
Heavy metal, organic matter, and disinfection byproduct release from drinking water pipe scales under stagnant conditions†
L.
Kurajica
 a,
M.
Ujević Bošnjak
*a,
A. S.
Kinsela
b,
J.
Štiglić
a and
T. D.
Waite
b
a,
M.
Ujević Bošnjak
*a,
A. S.
Kinsela
b,
J.
Štiglić
a and
T. D.
Waite
b
aCroatian Institute of Public Health, Rockefeller Street 7, 10000 Zagreb, Croatia. E-mail: magdalena.ujevic@hzjz.hr; Fax: +385 (0)1 46 83 009
bWater Research Centre, School of Civil and Environmental Engineering, University of New South Wales, Sydney, NSW 2052, Australia
First published on 30th November 2022
Abstract
Physico-chemical and microbiological processes occurring in water distribution systems (WDSs) can result in the formation of pipe scales which accumulate harmful metals. Irregular changes in either supply-water quality or physical disturbances can induce the mobilization and release of these metals. This study investigates morphological and physico-chemical characteristics of pipe scales from three different water supply zones in Croatia. Magnetite, siderite and goethite were the main constituents of pipe scales, with iron the most abundant metal across all three supply zones, followed by Al and Mn. Arsenic was present in only one pipe from the WDS supplied with elevated As-containing source water (30 to 50 μg L−1). Stagnation release experiments were used to explore the potential metal and organic matter (OM) release from pipe scales using water from the three water supply zones. Different release patterns were observed for the monitored metals (Fe, Al, Mn, As, Pb) with a close relationship with OM components indicative of the important role of OM in metal mobilization. Significant correlations enabled the division of pipe scales into three groups; with group 1 exhibiting a close relationship between Fe, Pb and As, group 2 exhibiting a close relationship between Fe, Al and Zn, and group 3 exhibiting a close relationship between Mn and sulfate. These relationships imply that the co-existing metals in scales were co-released into the water. Experiments involving the addition of humic acid to the pipe exhibiting high As showed an elevated release of As and Pb, implying a greater risk of their release if concentrations of humic-like OM in source waters increase over time.
Water impactHazardous materials like arsenic, lead, cadmium may accumulate in pipe scales. Changes in supply water quality could cause mobilization of scale materials into the system and cause water discoloration and pose a health risk to end users. This work investigates metal and disinfection byproducts release processes under different conditions. The aim was to help water supply utilities to better understand on-going issues associated with their systems and enhance water quality for end users. |
1. Introduction
Drinking water supply utilities are obligated to provide good quality water throughout the entirety of their water distribution system (WDS), including to the consumer's tap.1 However, treated water invariably contains particles, microorganisms, organic and inorganic matter.2 Physicochemical and microbiological process, often associated with the aforementioned components, can cause water quality deterioration within WDSs,3,4 especially over longer water retention times. Commonly occurring processes include nitrification,5 internal corrosion6 and biofilm growth,7 all of which can result in the formation of pipe scale. Pipe material, electrochemistry, chemical composition and microbiology of the bulk water are factors which have the highest influence on pipe scale formation.8 Although iron (Fe), manganese (Mn) and aluminium (Al) are the most common metal elements in such pipe scale deposits,1 more harmful components including arsenic (As), lead (Pb), copper (Cu), cadmium (Cd) and chromium (Cr) may also accumulate.9 All of the aforementioned materials can accumulate and stabilize in WDSs depending on antecedent water quality and environmental conditions.3,10–12However, irregular or periodic changes in supply-water quality (and velocity) can cause transitional effects, including the destabilization and mobilization of scale materials into the broader water supply system.1,13,14 Chemical destabilization, for example, may occur when pH, oxidation–reduction potential and ion composition in the supply water changes or, alternatively, when disinfection processes change.1 The release of certain metals can cause water discoloration12,15–17 and potentially pose a health risk to end users.9,18,19 Importantly, not all such transitional effects are detectable due to their periodic release profile, nor are they visible. For example, pathogenic microorganisms which are often detected in biofilms20–22 could present an undetected risk to public health upon scale/biofilm destabilization and release.
Organic matter (OM), which can also accumulate within WDS pipes, promotes biofilm growth and forms harmful disinfection byproducts (DBPs).23 Furthermore, the complexation of OM with inorganic components such as Fe, Mn, Al increases the solubility and mobility of oxidized forms of these metals in WDSs.24 Organic matter also plays an important role in the mobilization of As in groundwaters.25–30
The importance of controlling and monitoring water quality at both the exit of any water treatment plant (WTP) and at the consumers tap, to determine changes in the quality of water, is widely recognized. However, to obtain a more detailed understanding of WDS performance and safety, it is also necessary to investigate and characterize the presence of pipe scale. The scientific literature contains a range of detailed information on pipe scale characteristics, which is vital given their potential impact upon water quality for the consumer.8,31–35 However, it is equally important to determine how these pipe scales behave under different conditions and their potential to release harmful components into the water supply network. Indeed, release experiments are critical for determining the behavior of Fe, Mn, Al, As and other metals under differing/changing water conditions. An important component of this issue involves examination of the role of OM in metal release and DBP formation. While there have been some studies examining the effects of pH, alkalinity, chloride and sulfate concentrations on metals release,36–38 there have been only a limited number of studies of the impact of OM.38,39
This study investigates metal release processes from three pipe scales collected from different water supply zones in Croatia, each with specific source water characteristics. It also explores the role of OM on metal release and DBP formation in pipe scales. Critically, DBP formation potential stemming from biofilms which are associated with pipe scale is an emerging issue globally40,41 and it is important to explore their relationship under long retention time conditions. Additionally, attention is given to the co-release behavior of metals given the limited information currently available on this issue.36 The practical aim of this study was to help water supply utilities to better understand on-going issues associated with their WDSs and enhance water quality for end users.
2. Materials and methods
2.1. Study cases
Three pipe sections were collected from three different water supply zones in Croatia. Pipes 1 and 2 (Fig. 1a and b) were sourced from continental Croatia whereas pipe 3 was collected from coastal Croatia (Fig. 1c). Pipes 1 and 3 were composed of galvanized steel while pipe 2 was constructed from iron with all three pipes having been in service for approximately 30 years. Pipes 1 and 2 (P1 and P2) were collected from the WDS while pipe 3 (P3) was a part of a residential network. The pipe diameters were 5, 10 and 2 cm for P1, P2 and P3, respectively. Source waters for P1, P2 and P3 were groundwater, surface water, and brackish water, respectively. Traditional water treatment processes were used at all three sites, including coagulation, precipitation, filtration, and disinfection with sodium hypochlorite (NaClO). | ||
| Fig. 1 Appearance of corrosion scales on pipes internal surfaces, pipe 1 (a), pipe 2 (b) and pipe 3 (c). | ||
2.2. Scale sampling and preparation
The pipes were cut from the system and placed into sterile bags, before being transported to the laboratory and frozen. Pipe scales were collected by swabbing the inner pipe wall with a sterile spatula, after which the pipe scale samples were freeze dried (Labconco freeze dryer 7002030, Kansas City, MO, USA) and ground into fine particles using a mortar and pestle. The physicochemical characteristics of these powdered scale samples were determined as was their heavy metal content. Stagnation release experiments were also undertaken using the powdered scale samples with scale powders used for these experiments due to their relatively uniform morphology and particle size.362.3. Characterization of pipe scales
The X-ray diffraction (XRD) spectra of scale samples were measured using an X-ray diffractometer (Shimadzu XRD-6000, Kyoto, Japan) with a Cu-Kα source over a 2θ range from 3 °C to 70 °C at a scan rate of 2° min−1. The scanning electron microscope (SEM) images were obtained using a scanning electron microscope (Tescan Vega III Easyprobe, Brno, Czech Republic) coupled with a Bruker XFlash Detector 4010-M energy dispersive spectrometer (EDS) (Billerica, Massachusetts, USA). Samples were placed on Al-stubs using double-sided C-tape before being sputter coated with either Au or Pd prior to their analysis. Multiple particles were analyzed with representative images displayed in the manuscript.To determine total metal content, scale samples were digested in concentrated acid following the USEPA 3051A method,42 utilizing microwave digestion (Anton Paar multiwave go, Graz, Austria). The temperature of the samples was programmed to rise to 175 ± 5 °C in approximately 5.5 ± 0.25 min and remain at 175 ± 5 °C for another 4.5 min. Trace metals concentrations were determined using ICP-MS (Agilent 7900, Santa Clara, California, USA). Information on quality assurance and quality control can be found in the ESI† (Fig. S2).
2.4. Stagnation release experiments with and without humic acid addition
Tap water from the three WDSs where the pipes were collected, were sampled, analyzed and used for the laboratory experiments described below. Measured water quality parameters are presented in Tables S1 and S2.†To study the role of OM on pipe scale behavior, collected tap waters were amended with 2, 5, and 10 mg L−1 of humic acid (HA). These HA solutions were prepared from a 1 g L−1 stock solution using a commercial humic acid (FLUKA, Sigma Aldrich, lot: 1415-93-6), which involved dissolving 206.8 mg of humic acid in 1 L of aqueous solution of 4.0 g L−1 NaOH.43
Release experiments were initiated by weighing scale powder (1.7 g) into borosilicate glass beakers before adding 1 L of tap water from the corresponding WDS. In the HA-additive experiments, 1 L of corresponding tap water with an additional 2, 5, or 10 mg L−1 of HA was added. All beakers were protected from the light and all experiments were carried out in triplicate. HA-additive experiments were performed only with scale powder from pipes 1 and 2 as there was insufficient scale powder from pipe 3.
Preliminary experiments were performed to determine the mass of the scale powder, the volume of water and the release time required. The experimental duration was set at 96 hours, with samples collected after 0, 0.08 (5 min), 2, 4, 24, 72, and 96 h. The exception was the HA-additive experiment on pipe 2 which lasted for 240 h. Since water retention time can be longer than 96 h in some Croatian WDSs, we wanted to examine in at least one experiment whether retention time affects metal release significantly. Immediately prior to sampling, the beakers were gently mixed. To achieve representative and uniform samples, they were collected from the same position below the water surface.44 Samples for measuring “released dissolved” metals were filtered through 0.45 μm filter membranes and acidified to 1% v/v with high purity nitric acid. Conversely, samples for measurement of “released total” metals were not filtered, with the sampled aliquots subjected to microwave digestion as described previously.
2.5. Physico-chemical analysis of tap water and water from experiments
The parameters pH, oxidation–reduction potential and electrical conductivity (EC) were measured using a multi-parameter meter (HACH HQ 40D, Loveland, Colorado, USA). The concentrations of anions (F−, Cl−, NO3−, SO42−), cations (Na+, K+, Mg2+, Ca2+), chlorate (ClO3−) and chlorite (ClO2−) ions were determined using ion chromatography (Thermo Scientific Dionex DX 5000, Waltham, Massachusetts, USA) by HRN EN ISO 10304-4:2001. Alkalinity (HCO3−) measurements were performed by titration according to HRN EN ISO 9963-1:1998. Ion balances, calculated as 100 × (cations − anions)/(cations + anions), were usually better than ±5%. THMs (trichloromethane (TCM), tribromomethane (TBM), bromodichloromethane (BDCM), dibromochloromethane (DBCM)) were determined by gas chromatography (GC-MS Shimadzu TQ8040, Kyoto, Japan) according to HRN EN ISO 10301:2002 while the US EPA 552.3 method45 was used for determining HAAs (monochloroacetic acid (MCAA), dichloroacetic acid (DCAA), trichloroacetic acid (TCAA), monobromoacetic acid (MBAA) and dichloroacetic acid (DCAA)) (GC-ECD Thermo Fisher Scientific Trace 1300, Waltham, Massachusetts, USA). Further details on the methods used are available in Kurajica et al. (2020).46 A spectrofluorometer (Horiba Aqualog Jobin Yvonn, Kyoto, Japan) was used to characterize the OM. Dissolved organic carbon (DOC) was measured with a TOC Analyzer (Shimadzu TOC-LCSH FA E200) according to HRN EN 1484:2002. Total and dissolved trace metals concentrations were determined using ICP-MS (Agilent 7900, Santa Clara, California, USA). Samples for dissolved metals analysis were filtered using 0.45 μm PET filters (Chromafil, lot: 0.197).Fluorescence analysis was conducted on 0.45 μm filtered samples with all analyses carried out within 24 hours of sample collection. Excitation–emission matrices (EEMs) were obtained by scanning excitation wavelengths from 240 nm to 600 nm (5 nm increments) and emission wavelengths from 246.62 to 829.14 nm (5 nm increments) with 1.0 s integration times. Six models were built for the collected data using Eigenvector Solo software (Eigenvector Research Inc., Manson, WA, USA), one for every experiment and one for the samples of biofilm from three pipe scales. Components and maximum excitation and emission values for every model are presented in Table S3.† Components such as humic-like, tryptophan-like and tyrosine-like organic matter were variably present in the samples.47 The relative concentrations of all components were estimated from the measured fluorescence intensity.
Pipe scale samples for fluorescence measurements were prepared by adding scale powder into deoxygenated deionized water with the solid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :liquid ratio of 3 g:30 mL in serum bottles which were homogenized. Samples were filtered (0.45 μm Millipore PET) prior to being analyzed.29
:liquid ratio of 3 g:30 mL in serum bottles which were homogenized. Samples were filtered (0.45 μm Millipore PET) prior to being analyzed.29
2.6. Multivariate statistical analysis and thermodynamic equilibrium calculations
Statistica Version 9.1. (Stat.Sof.Inc., Tulsa, Oklahoma, USA) was used for statistical analysis. Spearman's correlation coefficient was used to analyze the correlation between the release of metals in the experiments. Principal component and classification analysis (PCCA) was used to assess the relationships between major ions concentrations, OM components and metals released from the corrosion scales. Thermodynamic equilibrium calculations were performed using the software package Visual MINTEQ ver. 3.1.48 The calculations were conducted to assess the propensity for mineral formation under differing conditions.3. Results
3.1. Physicochemical characterization of scales – morphology, elemental composition, and crystal structure
As evident in Fig. 1, deposits from pipes 1 and 3 had a red to brown color and were particularly thick. Conversely, deposits from pipe 2 were brown to black with the thinner layers. SEM/EDS analysis showed samples from pipe 1 were morphologically diverse (Fig. 2a), displaying a large range of particle sizes, with clearly defined edges and occasionally fine needle-like agglomerations on their surface with this latter material possibly dried OM. Additional SEM images of pipe scales 1, 2 and 3 are presented in Fig. S1.† Spectral analyses indicated that pipe 1 predominantly contained Fe, O, C, Si, Al and Zn (Fig. S2a†). Even though our EDS analysis did not detect As, which was unsurprising given the detection limit of the technique, analysis of the metal content by ICP-MS showed that this sample contained greater quantities of As (∼0.04%, refer to section 3.3) than samples from the other pipes. Needle-like structures in scales containing As(V) have been observed previously.35 | ||
| Fig. 2 SEM images of pipe scale 1 (a), pipe scale 2 (b) and pipe scale 3 (c). Note that the scale bars for pipes 1, 2 and 3 are 50, 100 and 50 μm, respectively. | ||
Samples from pipe 2 were morphologically more homogenous but exhibited varying degrees of agglomeration (Fig. 2b). The morphology was compact and did not exhibit any crystalline/angular features. EDS analysis showed that the sample consisted mostly of Fe, O, C, Si, Al and S (Fig. S2b†). A limited proportion of morphologically flat particles had different compositions, consisting mostly of Si and O with lesser amounts of C, Fe, Mg and Al, indicating the likely presence of silicates and minor carbonates.
The sample from pipe 3 was also morphologically homogenous and was, potentially, a combination of agglomerates or primary particles that were not crushed in the grinding process (Fig. 2c). The particles exhibited a mostly fluffy morphology, appearing to form from the agglomeration of needle-like particles. EDS analysis showed that the samples consisted mostly of Fe and O as well as lesser amounts of C and Zn and, in only one of the samples, Cl (Fig. S2c†).
XRD analysis of the pipe 1 scale (Fig. S3a†) showed that the sample predominantly contained magnetite (Fe3O4) and goethite (α-FeOOH) with the possible presence of small amounts of lepidocrocite (γ-FeOOH) and siderite (FeCO3). Pipe 2 was characterized by the presence of goethite, maghemite (γ-Fe2O3), lepidocrocite and silicon dioxide (Fig. S3b†). Pipe 3 predominantly contained magnetite, goethite and lepidocrocite as well as siderite in a greater amount compared to pipes 1 and 2 (Fig. S3c†). Semi-quantitative analysis showed that pipe scales 1 and 3 were magnetite dominated, while pipe scale 2 was goethite dominated. Similar phases have been identified in corrosion scales analyzed in the literature.32,35,37,49 Iron oxides including magnetite, goethite and lepidocrocite have strong affinities for trace metals whilst still being stable in WDSs.50 As previously mentioned, WDS-2 was fed with a surface water which typically contains high Cl and SO4 concentrations. However, the brackish water from WDS-3 contained the highest Cl and SO4 concentrations (Table S1†) and, as such, can be considered the most corrosive water source. Indeed, waters of this kind have been previously shown to form compact corrosion scales with high magnetite content.49 Mineral phases incorporating other metals such as As, Mn, Al and Pb were not detected, likely due to their lower crystallinity, non-structural incorporation (i.e., adsorbed only) and/or because of their lower abundance.
3.2. Metal content in pipe scales
The concentrations of metals in pipe scale samples are presented in Table 1. Iron was the most abundant element in all three pipe scale samples, accounting for 60, 51, and 56% of the scale mass in pipes 1, 2, and 3, respectively (Table 1). Iron is commonly observed to be the dominant metal in corrosion scales in WDSs, with trace heavy metals present as adsorption products or co-precipitates.32,51 Indeed, the next most abundant metal was Zn which accounted for a maximum of 4.4% of the scale mass. Greater concentrations of Zn were measured in pipes 1 and 3, which were composed of galvanized steel, indicating that the oxidation of zinc coatings may be responsible for the presence of Zn in the scale samples.51,52 Aluminium was the next most abundant metal after Fe in the non-galvanized iron pipe 2, where it accounted for 0.54% of the scale mass, whereas it was less abundant in pipes 1 and 3 at 0.21 and 0.03%, respectively. Aluminium in corrosion scales may be derived either from the source water or from Al-based coagulants used in water treatment processes.52,53 Given the elevated concentration of Al in the source water feeding WDS 2 (100.5 μg L−1), the Al-scale was not generated from water treatment processes in this instance. Aluminium has been shown to accumulate as both alumina (Al2O3) and gibbsite (Al(OH)3) in pipe scales.51 Manganese accounted for between 0.06 and 0.08% of the scale mass and was likely present as a result of Mn in source waters. Indeed, even very low concentrations in source waters can result in deposition on pipe wall surfaces as both disinfectants and microorganisms can oxidize dissolved Mn(II) to insoluble Mn(IV).12,54 Arsenic accounted for up to 0.04% of the scale mass in pipe 1 while Pb accounted for up to 0.05% of the scale mass in pipe 3. The higher abundance of Pb in pipe 3 is probably connected to the fact that this pipe was a part of the house network and had the smallest diameter. It could be that some part of the house network consisted of lead pipes. Concentrations of the remaining metals were generally lower, suggesting a lesser risk of their release to solution.| Metal | Pipe 1 | % | Pipe 2 | % | Pipe 3 | % |
|---|---|---|---|---|---|---|
| Fe (mg kg−1) | 604573 |
60 | 511197 |
51 | 557844 |
56 |
| As (mg kg−1) | 380 | 0.038 | 5 | 0.001 | 30 | 0.003 |
| Mn (mg kg−1) | 585 | 0.059 | 572 | 0.057 | 844 | 0.084 |
| Al (mg kg−1) | 2135 | 0.214 | 5382 | 0.538 | 291 | 0.029 |
| Zn (mg kg−1) | 4225 | 0.422 | 57 | 0.006 | 44351 |
4.435 |
| V (mg kg−1) | 40 | 0.004 | 23 | 0.002 | 14 | 0.001 |
| Cr (mg kg−1) | 94 | 0.009 | 52 | 0.005 | 191 | 0.019 |
| Co (mg kg−1) | 3.6 | 0.000 | 16 | 0.002 | 16 | 0.002 |
| Ni (mg kg−1) | 7.0 | 0.001 | 35 | 0.003 | 106 | 0.011 |
| Cu (mg kg−1) | 36 | 0.004 | 36 | 0.004 | 589 | 0.059 |
| Se (mg kg−1) | 0.1 | 0.000 | 0.1 | 0.000 | 1.4 | 0.000 |
| Sr (mg kg−1) | 13 | 0.001 | 9 | 0.001 | 51 | 0.005 |
| Mo (mg kg−1) | 17 | 0.002 | 10 | 0.001 | 23 | 0.002 |
| Cd (mg kg−1) | 0.5 | 0.000 | 0.05 | 0.000 | 0.5 | 0.000 |
| Sn (mg kg−1) | 1.0 | 0.000 | 2.1 | 0.000 | 19 | 0.002 |
| Sb (mg kg−1) | 2.9 | 0.000 | 0.6 | 0.000 | 10 | 0.001 |
| Ba (mg kg−1) | 98 | 0.010 | 53 | 0.005 | 474 | 0.047 |
| Pb (mg kg−1) | 170 | 0.017 | 6.3 | 0.001 | 462 | 0.046 |
| U (mg kg−1) | 3.4 | 0.0003 | 0.3 | 0.00003 | 3.8 | 0.0004 |
3.3. Organic matter characterization in pipe scales
PARAFAC modeling showed that OM in the three collected pipe scale samples consisted of humic substances (humic-like peak) and protein-like substances (tyrosine-like B1 and tyrosine-like B2 peaks).47,55 Tyrosine-like components were most prevalent in each sample (∼70%) while the humic-like component was present in smaller proportions (∼30%) (Fig. 3). Previous research of a similar nature has shown that the biofilm portion of pipe scale consists mostly of protein-like components (∼90%)40 however these measurements were not made on samples from real WDSs.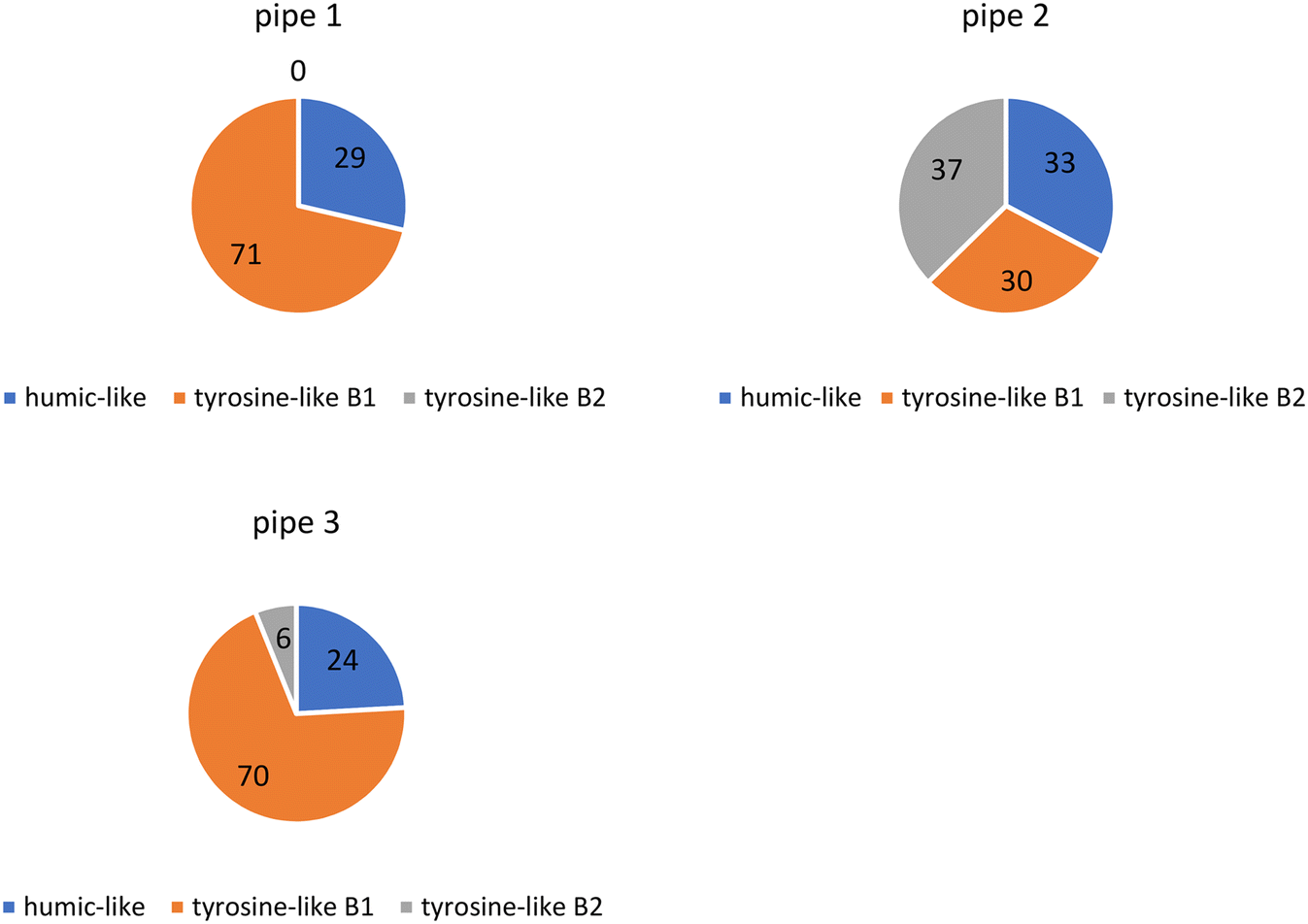 | ||
| Fig. 3 The percentage of PARAFAC components in three pipe scale samples. | ||
3.4. Release potential of metals and changes in composition of OM and DBP concentrations under stagnation water conditions
Basic physicochemical parameters, as well as DBP, OM, and metal concentrations were monitored under stagnation conditions across the 96 h experimental period (Table S4†). As iron was the most abundant metal in all three scales, it was also found to have the highest concentrations in the “released dissolved” samples, as expected. “Released dissolved” Fe concentrations increased slowly for the first 24 h of the experiment, after which they started to decrease (Fig. 4a). A similar observation was made by Tian et al. (2021).36 The initial increase could be the result of the gradual release of more recalcitrant forms of Fe in the scale. Additionally, released Fe can form secondary Fe(III) phases which could explain the subsequent decrease in Fe.57 Although XRD analyses were also conducted after the experiment, the diffractogram showed similar mineral content to that performed at the start of the experiment (Fig. S3d†) which, given the sensitivity of the instrument, was unsurprising. pH increased over the exposure time from 7.5 to 7.9 while ORP decreased from 495 to 403 mV (Table S4†). As observed in previous studies, iron was mainly released during the initial period of exposure.37,58,59 “Released dissolved” Al behaved similarly to Fe in the experiment with pipe 1 (Fig. 4b), increasing for the first 24 h, before declining. Previous research has identified that increased flow velocity and other hydraulic perturbations have significant impacts on the release and deposition of particulate Al.60 Our experiments showed that the release of Al also occurs under stagnant conditions. “Released dissolved” Mn concentrations increased slowly for the first 72 h, after which they appeared to reach quasi-equilibrium (Fig. 4c). Due to the elevated historical accumulation of As in WDS 1, As release was only evident from pipe scale 1 (Fig. 4d), which showed concentrations of dissolved As decreasing over time. Dissolved Pb concentrations rapidly decreased over the first two hours of the experiment (Fig. 4e), suggesting precipitation or adsorption of this metal. While many factors can influence dissolved and particulate Pb concentrations in WDSs, experimental data has shown that changes in water flow resulted in the highest increases in dissolved and particulate Pb.61 A slight (overall) increase in sulfate concentration was observed from pipe 1, as was the case for all three WDS scale materials (Fig. 4f).
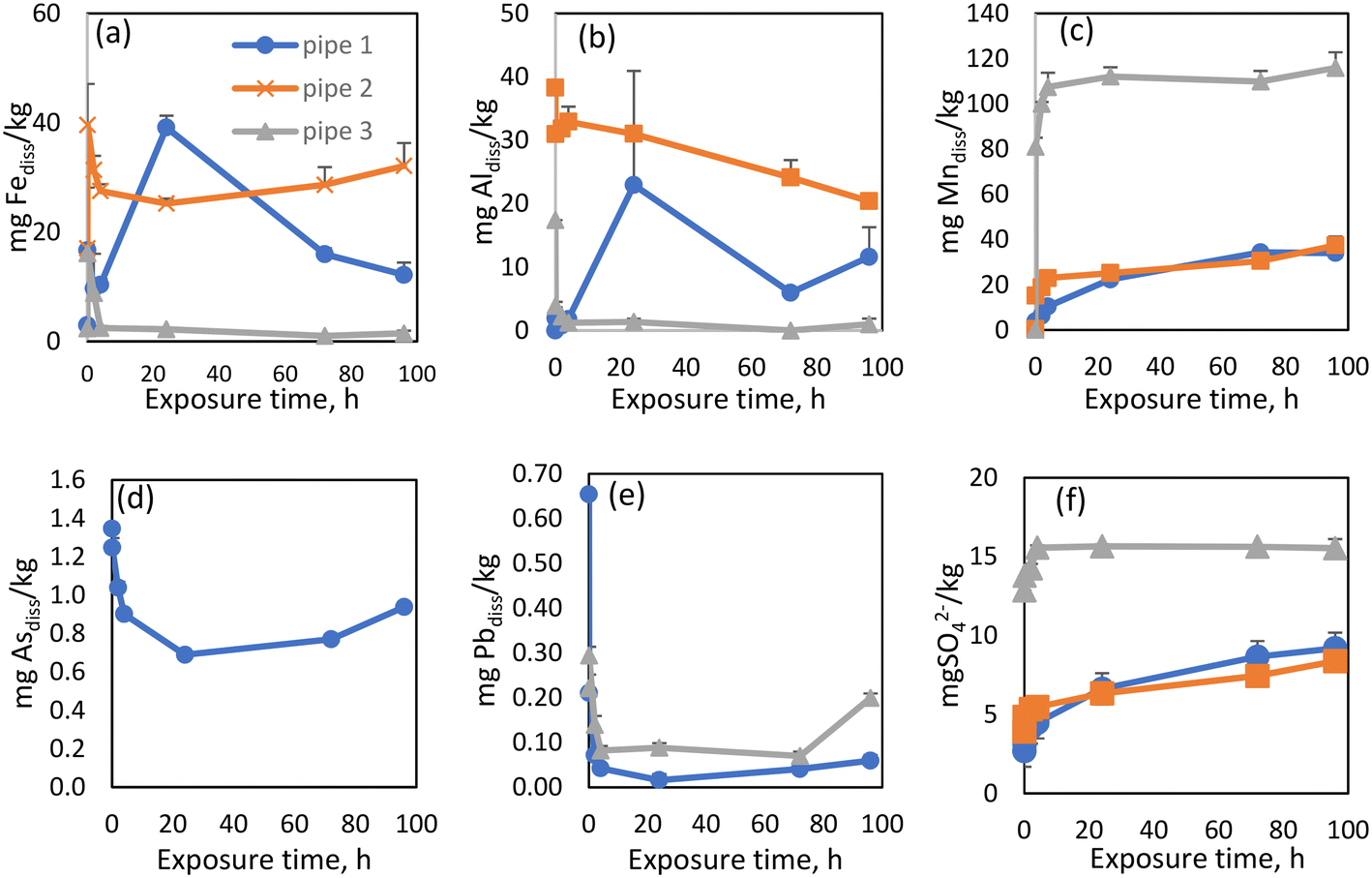 | ||
| Fig. 4 A comparative study of release rates between pipes for released dissolved Fe (a), Al (b), Mn (c) As (d), Pb (e) and SO4 ions (f). | ||
Monitoring the OM composition during the exposure time was performed as previous studies have shown that OM could play a role in controlling the biostability of water.62 The OM released from pipe 1 consisted of four humic-like components (Table S3†), whose intensities were stable during exposure time (Fig. S4a†). OM is a well-known precursor of DBPs such as trihalomethanes and haloacetic acids63,64 and a strong correlation was previously found between protein-like OM and THMs.65 Total trihalomethanes (TTHM) and total haloacetic acids (THAA) were measured at the beginning and end of the release experiments (Table S4†). Concentrations of TTHM and THAA in WDS 1 were initially 0.5 μg L−1 and 4 μg L−1, respectively. After the release experiment was complete, the TTHM concentrations were below the LOQ during, while THAA concentrations remained stable (∼3 μg L−1). The concentrations of both classes of DBPs were lower than expected based on our previous research in which we examined this WDS in greater detail.14 However, the tap water used for these current experiments was sampled during winter, when a lower concentration of DBPs was expected.66
The OM released from pipe scale 2 consisted of both humic-like and protein-like substances (tryptophan- and tyrosine-like) (Table S3†). Humic-like substances were more prevalent in this case with the intensities of all four components initially increasing after the start of the experiment, before decreasing within 4 h (Fig. S4†).
Concentrations of TTHM and THAA were below the LOQ and 33 μg L−1, respectively. During the experiment TTHM concentrations remained below LOQ while THAA concentrations increased marginally to 36 μg L−1 (Table S4†).
Previous research has demonstrated particulate Mn and Al co-release behavior and that these components were more labile with greater tendency than iron to be released back into the water.70 Our research showed the co-existence and linear co-release of Mn and SO4 in all three experiments (R2 = 0.98/0.89/0.86, p < 0.05 for WDS1/WDS2/WDS3) (Fig. S5†). We have also found the linear co-release of Fe and As (R2 = 0.99, p < 0.05), Al and As (R2 = 0.85, p < 0.05) and Pb and As (R2 = 0.88, p < 0.05) from pipe scale 1 (Fig. S6†).
The OM released from pipe 3 consisted of both humic (humic-like 1, 2, 3) and protein-like substances (tryptophan-like) (Table S3†) both of which demonstrated considerable variability over the experimental time course (Fig. S4c†). The WDS source water contained very low levels of all four components initially, before increasing upon exposure to the pipe scale, peaking from between 0.08 and 4 hours. The concentrations of TTHM and THAA were relatively stable across the release experiment, ranging between 2 and 3 μg L−1 (Table S4†). Even though greater quantities of OM were released from pipe scale 3 (compared to scale 2), the concentrations of HAAs were substantially lower, implying that other factors aside from OM concentration determines the quantities of DBPs generated.
Spearman's correlation coefficient was also used to identify the relationships between metals and to explore the co-release potential of these elements with stagnation time. Significant correlations were found between total As and Fe, dissolved As and dissolved and total Pb. Dissolved Al concentrations correlated with dissolved Fe and dissolved Zn concentrations, whereas dissolved Mn concentrations correlated with dissolved Pb and As concentrations.
The PCCA generated from all measured components revealed that two factors explained 80% of the total data set variance (Fig. 5a), with the different pipe scales positioned within discrete areas of the PCCA (Fig. 5b).
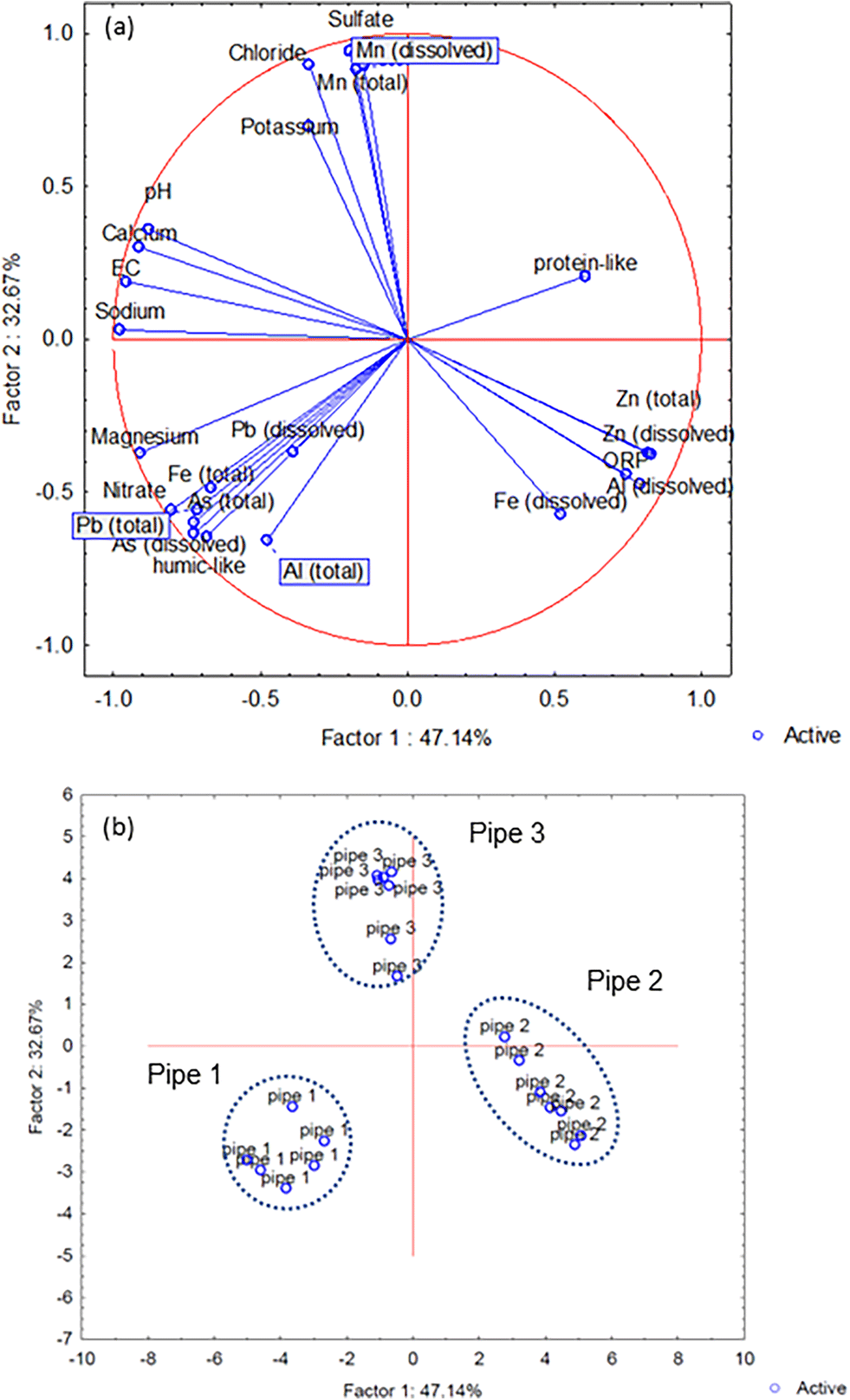 | ||
| Fig. 5 Principal component and classification analysis (PCCA) of metal concentrations, PARAFAC components, major ions, and physico-chemical parameters for three experiments together. Position of measuerd parameters (a) and pipe scales (b). | ||
3.5. Effect of humic acid on metal release
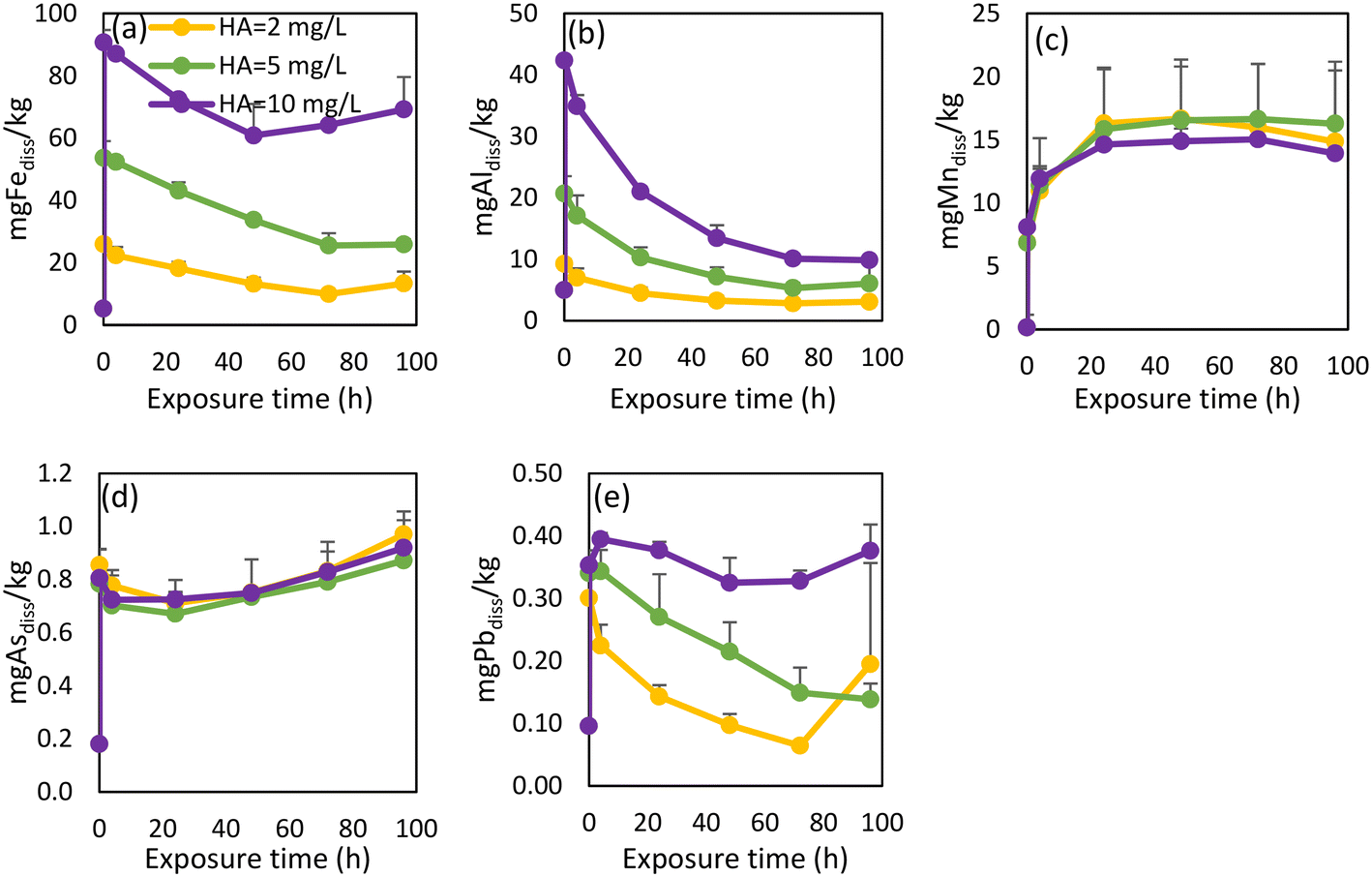 | ||
| Fig. 6 Release rates for released dissolved Fe (a), Al (b), Mn (c), As (d), Pb (e) from pipe scale 1 after additional humic acid concentrations were added to WDS 1 source water. | ||
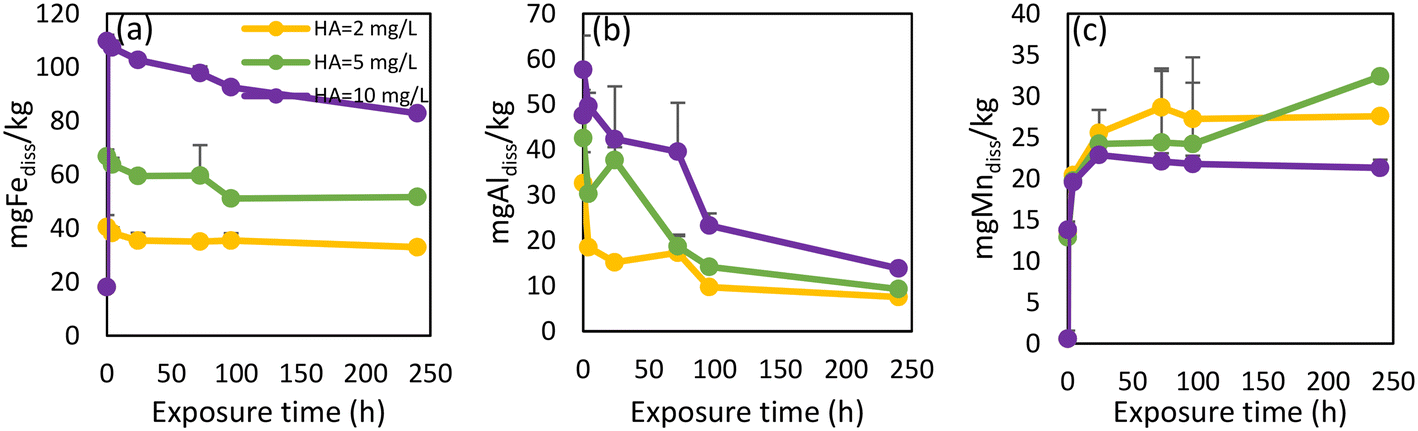 | ||
| Fig. 7 Release rates for released dissolved Fe (a), Al (b) and Mn (c) from pipe scale 2 after additional humic acid concentrations were added to WDS 2 source water. | ||
The intensities of OM PARAFAC humic-like components increased proportionally in response to the additional HA (Fig. S8†). An initial increase of intensities was observed, after which all component intensities started to decrease. A possible explanation for this gradual declining intensity was that the HA complexed with metals or adsorbed onto particulates,71 a point which would also explain the observed decrease of dissolved metal concentrations.
3.6. Comparison of the release amounts measured in the stagnation experiments to the total concentrations measured in pipe scales
The proportional release of individual elements from each pipe scale under stagnant conditions was calculated by comparing dissolved and total metal concentrations (measured by digestion), to give an approximate indication of element release susceptibility (Table S7†). Even though Fe was the most abundant metal present under stagnant conditions for all three pipe scales, its proportional release was relatively low, with only 0.05 to 0.19% of Fe in the scale released. Indeed, all other measured metals were released from the pipe scale in greater proportional quantities than Fe. Trace metals including V, Mo, Se and Cd, which have a low abundance in pipe scales, were released to a far greater degree under stagnant conditions. This would logically suggest that the adsorbed components of pipe scale are released to a greater extent compared to the components which make up the primary scale material (i.e., iron oxides).In a similar manner, the percentages of released metals under different HA concentrations were examined, with the results from pipe scale 2 presented in Table S8.† It was clear that the addition of HA increased the release of all metals from pipe scale 2, with the largest proportional increase evident for Fe.
Summary of the experiments major findings under stagnant conditions with and without addition of HA were presented in Fig. S9.†
4. Discussion
4.1. Release of metals, OM and DBPs from pipe scales under stagnation conditions with tap water
The three pipes examined here were comprised of similar materials (galvanized iron) and were of similar age (∼30 years). Despite their differing diameters (2–10 cm) and markedly different source water chemistry (surface, ground, and brackish origins), mineral phase analysis of the internal pipe scale showed very similar composition with iron oxides/oxyhydroxides predominating in all three pipes (Fig. S3†). Exploring mineral phases is critical to better understand the accumulation and release mechanisms occurring in WDSs. An analysis of the pipe scale metal content showed predictable results, with Fe having the highest content in all three pipes, followed by Al and Mn. Arsenic was present in higher concentrations only in scales from pipe 1 (Table 1), attributable to the supply by historical source waters containing elevated quantities of this harmful constituent. The differences in content of harmful substances in pipe scales is the likely result of interactive impact of water composition, pipe material and hydraulic conditions.51 One previous study found that pipe material had an important impact on the accumulation of Fe, Zn and Ca in pipe scales, while water composition influenced the content of Si, Al and S, whereas hydraulic conditions influenced the concentrations of C and Ca.51 In our study, pipe 1 and 2 composition (galvanized steel) likely contributed to the higher concentrations of Zn. Water from WDS 1 containing higher As concentrations caused higher As concentrations only in pipe 1. Water from WDS 2 contained the highest Al concentration and caused the highest Al concentrations in pipe scale 2.Iron was the most abundant metal released from all three pipe scales under stagnant conditions, which was expected considering their composition. Among all measured elements, only Mn (in pipe scale 3) reached quasi-equilibrium over the experimental time course. Three stages of Mn release were observed, including an initial rapid release, a subsequent slower release, followed by attainment of a quasi-equilibrium which was maintained until the experiment conclusion with this behavior similar to that observed by Tian et al. (2021).36 A similar trend of Mn concentrations under stagnant conditions was observed by Li et al. (2020)37 in studies of scale taken from pipes transporting riverine supplied waters. Such a marked increase in Mn concentrations suggests that longer stagnation times may translate to even greater increases in Mn release, causing on-going water quality and health issues for these WDSs.37 Previous research have shown that the seasonal variations in water temperature could influence metal release. For example, higher water temperature increased the release of Fe, Zn and Pb, while not affecting the release of Mn.36 Although the influence of water temperature on metal release was not monitored in this study, it should form part of future investigations.
A very similar increasing concentration trend was observed between Mn and SO4 ions, especially in WDSs 2 and 3. Anions such as SO4 tend to adsorb to iron oxides72 and could have an impact on the release of Mn and Fe from pipe scales.73,74 The impact of SO4 concentrations was also confirmed with statistical analysis, with linear correlation observed between Mn and SO4 concentrations in experiments from all three pipe scales (Fig. S5†).
The analysis of OM composition showed that water from the WDSs and that released from pipe scales under stagnant conditions consisted of humic-like and protein-like components (Table S3†). Research has shown that iron oxides/oxyhydroxides (including magnetite, goethite and maghemite) have the capacity to adsorb OM,62 which may occur via anion exchange, ligand exchange, surface complexation, hydrogen bonding and/or hydrophobic interactions.75 Previous research into biofilms (a component of pipe scale) showed that the OM was mostly comprised of protein-like material, with a lesser abundance of humic-like components,40 a point confirmed by our OM analysis of pipe scales. The composition of OM was very similar in all three pipe scale samples (Fig. 3). During the stagnation experiments, humic-like OM was preferentially released from scale material, despite being the minor component, indicative of its more facile release from pipe scales.
The analysis of source waters revealed that DBP concentrations were very low in WDSs 1 and 3 (Table S1†) with most insight to be gained from WDS 2. WDS 2 is supplied by surface water, which is comparatively rich in protein-like OM with these organic moieties known to produce larger amounts of DBPs.76–78 DCAA and TCAA were found to be the dominant HAAs (Table S1†), in support of our previous research which showed a strong correlation between the tryptophan-like OM component and TCAA concentrations.14 Li et al. (2020)40 have also found that tryptophan-like substances play a major role in producing DBPs derived from biofilms. The ∼10% higher HAAs concentrations measured during the experiment with pipe scale 2 (compared to HAAs levels in the tap water) were probably related to the release of OM from scale. The results suggest that OM from source waters could be continuously accumulated in biofilms and pipe scales through biological reactions including biotransformation and biosorption of OM.79,80
Interpretation of the Spearman's correlation coefficients and PCCA analyses indicated the metals released from different pipe scales under stagnant conditions could be divided into three groups: WDS 1 – Fe, Pb and As, WDS 2 – Fe, Al and Zn, and WDS 3 – Mn and SO4. Metals within these groups both co-existed in the WDSs and were co-released to the bulk water from scales under stagnation. The strongest co-release correlation was identified in group 3, between Mn and SO4. Previous research exploring the co-release of metals from pipe scales found the highest correlation between Mn and Ca (ref. 36) which we also identified in experiment 2 (R2 = 0.65) (Fig. S5†). Calcium often exists as calcite (CaCO3) in the WDSs and has been suggested to play a role in Mn release.36 The role of CaCO3 in regulating Mn mobility due to its reactive surface81 remains to be confirmed in our system with empirical data not identifying a presence of calcite.
Previous research has shown that OM plays a role in altering As mobility in groundwaters via the complexation of As by humic-like substances, competitive sorption and/or electron shuttling reactions.25,82 Arsenic has also been shown to form ternary complexes with Fe and OM in sediments which could also exert an influence on As enrichment in groundwaters.30 Aside from humic-like OM, protein-like OM could also participate in As mobilization by triggering reductive dissolution of Fe oxides by Fe-reducing bacteria.30 Our statistical analyses showed a strong relationship between As and humic-like OM, as well as similar release patterns between Fe and As. Collectively, these findings suggest that OM (humic substances), along with Fe, have important roles in the accumulation and release of As in this WDS. Direct evidence for this association is difficult to produce with the techniques used in this study. XRD analysis of pipe 1 did not show any As mineral phases, which was expected given the As content (0.4%). Arsenic was most likely adsorbed to iron-based corrosion products9 rather than (co-)precipitated as solid As minerals.
Despite Mn concentrations in all three WDS being very low (≤1 μg L−1, Table S2†), Mn was found to accumulate in the WDS pipe scale. Clearly, elevated Mn concentrations in source waters were not necessary for Mn buildup in pipe scale with scale in pipe 3 containing the most Mn despite having the lowest source concentration. Interestingly, the stagnation experiments also revealed that Mn in pipe scale 3 had the highest tendency to be released. One explanation for this phenomenon may relate to the OM form as recent research has shown that protein-like OM can mobilize Mn to a greater extent than humic forms.83 This may mean that Mn could pose a potential problem in WDS 3, especially at the end of the network, where the retention times are longer, and the consumption of water is lower, and protein-like OM is prevalent, as per the evidence from our pipe scale characterization from WDS3 (Fig. 3). As such, maintaining protein-like OM at lower levels in waters entering WDSs could control the unwanted accumulation and release of Mn. However, maintaining biological stability in the WDS could also have an important role in controlling Mn concentrations.
4.2. Effect of humic acid on metal release from pipe scales
The impact of increased OM on metal release from WDS pipe scales has not been widely explored. Since an increase in OM concentration and changes in OM composition are a possibility as a result of many different factors including climate change,77 it is important to understand how such conditions will affect metal mobilization in WDSs. Our findings show that even a small increase in HA concentration caused the release of As from pipe scale 1. Although the accumulation of As in pipe scale occurred through the supply of legacy elevated As-containing source water, contaminant release may still occur well after treatment processes have improved when alterations to the composition of water entering this WDS cause transitional effects.1 In our previous study where we monitored how different compositions of surface water and groundwater in this WDS affected metal concentrations, we noted that changes in source water composition caused such transitional effects, inducing the release of Fe, Mn, Al and As.14 However, these changes did not cause Pb release. Lead concentrations in source waters were very low (below 1 μg L−1) (Table S2†). Therefore, the elevated Pb content in pipe scale 1 (170 mg kg−1, Table 1) is surprising. Similar research on metal concentrations in 58 corrosion scale samples from the literature showed much lower Pb concentrations, with relative abundance lower than 0.0015%.49 Under stagnant conditions using water unamended with HA, a pulse of Pb was released initially, as has been noted previously.84 However, under higher HA concentrations, small amounts of Pb were released over longer periods (Fig. 6e). Both instances have unwanted safety implications. Although Pb leaching into drinking water is a greater danger in WDSs containing lead pipes, the transition to another water source with high chloride to sulfate mass ratios and/or elevated HA concentrations are alternative scenarios requiring consideration.Previous research has explored the influence of water temperature, pH, alkalinity and SO4 concentrations on Mn release from pipe scales, with SO4 ions and alkalinity having the most significant effects on the release of Mn.37,44 The impact of SO4 concentrations upon Mn release has also been demonstrated in this paper (Fig. S5†). However, the influence of OM on Mn mobilization or release from pipe scales has yet to be adequately addressed in the scientific literature. Based on our results, adding HA in different concentrations did not cause either more extensive or more rapid release of Mn (Fig. 6c and 7c). Abdelrady et al. (2020) explored the impact of OM on Fe, Mn and As mobilization during river bank filtration and found that humic substances increased the release of Fe and Mn into the filtrate water while elevated concentrations of tyrosine-like OM induced additional As release.83
Overall, these results, summarized at Fig. S9,† emphasize the significant role of HA-like OM in the mobilization of accumulated metals on the inner walls of pipes and highlight the importance of monitoring OM in WDSs.
5. Conclusions
Three pipes exhibiting the presence of corrosion scale were collected from different WDSs across Croatia with each of these WDSs supplied with markedly different source waters (i.e., ground, surface, and brackish waters).Pipe scale characterization showed that all three samples consisted predominantly of iron oxides and carbonates, specifically magnetite, goethite and siderite. Iron was the most abundant metal in all three pipe scales followed by Al and Mn. Arsenic was found in higher concentrations exclusively in pipe 1 due to the supply of elevated-As waters in preceding decades to WDS 1. Conversely, elevated quantities of Al and Mn were found in all three pipe scales despite these elements being present in low concentrations in WDS source waters. This confirms that certain metals, such as Al and Mn, have a propensity to accumulate in pipe scale irrespective of their feed concentration.
Under stagnant conditions, the metals Fe, Al, Mn, As, and Pb showed different release patterns in the absence of additional HA. Fe, Al, As and Pb showed co-release behavior from pipe scale 1, which was dominated by magnetite mineralogy. Manganese showed co-release behavior with SO4 ions in all three experiments confirming the strong association and mobilization potential between the two elements. The highest release of Mn occurred from pipe scale 3 with this observation implying a potential problem for this WDS, especially with longer stagnation times. This was also found to be the case for Pb in pipe scale 3.
While the OM present in all pipe scale samples was predominantly tyrosine-like, under stagnant conditions the more minor humic-like OM component was preferentially released. Furthermore, the intensities of both humic- and protein-like OM PARAFAC components increased after scale from pipes 2 and 3 was subjected to stagnant water conditions, showing the transience of multiple OM forms in WDSs. These results suggest that there is a low risk of DBP release from pipe scale in WDSs 1 and 3 in areas with longer retention times. However, an increase in DBP concentrations, especially HAAs, is more likely to occur in the WDS 2 network.
Numerous correlations were observed between OM and metal concentrations indicative of potential co-release and interactive behavior in pipe scale materials. More specifically, the correlations partitioned into three groups. The first group included Fe, Al and Zn, the second group Fe, Pb and As, and the third group Mn and SO4. These groups were specific to individual WDSs and were presumably a function of the source water composition, pipe material and hydraulics in the system.
The presence of additional HA (2–10 mg L−1) accelerated the release of all monitored metals from pipe scales 1 and 2. Most importantly, HA increased the release of the particularly harmful metals As and Pb from scale pipe 1 which contained elevated concentrations of these contaminants. These results imply that even slight changes in the composition of OM in source waters could cause the release of accumulated metals in pipe scale and lead to possible health problems for consumers. Tryptophan- and tyrosine-like OM may also play a role in metal release, and this is a topic certainly worthy of further study.
Although the number of individual pipes examined in this study was limited and further replication is required to confirm our findings, they nonetheless provide useful insight into a range of possible issues associated with the release of harmful components from pipe scale. It is evident from this study that the role of OM, particularly HA components, is an important factor in the kinetics of metal release from pipe scale and should be considered by water utilities when assessing possible changes to source water composition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Croatian Science Foundation under the project number [UIP-2017-05-3088]. The authors would like to thank their colleagues from the Croatian Institute of Public Health, Department for Water Safety and Water Supply and Unit for Metals and Metalloids for their help with sample collection and analysis. We would also like to thank colleagues from the water supply companies for their help with sampling, providing pipes and information about their water supply system.References
- G. Liu, Y. Zhang, W. J. Knibbe, C. Feng, W. Liu, G. Medema and W. van der Meer, Water Res., 2017, 116, 135–148 CrossRef CAS PubMed.
- E. I. Prest, F. Hammes, M. C. M. van Loosdrecht and J. S. Vrouwenvelder, Front. Microbiol., 2016, 7, 1–24 Search PubMed.
- I. J. H. G. Vreeburg and D. J. B. Boxall, Water Res., 2007, 41, 519–529 CrossRef CAS PubMed.
- G. Liu, J. Q. J. C. Verberk and J. C. Van Dijk, Appl. Microbiol. Biotechnol., 2013, 97, 9265–9276 CrossRef CAS PubMed.
- J. M. Regan, G. W. Harrington, H. Baribeau, R. De Leon and D. R. Noguera, Water Res., 2003, 37, 197–205 CrossRef CAS.
- R. Renner, Environ. Sci. Technol., 2008, 42, 4240 CrossRef PubMed.
- T. Schwartz, S. Hoffmann and U. Obst, J. Appl. Microbiol., 2003, 95, 591–601 CrossRef CAS PubMed.
- J. Liu, H. Chen, L. Yao, Z. Wei, L. Lou, Y. Shan, S. D. Endalkachew, N. Mallikarjuna, B. Hu and X. Zhou, J. Hazard. Mater., 2016, 317, 27–35 CrossRef CAS PubMed.
- D. A. Lytle, T. J. Sorg and C. Frietch, Environ. Sci. Technol., 2004, 38, 5365–5372 CrossRef CAS PubMed.
- G. Liu, G. L. Bakker, S. Li, J. H. G. Vreeburg, J. Q. J. C. Verberk, G. J. Medema, W. T. Liu and J. C. Van Dijk, Environ. Sci. Technol., 2014, 48, 5467–5476 CrossRef CAS PubMed.
- K. C. Makris, S. S. Andra and G. Botsaris, Crit. Rev. Environ. Sci. Technol., 2014, 44, 1477–1523 CrossRef CAS.
- G. H. Khoe and T. D. Waite, Environ. Technol. Lett., 1989, 10, 479–490 CrossRef CAS.
- L. Kurajica, M. Ujević Bošnjak, A. S. Kinsela, J. Štiglić, T. D. Waite, K. Capak and Z. Pavlić, Sci. Total Environ., 2021, 762, 144159 CrossRef CAS PubMed.
- L. Kurajica, M. Ujević Bošnjak, A. S. Kinsela, M. Bieroza, J. Štiglić, T. D. Waite, K. Capak and Ž. Romić, Chemosphere, 2022, 292, 133406 CrossRef CAS PubMed.
- D. Li, Z. Li, J. Yu, N. Cao, R. Liu and M. Yang, Appl. Environ. Microbiol., 2010, 76, 7171–7180 CrossRef CAS.
- F. Yang, B. Shi, Y. Bai, H. Sun, D. A. Lytle and D. Wang, Water Res., 2014, 59, 46–57 CrossRef CAS PubMed.
- M. Edwards, S. Jacobs and R. J. Taylor, J. - Am. Water Works Assoc., 2000, 92, 72–82 CrossRef CAS.
- D. A. Lytle, T. J. Sorg, M. Christy and W. Lili, J. - Am. Water Works Assoc., 2010, 102, 87–98 CrossRef CAS.
- M. Hanna-Attisha, J. LaChance, R. C. Sadler and A. C. Schnepp, Am. J. Public Health, 2016, 106, 283–290 CrossRef PubMed.
- M. Pryor, S. Springthorpe, S. Riffard, T. Brooks, Y. Huo, G. Davis and S. A. Sattar, Water Sci. Technol., 2004, 50, 83–90 CrossRef CAS.
- L. Chaves Simões and M. Simões, RSC Adv., 2013, 3, 2520–2533 RSC.
- J. O. Falkinham, A. Pruden and M. Edwards, Pathogens, 2015, 4, 373–386 CrossRef PubMed.
- J. Yu, D. Kim and T. Lee, Water Sci. Technol., 2010, 61, 163–171 CrossRef CAS PubMed.
- M. P. Ginige, J. Wylie and J. Plumb, Biofouling, 2011, 27, 151–163 CrossRef CAS PubMed.
- N. Mladenov, Y. Zheng, M. P. Miller, D. R. Nemergut, T. Legg, B. Simone, C. Hageman, M. M. Rahman, K. M. Ahmed and D. M. Mcknight, Environ. Sci. Technol., 2010, 44, 123–128 CrossRef CAS.
- H. M. Anawar, S. M. Tareq and G. Ahmed, Physics and Chemistry of the Earth, Parts A/B/C, 2013, 58–60, 49–56 CrossRef.
- P. Sharma, J. Ofner and A. Kappler, Environ. Sci. Technol., 2010, 44, 4479–4485 CrossRef CAS PubMed.
- H. V. Kulkarni, N. Mladenov, K. H. Johannesson and S. Datta, Appl. Geochem., 2017, 77, 194–205 CrossRef CAS.
- H. Guo, X. Li, W. Xiu, W. He, Y. Cao, D. Zhang and A. Wang, J. Hydrol., 2019, 571, 448–459 CrossRef CAS.
- W. Liu, Y. Wang, J. Li, K. Qian and X. Xie, Environ. Pollut., 2020, 262, 114305 CrossRef CAS PubMed.
- T. L. Gerke, J. B. Maynard, M. R. Schock and D. L. Lytle, Corros. Sci., 2008, 50, 2030–2039 CrossRef CAS.
- C. Y. Peng, G. V. Korshin, R. L. Valentine, A. S. Hill, M. J. Friedman and S. H. Reiber, Water Res., 2010, 44, 4570–4580 CrossRef CAS PubMed.
- H. Sun, B. Shi, D. A. Lytle, Y. Bai and D. Wang, Environ. Sci.: Processes Impacts, 2014, 16, 576–585 RSC.
- M. Li, Z. Liu and Y. Chen, Water, 2018, 10, 19–21 Search PubMed.
- N. He, Y. Tian, C. Liu, W. Zhao, R. Liu and J. Huang, Chemosphere, 2021, 269, 129396 CrossRef CAS PubMed.
- Y. Tian, J. Li, S. Jia and W. Zhao, Chemosphere, 2021, 267, 129270 CrossRef CAS PubMed.
- M. Li, Y. Wang, Z. Liu, Y. Sha, G. V. Korshin and Y. Chen, Water Res., 2020, 175, 115675 CrossRef CAS PubMed.
- L. Pan, G. Li, J. Li, J. Gao, Q. Liu and B. Shi, Sci. Total Environ., 2022, 806, 150549 CrossRef CAS PubMed.
- C. Y. Peng, J. F. Ferguson and G. V. Korshin, Water Res., 2010, 44, 4570–4580 CrossRef CAS PubMed.
- L. Li, Y. Jeon, H. Ryu, J. W. Santo Domingo and Y. Seo, Chemosphere, 2020, 246, 125745 CrossRef CAS PubMed.
- Z. Wang, L. Li, R. W. Ariss, K. M. Coburn, M. Behbahani, Z. Xue and Y. Seo, Sci. Total Environ., 2021, 753, 141606 CrossRef CAS PubMed.
- USEPA, 2007.
- A. Rodrigues, A. Brito, P. Janknecht, M. F. Proena and R. Nogueira, J. Environ. Monit., 2009, 11, 377–382 RSC.
- S. Zhang, Y. Tian, Y. Guo, J. Shan and R. Liu, Chemosphere, 2021, 262, 1–10 Search PubMed.
- US EPA, 2003.
- L. Kurajica, M. Ujević Bošnjak, M. Novak Stankov, A. S. Kinsela, J. Štiglić, D. T. Waite and K. Capak, J. Environ. Manage., 2020, 276, 111360 CrossRef CAS PubMed.
- P. G. Coble, Mar. Chem., 1996, 51, 325–346 CrossRef CAS.
- J. P. Gustafsson, Dep. L. Water Recources, Stock. Sweden, 2011, pp. 1–73 Search PubMed.
- F. Yang, B. Shi, J. Gu, D. Wang and M. Yang, Water Res., 2012, 46, 5423–5433 CrossRef CAS PubMed.
- P. Sarin, V. Snoeyink, J. Bebee, W. Kriven and J. Clement, Water Res., 2001, 35, 2961–2969 CrossRef CAS.
- M. Li, Z. Liu, Y. Chen and M. Zhang, Environ. Sci. Pollut. Res., 2019, 26, 19906–19914 CrossRef PubMed.
- J. Lin, M. Ellaway and R. Adrien, Corros. Sci., 2001, 43, 2065–2081 CrossRef CAS.
- E. J. Kim and J. E. Herrera, Environ. Sci. Technol., 2010, 44, 6054–6061 CrossRef CAS PubMed.
- G. Li, X. Ma, R. Chen, Y. Yu, H. Tao and B. Shi, Water Res., 2019, 163, 1–9 Search PubMed.
- M. Bieroza, A. Baker and J. Bridgeman, Educ. Chem. Eng., 2012, 7, e22–e31 CrossRef.
- M. Ujević, Ž. Duić, C. Casiot, L. Sipos, V. Santo, Ž. Dadić and J. Halamić, Appl. Geochem., 2010, 25, 1017–1029 CrossRef.
- P. Sarin, V. L. Snoeyink, D. A. Lytle and W. M. Kriven, J. Environ. Eng., 2004, 130, 364–373 CrossRef CAS.
- M. Li, Z. Liu, Y. Chen and Y. Hai, Water Res., 2016, 106, 593–603 CrossRef CAS PubMed.
- P. Sarin, V. L. Snoeyink, J. Bebee, K. K. Jim, M. A. Beckett, W. M. Kriven and J. A. Clement, Water Res., 2004, 38, 1259–1269 CrossRef CAS PubMed.
- Y. He, L. Pan, R. Chen and B. Shi, Chemosphere, 2021, 275, 130067 CrossRef CAS PubMed.
- Y. Xie and D. E. Giammar, Water Res., 2011, 45, 6525–6534 CrossRef CAS PubMed.
- V. Gauthier, B. Gérard, J. M. Portal, J. C. Block and D. Gatel, Water Res., 1999, 33, 1014–1026 CrossRef CAS.
- S. D. Richardson, M. J. Plewa, E. D. Wagner, R. Schoeny and D. M. DeMarini, Mutat. Res., Rev. Mutat. Res., 2007, 636, 178–242 CrossRef CAS PubMed.
- S. D. Richardson and C. Postigo, ACS Symp. Ser., 2015, 1190, 189–214 CrossRef CAS.
- C. Ma, H. Xu, L. Zhang, H. Pei and Y. Jin, Sci. Total Environ., 2018, 640–641, 609–618 CrossRef CAS PubMed.
- M. J. Rodriguez, J.-B. Sérodes and P. Levallois, Water Res., 2004, 38, 4367–4382 CrossRef CAS PubMed.
- S. Masters and M. Edwards, Environ. Eng. Sci., 2015, 32, 361–369 CrossRef CAS.
- A. D. Hulsmann, J. Inst. Water Environ. Manage., 1990, 4, 19–25 CrossRef CAS.
- B. F. Trueman and G. A. Gagnon, Environ. Sci. Technol., 2016, 50, 9053–9060 CrossRef CAS PubMed.
- G. Li, Y. Ding, H. Xu, J. Jin and B. Shi, Chemosphere, 2018, 197, 73–80 CrossRef CAS PubMed.
- Y. Ai, C. Zhao, L. Sun, X. Wang and L. Liang, Sci. Total Environ., 2020, 702, 135072 CrossRef CAS PubMed.
- F. Frau, R. Biddau and L. Fanfani, Appl. Geochem., 2008, 23, 1451–1466 CrossRef CAS.
- H. Sun, B. Shi, F. Yang and D. Wang, Water Res., 2017, 114, 69–77 CrossRef CAS PubMed.
- W. Wang, X. Zhang, H. Wang, X. Wang, L. Zhou, R. Liu and Y. Liang, Water Res., 2012, 46, 4063–4070 CrossRef CAS PubMed.
- R. M. Safiur, M. Whalen and G. A. Gagnon, Chem. Eng. J., 2013, 234, 149–157 CrossRef.
- S. W. Krasner, Philos. Trans. R. Soc., A, 2009, 367, 4077–4095 CrossRef CAS PubMed.
- J. P. Ritson, N. J. D. Graham, M. R. Templeton, J. M. Clark, R. Gough and C. Freeman, Sci. Total Environ., 2014, 473–474, 714–730 CrossRef CAS PubMed.
- Y. Hou, W. Chu and M. Ma, J. Environ. Sci., 2012, 24, 1204–1209 CrossRef CAS PubMed.
- A. K. Camper, Int. J. Food Microbiol., 2004, 92, 355–364 CrossRef CAS PubMed.
- Z. Wang, C. M. Hessler, Z. Xue and Y. Seo, Water Res., 2012, 46, 1052–1060 CrossRef CAS PubMed.
- J. M. Astilleros, C. M. Pina, L. Fernandez-Diaz and A. Putnis, Geochim. Cosmochim. Acta, 2002, 66, 3177–3189 CrossRef CAS.
- N. Mladenov, Y. Zheng, B. Simone, T. M. Bilinski, D. M. McKnight, D. Nemergut, K. A. Radloff, M. M. Rahman and K. M. Ahmed, Environ. Sci. Technol., 2015, 49, 10815–10824 CrossRef CAS PubMed.
- A. Abdelrady, S. Sharma, A. Sefelnasr and M. Kennedy, J. Environ. Manage., 2020, 258, 110003 CrossRef CAS PubMed.
- D. A. Lytle and M. R. Schock, J. Water Supply: Res. Technol.--AQUA, 2000, 49, 243–257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ew00537a |
| This journal is © The Royal Society of Chemistry 2023 |