
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyl-phenanthrenes in early life stage fish: differential toxicity in Atlantic haddock (Melanogrammus aeglefinus) embryos†
Carey E.
Donald
 *a,
Charlotte L.
Nakken
ab,
Elin
Sørhus
a,
Prescilla
Perrichon
c,
Kåre B.
Jørgensen
d,
Hege K.
Bjelland
d,
Christine
Stølen
d,
Sindhu
Kancherla
d,
Philipp
Mayer
e and
Sonnich
Meier
a
*a,
Charlotte L.
Nakken
ab,
Elin
Sørhus
a,
Prescilla
Perrichon
c,
Kåre B.
Jørgensen
d,
Hege K.
Bjelland
d,
Christine
Stølen
d,
Sindhu
Kancherla
d,
Philipp
Mayer
e and
Sonnich
Meier
a
aInstitute of Marine Research, 5817 Bergen, Norway. E-mail: carey.donald@hi.no
bDepartment of Chemistry, University of Bergen, 5020 Bergen, Norway
cInstitute of Marine Research, Austevoll Research Station, 5392 Storebø, Norway
dDepartment of Chemistry, Bioscience, and Environmental Engineering, Faculty of Science and Technology, University of Stavanger, 4036 Stavanger, Norway
eDepartment of Environmental and Resource Engineering, Technical University of Denmark, Bygningstorvet 115, 2800 Kgs. Lyngby, Denmark
First published on 2nd February 2023
Abstract
Tricyclic polycyclic aromatic hydrocarbons (PAHs) are believed to be the primary toxic components of crude oil. Such compounds including phenanthrene are known to have direct effects on cardiac tissue, which lead to malformations during organogenesis in early life stage fish. We tested a suite of 13 alkyl-phenanthrenes to compare uptake and developmental toxicity in early life stage haddock (Melanogrammus aeglefinus) embryos during gastrulation/organogenesis beginning at 2 days post fertilization via passive dosing. The alkyl-phenanthrenes were tested at their solubility limits, and three of them also at lower concentrations. Measured body burdens were linearly related to measured water concentrations. All compounds elicited one or more significant morphological defects or functional impairment, such as decreased length, smaller eye area, shorter jaw length, and increased incidence of body axis deformities and eye deformities. The profile of developmental toxicities appeared unrelated to the position of alkyl substitution, and gene expression of cytochrome 1 a (cyp1a) was low regardless of alkylation. Mortality and sublethal effects were observed below the expected range for baseline toxicity, thus indicating excess toxicity. Additionally, PAH concentrations that resulted in toxic effects here were far greater than when measured in whole crude oil exposures that cause toxicity. This work demonstrates that, while these phenanthrenes are toxic to early life stage fish, they cannot individually account for most of the developmental toxicity of crude oil, and that other compounds and/or mixture effects should be given more consideration.
Environmental significanceOf the thousands of chemicals in petroleum products, phenanthrene and other tricyclic compounds are believed to cause much of the toxicity. This study compares the toxic effects of a suite of alkylated-phenanthrenes in early life stages of a cold-water fish, the Atlantic haddock, a species that can be impacted by petroleum extraction in the Arctic. Some degree of toxicity resembling crude oil toxicity was observed for all tested compounds, albeit at high concentrations. The results suggest that additional compounds beyond PAHs and pollutant interactions play a larger role in the toxicity of petroleum products and environmental mixtures. |
Introduction
Crude oil is an immensely complex mixture known to cause developmental toxicity in fish early life stages. Despite this complexity, the primary cardiotoxicity of crude oil is coupled to the concentrations of tricyclic polycyclic aromatic hydrocarbons (PAHs) measured in both exposure water and tissue of exposed embryos.1–5 PAH exposure levels are therefore often measured and used as the metric basis to represent the degree of crude oil toxicity.6,7 When exposed individually, PAHs have been shown to reproduce many of the functional and morphological defects associated with exposure to the whole crude oil mixture.4–6 Of particular interest are the tricyclic PAHs, like the prototypical phenanthrene which are known to be cardiotoxic.2,3,8However, a major data gap lies in the fact that no single PAH acts at the same potency as in a PAH-containing mixture.5 Studies characterizing the mechanisms of toxicity often exceed the aqueous solubilities (which vary between <1 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg L−1) of the PAH compounds.3,9–14 Thus, there is still a large unresolved discrepancy between toxicity studies with single compounds and complex oil mixtures.5 Other components in oil likely contribute to mixture toxicity, possibly through activation of the aryl hydrocarbon receptor (AHR) which acts as a xenobiotic receptor.15,16 Upon activation, AHR initiates metabolizing enzymes like cytochrome P450 1A (cyp1a) and others in the CYP family, which can form toxic metabolites.17,18 In addition to suspected specific modes of action, a high accumulation of lipophilic compounds is believed to cause lethality and sublethal baseline toxicity by disrupting membrane fluidity and ion regulation.5,19 Without understanding the toxicities of alkyl-phenanthrenes, we cannot make predictions on how they contribute to mixture toxicity.
000 μg L−1) of the PAH compounds.3,9–14 Thus, there is still a large unresolved discrepancy between toxicity studies with single compounds and complex oil mixtures.5 Other components in oil likely contribute to mixture toxicity, possibly through activation of the aryl hydrocarbon receptor (AHR) which acts as a xenobiotic receptor.15,16 Upon activation, AHR initiates metabolizing enzymes like cytochrome P450 1A (cyp1a) and others in the CYP family, which can form toxic metabolites.17,18 In addition to suspected specific modes of action, a high accumulation of lipophilic compounds is believed to cause lethality and sublethal baseline toxicity by disrupting membrane fluidity and ion regulation.5,19 Without understanding the toxicities of alkyl-phenanthrenes, we cannot make predictions on how they contribute to mixture toxicity.
To further understand the role of tricyclic PAHs in crude oil toxicity, we aimed to characterize the differential uptake and toxicity of phenanthrene and 13 alkyl-phenanthrenes in Atlantic haddock (Melanogrammus aeglefinus), a cold-water species of high ecological and economical importance. Phenanthrene and derivative compounds were selected because of phenanthrene's documented cardiotoxicity. Recently, Sørhus, et al. 2021 (ref. 1) reported the sensitivity of haddock to crude oil by exposing haddock eggs to a micro-dispersion of weathered crude oil for three days and measuring an array of morphological endpoints after hatch. The single compound exposures in the present work were designed to be directly comparable to the whole oil toxicity study,1 in addition to being a developmental screen of yet-unstudied alkyl-phenanthrenes. The suite of compounds includes nine synthesized compounds in addition to those commercially available. We used passive dosing techniques to achieve a constantly renewed exposure level at each compound's water solubility limit without the need for cosolvents. Exposures at multiple sub-solubility levels were also performed for a selected subset of compounds. After hatching, multiple endpoints were measured to assess defects in the body axis, craniofacial structures, and heart form and function. The results provide further foundations on how tricyclic PAHs individually contribute to the developmental toxicity of crude oil, or if other compounds or mixture effects should be given more consideration.
Materials and methods
Materials
| Compound name | Abbreviation | Sourcea | Solubility, 25 °C, μg L−1 | logKow |
Exposuresf | |
|---|---|---|---|---|---|---|
|
a UiS indicates synthesized at the University of Stavanger, Norway.
b Estimated value, using KOWWIN v1.67 for log octanol–water coefficient (logKow); or WSKOW v1.41 for solubility.20
c Value is the experimental value reported in U.S. Environmental Protection Agency 2015.20
d Exposures performed in 2019.
e Value shown is for 9-propylphenanthrene as values for 3-PP were not available.
f
S indicates exposures at solubility limits, and S/X are dilutions of solubilities.
|
||||||
| C0 | Phenanthrene |
PHE | Sigma-Aldrich | 677b, 1150c | 4.46c | S, S/3, S/9, S/27, S/81 |
| S, S/3, S/9d | ||||||
| C1 | 1-Methylphenanthrene | 1-MP | ChemCruz™ | 171b, 269c | 5.08c | S |
| 2-Methylphenanthrene | 2-MP | UiS | 263b, 280c | 5.15c | S | |
| 3-Methylphenanthrene | 3-MP | ChemCruz™ | 263b, 280c | 5.15c | S | |
| 4-Methylphenanthrene | 4-MP | UiS | 171b, 269c | 5.08c | S, S/3, S/9, S/27 | |
| S, S/3d | ||||||
| 9-Methylphenanthrene | 9-MP | ChemCruz™ | 247b | 4.89b | S | |
| C2 | 1,4-Dimethylphenanthrene | 1,4-DMP | UiS | 71.3b | 5.44b | S, S/3, S/9 |
| S, S/3d | ||||||
| 1,7-Dimethylphenanthrene | 1,7-DMP | UiS | 71.3b | 5.44b | S | |
| 2,3-Dimethylphenanthrene | 2,3-DMP | UiS | 71.3b | 5.44b | S | |
| 2,7-Dimethylphenanthrene | 2,7-DMP | UiS | 71.3b | 5.44b | S | |
| 3,6-Dimethylphenanthrene | 3,6-DMP | UiS | 71.3b | 5.44b | S | |
| 3-Ethylphenanthrene | 3-EP | UiS | 79.7b | 5.38b | S | |
| C3 | 3-Propylphenanthrene | 3-PP | UiS | 25.6b,e | 5.87b,e | S |
| C4 | Retene (1-methyl-7-isopropylphenanthrene) | RET | ChemCruz™ | 8.48b | 6.35b | S |
Exposures
Three compounds, PHE, 4-MP, and 1,4-DMP were selected for additional exposures at several concentrations at dilutions of solubilities. These three were selected as they represent the non-substituted (C0), a methyl-substituted (C1), and a dimethyl-substituted phenanthrene (C2). Saturated methanol solutions were used for loading the silicone for the highest exposure (S), as described above. An aliquot of the saturated loading solution was filtered and diluted to sequential three-fold dilutions for loading lower levels: S/3, S/9, S/27, and S/81.
After reviewing preliminary data and making minor adjustments to the exposure system, the following exposures were repeated the subsequent year, 2019: PHE (S, S/3, S/9), 4-MP (S and S/3), 1,4-DMP (S and S/3) (Table 1). This time we increased the mass of silicone (from 1.2 g to between 8.6 and 12 g per jar), using a thicker, 3 mm diameter silicone rod, to yield more stable exposure concentrations. We also used fewer eggs, down from 120 to 100 eggs (from 0.26 g to 0.22 g). Exposure to one additional compound, 2-MP, was also conducted in 2019.
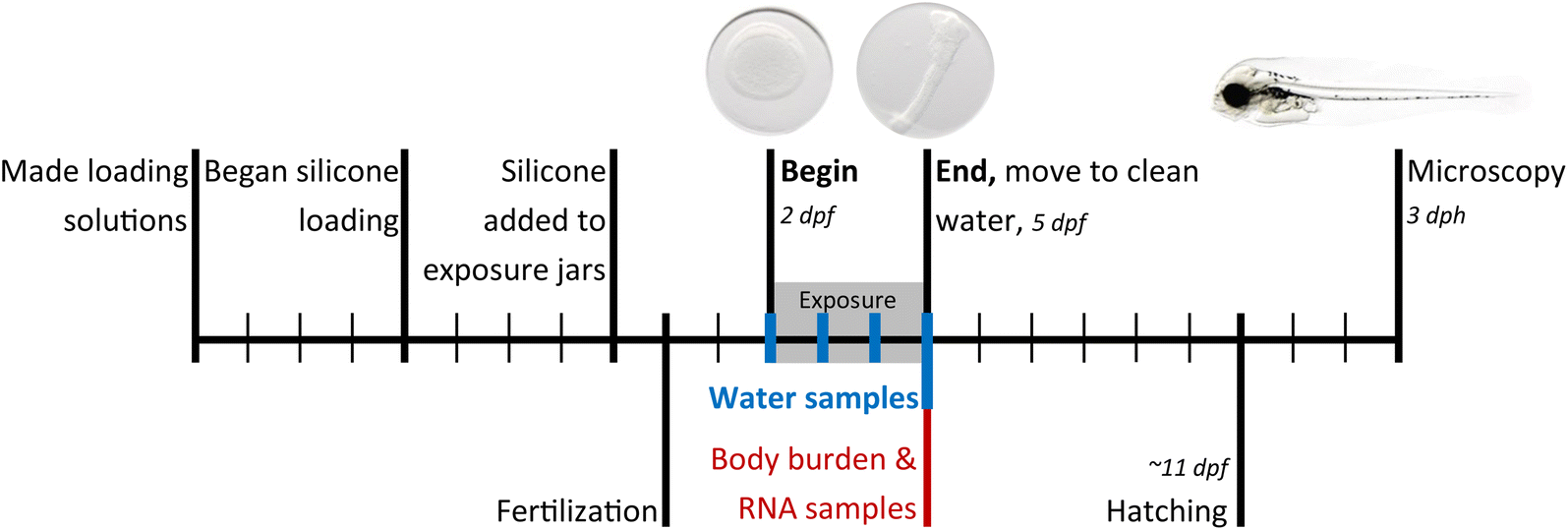 | ||
| Fig. 1 Experimental design, where each mark on the timeline represents a day. Exposures began at 2 dpf (beginning gastrulation) and ended at 5 dpf (20 somite or cardiac cone stage). dpf = days post fertilization, dph = days post hatching. | ||
Analysis
For analysis of PAH body burden, one sample of 10–24 eggs was collected from each triplicate jar at the conclusion of the exposures (72 h) and snap-frozen in liquid nitrogen until extracted. For several treatments, the number of replicate body burden samples was limited by the number of remaining living eggs in a jar; the number of body burden replicates is provided in Tables S2 and S3.† After adding internal standard, the samples were homogenized and extracted as given in Sørensen et al. (2016)26 using 1:1 n-hexane:dichloromethane, dried with 0.2 g Na2SO4, and cleaned up with Chromabond SiOH solid phase extraction columns (3 mL, 500 mg, Machery-Nagel, Germany) and eluted with 9:1 n-hexane:dichloromethane. Final extracts were reduced to 50–100 μL but diluted up to 1000 μL when necessary if the chromatographic response exceeded responses in the calibration curve.
Quantitative analysis of water and body burden samples was performed on an Agilent 7890 gas chromatograph coupled to an Agilent 7010c triple quadrupole mass detector (GC-MS/MS) based on the methods described in Sørensen, et al. 2016.27 Specific MS/MS parameters are provided in Table S1.† Body burden samples were converted to a wet weight basis using the average mass of one egg, 0.00218 g ± 0.00018 g (mean ± SD). This average was calculated from a concurrent study1 from 50 samples of ∼100 eggs counted under a microscope, and gives a low relative standard deviation of 8%. We used this average value derived from a concurrent study with more eggs because it was difficult to remove small amounts of excess water from these samples containing as few as 10 eggs.
Traditional morphological endpoints used in oil exposure1 were measured and are demonstrated in Fig. S1.† Morphometric measurements were processed using the ObjectJ package in ImageJ. Standard length (μm), finfold (μm2), eye areas (μm2), eye-to-nose length (ethmoid plate, μm), and jaw length (μm) and angle (°) were measured. Net yolk (area of the yolk sac, μm2) and total yolk (area of the yolk sac and adjacent edema, μm2) were measured. As a toxic endpoint, net yolk changes suggest over- or under-utilization of yolk sac nutrients. Yolk sac edema (%) was calculated: yolk sac edema = (total yolk − net yolk)/total yolk, where higher yolk sac edema indicates fluid dysregulation.
Three types of categorical phenotypes were assigned to each larva to describe the presence of deformities in the body axis, eye shape, and jaw shape. The presence of a body axis deformity (having spinal curvature or an arched or hunched spine) was noted. The incidence of eye shape deformity was scored according to 4 categories of phenotypes, severity dependent: (1) no effect, (2) eye bend (one protrusion from circular), (3) eye bulky (two or more protrusions from circular), and (4) lens out.28 Finally, jaw deformities were scored by severity from (1) no effect, (2) slight deformity, including a thicker, shortened lower jaw, (3) moderate deformity, including a twisted jaw structure (4) severe deformity with straight jaw hanging open, to (5) no lower jaw structures. In statistical pairwise comparisons to control, the eye and jaw shape phenotype sets were each reduced to presence/absence of a deformity.
Heart rate was determined by manually counting the number of cycles of atrial and ventricular contractions in each 20 s-video collected at 3 dph. The diastolic ventricular area (μm2), and the atrial and ventricular major axis during the systolic and diastolic events were outlined in still images from the videos. Fractional shortening (FS, %) of the atrium and ventricle was then calculated: FS = (diastole diameter − systole diameter)/diastole diameter. The phenotype “silent ventricle” was also scored if FS was less than 2%.
One-way hierarchical clustering was performed to find groupings of compounds with similar significant endpoints. Linear regressions were used to relate expected dilution to exposure concentrations and body burden, exposure concentrations to body burden, and bioconcentration to logKow. All statistics were performed using JMP 14 (SAS, Cary, NC, USA), R version 4.0.2, and Microsoft Excel 2016.
Results
Passive dosing
Exposures were well documented with quantitative analysis of water and body burden. Tables S2 and S3† include full details of each exposure treatment, dose level, and experiment year. The experiments with PHE, 4-MP, and 1,4-DMP were conducted at multiple exposure concentrations and corresponding body burdens The 3-fold dilutions series of the methanol loading solutions led to predictable exposures at dilutions of solubilities (Fig. 2). There was one exception; in 2018, microcrystals of the three test compounds were transferred from the saturated solution to their S/3 dilutions. This resulted in lower effective dilution factors between S and S/3, especially for PHE and 4-MP (Table S4†). In subsequent, 2019 experiments, the S loading solution was filtered before serial dilutions.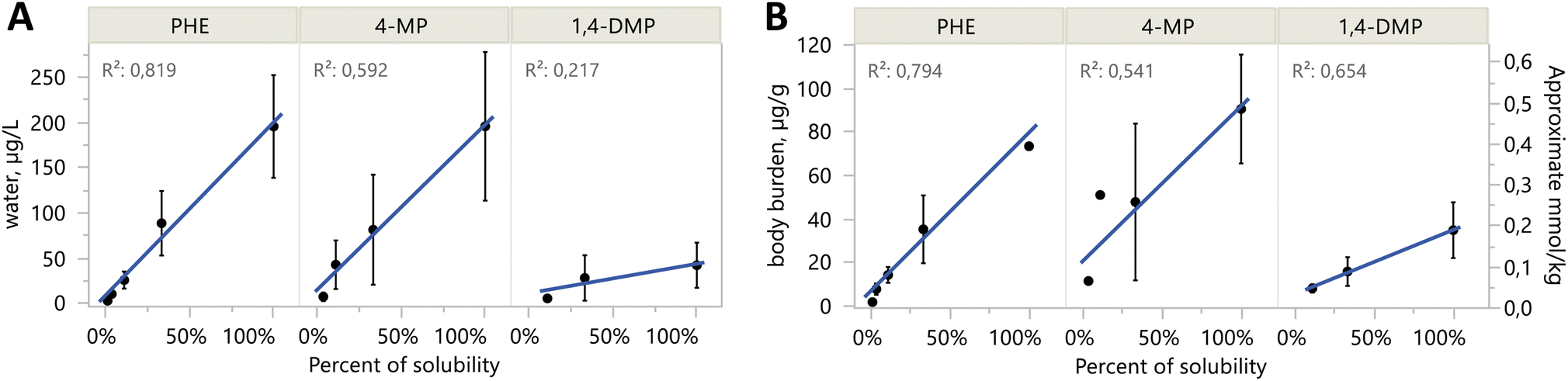 | ||
| Fig. 2 Aqueous concentrations (A) and body burden levels (B) of passive dosing exposures are linearly related to expected dilution levels. Percent of solubility (x-axis) is the expected percent of solubility for the different exposure levels: S = 100%, S/3 = 33%, S/9 = 11%, etc. Data points shown are mean ± SD, including replicates from both years, and from all days 0–4 for water in (A). The right y-axis for body burden (B) in mmol kg−1 is approximate because of different molar masses among compounds. | ||
Mean water concentrations achieved were not different between years in S treatments for PHE and 4-MP (two-sided t-tests, p-values = 0.30 and 0.26, respectively). Most water samples from 1,4-DMP in 2019 were lost due to laboratory error, so no such comparison could be made. For the purpose of presentation, results from both years' S treatments of PHE, 4-MP, and 1,4-DMP are combined in the main text, while complete results separated by year are in the ESI.† Water concentrations from the dilutions exposures do vary by year, and thus dilution results are only presented alongside all other treatments in Tables S2 and S3.†
Passive dosing can maintain constant test concentrations over time.29 This was not fully achieved in the 2018 experiments, where water concentrations dropped 28–74% after the addition of eggs between sampling on days 0 and 1 (Table S5†). Concentrations were then stable between days 1 and 3 (Fig. S3†). In the 2019 experiments, the phase ratio between silicone and eggs was increased, which resulted in a more stable exposure with concentrations decreases of 10–17% within the first days. This moderate decline in water concentrations was difficult to avoid because of limited amounts and high costs of several of the test compounds.
Developmental toxicity screen among alkyl-phenanthrene compounds
Passive dosing was used to establish and maintain concentrations of each substance at the saturation limit, which means that the diverse compounds were administered at their respective solubilities. Generally, concentrations of the compounds in the S treatments, i.e. at aqueous solubility, decreased with increasing alkyl substitution (Fig. 3A). Exposure concentrations of two C1 compounds (2-MP, 3-MP, and 4-MP) were comparable to PHE. Clear differences among isomers were noted, notably among the C1s, where exposures varied from 22.4 μg L−1 for 1-MP, to 197 μg L−1 for 2-MP. The lowest measured exposure concentration was for RET, at 2.30 μg L−1, which corresponded to the lowest measured body burden of 1.28 μg g−1. Full details of each exposure treatment, dose level, and experiment year are provided in Tables S2 and S3.†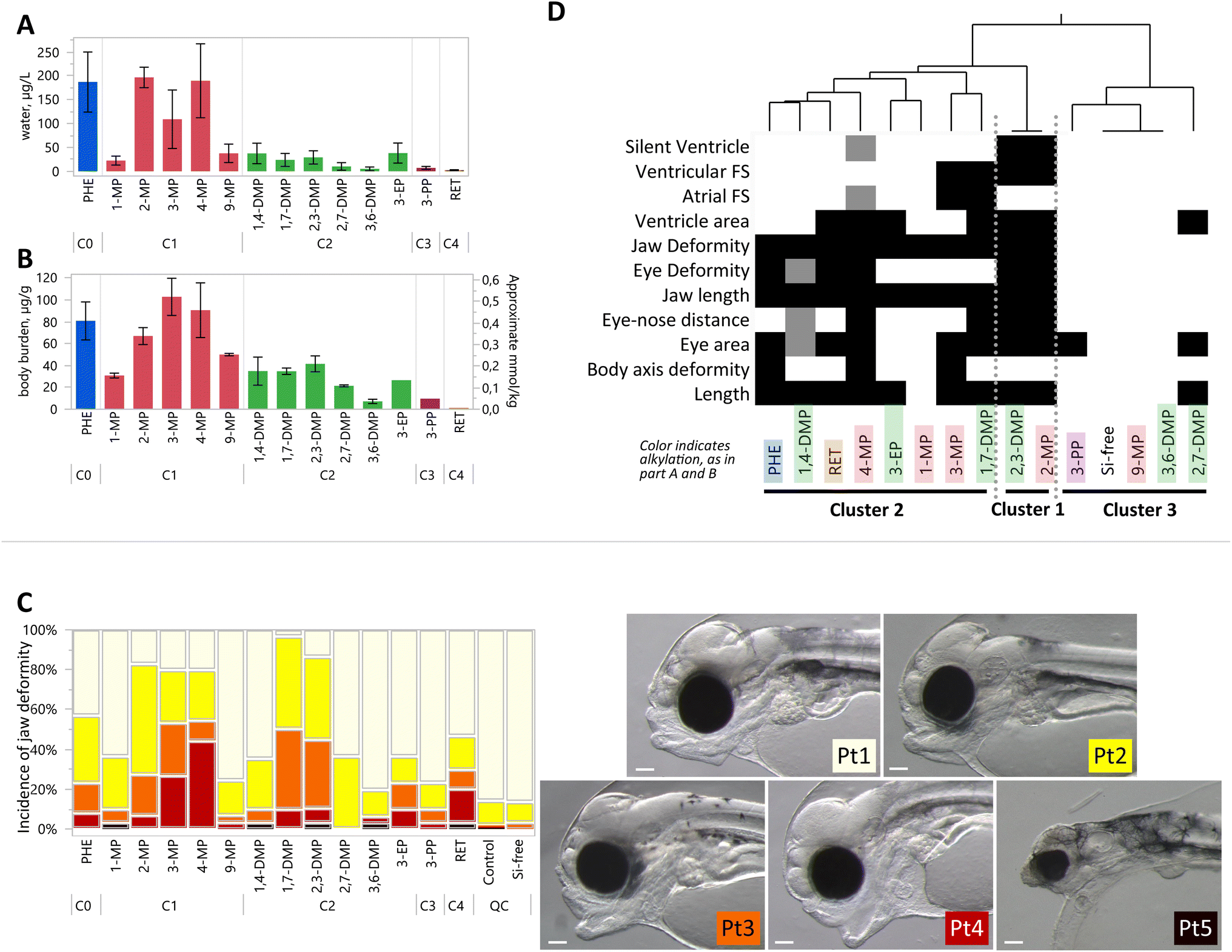 | ||
| Fig. 3 Comparative developmental toxicity of exposures at solubility in water. Full results for all exposure levels and years are provided in the ESI;† only S treatments are shown here. (A) Water concentrations of each tested compound. Bars represent mean ± SD, from all replicates on days 0–4, including replicates from both years where applicable. (B) Body burden concentrations at the end of the exposure. Bars represent the mean of S treatments ± SD, including replicates from both years where applicable. The right y-axis for mmol kg−1 is an approximate value due to molar mass differences among compounds. (C) Incidence of jaw phenotypes depending on a severity gradient from Pt1 (cream), Pt2 (yellow), Pt3 (orange), Pt4 (red) to Pt5 (black), demonstrated in pictures at right. Scale bar is 0.1 mm. Jaw phenotype is shown as an example of one of 16 sublethal endpoints measured in this study. All bars represent 30 observations, except for PHE, 4-MP, and 1,4-DMP which are 60 observations, and Si-control which is 90 observations, combined from both exposure years. (D) One-way hierarchical clustering of differential toxicities, where shading indicates an endpoint that was significantly different from control. Lighter shades indicate the endpoint that was significant in one, but not in both the repeated exposures (Si-control exposure was performed three times). Clusters of compounds are identified 1–3, where cluster 1 shows the greatest number of significant toxic endpoints. | ||
Body burden concentrations were also decreasing with alkylation number (Fig. 3B). PHE (81 μg g−1 ww) and the C1-PHEs had the highest average values (between 31 and 103 μg g−1 ww). Among the C2-PHEs, 3,6-DMP and 2,7-DMP had the lowest levels of body burden (<25 μg g−1 ww), and the lowest water concentrations (<15 μg L−1). The lowest body burden was measured in RET, at 1.28 μg g−1 ww. Among the three compounds with exposures at multiple concentrations, body burdens increased linearly with water concentrations for PHE (R2 = 0.94, p < 0.001), 4-MP (R2 = 0.69, p < 0.001), and 1,4-DMP (R2 = 0.56, p = 0.004) (Fig. S4†). Body burden levels for any compound in control treatments were low and averaged 0.019 μg g−1 ww.
Expression of cyp1a was generally low. The highest average relative expression measured in any treatment was less than 4-fold over control (Tables S2 and S3†), and is therefore not presented as a figure. Notably, however, 3-EP, 3-PP, and RET were not measured. Several of the treatments did have low, but significant induction compared to controls: PHE (S/9 in 2019), 4-MP (S/3 in 2019), 1,4-DMP (S and S/3 in 2019), and one of the 3 Si-free control treatments (2019) (Tables S2 and S3†). Expression of cyp1a was slightly negatively related to increasing body burden in each PHE (R2 = 0.14, p < 0.03), 4-MP (R2 = 0.38, p < 0.001), and 1,4-DMP (R2 = 0.16, p = 0.07).
Morphometric endpoints were collected to quantify the degree of developmental toxicity. The results from one endpoint, jaw malformation, are presented in Fig. 3C as an example. Six compounds induced jaw malformation in over 50% of larvae including PHE (57%), 2-MP (83%), 3-MP (80%), 4-MP (80%), 1,7-DMP (97%), and 2,3-DMP (85%). The majority of jaw deformities among the C1-PHEs were moderate and severe deformity phenotypes (Pt3 and Pt4). In comparison, many phenotypes in the C2-group, 1,7-DMP and 2,3-DMP were less severe, with slight and moderate deformity phenotypes (Pt2 and Pt3).
All compounds elicited one or more significant morphological defects or functional impairment, measured with an array of 16 endpoints (Fig. S5†). Four endpoints were significant in at least one of the 3 Si-free controls: finfold, net yolk, jaw angle, and heart rate. Data for these endpoints were therefore not considered further in the comparative screen but are provided in the ESI.† Yolk sac edema was the only endpoint that was not significant in any treatment. Significant effects were distributed among compounds in all the alkylation groups, and include decreased length, smaller eye area, shorter eye-nose distance, and jaw length, and increased incidence of body axis deformities and eye deformities (Fig. S6–S8†). Craniofacial and body axis phenotypes were observed to be similar to those described in crude oil exposure, i.e. reduced jaw length, eye deformities, and spinal curvature.1,3,30–33
Toxicological endpoints for the compounds exposed at their water solubilities are condensed and represented in a heatmap in Fig. 3D, where hierarchical clustering highlights similarities among toxic responses. The compounds with the greatest number of significant endpoints are sorted into cluster 1, which includes a C1-phenanthrene (2-MP) and a C2-phenanthrene (2,3-DMP). Each cluster contains phenanthrenes with different numbers of alkyl substitutions, and therefore exposure concentrations and body burden varied considerably among the compounds within clusters. For example, cluster 2 includes both PHE with the highest and RET with the lowest body burden.
Mortality was significant in 4 compounds (PHE, 3-EP, 3-PP, and RET) although data were not available for seven of the tested compounds (Fig. S6†).
The tested compounds, when exposed at their solubility limits, resulted in differing degrees of cardiac toxicity. Morphological (ventricle area) and/or functional heart defects (atrial and ventricular FS, silent ventricle) were present in 3 of 5 C1 compounds, and 5 of 6 C2 compounds (Fig. S8†). No cardiac effects were induced by 1-MP, 9-MP, 3,6-DMP, or 3-PP. Atrial FS was lower than controls for 4-MP and 3-MP treatments but was higher than controls with 2,3-DMP. Ventricular FS decreased with 4-MP, 1,7-DMP, and 2,3-DMP. The only treatments with a significant incidence of silent ventricle were 2-MP and 2,3-DMP.
Discussion
Passive dosing and comparing body burdens
The maximum exposure level (S) varies for each tested compound based on specific physiochemical properties. Water solubility, lipid-water partitioning, and biotransformation all affect the resulting internal dose, which was expected to vary greatly within this homologue series.31 Generally, higher alkylation led to lower water solubility (lower exposure concentration) and lower body burden. To be able to compare the compounds in this work, we consider responses elicited from the S exposures, which are exposure levels determined by each compound's limit of solubility. Indeed most of the compounds were exposed at only one level—a level limited by solubility that could theoretically occur in the environment. Differences in toxicity discussed here are then the combined outcomes of dose and potency. The purity of all compounds is expected to be at least 97%, although exact values are not available for the synthesized compounds (Table 1). While unlikely, even small amounts of impurities with high potencies could affect toxicities.The presented exposures were performed on a sensitive, cold-water marine fish, at concentrations within the limits of solubility. Mechanistic or high-throughput studies have often exceeded the aqueous solubility of the PAH compounds9–12 using co-solvents like dimethyl sulfoxide. The key advantages of using passive dosing techniques are that they (1) provide dissolved concentrations within solubility limits, (2) buffer concentrations against losses, and (3) avoid co-solvent addition. Both water concentrations and body burden were measured to thoroughly characterize the exposures. Comparisons to expected, published solubility values for most compounds are difficult because the present study was not conducted at standard conditions. Our experimental value for PHE, 187 μg L−1, was somewhat lower than 301 μg L−1, a temperature- and salinity-adjusted value derived from Whitehouse 1984 (ref. 34) (see comments in the ESI†).
The three days exposure time (72 h) used in the present study was selected to correspond to the experimental setup of oil exposure used in Sørhus, et al. 2021.1 It was observed therein that three days of oil exposure at embryo stages were sufficient to induce severe developmental damages in haddock larvae. PAH-uptake studies from a 9-day oil exposure of haddock and cod embryos have shown that the body burdens of phenanthrenes were peaking after three days of exposure and rapidly declined thereafter.32 This shows that PAH elimination has a major impact on the bioaccumulation of fish embryos during oil exposure.
Single compound exposures are likely to follow different uptake and depuration kinetics. In Atlantic cod (Gadus morhua), a close relative to haddock, phenanthrene, and other more hydrophobic compounds had not yet reached a steady state in larval exposures after 72 hours of exposures at 6.5 °C.35 Accordingly, we expected that the test compounds in the present study would also be in the uptake phase and would not yet have reached a steady-state equilibrium. More hydrophobic chemicals will have higher bioconcentration at equilibrium, ignoring metabolism; however, a steady state was not reached in this study. Instead, the more hydrophobic chemicals gave lower body burdens at a fixed exposure time because they require a longer time to reach a steady state. These findings are consistent with kinetically limited bioconcentration.36 The highest body burdens were found among the C0 and C1-phenanthrenes which have a shorter time to steady-state. Bioconcentration factor (BCF, L kg−1 ww, equal to body burden divided by exposure concentration) increased with alkylation and octanol–water coefficient, Kow (Fig. S9†). Importantly, BCF values in this study are not at equilibrium. Therefore, we avoid comparisons among compounds or with published literature. Nevertheless, RET appears to be a low outlier, with a lower body burden than would be predicted from its Kow. RET is known to be rapidly metabolized9,11,37 although cyp1a samples for RET were not collected in this study.
Increasing the mass and surface area of the passive dosing material can be used to more quickly counteract depletion from the water phase.25 Passive dosing has been used to effectively deliver constant concentrations over time,38,39 although deviations have been noted with increased presence of organic or microbial material late in the assay.40,41 In our 2018 experiments, the ratio of silicone to eggs was too low (both by mass and by surface area) and the rate of uptake into the eggs exceeded the release from silicone. This was observed through a concentration decrease after the addition of eggs. Therefore, in 2019 we repeated some experiments with a higher silicone mass (8–11 times, surface area 5–7 times) and reduced egg number (17%). Concentrations after day 0 showed a smaller drop in these experiments. The eventual mass of silicone was limited by two constraints, a higher mass of neat compound was needed to load more silicone, and 100 eggs were the minimum number of eggs to ensure enough animals survived until sampling and imaging.
A subset of three compounds were exposed both at solubility, and at subsequent dilutions below solubility: PHE, 4-MP, and 1,4-DMP. These exposures were repeated in two separate years for two purposes. First, the repeated year allowed us to refine the passive dosing technique, and we achieved more stable exposure concentrations by increasing the relative amount of silicone. Filtering the methanol loading solution yielded better control over dilutions in 2019, which were closer to the 3-fold dilutions we had been aiming for. Second, the repeated year provided replicate data for toxicity results. Because of the small changes to the passive dosing technique that led to different exposure concentrations in the dilutions, the toxicity results for the dilution exposures cannot be compared strictly between years. They can be compared more generally, however, because mean concentrations in water and body burden uptake were similar (read, most within a factor of 2) between the two years with repeated exposure levels (Fig. S10†). For example, 4-MP (S) exhibited 9 toxic endpoints in 2019 (Table S3†); these 9 along with 2 more had been observed in the 2018 experiment (Table S2†); water concentrations were similar in both years. As a counter example, PHE (S) had 6 sublethal endpoints in 2018, but only 3 in 2019, despite similar mean water concentrations (184 μg L−1 in 2018, and 194 μg L−1 in 2019). We theorize that differences between years might be attributed to developmental timing and the concentration profile over time. In 2018, the concentration was initially higher, then dropped by 28–74% after adding eggs, while concentrations were more stable over the three days in 2018 (Table S5 and Fig. S3†). We refrain from in-depth concentration–response analysis of PHE, 4-MP, and 1,4-DMP because of the inconsistencies in passive dosing between years, and also because the key objective of this work was to compare differential toxicities among the larger family of alkyl-phenanthrenes.
Static (single dose) and semi-static (daily solution renewal) toxicity tests are often criticized because test concentrations can decline significantly due to sorptive and evaporative losses.31 Passive dosing overcomes this problem by making freely dissolved test concentrations a controlled rather than dependent experimental variable. While passive dosing requires substantial preparation (washing, loading, equilibrium), the present and other related studies demonstrate that passive dosing can provide stable test concentrations for extended periods of time.42 We suggest finer filtration of loading solutions to remove microcrystals of the solid compound, and of water samples before extraction to remove small suspended organic material.
Differential toxicities among alkyl-phenanthrene compounds
The present work was conducted at one or only a few concentrations for each compound, thus it was not possible to calculate and compare median effective concentrations (EC50) values. Instead, the compounds' toxicities were compared at the concentrations determined by their physicochemical properties; the less-alkylated phenanthrenes were more water soluble and resulted in higher aqueous exposure concentrations. The profile of developmental toxicities appeared unrelated to the position of alkyl substitution, and expression of cyp1a was low regardless of alkylation.Evidence of differential mechanisms of toxicity is reported within the PAH homologue series, and even among alkyl isomers with the same Kow.31 Among phenanthrenes, the clearest example is PHE which acts directly on cardiac cells to reduce cardiac function.3,8,43–45 In contrast, RET responses are mediated by AHR but are cyp-independent, and first lead to morphological cardiac disruption with resulting impairment in cardiac function.12,31 RET strongly induces expression of cyp1a, unlike PHE, yet both are metabolized by CYP1A.9,37 Compounds with different modes of action, however, can lead to similar endpoints. For example, reduced eye size during development can arise from disruption of cyp-balance,28 or as a secondary effect from circulatory effects.3,46 Despite different known mechanisms of action, PHE and RET clustered together in the present analysis (Fig. 3D). The diversity of outcomes among the phenanthrene analogs exposed at their water solubilities supports the notion that effects of crude oil and PAHs are the result of multiple mechanisms of action.47
Induction of cyp1a is a sensitive biomarker for exposure to petroleum mixtures, and haddock embryos at this early age are indeed mature enough to metabolize.1 Following exposure to crude oil, Cyp enzymes play an overall protective role by initiating metabolism that ultimately leads to excretion of oil compounds.47 Paradoxically, phenanthrene is a poor inducer of cyp1a although it is readily metabolized by Cyp enzymes.37 Thus, when other Cyp-inducers are present, e.g., after oil exposure, PHE is a good substrate for Cyp1a and Cyp1b and is rapidly metabolized and eliminated.31,37 Schober, et al. 2010 (ref. 48) also demonstrated that phenanthrene is metabolized by human and mouse Cyp1a1, Cyp1a2, Cyp2e1, and human Cyp1b1. In the present study, cyp1a expression was low across all these tricyclic PAHs, despite high body burdens. Like PHE, our data suggest that these alkyl-phenanthrenes are also poor inducers of their own metabolism (excluding RET, 3-EP and 3-PP which were not tested). We observed weak, slightly negative correlations with cyp1a induction for each PHE, 4-MP, and 1,4-DMP, which suggest they may slightly impede their own metabolism. We theorize that the body burden levels achieved here are therefore higher than what could be expected in a worst-case environmental oil exposure that would include metabolism-stimulating compounds.
The location of the alkyl substitution on the phenanthrene molecule can affect metabolism and toxicity.31 Some alkyl-phenanthrenes, notably RET, are known to induce expression of cyp1a. Sun, et al. 2014 (ref. 49) reported that alkyl substitution on phenanthrenes increases potency in the AHR signaling pathway for xenobiotic metabolism, which includes CYP genes as targets. They also observed that equatorial substitutions (at the 1-, 2-, or 3- positions) have higher potencies than with substitutions at the 4- or 9-positions. Recent findings from Wang, et al. 2022 (ref. 50) show that alkyl substitutions in the 1- or 9-positions have higher mutagenicity compared to 2- and 3-positions. Wolinska, et al. 2013 (ref. 51) studied the cyp1a induction by 1-MP and 4-MP in the gills and liver of roach fish and found that expression was similarly low at <10-fold increase in induction following 2 or 7days exposure at 100 μg L−1. With only small changes in cyp1a expression overall, we observed no trends with the alkylation position.
Alkyl substitutions on phenanthrene can block or inhibit pathways and change the array of metabolites and degree of toxicity.52,53 Altered metabolism likely plays a role in toxicity among homologues as different metabolite pathways will yield different metabolites and toxicities. Metabolites can be more toxic than PAH itself.15,17,18,54 The metabolites of RET are strongly considered suspects for toxicity alongside RET.37,55 Hydroxylated phenanthrenes are found to be up to 4 times more toxic than their non-hydroxylated equivalents.17 Additionally, the presence of an alkyl group enables the formation of carboxylic acid metabolites.56 Differences in metabolism among the tested phenanthrenes (that either detoxify or bioactivate) may explain some differences in toxicity.
Several of the commercially available compounds herein have been tested in other early life stage fish assays. Turcotte, et al. 2011 (ref. 41) demonstrated a link between the chemical structure and toxicity in Japanese medaka (Oryzias latipes) in a series of alkyl-phenanthrenes that included 1-MP, 1,7-DMP, 2,7-DMP, and RET, where toxicity generally increased with alkylation and hydrophobicity when comparing the presence of deformities and edemas with EC50. Values for EC50 are not calculated in the present study as most compounds were exposed only at their solubility in water. Geier, et al. 2018 (ref. 10) performed a screen for over 100 s PAHs in embryonic zebrafish (Danio rerio). They observed no significant morphological or lethal effects for PHE and 3,6-DMP up to 50 μM nominal concentration (8900 μg L−1 for PHE and 10300 μg L−1 for 3,6-DMP). In contrast, we observed toxic morphological effects for PHE at concentrations hundreds of folds lower as low as 10.8 μg L−1 (reduced eye size). Differences in observed toxicity might in part be due to differences in exposure parameter (nominal versus measured concentration) and dosing regimen (passive dosing versus solvent spiking).
Similar to another study in early life stage marine medaka (Oryzias melastigma),9 we observed RET to be more toxic than PHE in haddock embryos. Five developmental phenotypic endpoints (shorter length and jaw, smaller eye and ventricle area, and more eye deformities) were significantly different after exposure to RET (2.30 μg L−1, SD = 0.974), but we observed no toxicity to a comparable level of exposure to PHE (3.40 ± 1.26 μg L−1, S/81, 2018). The resulting body burdens were similar between these two selected exposures: PHE was 2.11 ± 0.264 μg g−1 ww, and RET was 1.28 μg g−1 ww (no replicates). Previously, a comparatively lower RET body burden has been cited as evidence that RET is metabolized faster.9,11 RET body burden was also low in the present study, which we expect is a result of high metabolic turnover despite not being able to collect samples for cyp1a expression. Both PHE and 3,6-DMP were also tested in Japanese medaka (Oryzias latipes), but there were little effects for either in terms of hatching success or blue sac disease presence.57 Like differences in exposure techniques, other differences in experiments like removal of the chorion, temperature, and rate of embryonic development and metabolic capacity can complicate inter-study comparisons (even when two studies are designed to be comparable—see section Comparisons to crude oil exposures below).
Cardiotoxicity
The developmental screening study design did not allow for assessment of acute cardiotoxicity, as 8 days passed between exposure and microscopy. The test compound may have had time to depurate, and body burden content at the time of microscopy (3 dph) was not measured. PHE directly affects the electrophysiology of cardiomyocytes.8 Considering that the acute, functional effects of phenanthrene are transient,3 the significant changes in the developmental screen are either the result of irreversible cardiac damage incurred earlier, or poor elimination of components that have acute effects. However, downstream morphological effects can be caused by the acute, functional effects, even after those acute, functional effects have resolved with the removal of the compound.3 Sublethal effects of crude oil can affect aerobic activity and swimming performance2 and cause delayed mortality.58 No dose of PHE in the developmental screen resulted in significant changes in contractility (atrial or ventricular FS). The minimal degree of functional heart defects for PHE after the depuration period in the present study suggests that any transient effects on function did not result in significant, malformations in the heart at the conclusion of the study, three days after hatch.Exposure before heart development can lead to fluid and lipid dysregulation that is correlated with craniofacial skeletal defects1,3,58 and eye malformations.46 In a recent study with developing haddock, exposure to ∼200 μg per L PHE from 8 to 14 dpf46 resulted in a similar degree and incidence of craniofacial defects as the PHE exposures in the developmental screen in the present study. This suggests that craniofacial effects are at least similar whether exposed to phenanthrene at 2–5 dpf or 8–14 dpf. Additionally, comparable toxicities were seen with exposure to oil microdispersion from 7 to 10 dpf (“late exposure” in the comparable study by Sørhus, et al. 2021 (ref. 1)), yet the concentration of total PAHs was up to 20-fold less than levels in the present study, between 0.5 and 9.7 μg L−1. PHE and crude oil can result in similar phenotypes, but PHE appears individually much less potent than when exposed to a mixture. With the present study design, we cannot directly connect early, acute functional effects to the later craniofacial effects as have been documented in zebrafish.3
Baseline toxicity
Hydrophobic compounds like those in crude oil are purported to act by a nonspecific mode of action, baseline toxicity, in which a high accumulation of lipophilic compounds disrupts membrane fluidity and ion regulation.5,19 Critical body residue for mortality from baseline toxicity, often called narcosis, is 2–8 mmol kg−1 ww in small, aquatic organisms, or 40–160 mmol kg−1 lipid.5,19,59 Among the treatments with significant mortality, body burden ranged from 0.40 μmol g−1 ww (PHE S, 2018) down to 0.005 μmol g−1 ww (RET), all of which were below the suggested mortality range (Fig. 4A). This observed excess toxicity points to mode(s) of action other than baseline toxicity because mortality occurred at lower body burdens.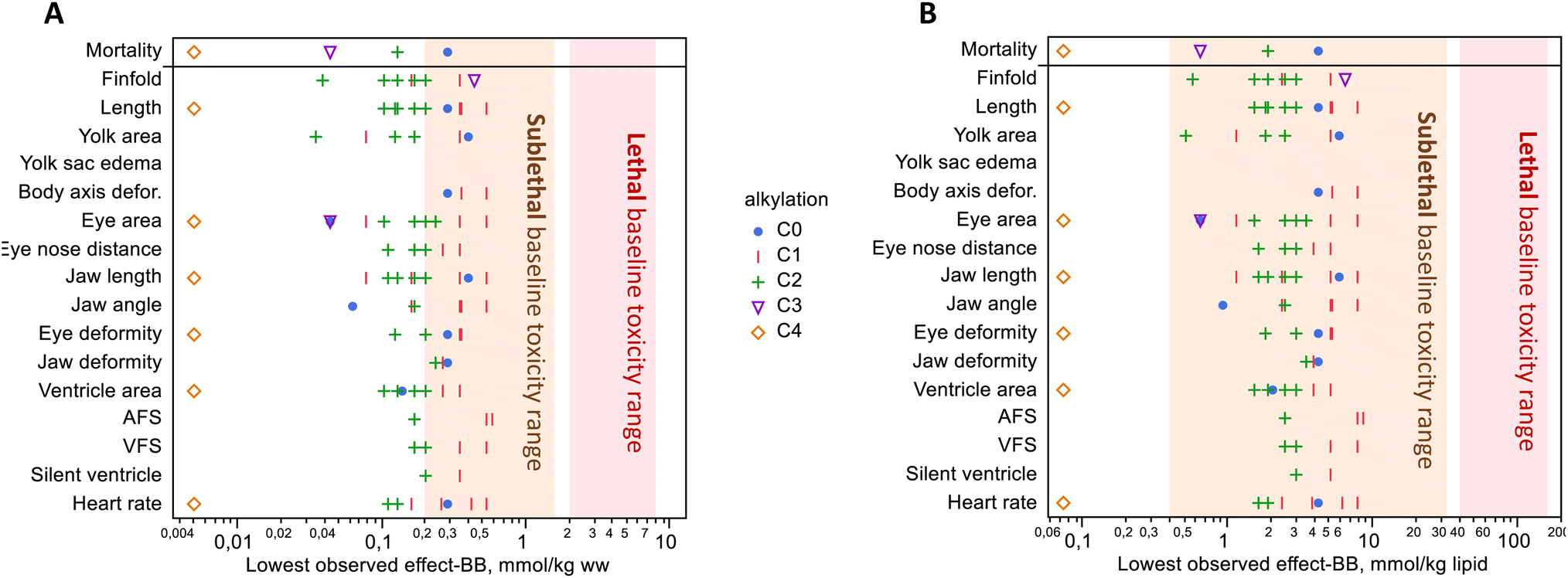 | ||
| Fig. 4 Lowest observed effect levels with significant toxicity versus control, with units in wet weight (A) and lipid weight (B). Mortality occurred at concentrations well below the range of lethal baseline toxicity. Significant mortality was observed in only 4 of the 7 compounds where mortality was measured. Sublethal effects were observed below and within the range of expected sublethal base toxicity. Compounds are grouped by the degree of alkylation; see Fig. S11† for a figure that shows individual compounds. No data are shown if the effect was not significant at any exposure level(s); refer to Table 1 for which compounds were exposed to only 1 or several levels. | ||
Sublethal baseline toxic effects on behavior, growth, and development occur in the concentration range of 0.2–1.6 mmol kg−1 ww (0.4–32 mmol kg−1 lipid), approximately 5–10 fold lower than mortality.19,59,60 Our results showed toxicity at concentrations that were around the lower limit of this range. Approximately half of the body burdens with the lowest observed sublethal effects occurred below this range when compared on a wet weight basis (Fig. 4A). A dose–response study is needed to establish the lowest observed effect concentrations, and we did test multiple exposure levels for 3 of the 14 PAHs in the present study. For the remaining compounds exposed at a single dose, we consider that concentration to be the lowest observed effect concentration if that effect was significant, for the purpose of comparing to sublethal baseline effects. When the lowest observed effect body burdens were adjusted for the lipid weight in haddock eggs (6.8 mg lipid per g ww),26 almost all sublethal endpoints fell within the baseline toxicity range (Fig. 4B). It should be noted that lipid content was not measured in this study, and conversion to these units is approximate just for the purpose of comparison. These findings are similar to recent zebrafish experiments using passive dosing with PHE, in which sublethal effects were generally within the expected range.60 Along with the excess mortality, the disparate profile of effects within this homologue series suggests that there are multiple other, more specific, and possibly yet-to-be-determined modes of action at play. Recent proteomic and metabolomic findings from exposure to a binary mixture of RET and fluoranthene in rainbow trout support this notion that mixture toxicity arises from multiple pathways rather than one.61
Comparisons to crude oil exposures in Sørhus, et al. 2021 (ref. 1)
Oil exposure leads to craniofacial deformities, functional and morphological cardiac abnormalities, and osmoregulatory disruptions (edema).4,5 Despite differential toxicities even within a homologue group, when exposed to the complex mixture of crude oil, the embryotoxic effects are consistent across species and geologically distinct oils.4 These common syndromes of developmental abnormalities were also observed following exposure to individual phenanthrene compounds in different severity levels in the present study, however, the concentrations needed were higher.The timing for the present study design was modeled after a recent study that exposed haddock to crude oil microdispersion for 3 days.1 In the high dose, early exposure concentration in the comparable study (H surf early, 300 μg oil per L, sum PAHs in water = 6.4 μg L−1), Sørhus, et al. 2021 (ref. 1) reported that body burden for total measured PAHs reached 7.8 μg g−1 ww (of which 24% were C0–C4-phenanthrenes). This dose resulted in numerous significant effects—jaw, eye, and heart deformities, edema, and reduced heartbeat and ventricle fractional shortening. By comparison, body burden in the present study reached as much as 10 times higher (103 μg g−1 ww, 3-MP), and only RET was below that level among all the treatments (1.28 μg g−1 ww).
These direct comparisons with whole exposures are complicated by the “stickiness” of haddock eggs. Atlantic haddock are particularly sensitive to crude oil exposures in the early egg phase because of a hydrophobic chorion membrane that attracts microdroplets of oil, making them approximately 10 times more susceptible to the toxic effects of oil than closely related cod (Gadus morhua).32,62 This effectively extends the exposure duration as oil droplets adhered to the chorion remain after exposure stops. Unlike our previous reports of toxicity from crude oil with microdroplet dispersions, the passive dosing system delivered the test compound exclusively in the dissolved phase, and the effective exposure period was not extended past 72 hours.
The hydrophobic surface of the haddock eggs allows for exposure to more oil components that are less water soluble. Another meaningful comparison of the present study to an oil mixture could be to the water-soluble fraction (WSF) where oil droplets were filtered out. However, in Sørhus, et al. 2021 (ref. 1) WSF high dose (H WSF early, 300 μg oil per L), the exposure, and body burden concentrations (sum PAHs in water = 3.7 μg L−1; body burden = 0.98 μg g−1 ww) were lower than in the present study. These embryos in the comparable study were nearly unaffected across a similar array of endpoints that were measured in the present study. In contrast, a degree of toxicity similar to the present study was observed in the low treatment of Sørhus, et al. 2021 (ref. 1) (microdroplets not filtered out, 30 μg oil per L, sum PAHs in water = 0.47 μg L−1). Body burdens in their low dose reached 0.18 μg g−1 ww (of which 25% were phenanthrenes) and 7-fold increased cyp1a expression, suggesting that comparatively lower doses of PAHs are associated with toxic effects when part of the whole oil.
Drastically different metabolic rates also complicate comparisons of body burden levels and toxicity between exposures to mixtures and single compounds. Metabolism also has the potential to bioactivate as well as detoxify. At 72 h of exposure for the unfiltered high dose, in the comparable study1cyp1a expression levels were nearly 200-fold higher than controls, suggesting efficient elimination. For WSF, cyp1a was elevated 30-fold, indicating a higher elimination rate than any treatment in the present study even though we were measuring substantially higher measured PAH body burden. In contrast, cyp1a levels in the present study were no more than 4-fold higher than controls, reflecting a lower metabolic turnover. In the absence of the complex mixture of the oil, the eggs in the individual compound exposures of the present study were able to accumulate to higher tissue concentrations that likely remained elevated longer. In a crude oil microdroplet exposure, the effects of the hydrophobic eggs (extending exposure duration) and the enhanced metabolic rate (reducing exposure duration and/or bioactivate) counteract each other to an unknown degree. The low dose resulted in higher toxicity than WSF despite lower exposure amounts,1 suggesting either that (a) more lipophilic compounds in the adhered oil microdroplets increase toxicity, (b) the adhered microdroplets effectively extended the exposure period, (c) water-soluble oil components are more easily eliminated by xenobiotic metabolism, or (d) a combination. As with exposures to only the water-soluble components of oil, the single phenanthrenes in the present study require greater concentrations to yield the same degree of toxic outcomes as oil.
Conclusions
All the tested compounds induced one or more toxic outcomes at concentrations at their solubility in water. Among the 14 tricyclic PAHs studied here, no clear trend emerged that links the chemical structure of the homologues with early life stage toxicity. Effects of metabolism and the hydrophobic layer on haddock eggs complicate direct comparisons of exposures between single compounds and complex mixtures.In this work, the exposure levels with significant toxic effects lie between two ranges. First, the body burdens with significant mortality are well below the expected range for lethal baseline toxicity, and the sublethal effects occurred at around the lower end of the expected range for sublethal baseline toxicity. The excess toxicity presented here, especially mortality, suggests that these compounds have more specific modes of action than nonpolar narcosis. Second, the effect concentrations in the present study are much higher than concentrations of PAHs in crude oil exposures that cause toxicity in Sørhus, et al. 2021,1 a comparable study. These results suggest that these individual phenanthrenes can indeed produce the array of morphological toxic effects seen with crude oil, but the required dose is much higher than when part of a complex mixture.
Recently, the notion that individual PAHs are the major, or even sole, responsible toxicants in crude oil mixtures is being challenged.5 Further mixture toxicity experiments are needed, including (a) a bottom-up approach using simple, low-concentration mixtures of PAHs, and (b) a top-down approach like effects-directed analysis combined with ever more advanced chemical analyses. The scope of the present study was limited to phenanthrenes, however, further studies should be aimed to understand the individual toxicities of other groups of PAHs that are suspected to be potent through specific mechanisms of action. Such studies will help narrow down and identify the causative individual toxicants or combinations in crude oil and petroleum products.
Ethical statement
The Austevoll Research Station has permits from the Norwegian Directorate of Fisheries to catch and maintain stocks of Atlantic Haddock (H-AV 77, H-AV 78, and H-AV 79). No permits are needed for work with embryos and yolk sac larvae.Author contributions
SM, CED, CLN, and PM designed and carried out the passive dosing experiments. KBJ, HKB, CS, and SK synthesized the compounds. CED, CLN, ES, PP, and SM collected and analyzed microscopy images. ES performed qPCR, and CLN and CED performed chemical and statistical analysis. CED, CLN, ES, and PP drafted the manuscript with the help of all authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work was funded by the Research Council of Norway (EGGTOX: Unraveling the mechanistic effects of crude oil toxicity during early life stages of cold-water marine teleosts, Project # 267820) and the Institute of Marine Research, Norway. The funders had no role in the study design, data collection, analysis, preparation of the manuscript, or the decision to publish. The authors acknowledge the help of Margareth Møgster for managing broodstock and providing haddock eggs, and to Anders Thorsen, Alysha Cypher, Grethe Tveit, Franziska Randers, Libe Aranguren-Abadía during exposures and analyses. We also appreciate the help of Hiwot M. Tiruye in synthesis, and Lisbet Sørensen for insightful discussions.References
- E. Sørhus, C. E. Donald, D. da Silva, A. Thorsen, Ø. Karlsen and S. Meier, Untangling mechanisms of crude oil toxicity: linking gene expression, morphology and PAHs at two developmental stages in a cold-water fish, Sci. Total Environ., 2021, 757, 143896, DOI:10.1016/j.scitotenv.2020.143896.
- C. E. Hicken, T. L. Linbo, D. H. Baldwin, M. L. Willis, M. S. Myers, L. Holland, M. Larsen, M. S. Stekoll, S. D. Rice, T. K. Collier, N. L. Scholz and J. P. Incardona, Sublethal exposure to crude oil during embryonic development alters cardiac morphology and reduces aerobic capacity in adult fish, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7086–7090, DOI:10.1073/pnas.1019031108.
- J. P. Incardona, T. K. Collier and N. L. Scholz, Defects in cardiac function precede morphological abnormalities in fish embryos exposed to polycyclic aromatic hydrocarbons, Toxicol. Appl. Pharmacol., 2004, 196, 191–205, DOI:10.1016/j.taap.2003.11.026.
- J. P. Incardona, Molecular Mechanisms of Crude Oil Developmental Toxicity in Fish, Arch. Environ. Contam. Toxicol., 2017, 73, 19–32, DOI:10.1007/s00244-017-0381-1.
- J. P. Meador and J. Nahrgang, Characterizing Crude Oil Toxicity to Early-Life Stage Fish Based On a Complex Mixture: Are We Making Unsupported Assumptions?, Environ. Sci. Technol., 2019, 53, 11080–11092, DOI:10.1021/acs.est.9b02889.
- M. G. Carls and J. P. Meador, A Perspective on the Toxicity of Petrogenic PAHs to Developing Fish Embryos Related to Environmental Chemistry, Hum. Ecol. Risk Assess., 2009, 15, 1084–1098, DOI:10.1080/10807030903304708.
- M. G. Barron, M. G. Carls, R. Heintz and S. D. Rice, Evaluation of fish early life-stage toxicity models of chronic embryonic exposures to complex polycyclic aromatic hydrocarbon mixtures, Toxicol. Sci., 2004, 78, 60–67, DOI:10.1093/toxsci/kfh051.
- F. Brette, H. A. Shiels, G. L. Galli, C. Cros, J. P. Incardona, N. L. Scholz and B. A. Block, A Novel Cardiotoxic Mechanism for a Pervasive Global Pollutant, Sci. Rep., 2017, 7, 41476, DOI:10.1038/srep41476.
- J. Mu, J. Wang, F. Jin, X. Wang and H. Hong, Comparative embryotoxicity of phenanthrene and alkyl-phenanthrene to marine medaka (Oryzias melastigma), Mar. Pollut. Bull., 2014, 85, 505–515, DOI:10.1016/j.marpolbul.2014.01.040.
- M. C. Geier, A. C. Chlebowski, L. Truong, S. L. Massey Simonich, K. A. Anderson and R. L. Tanguay, Comparative developmental toxicity of a comprehensive suite of polycyclic aromatic hydrocarbons, Arch. Toxicol., 2018, 92, 571–586, DOI:10.1007/s00204-017-2068-9.
- J. O. Honkanen, C. B. Rees, J. V. K. Kukkonen and P. V. Hodson, Temperature determines the rate at which retene affects trout embryos, not the concentration that is toxic, Aquat. Toxicol., 2020, 222, 105471, DOI:10.1016/j.aquatox.2020.105471.
- J. A. Scott, J. P. Incardona, K. Pelkki, S. Shepardson and P. V. Hodson, AhR2-mediated, CYP1A-independent cardiovascular toxicity in zebrafish (Danio rerio) embryos exposed to retene, Aquat. Toxicol., 2011, 101, 165–174, DOI:10.1016/j.aquatox.2010.09.016.
- V. McGruer, P. Tanabe, S. M. F. Vliet, S. Dasgupta, L. Qian, D. C. Volz and D. Schlenk, Effects of Phenanthrene Exposure on Cholesterol Homeostasis and Cardiotoxicity in Zebrafish Embryos, Environ. Toxicol. Chem., 2021, 40, 1586–1595, DOI:10.1002/etc.5002.
- J. Mu, F. Jin, J. Wang, Y. Wang and Y. Cong, The effects of CYP1A inhibition on alkyl-phenanthrene metabolism and embryotoxicity in marine medaka (Oryzias melastigma), Environ. Sci. Pollut. Res. Int., 2016, 23, 11289–11297, DOI:10.1007/s11356-016-6098-2.
- L. Kamelia, L. de Haan, B. Spenkelink, B. Bruyneel, H. B. Ketelslegers, P. J. Boogaard and I. Rietjens, The role of metabolism in the developmental toxicity of polycyclic aromatic hydrocarbon-containing extracts of petroleum substances, J. Appl. Toxicol., 2020, 40, 330–341, DOI:10.1002/jat.3906.
- S. M. Billiard, M. E. Hahn, D. G. Franks, R. E. Peterson, N. C. Bols and P. V. Hodson, Binding of polycyclic aromatic hydrocarbons (PAHs) to teleost aryl hydrocarbon receptors (AHRs), Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2002, 133, 55–68, DOI:10.1016/S1096-4959(02)00105-7.
- S. Fallahtafti, T. Rantanen, R. S. Brown, V. Snieckus and P. V. Hodson, Toxicity of hydroxylated alkyl-phenanthrenes to the early life stages of Japanese medaka (Oryzias latipes), Aquat. Toxicol., 2012, 106–107, 56–64, DOI:10.1016/j.aquatox.2011.10.007.
- J. E. Schrlau, A. L. Kramer, A. Chlebowski, L. Truong, R. L. Tanguay, S. L. M. Simonich and L. Semprini, Formation of Developmentally Toxic Phenanthrene Metabolite Mixtures by Mycobacterium sp. ELW1, Environ. Sci. Technol., 2017, 51, 8569–8578, DOI:10.1021/acs.est.7b01377.
- J. P. Meador, W. J. Adams, B. I. Escher, L. S. McCarty, A. E. McElroy and K. G. Sappington, The tissue residue approach for toxicity assessment: findings and critical reviews from a Society of Environmental Toxicology and Chemistry Pellston Workshop, Integr. Environ. Assess. Manage., 2011, 7, 2–6, DOI:10.1002/ieam.133.
- U.S. Environmental Protection Agency, Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.1.25, 2015 Search PubMed.
- H. M. Tiruye and K. B. Jørgensen, Oxidative synthesis of ortho-quinones from hydroxy-PAHs by stabilized formulation of 2-iodoxybenzoic acid (SIBX), Tetrahedron, 2022, 133144, DOI:10.1016/j.tet.2022.133144.
- T. Böhme, M. Lorentzen and K. B. Jørgensen, Regiospecific Synthesis of Dimethylphenanthrenes, Polycyclic Aromat. Compd., 2017, 37, 106–113, DOI:10.1080/10406638.2016.1179651.
- K. B. Jørgensen, Photochemical oxidative cyclisation of stilbenes and stilbenoids—the Mallory-reaction, Molecules, 2010, 15, 4334–4358, DOI:10.3390/molecules15064334.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, Improved methodology for photocyclization reactions, J. Org. Chem., 1991, 56, 3769–3775, DOI:10.1021/jo00012a005.
- R. Hammershøj, H. Birch, K. K. Sjøholm and P. Mayer, Accelerated Passive Dosing of Hydrophobic Complex Mixtures-Controlling the Level and Composition in Aquatic Tests, Environ. Sci. Technol., 2020, 54, 4974–4983, DOI:10.1021/acs.est.9b06062.
- L. Sørensen, M. S. Silva, A. M. Booth and S. Meier, Optimization and comparison of miniaturized extraction techniques for PAHs from crude oil exposed Atlantic cod and haddock eggs, Anal. Bioanal. Chem., 2016, 408, 1023–1032, DOI:10.1007/s00216-015-9225-x.
- L. Sørensen, S. Meier and S. A. Mjøs, Application of gas chromatography/tandem mass spectrometry to determine a wide range of petrogenic alkylated polycyclic aromatic hydrocarbons in biotic samples, Rapid Commun. Mass Spectrom., 2016, 30, 2052–2058, DOI:10.1002/rcm.7688.
- K. K. Lie, S. Meier, E. Sørhus, R. B. Edvardsen, Ø. Karlsen and P. A. Olsvik, Offshore Crude Oil Disrupts Retinoid Signaling and Eye Development in Larval Atlantic Haddock, Front. Mar. Sci., 2019, 6, 1–14, DOI:10.3389/fmars.2019.00368.
- K. E. Smith, N. Dom, R. Blust and P. Mayer, Controlling and maintaining exposure of hydrophobic organic compounds in aquatic toxicity tests by passive dosing, Aquat. Toxicol., 2010, 98, 15–24, DOI:10.1016/j.aquatox.2010.01.007.
- L. Sun, J. Ruan, M. Lu, M. Chen, Z. Dai and Z. Zuo, Combined effects of ocean acidification and crude oil pollution on tissue damage and lipid metabolism in embryo-larval development of marine medaka (Oryzias melastigma), Environ. Geochem. Health, 2019, 41, 1847–1860, DOI:10.1007/s10653-018-0159-z.
- P. V. Hodson, The Toxicity to Fish Embryos of PAH in Crude and Refined Oils, Arch. Environ. Contam. Toxicol., 2017, 73, 12–18, DOI:10.1007/s00244-016-0357-6.
- L. Sørensen, E. Sørhus, T. Nordtug, J. P. Incardona, T. L. Linbo, L. Giovanetti, O. Karlsen and S. Meier, Oil droplet fouling and differential toxicokinetics of polycyclic aromatic hydrocarbons in embryos of Atlantic haddock and cod, PLoS One, 2017, 12, e0180048, DOI:10.1371/journal.pone.0180048.
- E. Sørhus, J. P. Incardona, Ø. Karlsen, T. Linbo, L. Sørensen, T. Nordtug, T. van der Meeren, A. Thorsen, M. Thorbjørnsen, S. Jentoft, R. B. Edvardsen and S. Meier, Crude oil exposures reveal roles for intracellular calcium cycling in haddock craniofacial and cardiac development, Sci. Rep., 2016, 6, 31058, DOI:10.1038/srep31058.
- B. G. Whitehouse, The effects of temperature and salinity on the aqueous solubility of polynuclear aromatic hydrocarbons, Mar. Chem., 1984, 14, 319–332, DOI:10.1016/0304-4203(84)90028-8.
- G. I. Petersen and P. Kristensen, Bioaccumulation of lipophilic substances in fish early life stages, Environ. Toxicol. Chem., 1998, 17, 1385–1395, DOI:10.1002/etc.5620170724.
- J.-H. Kwon, S.-Y. Lee, H.-J. Kang, P. Mayer and B. I. Escher, Including Bioconcentration Kinetics for the Prioritization and Interpretation of Regulatory Aquatic Toxicity Tests of Highly Hydrophobic Chemicals, Environ. Sci. Technol., 2016, 50, 12004–12011, DOI:10.1021/acs.est.6b03942.
- S. Hawkins, S. Billiard, S. Tabash, R. Brown and P. Hodson, Altering cytochrome P4501A activity affects polycyclic aromatic hydrocarbon metabolism and toxicity in rainbow trout (Oncorhynchus mykiss), Environ. Toxicol. Chem., 2002, 21, 1845–1853, DOI:10.1002/etc.5620210912.
- H. Birch, V. Gouliarmou, H.-C. H. Lützhøft, P. S. Mikkelsen and P. Mayer, Passive Dosing to Determine the Speciation of Hydrophobic Organic Chemicals in Aqueous Samples, Anal. Chem., 2010, 82, 1142–1146, DOI:10.1021/ac902378w.
- F. Stibany, S. N. Schmidt, A. Schaffer and P. Mayer, Aquatic toxicity testing of liquid hydrophobic chemicals – passive dosing exactly at the saturation limit, Chemosphere, 2017, 167, 551–558, DOI:10.1016/j.chemosphere.2016.10.014.
- Y. Kiparissis, P. Akhtar, P. V. Hodson and R. S. Brown, Partition-Controlled Delivery of Toxicants: A Novel In Vivo Approach for Embryo Toxicity Testing, Environ. Sci. Technol., 2003, 37, 2262–2266, DOI:10.1021/es026154r.
- D. Turcotte, P. Akhtar, M. Bowerman, Y. Kiparissis, R. S. Brown and P. V. Hodson, Measuring the toxicity of alkyl-phenanthrenes to early life stages of medaka (Oryzias latipes) using partition-controlled delivery, Environ. Toxicol. Chem., 2011, 30, 487–495, DOI:10.1002/etc.404.
- A. Ribbenstedt, L. Mustajarvi, M. Breitholtz, E. Gorokhova, P. Mayer and A. Sobek, Passive dosing of triclosan in multigeneration tests with copepods – stable exposure concentrations and effects at the low mug/L range, Environ. Toxicol. Chem., 2017, 36, 1254–1260, DOI:10.1002/etc.3649.
- E. R. Vehniainen, J. Haverinen and M. Vornanen, Polycyclic Aromatic Hydrocarbons Phenanthrene and Retene Modify the Action Potential via Multiple Ion Currents in Rainbow Trout Oncorhynchus mykiss Cardiac Myocytes, Environ. Toxicol. Chem., 2019, 38, 2145–2153, DOI:10.1002/etc.4530.
- S. N. Kompella, F. Brette, J. C. Hancox and H. A. Shiels, Phenanthrene impacts zebrafish cardiomyocyte excitability by inhibiting IKr and shortening action potential duration, J. Gen. Physiol., 2021, 153, e202012733, DOI:10.1085/jgp.202012733.
- D. V. Abramochkin, S. N. Kompella and H. A. Shiels, Phenanthrene alters the electrical activity of atrial and ventricular myocytes of a polar fish, the Navaga cod, Aquat. Toxicol., 2021, 235, 105823, DOI:10.1016/j.aquatox.2021.105823.
- E. Sørhus, S. Meier, C. E. Donald, T. Furmanek, R. B. Edvardsen and K. K. Lie, Cardiac dysfunction affects eye development and vision by reducing supply of lipids in fish, Sci. Total Environ., 2021, 800, 149460, DOI:10.1016/j.scitotenv.2021.149460.
- J. P. Incardona, M. G. Carls, H. Teraoka, C. A. Sloan, T. K. Collier and N. L. Scholz, Aryl Hydrocarbon Receptor–Independent Toxicity of Weathered Crude Oil during Fish Development, Environ. Health Perspect., 2005, 113, 1755–1762, DOI:10.1289/ehp.8230.
- W. Schober, G. Pusch, S. Oeder, H. Reindl, H. Behrendt and J. T. Buters, Metabolic activation of phenanthrene by human and mouse cytochromes P450 and pharmacokinetics in CYP1A2 knockout mice, Chem.-Biol. Interact., 2010, 183, 57–66, DOI:10.1016/j.cbi.2009.09.008.
- Y. Sun, C. A. I. Miller, T. E. Wiese and D. A. Blake, Methylated phenanthrenes are more potent than phenanthrene in a bioassay of human aryl hydrocarbon receptor (AhR) signaling, Environ. Toxicol. Chem., 2014, 33, 2363–2367, DOI:10.1002/etc.2687.
- D. Wang, V. Schramm, J. Pool, E. Pardali, A. Brandenburg, I. Rietjens and P. J. Boogaard, The effect of alkyl substitution on the oxidative metabolism and mutagenicity of phenanthrene, Arch. Toxicol., 2022, 96, 1109–1131, DOI:10.1007/s00204-022-03239-9.
- L. Wolinska, P. Brzuzan, M. Wozny, M. K. Luczynski and M. Gora, CYP1A expression in liver and gills of roach (Rutilus rutilus) after waterborne exposure to two phenanthrene derivatives, 1-methylphenanthrene and 4-methylphenanthrene, Environ. Toxicol. Chem., 2013, 32, 1604–1610, DOI:10.1002/etc.2225.
- E. J. LaVoie, L. Tulley-Freiler, V. Bedenko and D. Hoffman, Mutagenicity, tumor-initiating activity, and metabolism of methylphenanthrenes, Cancer Res., 1981, 41, 3441–3447 CAS.
- K. Rudnicka, S. Tejs, K. A. Budzikur, D. Mielżyńska-Švach, E. Jakimiuk, A. Chachaj, M. Góra, K. Żelazna and M. K. Łuczyński, Assessment of mutagenic activity of methyl- and phenylphenanthrenes based on Salmonella test and micronucleus test, Environ. Biotechnol., 2014, 9, 65–71, DOI:10.14799/ebms193.
- G. Diamante, E. S. M. G. do Amaral, N. Menjivar-Cervantes, E. G. Xu, D. C. Volz, A. C. Dias Bainy and D. Schlenk, Developmental toxicity of hydroxylated chrysene metabolites in zebrafish embryos, Aquat. Toxicol., 2017, 189, 77–86, DOI:10.1016/j.aquatox.2017.05.013.
- P. V. Hodson, K. Qureshi, C. A. Noble, P. Akhtar and R. S. Brown, Inhibition of CYP1A enzymes by alpha-naphthoflavone causes both synergism and antagonism of retene toxicity to rainbow trout (Oncorhynchus mykiss), Aquat. Toxicol., 2007, 81, 275–285, DOI:10.1016/j.aquatox.2006.12.012.
- L. M. V. Malmquist, E. S. Boll, N. J. Nielsen and J. H. Christensen, Separation, detection and identification of phase I and phase II metabolites and their corresponding polycyclic aromatic compounds, Anal. Methods, 2017, 9, 3323–3328, 10.1039/c7ay00715a.
- S. Rhodes, A. Farwell, L. M. Hewitt, M. Mackinnon and D. G. Dixon, The effects of dimethylated and alkylated polycyclic aromatic hydrocarbons on the embryonic development of the Japanese medaka, Ecotoxicol. Environ. Saf., 2005, 60, 247–258, DOI:10.1016/j.ecoenv.2004.08.002.
- B. J. Laurel, L. A. Copeman, P. Iseri, M. L. Spencer, G. Hutchinson, T. Nordtug, C. E. Donald, S. Meier, S. E. Allan, D. T. Boyd, G. M. Ylitalo, J. R. Cameron, B. L. French, T. L. Linbo, N. L. Scholz and J. P. Incardona, Embryonic Crude Oil Exposure Impairs Growth and Lipid Allocation in a Keystone Arctic Forage Fish, iScience, 2019, 19, 1101–1113, DOI:10.1016/j.isci.2019.08.051.
- L. S. McCarty, P. F. Landrum, S. N. Luoma, J. P. Meador, A. A. Merten, B. K. Shephard and A. P. van Wezel, Advancing environmental toxicology through chemical dosimetry: external exposures versus tissue residues, Integr. Environ. Assess. Manage., 2011, 7, 7–27, DOI:10.1002/ieam.98.
- R. Massei, D. Knapen, A. Covaci, R. Blust, P. Mayer and L. Vergauwen, Sublethal Effect Concentrations for Nonpolar Narcosis in the Zebrafish Embryo, Environ. Toxicol. Chem., 2021, 40, 2802–2812, DOI:10.1002/etc.5170.
- A. N. M. Eriksson, C. Rigaud, A. Rokka, M. Skaugen, J. H. Lihavainen and E. R. Vehniainen, Changes in cardiac proteome and metabolome following exposure to the PAHs retene and fluoranthene and their mixture in developing rainbow trout alevins, Sci. Total Environ., 2022, 830, 154846, DOI:10.1016/j.scitotenv.2022.154846.
- E. Sørhus, R. B. Edvardsen, O. Karlsen, T. Nordtug, T. van der Meeren, A. Thorsen, C. Harman, S. Jentoft and S. Meier, Unexpected interaction with dispersed crude oil droplets drives severe toxicity in Atlantic haddock embryos, PLoS One, 2015, 10, e0124376, DOI:10.1371/journal.pone.0124376.
Footnote |
| † Electronic supplementary information (ESI) available: Data generated during this study are included: GC-MS/MS details, complete data for all treatments and plots for endpoints, water concentrations measured over exposure time, examples of biometric endpoints, significant endpoints sorted by compound alkylation, comparisons between repeated experiments, and chemical synthesis information. See DOI: https://doi.org/10.1039/d2em00357k |
| This journal is © The Royal Society of Chemistry 2023 |