Practical considerations for the electrochemical denitrification of real wastewater†
Received
7th May 2022
, Accepted 8th November 2022
First published on 8th November 2022
Abstract
High concentrations of nitrate (NO3−) in water is a serious threat to the environment and human health. Therefore, there is an urgent to explore and develop real wastewater treatment methods. In this study, the electrochemical reduction of NO3− using brass as the cathode and IrO2–RuO2/Ti as the anode was studied, and the influence of various conditions, including the current density, conductivity, initial pH, Cl− concentration and, coexisting matter (PO43−, CO32−, Ca2+, and organics), were systematically investigated. The optimized method was verified for the reduction of NO3− in simulated wastewater and real wastewater. It was found that in the presence of 500 mg L−1 Cl−, a reduction rate above 80% could be readily achievable for 50 mg L−1 NO3−–N in 100 min electrolytic treatment at a current density of 10 mA cm−2. The presence of CO32− and Ca2+ had no significant effect on NO3− reduction, but the presence of PO43− could substantially decrease the reduction rate of NO3−–N due to the competitive adsorption of HPO42− and H2PO4− on the brass electrode. The interference from PO43− could be largely alleviated by the addition of Ca2+ to the sample solution. The presence of organic matters also adversely influenced the reduction rate of NO3−–N, whereby the reduction rate of NO3−–N decreased to 71.1% in a solution with 200 mg L−1 COD. As the organic matter could be degraded in prolonged electrolytic treatment, our method showed good adaptability for real water treatment. The results obtained in the present study demonstrated that the electrolytic method with brass as the cathode and IrO2–RuO2/Ti as the anode not only had a satisfactory efficiency for NO3−–N removal but also exhibited good durability in real water treatment.
Water impact
Electrochemical denitrification is a promising technique for wastewater treatment; however, the factors that influence its practical application have seldom been addressed so far. In the present study, the influence of coexisting matter, including PO43−, CO32−, Ca2+, and organics, on the electrochemical reduction of NO3− was systematically investigated using commercially available brass as the cathode and IrO2–RuO2/Ti as the anode. The results obtained in this study should have great value in the practical application of clean methods for real water purification, and hence would be interesting to the wider academic and industrial community.
|
1. Introduction
The use of nitrogen fertilizers, agricultural runoff, and wastewater from food processing and industrial production results in a large amount of nitrogen effluent to the environment, which imposes a serious threat to the natural nitrogen cycle.1–3 According to the regulation of the World Health Organization, the maximum allowable concentration of NO3−–N in drinking water is 11.3 mg L−1.4,5 Therefore, efficient and clean technologies are urgently required to remove NO3− in polluted water. Various methods, such as electrochemical denitrification,6,7 adsorption,8 ion exchange,9,10 reverse osmosis,11 and biological denitrification,12 have been developed for the reduction of NO3−. Among these, electrochemical denitrification has the advantages of no secondary pollution, simple operation, cost-effectiveness, and environmental friendliness. Hence, it is generally recognized that electrochemical reduction has a great potential in the effective removal of NO3−.13–16
Many studies regarding the electrochemical reduction of NO3− have been published in recent years. In particular, the mechanistic aspects of NO3− reduction have been extensively investigated. It is now generally acknowledged that two major processes are involved in the electrochemical reduction of NO3−.17,18 In the first pathway, NO3−–N obtains electrons from the cathode and is directly reduced to N2 or NH3.19 In the second pathway, NO3−–N can be reduced stepwise by the reductive species formed on the cathode surface.17 As shown in eqn (1), H2O is initially reduced by the Volmer reaction to produce atomic hydrogen (H(ads)).20 At the same time, NO3− is adsorbed on the cathode surface (eqn (2)), where it is stepwise hydrogenated by H(ads) to form NO2(ads)−, NO(ads), and N(ads) (eqn (3)–(5)).18 Because the formation of the N–H bond mediated by H(ads) is faster than that of the N–N bond in terms of the kinetics, it results in the subsequent production of ammonia following eqn (6)–(8).21,22
| | | H2O + e− → H(ads) + OH− (Volmer) | (1) |
| | | NO3(ads)− + 2H(ads) → NO2(ads)− + 2H2O | (3) |
| | | NO2(ads)− + H(ads) → NO(ads) + OH− | (4) |
| | | NO(ads) + 2H(ads) → N(ads) + 2H2O | (5) |
| | | N(ads) + H(ads) → NH(ads) | (6) |
| | | NH(ads) + H(ads) → NH2(ads) | (7) |
| | | NH2(ads) + H(ads) → NH3(ads) | (8) |
Further studies have revealed that the cathode materials play an important role in nitrate reduction. Various cathode materials, including Cu,23 Fe,24 Pt,25 Zn,26 brass,27 Cu/Ni,28 TiO2,29 Co3O4,30 and boron-doped diamond,31 have been explored for the electrochemical reduction of NO3−. Among them, brass exhibited a good efficiency in NO3− reduction.32,33 Moreover, brass is commercially available at low cost and in a controllable size; hence, it is suitable for practical applications as the cathode.34
Although much research has been conducted, most the reported works are restricted to the electrochemical reduction of nitrate in simulated water solutions and experiments conducted at the laboratory scale, while the practical application of the electroreduction of NO3− in real water has not yet been comprehensively investigated. The different sources of nitrate effluent could produce large quantities of wastewater with complex compositions.30,35 Domestic sewage, for example, contains a lot of organic matter and inorganic salts, such as Cl−, SO42−, PO43−, CO32−, K+, Ca2+, and Na+.36–38 Understanding the influence of these coexisting substances on the electrochemical denitrification is critical for the application of this novel and clean method in real water purification.
Therefore, the influence of the most frequently encountered interferences in wastewater on the electrochemical reduction of NO3− was investigated in the present work. A brass (Cu: 62 wt%; Zn: 38 wt%) plate was used as the cathode and an IrO2–RuO2/Ti plate was chosen as the anode. After the preliminary optimization of the experimental conditions (current density, conductivity, initial pH, and Cl− concentration), the effects of various pollutants (PO43−, CO32−, Ca2+, and organics) in wastewater on the electrochemical reduction of NO3− were systematically investigated. Then, the optimized method was applied for the treatment of 2.5 L real wastewater for showing the promising applicability of the electrochemical reduction of NO3−–N in real water purifications.
2. Materials and methods
2.1 Apparatus and reagents
To optimize the electrochemical reduction system, a simple electrolytic setup was initially used. A 100 mL beaker served as the electrolytic cell to treat 50 mL solutions, in which an equal-sized anode and cathode (2 cm × 2 cm) with a fixed distance of 3 cm were inserted. A DC power supply (KXN-1005D, Shenzhen Zhaoxin Electronic Instrument Equipment Co., Ltd, China) with a current range of 0–5 Å and a voltage range of 0–100 V was adopted in the electrolytic experiment. An IrO2–RuO2/Ti plate (obtained from Qinghe Yunxuan Metal Material Co., LTD, China) was used as the anode. The brass plate was used as the cathode, which consisted of 62 wt% Cu and 38 wt% Zn, obtained from Taizhou Chunshi New Material Co., LTD (China). Before the electrolysis experiments, the brass plate was polished with 220 mesh sandpaper, and subsequently washed with ultrapure water and ethanol. For real water treatment, a large plastic box (ca. 3 L) was used as the electrolytic cell and an aliquot of 2.5 L real wastewater was treated in batch mode. Two pairs of cathodes and anodes (all 10 cm × 10 cm in size) were installed alternately with an interval of 3 cm. The conductivity and pH were measured by a multi-parameter analyzer (DZS-708L, Shanghai Precision Scientific Instrument Co., Ltd, China). UV-visible spectroscopy was used for the determination of the concentrations of the various nitrogen species (DH-2000-BAL, Ocean Optics, USA). All the reagents used were analytical grade and the specific information on them is given in the ESI.† All the solutions were prepared with ultrapure water.
2.2 Electrochemical nitrate reduction experiments
Unless otherwise specified, an aliquot of 50 mL electrolytic solution containing 50 mg L−1 NO3−–N was used for the electrochemical reduction experiments. The experiment parameters, including the current density, conductivity, initial pH, and Cl− concentration, were optimized by a control variable method. The initial parameters were set as follows: the current density was 10 mA cm−2, the initial pH was 7, the conductivity was 16.4 mS cm−1 controlled by 0.1 M Na2SO4, and the Cl− concentration was 0 mg L−1. The interference factors, including PO43−, CO32−, Ca2+, and organics, were systematically investigated. After the addition of an appropriate amount of interferences to the solution, the pH and the conductivity of the solution were adjusted to be consistent with the initial parameters so that the obtained results were comparable. To verify the optimized conditions, two kinds of simulated wastewater were prepared, both containing 50 mg L−1 NO3−–N, 100 mg L−1 PO43−, 100 mg L−1 CO32−, 500 mg L−1 Cl−, and 100 mg L−1 COD, and the conductivity of the samples was adjusted to 16.4 mS cm−1. The sample with Cl− added in the form of NaCl was termed simulated wastewater I and that in the form of CaCl2 was termed simulated wastewater II. The real wastewater was obtained from a local water treatment company, and the concentrations of interference species were determined to be 268.7 mg L−1 NO3−–N, 13.3 mg L−1 NO2−–N, 234.1 mg L−1 NH4+–N, 564.5 mg L−1 TN, 2249.9 mg L−1 Cl−, 340.2 mg L−1 COD, and 78.4 mg L−1 PO43−. The conductivity was 23.5 mS cm−1 and the pH was 3.2.
2.3 Analysis
The concentrations of NO3−–N, NO2−–N, NH4+–N, and Cu2+ were determined by conventional UV-visible spectrometric methods. Briefly, the NO3−–N concentration of the water samples taken during the experiment was directly monitored at 220 nm,39 while the NH4+–N concentration was determined by Nessler reagent,40 and the NO2−–N concentration at 540 nm by N-(1-naphthyl)-ethylenediamine spectrophotometry.39 Cu was monitored by the sodium diethyldithiocarbamate spectrophotometric method. The active chlorine concentrations were determined by iodimetry. The total nitrogen (TN) was determined by the potassium persulfate method.41 COD was measured by the potassium dichromate method.30 The calculations of the NO3−–N reduction rate, NO2−–N yield rate, NH4+–N yield rate, TN removal rate, electro-energy utilization efficiency, energy consumption, and the first-order kinetic equation are shown in the ESI.† The details of the density functional theory (DFT) calculations are provided in the ESI† material.
3. Results and discussion
3.1 Effect of current density
The current density has great influence on the reduction and oxidation performance of an electrochemical process. Hence, the effect of current density on electrochemical denitrification was initially studied, and the results are presented in Fig. 1. As can be seen, the reduction rate of NO3−–N increased with the increase in current density (Fig. 1a), i.e., when the current density was set at 5 mA cm−2, the NO3−–N reduction rate was 83.7% after 100 min, then when the current density was increased to 25 mA cm−2, the NO3−–N reduction increased by 11.5% ± 0.8%. For the different current densities, the electrocatalytic reduction rates were in agreement with the first-order kinetics equation (Fig. 1b), and the rate constant increased from 0.0176 (R2 = 0.9561) to 0.0306 (R2 = 0.9989) min−1. At the same time, it can be seen from Fig. 1c that with the increase in the current density, the NH4+–N yield rate also increased gradually. Different with the NO3−–N reduction, the yield rate of NO2−–N decreased with the increase in current density (Fig. S1†). This was because at high current density, more H(ads) was generated,42 which facilitated the direct electrochemical reduction of NO3−–N and the subsequent formation of NH4+–N.39 All these indicated that the reduction efficiency and rate can be improved by increasing the current density.
 |
| | Fig. 1 Effect of the current density on (a) NO3−–N reduction, (b) NO3−–N reduction kinetics, (c) NH4+–N yield, and (d) electro-energy utilization efficiency. | |
However, the electro-energy utilization efficiency should also be considered with the increase in current density. Based on the amount of NO3−–N reduction and the production of NO2−–N and NH4+–N, the electro-energy utilization efficiency was calculated under different current densities. As shown in Fig. 1d, though the NO3−–N reduction rate increased, the electro-energy utilization efficiency decreased from more than 46.8% to 10.4% when the current density was increased from 5 to 25 mA cm−2, i.e., most of the electronic energy was wasted for H2O electrolysis at the high current density.30 Therefore, a high current density is not recommended in the practical treatment of NO3−–N wastewater. The NO3−–N reduction rate, the yield of by-products, and the electro-energy utilization efficiency should be comprehensively considered., In the present work, 10 mA cm−2 was used in our subsequent experiments.
3.2 Effect of the electrical conductivity
The effect of different conductivities on NO3−–N reduction was studied using Na2SO4 as the electrolyte. Fig. S2a† shows that the NO3−–N reduction rate increased with the increase in conductivity. When the conductivity of the solution was 40.3 mS cm−1, a reduction rate of 91.5% was achieved after 100 min. A similar trend when studying the effect of the electrolyte concentration on NO3− reduction was also reported in the literature.43 This was mainly due to the increased conductivity of the solution, which facilitates the electron-transfer process of the electrochemical reaction, and hence the reaction rate and current efficiency are improved. Similarly, the NH4+–N yield also increased with the increase in conductivity (Fig. S2c†). In contrast, the NO2−–N yield gradually decreased with the increase in conductivity (Fig. S2b†).
3.3 Influence of the initial pH on NO3−–N reduction
In order to understand the effect of the initial pH on electrochemical denitrification when brass was used as the cathode, NO3− reduction was performed at different initial pH levels, and the data obtained are shown in Fig. S3a.† When the initial pH value was adjusted to 3.0, 5.0, 7.0, and 9.0, the NO3−–N reduction rate was 90.5%, 90.3%, 85.9%, and 82.7%, respectively, after electrolysis for 100 min. The NO3−–N reduction rate in the acidic environment was slightly higher than that in the neutral and alkaline conditions. A similar trend was observed for the yield rates of NO2−–N (Fig. S3b†) and NH4+–N (Fig. S3c†). In acidic solution, more H+ was adsorbed on the cathode surface to form H(ads), which was more conducive to the reduction of NO3−–N (eqn (1)–(8)).25 In the process of the reaction, the pH of the solution was increased gradually, generally ending up in alkaline conditions (Fig. S3d†). During this process, the deionization should take place for the weak acid or base products. For example, a greater proportion of NH4+ will be changed to NH3 in alkaline conditions. Considering that the reduction rate in neutral conditions was very close to that obtained in acidic conditions, our subsequent experiments were conducted in neutral conditions.
3.4 Effect of the Cl− concentration on the denitrification
It is well-known that ClO− generated by the oxidation of Cl− can react with NH4+ to generate N2, and hence could completely remove nitrogen from water. Fig. 2a shows that the addition of Cl− in the electrolytic system did not significantly change the NO3−–N reduction rate. However, the addition of Cl− had a great influence on the formation of NH4+–N. As shown in Fig. 2b, the NH4+–N yield rate decreased significantly with the increase in the Cl− concentration. In the absence of Cl−, the yield of NH4+–N accounted for 81.5% of the total nitrogen after 100 min treatment. When the Cl− concentration was increased to more than 1500 mg L−1, NH4+–N was not found. The yield rate of NO2−–N, however, increased at the beginning of the electrolytic treatment, and then gradually declined with the extension of the reaction time (Fig. S4†). As can be seen from Fig. 2c, the TN removal rate increased from 3.5% to 80.1% when the Cl− concentration was increased from 0 mg L−1 to 2000 mg L−1. This was because the Cl− was oxidized at the anode to generate Cl2, and then Cl2 dissolved in water to generate active chlorine (HClO, ClO−). The generated NH4+–N could be effectively oxidized to N2 with the help of ClO− (eqn (9)–(12)), which greatly improved the removal rate of TN.44 The active chlorine concentrations in the treated solution was determined, and the results are shown in Fig. 2d. The higher the concentration of Cl− in the solution, the more reactive chlorine was produced. Thus, the TN removal rate was better and this was consistent with the above analysis. Considering the practical cost and the NO3−–N reduction rate, the Cl− concentration of 500 mg L−1 was used in the subsequent studies.| | | Cl2 + H2O → HClO + Cl− + H+ | (10) |
| | | 2NH4+ + 3ClO− → N2 + 3Cl− + 2H+ + 3H2O | (12) |
 |
| | Fig. 2 Effect of Cl− concentration on (a) NO3−–N reduction, (b) NH4+–N yield, (c) TN removal efficiency, and (d) active chlorine. | |
3.5 Effect of other inorganic ions on the reduction of NO3−
Phosphate is widely present in various wastewaters; among some industrial and agricultural wastewaters, the phosphate concentration can reach 1000 mg L−1.45 Therefore, the influence of PO43− on the electrochemical reduction of NO3−–N was also investigated, and the results are presented in Fig. 3a. In the absence of PO43−, a reduction rate of 80.2% was obtained for NO3−–N, whereas in the presence of 50 mg L−1 PO43−, the rate decreased by 3.6%. When further increasing the PO43− concentration to 100 and 150 mg L−1, the reduction rate of NO3−–N decreased by 32.9% ± 1.8% and 43.7% ± 0.5%, respectively. Similarly, the yield rates of NO2−–N and NH4+–N also decreased gradually with the increase in the PO43− concentration (Fig. S5a and b†). These observations indicated that the electrolytic reactions were inhibited to some extent by the coexisting PO43−. Such inhibition was also observed by Fan et al., who reported that NO3− reduction on the electrode was decreased in the presence of PO43−.46 It was noted, however, that when the pH of the solution was kept constant at 3.0, 7.0, and 10.5 by continuously adding H2SO4 during the electrolytic process, the reduction rate of NO3−–N increased to 63.7%, 49.2%, and 31.9%, respectively (Fig. 3b), indicating that the protonation of PO43− could alleviate its interference toward NO3−–N reduction.
 |
| | Fig. 3 Effects of (a) different concentrations of PO43− and (b) constant pH on NO3−–N reduction. | |
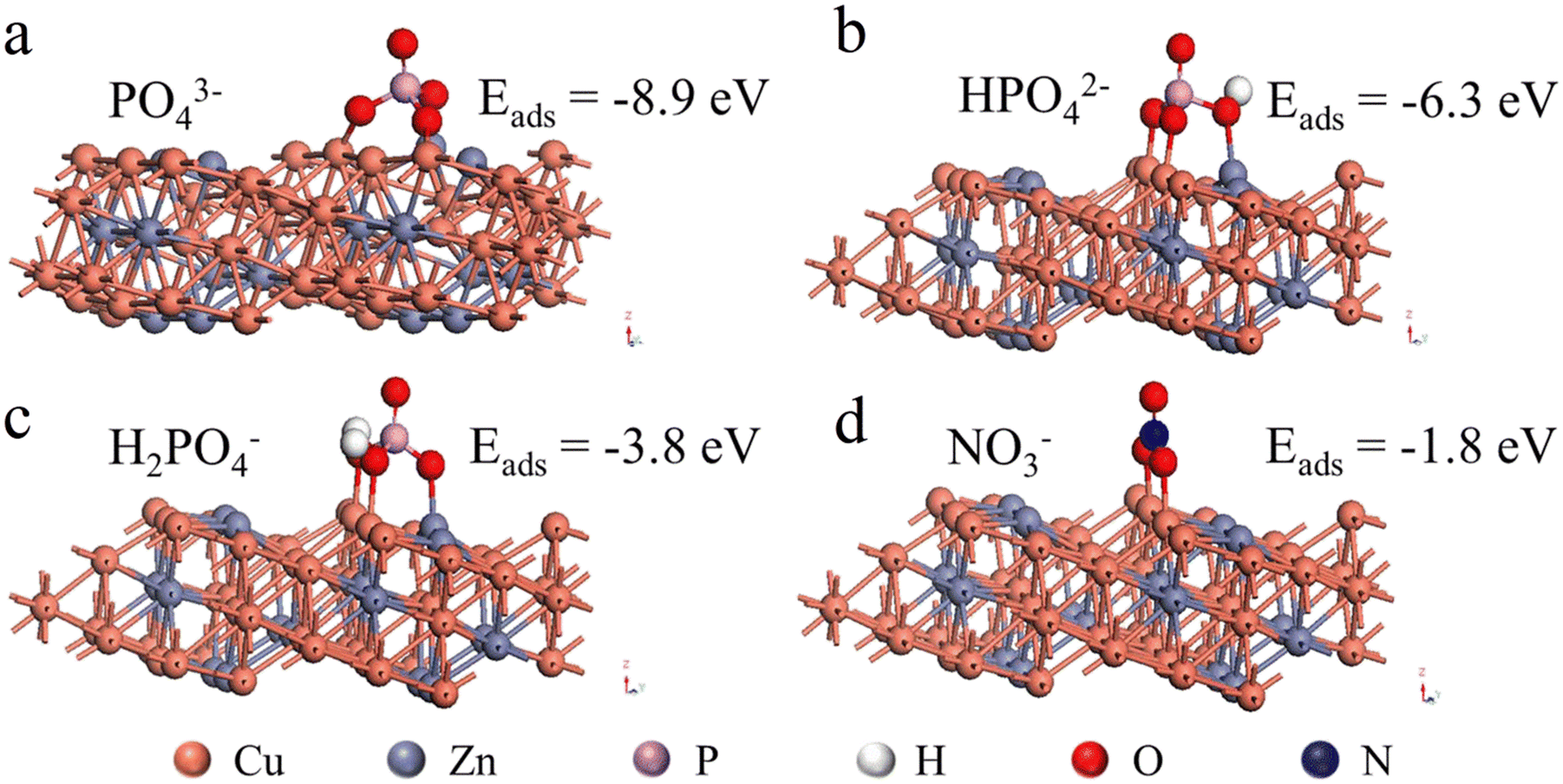
PO43− should be present in form of HPO42− and H2PO4− in the tested pH range, but it is not known how these species interfere with nitrate reduction. To better understand the observed phenomenon, the adsorption of PO43− on the electrode surface was further investigated by DFT calculations. The adsorption configurations of PO43−, HPO42−, H2PO4−, and NO3− on the brass surface and the corresponding adsorption energies are presented in Fig. 4. As can be seen, the adsorption of PO43−, HPO42−, H2PO4−, and NO3− on the surface of brass was thermodynamically favorable with energies of −8.9, −6.3, −3.8, and −1.8 eV, respectively. The relatively lower adsorption energy of PO43−, HPO42−, and H2PO4− suggested that these species could hinder the adsorption of NO3− on the electrode, which should account for the phosphate interference with nitrate reduction. Hence, this suggested conducting the electrolytic reduction of NO3− in slightly acidic conditions, so that the interferences from phosphate could be minimized.
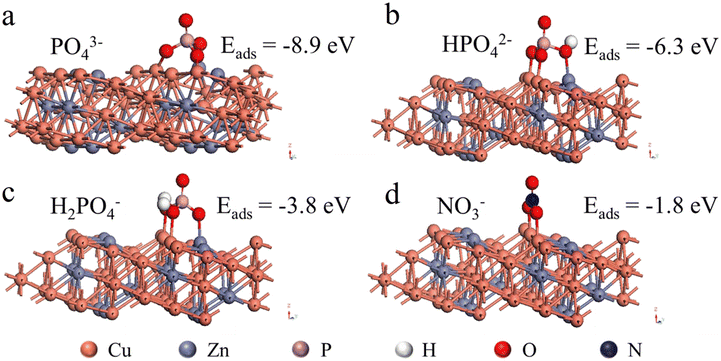 |
| | Fig. 4 Adsorption configurations of (a) PO43−, (b) HPO42−, (c) H2PO4−, and (d) NO3− on the brass surface and the corresponding adsorption energies. | |
Carbonates are ubiquitous in wastewater,7 thus the effect of CO32− on the electrolytic reduction of NO3−–N was also studied and the results are presented in Fig. S6.† As can be seen, with the increase in CO32− concentration, the NO3−–N reduction rate varied between 81.4–79.8% (Fig. S6a†), presumably due to the strong protonation nature of CO32−. However, the yield rates of NO2−–N and NH4+–N slightly decreased with the increase in CO32− concentration (Fig. S6b and c†). In general, the existence of carbonate has little effect on the reduction of NO3−–N.
The influence of cationic metals on NO3−–N reduction was investigated by adding Ca2+ as the representative species. Ca2+ was added in the form of CaCl2. When the Cl− concentration was controlled to be 500 mg L−1, the Ca2+ concentration was 282.4 mg L−1 in the solution. The NO3−–N reduction results in the presence of CaCl2 are shown in Fig. S7a.† As can be seen, the efficiency of NO3−–N reduction was similar to that obtained in the presence of the same concentration of NaCl. The NO2−–N and NH4+–N yield rates were also similar with those obtained in the absence of Ca2+ (Fig. S7b and c†), i.e., the presence of Ca2+ had little effect on the reduction of NO3−.
3.6 Effect of organics on the reduction of NO3−
Wastewater often contains different amounts of organic pollutants, which are usually judged by the COD values. In order to explore the influence of organics on NO3− reduction, glucose was used as the COD model compound in this study. It can be seen from Fig. 5a that with the increase in glucose concentration and the COD of the solution, the NO3−–N reduction rate slightly declined in the experiments, i.e., the NO3−–N reduction rate changed from 80.2% to 71.1% for 0 mg L−1 COD and 200 mg L−1 COD, respectively. As a kind of polyhydroxy aldehyde, glucose can be reduced on the cathode to form mannitol or sorbitol in slightly alkaline conditions, which should be responsible for the declined reduction rate of NO3−–N.47 Fig. S8a† shows that the NO2−–N yield rate was relatively low in the presence of glucose, presumably due to the consumption of the produced NO2− by glucose. With the increase in COD, the yield rate of NH4+–N gradually increased and the TN removal rate decreased (Fig. S8b and c†). This was because glucose could compete with NH4+–N to consume the ClO− that was produced at the anode, leading to a decline in the elimination of NH4+–N.48,49 The electrolytic oxidation of glucose could be evidenced by the decreased COD value after the reaction, and with the increase in initial COD concentration, the COD removal load also increased gradually (Table S1†). With prolonged electrolytic reaction, it is expected that the COD of the solution could be further reduced and its influence on NO3−–N reduction should be alleviated.
 |
| | Fig. 5 Effects of (a) COD on NO3−–N reduction; (b) the NO3−–N reduction efficiencies of simulated wastewater I and simulated wastewater II. | |
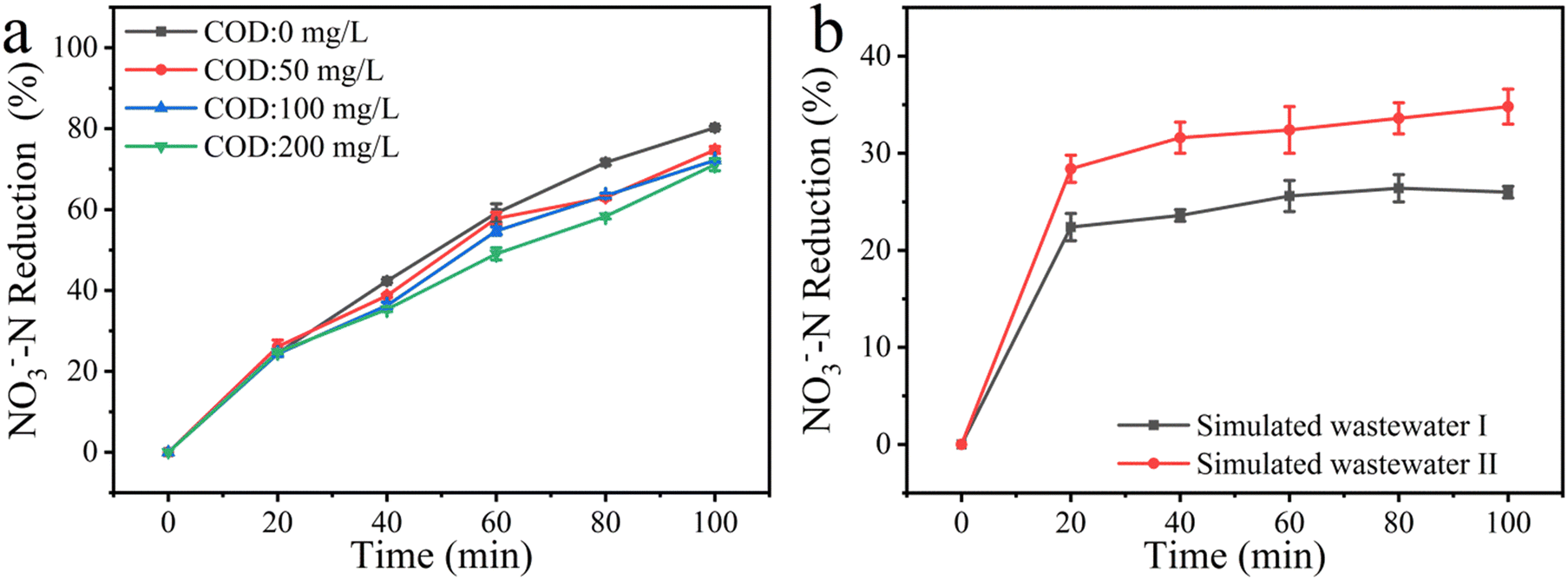
3.7 Treatment of simulated wastewater
Before the practical application of the electrolytic reduction of nitrate in real wastewater, the above optimized parameters were validated with two kinds of simulated wastewater. Both the simulated wastewaters contained a certain amount of interference species and 500 mg L−1 Cl−, but in wastewater I the chloride was added in the form of NaCl, whereas in wastewater II the chloride was added in the form of CaCl2, with an aim to see if the formation of calcium phosphate precipitate could alleviate the interference from phosphates. As shown in Fig. 5b, the NO3−–N reduction rate was fast in the first 20 min of the electrolytic reaction, then gradually slowed down, presumably due to the adsorption of PO43−, HPO42−, H2PO4−, and organic matter on the electrode covering up the available active sites, which hindered the reduction of NO3−–N. As expected, the NO3−–N reduction rate of simulated wastewater II was higher than that of simulated wastewater I. Flocs were observed in the beaker containing CaCl2, indicating the formation of calcium phosphate or calcium carbonate precipitates during the electrolytic reaction. The decrease in the PO43− concentration was conducive to the adsorption of NO3−, resulting in an increase in NO3− reduction. The yield rates of NO2−–N and NH4+–N in simulated wastewater II were higher than those in simulated wastewater I (Fig. S9a and b†). Moreover, the removal rate of TN from simulated wastewater II was more than that for simulated wastewater I (Fig. S9c†). Therefore, the presence of Ca2+ is conducive to the reduction of NO3−–N when there are many substances in the wastewater. In addition, the presence of Cu in the two treated solutions was detected after the reaction. Cu was not detected in the solution; hence the leakage of Cu may not be a serious problem in this method.
3.8 Treatment of real wastewater
Being an efficient, clean, and economical method, the electrochemical denitrification technology has great potential in wastewater treatment. However, the complex matrix and various interferences in real wastewater may compromise the efficiency of electrochemical denitrification. To prove the applicability of the present method for real water treatment, a heavily polluted water sample obtained from a local wastewater treatment plant was selected for the electrolytic treatment. The detailed information of the wastewater was provided in the Materials and methods section. The electrolytic experiment was carried out with four electrode plates, and anodes and cathodes were alternatively installed to allow the cathodic-produced NH4+–N to quickly reach the anodic area, where it could be oxidized by the formed ClO−.
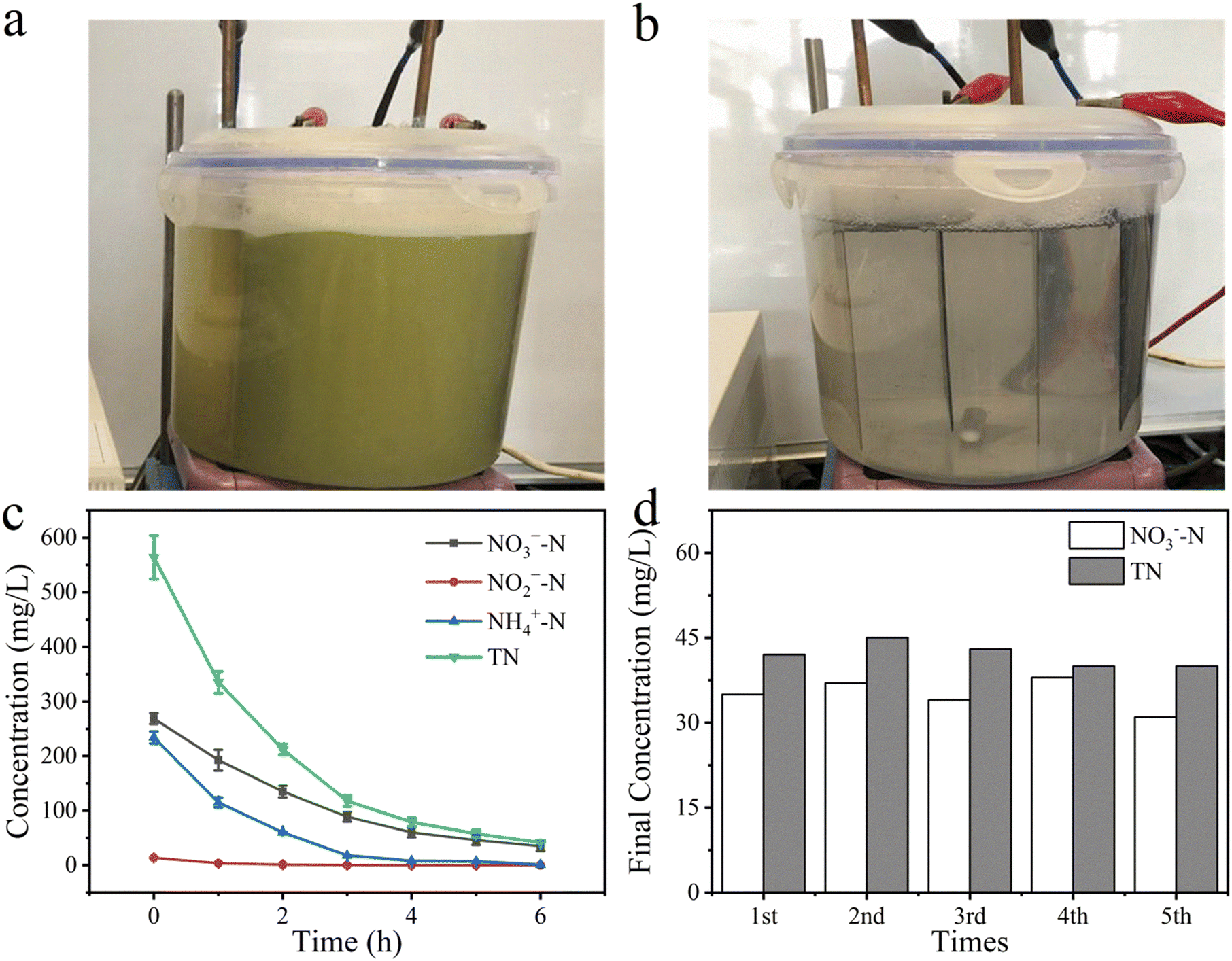
Fig. 6a shows the appearance of the wastewater sample, where it can be seen that the wastewater was green-yellow and virtually not transparent. As shown in Fig. 6b, after electrolytic treatment for 1 h, the water sample became transparent and the electrodes inside could be clearly visualized. During the reaction, many bubbles were generated in the reactor (Fig. S10a†) and upon the completion of electrolytic treatment, a large number of white flocs had accumulated above the solution (Fig. S10b†). This may be due to the presence of surfactants in the wastewater sample, leading to the yield of a large amount of foam during the high-speed mixing. It was observed that the residual amount of NO3−–N decreased with the increase in the duration of electrolytic treatment up to 6 h, and the monitored data are presented in Fig. 6c. As can be seen, the concentration of NO3−–N decreased from 268.7 mg L−1 to 35.1 mg L−1, whereas the concentrations of NO2−–N and NH4+–N also decreased from their initial values of 13.3 mg L−1 and 234.1 mg L−1, respectively, to undetectable levels. Also, TN decreased from 564.5 mg L−1 to 42.3 mg L−1. The high removal rates for TN and NH4+–N could be attributed to the presence of a high concentration of Cl− in the wastewater. The COD of the wastewater decreased from 340.2 mg L−1 to 72.6 mg L−1, which could also be ascribed to the direct and indirect oxidation of organic pollutants by the electrogenerated hydroxyl radical and ClO− during the 6 h electrolytic treatment.49 These observations suggested that the electrolytic method developed in the present work is not only effective for NO3− reduction and TN removal, but also exhibits great potential for the degradation of the COD of polluted water. In addition, the removal load of NO3−–N, NO2−–N, NH4+–N, and TN was less than 0.1 kg m−3 h−1. The above results indicate that the reactor has a fairly efficient performance (Fig. S11a†).
 |
| | Fig. 6 Picture of the real wastewater sample (a) before and (b) after treatment; (c) concentration of NO3−–N, NO2−–N, NH4+–N, and TN; (d) repeated experiments for the real wastewater denitrification. | |
The stability of the electrodes is of primary importance for their practical applications. Hence in this work, a number of repeated electrolytic experiments were conducted to evaluate the stability of the electrodes. To do this, two same pieces of brass cathode were repeatedly applied in the wastewater treatment, and during each cycle, the foams or other residues on the brass cathode were washed away with pure water. Similar NO3−–N concentrations, TN concentrations (Fig. 6d), and energy consumption (Fig. S11b†) were obtained in the five cycles, indicating that the brass cathode was stable and has promising feasibility for use in wastewater treatment. Similarly, two same pieces of IrO2–RuO2/Ti anode were used during all the experiments, and no obvious corrosion was found in up to 6 months of use. Therefore, the anode and cathode used in this work have excellent practical applicability in real water treatment.
The cost of the water treatment method is another important factor to be considered for its practical application. The electrolytic system reported here was quite simple, whereby only a DC power supply, a stirrer, and two pairs of cathode and anode were required. Based on our lab-scale experiment, an estimated cost for the treatment of 1 T wastewater was determined and is given in Table S2.† As can be seen, the total cost was only 0.012 US$ per gTN, which is very competitive in comparison with the costs obtained from other methods. Furthermore, the electrolytic treatment is simple in operation with a minimum requirement for duty workers.
4. Conclusions
In this work, the electrochemical reduction of NO3−–N was investigated by using IrO2–RuO2/Ti as the anode and brass as the cathode. Our experiment results proved that the electrolytic treatment is a promising technique for the removal of NO3−–N in polluted water, and that the electrodes used in this work were not only effective for the removal of TN and COD in real wastewater, but also exhibited good durability and low cost. Coexisting ions, such as Ca2+ and CO32−, had no significant influence on the electrochemical reduction of nitrate nitrogen. The presence of Cl− could effectively remove the electrogenerated NH4+–N, hence greatly improving the removal rate of TN. However, when the concentration of Cl− was too high (above 3000 mg L−1), it could cause corrosion of the electrodes in prolonged operation. The co-existence of organics and PO43− showed some negative effects on the reduction efficiency of nitrate nitrogen. We verified by DFT calculations that PO43− can compete with NO3− for the active sites of adsorption on the electrode surface, exhibiting an adverse influence on NO3− reduction, and such an adverse effect is particularly pronounced in alkaline conditions. For real water treatment, it is suggested that wastewater with a high COD should be degraded before the electrolytic treatment of the nitrates.
Author contributions
Miss Dehui Li has conducted all the experiments and data analysis. She also wrote the first draft of the manuscript. Mr. Cheng Fu performed DFT calculation. Dr. Chan Wang and Prof. Qijun Song have involved the result discussions and revised the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from National First-Class Discipline Program of Food Science and Technology (JUFSTR20180301) and the technical assistant from the Central Laboratory, School of Chemical and Material Engineering, Jiangnan University are gratefully acknowledged.
Notes and references
- Y. Zhang, Y. Zhao, Z. Chen, L. Wang, P. Wu and F. Wang, Electrochim. Acta, 2018, 291, 151–160 CrossRef CAS.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- J. Martínez, A. Ortiz and I. Ortiz, Appl. Catal., B, 2017, 207, 42–59 CrossRef.
- L. Fewtrell, Environ. Health Perspect., 2004, 112, 1371–1374 CrossRef PubMed.
- N. Fan, Z. Li, L. Zhao, N. Wu and T. Zhou, Chem. Eng. J., 2013, 214, 83–90 CrossRef CAS.
- X. Ma, M. Li, C. Feng and Z. He, J. Hazard. Mater., 2020, 388, 122085 CrossRef CAS PubMed.
- B. P. Dash and S. Chaudhari, Water Res., 2005, 39, 4065–4072 CrossRef CAS PubMed.
- K. M. Reddy and S. P. Singh, Int. J. Chem. React. Eng., 2020, 18, 0113 Search PubMed.
- F. Liu, M. Li, H. Wang, X. Lei, L. Wang and X. Liu, J. Electrochem. Soc., 2016, 163, E421–E427 CrossRef CAS.
- M. Alikhani and M. R. Moghbeli, Chem. Eng. J., 2014, 239, 93–104 CrossRef CAS.
- R. Epsztein, O. Nir, O. Lahav and M. Green, Chem. Eng. J., 2015, 279, 372–378 CrossRef CAS.
- F. Liu, Int. J. Electrochem. Sci., 2016, 11, 8308–8322 CrossRef CAS.
- X. Ma, M. Li, X. Liu, L. Wang, N. Chen, J. Li and C. Feng, Chem. Eng. J., 2018, 348, 171–179 CrossRef CAS.
- L. Szpyrkowicz, S. Daniele, M. Radaelli and S. Specchia, Appl. Catal., B, 2006, 66, 40–50 CrossRef CAS.
- Z. Liu, S. Dong, D. Zou, J. Ding, A. Yu, J. Zhang, C. Shan, G. Gao and B. Pan, Water Res., 2020, 173, 115596 CrossRef CAS PubMed.
- M. Guo, L. Feng, Y. Liu and L. Zhang, Environ. Sci.: Water Res. Technol., 2020, 6, 1095–1105 RSC.
- J. Gao, B. Jiang, C. Ni, Y. Qi, Y. Zhang, N. Oturan and M. A. Oturan, Appl. Catal., B, 2019, 254, 391–402 CrossRef CAS.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- M. R. G. de Chialvo and A. C. Chialvo, Electrochim. Acta, 1998, 44, 841–851 CrossRef.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Chem. Eng. J., 2020, 382, 123034 CrossRef CAS.
- C. Yang, Q. B. Zhang, M. Y. Gao, Y. X. Hua and C. Y. Xu, J. Electrochem. Soc., 2016, 163, D469–D475 CrossRef CAS.
- W. Li, C. Xiao, Y. Zhao, Q. Zhao, R. Fan and J. Xue, Catal. Lett., 2016, 146, 2585–2595 CrossRef CAS.
- C. Sun, F. Li, H. An, Z. Li, A. M. Bond and J. Zhang, Electrochim. Acta, 2018, 269, 733–741 CrossRef CAS.
- P. Kuang, C. Feng, M. Li, N. Chen, Q. Hu, G. Wang and R. Li, J. Electrochem. Soc., 2017, 164, E103–E112 CrossRef CAS.
- Z. Mácová, K. Bouzek and J. Šerák, J. Appl. Electrochem., 2007, 37, 557–566 CrossRef.
- D. Reyter, D. Belanger and L. Roue, Water Res., 2010, 44, 1918–1926 CrossRef CAS PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- L. Su, K. Li, H. Zhang, M. Fan, D. Ying, T. Sun, Y. Wang and J. Jia, Water Res., 2017, 120, 1–11 CrossRef CAS.
- M. Ghazouani, H. Akrout and L. Bousselmi, Desalin. Water Treat., 2015, 53, 1107–1117 CAS.
- Z. Mácová and K. Bouzek, J. Appl. Electrochem., 2005, 35, 1203–1211 CrossRef.
- C. Polatides and G. Kyriacou, J. Appl. Electrochem., 2005, 35, 421–427 CrossRef CAS.
- L. Mattarozzi, S. Cattarin, N. Comisso, R. Gerbasi, P. Guerriero, M. Musiani, L. Vázquez-Gómez and E. Verlato, J. Electrochem. Soc., 2015, 162, D236–D241 CrossRef CAS.
- X. Zhang, S. Chen, F. Ai, L. Jin, N. Zhu and X. Z. Meng, Environ. Sci. Pollut. Res., 2021, 29, 12377–12386 CrossRef PubMed.
- S. P. Munasinghe-Arachchige, I. S. A. Abeysiriwardana-Arachchige, H. M. K. Delanka-Pedige and N. Nirmalakhandan, Algal Res., 2020, 51, 102023 CrossRef.
- E. Wołejko, U. Wydro, A. Jabłońska-Trypuć, A. Butarewicz and T. Łoboda, Environ. Pollut., 2018, 232, 347–357 CrossRef.
- X. Shi, L. Sang, X. C. Wang and P. Jin, Chem. Eng. J., 2018, 351, 240–247 CrossRef CAS.
- X. Lei, F. Liu, M. Li, X. Ma, X. Wang and H. Zhang, Chemosphere, 2018, 212, 237–244 CrossRef CAS PubMed.
- W. Gao, L. Gao, D. Li, K. Huang, L. Cui, J. Meng and J. Liang, J. Electroanal. Chem., 2018, 817, 202–209 CrossRef CAS.
- M. Yang, J. Wang, C. Shuang and A. Li, Chemosphere, 2020, 255, 126970 CrossRef CAS.
- J. Fu, F. Yao, T. Xie, Y. Zhong, Z. Tao, S. Chen, L. He, Z. Pi, K. Hou, D. Wang, X. Li and Q. Yang, Sep. Purif. Technol., 2021, 276, 119329 CrossRef CAS.
- I. Katsounaros and G. Kyriacou, Electrochim. Acta, 2007, 52, 6412–6420 CrossRef CAS.
- M. Li, C. Feng, Z. Zhang and N. Sugiura, Electrochim. Acta, 2009, 54, 4600–4606 CrossRef CAS.
- F. Ogata, Y. Uematsu, M. Fukuda, C. Saenjum, M. Kabayama, T. Nakamura and N. Kawasaki, J. Environ. Chem. Eng., 2020, 8, 104385 CrossRef CAS.
- N. W. Fan, Z. K. Li, L. Zhao, N. M. Wu and T. Zhou, Chem. Eng. J., 2013, 214, 83–90 CrossRef CAS.
- T. C. Chou, W. J. Chen, H. J. Tien, J. J. Jow and T. Nonaka, Electrochim. Acta, 1985, 30, 1665–1674 CrossRef CAS.
- Y. Wang, M. Chen, C. Wang, X. Meng, W. Zhang, Z. Chen and J. Crittenden, Chem. Eng. J., 2019, 374, 626–636 CrossRef CAS.
- X. Li, D. Shao, H. Xu, W. Lv and W. Yan, Chem. Eng. J., 2016, 285, 1–10 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*