
Efficient method of arsenic removal from water based on photocatalytic oxidation by a plasmonic–magnetic nanosystem†
María Y.
Paredes
a,
Luciana P.
Martinez
 b,
Beatriz C.
Barja
cd,
M. Claudia
Marchi
ce,
Matías
Herran
f,
Gustavo
Grinblat
be,
Andrea V.
Bragas
be,
Emiliano
Cortés
f and
Alberto F.
Scarpettini
*ag
b,
Beatriz C.
Barja
cd,
M. Claudia
Marchi
ce,
Matías
Herran
f,
Gustavo
Grinblat
be,
Andrea V.
Bragas
be,
Emiliano
Cortés
f and
Alberto F.
Scarpettini
*ag
aGrupo de Fotónica Aplicada, Facultad Regional Delta, Universidad Tecnológica Nacional, 2804 Campana, Argentina. E-mail: ascarpettini@frd.utn.edu.ar
bUniversidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Física, 1428 Buenos Aires, Argentina
cUniversidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Inorgánica Análítica y Química Física, 1428 Buenos Aires, Argentina
dCONICET - Universidad de Buenos Aires, Instituto de de Química, Física de los Materiales, Medio Ambiente y Energía (INQUIMAE), 1428 Buenos Aires, Argentina
eCONICET - Universidad de Buenos Aires, Instituto de Física de Buenos Aires (IFIBA), 1428 Buenos Aires, Argentina
fNanoinstitute Munich, Faculty of Physics, Ludwig-Maximilians-Universität München, 80539 Munich, Germany
gConsejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
First published on 13th December 2022
Abstract
Arsenic is one of the most toxic elements in natural waters since prolonged exposure to this metalloid can cause chronic damage to health. Its removal from groundwater remains one of the greatest environmental challenges to be addressed nowadays. Here, we present core–satellite hybrid nanostructures formed by plasmonic gold satellites supported onto magnetic iron oxide cores for sunlight-driven remediation of arsenic-containing water. Our experimental results show that the gold nanoparticles catalyze the oxidation of arsenic to much less toxic species and that – upon illumination – the generated heat and hot carriers further enhance the reaction rate. The iron oxides act as an arsenic adsorbent, enabling the complete removal of the catalysts and the adsorbed oxidized arsenic species through a magnet. We quantified the different catalytic contributions, showing that the plasmonic one is of the same order as the surface one. This work highlights the synergy between plasmonic catalysts and iron oxides for light-assisted water remediation.
Environmental significanceArsenic (As) poisoning of drinkable water is currently a big challenge, especially because there exists a strong correlation between As and cancer in humans. Notably, As(III) is 60 times more toxic than As(V). Developing technologies for efficient As extraction is then needed. In our study we integrate Au nanoparticles – visible light photocatalysts – to iron oxides (FexOy) to realize a hybrid structure achieving remarkable As(III)-to-As(V) oxidation and As adsorption. While Au nanoparticles increases the reaction rate 260 times, oxidizing almost 70% of the initial [As(III)] within one hour, FexOy adsorb the As and also enable its magnetic removal. This study stresses how relevant it is to combine materials with complementary functionalities to solve high-impact social problems. |
1. Introduction
The presence of arsenic (As) in drinkable water has become one of the central concerns in environmental science.1 Long exposure times to this highly toxic compound is strongly related to cancer in human beings.2 The As toxicity varies with the chemical state, the pH and the redox potential.3 For instance, As(III) is considered to be 60 times more toxic than As(V).4 This is the reason why most of the removal methods focus on oxidizing As(III) to its less poisonous form As(V).5Towards long-term solutions, methodologies for As removal should aim for cheap, sustainable, scalable and accessible technologies for populations at risk. In most of current technologies simultaneous or sequential chemical processes are employed.6,7 Iron oxides – abundant compounds in the environment – present a relatively high As adsorption in water.8 The magnetic nature of this adsorbent facilitates the As removal by simply using an external magnet. However, for the arsenic left unadsorbed, a prior oxidative treatment must be conducted before the removal step.6
One remediation route that has been largely explored in recent years is the photocatalytic oxidation of As(III) driven by TiO2 nanoparticles under UV illumination.9 However, the most common structures of TiO2, anatase and rutile, are only effective under irradiation wavelengths lower than 411 nm, representing just 5% of the sunlight emission. Exploring novel methods to convert As(III) to As(V) using sunlight is an appealing, cheap and accessible route, and new photocatalytic materials should be explored to absorb visible wavelengths, where the sun emits more prominently.
Plasmonic metal nanoparticles (MNPs) sustain localized surface plasmon resonances (LSPRs), which are collective and coherent oscillations of the free electrons of the metal, in response to an applied electromagnetic field. Plasmon frequencies strongly depend on the size, shape, material, and chemical environment of the MNP.10 Tuning these parameters enables control of their scattering/absorption cross-section ratio as well as their resonance frequency all over the visible + NIR spectrum, where the sun emission peaks. The LSPR phenomenon is accompanied by interesting physical effects such as the enhancement of the optical near-field, and the generation of hot charge carriers and local heat.11 All these processes can contribute to promoting or enhancing photocatalytic reactions at the surface of resonantly illuminated MNPs.12–14 Plasmonic catalysis has been recently used in sunlight-to-chemical energy conversion for solar fuel production (H2 generation and CO2 reduction),15,16 plasmon-assisted chemistry of organic compounds,17–19 hot-carrier-driven redox reactions,20–22 and degradation of environmental pollutants.23,24 However, there are far fewer examples of the reduction of inorganic ions such as arsenic.25–28 Gold and iron oxide hybrid nanoparticles have been used as bifunctional plasmonic–magnetic systems in the last few years,29–32 with applications in SERS detection and photocatalysis. Magnetic properties are usually exploited for selective separation of tagged molecules or recyclable catalysts.33–36
In this work we use colloidal core–satellite nanoparticles, formed by a magnetic iron oxide core of magnetite (Fe3O4) and maghemite (γ-Fe2O3) surrounded by gold nanoparticle (AuNP) satellites for the remediation of arsenical water. While the AuNPs provide their catalytic and photocatalytic properties on the oxidation of As(III) to As(V) (see the schematic in Fig. 1), the magnetic cores act as vehicles for As adsorption and removal from water with external magnets. We show that AuNPs are capable of efficiently photo-catalyzing this process when illuminating at their plasmonic resonance. Moreover, we disentangle the different contributions to the (photo)catalysis due to the surface of the AuNPs, the temperature increment by resonant irradiation and the hot carriers' generation after the plasmon decay. We also analyze their catalytic efficiency when the AuNPs are free or adsorbed on magnetic cores. Finally, we show the adsorption isotherms and discuss the remediation process.
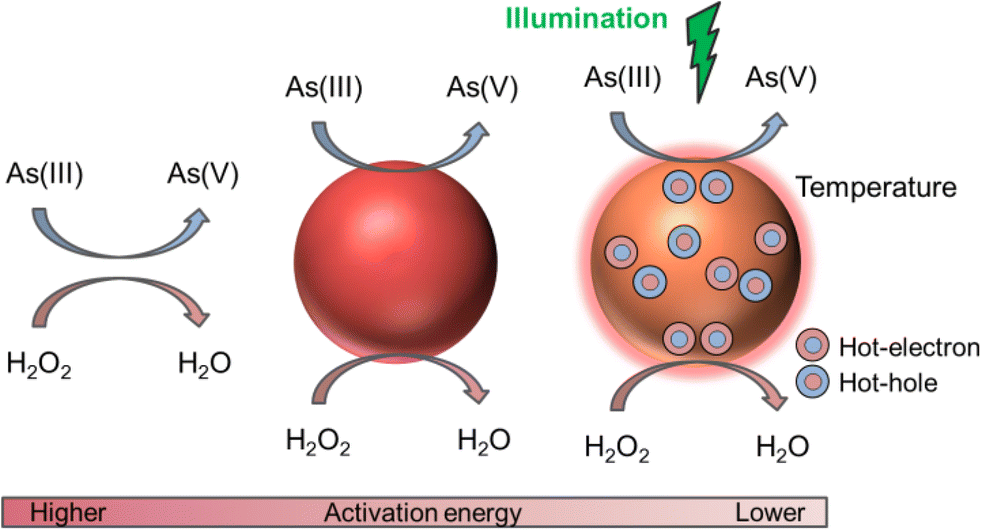 | ||
| Fig. 1 Schematic illustration of the mechanisms involved in the redox reaction between As(III) and H2O2 (left), assisted by the AuNP surface (middle), and under plasmon excitation (right), where the temperature increases and hot carriers reach the surface of the AuNP. | ||
2. Results and discussion
We start by studying the redox reaction kinetics with and without the plasmonic catalyst and evaluate the effect of different illumination and temperature conditions. The experimental data enabled us to decouple the mechanisms that accelerate the reaction and estimate their contributions. We then show an arsenic remediation method by using hybrid plasmonic–magnetic nanostructures to oxidize, adsorb and remove As. We study the performance of this nanosystem in the photocatalysis and adsorption processes.2.1. Homogeneous redox reaction
Conversion of arsenite As(III) to arsenate As(V) by the oxidant hydrogen peroxide H2O2 is a well-characterized reaction in the literature.37 The reaction consists of the following oxidation and reduction steps:| As(III) → As(V) + 2e− | (1) |
| H2O2 + 2H+ + 2e− → 2H2O | (2) |
Prior to our studies of the heterogeneous reaction with AuNPs, we characterized the kinetics of the homogeneous reaction using the molybdenum blue (MB) colorimetric method (see the Experimental methods section for details). We prepared samples of 1 mL volume with initial concentrations [As(III)]0 = 11 ppm and [H2O2]0 = 59 ppm and studied the As(III) to As(V) conversion (%) as a function of reaction time. The initial As(III) concentration was chosen to achieve complete conversion (see Fig. S3 in the ESI†). Fig. 2a shows the experimental data (gray triangles) of the As(III) to As(V) conversion as a function of time as well as the values calculated using the reaction rate constant determined by Pettine et al.37 (gray line) at the measured pH (7.3) and room temperature (24 °C). As can be seen in the graph, the experimental data agree very well with the redox kinetics. Note that for these [As(III)]0 and [H2O2]0 concentrations the kinetics is quite slow; it takes 3 hours to convert only 10% of the initial As(III) and the conversion is about 60% after four days. For the control sample without H2O2 (data not shown) the conversion rate – probably due to the oxidation of As(III) with the O2 of the environment – is less than 2% in 4 days.
 | ||
| Fig. 2 As(III) to As(V) conversion within a AuNP suspension, with initial concentrations [As(III)]0 = 11 ppm and [H2O2]0 = 59 ppm. (a) Arsenic conversion at 24 °C and pH = 7.3. Experimental data: homogeneous reaction (gray triangles), heterogeneous reaction with [50 nm AuNPs] = 6.0 × 1010 NP per mL (violet squares), [15 nm AuNPs] = 3.5 × 1011 NP per mL (blue diamonds) and [15 nm AuNPs] = 7.3 × 1011 NP per mL (blue circles). The concentrations [50 nm AuNPs] and the highest value of [15 nm AuNPs] have the same total exposed area of AuNPs, which is 4.8 cm2 mL−1. The solid lines are the fits using the heterogeneous reaction equation (see the ESI†) with E = p·Ea, p < 1, where Ea is the activation energy of the homogeneous reaction. (b) As(III) to As(V) conversion over 5 min as a function of [15 nm AuNPs]. (c) As(III) to As(V) conversion after 5 min of reaction under irradiation as a function of the light irradiance at 532 nm wavelength, for two different concentrations of 15 nm AuNPs. (d) Sample temperature evolution when illuminating with 532 nm wavelength at 1.9 W cm−2 light irradiance, for two different concentrations of AuNPs, measured inside the solution with a type K thermocouple. | ||
In this work we propose the use of AuNPs to accelerate this slow conversion rate. Under resonant illumination at the plasmon frequency, the AuNPs are the sources of heat and hot carriers which will further enhance the conversion of As(III) to As(V), as we will show in the following sections.
2.2. Heterogeneous redox catalysis
In this section, we analyze the reaction kinetics in the presence of AuNPs and compare it with the results of the homogeneous catalysis (without AuNPs). To understand how AuNPs assist the As oxidation under resonant irradiation, we first performed control experiments in the absence of light using 15 and 50 nm diameter AuNPs (see the Experimental methods section for synthesis details). Fig. 2a shows the As(III) to As(V) conversion as a function of time with the same initial concentrations ([As(III)]0 = 11 ppm and [H2O2]0 = 59 ppm) for the three systems: homogeneous, with 15 and 50 nm AuNPs, for concentrations [50 nm AuNPs] = 6.0 × 1010 NP per mL (violet squares), [15 nm AuNPs] = 3.5 × 1011 NP per mL (blue diamonds) and 7.3 × 1011 NP per mL (blue circles). The concentration [50 nm AuNPs] was chosen to have the same total area of AuNPs per mL (4.8 cm2 mL−1) as the higher value of [15 nm AuNPs]. We quantify the photocatalysis through an effective activation energy assigned to each heterogeneous system, defined as E = p·Ea, with p < 1, where Ea corresponds to the activation energy of the homogeneous reaction. The same pre-exponential factor is considered in all cases (see reaction equations in the ESI†). The obtained effective activation energies are indicated next to each curve (solid lines in the figure).We find that the presence of the NPs accelerates the redox reaction. The oxidation of As(III) is completed in one hour for 15 nm AuNPs and in less than one day for 50 nm AuNPs, significantly faster than the homogeneous redox process, even without illumination. Moreover, the smaller NPs are found to be the most efficient, as can be deduced by comparing the cases of [15 nm AuNPs] and [50 nm AuNPs] with the same total exposed area per mL. The different reaction rates may be due to the distinct surface curvatures of the AuNPs – more uncoordinated atoms in the smallest size – and their different colloidal mobilities. When the effective activation energy is E = 0.86·Ea (15 nm AuNP case with higher concentration), the rate of conversion is approximately 190 times greater than that of the homogeneous reaction at 24 °C. This increment in the rate of the reaction can be explained by the presence of active sites at the AuNP surfaces, which is consistent with a heterogeneous catalysis process. Note that the conversion rate increases with the AuNP concentration at a fixed AuNP size, as can be seen in Fig. 2b, due to the increment in the total exposed surface.
2.3. Heterogeneous redox catalysis assisted by localized surface plasmon resonance
In this section we study the reaction in the presence of 15 nm AuNPs, i.e., the most efficient catalyst, under resonant laser excitation at 532 nm. In the first experiment, samples with the same [As(III)]0 and [H2O2]0 initial concentrations – as in the previous cases – and two different AuNP concentrations (3.5 × 1011 NP per mL and 7.3 × 1011 NP per mL) were irradiated with different irradiances along the same reaction time (5 min). Fig. 2c reveals that the heterogeneous catalysis increases the conversion rate under light irradiation. As the light irradiance is increased, the conversion increases for both AuNP concentrations. Every time a sample is irradiated, an increase in the solution temperature is also observed, as can be seen in Fig. 2d, until an asymptotic value is reached in about 30 minutes (approximately 3 °C and 5 °C above room temperature for 3.5 × 1011 NP per mL and 7.3 × 1011 NP per mL colloid concentration, respectively, with 1.9 W cm−2 irradiance). We attribute this behavior to multiple scattering events in concentrated colloid media, which can enlarge the absorption cross-section by several orders of magnitude, as has been shown in the literature.38,39 Indeed, in the absence of thermal coupling, the temperature increase is predicted to be negligible under these illumination conditions (see the ESI†).40 In this scenario, a steady state involving NP absorption, heating and dissipation to the surrounding media is achieved after a few minutes. Accordingly, a higher final temperature was observed for the more concentrated sample. Control measurements were performed with out-of-resonance illumination, obtaining negligible temperature increments and As(III) to As(V) conversions close to the no irradiation case.In the second stage, we investigated the time evolution of the As(III)-to-As(V) oxidation at a fixed AuNP concentration (7.3 × 1011 NP per mL) and a fixed irradiance (1.9 W cm−2). We measured oxidized arsenic at 6 different times (2, 5, 15, 30, 45 and 60 minutes) under 4 different conditions (see the schematic in Fig. 3a): without irradiation at room temperature (H, heterogeneous), with irradiation and the consequent increase in temperature (HIT, heterogeneous-irradiation-thermal), without irradiation in a bath that reproduces the HIT heating curve (HT, heterogeneous-thermal), and with irradiation but in a bath kept at room temperature (HI, heterogeneous-irradiation). Thus, we sought to identify and quantify the heterogeneous, thermal and hot electron contributions to catalysis.
 | ||
| Fig. 3 (a) Diagrams of the redox experiments performed with AuNPs: heterogeneous catalysis (H), heterogeneous-thermal (HT), heterogeneous-irradiation (HI) and heterogeneous-irradiation-thermal (HIT). The cuvette is immersed in a water bath to keep it close to room temperature TR in HI, or to reproduce the HIT temperature increase but without irradiation in HT. (b) Arsenic conversion in a solution of AuNP colloids with arsenite and H2O2 after different reaction times at room temperature (H), with 1.9 W cm−2 plasmon-resonant light irradiation (HIT), without irradiation but in a bath that reproduces the HIT temperature increase (HT), and with irradiation in a bath that forces the system to remain at room temperature (HI). Error bars are below 3% in all cases. In the inset, the left column for each time shows the result of the heterogeneous catalysis (blue bar), added to the effective contributions of the temperature increase (red) and hot carriers generated by the light irradiation in resonance with the plasmon energy (green). The orange bars correspond to arsenic conversion due to all the contributions together under the HIT conditions, which nearly match the sum of individual effective contributions. (c) Characteristic times τ of the reactions plotted in (b), from which it follows that the oxidation in the HIT case is 2.2 times faster than that in the H case. | ||
Fig. 3b shows the arsenic conversion over one hour for the 4 mentioned cases, all of them tending towards complete oxidation. Experiments with irradiation and temperature always maintain conversion values above the heterogeneous catalysis (H). The HT case matches H at short times and reaches higher conversion values at longer times when the temperature is higher. In the irradiation cases (HI and HIT), a significant increase in the conversion is observed during the first 10 minutes, suggesting an important plasmonic contribution to the catalysis. Furthermore, HIT conversion values remain above HI values at all times. Fig. 3c shows the corresponding reaction times obtained from fitting the experimental data, revealing that the HIT reaction rate is 2.2 times faster than that of the H condition.
The different (effective) contributions to the catalysis at times prior to saturation can be visualized in the inset of Fig. 3b. The blue bars show the arsenic conversion by heterogeneous catalysis only (H), while the red and green segments correspond to the effective thermal (HT-H, i.e., we subtract H from HT) and hot carriers' (HI-H) contributions, respectively. We emphasize that these are effective contributions, which by construction will have a dominant heterogeneous component when approaching saturation. Moreover, the effective hot carriers' component is an estimated maximum value as there could be an additional local thermal contribution to photocatalysis around the AuNPs in the steady-state dissipation regime that cannot be separated. In the bar chart, the main incidence of the temperature increase is observed again at long times, while the hot carriers' contribution is more prominent at the beginning of the process. The orange bars show, for the same times, the conversion obtained in the HIT case, with all the contributions present (heterogeneous, thermal and irradiation). Notably, there is a great coincidence between the sum of the effective contributions determined separately and the HIT measurements, supporting this analysis. The small differences are below the colorimetric method error. A similar behavior can be observed when decreasing the initial concentration of As(III) to 4 and 2 ppm, as can be seen in the ESI.†
All these mechanisms in plasmon-induced photocatalysis not only halve the reaction time compared to heterogeneous catalysis, but also enable conversion values closer to total oxidation in the saturation regime. Reducing the concentration of As(III), the most toxic species, to the lowest possible value is a crucial aspect.
It should be noted that irradiance values used in our experiments are 2 to 3 orders of magnitude greater than the solar irradiance at the same wavelength. Hot spots in nanostructured photocatalysts could be used to achieve similar reaction times with sunlight. Optical and chemical hot spots are widely used in plasmonics to enhance the near-field, increase the light absorption, and optimize the reactivity of carriers.14 Sunlight concentrators and light-emitting diodes could also be used to increase the optical power and to further reduce the activation barrier of the redox reaction.41
2.4. Redox catalysis with core–satellite nanoparticles
The synthesized 15 nm diameter AuNPs were then adsorbed onto Fe3O4/γ-Fe2O3 magnetic nanoparticles in a core–satellite configuration, as described in the Experimental methods. This novel hybrid nanostructure not only allows for the oxidative catalysis of arsenic on AuNPs through the plasmonic effect under resonant irradiation, but also enables the adsorption of the arsenic species in water and finally its easy removal with a magnet. We performed the same H, HT, HI and HIT experiments (with the same H2O2 and As(III) initial concentrations) over the same times as the previous measurements plus 2 minutes, which is the time needed to magnetically remove the core–satellite NPs before measuring the arsenic concentration. The core–satellite NP concentration was adjusted such that it had the same AuNP concentration as in the preceding experiments (7.3 × 1011 AuNP per mL). We observed that the evolution of the measured temperature under the HIT conditions was the same as in the previous case without magnetic nanoparticles, reaching an increase of 5 °C with respect to room temperature in a few minutes for the same irradiance of 1.9 W cm−2. This fact confirms that only AuNPs absorb light, heat up and dissipate to the environment. Then, the same temperature curve of Fig. 2d (orange curve) was reproduced in the thermal bath for the HT experiments.Fig. 4a–d show the results obtained after different times of heterogeneous catalysis (H), with resonant irradiation and consequent heating (HIT), with a thermal bath following the heating curve but without irradiation (HT), and with a bath at room temperature under irradiation (HI). As(V) (circles) and unoxidized As(III) (squares) present in solution, and the arsenic adsorbed in the core–satellite NPs (triangles) were measured using the MB colorimetric method and known initial arsenic. The point-to-point sum of the 3 curves reaches 100% of the initial arsenic. In all cases, a growth of oxidized arsenic is observed over time, together with a decrease in non-oxidized arsenic, while arsenic adsorption on NPs rapidly saturates within a few minutes. If we compare these conversion values with those obtained with the previous system (only AuNPs), it is evident that iron oxides reduce the catalytic activity of AuNPs. The reaction rate in the H case is 120 times faster than that in the homogeneous case, and 1.6 times slower than with only AuNPs under the same conditions. The reason could be both a partial blocking of the exposed surface of the catalyst due to the AuNPs being attached to a silanized surface and a decrease in available arsenic due to its adsorption on iron oxides. In any case, this could be addressed by simply increasing the concentration of NPs. Comparing the H and HT measurements, we observe that there was a higher arsenic conversion when the thermal contribution was present, as expected from the previous analysis. In the same way, there was a greater conversion in the HI and HIT cases with irradiation (being even greater in the latter due to temperature), evidencing a large plasmonic contribution to catalysis. The reaction rate in the HIT case is 2.2 times faster than that in the H case (see Fig. 5b), the same factor as obtained with only AuNPs. In the cases without light irradiation, a little more than 10% of the arsenic is adsorbed, while in the cases with irradiation this value is less than 10%. This difference may be due to a higher local temperature around the AuNPs that attenuates the arsenic adsorption process.
 | ||
| Fig. 4 (a–d) Temporal evolution of As(III) (squares), As(V) (circles) and adsorbed As (triangles) on core–satellite NPs during arsenic oxidation and adsorption in the presence of [H2O2]0 = 59 ppm with initial [As(III)]0 = 11 ppm, under the 4 experimental conditions H, HT, HI and HIT, to distinguish the different contributions to catalysis. | ||
 | ||
| Fig. 5 (a) Comparison of the sum of heterogeneous (H, blue), thermal (HT-H, red), and plasmonic (HI-H, green) contributions to arsenic conversion with core–satellite NPs over time, with all contributions together in measurements under plasmon-resonant light (HIT, orange). A great catalytic contribution of the hot carriers is observed. (b) Characteristic times τ of the reactions plotted in (a), from which it follows that the oxidation in the HIT case is 2.2 times faster than that in the H case. (c) Snapshots of the core–satellite NP separation process using a magnet placed next to the cuvette. In the left panel, a homogeneous coloration is observed at the beginning, and after one minute the NPs are located against the wall of the container (right panel). Through UV-vis spectroscopy we ensured a complete separation after 2 minutes. | ||
We can now calculate the effective thermal (HT-H) and hot carriers' (HI-H) contributions at each time and add them to the heterogeneous catalysis (H), and then compare all of them with the HIT measurements, as performed before with the case of bare AuNPs as shown in Fig. 3b. In Fig. 5a we observe the accumulated bars of these 3 contributions separately: heterogeneous (blue), thermal (red) and hot carriers (green), in excellent agreement with all these contributions integrated in HIT (orange). It can be noticed that the effective hot carriers' contribution to catalysis is of the same order as the surface contribution, each one responsible for about half of arsenic oxidation over time and both greater than the thermal component. Experiments carried out with lower initial arsenic concentrations ([As(III)]0 = 2 and 4 ppm) can be found in the ESI.†
Finally, the complete removal of the catalyst and adsorbed arsenic is achieved by using the magnetic character of the hybrid nanosystem. A magnet is placed against the container wall and within two minutes the core–satellite NPs accumulate next to it. Fig. 5c shows two snapshots of the magnetic separation process from a video available in the ESI.† We verified by UV-vis spectroscopy that there were no detectable traces of the nanocatalyst in the liquid after extraction from the beaker.
2.5. Arsenic adsorption
As can be observed in Fig. 4a–d, the arsenic adsorption in the core–satellite NPs is around 10%. To gain further insight into the adsorption process, Fig. 6a and b show the adsorption isotherms of (a) As(III) and (b) As(V) on magnetic NPs (violet circles), on magnetic NPs functionalized with APTMS (green squares) and on core–satellite NPs (brown triangles) measured at room temperature. The representative SEM image of core–satellite NPs showing AuNPs attached on the APTMS functionalized surface of iron oxide NPs can be seen in Fig. 6c. Each point in Fig. 6a and b corresponds to a different initial arsenic concentration (up to 18 ppm), while the adsorbent mass remained constant. The total arsenic in equilibrium [As]e was measured after 24 hours, the difference with the initial arsenic concentration is the adsorbed fraction. At low initial arsenic concentrations, the isotherms follow the Langmuir model (with total adsorption). However, at higher concentrations no saturation is observed, probably indicating a multilayer adsorption process. It can be observed that iron oxides without silanes or AuNPs are a better adsorbent and that both arsenic species are adsorbed similarly, consistent with literature reports.42–44 It is interesting to note that for higher initial arsenic concentrations, the presence of AuNPs (brown triangles) improves its adsorption compared to magnetic NPs with the APTMS layer (green squares). This result suggests that As species can also be adsorbed on the gold surface,45 as also confirmed by EDX analysis (energy dispersive X-ray spectroscopy) performed on one of the samples used in the oxidation processes under HIT conditions. As can be observed from the TEM (Fig. 6d) and HAADF-STEM images (Fig. 6e), and the sequence of EDX images showing the spatial location of iron (Fig. 6f, pink), gold (Fig. 6g, yellow) and arsenic (Fig. 6h, blue) in the same area, the presence of arsenic is detected not only in the iron oxide areas, but also in the AuNPs. To adsorb all the arsenic present in solution, oxidized or not, and thus have a good water remediation method, an extra amount of iron oxide magnetic NPs (without AuNPs) must be added. Any small traces of unadsorbed arsenic (below health standards) would be harmless owing to the previous photocatalytic oxidation process.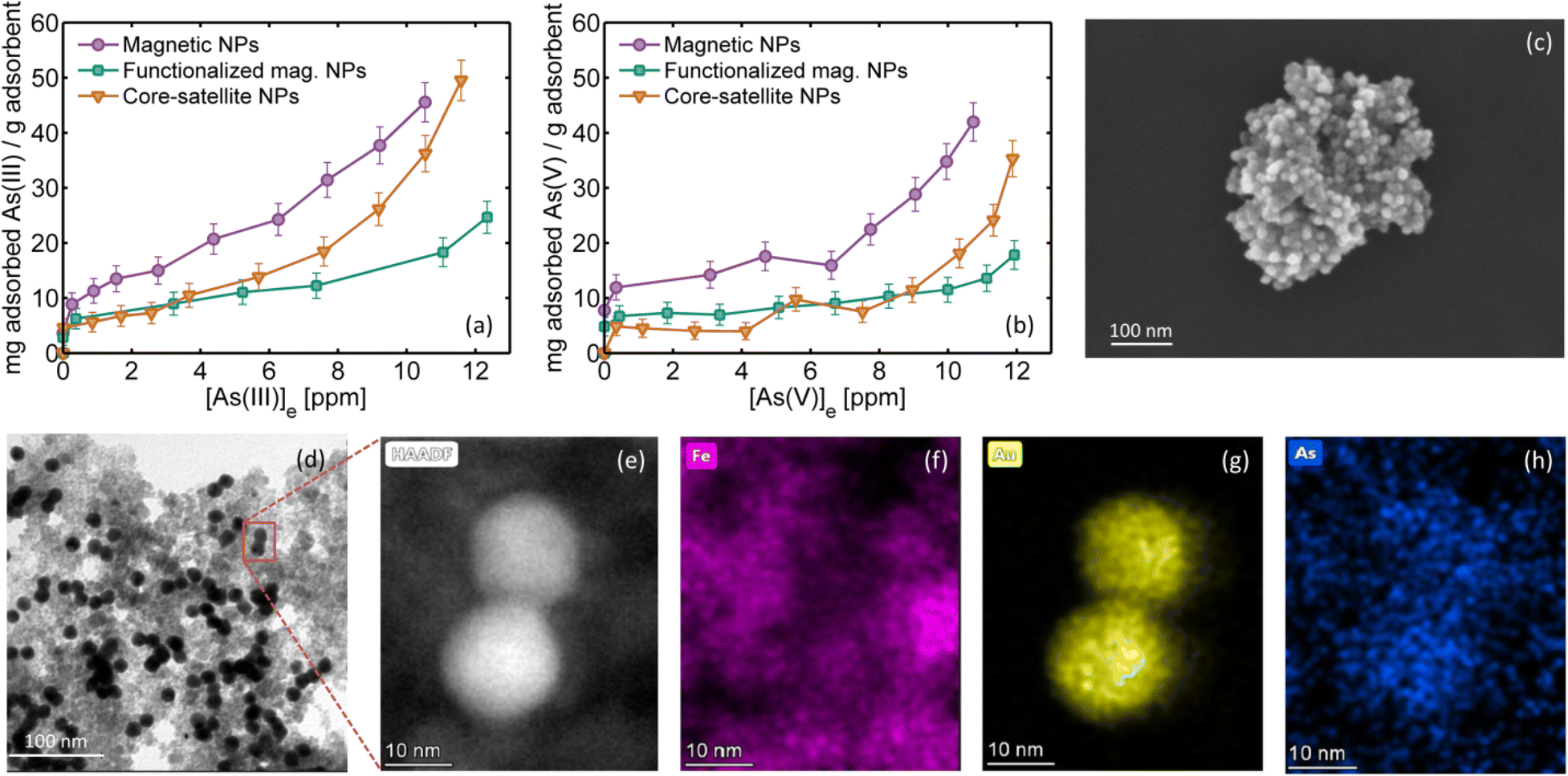 | ||
| Fig. 6 Adsorption isotherms at room temperature for (a) As(III) and (b) As(V) on magnetic (iron oxides) NPs (violet circles), on APTMS functionalized magnetic NPs (green squares) and on core–satellite NPs (brown triangles). (c) SEM and (d) TEM images of core–satellite NPs. Gold NPs can be clearly seen attached to iron oxide surfaces. (e) HAADF-STEM image of two AuNPs adsorbed on magnetic NPs. (f–h) EDX images of the same region with the spatial localization of iron (pink), gold (yellow) and arsenic (blue). | ||
Contaminant desorption studies must be performed to ensure the regeneration and reusability of the nanosystem, and how its efficiency evolves over successive remediation cycles. Furthermore, natural waters contaminated with arsenic are complex systems where other ions and organic molecules compete for adsorption sites. Experiments have been performed to understand the competition between arsenic and phosphates on the surface of iron oxides.42,46 Finally, photocatalytic tests should also be performed with nanosystems using less expensive plasmonic materials to compare their efficiency with gold and to assess the most suitable catalyst.47,48
3. Conclusions
This work is the first study on the remediation of arsenic-containing water using core–satellite nanoparticles. The magnetic–plasmonic hybrid catalysts oxidize As(III) to the less toxic As(V), and also enable magnetic removal of the catalyst and the adsorbed contaminant. The inclusion of 15 nm AuNPs led to a 190 times larger oxidation rate. Moreover, when resonantly illuminating with an irradiance of 1.9 W cm−2, the redox process is even 2.2 times faster. The same AuNPs, supported on iron oxides in the core–satellite nanosystem, reach a reaction rate 120 times higher than the homogeneous case and, when illuminated, 2.2 times higher than the heterogeneous catalysis. Our results indicate that an improved performance in As removal is accomplished once materials with complementary functionalities are engineered. This study brings insight into the design rules for more efficient technologies for water remediation.4. Experimental methods
4.1. Gold nanoparticles
Gold nanoparticles were synthesized by citrate reduction in boiling water. The final size of the colloids was controlled by the initial molar ratio between tetrachloroauric(III) acid (HAuCl4) and trisodium citrate (Na3Ct) following the traditional Frens's synthesis.49–51 Nearly monodisperse AuNPs with diameters and standard deviation of (14.4 ± 1.0) nm and (50.2 ± 5.5) nm were obtained with 0.19 and 0.78 HAuCl4/Na3Ct ratios, respectively, denoted as 15 and 50 nm AuNPs. The extinction spectra of the aqueous colloid solutions, SEM images and diameter histograms are shown in Fig. S1 of the ESI.†4.2. Magnetic–plasmonic core–satellite nanoparticles
Fe3O4/γ-Fe2O3 magnetic nanoparticles were prepared by coprecipitation of iron precursors according to the Massart method.52,53 4 mL of an aqueous solution of FeCl3 (1 M) and 1 mL of an acidic solution of FeSO4 (2 M) in HCl (2 M) were mixed. This solution was added to a NH4OH solution (0.7 M, 50 mL) under rapid stirring, and it was kept for 30 minutes. The precipitate was allowed to settle and most of the supernatant was discarded. Then, the dispersion was washed 3 times magnetically (NPs were separated from the suspension by holding a magnet against the beaker) with 60 mL of NH4OH 0.7 M (pH: 11), final volume.To functionalize the surface, 15 mL of Fe3O4/γ-Fe2O3 nanoparticle suspension was dispersed in 50 mL of NH4OH solution (pH: 11). A solution of (3-aminopropyl)trimethoxysilane, APTMS (0.6 M) was added in excess to cover the entire surface of the particles, and it was kept in an ultrasonic bath at 30 °C in the dark for 2 hours. The functionalized nanoparticles were magnetically washed 3 times to remove excess reagents and dispersed in 30 mL of pure water.
The formation of core–satellite nanostructures was achieved through electrostatic attraction between the particles by adding a solution of 15 nm AuNPs to the aqueous dispersion of silanized magnetic nanoparticles in a ratio of 1.7 × 1015 AuNPs per gram of magnetic NPs.54 The resulting solution was kept under stirring for one hour and then at rest for 24 hours. Then, it was purified twice with a magnet, discarding the supernatant, and dispersing in the same volume of pure water. The assembly was monitored by UV-vis spectroscopy. We ensured the completeness of AuNPs adsorption on the magnetic NPs, since no LSPR peak was observed at the supernatant in each purification.
4.3. Sample preparations
For the studies of the redox reaction, two stock aqueous solutions of 100 ppm sodium arsenite (NaAsO2 salt analytical grade, Carlo-Erba) and 3000 ppm hydrogen peroxide (H2O2) were prepared. The samples were obtained by diluting aliquots of the stock solutions to a final volume of 1.00 mL in a glass cuvette with ultrapure water. The total initial As(III) sample concentrations, [As(III)]0, were controlled in the 1–20 ppm range while the total initial H2O2 sample concentrations [H2O2]0 were fixed at 59 ppm.4.4. Detection of arsenite oxidation
The colorimetric method of molybdenum blue (MB)55,56 is a very selective, sensitive and accessible method for detecting As(V), which simply consists in the addition of a colored reagent to the samples. The detection relies on the formation of the arsenic-molybdic complex between arsenate As(V) and ammonium molybdate (in the presence of a reducing agent), turning the solution bluer with the increasing As(V) concentration. This enables quantifying the As(V) concentration through absorbance measurements around 900 nm.56 The addition of the color reagent lowers the pH and stops the redox reaction.In this work, we used the MB colorimetric method to determine the conversion from As(III) to As(V) on a redox reaction. First, we prepared samples with known [As(III)]0 in presence of [H2O2]0 and nanoparticles, and then left them for a controlled period of time during which the oxidation happened. Finally, we added the color reagent to the samples and measured the absorbance of the arsenic–molybdic complex. With a calibration curve for the MB colorimetric method (Fig. S2 in the ESI†) we determined the final [As(V)] on the samples. In this way, we quantified the percentage of As(III) to As(V) conversion (%) as a function of reaction time, under different experimental conditions, with an error below 3%. For further details, see the ESI.†
In the case of samples with core–satellite NPs, which oxidize and adsorb arsenic, an additional step must be taken into account to quantify the As(III) to As(V) conversion rate. Once the photocatalysis was performed, a first aliquot was extracted after the reaction time and the amount of As(V) present in solution was measured using the MB colorimetric method. Then, in a second aliquot the remaining As(III) was first oxidized to As(V) using potassium permanganate (KMnO4 0.21 mM), thus quantifying the total As content in solution. The unreacted As(III) in solution was then calculated by subtracting the As(V) measured in the first aliquot to the latter value. The difference with the known initial arsenic present in each sample gave us the arsenic adsorbed in the core–satellite NPs.
4.5. Redox experiments
The catalytic performance of AuNPs was studied through four types of experiments to evaluate the heterogeneous (H), thermal (T) and irradiation (I) contributions. These experiments are named and designed as follows (see Fig. 3a):(1) H, heterogeneous catalysis. The solution is kept at room temperature in darkness.
(2) HIT, heterogeneous-irradiation-thermal catalysis. The solution is irradiated with a laser in the absence of an external temperature controller, thus allowing the temperature to rise and stabilize.
(3) HT, heterogeneous-thermal. The solution is immersed in a thermal bath that reproduces the temperature evolution in HIT but is kept in darkness.
(4) HI, heterogeneous-irradiation. The solution is irradiated with a laser and immersed in a thermal bath kept at room temperature.
First, we assessed the heterogeneous catalysis (H): AuNPs are added to the sample solution with As(III) and the oxidizing agent H2O2, the solution is kept at room temperature (24 °C) in dark conditions for a certain time, and then the amount of oxidized arsenic is measured with the MB colorimetric method. The procedure is also performed with core–satellite NPs, where in addition to the previous measurement, the quantity of unreacted As(III) and adsorbed arsenic on the NPs were estimated from a second aliquot. By comparing these experiments, we were not only able to determine changes in the AuNPs catalytic activity once they were embedded in the iron oxide matrix, but also the As removal efficiency in the absence of green light.
Second, to investigate the photocatalytic performance of the system, the same experiment was conducted under identical conditions but now under green light illumination along the reaction course. A 532 nm wavelength laser at 0.75 W power (1.9 W cm−2 irradiance) was employed as the light source. We observe that during this irradiation process the temperature of the sample increases a few degrees and stabilizes in about 30 minutes (see Fig. 2d for temporal evolution of sample temperature), due to light absorption of AuNPs and their consequent heat dissipation towards the aqueous medium. Through these experiments, we evaluated the thermal and hot carriers' contribution to the catalysis, in addition to the heterogeneous contribution (HIT: heterogeneous-irradiation-thermal).
Third, to disentangle the thermal component from the hot carriers' contribution, the first experiment (no irradiation) was performed employing now a thermal bath (HT) reproducing the temperature increase in the previous experiment, where the sample was irradiated with green light.
Last, the light contribution (HI) was evaluated by illuminating with green light and controlling the overall temperature of the sample by means of an external water bath with fixed temperature at 24 °C. This last scenario is equivalent to adding a cold source to quickly dissipate the heat accumulated in the solution due to the plasmonic heating of the nanoparticles. The temperature gradient from the AuNPs surface to the medium is still present along with a hot carriers' contribution, both are plasmonic and catalytic effects that contribute to accelerating the arsenic oxidation.
These catalytic tests were performed during times of 2, 5, 15, 30, 45 and 60 minutes, with additional 2 minutes for each time in the case of magnetic NPs, since this is the time that takes to separate them with a magnet before adding the color reagent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by PICT 2017-2534, UBACyT Proyecto 20020170100432BA, PID-UTI4836 and PID-UTI6567. The authors also acknowledge funding and support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under grant numbers EXC 2089/1-390776260 (Germany's Excellence Strategy), the Bavarian program Solar Energies Go Hybrid (SolTech), the Center for NanoScience (CeNS), the DAAD Programme for Project-Related Personal Exchange (PPP) 57573042, the European Commission for the ERC-STG Catalight 802989 and the Royal Society for the Challenge Grants CH-2016, CH160100.References
- B. K. Mandal and K. T. Suzuki, Arsenic Round the World: A Review, Talanta, 2002, 58(1), 201–235, DOI:10.1016/S0039-9140(02)00268-0
.
- World Health Organization, International Programme on Chemical Safety (Environmental Health Criteria 224), Arsenic and arsenic compounds, World Health Organization, Geneva, 2nd edn, 2001. https://whqlibdoc.who.int/ehc/WHO_EHC_224.pdf Search PubMed.
- D. K. Nordstrom, J. Majzlan and E. Königsberger, Thermodynamic Properties for Arsenic Minerals and Aqueous Species, Rev. Mineral. Geochem., 2014, 79(1), 217–255, DOI:10.2138/rmg.2014.79.4
-
J. Bundschuh, A. Pérez Carrera and M. I. Litter, Distribución del Arsénico en las Regiones Ibérica e Iberoamericana, CYTED, Ciencia y Tecnología para el Desarrollo, 2008 Search PubMed
- E. A. Rangel Montoya, L. E. Montañez Hernández, M. P. Luévanos Escareño and N. Balagurusamy, Impacto del Arsénico en el Ambiente y su Transformación por Microorganismos, Terra Latinoam., 2015, 33(2), 103–118 Search PubMed
-
M. I. Litter, A. M. Sancha and A. M. Ingallinella, Tecnologías Económicas para el Abatimiento de Arsénico en Aguas, CYTED, Ciencia y Tecnología para el Desarrollo, 2010 Search PubMed
-
M. de Boggio, I. Levy, M. Mateu, J. M. Meichtry, S. S. Farias, G. Lopez, D. Bahnemann, R. Dillert and M. I. Litter, Low-Cost Solar Technologies for Arsenic Removal in Drinking Water: Challenges for Safe Water Production, in The Global Arsenic Problem, CRC Press, 2010, pp. 209–218. DOI:10.1201/b10537-21
- M. Martínez-Cabanas, M. López-García, J. L. Barriada, R. Herrero and M. Sastre de Vicente, Green Synthesis of Iron Oxide Nanoparticles, Development of Magnetic Hybrid Materials for Efficient As(V) Removal, Chem. Eng. J., 2016, 301, 83–91, DOI:10.1016/j.cej.2016.04.149
- P. M. Jayaweera, P. I. Godakumbura and K. A. S. Pathiratne, Photocatalytic Oxidation of As(III) to As(V) in Aqueous Solutions: A Low Cost Pre-Oxidative Treatment for Total Removal of Arsenic from Water, Curr. Sci., 2003, 84(4), 541–543 CAS
-
S. A. Maier, Plasmonics: Fundamentals and Applications, Springer, 2007, p. 245 Search PubMed
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-Induced Hot Carrier Science and Technology, Nat. Nanotechnol., 2015, 10(1), 25–34, DOI:10.1038/nnano.2014.311
- E. Cortés, Efficiency and Bond Selectivity in Plasmon-Induced Photochemistry, Adv. Opt. Mater., 2017, 5(15), 1–13, DOI:10.1002/adom.201700191
- E. Cortés, R. Grzeschik, S. A. Maier and S. Schlücker, Experimental characterization techniques for plasmon-assisted chemistry, Nat. Rev. Chem., 2022, 6, 259–274, DOI:10.1038/s41570-022-00368-8
- J. Gargiulo, R. Berté, Y. Li, S. A. Maier and E. Cortés, From Optical to Chemical Hot Spots in Plasmonics, Acc. Chem. Res., 2019, 52(9), 2525–2535, DOI:10.1021/acs.accounts.9b00234
- M. Herran, A. Sousa-Castillo, C. Fan, S. Lee, W. Xie, M. Döblinger, B. Auguié and E. Cortés, Tailoring Plasmonic Bimetallic Nanocatalysts Toward Sunlight-Driven H2 Production, Adv. Funct. Mater., 2022, 2203418, DOI:10.1002/adfm.202203418
- S. Ezendam, M. Herran, L. Nan, C. Gruber, Y. Kang, F. Gröbmeyer, R. Lin, J. Gargiulo, A. Sousa-Castillo and E. Cortés, Hybrid Plasmonic Nanomaterials for Hydrogen Generation and Carbon Dioxide Reduction, ACS Energy Lett., 2022, 7(2), 778–815, DOI:10.1021/acsenergylett.1c02241
- A. Gellé, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Applications of Plasmon-Enhanced Nanocatalysis to Organic Transformations, Chem. Rev., 2020, 120(2), 986–1041, DOI:10.1021/acs.chemrev.9b00187
- A. Stefancu, L. Nan, L. Zhu, V. Chis, I. Bald, M. Liu, N. Leopold, S. A. Maier and E. Cortes, Controlling Plasmonic Chemistry Pathways through Specific Ion Effects, Adv. Opt. Mater., 2022, 10, 2200397, DOI:10.1002/adom.202200397
- A. Stefancu, O. M. Biro, O. Todor-Boer, I. Botiz, E. Cortés and N. Leopold, Halide–Metal Complexes at Plasmonic Interfaces Create New Decay Pathways for Plasmons and Excited Molecules, ACS Photonics, 2022, 9(3), 895–904, DOI:10.1021/acsphotonics.1c01714
- E. Cortés, W. Xie, J. Cambiasso, A. S. Jermyn, R. Sundararaman, P. Narang, S. Schlücker and S. A. Maier, Plasmonic hot electron transport drives nano-localized chemistry, Nat. Commun., 2017, 8, 14880, DOI:10.1038/ncomms14880
- E. Pensa, J. Gargiulo, A. Lauri, S. Schlücker, E. Cortés and S. A. Maier, Spectral Screening of the Energy of Hot Holes over a Particle Plasmon Resonance, Nano Lett., 2019, 19(3), 1867–1874, DOI:10.1021/acs.nanolett.8b04950
- W. Xie and S. Schlücker, Hot electron-induced reduction of small molecules on photorecycling metal surfaces, Nat. Commun., 2015, 6, 7570, DOI:10.1038/ncomms8570
- H. Wei, S. K. Loeb, N. J. Halas and J. H. Kim, Plasmon-enabled degradation of organic micropollutants in water by visible-light illumination of Janus gold nanorods, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(27), 15473–15481, DOI:10.1073/pnas.2003362117
- M. E. King, C. Wang, M. V. Fonseca Guzman and M. B. Ross, Plasmonics for environmental remediation and pollutant degradation, Chem. Catalysis, 2022, 2, 1–13, DOI:10.1016/j.checat.2022.06.017
- N. Ortiz, B. Zoellner, S. J. Hong, Y. Ji, T. Wang, Y. Liu, P. A. Maggard and G. Wang, Harnessing Hot Electrons from Near IR Light for Hydrogen Production Using Pt-End-Capped-AuNRs, ACS Appl. Mater. Interfaces, 2017, 9(31), 25962–25969, DOI:10.1021/acsami.7b05064
- M. D. Bordenave, A. F. Scarpettini, M. V. Roldán, N. Pellegri and A. V. Bragas, Plasmon-induced photochemical synthesis of silver triangular prisms and pentagonal bipyramids by illumination with light emitting diodes, Mater. Chem. Phys., 2013, 139(1), 100–106, DOI:10.1016/j.matchemphys.2012.12.061
- Z. Zhai, J. S. DuChene, Y. C. Wang, J. Qiu, A. C. Johnston-Peck, B. You, W. Guo, B. DiCiaccio, K. Qian, E. W. Zhao, F. Ooi, D. Hu, D. Su, E. A. Stach, Z. Zhu and W. D. Wei, Polyvinylpyrrolidone-induced anisotropic growth of gold nanoprisms in plasmon-driven synthesis, Nat. Mater., 2016, 15, 889–895, DOI:10.1038/nmat4683
- R. Kamarudheen, G. W. Castellanos, L. P. J. Kamp, H. J. H. Clercx and A. Baldi, Quantifying Photothermal and Hot Charge Carrier Effects in Plasmon-Driven Nanoparticle Syntheses, ACS Nano, 2018, 12(8), 8447–8455, DOI:10.1021/acsnano.8b03929
- K. C.-F. Leung, S. Xuan, X. Zhu, D. Wang, C.-P. Chak, S.-F. Lee, W. K.-W. Hob and B. C.-T. Chung, Gold and iron oxide hybrid nanocomposite materials, Chem. Soc. Rev., 2012, 41, 1911–1928, 10.1039/C1CS15213K
- D. Wang and D. Astruc, Fast-Growing Field of Magnetically Recyclable Nanocatalysts, Chem. Rev., 2014, 114(14), 6949–6985, DOI:10.1021/cr500134h
- S. Moraes Silva, R. Tavallaie, L. Sandiford, R. D. Tilley and J. J. Gooding, Gold coated magnetic nanoparticles: from preparation to surface modification for analytical and biomedical applications, Chem. Commun., 2016, 52, 7528–7540, 10.1039/C6CC03225G
- J. Canet-Ferrer, P. Albella, A. Ribera, J. V. Usagre and S. A. Maier, Hybrid magnetite–gold nanoparticles as bifunctional magnetic–plasmonic systems: three representative cases, Nanoscale Horiz., 2017, 2, 205–216, 10.1039/C6NH00225K
- P. Quaresma, I. Osório, G. Dória, P. A. Carvalho, A. Pereira, J. Langer, J. P. Araújo, I. Pastoriza-Santos, L. M. Liz-Marzán, R. Franco, P. V. Baptistac and E. Pereira, Star-shaped magnetite@gold nanoparticles for protein magnetic separation and SERS detection, RSC Adv., 2014, 4, 3659–3667, 10.1039/C3RA46762G
- Y. Li, J. Zhao, W. You, D. Chenga and W. Ni, Gold nanorod@iron oxide core–shell heterostructures: synthesis, characterization, and photocatalytic performance, Nanoscale, 2017, 9, 3925–3933, 10.1039/C7NR00141J
- M. Melchionna, A. Beltram, A. Stopin, T. Montini, R. W. Lodge, A. N. Khlobystov, D. Bonifazi, M. Prato and P. Fornasiero, Magnetic shepherding of nanocatalysts through hierarchically-assembled Fe-filled CNTs hybrids, Appl. Catal., B, 2018, 227, 356–365, DOI:10.1016/j.apcatb.2018.01.049
- F. V. Guzman, P. A. Mercadal, E. A. Coronado and E. R. Encina, Near-Field Enhancement Contribution to the Photoactivity in Magnetite–Gold Hybrid Nanostructures, J. Phys. Chem. C, 2019, 123(49), 29891–29899, DOI:10.1021/acs.jpcc.9b09421
- M. Pettine, L. Campanella and F. J. Millero, Arsenite Oxidation by H2O2 in Aqueous Solutions, Geochim. Cosmochim. Acta, 1999, 63(18), 2727–2735, DOI:10.1016/S0016-7037(99)00212-4
- N. J. Hogan, A. S. Urban, C. Ayala-Orozco, A. Pimpinelli, P. Nordlander and N. J. Halas, Nanoparticles Heat through Light Localization, Nano Lett., 2014, 14(8), 4640–4645, DOI:10.1021/nl5016975
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, Challenges in Plasmonic Catalysis, ACS Nano, 2020, 14(12), 16202–16219, DOI:10.1021/acsnano.0c08773
- G. Baffou and H. Rigneault, Femtosecond-Pulsed Optical Heating of Gold Nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 035415, DOI:10.1103/PhysRevB.84.035415
- E. Cortés, Activating plasmonic chemistry, Science, 2018, 362(6410), 28–29, DOI:10.1126/science.aav1133
- S. Dixit and J. G. Hering, Comparison of Arsenic(V) and Arsenic(III) Sorption onto Iron Oxide Minerals: Implications for Arsenic Mobility, Environ. Sci. Technol., 2003, 37(18), 4182–4189, DOI:10.1021/es030309t
- C. H. Liu, Y. H. Chuang, T. Y. Chen, Y. Tian, H. Li, M. K. Wang and W. Zhang, Mechanism of arsenic adsorption on magnetite nanoparticles from water: thermodynamic and spectroscopic studies, Environ. Sci. Technol., 2015, 49(13), 7726–7734, DOI:10.1021/acs.est.5b00381
- J. T. Mayo, C. Yavuz, S. Yean, L. Cong, H. Shipley, W. Yu, J. Falkner, A. Kan, M. Tomson and V. L. Colvin, The effect of nanocrystalline magnetite size on arsenic removal, Sci. Technol. Adv. Mater., 2007, 8, 71–75, DOI:10.1016/j.stam.2006.10.005
- C. Zong, Z. Zhang, B. Liu and J. Liu, Adsorption of arsenite on gold nanoparticles studied with DNA oligonucleotide probes, Langmuir, 2019, 35(22), 7304–7311, DOI:10.1021/acs.langmuir.9b01161
- H. J. Shipley, S. Yean, A. T. Kan and M. B. Tomson, A Sorption Kinetics Model for Arsenic Adsorption to Magnetite Nanoparticles, Environ. Sci. Pollut. Res., 2010, 17(5), 1053–1062, DOI:10.1007/s11356-009-0259-5
- S. Rej, L. Mascaretti, E. Y. Santiago, O. Tomanec, S. Kment, Z. Wang, R. Zbořil, P. Fornasiero, A. O. Govorov and A. Naldoni, Determining Plasmonic Hot Electrons and Photothermal Effects during H2 Evolution with TiN–Pt Nanohybrids, ACS Catal., 2020, 10(9), 5261–5271, DOI:10.1021/acscatal.0c00343
- A. Naldoni, Z. A. Kudyshev, L. Mascaretti, S. P. Sarmah, S. Rej, J. P. Froning, O. Tomanec, J. E. Yoo, D. Wang, S. Kment, T. Montini, P. Fornasiero, V. M. Shalaev, P. Schmuki, A. Boltasseva and R. Zbořil, Solar Thermoplasmonic Nanofurnace for High-Temperature Heterogeneous Catalysis, Nano Lett., 2020, 20(5), 3663–3672, DOI:10.1021/acs.nanolett.0c00594
- G. Frens, Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, Turkevich Method for Gold Nanoparticle Synthesis Revisited, J. Phys. Chem. B, 2006, 110(32), 15700–15707, DOI:10.1021/jp061667w
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, Size Control of Gold Nanocrystals in Citrate Reduction: The Third Role of Citrate, J. Am. Chem. Soc., 2007, 129(45), 13939–13948, DOI:10.1021/ja074447k
- R. Massart, Preparation of aqueous magnetic liquids in alkaline and acid media, IEEE Trans. Magn., 1981, MAG-17(2), 1247–1248 CrossRef CAS
- V. Salgueiriño-Maceira, M. A. Correa-Duarte, M. Farle, A. López-Quintela, K. Sieradzki and R. Diaz, Bifunctional Gold-Coated Magnetic Silica Spheres, Chem. Mater., 2006, 18(11), 2701–2706, DOI:10.1021/cm0603001
- A. F. Scarpettini and A. V. Bragas, Coverage and aggregation of gold nanoparticles on silanized glasses, Langmuir, 2010, 26(20), 15948–15953, DOI:10.1021/la102937b
- D. L. Johnson and M. E. Q. Pilson, Spectrophotometric Determination of Arsenite, Arsenate, and Phosphate in Natural Waters, Anal. Chim. Acta, 1972, 58(2), 289–299, DOI:10.1016/S0003-2670(72)80005-9
- V. Lenoble, V. Deluchat, B. Serpaud and J. C. Bollinger, Arsenite Oxidation and Arsenate Determination by the Molybdene Blue Method, Talanta, 2003, 61(3), 267–276, DOI:10.1016/S0039-9140(03)00274-1
Footnote |
| † Electronic supplementary information (ESI) available: Details about the characterization of the AuNPs, the arsenic detection in water by the colorimetric method of molybdenum blue (MB), redox reaction equations and experimental results supporting the catalysis of the oxidation of As(III) by AuNPs, theoretical calculations of the temperature increase in AuNP solutions induced by light, experimental results with lower arsenic concentrations, and a video of the magnetic separation of the nanosystem from water. See DOI: https://doi.org/10.1039/d2en01082h |
| This journal is © The Royal Society of Chemistry 2023 |