Decomposition and mineralization of glyphosate herbicide in water by radical and non-radical pathways through peroxymonosulfate activation using Co3O4/g-C3N4: a comprehensive study†
Received
6th September 2022
, Accepted 17th November 2022
First published on 18th November 2022
Abstract
In this study, Co3O4 nanoparticles were deposited on g-C3N4 nanoplates to produce a Co3O4/g-C3N4 composite with high activity for peroxymonosulfate (PMS) activation. The materials were characterized by SEM, BET, FTIR, XRD, Raman, TGA, and EDX to explore their properties. In herbicide removal tests, the Co3O4/g-C3N4 ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 gave the strongest PMS activation effectiveness. Glyphosate (Gly) was decomposed rapidly in the first 30 s of reaction with a decomposition efficiency of 87.18% and a mineralization efficiency of 73% under optimum conditions (e.g., 50 mg L−1 catalyst, 200 mg L−1 PMS, 50 mg L−1 Gly, pH 11, and temperature of 25 °C). The Gly decomposition rate by the Co3O4/g-C3N4(10%)/PMS system was 3, 1.6, and 1.45 times higher than those of g-C3N4/PMS, Co3O4/PMS, and ZIF-67/g-C3N4/PMS systems, respectively. The effects of catalyst dosage, PMS content, oxidants, initial glyphosate concentration, and the existence of anions on glyphosate decomposition were also investigated. The presence of g-C3N4 not only enhances the glyphosate decomposition but also reduces the dissolution of Co in water by around 3.2 times as compared to pure Co3O4. In the presence of anions, the Gly decomposition decreased in the order SO42− < NO3− < Cl− < HCO3−. The radical scavenging test showed that 1O2 and O2˙− are the key reactive oxygen species, and the glyphosate decomposition mechanism was proposed for Co3O4/g-C3N4via heterogeneous catalytic activation of PMS. The mass spectrometry results indicate that the decomposition pathway of glyphosate occurs by cleaving the C–N bond and forming major intermediates such as methyl phosphonic acid, glycine, and inorganic ions (e.g., phosphate, nitrate, and ammonium). In durability tests, the glyphosate treatment efficiency decreased by only around 11% after five consecutive test cycles, implying its great potential application for the treatment of hazardous pollutants in wastewater.
:1 gave the strongest PMS activation effectiveness. Glyphosate (Gly) was decomposed rapidly in the first 30 s of reaction with a decomposition efficiency of 87.18% and a mineralization efficiency of 73% under optimum conditions (e.g., 50 mg L−1 catalyst, 200 mg L−1 PMS, 50 mg L−1 Gly, pH 11, and temperature of 25 °C). The Gly decomposition rate by the Co3O4/g-C3N4(10%)/PMS system was 3, 1.6, and 1.45 times higher than those of g-C3N4/PMS, Co3O4/PMS, and ZIF-67/g-C3N4/PMS systems, respectively. The effects of catalyst dosage, PMS content, oxidants, initial glyphosate concentration, and the existence of anions on glyphosate decomposition were also investigated. The presence of g-C3N4 not only enhances the glyphosate decomposition but also reduces the dissolution of Co in water by around 3.2 times as compared to pure Co3O4. In the presence of anions, the Gly decomposition decreased in the order SO42− < NO3− < Cl− < HCO3−. The radical scavenging test showed that 1O2 and O2˙− are the key reactive oxygen species, and the glyphosate decomposition mechanism was proposed for Co3O4/g-C3N4via heterogeneous catalytic activation of PMS. The mass spectrometry results indicate that the decomposition pathway of glyphosate occurs by cleaving the C–N bond and forming major intermediates such as methyl phosphonic acid, glycine, and inorganic ions (e.g., phosphate, nitrate, and ammonium). In durability tests, the glyphosate treatment efficiency decreased by only around 11% after five consecutive test cycles, implying its great potential application for the treatment of hazardous pollutants in wastewater.
Water impact
Glyphosate is one of the most popular herbicides worldwide, and its excessive use causes many environmental and ecological issues. Among the treatment methods, advanced oxidation processes (e.g., peroxymonosulfate activation by nanomaterials) are one of the most promising technologies. Thus, a comprehensive and insightful study could provide a fundamental and practical basis for the application of this technology.
|
1. Introduction
In developing countries, specifically Vietnam, agricultural activities play a significant role in economic development, and using agro-chemicals is an almost mandatory solution in crop cultivation. Glyphosate (denoted as Gly) is one of the most popular herbicides worldwide, which was once considered non-toxic. However, its excessive use in agricultural lands has polluted the soil and water environment. Today, its residues are detected in soil, water, flora, food, and even in human urine.1 According to the US EPA, the maximum allowable limit of Gly in water is 700 μg L−1. However, Gly concentrations range from 50 to 76 mg L−1 as a result of improper rinsing of the herbicide container.2 This causes severe effects on many organisms, causing genotoxicity, cytotoxicity, nuclear aberrations, hormonal disruption, chromosomal aberrations, and DNA damage. Its toxicity occurs in unicellular and multicellular organisms and has also been observed in many vertebrates, including humans.3
In 2005, the Food and Agriculture Organization (FAO) reported that Gly and its major metabolite, aminomethylphosphonic acid (AMPA), are potentially toxic, mainly due to the residue accumulation in the food chain.4 The WHO IARC has classified Gly as “possibly carcinogenic to humans” (category 2A). Its residues in grapes, soil, and groundwater at a grape farm in Binh Thuan Province (Vietnam) have exceeded 5–200 times the European allowable limit, especially in groundwater.5 Therefore, several methods for controlling Gly residues in an aqueous medium have been reported. The methods for removing Gly from pollution sources are mainly adsorption6,7 and oxidation processes, such as wet air oxidation conditions,8 catalytic wet air oxidation under atmospheric conditions,2,9 catalytic activation,10,11 and photocatalysis.12 Among them, advanced oxidation processes (AOPs) are more promising than the other methods since AOPs can mineralize toxic and hard-biodegradable pollutants or partially decompose them into simple and less toxic compounds. Besides H2O2, AOPs are capable of generating more reactive oxygen species (ROS) with stronger oxidizing properties when using composites from transition metal oxides such as Co, Ni, and Mn as catalysts to activate some strong oxidizing agents such as hydrogen peroxide (H2O2), peroxymonosulfate (PMS), and persulfate (PS). In particular, PMS (with the trade name of Oxone) is a strong, durable, non-toxic, and inexpensive oxidant, which is often used as an oxidizing agent. Furthermore, PMS has an asymmetric structure and a redox potential of 1.82 V and performs well over a wide pH range from 4 to 12.13 PMS can be activated by different agents such as ultrasound, ultraviolet irradiation, alkalis, carbon materials, and heterogeneous catalysts.14,15 AOPs based on sulfate radicals (SO4˙−) formed from PMS activation by heterogeneous catalysis are increasingly used because SO4˙− has a higher redox potential (Eo = 2.5–3.1 V) than HO˙ (Eo = 1.8–2.7 V), better stability, and longer half-life (30–40 s as compared to less than 1 s for HO˙) and exhibits great oxidizing capacity over a wide pH range from 3 to 9.16,17 Therefore, it has been widely studied and applied in sulfate-based AOPs, which could be suitable for removing Gly in water.
Zeolitic imidazolate frameworks (e.g. ZIF-67), a type of metal–organic framework (MOF), is a zeolite structure formed from the tetrahedral metal ion Co and the imidazolate ligand. Compared with conventional heterogeneous catalysts, ZIF-67 has high thermal stability, large specific surface area, tunable porosity, various nanoscale pore sizes, and good metal dispersion, which can enhance the mass transport of pollutants and provide abundant active sites for catalytic reactions. However, when using ZIF-67 directly as a catalyst, the imidazole ligands in the ZIF-67 structure are easily oxidized in some AOPs such as Fenton and Fenton-like processes. Under such strong oxidation conditions, the structure of ZIF-67 can be destroyed, thus enhancing the metal dissolution ability in the aquatic environment and causing secondary pollution. Among the metals, Co3O4 derived from ZIF-67 has many advantages such as a simple synthesis process by heat treatment; thus, the synthesis process can be well-controlled. In addition, synthesizing Co3O4 from ZIF-67 can retain the key characteristics of ZIF-67 such as maintaining the structural polyhedra of ZIF-67, which makes better Co3O4 dispersion and enhances the Co3+/Co2+ ratio on the surface with diverse active sites (Co–O and Co–OH) for catalytic reactions.18,19 Co3O4 derived from ZIF-67 has been used in environmental treatments such as toluene oxidation,19 soot oxidation,20,21 bromate reduction,22 photocatalysis,23,24 and catalytic activation.25,26 However, Co3O4 is unstable and easily dissolves in solution; thus, the use of single Co3O4 in environmental catalysis is limited by its secondary pollution. Therefore, to improve its catalytic activity and stability, Co3O4 was synthesized on different substrates for removing organics via PMS activation, such as Co3O4/graphene for orange II,27 Co3O4/g-C3N4 for diclofenac sodium,28 Co3O4/C for bisphenol A,29 Co3O4/δ-FeOOH for lomefloxacin,30 and CuO–Co3O4–CeO2 for 2,4-D.31
On the other hand, graphite carbon nitride (g-C3N4) consists of layers stacked along the axis to graphite layers composed of hexagonal rings of triazine (C3N3) and tri-s-triazine (heptazine, C6N7) structural units. It is a potential non-metallic photocatalyst due to its low bandgap energy for visible light use (2.63–2.72 eV), easy synthesis, thermal stability (up to 600 °C), low production cost, and environmental friendliness. Thus it was used as an effective support for Co3O4 in several environmental applications. This is due to the six nitrogen lone pairs in g-C3N4 acting as electron donors, thereby providing sites for transition metal ions, which reduces the leaching of metal ions into the water environment. In addition, the abundant pyrrolic N atoms present in g-C3N4 offer adsorption sites for organic pollution on the surface of the material.32,33 Co3O4/g-C3N4 was used for photocatalytic oxidation of methyl orange,34 tetracycline hydrochloride,35 rhodamine B,36 and NO in air,37 gas-phase Hg0 removal,38 and catalytic oxidation of CO.39,40 In particular, the PS/PMS activation by Co3O4/g-C3N4 materials was applied for the removal of diclofenac sodium,28 tetracycline,41 and acid orange 7,42 or in combination with photocatalysis for treatment of tetracycline,43,44 iohexol,45 and atrazine,46 and degradation of pesticides by other heterogeneous catalysts such as diazinon (ZnO@SiO2@Fe3O4/PMS/UV),47 malathion (g-C3N4/PMS/vis),48 and imidacloprid (OCN/PMS/vis).49 However, the above publications face the disadvantages of the low surface area of Co3O4/g-C3N4 materials, the requirement of energy supply (UV, visible light) for PMS activation, and a large amount of catalyst used, thus resulting in low efficiency of organic pollutant decomposition and long pollutant removal time. Meanwhile, the role of g-C3N4 and the synergistic effect of materials and PMS have not been elucidated. Furthermore, there were no comparisons for the activation of oxidants such as peroxymonosulfate, persulfate, and hydrogen peroxide using Co3O4/g-C3N4 as a heterogeneous catalyst.
In this work, Co3O4/g-C3N4 composites were synthesized from ZIF-67/g-C3N4via one-step pyrolysis. The detailed formation mechanisms of Co3O4/g-C3N4 derived from ZIF-67/g-C3N4 were proposed. The Co3O4/g-C3N4 composites were then used for activating PMS to treat Gly in water. In addition, some factors that affect the catalytic activation (i.e., catalyst dose, PMS content, Gly concentration, pH, type of oxidant, temperature, and the presence of anions) were investigated. The decomposition pathway of glyphosate was also determined. The major role of reactive oxygen species in the Gly decomposition was determined and the reaction mechanism was proposed. The durability and reusability tests for the Co3O4/g-C3N4 material were also performed.
2. Materials and methods
2.1. Chemicals
All chemicals used in this work are lab-grade and directly used without purification. C3H6N6 (melamine), Co(NO3)2·6H2O, 2KHSO5·KHSO4·K2SO4 (Oxone or PMS), C4H6N2 (2-methylimidazole, 2-MID), and C3H8NO5P (glyphosate, Gly) were bought from Shanghai Macklin Biochemical Co., Ltd, China. NaNO3, Na2SO4, NaHCO3, SnCl2·2H2O, C3H8O3, (NH4)2MoO4·7H2O, CH3OH, (CH3)3COH (TBA), C2H5OH (EtOH), C6H5OH (PheOH), C6H4O2 (p-BQ), C5H6O2 (FFA), NaCl, NaOH, H2SO4, and HCl were purchased from Xilong Scientific Co., Ltd, China.
2.2. Synthesis and characterization of the Co3O4/g-C3N4 composite
The synthesis procedure of the Co3O4/g-C3N4 material is shown in Fig. S1 of the ESI.† The g-C3N4 support was prepared from the melamine precursor by the thermal condensation method reported in previous work.50,51 Co3O4 and Co3O4/g-C3N4 materials were then synthesized by the solvent diffusion method followed by pyrolysis.52 Typically, 25 mL of methanol was used to dissolve a desired amount of g-C3N4 and sonicated for 1 h to obtain solution A with a light yellow color. Meanwhile, 100 mL of methanol was used to dissolve 2.925 g of cobalt nitrate hexahydrate and stirred for 30 min to obtain solution B with a pink color. Solution C was formed after mixing solution A and solution B and stirring for 30 min. Besides, 50 mL of methanol was used to dissolve 3.28 g of 2-methylimidazole and stirred for 30 min to obtain solution D. Solution D was then slowly added by using a burette into solution C, stirred for 30 min, and left for 24 h of reaction. After that, the suspension was centrifuged and washed with methanol, followed by drying at 60 °C for 12 h to form a purple solid of the ZIF-67/g-C3N4 precursor. The Co3O4/g-C3N4 composite was finally attained by heating the ZIF-67/g-C3N4 precursor at 350 °C for 3 h with a temperature rate of 2 °C min−1. The mass of g-C3N4 in the material varies in the range of 1–30% by changing the g-C3N4/Co3O4 ratio during the material synthesis. The fabrication of pure Co3O4 was conducted under the same procedure and conditions but without the addition of g-C3N4.
Fourier transform infrared spectroscopy (FTIR, Perkin Elmer BX) was used to determine the surface chemical properties of the materials. X-ray diffraction (XRD, Bruker D8 Advance) was employed to analyze the crystal phase and crystallinity of the materials. Energy-dispersive X-ray spectroscopy (EDX, Horiba EMAX Energy) was applied for analyzing the surface chemical composition. Scanning electron microscopy (SEM, Hitachi SU810) was used to examine the surface morphology. N2 adsorption and desorption isotherms (Micromeritics TriStar II Plus 2.03) were employed for evaluating the specific surface area (BET) and pore structures. The Raman spectra of the samples were recorded at 785 nm laser excitation by using a DXR Raman spectrophotometer (Horiba LabRam Evolution). Inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7900) was used to determine the Co content dissolved in the solution after the reaction when using ZIF-67/g-C3N4, Co3O4, and Co3O4/g-C3N4.
2.3. PMS activation for glyphosate decomposition
The PMS activation by the Co3O4/g-C3N4 catalyst was performed using a batch reactor. At first, 100 mL of 50 mg L−1 Gly solution was taken into a 250 mL beaker at room temperature (25 ± 2 °C). The solution was then mixed with 0.005 g of Co3O4/g-C3N4 catalyst and sonicated for 10 min to homogenize the mixture. The mixture was then stirred magnetically for 30 min to reach an equilibrium of adsorption and desorption before being mixed with 4 mL of 5000 mg L−1 PMS solution for reaction. During the test, 2 mL of the water sample was regularly taken and centrifuged to remove the catalyst and sent for determining the phosphate concentration as reported in previous work.11 The decomposition efficiency of Gly was evaluated indirectly via the concentration of phosphate (PO43−) formed in solution by the following equation.where Ct (mM) is the phosphate concentration at time t and Co (mM) is the initial concentration of Gly.
The phosphate (the product from the decomposition of Gly) concentration was analyzed by SMEWW 4500-P D (stannous chloride method). In an acidic medium, PO43− ions will react with an ammonium molybdate reagent to yield a yellow ammonium phosphomolybdate complex. The complex is reduced by reducing agents such as SnCl2 to a blue molybdenum complex. The intensity of the color is dark or light depending on the content of phosphate ions present in the water sample. Ammonium and nitrate were analyzed by SMEWW 4500-B F: 2017 and SMEWW 4500-D, respectively.
0.1 M NaOH solution was used for adjusting the pH value of the solution. For determining the key reactive oxygen species (ROS) during the reaction, some radical scavenging agents were used, including tert-butyl alcohol (TBA), ethanol (EtOH), phenol (PheOH), and furfuryl alcohol (FFA), and were added into the solution before adding PMS to quench the radicals. The reusability and the stability of the material were also studied by doing the reaction and regeneration for five consecutive cycles.
Mass spectrometry (MS, Waters, USA) was used to detect the intermediates and products of Gly decomposition. A total organic carbon (TOC) analyzer (multi-N/C 2100S, Analytik Jena, Germany) was used to evaluate the mineralization of Gly.
3. Results and discussion
3.1. Material characterization
The SEM images of g-C3N4, Co3O4, and Co3O4/g-C3N4(10%) are shown in Fig. 1. g-C3N4 has an irregular layered structure with several stacked layers (Fig. 1(a)), which can be produced by the release of gases (e.g., CO2, H2O, and NH3) during the calcination process.53 Co3O4 has a nanoparticle morphology agglomerated in porous and uniform clusters (Fig. 1(b)). The morphology of Co3O4/g-C3N4 is similar to that of Co3O4; however, the layered structure of g-C3N4 is also observed, showing that g-C3N4 serves as the substrate for Co3O4 to grow on the surface (Fig. 1(c)). The EDX results of Co3O4, g-C3N4, and Co3O4/g-C3N4(10%) are presented in Fig. S2.† The Co and O elements were detected in the spectrum of Co3O4 (Fig. S2(a)†) while C and N were in the spectrum of g-C3N4 (Fig. S2(b)†). Furthermore, the Co3O4/g-C3N4 composite spectrum contains C, O, N, and Co elements (Fig. S2(c)†). There is no signal of other impurities, proving that the materials Co3O4, g-C3N4, and Co3O4/g-C3N4(10%) were successfully synthesized with high purity.54,55
 |
| | Fig. 1 SEM images of (a) g-C3N4, (b) Co3O4, and (c) Co3O4/g-C3N4(10%). | |
The mechanism for the formation of the g-C3N4 and Co3O4/g-C3N4 materials from melamine and ZIF-67/g-C3N4 precursors, respectively, is explained as follows. Available and inexpensive nitrogenous precursors (e.g., melamine, urea, cyanimide, thiourea, and dicyanamide) were easily used to prepare the g-C3N4 material via the thermal condensation method. The g-C3N4 synthesis mainly involves nucleophilic addition, polycondensation, and polymerization.56 With melamine as a precursor, when the temperature increases, ammonia removal and polycondensation occur to form melem – a base unit of g-C3N4,57 which then undergoes rearrangement and multi-condensation to form g-C3N4 with subsequent heat treatment.58
The abundance of nitrogen coordination sites on the g-C3N4 surface facilitates the fixation of Co2+ into tri-s-triazine units by electrostatic interaction. When 2-MID is added to the solution, a complexation process occurs between the central metal ion Co2+ and the N atom of the N heterocycle of 2-methylimidazole, followed by the deprotonation of 2-methylimidazole in the cluster. Finally, there is oligomerization by inter-cluster binding via Co2+ by deprotonated 2-methylimidazole ligands.59 The clusters then grow into crystal nuclei with rapid nucleation, and the ZIF-67 nuclei continued to grow into nano-ZIF-67.60 The in situ growth of ZIF-67 allows ZIF-67 to bind tightly to g-C3N4 by π–π interactions and electrostatic attraction.52 Finally, the oxidation was carried out at 350 °C under atmospheric conditions to decompose ZIF-67 into Co3O4 and convert ligands into carbon oxides, nitrogen oxides, and water vapor, thus increasing the porosity of the materials.61 Co3O4 crystals surrounded by g-C3N4 were obtained by calcining the composite in air.52,62
The XRD pattern of g-C3N4 in Fig. 2(a) has two diffraction peaks at 2θ of 13.15° for the cyclic arrangement of tri-s-triazine units placed in the (100) plane and 27.25° for the link of the corresponding plane (002) aromatic conjugated systems (JCPDS card no. 87-1526).41,63 The average crystal size of g-C3N4 calculated from the (002) plane is 9.57 nm. Co3O4 diffraction peaks are observed at 2θ of 18.96, 31.26, 36.84, 38.59, 44.89, 55.58, 59.53, and 65.23° which can be assigned to the planes of (111), (220), (311), (222), (400), (422), (511), and (440), respectively (JCPDS card no. 42-1467).19 No other diffraction peaks were detected, indicating that the Co3O4 prepared from the ZIF-67 precursor has high purity and good crystallinity with an average crystal size calculated from the (311) plane of 45.32 nm. In addition, no other impurity peaks were detected, indicating that the synthesized g-C3N4 and Co3O4 had relatively high purity and had a well-crystalline structure. For the Co3O4/g-C3N4(10%) composite, peaks at 2θ of 19.00, 31.26, 36.85, 38.64, 44.87, 55.57, 59.39, and 65.19° are similar to those observed in the Co3O4 material. However, the characteristic peaks of g-C3N4 were not observed in Co3O4/g-C3N4, which is because of the small percentage of g-C3N4 (10%) in the composite and the uniform dispersion and relatively complete coverage of Co3O4 on the g-C3N4 support. In brief, the crystal structure of Co3O4 was not changed after being modified with g-C3N4, and all the materials were successfully synthesized with relatively high purity.
 |
| | Fig. 2 (a) XRD patterns and (b) FT-IR spectra of g-C3N4, Co3O4, and Co3O4/g-C3N4(10%). | |
As seen in the FTIR spectrum of g-C3N4 (Fig. 2(b)), a peak at 803.56 cm−1 corresponds to the characteristic vibration of C–N bonds in the aromatic ring. The strong peaks in the range of 1131–1454 cm−1 are attributed to valence vibrations of the C–N bond outside the aromatic rings and the peak at 1626.7 cm−1 is the valence vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. The broad absorption bands at 3159.62–3500 cm−1 are oscillations of the primary and secondary amines due to their intermolecular hydrogen bond formation. Here, the hydrogen atoms are also bonded to CN in the aromatic ring as well as to the C–NH2 and C–NH–C groups in the graphene-type structure of g-C3N4. In addition, the peak at 805.39 cm−1 is associated with the s-triazine ring.50,52 For Co3O4, the bands at 554.54 and 669.06 cm−1 represent the vibration of the octahedral and tetrahedral coordinating Co3+ and Co2+ with oxygen in the spinel crystal lattice. These results confirm that the pure cubic Co3O4 spinel was obtained by pyrolysis from the precursor ZIF-67.37 In the Co3O4/g-C3N4(10%) composite, most of the peaks and bands coincide with the Co3O4 material and the characteristic bands of g-C3N4 are observed with lower peak intensity than those of g-C3N4, possibly because of the low amount of g-C3N4 in the composite and the cover of Co3O4 on its surface.
N bond. The broad absorption bands at 3159.62–3500 cm−1 are oscillations of the primary and secondary amines due to their intermolecular hydrogen bond formation. Here, the hydrogen atoms are also bonded to CN in the aromatic ring as well as to the C–NH2 and C–NH–C groups in the graphene-type structure of g-C3N4. In addition, the peak at 805.39 cm−1 is associated with the s-triazine ring.50,52 For Co3O4, the bands at 554.54 and 669.06 cm−1 represent the vibration of the octahedral and tetrahedral coordinating Co3+ and Co2+ with oxygen in the spinel crystal lattice. These results confirm that the pure cubic Co3O4 spinel was obtained by pyrolysis from the precursor ZIF-67.37 In the Co3O4/g-C3N4(10%) composite, most of the peaks and bands coincide with the Co3O4 material and the characteristic bands of g-C3N4 are observed with lower peak intensity than those of g-C3N4, possibly because of the low amount of g-C3N4 in the composite and the cover of Co3O4 on its surface.
The N2 adsorption–desorption isotherms of Co3O4, g-C3N4, and Co3O4/g-C3N4(10%) belong to type IV according to the BJH classification (Fig. S3(a)†) and the mesoporous nanomaterial (Fig. S3(b)†). The surface area of the Co3O4 material was 34.04 m2 g−1 with a total pore volume of 0.057 cm3 g−1 and an average pore size of 7.68 nm. This surface area is similar to the other reported values of 16.66 m2 g−1 (ref. 37) or 20 to 50.9 m2 g−1.24 The Co3O4 material has a porous structure that is produced by the release of gas during the pyrolysis of ZIF-67 and the oxidation of the 2-MID ligands in the ZIF-67 structure.61 The surface area of the g-C3N4 material was 12.35 m2 g−1, similar to those in the literature of 3.6–39.5 m2 g−1 (ref. 50) with a pore volume of 0.08 cm3 g−1 and a pore size of 45.2 nm. The surface area of the Co3O4/g-C3N4(10%) material was 58.63 m2 g−1 with a pore volume of 0.12 cm3 g−1 and a pore size of 8.1 nm, in which the surface area is higher than those of published g-C3N4/Co3O4 of 17.02 m2 g−1 (ref. 35) and 28.49 m2 g−1.64 When forming composites, the surface area significantly increased as compared to the single materials of g-C3N4 and Co3O4. Catalysts with a highly porous structure and high surface area would facilitate the diffusion of the reactants, thereby providing high adsorption capacity and more catalytic active sites for pollutant treatment.
The TGA curves of the ZIF-67, Co3O4, g-C3N4, g-C3N4/ZIF-67, and Co3O4/g-C3N4 materials in the temperature range of 50–800 °C in air are given in Fig. S4.† The weight loss of ZIF-67 exhibits three phases of (i) 3.1% during 50–350 °C of methanol in the pores of ZIF-67,65 (ii) 65% during 350–420 °C due to the breakdown of the metal–ligand coordination bonds, framework collapse, and organic ligand decomposition, and (iii) almost no further loss at above 420 °C for stable Co3O4.19 For g-C3N4, the slight weight loss below 570 °C could be due to the evaporation of volatile impurities and the removal of adsorbed water, indicating its high thermal stability. The main weight loss after 570–670 °C is because 100% of g-C3N4 was decomposed after 670 °C to form small molecules such as CO2 and NH3.56 ZIF-67/g-C3N4 has a similar TGA curve to ZIF-67, where g-C3N4/ZIF-67 is stable thermally up to 400 °C, which is then rapidly decomposed in the range of 400–450 °C (for both g-C3N4 and ZIF-67), and finally stable after 450 °C, with a total weight loss of about 60%. Thus, the durability of the ZIF-67/g-C3N4 composite was increased as compared to ZIF-67. In addition, the TGA curve of Co3O4 did not change during the calcination time while Co3O4/g-C3N4 decreased slightly at temperatures below 485 °C. The mass of Co3O4 remained at 92% after 575 °C, showing an actual content of g-C3N4 in the composite of about 8%.
Raman spectroscopy was used to confirm the presence of g-C3N4 in the Co3O4/g-C3N4 composites (Fig. S5†). Strong absorption peaks at 478.47, 517.66, 611.36, and 681.17 cm−1 are consistent with the values for pure spinel Co3O4. The peak at 681.17 cm−1 is attributed to the characteristics of the octahedral A1g sites. In addition, the Eg (478.47 cm−1) and F2g (517.66 and 611.36 cm−1) sites are related to the combined oscillations of the tetrahedral position and octahedral oxygen motion.66,67 In the Raman spectrum of g-C3N4, the characteristic peaks of the extended lattice at 471.00, 709.17, 976.44, 1230.28, and 1308.29 cm−1 are in agreement with the values obtained from pure g-C3N4.68 All the characteristic peaks of Co3O4 are observed in the Co3O4/g-C3N4 spectrum. However, the intensity of the characteristic peaks of g-C3N4 was significantly reduced and some peaks even disappeared in the composites. This indicates the reduction of g-C3N4 during the preparation of the Co3O4/g-C3N4 composite.67,68
3.2. Glyphosate decomposition tests
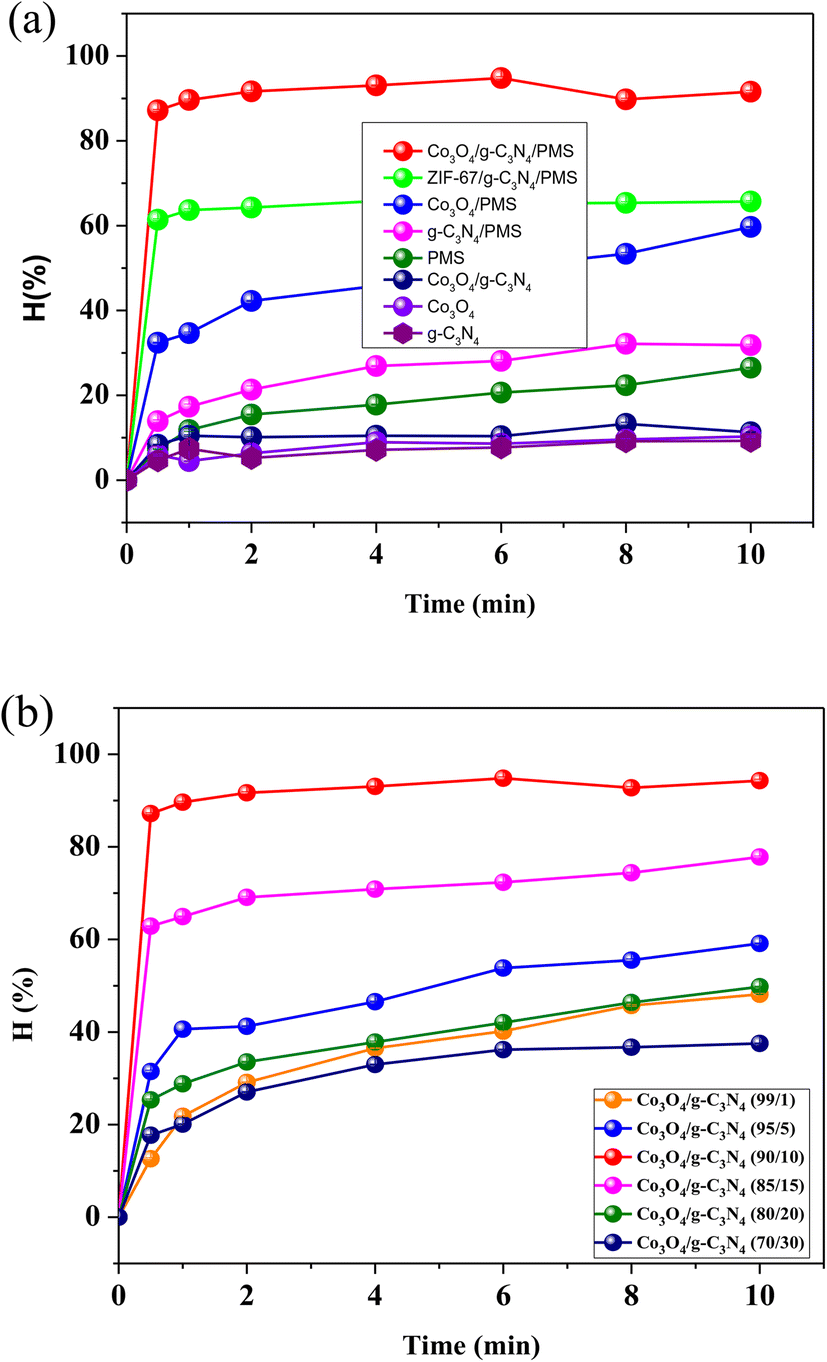
The PMS activation was applied for Gly removal in water and the tests were first set up with various adsorption/reaction systems using Co3O4, g-C3N4, Co3O4/g-C3N4, PMS, Co3O4/PMS, g-C3N4/PMS, and Co3O4/g-C3N4(10%)/PMS. As seen in Fig. 3(a), the Gly decomposition efficiency by g-C3N4, Co3O4, and Co3O4/g-C3N4 reached 9.28%, 10.3%, and 13.2%, respectively. Without PMS and light, the materials (i.e., g-C3N4, Co3O4, Co3O4/g-C3N4) have a limited ability to remove Gly, which mainly takes place based on the adsorption mechanism on the surface of the materials. This shows that glyphosate removal by adsorption is negligible and glyphosate degradation is the dominant mechanism for glyphosate removal.32
 |
| | Fig. 3 Glyphosate decomposition efficiency by (a) different reaction systems and (b) mass ratios of g-C3N4/Co3O4 (reaction conditions: 50 mgGly L−1, 50 mgcatalyst L−1, 200 mgPMS L−1, pH 11, 25 °C). | |
In addition, the chemical activity of PMS for Gly removal is also low with an efficiency of 26.5%, which can be explained by the weak oxidizing properties of PMS (Eo = 1.82 V). In a strongly alkaline environment, PMS can be converted into sulfate radicals with strong oxidizing properties;69 however, the activity of this form on Gly is still limited. In the presence of both materials and PMS, the Gly decomposition efficiency increases significantly. In particular, the Co3O4/g-C3N4(10%)/PMS system gave a Gly decomposition efficiency of 94.28%, which was 3, 1.6, and 1.45 times higher than that of the g-C3N4/PMS, Co3O4/PMS, and ZIF-67/g-C3N4/PMS systems, respectively. These results indicate that the combination of Co3O4/g-C3N4 and the presence of PMS greatly enhanced the activity of both single materials thanks to their synergistic effect on PMS activation, which was also observed for Fe3O4/CeO2,70 Co/Bi25FeO40,71 ZnO@AC@FeO,72 and SCF@GCN.73
The effect of the g-C3N4:Co3O4 ratio on the Gly decomposition is presented in Fig. 3(b). At all ratios, Gly decomposed rapidly in the first 30 s of reaction, and then slowly increased during the remaining time of reaction. The decomposition efficiency rises sharply with the increase of the g-C3N4:Co3O4 ratio from 1 to 10%, and then decreases with further increases of the ratio from 10 to 30%. When the g-C3N4 content is greater than 10%, the excess g-C3N4 on the Co3O4 surface can reduce the material surface area and therefore the surface active sites, thereby reducing its PMS activation ability. Thus, the g-C3N4:Co3O4 ratio of 10:90 was chosen for further investigation.
The effects of operating conditions during PMS activation for Gly decomposition in water were investigated with various conditions of catalyst dose, Gly concentration, some oxidants (H2O2, PMS, Na2S2O8), PMS content, pH, and the presence of anions in solution (Fig. 4). As described in Fig. 4(a), the decomposition efficiency increased with increasing catalyst content from 0 to 200 mg L−1 and then slightly decreased with a further increase up to 500 mg L−1. An increase in catalyst dose would increase the active sites, which increases the PMS activation ability to produce more reactive oxygen species, thus leading to the increase in Gly decomposition.70,74 However, with the excess catalyst content, the radical decomposition reaction by Co2+ can occur (reactions (2) and (3)), thus reducing the efficiency of Gly decomposition. At the catalyst dose of 50 mg L−1, the Gly decomposition efficiency reached 94.28%, a value that is relatively high and not too different from those at 100 and 200 mg L−1. Furthermore, a lower catalyst concentration reduces the dissolved concentration of Co ions in the solution, thereby reducing secondary pollution. Therefore, 50 mg L−1 was chosen as a suitable catalyst content for further investigations.
| | ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co2+ + SO4˙− → Co3+ + SO42− Co2+ + SO4˙− → Co3+ + SO42− | (2) |
| | | Co2+ + HO˙ → Co3+ + HO− | (3) |
Regarding the effect of the initial Gly concentration,
Fig. 4(b) shows that when the Gly concentration increased from 10 to 50 mg L
−1, the Gly decomposition efficiency after 10 min increased gradually from 45.8 to 94.28%. This may be because at low concentrations the diffusion of Gly to and from the catalyst surface is facilitated by mass transfer at low concentrations and unsaturated adsorption of Gly on the active surface of Co
3O
4/g-C
3N
4.
75,76 With a further increase to 75 and 100 mg L
−1, the decomposition efficiency decreased to 86.2 and 74.2%, respectively. The higher the initial Gly concentration, the more free radicals are required for complete oxidation. However, the activation process uses the same dose of PMS catalyst and oxidant, thus the decomposition efficiency is reduced. In addition, a high concentration increases the Gly adsorption on the material surface, which can reduce the material and PMS contact. This is not favorable for the activation and generation of ROS and therefore reduces the efficiency of the material for Gly decomposition. Therefore, the Gly concentration was selected to be 50 mg L
−1, since it can meet the requirements of time and efficiency for evaluation of other parameters.
 |
| | Fig. 4 Effects of (a) catalyst dose, (b) Gly concentration, (c) temperature, (d) type of oxidant, (e) PMS concentration, (f) initial solution pH, and (g) co-existing ions (reaction conditions: 10–100 mgGly L−1, 0–500 mgcatalyst L−1, 0–300 mgPMS L−1, pH 3.6–11, 0–45 °C). | |
The Gly decomposition ability in the temperature range from 0–45 °C (Fig. 4(c)) shows that Gly has a high removal efficiency during the first 30 s of reaction at different temperatures. At low temperatures of 0 and 10 °C, the decomposition efficiencies were not significant and reached 12.66 and 22.02%, respectively, with similar efficiency in the first 2 min of reaction. At these low temperatures, the collision rate between the reactants decreases, thereby reducing the activation efficiency. At a temperature of 15 °C, the efficiency increased significantly, reaching 62.68% after 10 min of the reaction. In the temperature range of 25–35–45 °C, the efficiency was not significantly different and reached 94.28, 96.87, and 99.83%, respectively. As the temperature increases, the Gly decomposition efficiency increases, which may be due to the thermal decomposition of the O–O bond in HSO5− to yield more ROS.77,78
Oxidants such as PMS, H2O2, and Na2S2O8 are often used as oxidizing agents in advanced oxidation processes. As shown in Fig. 4(d), the Gly decomposition efficiency by these oxidants is in the order PMS > H2O2 > Na2S2O8. The Gly decomposition efficiency of the activation of PMS reached 94.28%, much higher than those with H2O2 (17.4%) and Na2S2O8 (7.4%), which is consistent with Qi et al.69 The asymmetric structure of PMS would be more easily activated than H2O2 and Na2S2O8 and would have high reactivity with transition metal ions over a wide pH range. PMS can be activated by Co2+ to produce SO4˙− and SO5˙− while only SO4˙− is generated through the activation of the remaining oxidants. In addition, the standard redox potential of HO˙ is lower than that of SO4˙− since H2O2 activation usually occurs under acidic conditions; thus, the Gly decomposition efficiency by H2O2 is less than that by PMS. Meanwhile, H2O2 is less stable in a strongly alkaline environment (reaction (4)), thus reducing the activation of H2O2.
For persulfate, the activation process in an alkaline medium occurs weakly because (i) the existence of persulfate anions with a large negative charge enhances repulsion with the surface of the negatively charged catalyst, and (ii) the sulfate radical is converted to a weak oxidizing hydroxyl radical (
Eo = 1.78 V).
69 The alkaline activation of persulfate takes place by its hydrolysis in an alkaline medium to produce hydroperoxide and sulfate anions (
reaction (5)), followed by the reduction of another persulfate molecule by hydroperoxide to form sulfate and superoxide radicals (
reaction (6)). Under an alkaline environment, sulfate radicals react with hydroxide ions to form hydroxyl radicals (
reaction (7)).
| | | S2O82− + 2H2O → HO2− + 2SO42− + 3H+ | (5) |
| | | S2O82− + HO2− → SO4˙− + O2˙− + SO42− + H+ | (6) |
| | | SO4˙− + HO− → HO˙ + SO42− | (7) |
Fig. 4(e) shows the effect of the PMS content (0–300 mg L
−1) on the Gly decomposition efficiency. As the PMS content increases (0–200 mg L
−1), the Gly decomposition efficiency remarkably increases from 10.3 to 94.28% and the treatment time significantly decreases. At the fixed pollutant concentration and catalyst dose, more PMS will produce more radicals (
e.g., sulfate and hydroxyl radicals) during the activation by the Co
3O
4/g-C
3N
4 catalyst. When the PMS content increased to 200 mg L
−1, the Gly decomposition efficiency increased slowly and did not change much as compared to higher PMS content. This can be explained that too much PMS causes quenching processes of ROS by HSO
5−, SO
52−, and ROS themselves (
reactions (8)–(11));
70,79 therefore, 200 mg L
−1 was chosen as suitable PMS content for further experiments.
| | | HSO5− + SO4˙− → HSO4− + SO5˙− | (8) |
| | | HSO5− + HO˙ → SO5˙− + H2O | (9) |
| | | SO4˙− + SO4˙− → S2O82− | (10) |
The pH solution usually plays a significant role in advanced oxidation processes. As shown in
Fig. 4(f), the initial pH has a strong influence on the Gly decomposition efficiency, where the performance increased gradually with increasing pH from 3.6 to 11. The decomposition of Gly was very fast during the first 30 s and did not change significantly after 2 min. From Fig. S6(a),
† the pH
pzc value of the Co
3O
4/g-C
3N
4 material was determined to be 7.25. At pH > 7.25, the catalyst surface is negatively charged while at pH < 7.25, its surface is positively charged. At pH = 3.6 (
e.g., unadjusted pH), the Gly decomposition was only 17%, which can be explained as follows. In a strongly acidic environment, the strong hydrogen bonding between H
+ and O–O in PMS inhibits the interaction between PMS and the positively charged surface of the catalyst. In addition, H
+ ions also react with SO
4˙
− and HO˙ to produce HSO
4˙ and H
2O (
reactions (12) and
(13)), leading to a low Gly decomposition efficiency. When increasing the pH from 3.6 to 7, the Gly decomposition efficiency increased gradually from 17 to 44.5%. In this pH range, PMS is in the form of anion HSO
5− and Gly exists in the form of anions HL
− and HL
2−, thus increasing the electrostatic attraction between these anions and the positively charged surface. This enhances the PMS adsorption and activation to form more sulfate radicals, which are stable in this pH range.
Gly decomposition reached the highest efficiencies in strongly alkaline environments (
e.g., 67.74 and 94.28% at pH 9 and 11, respectively). Under these conditions, the catalyst surface carries a negative charge (pH > pH
pzc) while PMS exists mainly in form of SO
52− and Gly in the forms of HL
2− and L
3− anions.
11 Although these conditions reduce the adsorption capacity of SO
52−, HL
2−, and L
3− on the catalyst surface, this results in an increase in Gly decomposition, which can be explained as follows. In strongly alkaline environments (
e.g. pH 11), the main existing form of PMS is SO
5;
2 thus, the reactions can take place to form reactive oxygen species (
e.g.,
1O
2, O
2˙
−, and HO˙) (
reactions (7) and
(14)–(18)).
80 Both O
2˙
− and
1O
2 have high oxidizing properties with redox potentials of 2.4 and 2.2 V, respectively, with a long lifetime in solution (
e.g. 4 μs); thus, they can selectively react with electron-rich organic pollutants. The high decomposition efficiency of organic pollutants at pH 11 was also observed for Co/g-C
3N
4/PMS,
16 CuCoFe-LDH/PMS,
11 Co/Bi
25FeO
40/PMS,
71 and MnFe
2O
4/α-MnO
2/PMS.
78 However, when the pH increased from 11 to 12, the efficiency decreased sharply from 94.28 to 30.46%. At pH ≥ 12, there is a loss of the HSO
5− form for PMS; thus, it is not effective for the formation of
1O
2. Instead, the main radical in the solution is the hydroxyl radical (HO˙) with weaker oxidizing activity (
e.g., redox potential 1.8 V); thus, it reduces the Gly decomposition efficiency. In addition, cobalt hydroxide can be formed at a high pH of 12, causing a decrease in catalytic activity.
16| | | HSO5− + SO52− → 1O2 + HSO4− + SO42− | (14) |
| | | 2SO5˙− + H2O → 1.51O2 + 2HSO4− | (15) |
| | | SO52− + H2O → H2O2 + SO42− | (16) |
| | | HO˙ + H2O2 → HO2˙ + H2O | (17) |
In addition, the change in solution pH during Gly decomposition was also investigated (Fig. S6(b)
†). Notably, in the pH range of 3.6–7, the solution pH decreased rapidly after the first minute of the reaction and was stable at pH from 3.0 to 4.0. At pH = 9, the change in pH was most pronounced compared to the other pH values with a pH decrease to 5.62 after 10 min of reaction. At pH = 11 and 12, the solution pH insignificantly decreased to 8.98 and 11.7, respectively, after the reaction, which could be due to the formation of PO
43− during the oxidation of Gly. In brief, although Co
3O
4/g-C
3N
4 can activate PMS over a wide pH range from acidic, neutral, and alkaline media, the initial solution pH of 11 was chosen for further investigation due to its highest Gly decomposition.
Wastewater contains large amounts of anions such as SO42−, NO3−, Cl−, and HCO3−, which affect the Gly decomposition efficiency of the Co3O4/g-C3N4/PMS system (Fig. 4(g) and S7†). In the presence of anions, a slight inhibition of Gly decomposition occurred at the anion concentration of 5 mM, and the difference in Gly decomposition efficiency between anions was negligible. Specifically, the decomposition efficiency of Gly ranges from 83–89.15% in the presence of HCO3− and NO3− anions, respectively (Fig. S7†). When the anion concentration increased to 50 mM, the inhibition of Gly decomposition by anions increased obviously and the effect of anions on the Gly decomposition efficiency is in the following order: SO42− < NO3− < Cl− < HCO3− (Fig. 4(g)). While the SO42− anion almost does not react with sulfate radical SO4˙−, the NO3− anion can react with SO4˙− and HO˙ to form NO3˙, which has weaker oxidizing properties (Eo = 2.3–2.5 V) (reactions (19) and (20)).81 Regarding the Cl− anion at high concentrations, it can scavenge other radicals in AOPs, consume PMS and sulfate radicals, react with other radicals, and lead to the formation of Cl˙ (Eo = 2.47 V) and Cl2˙− (Eo = 2.09 V), which are weaker oxidizing agents than SO4˙− (reactions (21) and (22)).82 Similarly, HCO3− could react with SO4˙− and HO˙ to quench these radicals to form HCO3˙ (Eo = 1.65 V) and CO3˙− (Eo = 1.78 V) (reactions (23) and (24)). Therefore, in practice, it is necessary to have an anion pretreatment step before decomposing Gly in water by the Co3O4/g-C3N4/PMS system.
| | | SO4˙− + NO3− → SO42− + NO3˙ | (19) |
| | | HO˙ + NO3− → HO− + NO3˙ | (20) |
| | | SO4˙− + Cl− → SO42− + Cl˙ | (21) |
| | | HCO3− + SO4˙− → SO42− + HCO3˙ | (23) |
| | | HCO3− + HO˙ → CO3˙− + H2O | (24) |
Reusability and high stability are essential factors for practical applications of a catalyst in industrial use. After each reaction, the solid material was separated by centrifugation, washed 3 times with methanol, and dried for 3 h at 70 °C before being used in the subsequent cycle. The Gly decomposition efficiency after 10 min was still high after 5 cycles of recovery and reuse, which only decreased by 10.72% (Fig. S8
†). After the first 3 cycles, the Gly decomposition efficiency just decreased slightly, and there was a significant decrease after the 5th cycle. In general, the Co
3O
4/g-C
3N
4 material has a relatively stable structure, which could provide relatively stable operation in practical applications. The XRD analysis (Fig. S9
†) also illustrates that the Co
3O
4/g-C
3N
4 crystalline structure did not change after the activation reaction. Therefore, the Co
3O
4/g-C
3N
4 composite has excellent stability for organic pollutant decomposition by sulfate-based AOPs. On the other hand, the leaching of Co
2+ into the solution when using the ZIF-67/g-C
3N
4 precursor directly to activate PMS is very high at 3.78 mg L
−1 because of the rapid collapse of the ZIF-67 structure.
17 The concentrations of cobalt in the solution of the Co
3O
4/PMS and Co
3O
4/g-C
3N
4(10%)/PMS systems are 1.36 and 0.42 mg L
−1, respectively, indicating that g-C
3N
4 not only enhances the catalytic activity of Co
3O
4 but also reduces the solubility of Co metals by about 3.23 times. The content of dissolved Co
2+ in the Co
3O
4/g-C
3N
4(10%)/PMS system is lower than that in some reported systems of MnCo
2O
4/g-C
3N
4 (0.65 mg L
−1)
83 and NiCo
2O
4/g-C
3N
4 (5.8 mg L
−1).
84
3.3. Glyphosate decomposition mechanism
The activation of PMS by the Co3O4/g-C3N4 catalyst can form various reactive oxygen species (ROS) of SO4˙−, HO˙, SO5˙−, and 1O2, which can then participate in the decomposition reactions of Gly. At pH 11, the SO5˙− radical has a redox potential of 1.1 V, which is much lower than those of SO4˙− and HO˙. Therefore, the possibility of participating in the Gly decomposition of the SO5˙− radical is negligible. To determine the major types and roles of ROS in the Gly decomposition by the Co3O4/g-C3N4/PMS system, radical quenching experiments were conducted by adding appropriate organic solvents69 such as TBA, EtOH, PheOH, and FFA at concentrations of 10 mM and p-BQ at a concentration of 1 mM (Fig. 5). TBA is used to quench HO˙ (kHO˙ = 3.8–7.6 × 108 M−1 s−1, 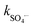 = 4.0–9.1 × 105 M−1 s−1), EtOH for both SO4˙− and HO˙ (
= 4.0–9.1 × 105 M−1 s−1), EtOH for both SO4˙− and HO˙ (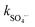 = 1.6–7.7 × 107 M−1 s−1; kHO˙ = 1.2–2.8 × 109 M−1 s−1), PheOH for both SO4˙− and HO˙ (
= 1.6–7.7 × 107 M−1 s−1; kHO˙ = 1.2–2.8 × 109 M−1 s−1), PheOH for both SO4˙− and HO˙ ( = 8.8 × 109 M−1 s−1; kPheOH–HO˙ = 6.6 × 109 M−1 s−1), FFA for both 1O2 and HO˙ (kFFA-1O2 = 1.2 × 108 M−1 s−1; kFFA-HO˙ = 1.5 × 1010 M−1 s−1), and p-BQ for O2˙− (
= 8.8 × 109 M−1 s−1; kPheOH–HO˙ = 6.6 × 109 M−1 s−1), FFA for both 1O2 and HO˙ (kFFA-1O2 = 1.2 × 108 M−1 s−1; kFFA-HO˙ = 1.5 × 1010 M−1 s−1), and p-BQ for O2˙− ( = 109 M−1 s−1).85 When adding EtOH, TBA, and PheOH, the Gly decomposition efficiency decreased to 66.9, 61.3%, and 56.28%, respectively. For p-BQ and FFA, the efficiencies decreased significantly to 43.78% and 38.27%, respectively, showing that p-BQ and FFA have a strong inhibitory effect on the PMS activation by Co3O4/g-C3N4. Thus, the role of ROS is in the order 1O2 > O2˙− > SO4˙− > HO˙, in which 1O2 and O2˙− play a major role in Gly decomposition by the Co3O4/g-C3N4/PMS system.
= 109 M−1 s−1).85 When adding EtOH, TBA, and PheOH, the Gly decomposition efficiency decreased to 66.9, 61.3%, and 56.28%, respectively. For p-BQ and FFA, the efficiencies decreased significantly to 43.78% and 38.27%, respectively, showing that p-BQ and FFA have a strong inhibitory effect on the PMS activation by Co3O4/g-C3N4. Thus, the role of ROS is in the order 1O2 > O2˙− > SO4˙− > HO˙, in which 1O2 and O2˙− play a major role in Gly decomposition by the Co3O4/g-C3N4/PMS system.
 |
| | Fig. 5 Effect of adding radical scavengers into the g-C3N4/Co3O4/PMS system (reaction conditions: 50 mgGly L−1, 50 mgcatalyst L−1, 200 mgPMS L−1, pH 11, 25 °C). | |
Based on relevant previous reports,69,80 experimental results on the pH effect with optimal pH 11, and the identification of ROS, the Gly decomposition mechanism by the Co3O4/g-C3N4/PMS system is proposed as shown in Fig. 6. In Co3O4/g-C3N4, g-C3N4 acts as an adsorbent of pollutants on the surface by strong intermolecular forces such as hydrogen bonding and π–π interactions between pollutant molecules and the remaining amine groups in g-C3N4, which can promote adsorption.16 At pH 11, after adding PMS, surface Co(II) and Co(III) sites activate HSO5− to produce Co(III) and SO4˙− (reaction (25)), and Co(II) and SO5˙− (reaction (26)), respectively.25 On the other hand, g-C3N4 has an electron-rich structure, containing heptazine rings with pyridine nitrogen groups. The six single electron pairs in nitrogen act as electron donors, thereby inducing the reduction of PMS. The C atoms adjacent to the N atoms can participate as an electron acceptor and PMS can act as an electron donor due to the presence of the peroxy bond in PMS, thus leading to oxidation of PMS to form SO5˙− (reaction (27)).33 In an alkaline environment, the sulfate radical and peroxymonosulfate are converted to HO˙ (reactions (7) and (28)), and the hydrolysis of HSO5− and SO52− anions forms H2O2 (reactions (29) and (16)).86 Then, the dissociation and decomposition reactions of H2O2 create HO2− (reactions (30) and (31)) in alkaline environments.87 The formation of 1O2 can be from the reaction between HSO5− and HO2−, HSO5− and SO52− or HO˙ and O2˙ (reactions (32)–(34), (14), and (15)). The formation of O2˙− is from the decomposition of HO2˙ (reactions (17) and (18)).11,78 These 1O2, O2˙, SO4˙−, and HO˙ radicals will then oxidize Gly on the catalyst surface to intermediates and mineralize them into inorganic ions (reactions (35) and (36)).
| | | Co(II) + HSO5− → Co(III) + SO4˙− + OH− | (25) |
| | | Co(III) + HSO5− → Co(II) + SO5˙− + H+ | (26) |
| | | g-C3N4 + HSO5− → g-C3N4− + SO5˙− + H+ | (27) |
| | | 2SO5˙− + 2OH− → 2SO42− + 2HO˙ + O2 | (28) |
| | | H2O2 + HO− → H2O + HO2− | (31) |
| | | HSO5− + HO2− → H2O + SO4˙− + 1O2 | (32) |
| | | 4SO4˙− + 2H2O → 4HSO4− + 1O2 | (33) |
| | | HO˙ + O2˙− → 1O2 + HO− | (34) |
| | | [1O2, O2˙, SO4˙−, HO˙] + Gly → intermediates + SO42− | (35) |
| | | [1O2, O2˙, SO4˙−, HO˙] + intermediates → H2O + CO2 + NH4+ + NO3− + PO43− | (36) |
The MS spectra of the samples collected at a certain time during Gly decomposition are shown in Fig. S10.
† Previous studies showed that the two decomposition pathways of Gly by AOPs are the cleavages of the C–N bond and the C–P bond. In the first pathway, Gly is converted to aminomethylphosphonic acid (AMPA) through the C–N bond cleavage reaction, followed by the further oxidation of AMPA to methylamine, formaldehyde, ammonium, nitrate, and phosphate. In the second one, Gly is converted to sarcosine through direct cleavage of the C–P bond, after which sarcosine can be oxidized to glycine, formaldehyde, and ammonium.
88,89 Glyphosate was recognized at
m/
z = 168 (ref.
90), while the intermediates methyl-phosphonic acid (MPA) and glycine were at
m/
z values of 97 and 95, respectively,
88,91 proving that the ROS such as
1O
2 and O
2˙
− attack the Gly molecules and cleave the C–N bonds. These intermediates are then mineralized to water, carbon dioxide, nitrate, and phosphate (
Fig. 7). On the other hand, the unobserved C–P cleavage products may be due to the rapid oxidation of these products to carbon dioxide and inorganic ions.
92 There was a rapid increase in the concentrations of PO
43−, NO
3−, and NH
4+ ions in the first 30 s of the reaction (Fig. S11
†), indicating that the Gly structure was rapidly destroyed. A low concentration of ammonium of 0.58 mg L
−1 was observed, probably due to it being oxidized to nitrate with a concentration of 5.24 mg L
−1 after 10 min.
8,92 For phosphate, the rapid cleavage of the C–P bond of methyl-phosphonic acid led to a sharp increase in phosphate concentration (24.46 mg L
−1) after the first 30 s of reaction and reached 26.42 mg L
−1 after 10 min of reaction.
 |
| | Fig. 6 Proposed mechanism of ROS formation and utilization in glyphosate decomposition by the g-C3N4/Co3O4/PMS system. | |
 |
| | Fig. 7 Proposed decomposition pathway of glyphosate by the g-C3N4/Co3O4/PMS system. | |
The mineralization efficiency of Gly during the reaction time was evaluated via its TOC value, which was 73% after 30 s and 88% after 10 min of reaction. The mineralization efficiency (88%) is lower than the decomposition efficiency of Gly (94.28%), which is attributed to the formation of low molecular weight and stable intermediates that remain in the solution and are not oxidized.93 The mineralization efficiency of Gly by the Co3O4/g-C3N4/PMS system is much higher than many other AOP processes such as the MWCNT-Al/O3 system (64% after 120 min),94 photoelectrocatalysis with TiO2/BDD as the anode (85.3% after 5 h),93 and photocatalysis with the Aeroxide-TiO2-P25/UV-A system (100% after 240 min).95
The decomposition of Gly by AOPs using different catalysts is summarized in Table S1.† The photocatalysis has some disadvantages such as low quantum efficiency and fast recombination of electron–hole pairs from 10−9–10−12 s, and thus it is usually applied for Gly decomposition at low concentrations of below 50 mg L−1 and has found many difficulties in practical application.12,96 The electro-Fenton method has a high Gly decomposition efficiency, but it also has some disadvantages such as high electrical energy consumption and strict pH of around 3.92,97 The wet oxidation method has the advantage of treating Gly at high content (e.g., 100 mg L−1) but its disadvantage is that the reaction conditions occur at high temperature and high pressure with relatively low decomposition efficiency.2,94 The comparison results show that our study has some advantages such as low doses of catalyst (50 mg L−1) to activate PMS (200 mg L−1), which can decompose Gly at a very high efficiency of 87.18% after a short reaction time of 30 s. The decomposition of Gly is mainly caused by oxygen species of 1O2 and O2˙−. Meanwhile, the activation of PMS by heterogeneous catalysis using Co3O4/g-C3N4 does not require an energy supply (e.g. light or electricity) and takes place in an alkaline environment, thus limiting the dissolution of Co which harms the aquatic environment. Compared to other homogeneous systems using Cu2+ and Fe2+,10,70 the Co3O4/g-C3N4 heterogeneous system can be recycled and applied to decompose Gly with a high efficiency of 83.56% after 5 cycles of reaction.
4. Conclusions
In this study, Co3O4/g-C3N4 materials were prepared successfully by the thermally decomposed solvent diffusion method. The synthesized material successfully activated PMS to treat more than 85% of glyphosate in water with rapid decomposition within the first 30 s of reaction. The activity of the Co3O4/g-C3N4/PMS catalyst is dependent on several operating factors (catalyst dose, Gly concentration, PMS content). However, it can work well over a wide pH range, tolerant of the influence of small amounts of some anions in water. Oxygen radicals in the forms of 1O2 and O2˙− play a major role in the breakdown of glyphosate, where they cleave the C–N bonds of glyphosate to form products such as methyl phosphonic acid, glycine, and inorganic ions (e.g. phosphate, nitrate, and ammonium). After 5 consecutive operating cycles, the catalyst system showed relative reusability and durability. This alternative is proposed for wastewater containing organic pollutants with high alkalinity. However, the by-products generated during and after the catalysis by the Co3O4/g-C3N4/PMS system to decompose pollutants need to be studied and evaluated further.
Conflicts of interest
There are no conflicts to declare.
References
- M. Krüger, P. Schledorn, W. Schrödl, H.-W. Hoppe, W. Lutz and A. A. Shehata, J. Environ. Anal. Toxicol., 2014, 4, 1–5 CrossRef.
- P. Gupta, K. Pandey and N. Verma, Chem. Eng. J., 2022, 428, 132008 CrossRef.
- J. P. K. Gill, N. Sethi, A. Mohan, S. Datta and M. Girdhar, Environ. Chem. Lett., 2018, 16, 401–426 CrossRef.
- S. H. Bai and S. M. Ogbourne, Environ. Sci. Pollut. Res., 2016, 23, 18988–19001 CrossRef PubMed.
-
T. Van Nam, T. T. Nguyen and P. T. X. Thu, Proceedings of the 5th International Conference on Low Carbon Asia, 2019, pp. 103–108 Search PubMed.
- K. Sen, J. K. Datta and N. K. Mondal, Appl. Water Sci., 2019, 9, 1–12 CrossRef CAS.
- R. C. Pereira, P. R. Anizelli, E. Di Mauro, D. F. Valezi, A. C. S. da Costa, C. T. B. Zaia and D. A. Zaia, Geochem. Trans., 2019, 20, 1–14 CrossRef CAS.
- D. Feng, L. Malleret, A. Soric and O. Boutin, Chemosphere, 2020, 247, 125930 CrossRef CAS PubMed.
- E. G. Vaschetto, M. I. Sicardi, V. R. Elías, G. O. Ferrero, P. M. Carraro, S. G. Casuscelli and G. A. Eimer, Adsorption, 2019, 25, 1299–1306 CrossRef CAS.
- L. Chen, X. Huang, M. Tang, D. Zhou and F. Wu, Environ. Chem. Lett., 2018, 16, 1507–1511 CrossRef CAS.
- N. T. Dung, V. D. Thao and N. N. Huy, Vietnam J. Chem., 2021, 59, 813–822 CAS.
- Q.-Y. Tang, W.-F. Chen, Y.-R. Lv, S.-Y. Yang and Y.-H. Xu, Sep. Purif. Technol., 2020, 236, 116243 CrossRef CAS.
- S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552–558 CrossRef CAS PubMed.
- B. Kakavandi, S. Alavi, F. Ghanbari and M. Ahmadi, Chemosphere, 2022, 287, 132024 CrossRef CAS PubMed.
- B. Kakavandi, E. Dehghanifard, P. Gholami, M. Noorisepehr and B. MirzaHedayat, Appl. Surf. Sci., 2021, 570, 151145 CrossRef CAS.
- M. Xie, J. Tang, L. Kong, W. Lu, V. Natarajan, F. Zhu and J. Zhan, Chem. Eng. J., 2019, 360, 1213–1222 CrossRef CAS.
- Y. Liu, X. Chen, Y. Yang, Y. Feng, D. Wu and S. Mao, Chem. Eng. J., 2019, 358, 408–418 CrossRef CAS.
- H. Chen, Z. Zhang, D. Hu, C. Chen, Y. Zhang, S. He and J. Wang, Chemosphere, 2021, 265, 129047 CrossRef CAS.
- K. Chen, S. Bai, H. Li, Y. Xue, X. Zhang, M. Liu and J. Jia, Appl. Catal., A, 2020, 599, 117614 CrossRef CAS.
- Y.-C. Tsai, N. N. Huy, D. C. W. Tsang and K.-Y. A. Lin, J. Colloid Interface Sci., 2020, 561, 83–92 CrossRef CAS PubMed.
- Y.-C. Tsai, N. Nhat Huy, J. Lee, Y.-F. Lin and K.-Y. A. Lin, Chem. Eng. J., 2020, 395, 124939 CrossRef CAS.
- D. D. Tuan, H. Yang, N. N. Huy, E. Kwon, T. C. Khiem, S. You, J. Lee and K.-Y. A. Lin, J. Environ. Chem. Eng., 2021, 9, 105809 CrossRef CAS.
- J. Guo, Y. Zhang, Y.-C. He and J. Shan, Polyhedron, 2020, 175, 114215 CrossRef.
- M. Yang, C. Zhang, Y. Fan, T. Lin, X. Chen, Y. Lu, H. Wang, L. Zhong and Y. Sun, Mater. Lett., 2018, 222, 92–95 CrossRef CAS.
- M. A. N. Khan, P. Kwame Klu, C. Wang, W. Zhang, R. Luo, M. Zhang, J. Qi, X. Sun, L. Wang and J. Li, Chem. Eng. J., 2019, 363, 234–246 CrossRef.
- W.-J. Liu, E. Kwon, N. N. Huy, T. C. Khiem, G. Lisak, T. Wi-Afedzi, C.-C. Wu, F. Ghanbari and K.-Y. A. Lin, J. Taiwan Inst. Chem. Eng., 2022, 133, 104253 CrossRef CAS.
- C. Wang, P. Shi, X. Cai, Q. Xu, X. Zhou, X. Zhou, D. Yang, J. Fan, Y. Min and H. Ge, J. Phys. Chem. C, 2016, 120, 336–344 CrossRef CAS.
- H. Shao, X. Zhao, Y. Wang, R. Mao, Y. Wang, M. Qiao, S. Zhao and Y. Zhu, Appl. Catal., B, 2017, 218, 810–818 CrossRef CAS.
- M. A. N. Khan, P. K. Klu, C. Wang, W. Zhang, R. Luo, M. Zhang, J. Qi, X. Sun, L. Wang and J. Li, Chem. Eng. J., 2019, 363, 234–246 CrossRef.
- H. Zhang, J. Wang, X. Zhang, B. Li and X. Cheng, Chem. Eng. J., 2019, 369, 834–844 CrossRef CAS.
- W. Li, Y. Li, D. Zhang, Y. Lan and J. Guo, J. Hazard. Mater., 2020, 381, 121209 CrossRef CAS.
- Y. Shen, M. J. Martín de Vidales, G. Gorni, A. Gómez-Herrero, F. Fernández-Martínez and A. J. Dos Santos-García, Chem. Eng. J., 2022, 444, 136610 CrossRef CAS.
- Y. Li, J. Li, Y. Pan, Z. Xiong, G. Yao, R. Xie and B. Lai, Chem. Eng. J., 2020, 384, 123361 CrossRef CAS.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546–553 CrossRef CAS.
- H. Wu, C. Li, H. Che, H. Hu, W. Hu, C. Liu, J. Ai and H. Dong, Appl. Surf. Sci., 2018, 440, 308–319 CrossRef CAS.
- I. Rabani, R. Zafar, K. Subalakshmi, H.-S. Kim, C. Bathula and Y.-S. Seo, J. Hazard. Mater., 2021, 407, 124360 CrossRef CAS PubMed.
- N. Liu, M. Tang, C. Jing, W. Huang, P. Tao, X. Zhang, J. Lei and L. Tang, J. Sol-Gel Sci. Technol., 2018, 88, 163–171 CrossRef CAS.
- Z. Zhang, J. Wu and D. Liu, Processes, 2019, 7, 279 CrossRef CAS.
- H. Yang, K. Lv, J. Zhu, Q. Li, D. Tang, W. Ho, M. Li and S. A. C. Carabineiro, Appl. Surf. Sci., 2017, 401, 333–340 CrossRef CAS.
- Y. Wang, J. Huang, J. Cao, G. Li and Z. Zhang, Surf. Rev. Lett., 2017, 24, 1750058 CrossRef CAS.
- H. Gao, H. Yang, J. Xu, S. Zhang and J. Li, Small, 2018, 14, 1801353 CrossRef.
- V. Ivanova-Kolcheva and M. Stoyanova, Bulg. Chem. Commun., 2019, 51, 158–164 Search PubMed.
- C. Jin, M. Wang, Z. Li, J. Kang, Y. Zhao, J. Han and Z. Wu, Chem. Eng. J., 2020, 398, 125569 CrossRef CAS.
- W. Zhang, W. Shi, H. Sun, Y. Shi, H. Luo, S. Jing, Y. Fan, F. Guo and C. Lu, J. Chem. Technol. Biotechnol., 2021, 96, 1854–1863 CrossRef CAS.
- W. Xiang, Q. Ji, C. Xu, Y. Guo, Y. Liu, D. Sun, W. Zhou, Z. Xu, C. Qi, S. Yang, S. Li, C. Sun and H. He, Appl. Catal., B, 2021, 285, 119847 CrossRef CAS.
- Q. Yang, J. An, Z. Xu, S. Liang and H. Wang, Colloids Surf., A, 2021, 614, 126161 CrossRef CAS.
- S. S. Rezaei, E. Dehghanifard, M. Noorisepehr, K. Ghadirinejad, B. Kakavandi and A. R. Esfahani, J. Environ. Manage., 2019, 250, 109472 CrossRef CAS.
- A. Sudhaik, P. Raizada, S. Thakur, A. K. Saini, P. Singh and A. Hosseini-Bandegharaei, Mater. Lett., 2020, 277, 128277 CrossRef CAS.
- J. Tan, Z. Li, J. Li, Y. Meng, X. Yao, Y. Wang, Y. Lu and T. Zhang, J. Hazard. Mater., 2022, 423, 127048 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- N. T. Dung, N. Van Hiep, M. B. Nguyen, V. D. Thao and N. N. Huy, Korean J. Chem. Eng., 2021, 38, 2034–2046 CrossRef CAS.
- D. Liu, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Small, 2019, 15, 1902291 CrossRef.
- Z. Jin, Q. Zhang, S. Yuan and T. Ohno, RSC Adv., 2015, 5, 4026–4029 RSC.
- M. Qiao, X. Wu, S. Zhao, R. Djellabi and X. Zhao, Appl. Catal., B, 2020, 265, 118587 CrossRef CAS.
- C. Guan, J. Jiang, S. Pang, X. Chen, R. D. Webster and T.-T. Lim, Chem. Eng. J., 2020, 387, 123726 CrossRef CAS.
- Z. Chen, S. Zhang, Y. Liu, N. S. Alharbi, S. O. Rabah, S. Wang and X. Wang, Sci. Total Environ., 2020, 731, 139054 CrossRef CAS.
- C. Hu, Y.-C. Chu, M.-S. Wang and X.-H. Wu, J. Photochem. Photobiol., A, 2017, 348, 8–17 CrossRef CAS.
- G. Mamba and A. K. Mishra, Appl. Catal., A, 2016, 198, 347–377 CrossRef CAS.
- E. L. Ribeiro, S. A. Davari, S. Hu, D. Mukherjee and B. Khomami, Mater. Chem. Front., 2019, 3, 1302–1309 RSC.
- Z. Öztürk, M. Filez and B. M. Weckhuysen, Chem. – Eur. J., 2017, 23, 10915–10924 CrossRef.
- N. Liu, P. Tao, C. Jing, W. Huang, X. Zhang, M. Wu, J. Lei and L. Tang, J. Mater. Sci., 2018, 53, 15051–15063 CrossRef CAS.
- T.-Y. Chen, L.-Y. Lin, D.-S. Geng and P.-Y. Lee, Electrochim. Acta, 2021, 376, 137986 CrossRef CAS.
- A. A. Yadav, S.-W. Kang and Y. M. Hunge, J. Mater. Sci.: Mater. Electron., 2021, 32, 15577–15585 CrossRef CAS.
- L. Yang, J. Liu, L. Yang, M. Zhang, H. Zhu, F. Wang and J. Yin, Renewable Energy, 2020, 145, 691–698 CrossRef CAS.
- C. Zhang, W. Chu, R. Jiang, L. Li, Q. Yang, Y. Cao and J. Yan, Catal. Lett., 2019, 149, 3058–3065 CrossRef CAS.
- A. Diallo, A. C. Beye, T. B. Doyle, E. Park and M. Maaza, Green Chem. Lett. Rev., 2015, 8, 30–36 CrossRef CAS.
- Y. Sun, J. Jiang, Y. Liu, S. Wu and J. Zou, Appl. Surf. Sci., 2018, 430, 362–370 CrossRef CAS.
- L. Liu, Y. Qi, J. Lu, S. Lin, W. An, Y. Liang and W. Cui, Appl. Catal., B, 2016, 183, 133–141 CrossRef CAS.
- C. Qi, X. Liu, J. Ma, C. Lin, X. Li and H. Zhang, Chemosphere, 2016, 151, 280–288 CrossRef CAS PubMed.
- L. Xue, L. Hao, H. Ding, R. Liu, D. Zhao, J. Fu and M. Zhang, Environ. Res., 2021, 201, 111618 CrossRef CAS.
- W. Li, Y. Zhang, Y. Liu, X. Cheng, W. Tang, C. Zhao and H. Guo, J. Taiwan Inst. Chem. Eng., 2019, 100, 56–64 CrossRef CAS.
- F. Hayati, S. Moradi, S. Farshineh Saei, Z. Madani, S. Giannakis, A. A. Isari and B. Kakavandi, J. Environ. Manage., 2022, 321, 115851 CrossRef CAS.
- M. Moradi, B. Kakavandi, A. Bahadoran, S. Giannakis and E. Dehghanifard, Sep. Purif. Technol., 2022, 285, 120313 CrossRef CAS.
- B. Liu, W. Song, H. Wu, Z. Liu, Y. Teng, Y. Sun, Y. Xu and H. Zheng, Chem. Eng. J., 2020, 398, 125498 CrossRef CAS.
- L. Zeng, S. Li, X. Li, J. Li, S. Fan, X. Chen, Z. Yin, M. Tadé and S. Liu, Chem. Eng. J., 2019, 378, 122039 CrossRef CAS.
- G. Fan, S. Yang, B. Du, J. Luo, X. Lin and X. Li, Environ. Res., 2022, 204, 112032 CrossRef CAS PubMed.
- S. Fadaei, M. Noorisepehr, H. Pourzamani, M. Salari, M. Moradnia, M. Darvishmotevalli and N. Mengelizadeh, J. Environ. Chem. Eng., 2021, 9, 105414 CrossRef CAS.
- L. T. Thao, T. V. Nguyen, V. Q. Nguyen, N. M. Phan, K. J. Kim, N. N. Huy and N. T. Dung, J. Environ. Sci., 2023, 124, 379–396 CrossRef.
- A. A. Isari, S. Moradi, S. S. Rezaei, F. Ghanbari, E. Dehghanifard and B. Kakavandi, Sep. Purif. Technol., 2021, 275, 119163 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- R. Yuan, L. Hu, P. Yu, H. Wang, Z. Wang and J. Fang, Chemosphere, 2018, 198, 204–215 CrossRef CAS PubMed.
- C. Cai, S. Kang, X. Xie and C. Liao, J. Hazard. Mater., 2020, 385, 121519 CrossRef CAS.
- X. Wang, J. Jiang, Y. Ma, Y. Song, T. Li and S. Dong, J. Colloid Interface Sci., 2021, 600, 449–462 CrossRef CAS.
- J. Jiang, X. Wang, C. Yue, S. Liu, Y. Lin, T. Xie and S. Dong, J. Hazard. Mater., 2021, 414, 125528 CrossRef CAS PubMed.
- N. T. Dung, N. H. Duc, V. T. Binh, V. D. Thao, M. B. Nguyen, L. V. Ngan and N. N. Huy, Sep. Purif. Technol., 2022, 285, 120358 CrossRef CAS.
- J. Luo, Y. Dai, X. Xu, Y. Liu, S. Yang, H. He, C. Sun and Q. Xian, J. Colloid Interface Sci., 2022, 610, 280–294 CrossRef CAS.
- N. T. Dung, L. T. Duong, N. T. Hoa, V. D. Thao, L. V. Ngan and N. N. Huy, Chemosphere, 2022, 287, 132141 CrossRef CAS.
- D. Feng, A. Soric and O. Boutin, Sci. Total Environ., 2020, 742, 140559 CrossRef CAS.
- P. Gupta and N. Verma, Chem. Eng. J., 2021, 417, 128029 CrossRef CAS.
- Y.-R. Lv, R.-K. He, Z.-Y. Chen, X. Li and Y.-H. Xu, J. Colloid Interface Sci., 2020, 560, 293–302 CrossRef CAS.
- S. Singh, V. Kumar and J. Singh, J. Environ. Chem. Eng., 2019, 7, 103098 CrossRef CAS.
- H. Lan, W. He, A. Wang, R. Liu, H. Liu, J. Qu and C. P. Huang, Water Res., 2016, 105, 575–582 CrossRef CAS.
- P. Alulema-Pullupaxi, L. Fernández, A. Debut, C. P. Santacruz, W. Villacis, C. Fierro and P. J. Espinoza-Montero, Chemosphere, 2021, 278, 130488 CrossRef CAS PubMed.
- N. Tan, Z. Yang, X.-B. Gong, Z.-R. Wang, T. Fu and Y. Liu, Sci. Total Environ., 2019, 650, 2567–2576 CrossRef CAS.
- P. Garcia-Muñoz, W. Dachtler, B. Altmayer, R. Schulz, D. Robert, F. Seitz, R. Rosenfeldt and N. Keller, Chem. Eng. J., 2020, 384, 123315 CrossRef.
- Q.-Y. Tang, M.-J. Yang, S.-Y. Yang and Y.-H. Xu, J. Hazard. Mater., 2021, 407, 124798 CrossRef CAS.
- M. H. Tran, H. C. Nguyen, T. S. Le, V. A. D. Dang, T. H. Cao, C. K. Le and T.-D. Dang, Environ. Technol., 2021, 42, 1155–1164 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Phung Thi Hong
Hanh
a,
Vu Dinh
Thao
a,
Le Viet
Ngan
*a,
Phung Thi Hong
Hanh
a,
Vu Dinh
Thao
a,
Le Viet
Ngan



 = 4.0–9.1 × 105 M−1 s−1), EtOH for both SO4˙− and HO˙ (
= 4.0–9.1 × 105 M−1 s−1), EtOH for both SO4˙− and HO˙ ( = 1.6–7.7 × 107 M−1 s−1; kHO˙ = 1.2–2.8 × 109 M−1 s−1), PheOH for both SO4˙− and HO˙ (
= 1.6–7.7 × 107 M−1 s−1; kHO˙ = 1.2–2.8 × 109 M−1 s−1), PheOH for both SO4˙− and HO˙ ( = 8.8 × 109 M−1 s−1; kPheOH–HO˙ = 6.6 × 109 M−1 s−1), FFA for both 1O2 and HO˙ (kFFA-1O2 = 1.2 × 108 M−1 s−1; kFFA-HO˙ = 1.5 × 1010 M−1 s−1), and p-BQ for O2˙− (
= 8.8 × 109 M−1 s−1; kPheOH–HO˙ = 6.6 × 109 M−1 s−1), FFA for both 1O2 and HO˙ (kFFA-1O2 = 1.2 × 108 M−1 s−1; kFFA-HO˙ = 1.5 × 1010 M−1 s−1), and p-BQ for O2˙− ( = 109 M−1 s−1).85 When adding EtOH, TBA, and PheOH, the Gly decomposition efficiency decreased to 66.9, 61.3%, and 56.28%, respectively. For p-BQ and FFA, the efficiencies decreased significantly to 43.78% and 38.27%, respectively, showing that p-BQ and FFA have a strong inhibitory effect on the PMS activation by Co3O4/g-C3N4. Thus, the role of ROS is in the order 1O2 > O2˙− > SO4˙− > HO˙, in which 1O2 and O2˙− play a major role in Gly decomposition by the Co3O4/g-C3N4/PMS system.
= 109 M−1 s−1).85 When adding EtOH, TBA, and PheOH, the Gly decomposition efficiency decreased to 66.9, 61.3%, and 56.28%, respectively. For p-BQ and FFA, the efficiencies decreased significantly to 43.78% and 38.27%, respectively, showing that p-BQ and FFA have a strong inhibitory effect on the PMS activation by Co3O4/g-C3N4. Thus, the role of ROS is in the order 1O2 > O2˙− > SO4˙− > HO˙, in which 1O2 and O2˙− play a major role in Gly decomposition by the Co3O4/g-C3N4/PMS system.


