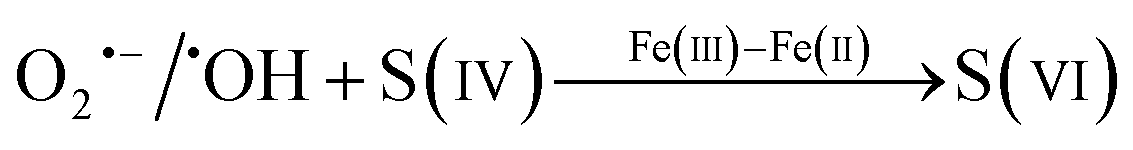Atmospheric fates of SO2 at the gas–solid interface of iron oxyhydroxide (FeOOH) minerals: effects of crystal structure, oxalate coating and light irradiance†
Received
22nd September 2022
, Accepted 18th November 2022
First published on 19th November 2022
Abstract
Iron oxyhydroxide (FeOOH) is one of the most abundant atmospheric iron oxides. Considering their diverse emission features and reaction processes, airborne FeOOH minerals present distinct crystal structures and coating compositions. Nevertheless, little attention has been paid to the atmospheric fates of SO2 over the gas–solid interface of FeOOH, let alone the impacts of sunlight and its irradiance. Herein, by employing in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), the heterogeneous reaction of SO2 was investigated on nanoscale FeOOH with different crystal structures, followed by a discussion on the effects of oxalate coating and light irradiance. Results indicated that the heterogeneous uptake capacity varies with crystal structure (α-FeOOH > β-FeOOH > γ-FeOOH), probably due to the different structural properties of the exposed crystal structures. The presence of stimulated solar irradiation facilitates the conversion of SO2 on β-FeOOH and γ-FeOOH, whereas it inhibits the heterogeneous uptake on α-FeOOH. The addition of 5 wt% oxalate was discovered to significantly increase the reactive uptake coefficients of irradiated α-FeOOH and β-FeOOH by a factor of 3 and 2, which can be explained by the complex nature of iron with oxalate. The kinetic synergism between oxalate and sunlight was, for the first time, investigated in the heterogeneous regime of sulfate formation. The heterogeneous reactivity of FeOOH is nonlinearly correlated to light intensity and the exact dependence varies with crystal structure. This study contributes to a better understanding of atmospherically relevant heterogeneous reactions and haze formation, thus promoting laboratory studies and model simulations concerning atmospheric chemistry.
Environmental significance
SO2 is one of the most important precursors of atmospheric fine particulate matter, thus playing a crucial role in haze formation. FeOOH, one of the most abundant atmospheric iron oxides, not only provides numerous reactive sites for the removal of airborne SO2 but also supports the rapid formation of secondary sulfate aerosols. This study for the first time investigates the atmospheric fate of SO2 on pristine and oxalate-containing FeOOH nanoparticles with different crystal structures. Experimental results indicate that the crystal factor affects the heterogeneous sulfate formation in the order α-FeOOH > β-FeOOH > γ-FeOOH, in relation to their inherent crystal characteristics. In addition, the synergism between oxalate coating and sunlight irradiation is for the first time discovered on airborne gas–solid interfaces. This study reveals the atmospheric relevance of FeOOH in SO2 oxidation and is helpful for better understanding dust-related heterogeneous reactions.
|
1. Introduction
Sulfur dioxide (SO2), one of the typical air pollutants emitted from the combustion of fossil and biomass fuels, contributes greatly to atmospheric sulfate aerosols.1,2 Globally, field-based observations have suggested that nearly half of the emitted SO2 is converted to sulfate, mainly through gas-phase oxidation by hydroxyl radicals, aqueous-phase oxidation by dissolved hydrogen peroxide, nitrogen dioxide, ozone, and the O2 catalyzed by transition metal ions, and heterogeneous oxidation on aerosol surfaces.1,3–5 However, traditional models cannot capture well the key feature of sulfate concentration derived from field measurements, indicating that the sulfate generated from gas- and aqueous-phase oxidation is not able to bridge the gap on a global scale.3,6 Atmospheric observations discovered that the surfaces of atmospheric particles serve as efficient media for the rapid formation of sulfate.7,8 However, the influential factors for heterogeneous chemistry and the exact reaction mechanism remain poorly understood.
Mineral dust,9 constituting ∼36 wt% of primary aerosol emissions, is the most widespread and concentrated aerosol in the troposphere.10,11 These nanoscale and microscale particles can be lifted from the ground and transported over thousands of kilometers due to wind force, leaving a rather long lifetime of up to 1 to 2 weeks depending on the specific geographical and meteorological conditions.12 Hence, these particles provide abundant reactive sites for the heterogeneous conversion of atmospheric trace gases within their lifespan.13 Hoffmann and co-workers14 found that the total iron concentrations in urban fogs and clouds were unusually high. Iron was estimated to account for 7 wt% of an urban aerosol sample.14 Given the abundance of iron oxides in aerosols,15 a deep insight into SO2 heterogeneous oxidation on the surface of iron oxides is warranted. The mass fractions of α-Fe2O3, α-FeOOH, and Fe3O4 in the community of Fe-bearing minerals were determined to be 7.5%, 60.8%, and 9.8%, respectively.2 Previous studies showed the important role of Fe2O3 and Fe3O4 in SO2 conversion, but few of them examined the contributions of the more abundant FeOOH.2,16–20 Previous studies suggested that FeOOH widely exists in atmospheric particulate matter, especially mineral dusts.21–24 In addition, the abundance of FeOOH is comparable to or even higher than that of hematite.25 Therefore, FeOOH can be used as a dust proxy to investigate the heterogeneous SO2 conversion. It was shown that the heterogeneous oxidation of SO2 depends largely on the crystalline phases of MnO2.26 Motivated by this finding, crystal structure may act as an important influential factor for the heterogeneous reaction of SO2 on FeOOH, with various crystalline forms in nature. FeOOH comprises goethite (α-FeOOH), akageneite (β-FeOOH) and lepidocrocite (γ-FeOOH), which have different crystal system types and crystalline surface structures, and may thus present different SO2 chemisorption processes.27 In addition, considering the formation process of FeOOH,28 they are widely present in aerosol particles with varied proportions.29–31
Solar irradiation was frequently considered in laboratory research. Irradiated Fe-containing dust may present higher heterogeneous reactivity toward atmospheric trace gases due to the photocatalytic effect of the transition metal oxides therein or the photochemical effects induced by the specific dust coatings.4,18 However, the relevant studies concerned the photocatalytic and/or photochemical impacts of the irradiated particles rather than the irradiation intensity dependence of a series of reactions. The intensity of sunlight irradiation should be emphasized by laboratory stimulations because it changes with longitude, latitude, altitude, season, and time of day.32,33 Such a variable was recently discovered to present significant impacts on the heterogeneous SO2 oxidation on Fe2O3 and ZnO,4,16 while no study of light irradiance has been performed for FeOOH.
Oxalate is one of the most abundant low molecular weight carboxylates in atmospheric aerosols,34 which is mainly formed via atmospheric oxidation of its higher homologues of long-chain diacids and other pollution-derived organic precursors (e.g., olefins and aromatic hydrocarbons).35 Oxalate can effectively form chelates with Fe(III) (Fe(III)–oxalate chelates), which is photochemically active and can initiate free radical chain reactions. Fe(III)–oxalate complexes are generally more photoactive than unchelated Fe(III) ions because the complexation produces a stronger absorbing chromophore, which comes from the transfer of electrons from the oxalate ligand to Fe(III).36 The photoreactions of Fe(III)–oxalate complexes have significant impacts on the redox cycle of iron and may be an important source of oxidizing species in the atmosphere, thus contributing to a profound effect on atmospheric SO2 photooxidation.37,38 Thus, it is highly desirable to investigate the interaction between oxalate and iron on typical mineral surfaces.
To the best of our knowledge, only Fu et al.2 discussed the heterogeneous sulfate formation on pristine FeOOH under dark conditions, while the effects of various variables were not considered. Herein, we discussed the effects of crystal structure, oxalate coating and light irradiance based on high-resolution infrared spectral data. The promotion effect of oxalate and its synergism with solar irradiation were for the first time reported on airborne gas–solid interfaces, followed by the illustration of reaction mechanisms varying with reaction conditions and particle types. This study is expected to complement the role of FeOOH in SO2 conversion and provide a new approach for bridging the gap between field observation and model simulation.
2. Experimental
2.1 Materials
All chemicals are of analytical grade and used as received without further purification. Deionized (DI) water (specific resistance ≧18.2 MΩ cm) was used throughout the experiment. High-purity air (79% N2 and 21% O2, 99.999% purity) and SO2 (100 ppm, balanced by N2) were supplied by Shanghai Yunguang Specialty Gases Inc. Before introducing into the reaction system, the high-purity air was dried and purified using silica gel and a molecular sieve. A xenon lamp (CEL-TCX250, Beijing Ceaulight Co., Ltd., China) was used as a simulated sunlight source, and its spectrum is shown in Fig. S1.† To ensure the stability of light irradiance during the whole test process, the lamp was turned on for 30 min before irradiating the sample.
Three types of iron oxyhydroxide with different crystal structures, goethite (α-FeOOH), akageneite (β-FeOOH) and lepidocrocite (γ-FeOOH), were synthesized according to previous reports.39–41 Sodium oxalate (Na2C2O4) was used as a proxy for oxalate, representative of the abundant dicarboxylic compounds in coarse aerosol mode. The FeOOH–oxalate mixtures were prepared by mechanically mixing Na2C2O4 with the as-prepared FeOOH powder (5% mass fraction of Na2C2O4 for all samples), followed by manual grinding in an agate mortar for 15 min.
2.2 Sample characterization
The crystal structure of FeOOH was characterized using an X-ray diffractometer (XRD, Bruker D8 Advance) with Cu Kα radiation (λ = 1.54 Å) ranging from 10° to 80° (2θ) with a scan speed of 2° min−1. Field emission scanning electron microscopy (FE-SSEM, Hitachi S-4800, Japan) was used to investigate the microscopic morphology of samples. The Brunauer–Emmett–Teller (BET) specific surface area, pore volume and pore size were analyzed by nitrogen adsorption–desorption isotherms measured at 78 K on a Micromeritics Tristar 3000 analyzer.
2.3
In situ DRIFTS experiments
In situ DRIFTS experiments were performed on an FTIR spectrometer (Tracer-100, Shimadzu, Japan) equipped with a high-sensitivity and liquid-nitrogen-cooled mercury–cadmium–telluride (MCT) detector. The reaction temperature was maintained at 25 °C using a temperature controller. More details on the experimental equipment can be found in our previous reports.4,16,17,42,43
First, the prepared particles were placed in a ceramic crucible in the reaction chamber. Before reaction, the samples were purged by high-purity air with a flow rate of 150 mL min−1 for 30 min to remove the surface absorbed water and impurities. Then, a single-beam spectrum was collected as the background spectrum. Next, a mixture of high-purity air and SO2 at stable RH was introduced into the reaction chamber at a total flow rate of 110 mL min−1. The SO2 concentration of the reactant gas flowing into the DRIFTS chamber was 8 ppm (1.97 × 1020 molecules per m3), which is comparable to the experimental settings in previous studies.4 The humidity of the gas mixture is maintained within 68 ± 5% at all times. In order to investigate the effects of sunlight and its light irradiance on the heterogeneous conversion of SO2, light irradiation with a pre-designed power density (15, 45 and 80 mW cm−2, denoted as P15, P45 and P80, respectively) was turned on at the same time. DRIFTS spectra ranging from 4500 to 700 cm−1 were collected automatically every 2 min with 100 scans averaged for each spectrum and a resolution of 4 cm−1. The entire reaction process took 60 min. Every experiment was repeated at least three times.
2.4 Ion analysis
The S(VI) species formed via a heterogeneous reaction can be quantified by ion chromatography (IC). As previously performed, the samples were extracted by a 10 min oscillation in 5 mL H2O containing 5 vol% formaldehyde to inhibit the oxidation of S(IV) species.1 Then, the leaching solution was filtered through a 0.22 μm polytetrafluoroethylene (PTFE) membrane filter for analysis by IC (Basic 883, Metrohm, Switzerland). The detection was performed with a weak base eluent (3.5 mM Na2CO3/1.0 mM NaHCO3) at a stable flow rate of 0.7 mL min−1. Sulfite was not analyzed due to the limitations of the analytical column. The quantitative analysis was guaranteed by reference to the reference materials produced by the National Research Center for Reference Materials.
2.5 Uptake coefficient estimations
The reactive uptake coefficient γ is defined as the number of reactive collisions between reactant gas molecules and particle surfaces per unit time (−d[SO2]/dt) divided by the theoretical total number of collisions per unit time (Z). The heterogeneous reaction on the surface of FeOOH is most probably irreversible as the generated products ((bi)sulfate and (bi)sulfite) are in the solid phase while the reactants (SO2 and O2) are gaseous. Since the concentration of O2 is very abundant compared to that of SO2, it can be assumed that the concentration of O2 remains constant. Furthermore, the concentration of active sites on FeOOH for SO2 adsorption in the initial period is also constant. In addition, it is reported that the correlation coefficients of ln[C/C0] versus time (t) (where C0 is the initial concentration of SO2 and C is the concentration of SO2) for metal oxides under different initial concentrations of SO2 were greater than 0.95 from some previous studies.1,44,45 Thus, the heterogeneous conversion of SO2 was consistent with pseudo-first-order reaction kinetics, i.e. −d[SO2]/dt = d[SO42−]/dt = KCSO2, where K is the rate constant and CSO2 is the concentration of SO2 at certain reaction times. The reactive uptake coefficient (γ) based on pseudo-first-order kinetics is calculated by the following equations (eqn (1)–(4)):45,46| |  | (1) |
| | 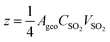 | (2) |
| |  | (3) |
| | | γBET = Ageo × γgeo/ABET | (4) |
where γgeo represents the geometric uptake coefficient, γBET is the BET uptake coefficient, (d[SO42−])/dt represents the formation rate of sulfates (ions per second), vSO2 is the mean molecular velocity of SO2 (m s−1), Ageo is the geometric surface area of FeOOH (m2), CSO2 is the concentration of SO2 (molecules per m3), R is the gas constant (J mol−1 K−1), T is the temperature (K), MSO2 is the molecular weight of SO2 (kg mol−1), and ABET is the BET surface area (m2). Detailed information on these parameters is given in Table S1.† The upper and lower limits of the uptake coefficients are γgeo and γBET depending on the reaction probability, respectively.1
3. Results and discussion
3.1 Particle characterization
According to Fig. 1a–c displaying the crystal structure results, all the diffraction peaks of the samples matched well with the corresponding standard cards for α-FeOOH (JCPDS29-0713), β-FeOOH (JCPDS34-1266), and γ-FeOOH (JCPDS44-1415). In addition, there was no other peak observed in the XRD pattern, suggesting that the prepared samples were of high purity. The XRD characteristic peaks are narrow and sharp, indicating the high crystallinity of the prepared FeOOH nanoparticles. The as-prepared FeOOH samples showed obviously different crystal forms, which would be appropriate for studying the effect of crystal structure on the heterogeneous conversion of SO2.
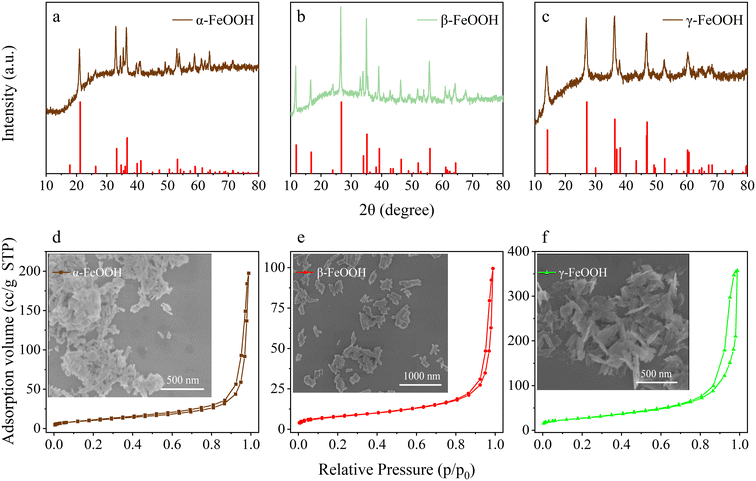 |
| | Fig. 1 XRD patterns, N2 adsorption–desorption isotherms, and SEM images for the three FeOOH samples: (a and d), α-FeOOH, (b and e), β-FeOOH, (c and f), γ-FeOOH. | |
In order to investigate the morphology structure of FeOOH, the FE-SEM images and BET data were obtained, shown in Fig. 1d–f. The FE-SEM images show that α-FeOOH (Fig. 1d) and β-FeOOH (Fig. 1e) were mainly composed of nanorod particles, but the length of α-FeOOH nanorods (∼100 nm) was about one-half that of β-FeOOH (∼210 nm). γ-FeOOH (Fig. 1f) presented a nanosheet structure. The close morphology and size of nano-FeOOH could minimize the effects of morphology and particle size on the heterogeneous SO2 uptake.
The N2 adsorption–desorption isotherms of the three samples (Fig. 1d–f) could be classified as type IV in the IUPAC classification with H3 hysteresis loops in the relative pressure range of 0.8 < p/p0 < 1.0, indicating the presence of large secondary mesoporous architectures (pore sizes are in the range of 2 to 50 nm). The BET specific surface areas of γ-FeOOH, α-FeOOH, and β-FeOOH were 96.20, 38.56 and 28.96 m2 g−1, respectively.
3.2 Effects of crystal structure and oxalate
3.2.1. Surface sulfur-containing species.
DRIFTS spectra are able to provide valuable information on adsorbed surface species by the vibrational modes. The assignments of the vibrational bands of adsorbed sulfur-containing species were analyzed in previous studies (Table S2†).2,5,46 In detail, the vibrational bands in the range of 1000–900 cm−1 correspond to chemisorbed (bi)sulfite, whereas the peaks at 1050 and 1010 cm−1 are assigned to bridging (bi)sulfate. In addition, the peaks at 1150 and 1250 cm−1 are attributed to adsorbed bidentate sulfate and water-soluble sulfate, respectively. As shown in Fig. 2, the spectra (1300–800 cm−1) of surface-bound products were recorded as a function of time. It is evident that new vibrational bands could be observed after SO2 exposure, and the intensities of these peaks rapidly increased in the first 10 min and then almost reached saturation with increasing time probably due to the complete consumption of surface active sites. Clearly, the DRIFTS spectra were distinct by the nanoscale FeOOH samples with different crystal structures, as discussed below.
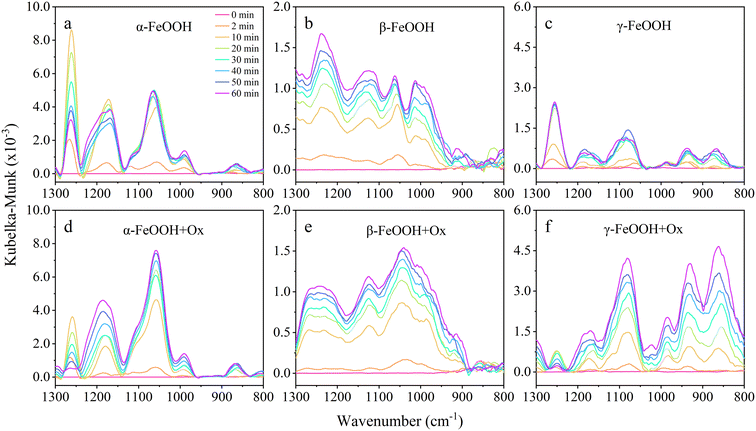 |
| | Fig. 2
In situ DRIFTS spectra collected for the three (a–c) pristine and (d–f) oxalate-coated samples during dark reactions: (a and d) α-FeOOH, (b and e) β-FeOOH, and (c and f) γ-FeOOH. | |
Fig. 2a displays the DRIFTS spectra of surface species on α-FeOOH as the reaction proceeded. There is a weak peak at 990 cm−1 and two main peaks at 1062 and 1170 cm−1, which could be attributed to (bi)sulfite, bridging (bi)sulfate and bidentate sulfate, respectively. Such spectral characteristics indicate that the prominent surface species were sulfates produced from SO2 conversion on α-FeOOH. The peak at 1260 cm−1 is not a real peak of surface generated species because it is obtained from the negative peak through the Kubelka–Munk transformation (Fig. S4†). The spectra collected for the SO2 conversion on β-FeOOH (Fig. 2b) indicate four surface products: (bi)sulfite (980 cm−1), bridging (bi)sulfate (1012 and 1060 cm−1), bidentate sulfate (1125 cm−1) and water-soluble sulfate (1235 cm−1). Similarly, the dominant generated surface species on β-FeOOH were sulfates with negligible production, suggesting that the pristine β-FeOOH is not conducive to the heterogeneous oxidation of SO2. Fig. 2c shows the spectra collected on γ-FeOOH, and the assignments of the peaks at 940, 1070 and 1175 cm−1 could be referenced to that of β-FeOOH. It is not an accurate vibrational band of surface species at 1250 cm−1 like that mentioned in α-FeOOH (Fig. S4†). Notably, on γ-FeOOH, the dominant sulfur-containing species are (bi)sulfite rather than the (bi)sulfate although it has a higher BET specific surface area, implicating the relatively weak capacity of γ-FeOOH for SO2.
Analogous to the previously studied Fe2O3 and Fe3O4, FeOOH may also serve as an important sink of airborne SO2 and source of sulfate aerosols. The nano-FeOOH samples displayed different DRIFTS spectra (vibrational bands and intensity), indicating that the crystal structure of mineral dusts could influence their heterogeneous reaction activities and product coordination structures. Considering the proportion of S(VI) species, α-FeOOH could be more efficient than β-FeOOH and γ-FeOOH in the oxidative conversion of S(IV) species.
The effects of oxalate on the heterogeneous uptake of SO2 under dark conditions were further investigated (Fig. 2d–f). There are obvious enhancements of three samples at the peaks ranging from 1100 to 1000 cm−1 and an extra enhancement of γ-FeOOH at 1000–900 cm−1. In other words, the oxalate facilitated the formation of bridging (bi)sulfate for all the three particles as well as the SO2 adsorption on the surface of γ-FeOOH. Furthermore, surface sulfur-containing species on pristine FeOOH under light irradiation were also investigated (Fig. S5†). Compared to the dark process, the dominant species of sulfur-containing products have changed under light irradiation. For α-FeOOH (Fig. 2a and S5a†), although the dominant species are still bridging sulfate and bidentate sulfate, there is a tendency for the peak at 1170 cm−1 to shift to higher wavenumbers, indicating the formation of water-soluble sulfate. β-FeOOH has a more pronounced response to light (Fig. 2b and S5b†), with bidentate sulfate as the new dominant product under light irradiation, and there is an overall increase in peak intensity, representing higher sulfate production. The prominent species on γ-FeOOH (Fig. 2c and S5c†) has converted from bridged sulfate in the dark reaction to water-soluble sulfate in the photoreaction, indicating that light greatly enhances the oxidation capacity of γ-FeOOH for SO2. Moreover, comparison of the spectra before and after the addition of oxalate under the dark and light conditions shows that the promotion effect of oxalate is more drastic in the presence of solar irradiation.
3.2.2. Kinetics assessment.
The different crystal forms and the presence or absence of oxalate result in not only different surface product types but also kinetic differences. To further discuss how crystal structure and oxalate coating affect the heterogeneous reactivity, we also calculated the uptake coefficient for sulfate formation on different FeOOH samples. It has been reported that the concentration of surface products has a positive linear relationship with the integrated absorbance (the integrated area of the characteristic infrared peaks of sulfur-containing species) based on DRIFTS spectra.1,3 Therefore, the amount of surface sulfate species, including water-soluble sulfate, bidentate sulfate, bridging sulfate, and bisulfate, was considered for quantitative analysis. The calibration curves of sulfate ions versus the integrated absorbance were obtained from IC analysis of a series of DRIFTS experiments with different reaction times. The calibration plots of conversion factors for α-FeOOH and β-FeOOH under light irradiation are shown in Fig. S2 and S3,† respectively. γ-FeOOH produced too little sulfate to calculate its conversion factor. In addition, according to a previous study,1 the addition of 5 wt% oxalate has a negligible effect on the conversion factors; thus we use the conversion factor of pure FeOOH particles as an alternative to the conversion factor of mixed particles. The conversion factors (f) for α-FeOOH (fα) and β-FeOOH (fβ) are 4.07 × 1015 and 1.89 × 1014 ions per absorbance unit (ABU), respectively. As a result, the number of sulfate ([SO42−]) can be calculated according to the following formula (eqn (5)):| | | [SO42−] = (integrated absorbance) × f | (5) |
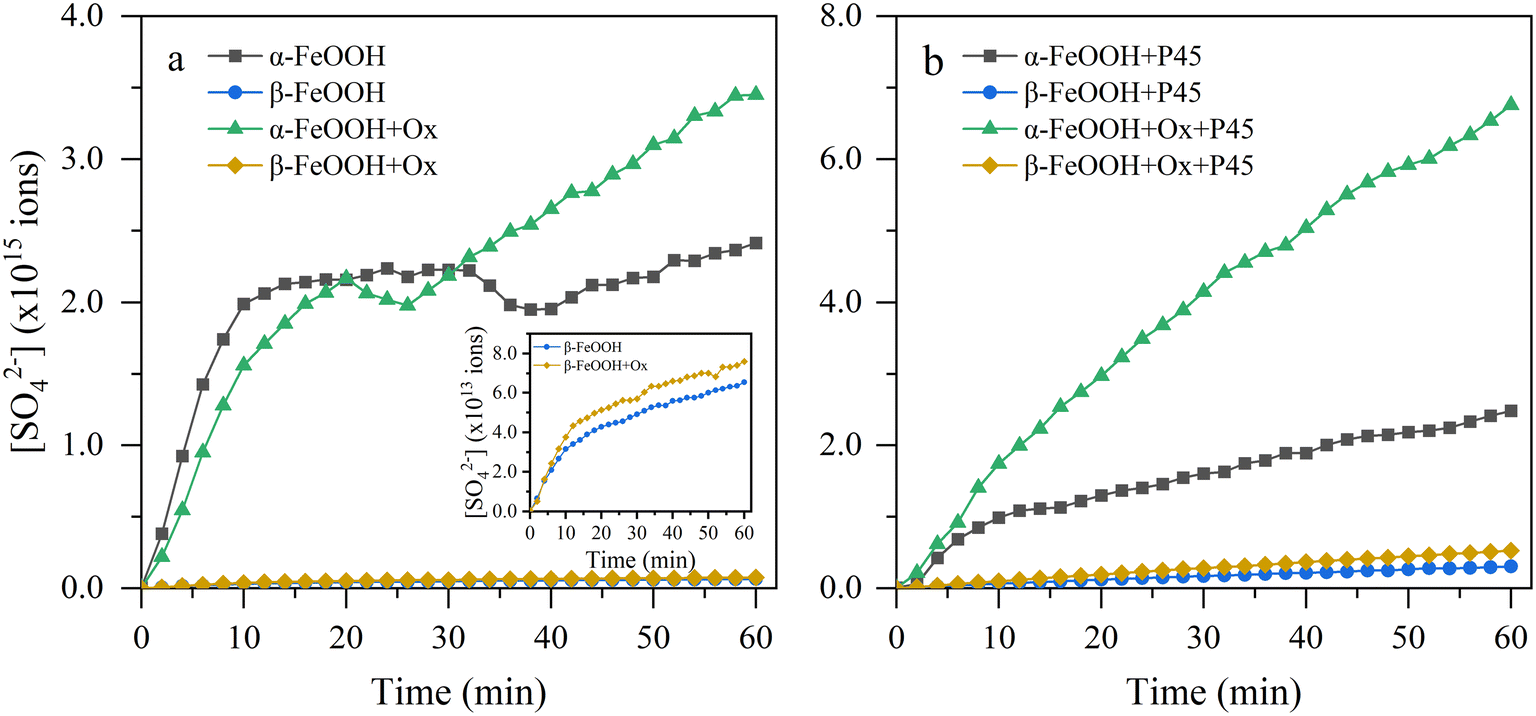
Fig. 3 displays the S(VI) species produced on different FeOOH samples as a function of time. The S(VI) species increased rapidly at first (initial stage) and then kept steady (stable stage) as the reaction proceeded, since the integrated area of the infrared peak is proportional to the species content. This was mainly because more surface reactive sites existed in the initial stage for the adsorption and oxidation of SO2. With increasing reaction time, the number of active sites decreased as the generated species covered the active sites, thus inhibiting further SO2 conversion.2,47 In addition, it was clear that the sulfates generated on different FeOOH samples were obviously distinct. The number of total sulfate species decreased following the order α-FeOOH > β-FeOOH > γ-FeOOH. It is worth mentioning that although the BET specific surface area of γ-FeOOH (96.20 m2 g−1) was much higher than that of α-FeOOH (38.56 m2 g−1), the reactivity of γ-FeOOH was much lower than that of α-FeOOH. It is possible that the density of active sites on the flake particles is very low, thus resulting in fewer active sites on γ-FeOOH.48 Such a result suggested that the particle crystal structure is a significant impact factor for the heterogeneous conversion of SO2 and will inevitably affect the atmospheric heterogeneous reaction, which should be paid more attention in further studies.
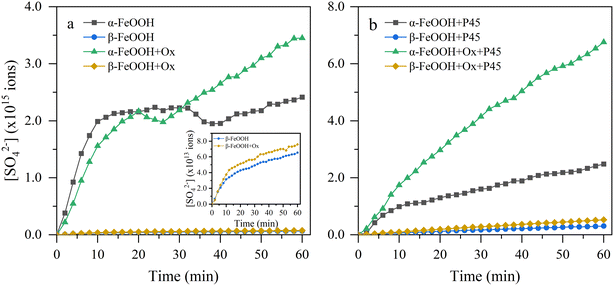 |
| | Fig. 3 Calculated ions of the sulfates produced on different FeOOH samples under (a) dark and (b) light conditions. Inset: local magnified image of pristine and oxalate-coating β-FeOOH. | |
The calculated γgeo and γBET for the nano-FeOOH samples are shown in Table 1. The number of sulfate ions produced by γ-FeOOH is below the detection limit, reflecting its weakest reactivity. It is evident that the uptake coefficients were distinct, which could be attributed to the differences in surface and structure properties. Taking the BET surface area as the reaction surface area, the activities of these oxides towards SO2 were in the order α-FeOOH > β-FeOOH > γ-FeOOH under all conditions of this experiment. The γBET values of α-FeOOH and β-FeOOH under light irradiation were 3.65 × 10−11 and 0.70 × 10−11, respectively, which were comparable with previously documented results.2,49 Overall, it was obvious that the uptake capacity of SO2 varies with the nanoscale FeOOH samples with different crystal structures, probably due to their various BET surface areas, particle sizes, densities of surface reactive sites and structure properties.
Table 1 Reactive surface areas and uptake coefficients of SO2 on different FeOOH particle samplesa
| Sample |
A
BET (m2 g−1) |
A
geo (×10−5 m2) |
γ
BET (×10−11) |
γ
geo (×10−6) |
|
The amounts of products are too small to be quantified by IC, so the results of γ-FeOOH are not displayed.
The oxalate addition was 5 wt% for all samples.
|
| α-FeOOH |
1.05 |
1.96 |
2.03 |
1.09 |
| α-FeOOH + Oxb |
1.05 |
1.96 |
4.84 |
2.59 |
| α-FeOOH + P45 |
1.05 |
1.96 |
3.65 |
1.95 |
| α-FeOOH + Ox + P45 |
1.05 |
1.96 |
11.20 |
5.99 |
| β-FeOOH |
0.78 |
1.96 |
0.12 |
0.05 |
| β-FeOOH + Ox |
0.78 |
1.96 |
0.14 |
0.06 |
| β-FeOOH + P45 |
0.78 |
1.96 |
0.70 |
0.28 |
| β-FeOOH + Ox + P45 |
0.78 |
1.96 |
1.21 |
0.48 |
As shown in Table 1, the presence of 5 wt% oxalate slightly increased the γBET of α-FeOOH under dark conditions, while there was no significant enhancement for β-FeOOH. By the presence of solar irradiation, the γBET of α-FeOOH and β-FeOOH were enhanced by a factor of 3.0 and 1.7 in the presence of oxalate. Thus, a more pronounced promotion effect under light irradiation can be concluded. Such a conclusion is related to the chelation of oxalate with iron, forming a chelate with a strong photoactivity, which would be expounded later in the reaction mechanism section. Moreover, compared with the slight decrease in γBET of α-FeOOH, the γBET of β-FeOOH during the photoreaction increased by 6-fold, indicating that β-FeOOH has a more pronounced response to solar irradiation.
3.3 Effect of light irradiance
In the atmosphere, solar irradiation will inevitably influence many complex physiochemical reactions. Nevertheless, previous experimental studies of heterogeneous reactions have to date largely been carried out under a single light intensity.26,50 In this study, we investigated the effect of multiple levels of light irradiance on the heterogeneous uptakes of SO2 on the samples. Considering that the annual averaged solar irradiation intensity reaching the mid-latitude region is about 100 mW cm−2,51,52 three different light intensities of 15, 45 and 80 mW cm−2 were selected in this study.
The final spectra (1300–800 cm−1) collected for pristine α-FeOOH under the four light intensities are shown in Fig. 4a. With irradiance increasing, (bi)sulfite (971, 923, 886 cm−1) and bridging sulfate (1062 cm−1) decreased continuously and the predominant species gradually changed from bidentate sulfate (1158, 1125 cm−1) to water-soluble sulfate (1238, 1220 cm−1), implying the oxidation of S(IV) species. To further investigate the effect of light intensity on pristine α-FeOOH, the integrated area of the sulfate peaks was calculated as a function of reaction time (Fig. 4c). The integrated area decreased at first and then increased with increasing light intensity. However, within the light intensity range we studied, the integrated area of sulfate under light irradiation was generally lower than that under dark conditions. Such results indicated that the light irradiation may inhibit the heterogeneous oxidation of SO2, which may be caused by the saturated iron atoms42,53,54 and/or the photoinduced reductive dissolution of α-FeOOH.4,55
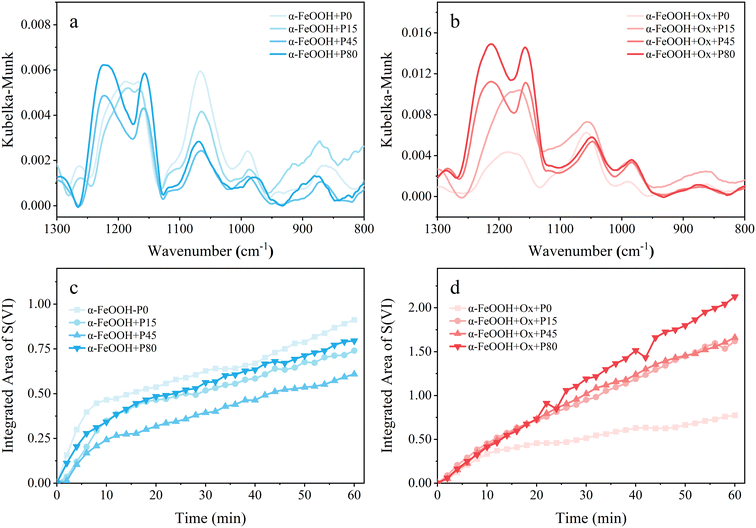 |
| | Fig. 4 (a and b) Final DRIFTS spectra after 60 min of SO2 exposure and (c and d) the temporal variations of the integrated area of sulfate peaks under four irradiance power densities (P0, P15, P45, P80). Both (a and c) pristine (light cyan) and (b and d) oxalate-containing (light LT red) α-FeOOH nanoparticles are considered. | |
The effect of light intensity on oxalate-coated α-FeOOH was further investigated. The spectra collected after 60 min of SO2 exposure and the temporal variations of the integrated area of sulfate peaks are displayed in Fig. 4b and d. Like the process on α-FeOOH, the dominant species gradually transformed from bidentate sulfate to water-soluble sulfate. However, in the presence of oxalate, there was only a slight decrease of sulfite and bridging sulfate with increasing light intensity, along with a continuous increase of bidentate and water-soluble sulfates. Such results indicated that sunlight could enhance the formation and solvation of sulfate on oxalate-coated α-FeOOH (Fig. 4d).
Noticeably, the presence of oxalate could greatly accelerate the conversion of SO2 under light conditions as mentioned above. Further calculations revealed that the integrated area of sulfate under the combined effect of oxalate and light was higher than the sum of that under the effect of oxalate and light irradiation alone (Fig. S6a and d†), indicating that there was a synergistic effect of light and oxalate in the heterogeneous conversion of SO2 on α-FeOOH. Considering the wide availability of oxalate and FeOOH in the atmosphere, such synergistic effects may exist in the atmospheric SO2 photooxidation and explain the stronger promotion effect of oxalate under sunlight.
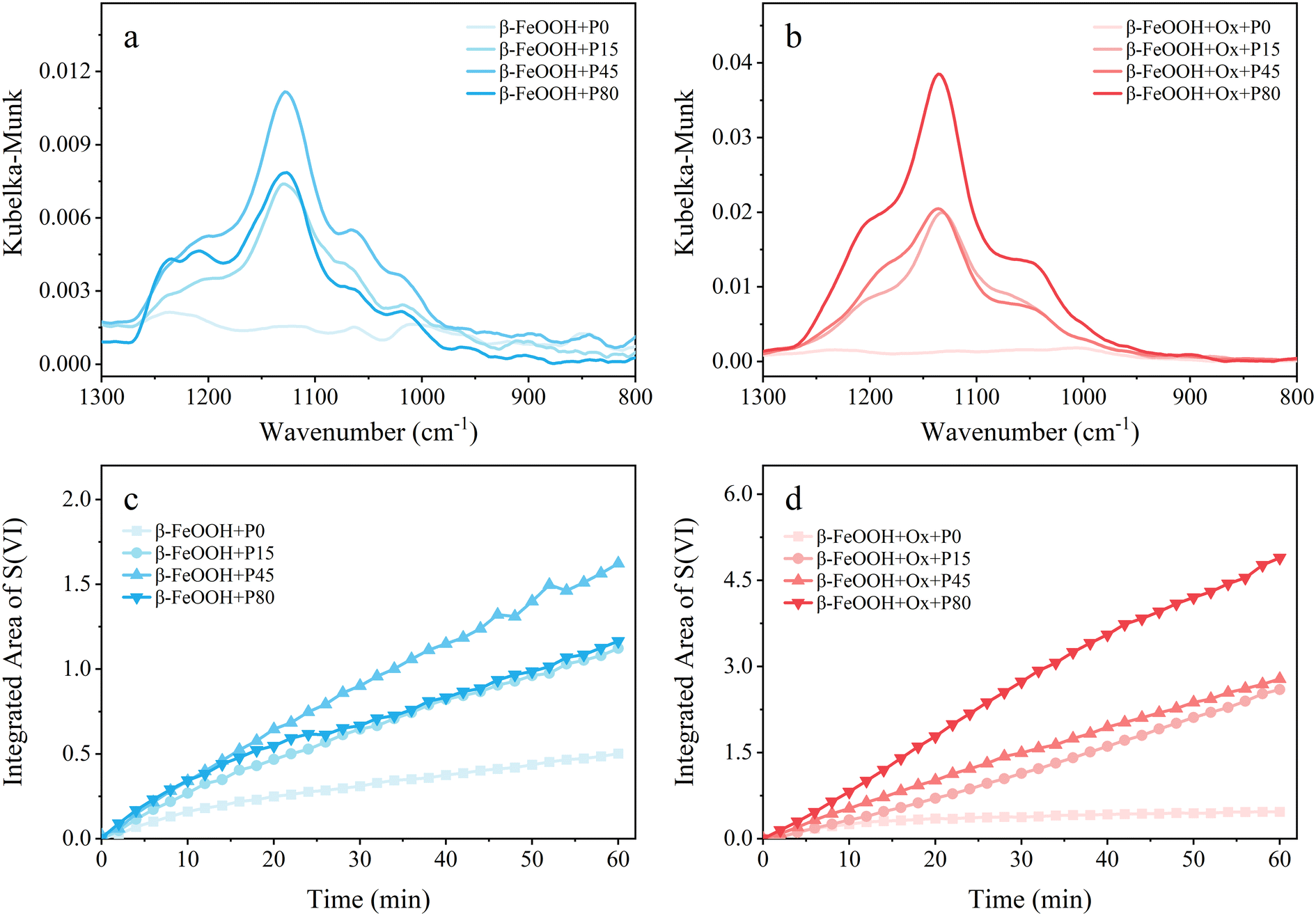
Fig. 5 shows the DRIFTS spectra and the sulfate integrated areas of β-FeOOH under the same conditions of α-FeOOH. On pristine β-FeOOH, (bi)sulfite, bridging sulfate and bidentate sulfate increased at first and then decreased with the increased light intensity, which could be explained by the photoinduced reductive dissolution of β-FeOOH under strong sunlight.4,55 However, the water-soluble sulfate kept increasing within the range of light irradiance in the experiments. The total sulfate production on pristine β-FeOOH increased initially in a low light intensity range and then decreased when the light irradiance exceeded the range, and the amount of sulfates in the presence of light irradiation was always higher than that under dark conditions. This conclusion was validated in Fig. 5c, showing the integrated areas of the characteristic peaks of sulfates. Nevertheless, on the oxalate-coated β-FeOOH nanoparticles, the peak intensities of all kinds of sulfates increased with increasing light intensity, and the corresponding integrated area increased as well, as shown in Fig. 5b and d, indicating that the oxalate increased the saturation light intensity of this reaction. It is worth noting that the dominant species was bidentate sulfate in the presence of light irradiation, which was different from the water-soluble sulfate under dark conditions (Fig. 2e). This is one point where the conversion of SO2 on the surface of β-FeOOH was not well understood.2
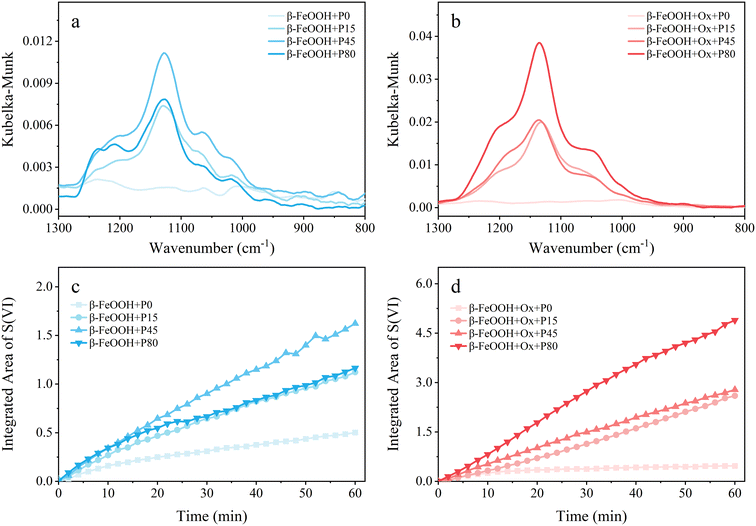 |
| | Fig. 5 (a and b) Final DRIFTS spectra after 60 min of SO2 exposure and (c and d) the temporal variations of the integrated area of sulfate peaks under four irradiance power densities (P0, P15, P45, P80). Both (a and c) pristine (light cyan) and (b and d) oxalate-containing (light LT red) β-FeOOH nanoparticles are considered. | |
In addition, according to the integrated area of sulfate in the absence or presence of oxalate shown in Fig. 5c and d, it could be concluded that the promotion effect of light on SO2 oxidation was remarkable on β-FeOOH whether the oxalate coating was present or absent. In addition, based on the calculated uptake coefficients shown in Table 1, under P45 irradiance, the γBET of β-FeOOH was enhanced by a factor of 5.8 and 8.6 in the absence and presence of oxalate, respectively, further demonstrating the remarkable promotion of light irradiation on β-FeOOH. The weak enhancement of oxalate on γBET suggested that the reaction is more susceptible to solar irradiation than to oxalate on β-FeOOH. Likewise, there is also a synergism between light irradiation and oxalate for the SO2 conversion on β-FeOOH, since the integrated area of sulfate on oxalate-coated β-FeOOH with light is higher than the sum of that with oxalate addition and light irradiation alone (Fig. S6b and e†).
For γ-FeOOH, Fig. 6(a and d) show that the light intensity promotes the conversion of SO2 on pristine and oxalate-coated samples, including bidentate sulfate and water-soluble sulfate. Moreover, in the experimental light intensity range, the generated sulfate kept increasing with increasing light intensity (Fig. 6b and e). In contrast to α-FeOOH and β-FeOOH, no saturation light effect was observed on γ-FeOOH. This may be due to its huge specific surface area or lower activity resulting in a greater dependence on light irradiation. In addition, to account for the higher SO2 adsorption on γ-FeOOH (Fig. 2c and f), we analyzed the (bi)sulfite chemisorbed on γ-FeOOH separately (Fig. 6c and f). It could be seen that the addition of oxalate increases the (bi)sulfite content significantly at any light intensity. As shown in Fig. 6c, the content of (bi)sulfite under different light intensities tended to be the same (P15 was an exception), indicating that light irradiation may not change the adsorption capacity of SO2 on the surface of pristine γ-FeOOH. In the presence of oxalate, the sulfite content under light irradiation was always lower than that under dark conditions but increased with increasing light irradiance (Fig. 6f). Such results may be mainly due to the conversion of S(IV) to S(VI) and more dissolved iron under stronger light irradiation.
 |
| | Fig. 6 (a and d) Final DRIFTS spectra after 60 min of SO2 exposure and (b, c, e and f) the temporal variations of the integrated area of sulfate and sulfite peaks under four irradiance power densities (P0, P15, P45, P80). Both (a–c) pristine (light cyan) and (d–f) oxalate-containing (light LT red) γ-FeOOH nanoparticles are considered. | |
3.4 Proposed mechanism for SO2 uptake on FeOOH
As discussed above, influenced by crystal structure, light irradiance and oxalate coating, there are diverse atmospheric fates for airborne SO2 over the gas–solid interface of FeOOH. Combining the documented information and the current experimental results, the following sections are expected to illustrate the primary physical–chemical processes on the studied nanoscale minerals.
3.4.1. Pristine FeOOH.
The first step of the studied heterogeneous event is the adsorption of gas-phase SO2 onto the absorbed state (reaction (1)). On the one hand, the interaction between SO2 and H2O, followed by the ionization procedures, leads to the formation of S(IV) species: H2SO3, HSO3− and SO32−.56 On the other hand, SO2 adsorbed on Lewis acid sites (coordinately unsaturated iron atoms) or Lewis basic sites (exposed oxygen atoms) could form physisorbed SO2 or chemisorbed sulfite, respectively.42,53 The reactions are shown as reaction (2) below, where O2− (lattice) refers to lattice oxygen on particle surfaces.| | | SO2(g) + O2− (lattice) → SO32− | (R2) |
Meanwhile, surface active oxygen (O−) is derived from the combination of molecular oxygen with the vacant oxygen sites viareactions (3) and (4). Subsequently, the chemisorbed sulfites are further oxidized to sulfate by O−, as described by reaction (5).17,57,58
| | | O(vacancy) + e− + O2 → O2− (ads) | (R3) |
| | | SO32− + O− → SO42− + e− | (R5) |
where e
− represents a conductive electron trapped in the vacant oxygen site.
57
Surface-adsorbed hydroxyl groups participate in the heterogeneous reactions.4 SO2 could react with one or two surface hydroxyl groups to form bisulfite or sulfite as described by reaction (6) or (8), respectively.42,59,60 The consumption of surface hydroxyl groups seems to be fast at the initial stage of the reaction and drops to a slower trend later (Fig. 3). Such variations indicate that the relevant process would be controlled by reaction (8) at the beginning and then by reaction (6). Subsequently, the generated (bi)sulfite would be oxidized to (bi)sulfate by reactive oxygen species (ROS) and hydroxyls. In addition, a Fe(III)–Fe(II) redox cycle could participate in the formation of (bi)sulfate as well. As shown in reaction (10), Fe(III) receives an electron from HSO3− and is deoxidized to Fe(II). According to previous studies,2,42,57 Fe(II) could be oxidized to Fe(III) by SO5˙−, HSO5˙−, and SO4˙− and would in turn catalyze the conversion of S(IV) species to S(VI) products. As shown in Fig. 2, under dark conditions, α-FeOOH presented the greatest sulfate products, followed by γ-FeOOH, with β-FeOOH producing the least. In addition, bridging and bidentate sulfates were mainly formed on the former two nanoscale samples, while water-soluble and bidentate sulfates were the predominant products on β-FeOOH.
| | | ◇ − FeIII(OH) + ◇ − H2O + SO2 → ◇ − FeIII(OH)·HSO3− + H+ | (R6) |
| | | ◇ − FeIII(OH)·HSO3− → ◇ − FeIIIOSO2 + H2O | (R7) |
| | | 2 ◇ − OH + SO2 → ◇ − HSO3 + H2O | (R8) |
| | | ◇ − HSO3 + O− → ◇ − HSO4− → ◇ − SO42− + H+ | (R9) |
| |  | (R10) |
The effect of solar irradiation can be proved by the photoinduced variation of the infrared signals (Fig. S5a–c†), which is attributed to ROS formation in the presence of sunlight irradiation. Irradiated semiconductor FeOOH can be photo-excited to produce electron–hole pairs. As well acknowledged, h+ can oxidize OH− and H2O to ˙OH (h+ + OH−/H2O → ˙OH) and e− can reduce O2 to O2˙− (e− + O2 → O2˙−). In addition, the iron hydroxy complexes can absorb in the near-UV region (290–400 nm), undergoing efficient photo-dissociation to form Fe(II) and ˙OH (reactions (11) and (12)).38,61 The generated reactive species can participate in the Fe(III)–Fe(II) redox cycle and the oxidation of S(IV) species to S(VI) products, resulting in a facilitation of SO2 conversion relative to the dark reaction. It is worthwhile to mention that the negative impact of solar irradiation on the conversion of SO2 can be explained by the saturation of iron atoms and photoinduced reductive dissolution, thus hindering the process on α-FeOOH described in Fig. 4a.
| | | Fe(III) + H2O → [Fe(III)(OH)]2+ + H+ | (R11) |
| | | [Fe(III)(OH)]2+ + hν → Fe(II) + ˙OH | (R12) |
The proposed mechanism (Scheme 1) summarizes the main pathway involved in the heterogeneous reactions of SO2 on pristine nano-FeOOH. Briefly, surface-adsorbed hydroxyl groups and ROS are involved in the formation of sulfate species, and the Fe(III)–Fe(II) redox cycle also contributes to the heterogeneous oxidation. The results from this study indicated that the heterogeneous uptake capacities are different between FeOOH samples with significantly different crystal structures, suggesting that crystal structure is an important influential factor for the heterogeneous conversion of SO2. The different crystal planes, resulting from various crystal structures, possess diverse surface active sites and disparate distribution properties, including the density of surface active sites and SO2 adsorption capacity. This may also explain the differences in heterogeneous reactivity between the particles with different morphologies.4
 |
| | Scheme 1 Proposed mechanism illustrating the heterogeneous reaction of SO2 on FeOOH surfaces. The blue arrows represent the primary pathways under dark conditions, and the red ones highlight the processes occurring merely in the presence of solar irradiation. | |
3.4.2. Oxalate-containing FeOOH.
As discussed in section 3.2.1, the promotion effect of oxalate coating is much more significant in the presence of solar irradiation relative to that under dark conditions due mainly to the fact that the reaction process and mechanism are basically consistent in the absence of solar irradiation regardless of the oxalate addition. Under dark conditions, the oxalate coating greatly enhanced the adsorption and oxidation of SO2 on FeOOH (Fig. 2), and the weak promoting effect of oxalate on sulfate formation could be mainly explained by the greatly enhanced dissolution of FeOOH resulting from the complexation of oxalate with iron,23,62,63 hindering the transformation of bridging sulfate to bidentate and water-soluble sulfates. In the presence of solar irradiation, more pathways are probable in the course of the SO2 heterogeneous reactions than those discussed in section 3.4.1. The proposed photochemical mechanism for the production of oxidizing species from Fe(III)–oxalate complexes is shown in the following reactions.38,64,65| | | Fe(III) + 3C2O42− → [Fe(III)(C2O4)3]3− | (R13) |
| | | [Fe(III)(C2O4)3]3− + hν → [Fe(II)(C2O4)2]2− + C2O4·− | (R14) |
| | | C2O4˙− → CO2˙− + CO2 | (R15) |
| | | CO2˙− + O2 → O2˙− + CO2 | (R16) |
| | | Fe(II) + O2˙−/HO2˙ + H+ → Fe(III) + H2O2 | (R18) |
| | | [Fe(II)C2O4] + H2O2 → [Fe(III)C2O4]+ + ˙OH + OH− | (R19) |
| |  | (R20) |
After the excitation of [Fe(III) (C2O4)3]3− (reaction (13)), the major photochemical process is the intramolecular electron transfer from oxalate to Fe(III), forming C2O4˙− as the primary intermediate (reaction (14)). The formed C2O4˙− radical would further dissociate into carbon dioxide anion radical (CO2˙−) and CO2 (reaction (15)).66–68 Although the photolysis mechanism for [Fe(III)(C2O4)2]− and [Fe(III)(C2O4)]+ has not been extensively studied, it is assumed that the reactions produce Fe(II) and CO2˙− eventually.69 The subsequent reaction of CO2˙− with O2 leads to the formation of intermediate superoxide ions (O2˙−) and hydroperoxyl radicals (HO2˙), which react with Fe(II) to produce H2O2 in acidic medium (reactions (16)–(18)). Furthermore, H2O2 will interact with Fe(II) (reaction (19)) to form ˙OH. These ROS (O2˙−, HO2˙ and ˙OH) greatly accelerate the oxidation of S(IV) (reaction (20)) along with accelerated iron dissolution.
As discussed above, the presence of illumination enhanced the formation of sulfate species on the oxalate-coated FeOOH, as proved by Fig. S5 and S6.† On α-FeOOH and γ-FeOOH, the proportions of bidentate and water-soluble sulfates further increased in the presence of oxalate coating, highlighting the crucial effects of the photoinduced ROS. On β-FeOOH, the predominant species was still bidentate sulfate and possessed a higher proportion. Sulfate weakly bound as outer sphere complexes by electrostatic attraction may serve as the main reason.2
The proposed mechanisms are summarized in Scheme 2. In summary, the addition of oxalate greatly facilitated the heterogeneous transformation of SO2 to sulfate on FeOOH during photoreaction. However, there are different enhancements of the irradiated oxalate coating on the FeOOH samples (Fig. S5†), which can be explained by the different affinities proposed in previous studies.55,70 In other words, the different affinities of oxalate and various crystalline types of FeOOH lead to its diverse promoting effects on the three samples, whose mechanisms remain to be further investigated.
 |
| | Scheme 2 The proposed mechanism of SO2 heterogeneous oxidation on oxalate-containing nano-FeOOH under solar irradiation. | |
4. Conclusions and implications
This study investigated the effects of crystal structure, oxalate coating, and light irradiance on the heterogeneous conversion of SO2 on nano-FeOOH minerals. Experimental results indicated that the studied nano-FeOOH samples displayed different reactivities (α-FeOOH > β-FeOOH > γ-FeOOH) in relation to their different specific surface areas, surface reactive sites and intrinsic structure properties. Simulated sunlight irradiation inhibited the heterogeneous uptake on α-FeOOH and strong light intensity also hindered the reaction on β-FeOOH, mainly limited by the reactive sites and the photoinduced reductive dissolution. The light intensity within the experimental range was conducive to the heterogeneous reaction on γ-FeOOH. The addition of oxalate could facilitate the heterogeneous oxidation of SO2 on nano-FeOOH especially under light irradiation, largely caused by the photoinduced ROS formation by iron–oxalate complexes.
Furthermore, to assess the importance of the heterogeneous reaction on FeOOH, the lifetime for removal of SO2 was estimated according to the formula given in previous reports:71,72
| |  | (6) |
where
A is the surface area density of FeOOH (cm
2 cm
−3),
ω is the mean molecular velocity (m s
−1), and
γ is the uptake coefficient. The mass loading of mineral dust is set to be 55 μg m
−3, representative of the averaged dust concentration in North China.
73,74 The atmospheric loading of goethite can be estimated by the hematite/goethite ratio ranging between 0.5 and 2.0,
75 along with the mass proportion of hematite (6.5%) in dust.
25,76 Hence, the
A of goethite ranges from 6.89 × 10
−7 cm
2 cm
−3 to 2.76 × 10
−6 cm
2 cm
−3. Based on the
γ of 1.95 × 10
−6 for pristine irradiated goethite (see
Table 1), the SO
2 lifetime with respect to the studied heterogeneous process ranges from 0.75 to 3.0 years. In comparison, the lifetime of SO
2 with respect to the gas-phase reaction with OH is 20 days if [OH] = 1.0 × 10
6 molecular cm
−3.
71 In comparison, the heterogeneous uptake of SO
2 on goethite is negligible under common atmospheric conditions. However, during dust storms, the concentration of dust can be up to 10
3–10
4 μg m
−3,
77,78 thus leading to a shorter SO
2 lifetime of 2.5–15 days. Considering the presence of oxalate coating, such a lifetime range may decrease to 12 h to 4.9 days. Accordingly, the heterogeneous reaction of SO
2 on FeOOH minerals plays a crucial role in the removal of SO
2 in the atmosphere.
As well acknowledged, FeOOH acts as the predominant Fe-containing airborne mineral in the atmosphere, thus making the kinetic results from this work more appropriate than those relevant to Fe2O3 and Fe3O4 for the modeling studies on the rapid formation of sulfate aerosols. In future laboratory research, FeOOH is recommended as a suitable mineral dust proxy for exploring the varied fates of airborne SO2 through dust heterogeneous chemistry. On the other hand, the various variables, including the crystal structure, oxalate coating and light intensity considered discussed above, present great influences on the heterogeneous sulfate formation, coupled with the distinct heterogeneous chemical mechanisms. Extensive discussions on the variables are needed in the following research on dust-related sulfate formation, thus providing alternative kinetic parameter selections for chemical transport models.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The research was financially supported by the National Natural Science Foundation of China (22176036, 21976030, 22006020, 42205099) and the Natural Science Foundation of Shanghai (19ZR1471200).
References
- L. D. Kong, X. Zhao, Z. Y. Sun, Y. W. Yang, H. B. Fu, S. C. Zhang, T. T. Cheng, X. Yang, L. Wang and J. M. Chen, The effects of nitrate on the heterogeneous uptake of sulfur dioxide on hematite, Atmos. Chem. Phys., 2014, 14, 9451–9467 CrossRef CAS.
- H. Fu, X. Wang, H. Wu, Y. Yin and J. Chen, Heterogeneous Uptake and Oxidation of SO2 on Iron Oxides, J. Phys. Chem. C, 2007, 111, 6077–6085 CrossRef CAS.
- C. Liu, Q. Ma, Y. Liu, J. Ma and H. He, Synergistic reaction between SO2 and NO2 on mineraloxides: a potential formation pathway of sulfate aerosol, Phys. Chem. Chem. Phys., 2012, 14, 1668–1676 RSC.
- K. Li, L. Kong, A. Zhanzakova, S. Tong, J. Shen, T. Wang, L. Chen, Q. Li, H. Fu and L. Zhang, Heterogeneous conversion of SO2 on nano α-Fe2O3: the effects of morphology, light illumination and relative humidity, Environ. Sci.: Nano, 2019, 6, 1838–1851 RSC.
- C. E. Nanayakkara, J. Pettibone and V. H. Grassian, Sulfur dioxide adsorption and photooxidation on isotopically-labeled titanium dioxide nanoparticle surfaces: roles of surface hydroxyl groups and adsorbed water in the formation and stability of adsorbed sulfite and sulfate, Phys. Chem. Chem. Phys., 2012, 14, 6957 RSC.
- P. Kasibhatla, W. L. Chameides and J. S. John, A three-dimensional global model investigation of seasonal variations in the atmospheric burden of anthropogenic sulfate aerosols, J. Geophys. Res.: Atmos., 1997, 102, 3737–3759 CrossRef CAS.
- R. M. Kirpes, A. L. Bondy, D. Bonanno, R. C. Moffet, B. Wang, A. Laskin, A. P. Ault and K. A. Pratt, Secondary sulfate is internally mixed with sea spray aerosol and organic aerosol in the winter Arctic, Atmos. Chem. Phys., 2018, 18, 3937–3949 CrossRef CAS.
- R.-J. Huang, Y. Zhang, C. Bozzetti, K.-F. Ho, J.-J. Cao, Y. Han, K. R. Daellenbach, J. G. Slowik, S. M. Platt, F. Canonaco, P. Zotter, R. Wolf, S. M. Pieber, E. A. Bruns, M. Crippa, G. Ciarelli, A. Piazzalunga, M. Schwikowski, G. Abbaszade, J. Schnelle-Kreis, R. Zimmermann, Z. An, S. Szidat, U. Baltensperger, I. E. Haddad and A. S. H. Prévôt, High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 2014, 514, 218–222 CrossRef CAS PubMed.
- M. Tang, D. J. Cziczo and V. H. Grassian, Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- L. Wu, S. Tong and M. Ge, Heterogeneous Reaction of NO2 on Al2O3: The Effect of Temperature on the Nitrite and Nitrate Formation, J. Phys. Chem. A, 2013, 117, 4937–4944 CrossRef CAS.
- L. Wu, S. Tong, L. Zhou, W. Wang and M. Ge, Synergistic Effects between SO2 and HCOOH on α-Fe2O3, J. Phys. Chem. A, 2013, 117, 3972–3979 CrossRef CAS PubMed.
- S. Gassó, A. Stein, F. Marino, E. Castellano, R. Udisti and J. Ceratto, A combined observational and modeling approach to study modern dust transport from the Patagonia desert to East Antarctica, Atmos. Chem. Phys., 2010, 10, 8287–8303 CrossRef.
- M. Tang, X. Huang, K. Lu, M. Ge, Y. Li, P. Cheng, T. Zhu, A. Ding, Y. Zhang, S. Gligorovski, W. Song, X. Ding, X. Bi and X. Wang, Heterogeneous reactions of mineral dust aerosol: implications for tropospheric oxidation capacity, Atmos. Chem. Phys., 2017, 17, 11727–11777 CrossRef CAS.
- P. Hoffmann, A. N. Dedik, J. Ensling, S. Weinbruch, S. Weber, T. Sinner, P. Gütlich and H. M. Ortner, Speciation of iron in atmospheric aerosol samples, J. Aerosol Sci., 1996, 27, 325–337 CrossRef CAS.
- Q. Lin, X. Bi, G. Zhang, Y. Yang, L. Peng, X. Lian, Y. Fu, M. Li, D. Chen, M. Miller, J. Ou, M. Tang, X. Wang, P. Peng, G. Sheng and Z. Zhou, In-cloud formation of secondary species in iron-containing particles, Atmos. Chem. Phys., 2019, 19, 1195–1206 CrossRef CAS.
- T. Wang, Y. Liu, Y. Deng, H. Cheng, Y. Yang, K. Li, X. Fang and L. Zhang, Irradiation intensity dependent heterogeneous formation of sulfate and dissolution of ZnO nanoparticles, Environ. Sci.: Nano, 2020, 7, 327–338 RSC.
- T. Wang, Y. Liu, Y. Deng, H. Fu, L. Zhang and J. Chen, The influence of temperature on the heterogeneous uptake of SO2 on hematite particles, Sci. Total Environ., 2018, 644, 1493–1502 CrossRef CAS.
- X. He, Z. Ma, X. Xi, A. Kudesi and J. Wang, Heterogeneous reaction of toluene/NO2/O3 on α-Fe2O3 nanoparticles: the impacts of O3, light illumination, and relative humidity on the formation of N-containing organic compounds (NOC), Environ. Sci.: Nano, 2022, 9, 3318–3330 RSC.
- X. He, J. Wu, Z. Ma, X. Xi and Y. Zhang, NH3-promoted heterogeneous reaction of SO2 to sulfate on α-Fe2O3 particles with coexistence of NO2 under different relative humidities, Atmos. Environ., 2021, 262, 118622 CrossRef CAS.
- R. Li, X. Jia, F. Wang, Y. Ren, X. Wang, H. Zhang, G. Li, X. Wang and M. Tang, Heterogeneous reaction of NO2 with hematite, goethite and magnetite: Implications for nitrate formation and iron solubility enhancement, Chemosphere, 2020, 242, 125273 CrossRef CAS.
- Z. Wang, T. Wang, H. Fu, L. Zhang, M. Tang, C. George, V. H. Grassian and J. Chen, Enhanced heterogeneous uptake of sulfur dioxide on mineral particles through modification of iron speciation during simulated cloud processing, Atmos. Chem. Phys., 2019, 19, 12569–12585 CrossRef CAS.
- X. Y. Zhang, G. S. Zhuang, J. M. Chen and H. X. Xue, Speciation of the elements and compositions on the surfaces of dust storm particles: The evidence for the coupling of iron with sulfur in aerosol during the long-range transport, Chin. Sci. Bull., 2005, 50, 738–744 CAS.
- Z. Wang, H. Fu, L. Zhang, W. Song and J. Chen, Ligand-Promoted Photoreductive Dissolution of Goethite by Atmospheric Low-Molecular Dicarboxylates, J. Phys. Chem. A, 2017, 121, 1647–1656 CrossRef CAS.
- G. Rubasinghege, R. W. Lentz, M. M. Scherer and V. H. Grassian, Simulated atmospheric processing of iron oxyhydroxide minerals at low pH: Roles of particle size and acid anion in iron dissolution, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6628–6633 CrossRef CAS PubMed.
- S. Go, A. Lyapustin, G. L. Schuster, M. Choi, P. Ginoux, M. Chin, O. Kalashnikova, O. Dubovik, J. Kim, A. Da Silva, B. Holben and J. S. Reid, Inferring iron-oxide species content in atmospheric mineral dust from DSCOVR EPIC observations, Atmos. Chem. Phys., 2022, 22, 1395–1423 CrossRef CAS.
- W. Yang, J. Zhang, Q. Ma, Y. Zhao, Y. Liu and H. He, Heterogeneous Reaction of SO2 on Manganese Oxides: the Effect of Crystal Structure and Relative Humidity, Sci. Rep., 2017, 7, 4550 CrossRef PubMed.
- K. Kaneko and K. Inouye, The mechanism of chemisorption of SO2 on iron (III) hydroxide oxides, Corros. Sci., 1981, 21, 639–646 CrossRef CAS.
- Z. Shu, L. Liu, W. Tan, S. L. Suib, G. Qiu, X. Yang, L. Zheng and F. Liu, Solar Irradiation Induced Transformation of Ferrihydrite in the Presence of Aqueous Fe2+, Environ. Sci. Technol., 2019, 53, 8854–8861 CrossRef CAS.
- D. M. Cwiertny, G. J. Hunter, J. M. Pettibone, M. M. Scherer and V. H. Grassian, Surface Chemistry and Dissolution of α-FeOOH Nanorods and Microrods: Environmental Implications of Size-Dependent Interactions with Oxalate, J. Phys. Chem. C, 2009, 113, 2175–2186 CrossRef CAS.
- S. O. Pehkonen, R. Siefert, Y. Erel, S. Webb and M. R. Hoffmann, Photoreduction of iron oxyhydroxides in the presence of important atmospheric organic compounds, Environ. Sci. Technol., 1993, 27, 2056–2062 CrossRef CAS.
- J. Mejia, E. E. Roden and M. Ginder-Vogel, Influence of Oxygen and Nitrate on Fe (Hydr)oxide Mineral Transformation and Soil Microbial Communities during Redox Cycling, Environ. Sci. Technol., 2016, 50, 3580–3588 CrossRef CAS PubMed.
- A. El Zein and Y. Bedjanian, Interaction of NO2 with TiO2 surface under UV irradiation: measurements of the uptake coefficient, Atmos. Chem. Phys., 2012, 12, 1013–1020 CrossRef CAS.
- T. Wang, Y. Liu, Y. Deng, H. Cheng, Y. Yang and L. Zhang, Photochemical reaction of NO2 on photoactive mineral dust: Mechanism and irradiation intensity dependence, J. Photochem. Photobiol., A, 2021, 416, 113319 CrossRef CAS.
- G. Zhang, Q. Lin, L. Peng, Y. Yang, F. Jiang, F. Liu, W. Song, D. Chen, Z. Cai, X. Bi, M. Miller, M. Tang, W. Huang, X. Wang, P. A. Peng and G. Sheng, Oxalate Formation Enhanced by Fe-Containing Particles and Environmental Implications, Environ. Sci. Technol., 2019, 53, 1269–1277 CrossRef CAS PubMed.
- K. Kawamura and S. Bikkina, A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 2016, 170, 140–160 CrossRef CAS.
- D. M. Mangiante, R. D. Schaller, P. Zarzycki, J. F. Banfield and B. Gilbert, Mechanism of Ferric Oxalate Photolysis, ACS Earth Space Chem., 2017, 1, 270–276 CrossRef CAS.
- D. A. Thomas, M. M. Coggon, H. Lignell, K. A. Schilling, X. Zhang, R. H. Schwantes, R. C. Flagan, J. H. Seinfeld and J. L. Beauchamp, Real-Time Studies of Iron Oxalate-Mediated Oxidation of Glycolaldehyde as a Model for Photochemical Aging of Aqueous Tropospheric Aerosols, Environ. Sci. Technol., 2016, 50, 12241–12249 CrossRef CAS PubMed.
- H. Pang, Q. Zhang, H. Wang, D. Cai, Y. Ma, L. Li, K. Li, X. Lu, H. Chen, X. Yang and J. Chen, Photochemical Aging of Guaiacol by Fe(III)-Oxalate Complexes in Atmospheric Aqueous Phase, Environ. Sci. Technol., 2019, 53, 127–136 CrossRef CAS PubMed.
- B. Tang, G. Wang, L. Zhuo, J. Ge and L. Cui, Facile Route to α-FeOOH and α-Fe2O3 Nanorods and Magnetic Property of α-Fe2O3 Nanorods, Inorg. Chem., 2006, 45, 5196–5200 CrossRef CAS PubMed.
- X. Huang, X. Hou, F. Wang, B. Guo, F. Song, L. Ling, J. Zhao and L. Zhang, Molecular-scale structures of uranyl surface complexes on hematite facets, Environ. Sci.: Nano, 2019, 6, 892–903 RSC.
- L. Shen, Y. Cao, Z. Du, W. Zhao, K. Lin and L. Jiang, Illuminate the active sites of γ-FeOOH for low-temperature desulfurization, Appl. Surf. Sci., 2017, 425, 212–219 CrossRef CAS.
- T. Wang, Y. Liu, Y. Deng, H. Fu, L. Zhang and J. Chen, Emerging investigator series: heterogeneous reactions of sulfur dioxide on mineral dust nanoparticles: from single component to mixed components, Environ. Sci.: Nano, 2018, 5, 1821–1833 RSC.
- T. Wang, Y. Y. Liu, Y. Deng, H. Y. Cheng, Y. Yang, Y. Q. Feng, L. W. Zhang, H. B. Fu and J. M. Chen, Photochemical Oxidation of Water-Soluble Organic Carbon (WSOC) on Mineral Dust and Enhanced Organic Ammonium Formation, Environ. Sci. Technol., 2020, 54, 15631–15642 CrossRef CAS PubMed.
- X. Zhang, G. Zhuang, J. Chen, Y. Wang, X. Wang, Z. An and P. Zhang, Heterogeneous Reactions of Sulfur Dioxide on Typical Mineral Particles, J. Phys. Chem. B, 2006, 110, 12588–12596 CrossRef CAS.
- Q. Ma, Y. Liu and H. He, Synergistic Effect between NO2 and SO2 in Their Adsorption and Reaction on γ-Alumina, J. Phys. Chem. A, 2008, 112, 6630–6635 CrossRef CAS.
- L. Li, Z. M. Chen, Y. H. Zhang, T. Zhu, J. L. Li and J. Ding, Kinetics and mechanism of heterogeneous oxidation of sulfur dioxide by ozone on surface of calcium carbonate, Atmos. Chem. Phys., 2006, 6, 2453–2464 CrossRef CAS.
- Y. Zhao, Y. Liu, J. Ma, Q. Ma and H. He, Heterogeneous reaction of SO2 with soot: The roles of relative humidity and surface composition of soot in surface sulfate formation, Atmos. Environ., 2017, 152, 465–476 CrossRef CAS.
- J. Li, C. Xiao, K. Wang, Y. Li and G. Zhang, Enhanced Generation of Reactive Oxygen Species under Visible Light Irradiation by Adjusting the Exposed Facet of FeWO4 Nanosheets To Activate Oxalic Acid for Organic Pollutant Removal and Cr(VI) Reduction, Environ. Sci. Technol., 2019, 53, 11023–11030 CrossRef CAS PubMed.
- M. Ullerstam, M. S. Johnson, R. Vogt and E. Ljungström, DRIFTS and Knudsen cell study of the heterogeneous reactivity of SO2 and NO2 on mineral dust, Atmos. Chem. Phys., 2003, 3, 2043–2051 CrossRef CAS.
- J. Park, M. Jang and Z. Yu, Heterogeneous Photo-oxidation of SO2 in the Presence of Two Different Mineral Dust Particles: Gobi and Arizona Dust, Environ. Sci. Technol., 2017, 51, 9605–9613 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- P. D. Wade, J. Taylor and P. Siekevitz, Mammalian cerebral cortical tissue responds to low-intensity visible light, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9322–9326 CrossRef CAS.
- L. Y. Wu, S. R. Tong, W. G. Wang and M. F. Ge, Effects of temperature on the heterogeneous oxidation of sulfur dioxide by ozone on calcium carbonate, Atmos. Chem. Phys., 2011, 11, 6593–6605 CrossRef CAS.
- G. Pacchioni, A. Clotet and J. M. Ricart, A theoretical study of the adsorption and reaction of SO2 at surface and step sites of the MgO(100) surface, Surf. Sci., 1994, 315, 337–350 CrossRef CAS.
- M. Huang, W. Xiang, T. Zhou, J. Mao, X. Wu and X. Guo, The critical role of the surface iron-oxalate complexing species in determining photochemical degradation of norfloxacin using different iron oxides, Sci. Total Environ., 2019, 697, 134220 CrossRef CAS.
- A. P. Prince, P. Kleiber, V. H. Grassian and M. A. Young, Heterogeneous interactions of calcite aerosol with sulfur dioxide and sulfur dioxide–nitric acid mixtures, Phys. Chem. Chem. Phys., 2007, 9, 3432 RSC.
- W. Yang, H. He, Q. Ma, J. Ma, Y. Liu, P. Liu and Y. Mu, Synergistic formation of sulfate and ammonium resulting from reaction between SO2 and NH3 on typical mineral dust, Phys. Chem. Chem. Phys., 2016, 18, 956–964 RSC.
- J. Baltrusaitis, D. M. Cwiertny and V. H. Grassian, Adsorption of sulfur dioxide on hematite and goethite particle surfaces, Phys. Chem. Chem. Phys., 2007, 9, 5542 RSC.
- T. Wang, Y. Liu, Y. Deng, H. Fu, L. Zhang and J. Chen, Adsorption of SO2 on mineral dust particles influenced by atmospheric moisture, Atmos. Environ., 2018, 191, 153–161 CrossRef CAS.
- K. Li, X. Fang, T. Wang, K. Gong, M. Ali Tahir, W. Wang, J. Han, H. Cheng, G. Xu and L. Zhang, Atmospheric organic complexation enhanced sulfate formation and iron dissolution on nano α-Fe2O3, Environ. Sci.: Nano, 2021, 8, 698–710 RSC.
- W. Feng and D. Nansheng, Photochemistry of hydrolytic iron (III) species and photoinduced degradation of organic compounds. A minireview, Chemosphere, 2000, 41, 1137–1147 CrossRef CAS PubMed.
- N. Bhandari, D. B. Hausner, J. D. Kubicki and D. R. Strongin, Photodissolution of Ferrihydrite in the Presence of Oxalic Acid: An In Situ ATR-FTIR/DFT Study, Langmuir, 2010, 26, 16246–16253 CrossRef CAS PubMed.
- P. Borer, B. Sulzberger, S. J. Hug, S. M. Kraemer and R. Kretzschmar, Photoreductive Dissolution of Iron(III) (Hydr)oxides in the Absence and Presence of Organic Ligands: Experimental Studies and Kinetic Modeling, Environ. Sci. Technol., 2009, 43, 1864–1870 CrossRef CAS PubMed.
- Y. Zuo and J. Hoigne, Formation of hydrogen peroxide and depletion of oxalic acid in atmospheric water by photolysis of iron(III)-oxalato complexes, Environ. Sci. Technol., 1992, 26, 1014–1022 CrossRef CAS.
- Y. Zuo, Kinetics of photochemical/chemical cycling of iron coupled with organic substances in cloud and fog droplets, Geochim. Cosmochim. Acta, 1995, 59, 3123–3130 CrossRef CAS.
- Z. Wang, C. Chen, W. Ma and J. Zhao, Photochemical Coupling of Iron Redox Reactions and Transformation of Low-Molecular-Weight Organic Matter, J. Phys. Chem. Lett., 2012, 3, 2044–2051 CrossRef CAS.
- J. Chen, H. Zhang, I. V. Tomov and P. M. Rentzepis, Electron Transfer Mechanism and Photochemistry of Ferrioxalate Induced by Excitation in the Charge Transfer Band, Inorg. Chem., 2008, 47, 2024–2032 CrossRef CAS PubMed.
- I. P. Pozdnyakov, O. V. Kel, V. F. Plyusnin, V. P. Grivin and N. M. Bazhin, New Insight into Photochemistry of Ferrioxalate, J. Phys. Chem. A, 2008, 112, 8316–8322 CrossRef CAS.
- C. Weller, S. Horn and H. Herrmann, Effects of Fe(III)-concentration, speciation, excitation-wavelength and light intensity on the quantum yield of iron(III)-oxalato complex photolysis, J. Photochem. Photobiol., A, 2013, 255, 41–49 CrossRef CAS.
- M. Huang, T. Zhou, X. Wu and J. Mao, Distinguishing homogeneous-heterogeneous degradation of norfloxacin in a photochemical Fenton-like system (Fe3O4/UV/oxalate) and the interfacial reaction mechanism, Water Res., 2017, 119, 47–56 CrossRef CAS PubMed.
- L. Huang, Y. Zhao, H. Li and Z. Chen, Kinetics of Heterogeneous Reaction of Sulfur Dioxide on Authentic Mineral Dust: Effects of Relative Humidity and Hydrogen Peroxide, Environ. Sci. Technol., 2015, 49, 10797–10805 CrossRef CAS.
- Q. Ma, L. Wang, B. Chu, J. Ma and H. He, Contrary Role of H2O and O2 in the Kinetics of Heterogeneous Photochemical Reactions of SO2 on TiO2, J. Phys. Chem. A, 2019, 123, 1311–1318 CrossRef CAS.
- X. Y. Zhang, Y. Q. Wang, T. Niu, X. C. Zhang, S. L. Gong, Y. M. Zhang and J. Y. Sun, Atmospheric aerosol compositions in China: spatial/temporal variability, chemical signature, regional haze distribution and comparisons with global aerosols, Atmos. Chem. Phys., 2012, 12, 779–799 CrossRef CAS.
- T. Wang, Y. Y. Liu, H. Y. Cheng, Z. Z. Wang, H. B. Fu, J. M. Chen and L. W. Zhang, Significant formation of sulfate aerosols contributed by the heterogeneous drivers of dust surface, Atmos. Chem. Phys., 2022, 22, 13467–13493 CrossRef CAS.
- F. J. Lazaro, L. Gutierrez, V. Barron and M. D. Gelado, The speciation of iron in desert dust collected in Gran Canaria (Canary Islands): Combined chemical, magnetic and optical analysis, Atmos. Environ., 2008, 42, 8987–8996 CrossRef CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Reactions on Mineral Dust, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS.
- X. J. Zhao, G. S. Zhuang, Z. F. Wang, Y. L. Sun, Y. Wang and H. Yuan, Variation of sources and mixing mechanism of mineral dust with pollution aerosol - revealed by the two peaks of a super dust storm in Beijing, Atmos. Res., 2007, 84, 265–279 CrossRef CAS.
- G. S. Zhuang, J. H. Guo, H. Yuan and C. Y. Zhao, The compositions, sources, and size distribution of the dust storm from China in spring of 2000 and its impact on the global environment, Chin. Sci. Bull., 2001, 46, 895–901 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Kedong
Gong
a,
Qiuyue
Ge
a,
Longqian
Wang
a,
Tao
Wang
*a and
Liwu
Zhang
a,
Kedong
Gong
a,
Qiuyue
Ge
a,
Longqian
Wang
a,
Tao
Wang
*a and
Liwu
Zhang