13 The study indicated that miR-128-mediated TFEB inhibition can increase the formation of α-syn oligomers and significantly increase axonal swelling by blocking autophagic flux.14 More recently, hepatic TFEB overexpression has been shown to induce the PGC1α/PPARα pathway to promote fatty acid oxidation and prevent chronic high-fat diet-induced hepatic steatosis in mice.15 The study demonstrated that LO2 challenged with palmitic acid showed a significant decrease in TFEB protein levels.16 To date, although the inhibition of TFEB mediated by miR-128 has been confirmed to cause lysosomal dysfunction, it has not been widely studied as a therapeutic target, especially in the field of liver injury.
Ginseng is a traditional Chinese herbal medicine and is widely used as food in Asia.17 It contains more than 60 kinds of active ingredients such as saponins, polysaccharides, and fatty acids.17 Ginsenoside is the main active ingredient, among which Gs-Rb1 is the highest. Recent studies have proved that Gs-Rb1 could protect the cardiovascular system, improve insulin resistance, resist lipid peroxidation and scavenge free radicals.18–20 There have also been a few reports on regulating lipid metabolism: Yu's research team found that after ginsenoside Gs-Rb1 treatment, the liver weight and TG content in the livers of DB/DB obese diabetic mice were significantly reduced;21 Lin et al. found that intraperitoneal injection of Gs-Rb1 into obese mice induced by a high-fat diet for three weeks could reduce their body weights and blood lipids.22 These reports confirmed that Gs-Rb1 has a certain effect on reducing the weight and alleviating fatty liver in obese mice, but there is still a lack of research on Gs-Rb1 in the field of autophagy, especially in the field of lipophagy.23 To date, there are few studies on the activity of Gs-Rb1 in lipophagy, and the specific mechanism is not clear. This paper aims to explore the molecular mechanism of Gs-Rb1 alleviating mice and hepatocyte lipid accumulation from the perspective of autophagy. This may provide a new research direction and target for alleviating lipid metabolism disorders using the active components of natural products.
2 Materials and methods
2.1 Reagents
The purity of Gs-Rb1 is 98% or above, as purchased from DeSiTe Biological Technology Co., Ltd (Chengdu, China). PA was purchased from Sigma Corporation (Missouri, USA). Bodipy 493/503 was purchased from Invitrogen (Carlsbad, CA). All the antibodies used in the experiment were from Proteintech (Chicago, USA). Adenovirus vectors were purchased from Hanheng Technology co., Ltd (Shanghai, China), and the hsa-miR-128 inhibitor/mimic and corresponding negative controls (NC) were purchased from Sangon Biotech Co., Ltd (Shanghai, China). All mRNA primers were purchased from Sangon Biotech Co., Ltd (Shanghai, China), and miR-128 and U6 primers were purchased from Applied Biological Materials Inc. (Vancouver, Canada).2.2 Animals and treatment
Male mice (C57BL/6J) aged 6–8 weeks were obtained from the Laboratory Animal Center of Jilin University (Jilin, China). All protocols of animal studies were approved by the Animal Care and Use Committee of Jilin University. All mice were randomly assigned to five groups (n = 5 per group), were housed under 12 h light/dark conditions and then were fed with a standard chow diet (SCD). The whole experiments were performed continually for 14 weeks. In the first 12 weeks, the mice were fed either with a SCD or HFD (D12492, xiaoshuyouli, Beijing, China). At week 13, the SCD-fed mice were further divided into 2 groups randomly, including the control group and the high Gs-Rb1 (40 mg kg−1) group; the HFD-fed mice were further divided into 3 groups randomly, including the HFD group, the HFD plus high Gs-Rb1 (40 mg kg−1) group and the HFD plus high Gs-Rb1 (40 mg kg−1) group plus CQ (50 mg kg−1) group. Gs-Rb1 was administered by gavage and CQ was injected for two weeks. Mice were weighed weekly until sacrifice. Liver tissues were obtained for subsequent experiments.2.3 Cell culture and treatment
HepG2 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and maintained in DMEM (HyClone, USA) containing 10% fetal bovine serum (Clark Bioscience, USA) at 37 °C in a culture chamber with 5% CO2. For passaging cultures, the cells were detached by digestion treatment with 0.25% trypsin (w/v) (Macgene, China). Rapamycin was pretreated for 3 h, and then the fresh medium was replaced for further treatment. CQ was added in the last 3 h before the end of the experiment.2.4 Histopathology of the liver
The liver tissue samples were fixed in 10% buffered formalin overnight, embedded in wax, and stained with hematoxylin and eosin stain (HE). The pictures of liver tissues were imaged using a microscope (Olympus IX71).2.5 Cell viability assays
HepG2 cells were incubated in 96-well plates with 8 × 103 cells per well. After 20 h, 20 μL of 5 mg mL−1 MTT was added to each well, and the cells were cultured for 4 h. The supernatant was discarded after centrifugation and 150 μL of DMSO was added to each well. Finally, the OD value (at 570 nm) was measured with a microplate reader (TECAN, Switzerland).2.6 Oil red O staining
HepG2 cells were incubated in 24-well plates with 1 × 105 cells per well and treated for 24 h according to the experimental scheme. First, the cells were washed three times with 1× PBS and then fixed in 4% paraformaldehyde at 37 °C for 40 min. Then, the cells were washed three times with 1× PBS again and incubated with Oil Red O solution (0.3% Oil Red O) for 1 h at 37 °C in the dark. The cryosections (5 μm) of the mice livers were directly incubated with Oil Red O solution for 1 h at 37 °C in the dark and then counterstained with hematoxylin. Cells and cryosections of the mice livers were then rinsed thoroughly with 1× PBS to remove free Oil Red O not bound to the lipid droplets. Finally, the stained lipid droplets were photographed with an inverted light microscope (Olympus, Tokyo, Japan).2.7 Measurement of the total cholesterol (TC) and triglyceride (TG) contents
HepG2 cells were incubated in 6-well plates with 5 × 105 cells per well and treated for 24 h according to the experimental scheme. The TG and TC contents in the liver of mice or HepG2 cells were measured using TC and TG assay kits (Applygen Technologies Inc., Beijing, China). The results were standardized by the protein content, measured in μM mg−1 protein.2.8 MDC staining
HepG2 cells were incubated in 24-well plates with 1 × 105 cells per well and treated for 24 h according to the experimental scheme. First, the cells were washed two times with 1× PBS and then stained with 300 μL of MDC solution (0.05 mmol L−1) for 45 min at 37 °C in the dark. Subsequently, the cells were washed twice again with 1× PBS and observed under a fluorescence microscope (BX53, Olympus, Japan).2.9 Transmission electron microscopy (TEM)
After 24 h of treatment according to the experimental scheme, HepG2 cells or liver were treated with 2.5% glutaraldehyde in sodium vanadate buffer (0.05 M) for more than 2 h, and then fixed with 1% phosphorus tetroxide and stained with uranyl acetate and lead citrate. Section samples were prepared and imaged under a TEM (H7650, Hitachi, Japan).2.10 Western blot analysis
HepG2 cells or liver were lysed and centrifuged to collect total cell protein, or cell nucleus and cytoplasmic protein were separated using a cytoplasmic protein and nuclear protein extraction kit (Beyotime, China), and the protein content in the supernatant was detected using a BCA protein assay kit (Beyotime, China). Equal amounts of protein (60–80 μg) were added to 8%, 10% and 12% gels for electrophoresis. Then, the protein on the gel was transferred to the polyvinylidene fluoride (PVDF) membrane using a Trans-Blot system (Bio-Rad, USA). The PVDF membrane was blocked and incubated with primary and secondary antibodies. Finally, an ECL chemiluminescence instrument (Azure C300, USA) was used for imaging.2.11 Transiently transfected with recombinant adenovirus
HepG2 cells were incubated on a special culture dish (with glass substrates). The cells in the dishes were transfected with adenovirus (MOI = 20) and replaced with fresh medium 6–8 h after transfection. The cells were treated for 24 h according to the experimental protocol. After treatment, images were obtained under a confocal laser scanning microscope.2.12 Double immunofluorescence
HepG2 cells were incubated on a special culture dish (with glass substrates) and treated for 24 h according to the experimental scheme. The liver tissues and the cells were fixed in 4% paraformaldehyde for 30 min, and then they were incubated with 0.3% Triton X-100 for 10 min and blocked with 5% goat serum to incubate the primary antibody (primary antibody against LC3 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) and secondary antibody (Alexa Fluor 594 goat anti-rabbit secondary antibody 1:500). The LDs and nucleus were then transfected with BODIPY 493/503 and DAPI, respectively. Finally, the liver tissues and the cells were imaged with a confocal laser scanning microscope (FV3S-SW, Olympus, Japan), in which the excitation wavelength of the red light channel was 594 nm, those of the green channel were 493 nm and 503 nm, and that of the blue channel was 440 nm.
:100) and secondary antibody (Alexa Fluor 594 goat anti-rabbit secondary antibody 1:500). The LDs and nucleus were then transfected with BODIPY 493/503 and DAPI, respectively. Finally, the liver tissues and the cells were imaged with a confocal laser scanning microscope (FV3S-SW, Olympus, Japan), in which the excitation wavelength of the red light channel was 594 nm, those of the green channel were 493 nm and 503 nm, and that of the blue channel was 440 nm.
2.13 Real-time PCR
According to the instructions provided by the manufacturer, the total RNA of HepG2 cells and the liver was extracted using an RNA extraction kit (Applied Biomaterials, Canada) and the first strand of cDNA was synthesized using a cDNA synthesis kit (Applied Biomaterials, Canada). A 20 μL qPCR reaction was established using a qPCR kit (Applied Biomaterials, Canada) and the parameters were set in a CFX96 real-time PCR system (Bio-Rad, USA) as follows: 95 °C per 10 min, followed by 40 cycles of 95 °C per 15 s and 60 °C per 1 min. GAPDH and U6 were used as housekeeping genes for the quantification of inter-group differences. The primer sequences are shown in Table 1.| Gana name | Primers | Annealing temperature (°C) |
|---|---|---|
| TFEB-F | AAGGTTCGGGAGTATCTGTCTG | 60 |
| TFEB-R | GGGTTGGAGCTGATATGTAGCA | 60 |
| ATP6V1A-F | GCATTGTAGGAGCAGTTTC | 60 |
| ATP6V1A-R | GAGCCAGTTGACAGAGGG | 60 |
| ATP6V1B2-F | GAAAGCCGTGGTGGGAGAG | 60 |
| ATPV1B2-R | CAGCCAATGTCCAAAGTC | 60 |
| CTSB-F | TGTGGTGGTCCTTGATCCTT | 60 |
| CTSB-R | AATCTGTCCAATGGTCGGGC | 60 |
| LAMP1-F | AGCATACCGGTGTGTCAGTG | 60 |
| LAMP1-R | GTTGGGGAAGGTCCATCCTG | 60 |
| GAPDH-F | AAAGGTGGAGGTCCATCCTG | 60 |
| GAPDH-R | GGGAAACTGTGGCGTGAT | 60 |
2.14 Statistical analysis
All data were analyzed by one-way analysis of variance (ANOVA) in SPSS 19.0 (SPSS Inc., Chicago, IL, USA). If the differences between groups were statistically significant, the Tukey test was used to compare the differences between groups in pairs. Dunnett's test was used to compare the differences between each group and the control group.3 Results and discussion
3.1 Effects of Gs-Rb1-induced autophagy, TFEB and miR-128 on HFD-induced lipid accumulation in mice
We explored the effects of a HFD on lipid accumulation in the liver of mice and found that as compared to controls, HFD exposure increased the relative liver weight and the TG and TC contents measured in the liver of mice (Fig. 1A). Consistently, HFD-fed increased the number of LDs in the liver of mice, assessed by Oil red O staining and HE staining (Fig. 1B). We found that compared with the control group, the liver of the HFD group had a light red appearance. Compared with the HFD group, HFD + Gs-Rb1 had a dark brown-red appearance, while the liver of the HFD + Gs-Rb1 + CQ group had a pale yellow color (Fig. 1B).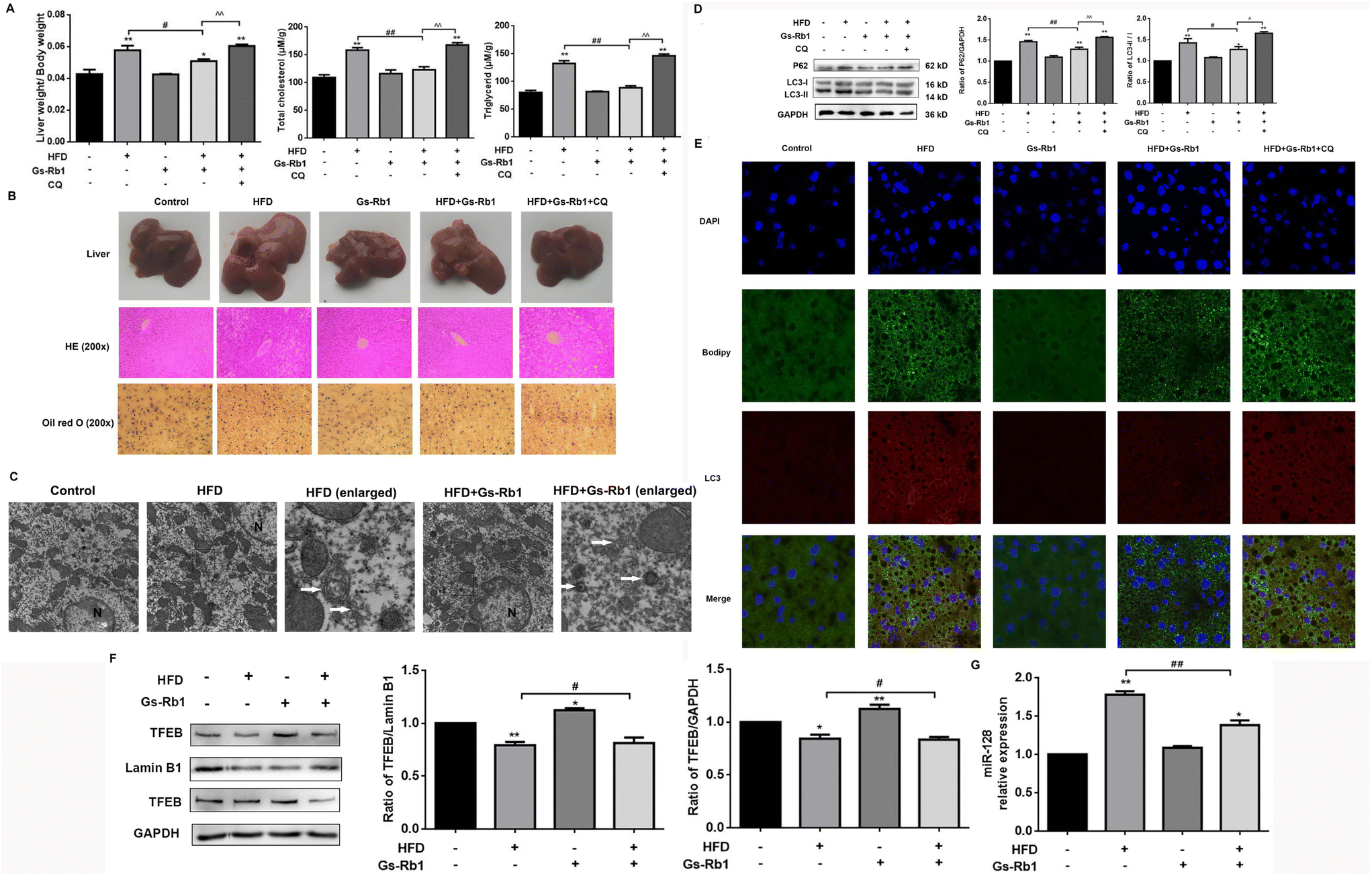 | ||
| Fig. 1 Effects of Gs-Rb1-induced autophagy, TFEB and miR-128 on HFD-induced lipid accumulation in mice. (A) Relative liver weight, TG and TC levels in the liver were detected. (B) Gross appearance of the liver was shown, and LDs of the liver were observed using HE staining (magnification ×200) and Oil Red O staining (red) (magnification ×200). (C) Autophagosome formation was detected in C57BL mice by TEM (original magnification, ×12000, enlarged magnification, ×50000). The representative autophagosomes were indicated by white arrows. N, nucleus. (D) The protein expressions of LC3 and P62 in HFD-fed mice treated Gs-Rb1 were detected by western blot (autophagy regulators CQ were added). Mean ± SD, n = 5. *P < 0.05 vs. control, **P < 0.01 vs. control, #P < 0.05 vs. HFD group, ##P < 0.01 vs. HFD group, ^^ P < 0.01 vs. HFD and Gs-Rb1 co-treatment group. (E) Representative images showed the colocalization of LC3 with LDs (magnification 1000×). (F) The protein expression of TFEB in the C57BL mice cell nucleus and cytoplasm was detected by western blot. (G) The effect of HFD administration and Gs-Rb1 treatment on the expression of miR-128 in C57BL mice was detected by qPCR. | ||
An autophagy inhibitor CQ was used to detect the changes in lipid accumulation in mice fed with a HFD to investigate whether autophagy plays a role in hepatic lipid accumulation. In contrast to the HFD, the HFD + Gs-Rb1 group showed decreased liver weight, TG and TC levels, and LDs, while CQ further increased the lipid contents in the liver (Fig. 1A and B). Furthermore, autophagosomes were observed by TEM, which is a standard method to measure the autophagic process. As shown in Fig. 1C, TEM images also showed more autophagosomes in the HFD and Gs-Rb1 group than in the control group. These results indicated that both a HFD and Gs-Rb1 could induce autophagosome accumulation. The western blot results showed that compared with the control group, a HFD alone significantly increased LC3-II and P62 protein expression (Fig. 1D). However, the above changes were significantly reduced in the liver of C57BL/6 mice given Gs-Rb1 by intragastric administration for 30 days. When CQ was added to the HFD and Gs-Rb1 co-treated group, the above changes were enhanced again. LD and LC3 colocalization was assessed by immunofluorescence to determine whether lipophagy was involved in lipid metabolism mediated by a HFD. As shown in Fig. 1E, compared with the control group, the green fluorescence and red fluorescence in the HFD group were simultaneously enhanced and well co-located, proving that the HFD induced the simultaneous increase of LDs and autophagosomes, and a large number of LDs were wrapped by autophagosomes. Conversely, the co-localization of LDs with LC3 in the Gs-Rb1-treated group was reduced. When CQ was added, the co-localization of LDs with LC3 was enhanced. This evidence suggested that the alleviating effect of Gs-Rb1 on HFD-induced lipid accumulation is related to autophagy.
TFEB is a major transcriptional regulator of autophagy.9–11 TFEB has been confirmed to be a target of miR-128 by the dual luciferase assay. It has been proven that Gs-Rb1 alleviated hepatotoxicity induced by foodborne 3-MCPD by stimulating autophagic flux via miR-128-targeted TFEB.24 Thus we detected the level of TFEB protein expression. The results showed that compared with the control group, a HFD alone could reduce the level of TFEB protein in the nucleus and cytoplasm. In comparison with the HFD group, HFD and Gs-Rb1 co-treatment significantly increased TFEB nucleus translocation (Fig. 1F). To investigate whether miR-128 is involved in Gs-Rb1 promoting lipid degradation through TFEB, we measured the level of miR-128 in C57BL mice (Fig. 1G). The results showed that Gs-Rb1 could significantly inhibit the increase of miR-128 induced by a HFD.
3.2 Effects of Gs-Rb1 on PA-induced lipid accumulation in HepG2 cells
We detected the effects of different doses of PA and Gs-Rb1 on the viability of HepG2 cells by MTT colorimetry. As shown in Fig. 2A, the maximum non-toxic doses of PA and Gs-Rb1 were 250 μM and 100 μM, respectively. To determine whether Gs-Rb1 has an alleviating effect on PA-induced lipid accumulation in HepG2 cells, we first stained the LDs with Oil Red O and detected the contents of TC and TG (Fig. 2B and C). The results showed that 250 μM PA treatment could lead to significant accumulation of LDs in HepG2 cells. Gs-Rb1 at 25 μM, 50 μM and 100 μM could alleviate the abnormal accumulation of LDs. Similar to the results of Oil Red O staining, 250 μM PA could significantly increase TC and TG contents in HepG2 cells, and different doses of Gs-Rb1 could alleviate PA-induced lipid accumulation, of which 50 μM Gs-Rb1 had the most significant effect. Therefore, a 250 μM PA-induced lipid accumulation model was used in the subsequent experiments, and a 50 μM Gs-Rb1 was used as the restorer.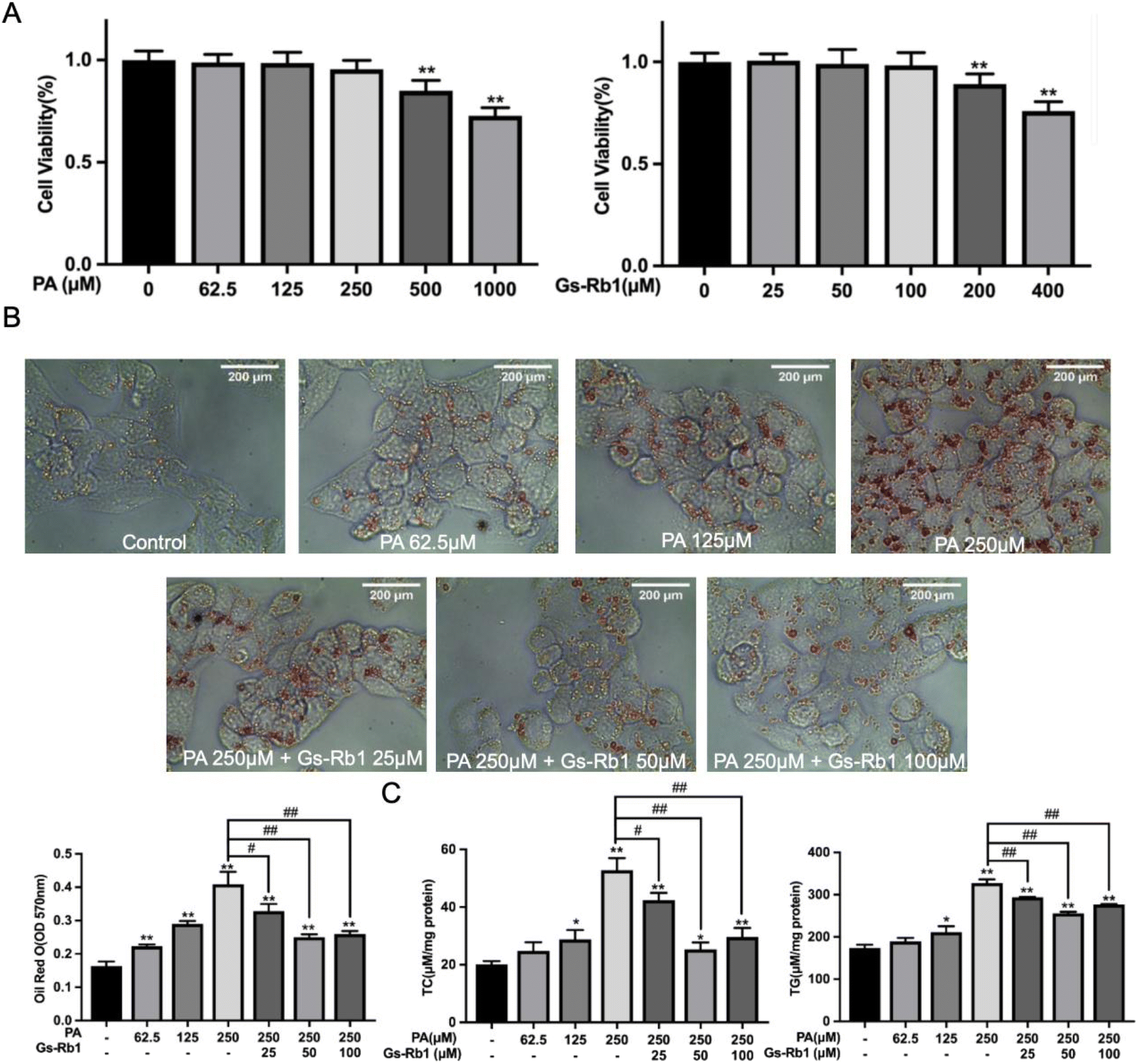 | ||
| Fig. 2 Effects of Gs-Rb1 on PA-induced lipid accumulation. (A) HepG2 cells were treated with different concentrations of PA (62.5, 125, 250, 500 and 1000 μM) or Gs-Rb1 (25, 50, 100, 200 and 400 μM) for 24 h, and the viability of HepG2 cells was detected by MTT colorimetry. (B) HepG2 cells were treated with non-toxic doses of PA and Gs-Rb1 for 24 h and stained with Oil Red O and (C) the contents of TC and TG were detected. Mean ± SD, n = 6. *P < 0.05 vs. control, **P < 0.01 vs. control, #P < 0.05 vs. 250 μM PA group, ##P < 0.01 vs. 250 μM PA group. | ||
3.3 Effects of Gs-Rb1 on the autophagic flux in PA-treated HepG2 cells
To further investigate whether the alleviating effect of Gs-Rb1 on lipid accumulation was associated with autophagy, we observed the number of autophagosomes and LDs in HepG2 cells by MDC staining and TEM. As shown in Fig. 3A, the fluorescence intensity and the number of autophagosomes in the PA group were stronger than those in the control group, and those in the PA and Gs-Rb1 co-treatment group were close to those in the PA group. Furthermore, we found that many LDs accumulated in PA group, and a certain number of autophagosomes were observed in the cytoplasm (Fig. 3B). However, the LDs in the cells decreased significantly in the PA and Gs-Rb1 co-treatment group. There are generally two reasons for the increase in the number of autophagosomes, one is that its production is promoted, and the other is that its degradation is inhibited. However, the PA-induced increase in autophagosomes was accompanied by the accumulation of LDs. Thus we have reason to suspect that this is due to the blocking of autophagic flux, which leads to the inhibition of the degradation of the autophagosome containing LDs. Furthermore, we suspect that Gs-Rb1 alleviates PA-induced lipid accumulation, probably because it stimulates autophagic flux. Therefore, we need further experiments to verify the conjecture.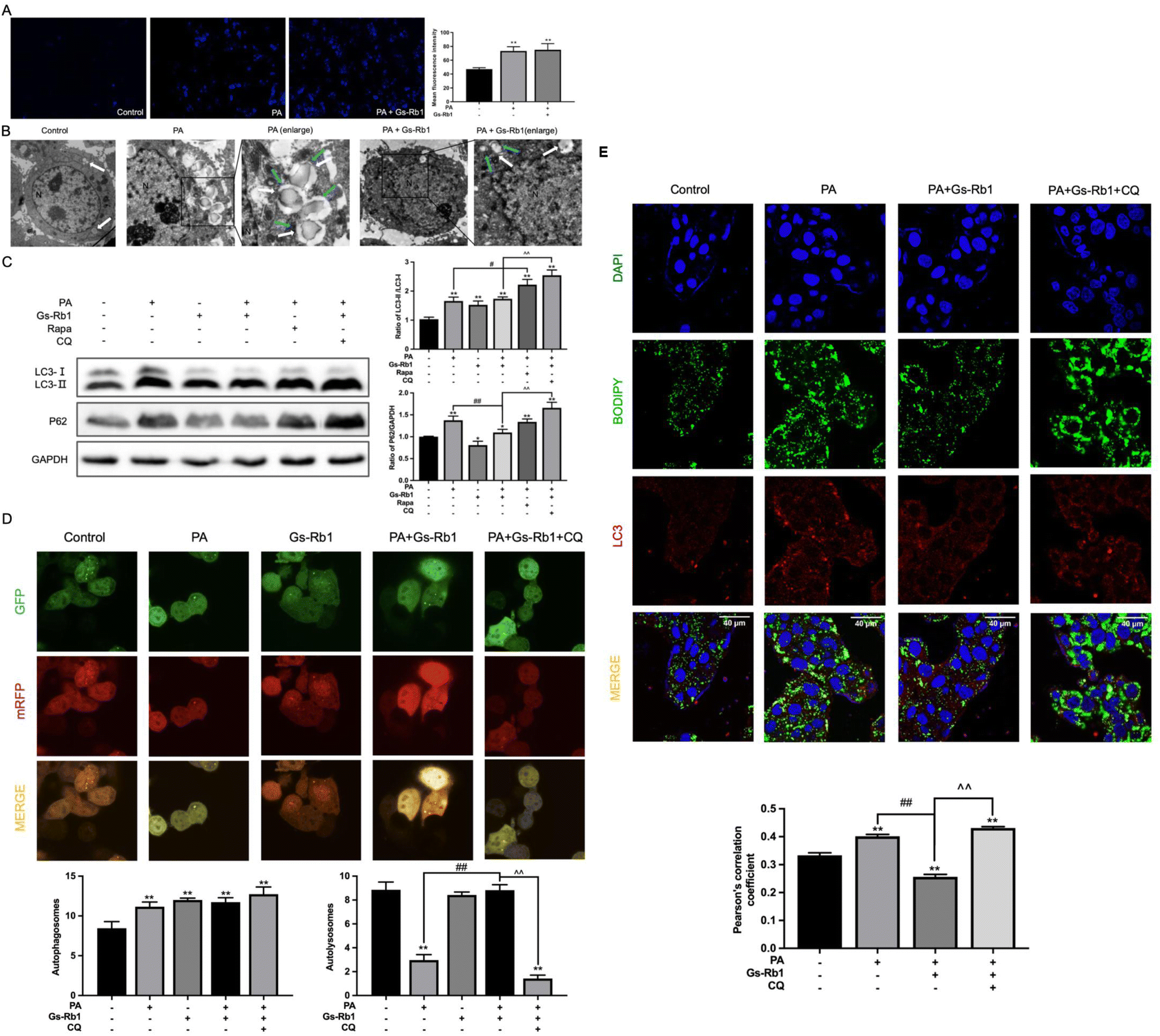 | ||
| Fig. 3 Effects of Gs-Rb1 on the autophagic flux in PA-treated HepG2 cells. HepG2 cells were treated with PA (250 μM) and Gs-Rb1 (50 μM) for 24 h, stained with MDC (magnification 200×) (A) or observed under TEM (magnification 1000× and 3000×) (B) the autophagosomes and LDs were marked with blue and white arrows, respectively. (C) The protein expressions of LC3 and P62 in HepG2 cells treated with PA and Gs-Rb1 were detected by western blot. (D) Autophagic flux was detected by infection with mRFP-GFP-LC3 autophagy double-labelled adenovirus. (E) Gs-Rb1 on LC3-BODIPY co-localization in PA-treated HepG2 cells. At least 50 cells were analyzed by Pearson's correlation coefficient. Mean ± SD, n = 3. *P < 0.05 vs. control, **P < 0.01 vs. control, #P < 0.05 vs. PA group, ##P < 0.01 vs. PA group, ^^ P < 0.01 vs. PA and Gs-Rb1 co-treatment group. | ||
To further judge whether Gs-Rb1 alleviates PA-induced lipid accumulation by stimulating autophagic flux, we first detected autophagic flux. LC3 protein is a marker protein for autophagy, and the expression of LC3-II can directly reflect the number of autophagosomes. We combined it with the autophagic degradation substrate P62 to analyze the dynamic process of autophagic flux. Western blot results showed the same results as in vivo results (Fig. 3C). This proves that Gs-Rb1 could alleviate PA-induced autophagic flux blockade. In addition to promoting autophagosome degradation, Gs-Rb1 also promoted the production of autophagosomes, which is also why the LC3-II protein did not decrease simultaneously with the P62 protein. Subsequently, we added two well-established tools for autophagy regulation, Rapa and CQ. The results showed that PA-induced autophagic flux blockade was not alleviated by Rapa and the activation effect of Gs-Rb1 on autophagic flux was inhibited by CQ.
Subsequently, the western blot results were confirmed by infecting mRFP-GFP-LC3 autophagy double-labeled adenoviruses. As shown in Fig. 3D, compared to the control group, the yellow puncta in the PA group increased significantly, while almost no red puncta were observed, indicating that PA inhibits autophagosome degradation and preserves green fluorescence. Compared with the PA group, there were obvious red puncta in the PA and Gs-Rb1 co-treatment group, indicating that Gs-Rb1 could alleviate the blocking of autophagic flux caused by PA. After CQ was added, the red puncta disappeared again and Gs-Rb1's promoter effect on autophagic flux was reversed. The above results showed that Gs-Rb1 could alleviate PA-induced autophagic flux blockade by promoting autophagosome degradation. Furthermore, the results of colocalization of LDs and LC3 were the same as the in vivo results (Fig. 3E).
This evidence suggested that the alleviating effect of Gs-Rb1 on PA-induced lipid accumulation is related to autophagy, and Gs-Rb1 enhances LD degradation by stimulating autophagic flux.
3.4 Effects of Gs-Rb1 on TFEB and miR-128 in PA-treated HepG2 cells
We have found that the key for Gs-Rb1 to alleviate PA-induced lipid accumulation is to promote the degradation of autophagosomes. As an important site of autophagosome degradation, TFEB is a major transcriptional regulator of lysosomal biosynthesis and has been demonstrated to be involved in regulating at least 73 genes associated with the autophagy–lysosomal pathway.25 According to previous studies, TFEB silencing mediated by miR-128 is associated with the downregulation of 18 genes associated with the lysosomal function.26,27The results showed that compared with the control group, PA treatment alone could reduce the level of TFEB protein in the nucleus and cytoplasm. In comparison with the PA group, PA and Gs-Rb1 co-treatment significantly increased TFEB nucleus translocation (Fig. 4A). Then we detected the expression of TFEB and its downstream target genes ATP6V1A, ATP6V1B2, CTSB and LAMP1 by PCR. They were involved in coding the vacuolar proton pump (V-ATPase), lysosomal hydrolase and lysosomal membrane, respectively (Fig. 4B). The results indicated the alleviating effect of Gs-Rb1 on lipid accumulation is related to the promotion of lysosomal function mediated by TFEB.
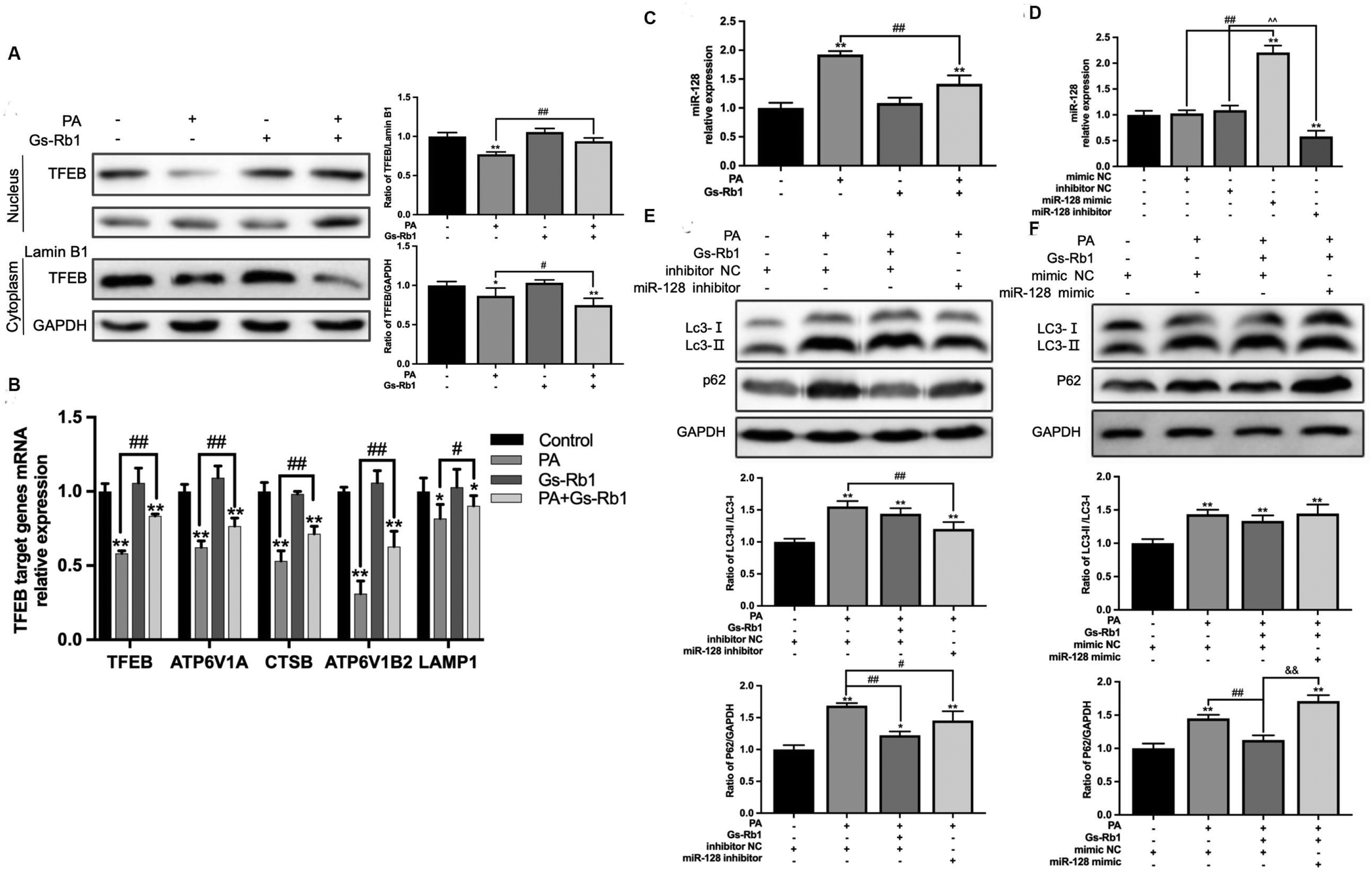 | ||
| Fig. 4 Effects of Gs-Rb1 on TFEB and miR-128 on autophagic flux in PA-treated HepG2 cells. (A) The protein expression of TFEB in the HepG2 cell nucleus and cytoplasm was detected by western blot. (B) The mRNA levels of TFEB and its downstream lysosomal related target genes were detected by qPCR. (C) The effect of PA and Gs-Rb1 treatment on the expression of miR-128 in HepG2 cells was detected by qPCR. (D) The expression of miR-128 after transfection with the hsa-miR-128 inhibitor/mimic was detected by qPCR to verify the transfection effect. (E and F) The protein expressions of LC3 and P62 in hsa-miR-128 inhibitor/mimic-transfected HepG2 cells treated with PA and Gs-Rb1 were detected by western blot. Mean ± SD, n = 3. *P < 0.05 vs. control, **P < 0.01 vs. control, #P < 0.05 vs. PA group, ##P < 0.01 vs. PA group, ^^P < 0.01 vs. inhibitor NC group, && P < 0.01 vs. mimic NC + PA + Gs-Rb1. | ||
To investigate whether miR-128 is involved in Gs-Rb1 promoting lipid degradation through TFEB, we measured the level of miR-128 in HepG2 cells (Fig. 4C and D). The results showed that Gs-Rb1 could significantly inhibit the increase of miR-128 induced by PA. Then we transfected the hsa-miR-128 inhibitor/mimic, and detected the autophagic flux and lipid content. The above results further verified the target role of miR-128 in Gs-Rb1 promoting lipid degradation by stimulating autophagy. We first detected the contents of LC3 and P62 proteins in HepG2 cells and found that the hsa-miR-128 inhibitor could significantly inhibit the increase of LC3-II and P62 caused by PA and alleviate the block of autophagic flux. The effect was similar to that of Gs-Rb1 (Fig. 4E). The Hsa-miR-128 mimic could counteract the alleviating effect of Gs-Rb1 on autophagic flux blockade induced by PA (Fig. 4F). These results indicated that miR-128 is involved in the remission of PA-induced autophagic flux blockade by Gs-Rb1.
3.5 Effects of miR-128 on lipid accumulation in PA-treated HepG2 cells
To determine the effect of miR-128 on lipid accumulation in HepG2 cells, hsa-miR-128 inhibitor/mimic-transfected HepG2 cells were stained with Oil Red O, and the contents of TC and TG in the cells were detected. By Oil Red O staining, we could observe that after transfection with the hsa-miR-128 inhibitor, the lipid accumulation caused by PA decreased significantly, while the hsa-miR-128 mimic inhibited the degradation of lipids by Gs-Rb1, and LDS accumulated again (Fig. 5A). The detection of TC and TG also confirmed this result (Fig. 5B).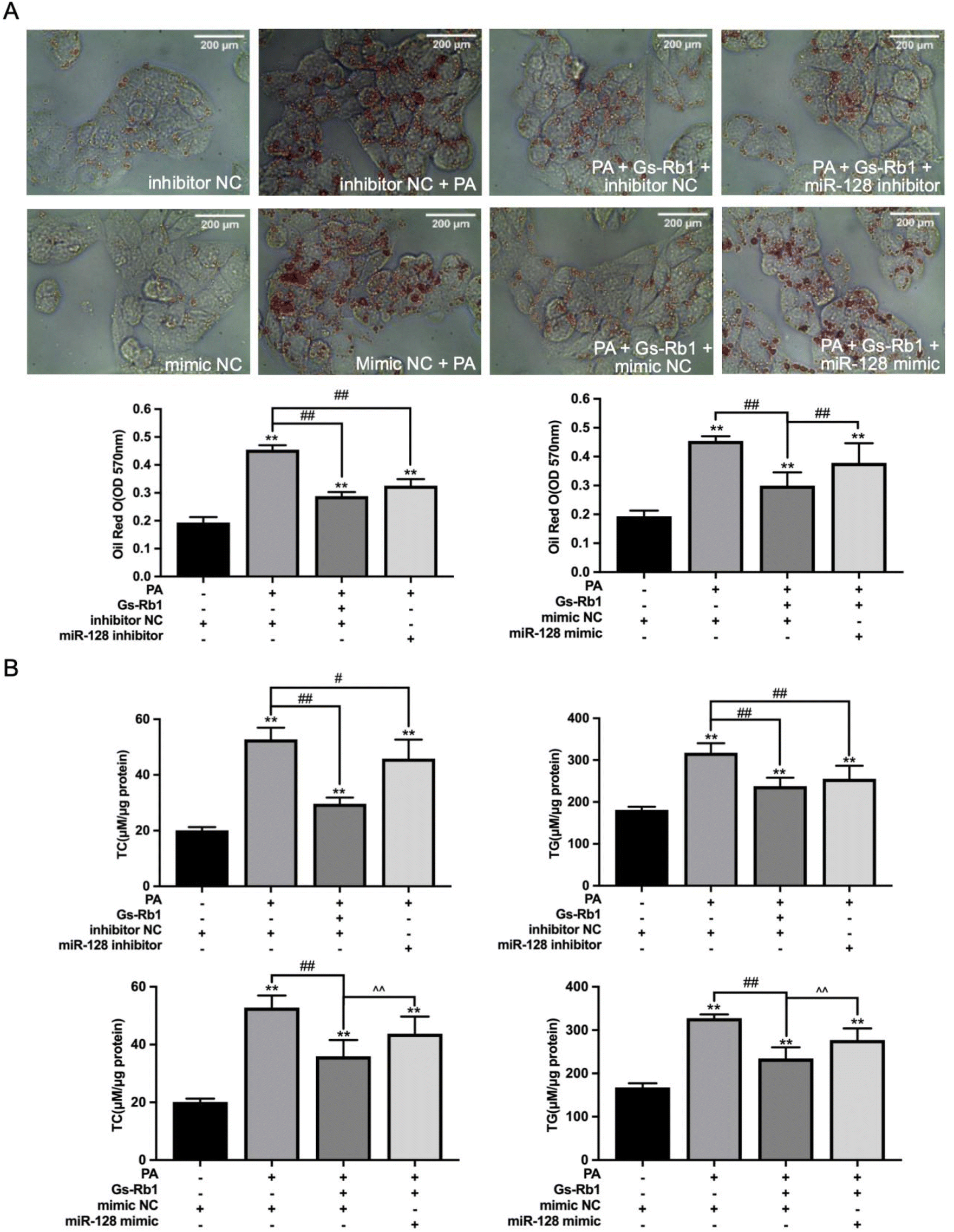 | ||
| Fig. 5 Effects of miR-128 on lipid accumulation in PA-treated HepG2 cells. (A) Hsa-miR-128 inhibitor/mimic-transfected HepG2 cells were treated with PA and Gs-Rb1 and stained with Oil Red O (A) and the contents of TC and TG were detected (B). Mean ± SD, n = 6. **P < 0.01 vs. control, #P < 0.05 vs. PA group, ##P < 0.01 vs. PA group. ^^ P < 0.01 vs. mimic NC + PA + Gs-Rb1 group. | ||
4 Conclusion
In this study, we explored that Gs-Rb1 could alleviate HFD and PA-induced lipid accumulation by stimulating autophagic flux. The specific mechanism might be that Gs-Rb1 enhances the transcription of TFEB and its downstream lysosome-related genes by inhibiting miR-128, improves the degradation ability of lysosomes to autophagosomes, and then promotes autophagic lipid degradation.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This study was financed by the National Natural Science Foundation of China (No. 32072315) and the Scientific Research Project of the Jilin Provincial Department of Education (No. JJKH20221032KJ).References
- M. Shibata, K. Yoshimura, N. Furuya, M. Koike, T. Ueno, M. Komatsu, H. Arai, K. Tanaka, E. Kominami and Y. Uchiyama, The MAP1-LC3 conjugation system is involved in lipid droplet formation, Biochem. Biophys. Res. Commun., 2009, 382, 419–423 CrossRef CAS PubMed.
- R. Singh, S. Kaushik, Y. Wang, Y. Xiang, M. Komatsu, K. Tanaka, A. M. Cuervo and M. J. Czaja, Autophagy regulates lipid metabolism, Nature, 2010, 458, 1131–1135 CrossRef PubMed.
- M. Ouimet, V. Franklin, E. Mak, X. Liao, I. Tabas and Y. L. Marcel, Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase, Cell Metab., 2011, 13, 655–667 CrossRef CAS PubMed.
- M. Martinez-Vicente, Z. Talloczy, E. Wong, G. Tang, H. Koga, S. Kaushik, R. De Vries, E. Arias, S. Harris, D. Sulzer and A. M. Cuervo, Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease, Nat. Neurosci., 2010, 13, 567–576 CrossRef CAS PubMed.
- L. Shi, K. Wang, Y. Deng, Y. Wang, S. Zhu, X. Yang and W. Liao, Role of lipophagy in the regulation of lipid metabolism and the molecular mechanism, Nanfang Yike Daxue Xuebao, 2019, 39, 867–874 CAS.
- Y. Zhang, J. Sun, X. Yu, L. Shi, W. Du, L. Hu, C. Liu and Y. Cao, SIRT1 regulates accumulation of oxidized LDL in HUVEC via the autophagy-lysosomal pathway, Prostaglandins Other Lipid Mediators, 2016, 122, 37–44 CrossRef CAS PubMed.
- N. Martinez-Lopez and R. Singh, Autophagy and Lipid Droplets in the Liver, Annu. Rev. Nutr., 2015, 35, 215–237 CrossRef CAS PubMed.
- Y. Huang, F. Liu, F. Zhang, P. Liu, T. Xu and W. Ding, Vanadium(IV)-chlorodipicolinate alleviates hepatic lipid accumulation by inducing autophagy via the LKB1/AMPK signaling pathway in vitro and in vivo, J. Inorg. Biochem., 2018, 183, 66–76 CrossRef CAS PubMed.
- C. Settembre, C. Di Malta, V. A. Polito, M. Garcia, F. Vetrini, S. U. S. Erdin, S. U. S. Erdin, T. Huynh, D. Medina, P. Colella, M. Sardiello, D. C. Rubinsztein and A. Ballabio, TFEB Links Autophagy to Lysosomal Biogenesis, Science, 2011, 332, 1429–1433 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina, A. Ballabio and T. Children, Signals for the lysosome: a control center for cellular clearance and energy metabolism, Nat. Rev. Mol. Cell Biol., 2015, 14, 283–296 CrossRef PubMed.
- C. S. Peskin, D. Tranchina, D. Y. Vargas, S. Tyagi, A. Van Oudenaarden, E. K. O. Shea, J. W. Hong, J. Zeitlinger, D. S. Rokhsar and A. N. Boettiger, A Gene Network Regulating Lysosomal Biogenesis and Function, Science, 2009, 473–477 Search PubMed.
- G. Stefani and F. J. Slack, Small non-coding RNAs in animal development, Nat. Rev. Mol. Cell Biol., 2008, 9, 219–230 CrossRef CAS PubMed.
- V. A. Gennarino, M. Sardiello, M. Mutarelli, G. Dharmalingam, V. Maselli, G. Lago and S. Banfi, HOCTAR database: A unique resource for microRNA target prediction, Gene, 2011, 480, 51–58 CrossRef CAS PubMed.
- M. Decressac, B. Mattsson, P. Weikop, M. Lundblad, J. Jakobsson and A. Björklund, TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1817–E1826 CrossRef CAS PubMed.
- Y. Wang, S. Gunewardena, F. Li, D. J. Matye, C. Chen, X. Chao, T. Jung, Y. Zhang, M. Czerwiński, H. M. Ni, W. X. Ding and T. Li, An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis, Nat. Commun., 2020, 11, 3612 CrossRef CAS PubMed.
- X. Chen, H. Chan, L. Zhang, X. Liu, I. H. T. Ho, X. Zhang, J. Ho, W. Hu, Y. Tian, S. Kou, C. S. Chan, J. Yu, S. H. Wong, T. Gin, M. T. V. Chan, X. Sun and W. K. K. Wu, The phytochemical polydatin ameliorates non–alcoholic steatohepatitis by restoring lysosomal function and autophagic flux, J. Cell. Mol. Med., 2019, 23, 4290–4300 CrossRef CAS PubMed.
- Y. M. Cai, Effect of ginsenoside Rg1, Rh2 on the expression of IL-1β, iNOS and TNF-α in activated microglia by Aβ, Chengdu University of Chinese Medicine, 2008, (In Chinese) Search PubMed.
- T. H. Lan, D. P. Xu, M. T. Huang, J. X. Song, H. L. Wu and M. Li, Ginsenoside Rb1 prevents homocysteine-induced EPC dysfunction via VEGF/p38MAPK and SDF-1/CXCR4 activation, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- D. H. Liu, Y. M. Chen, Y. Liu, B. S. Hao, B. Zhou, L. Wu, M. Wang, L. Chen, W. K. Wu and X. X. Qian, Ginsenoside Rb1 reverses H 2O 2-induced senescence in human umbilical endothelial cells: Involvement of eNOS pathway, J. Cardiovasc. Pharmacol., 2012, 59, 222–230 CrossRef CAS PubMed.
- J.-M. Lu, S. M. Weakley, Z. Yang, M. Hu, Q. Yao and C. Chen, Ginsenoside Rb1 Directly Scavenges Hydroxyl Radical and Hypochlorous Acid, Curr. Pharm. Des., 2012, 18, 6339–6347 CrossRef CAS PubMed.
- X. Yu, L. Ye, H. Zhang, J. Zhao, G. Wang, C. Guo and W. Shang, Ginsenoside rb1 ameliorates liver fat accumulation by upregulating perilipin expression in adipose tissue of db/db obese mice, J. Ginseng Res., 2015, 39, 199–205 CrossRef PubMed.
- N. Lin, D. L. Cai, D. Jin, Y. Chen and J. J. Shi, Ginseng Panaxoside Rb1 Reduces Body Weight in Diet-Induced Obese Mice, Cell Biochem. Biophys., 2014, 68, 189–194 CrossRef CAS PubMed.
- R. Guo, L. Wang, X. Zeng, M. Liu, P. Zhou, H. Lu, H. Lin and M. Dong, Aquaporin 7involved in GINSENOSIDE-RB1-mediated anti-obesity via peroxisome proliferator-activated receptor gamma pathway, Nutr. Metab., 2020, 17, 1–11 CrossRef PubMed.
- J. Lu, J. Lu, X. Bu, Y. Li, G. Ge and S. Guan, Ginsenoside Rb1 alleviates liver injury induced by 3-chloro-1,2-propanediol by stimulating autophagic flux via miR-128-targeted TFEB, J. Food Sci., 2021, 86, 1–13 CrossRef.
- X. Chao, S. Wang, K. Zhao, Y. Li, J. A. Williams, T. Li, H. Chavan, P. Krishnamurthy, X. C. He, L. Li, A. Ballabio, H. M. Ni and W. X. Ding, Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice, Gastroenterology, 2018, 155, 865–879 CrossRef CAS PubMed.
- M. Sardiello, M. Palmieri, A. di Ronza, D. L. Medina and M. Valenza, A Gene Network Regulating Lysosomal Biogenesis and Function, Science, 2009, 473–478 CrossRef CAS PubMed.
- S. Bala and G. Szabo, TFEB, a master regulator of lysosome biogenesis and autophagy, is a new player in alcoholic liver disease, Dig. Med. Res., 2018, 1, 16–16 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |