
High-loading As single-atom catalysts harvested from wastewater towards efficient and sustainable oxygen reduction†
Yangjun
Luo‡
a,
Yanwei
Wang‡
a,
Huijuan
Zhang‡
ab,
Youyuan
Wang‡
 a,
Jin
Wan
a,
Chuanzhen
Feng
a,
Lingmei
Liu
a,
Zaiping
Guo
*c,
Jian
Li
a and
Yu
Wang
*ab
a,
Jin
Wan
a,
Chuanzhen
Feng
a,
Lingmei
Liu
a,
Zaiping
Guo
*c,
Jian
Li
a and
Yu
Wang
*ab
aState Key Laboratory of Power Transmission Equipment Technology, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400044, P. R. China. E-mail: wangy@cqu.edu.cn
bCollege of Chemistry and Environmental Science, Inner Mongolia Normal University, Huhehaote, 010022, P. R. China
cSchool of Chemical Engineering and Advanced Materials, University of Adelaide, Adelaide 5005, Australia. E-mail: zaiping.guo@adelaide.edu.au
First published on 7th November 2023
Abstract
Arsenic (As) is a common element in groundwater contamination with similar chemical properties to nitrogen, exhibiting potential activity towards oxygen reduction reaction (ORR). However, the practical application of recovering arsenic-containing contaminants for ORR faces the formidable challenge of a trade-off between high activity and stability. Herein, we first report universal strategies to synthesize high-loading (up to 13.78 wt%) non-metal monoatoms on carbon and prepare As monoatomic catalysts as a demonstration. The dispersed zinc ions chelated by α-D-glucose significantly increase the loading of As monoatoms. Moreover, topological defects constructed by zinc evaporation enhance the intrinsic activity of adjacent As. The catalyst exhibits a much better half-wave potential (0.901 V) than 20% Pt/C (0.856 V). Re-calcination is further proposed to overcome the poor oxidation resistance of catalysts with abundant carbon defects. The catalyst treated by re-calcination demonstrates unprecedented stability, with only 9.86% deterioration in current density after 590 hours of operation in the fuel cell, outperforming the most advanced carbon-based catalysts. Our discoveries facilitate the practical application of high-loading, high-activity and high-stability non-metal catalysts originating from pollutants.
Broader contextH2/O2 fuel cells and metal–air batteries have gained widespread attention due to their high efficiency and environmental friendliness. ORR is the key process in fuel cells. Arsenite and arsenate in groundwater are serious health hazards, and appropriate recycling of As is conducive to achieving a virtuous cycle of environmental protection and economy. However, the large-scale application of As-based catalysts in fuel cells faces a formidable challenge of high activity and stability trade-off. Specifically, the lack of overall performance of non-metal single-atom catalysts (SACs) with low loadings limits their viable applications. Although topological defects can significantly enhance the catalytic activity of non-metals, carbon with abundant topological defects is usually more susceptible to oxidation, resulting in the deterioration of catalyst stability. Here, we first propose a general strategy to synthesize high-loading (up to 13.78 wt%) non-metal monoatoms on carbon, and further construct topological defects to modulate the activity of non-metal monoatoms. Re-calcination is further proposed to reduce the number of oxygen-containing functional groups associated with long-term operations and to restore the valence of the single-atom As. This work improves the activity of high-loading non-metal SACs and simultaneously suppresses their activity degradation, facilitating the application of non-metal SACs in fuel cells. |
1. Introduction
Elemental As is primarily present in an inorganic form in natural waters.1 Typically, arsenite(III) is predominantly distributed in groundwater, whereas arsenate(V) is dominantly distributed in surface water.2 In particular, arsenite is a serious health hazard, and appropriate recycling of arsenite is conducive to achieving a virtuous cycle of environmental protection and economy.3,4 Due to the low activity and susceptibility to oxidation of As, strategies to recycle As-containing contaminants for the synthesis of efficient and stable catalysts are almost absent.5 Encouragingly, the physical and chemical properties of non-metal SACs may be significantly altered due to their unique coordination environment and easily tunable electronic structure.6–8 Both single-atom phosphorus loaded on Mo2C in our previous work and single-atom iodine on nickel iodide have been successfully applied in alkaline hydrogen evolution reactions.7,8 Monoatomic selenium embedded in nitrogen-doped carbon surfaces also exhibits attractive alkaline ORR performance.6 However, the corresponding research ignores the possible effects of topological defects and doped nitrogen on the activity of non-metal monoatoms. Studies have shown that carbon topological defects can effectively modulate the electronic structure of non-metals (e.g. nitrogen) and thus significantly enhance their catalytic activity.9–16 Frustratingly, the lack of overall performance of non-metal SACs with low loadings limits their viable applications. Consequently, developing a general method to synthesize high-loading non-metal SACs and constructing topological defects to enhance the intrinsic activity are crucial for the practical application of As-based SACs.The large-scale application of catalysts in fuel cells faces a formidable challenge of low cost, high activity and stability trade-off.17–22 Various carbon defective materials and carbon materials doped/loaded with other heteroatoms show promising application prospects in ORR.6,9–12,23–27 Studies have shown that the small carbon plane rich in defects contributes to the breaking of O–O bonds, which accelerates the ORR process.28,29 Accordingly, As-based SACs on defect-rich carbon substrates probably exhibit promising catalytic activity. Unfortunately, the defect-rich carbon domains exhibit poor oxidation resistance and corrosion resistance.23,29 For carbon-based metal catalysts, such as single-atom iron embedded on nitrogen-doped carbon (Fe–NC), the oxidation of defect-rich carbon domains may trigger the demetallization of FeN4 sites from the carbon substrate during ORR. Structural disruption of the active site usually results in irreversible performance degradation.18,23,29,30 For carbon-based non-metal catalysts, oxidation of the carbon domains mainly affects the electronic structure of the active site, which may reduce the activity and stability of the catalyst.12,23,31 Therefore, developing a practicable strategy to inhibit oxidation of the carbon substrate prior to structural disruption of the active site would effectively improve the stability of the As-based catalyst on carbon.
Here, we first report a general methodology to synthesize high-loading (up to 13.78 wt%) non-metal monoatoms on carbon, which is used to successfully synthesize As SACs. Zinc (Zn) ions are first anchored to carbon after being chelated by α-D-glucose, with the excess glucose physically isolating the Zn ions and avoiding their large-scale agglomeration. The decomposition products of sodium arsenite (NaAsO2) are then captured in large quantities by Zn species via chemical vapour deposition (CVD) methods to increase the non-metallic loading. Finally, Zn evaporates to construct topological defects for modulating the activity of adjacent As. The SAC exhibits high activity with a half-wave potential of 0.901 V in 0.1 M KOH and maximum power densities of 701.9 mW cm−2 and 179.8 mW cm−2 in the H2/O2 fuel cell and Zn–air battery, respectively. Re-calcination is further proposed to reduce the number of oxygen-containing functional groups associated with long-term operations and to restore the valence of the single-atom As. Accordingly, the SAC treated by re-calcination exhibits a degradation of just 4 mV in half-wave potential after 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and only 9.86% deterioration in current density after 590 h of operation in fuel cells.
000 cycles and only 9.86% deterioration in current density after 590 h of operation in fuel cells.
2. Results and discussion
Synthesis and characterization of arsenic-based catalysts from wastewater
To evaluate the feasibility of utilizing arsenite for the synthesis of efficient ORR catalysts, NaAsO2 was investigated as a typical arsenic contaminant.4,32,33 As-based non-metal catalysts were synthesized as shown in Fig. 1 (see the Methods section for details). Firstly, regular carbon (RC) support, zinc nitrate and α-D-glucose (chelating agent) were dispersed in deionized water. Secondly, the resulting powder and NaAsO2 were placed on separate combustion boats and transferred downstream and upstream of the tube furnace, respectively. Finally, As species were loaded adjacent to the defects formed by the evaporation of zinc via the CVD method. Notably, a staged temperature increase was employed during the CVD process. In the first stage, NaAsO2 decomposed at 820 °C to produce As2O3, and the zinc species located downstream could capture the gaseous As2O3. Next, the temperature was further increased to 910 °C. Volatilization of Zn above 900 °C contributed to various carbon defects such as pentagonal, heptagonal, etc.10,11,34 (Fig. S1, ESI†). Simultaneously, As species were anchored in the vicinity of the defects. Finally, the temperature was raised to 1050 °C and the As species were further reduced by defective carbon (DC) to form As-DC1-1050 (Fig. S2, ESI†). The obtained samples were thoroughly washed with hydrochloric acid and deionized water to ensure the removal of possible residual metal effects. Elemental analysis shows that As-DC1-1050 contains 9.17 wt% As species and no observable N or Zn species are detected, which rules out their effect on ORR activity (Table S1, ESI†). Similarly, As-RC1-1050 and As-DC2-1050 were synthesized without the addition of zinc nitrate and α-D-glucose, respectively. The DC support was also synthesized without the addition of α-D-glucose and NaAsO2. | ||
| Fig. 1 The synthesis process, carbon oxidation (oxygen and hydroxyl groups) associated with long-term operations and the recession suppression strategy (re-calcination) of the As-DC1-1050 SAC. | ||
The X-ray diffraction (XRD) pattern reveals that As-DC1-1050, As-RC1-1050 and As-DC2-1050 all display only characteristic peaks similar to RC, and that no characteristic peaks of NaAsO2, As2O3 or others are observed (Fig. S3, ESI†). Transmission electron microscopy (TEM) characterization reveals that both As-DC1-1050 and As-RC1-1050 are porous materials (Fig. 2a and b). As-DC1-1050 is further confirmed to be porous by N2 adsorption/desorption, with a mean pore diameter of 1.96 nm, a pore volume of 0.62 cm3 g−1 and a specific surface area of 1263.7 m2 g−1 (Fig. 2c). High-angle annular dark-filed scanning TEM (HAADF-STEM) images show that the surfaces of As-DC1-1050 and As-RC1-1050 are scattered with atomic-sized white spots attributed to the arsenic species (Fig. 2d and f). X-ray energy dispersive spectroscopy (EDS) mappings demonstrate that elemental As is uniformly dispersed on the surface of As-DC1-1050 and As-RC2-1050 without significant aggregation (Fig. 2e and g). However, the surface of As-DC2-1050 is distributed with scattered As atoms and aggregated As clusters, indicating that α-D-glucose chelating zinc ions is beneficial to the dispersion of As species (Fig. 2h). The Raman spectra show an ID/IG ratio of 1.28 for As-DC1-1050, which is greater than that of 1.10 for As-RC1-1050 (Fig. 2i). This suggests that the evaporation of Zn species is more favorable to the formation of defects in As-DC1-1050. The abundance of defects may contribute to enhanced electron transfer in ORR.6,12
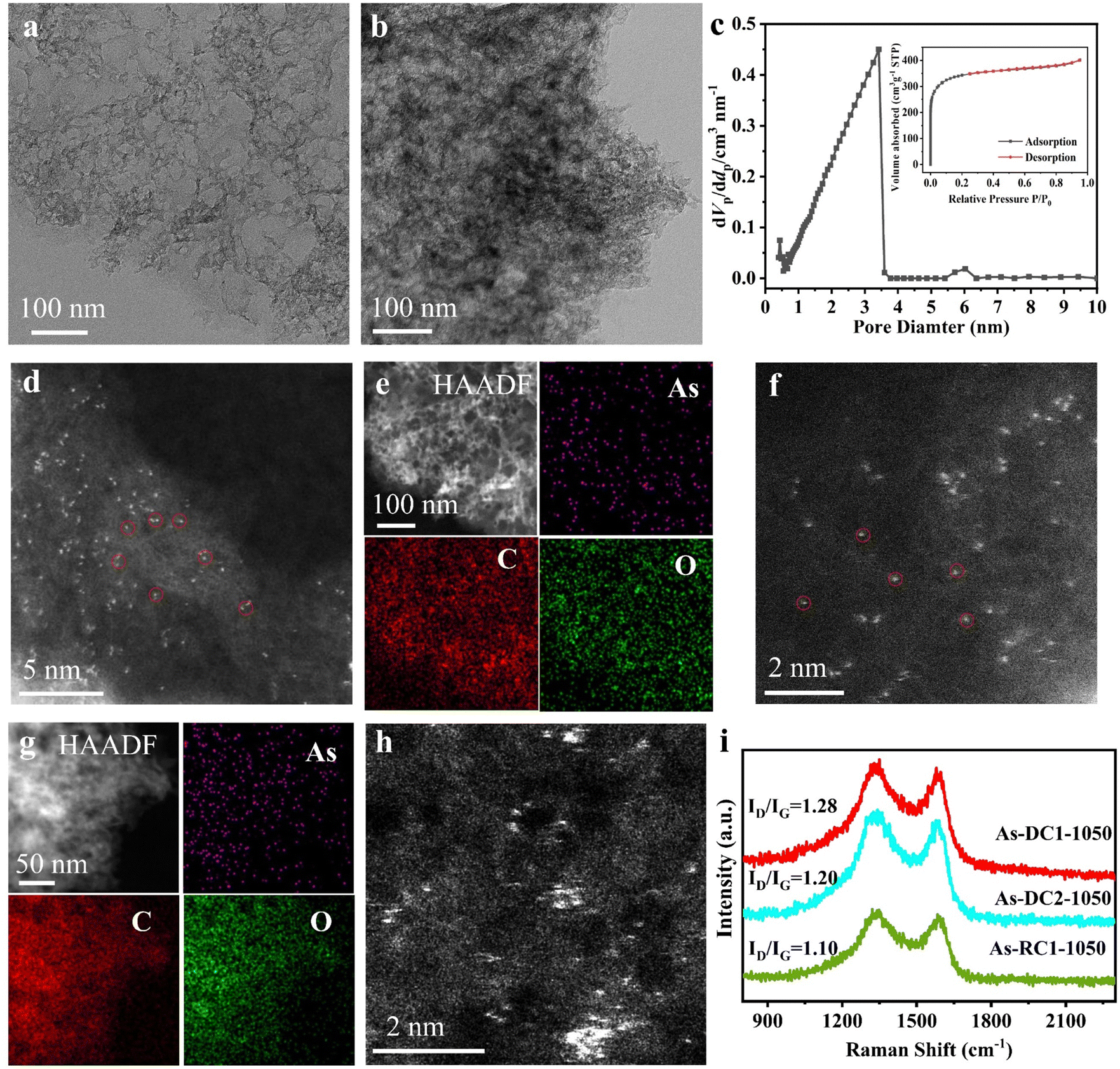 | ||
| Fig. 2 Characterization of the As-based catalysts. TEM images of (a) As-DC1-1050 and (b) As-RC1-1050. (c) Pore size distribution curve of As-DC1-1050 (inset: the adsorption/desorption isotherm). (d) HAADF-STEM and (e) EDS images of As-DC1-1050. (f) HAADF-STEM and (g) EDS images of As-RC1-1050. (h) HAADF-STEM image of As-DC2-1050. (i) Raman spectra of As-based catalysts. | ||
X-ray absorption spectroscopy (XAS) was performed to confirm the dispersion of As species in As-DC1-1050 and As-RC1-1050. As K-edge X-ray absorption near edge spectroscopy (XANES) shows that the intensities of As-DC1-1050 and As-RC1-1050 are located between those of the As foil and NaAsO2 samples, indicating that the valence state of As lies between 0 and +33,32 (Fig. 3a). Remarkably, the valence state of As-DC1-1050 is slightly lower than that of As-RC1-1050, which may stem from the abundance of defects in As-DC1-105035,36 (Fig. 2i). The k2-weighted Fourier transform (FT) of the extended X-ray absorption fine structure (EXAFS) spectra indicates that no As–As scattering paths are observed on As-DC1-1050 or As-RC1-1050, confirming that As is essentially not aggregated on the carbon surface (Fig. 3b). The dominant peak observed at ∼1.26 Å is quite close to the position of the As–O shell in Na3AsO4, so it can be attributed to the As–C/O scattering path.33 The fitting results elucidate that the As site in As-DC1-1050 is coordinated to 2.1 C atoms. Based on the experimentally observed images and fitting results, the structure model for As-DC1-1050 is shown in the inset of Fig. 3c (Fig. S1, S4 and Table S2, ESI†). Wavelet transform (WT) of the As k2-weighted EXAFS was further performed to distinguish the local As bonding environment in the As-DC1-1050 and As-RC1-1050 samples37 (Fig. S5 and S6, ESI†). As-DC1-1050 and As-RC1-1050 exhibit maximum intensity at ∼4.0 Å−1 and 4.3 Å−1, respectively, which originates from the As–C/O shell contribution (Fig. 3d and e).4 The subtle differences in k space may be due to variations in the coordination structure of the two (bond length, number of coordination sites, etc.).31,38 The As foil exhibits a maximum intensity at ∼8.8 Å−1, which is associated with the As–As shell (Fig. 3f). Thus, both HAADF-STEM images and XAS results demonstrate that the As species in As-DC1-1050 and As-RC1-1050 are monoatomically dispersed. Notably, the content of As in As-DC1-1050 (9.17 wt%) is much higher than that of As-RC1-1050 (2.26 wt%), indicating that Zn metal can effectively adsorb As species during the CVD process, thus increasing the loading of single-atom As (Table S1, ESI†).
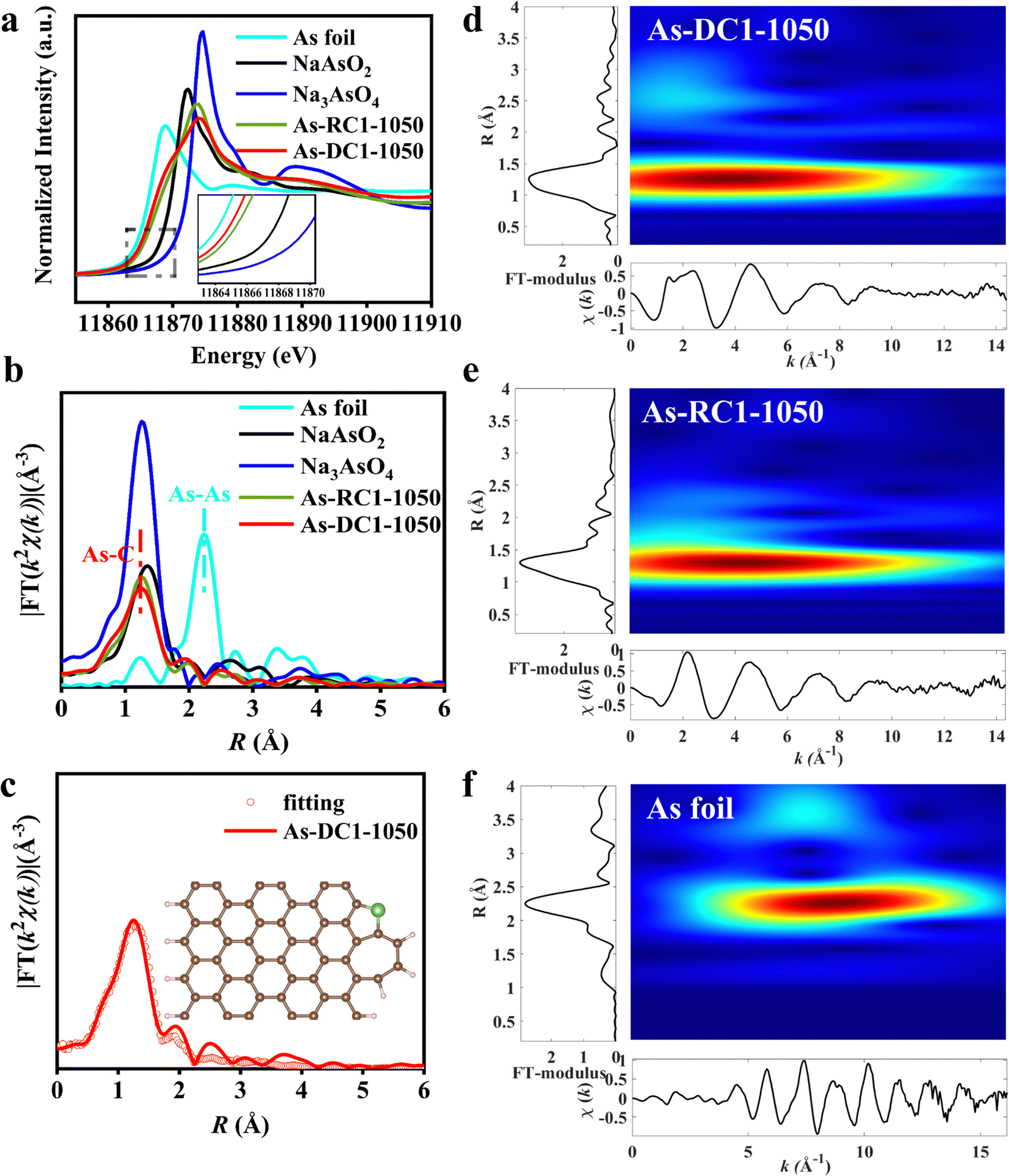 | ||
| Fig. 3 Atomic structure characterization of As-based catalysts. (a) As K-edge XANES spectra. (b) FT-EXAFS spectra of As-DC1-1050, As-RC1-1050, As foil, NaAsO2 and Na3AsO4. (c) FT-EXAFS fitting curves of As K-edge for As-DC1-1050 (inset: constructed structural model of As-DC1-1050). WT of the k2-weighted EXAFS data of (d) As-DC1-1050, (e) As-RC1-1050 and (f) As foil. | ||
ORR performance and recession suppression strategies
To evaluate the prospective application of the synthesized As-based non-metal catalysts, their ORR performance was conducted in 0.1 M KOH. As shown in Fig. 4a, As-DC2-1050 and As-RC1-1050 exhibit half-wave potentials (E1/2) of 0.802 V and 0.781 V, respectively. Intriguingly, As-DC1-1050 exhibits an onset potential and E1/2 of 1.016 V and 0.901 V, respectively, even surpassing the commercial 20% Pt/C (E1/2 of 0.856 V). To demonstrate the true active sites in As-DC1-1050, the DC support was also synthesized without the addition of NaAsO2. As shown in Fig. S7 (ESI†), the DC exhibits a similar LSV curve to the RC, indicating that the effect of Zn on the carbon defect activity is minimal. This proves that topological defects do significantly improve the activity of monatomic As.39 The results of linear sweep voltammetry (LSV) curves at different speeds indicate that the number of transferred electrons (n) of As-DC1-1050 is close to 4, which is also consistent with the rotating ring-disk electrode (RRDE) test results (3.90–3.93, Fig. 4b and c). The n of As-RC1-1050 is in the range of 3.74–3.88 and its H2O2 yield is up to 12.9%. The competition reactions between the two-electron and four-electron pathways inhibit its four-electron activity, resulting in a lower E1/2.17,34 In addition, As-DC1-1050 exhibits a Tafel slope of 65 mV dec−1 which is less than the 76 mV dec−1 of 20% Pt/C, indicating that As-DC1-1050 demonstrates comparable ORR kinetics to noble metal catalysts (Fig. 4d). The E1/2 of As-DC1-1050 degrades by only 12 mV and 20 mV after 10000 and 20000 cycles, respectively (Fig. 4e).
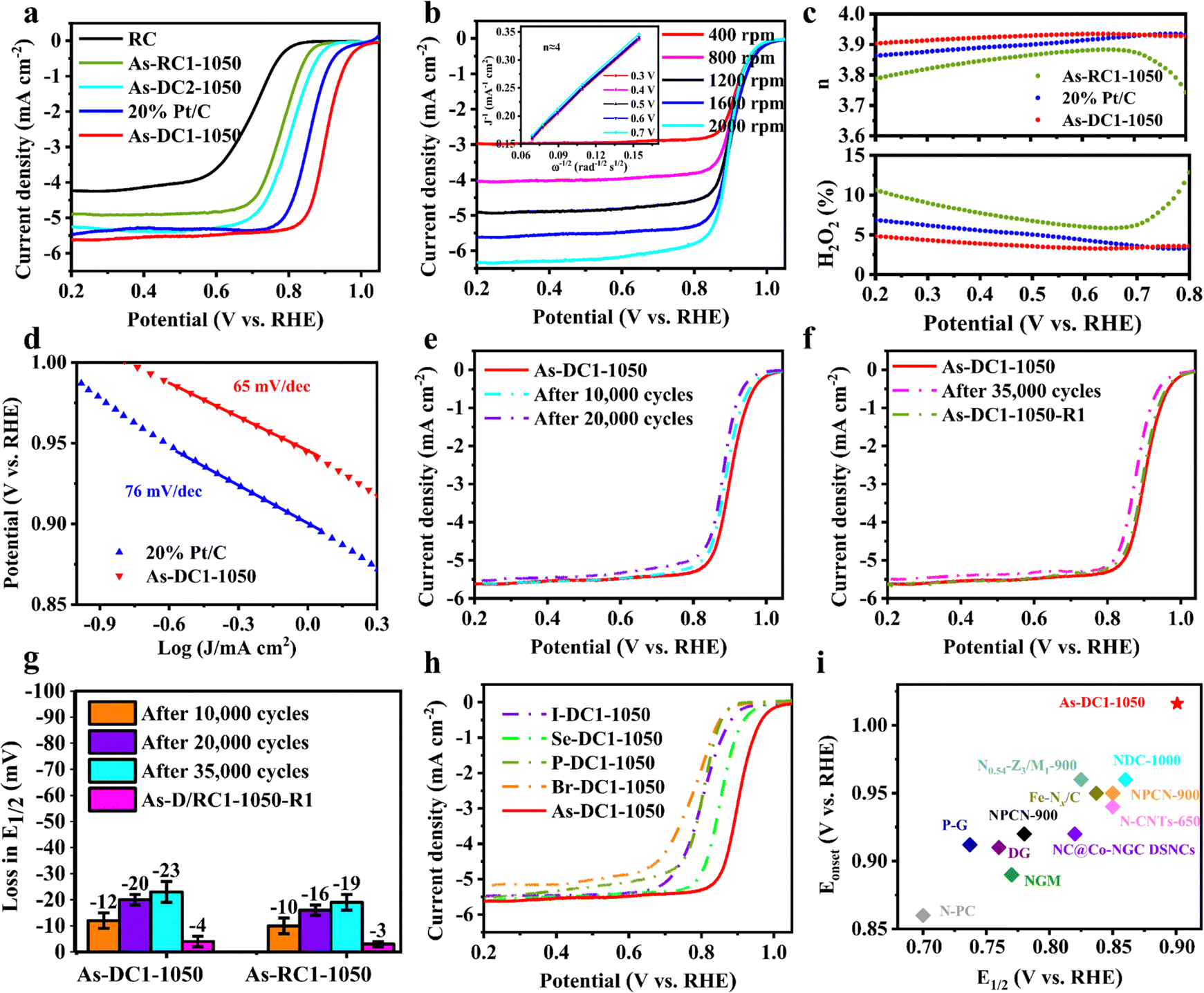 | ||
| Fig. 4 ORR performance in 0.1 M KOH of As-DC1-1050 and reference samples. (a) LSV curves obtained at 1,600 rpm. (b) LSV curves of the As-DC1-1050 catalyst from 400 to 2000 rpm. (inset: Koutecky–Levich plots). (c) Electron transfer number (n) and calculated H2O2 selectivity. (d) Tafel plots of As-DC1-1050 and 20% Pt/C. (e) ORR polarization curves of As-DC1-1050 before and after 10000 and 20000 cycles. (f) ORR polarization curves of As-DC1-1050 after 35000 cycles and after re-calcination (As-DC1-1050-R1). (g) Loss in E1/2 of As-DC1-1050 and As-RC1-1050 after ADT and after re-calcination. (h) LSV curves of Se-DC1-1050, I-DC1-1050, P-DC1-1050 and Br-DC1-1050. (i) Performance comparison of As-DC1-1050 with representative non-precious metal and metal-free catalysts. | ||
Detachment of metal ions and oxidation of the carbon substrate dominate the performance degradation of Fe–NC SACs.23,29,35 To investigate the degradation causes, As-DC1-1050 was further subjected to an accelerated durability test (ADT).30,40 According to our previous work, to collect an adequate amount of As-DC1-1050 after the ADT, the glassy carbon (GC) electrode was replaced by a platinum sheet (PS) electrode with an effective area of 2 cm × 2 cm.41 As-DC1-1050-GC and As-DC1-1050-PS show essentially the same ORR curves after 10000 cycles, indicating that the effect of the electrode on the catalyst can be neglected (Fig. S8, ESI†). The E1/2 of As-DC1-1050 decreases by 23 mV after 35000 cycles (Fig. 4f). Remarkably, the ICP results reveal that the dissolved As content of As-DC1-1050 is only 0.16 wt% after 35000 cycles, which is much less than its loading of 9.17 wt% (Table S3, ESI†). However, the dissolved As content of As-DC2-1050 is up to 0.40 wt% after 35000 cycles. Crucially, high concentrations of dissolved As are toxic to the groundwater environment. This indicates that As-DC1-1050 exhibits higher structural stability in 0.1 M KOH and does not show significant active site shedding.
X-ray photoelectron spectroscopy (XPS) was performed to analyse the surface elemental composition of As-DC1-1050 before and after the ADT tests. XPS results reveal that As-DC1-1050 exhibits a carbon-to-oxygen ratio of 95.51:4.49 in its initial state (Fig. S9a, ESI†). The carbon-to-oxygen ratio on the As-DC1-1050 surface transforms to 92.88:7.12 after 35000 cycles, indicating an increase in oxygen content on the As-DC1-1050 surface after ADT. Correspondingly, the C 1s spectrum of As-DC1-1050 after 35000 cycles shows that carbon bonds to oxygen at 286.53 eV and 288.13 eV to form C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively (Fig. S9b, ESI†).24,36 Fourier-transform infrared (FT-IR) spectroscopy further confirms that oxygen-containing functional groups (C–OH and C–O) are prone to arise on the surface of As-DC1-1050, and this problem has always limited the long-term stability of Fe–NC SACs (Fig. S10, ESI†).19,29,42,43
O, respectively (Fig. S9b, ESI†).24,36 Fourier-transform infrared (FT-IR) spectroscopy further confirms that oxygen-containing functional groups (C–OH and C–O) are prone to arise on the surface of As-DC1-1050, and this problem has always limited the long-term stability of Fe–NC SACs (Fig. S10, ESI†).19,29,42,43
The performance degradation caused by serious demetallization of Fe–NC SACs is usually irreversible.23,44,45 Since As-DC1-1050 does not show significant active site shedding, the performance degradation caused by carbon substrate oxidation is likely to be reversible. Therefore, we heated As-DC1-1050 after 35000 cycles to 400 °C for 30 min in a H2/Ar atmosphere to furnish As-DC1-1050-R1 (Fig. 1). As-DC1-1050-R1 exhibits a 19 mV increase in E1/2 compared to As-DC1-1050 after 35000 cycles (Fig. 4g). XPS and FT-IR spectra show that the oxygen-containing functional groups on the surface of As-DC1-1050-R1 decrease after re-calcination (Fig. S9 and S10, ESI†). As expected, As-RC1-1050 exhibits a similar performance recovery to As-DC1-1050 (Fig. 4g). Compared to As-RC1-1050 after 35000 cycles, As-RC1-1050-R1 demonstrates a 16 mV increase in E1/2. Since re-calcination does not restore the amount of As dissolved in the electrolyte, this further confirms that oxidation of the carbon substrate rather than detachment of non-metal As mainly dominates the performance degradation of As-DC1-1050. The re-calcination process refreshes the carbon defect environment, with an unprecedented suppression of reversible recession dominated by carbon oxidation associated with long-term operations.
Additionally, the applicability of the method to synthesize different non-metal SACs was further validated. Similarly, NaAsO2 was also replaced with SeO2, I2, NaH2PO2 and Br2 to synthesize Se-DC1-1050, I-DC1-1050, P-DC1-1050 and Br-DC1-1050. HAADF-STEM and EDS images show no significant aggregation of Se atoms on the surfaces of Se-DC1-1050 (Fig. S11, ESI†). Similar states are also observed on the surfaces of I-DC1-1050, P-DC1-1050 and Br-DC1-1050. Notably, the I content of I-DC1-1050 is up to 13.78 wt% (Table S1, ESI†). It demonstrates the universality of this methodology for loading high contents of non-metal single atoms on carbon substrates. Despite the lower single-atom loading of As-DC1-1050, it still exhibits a larger half-wave potential than that of Se-DC1-1050 (0.852 V) and I-DC1-1050 (0.803 V) (Fig. 4h), confirming the high activity of the As single atom. Additionally, As-DC1-1050 exhibits better methanol tolerance than Pt/C (Fig. S12, ESI†).20,21,46 More pleasingly, the E1/2 and onset potential of As-DC1-1050 in 0.1 M KOH are comparable to those of the most advanced non-metal catalysts (Fig. 4i and Table S4, ESI†). Alternatively, As-DC1-1050 exhibits better charge transfer resistance than other As-based non-metal catalysts in 0.1 M HClO4 (Fig. S13, ESI†).6 Most non-precious metal catalysts exhibit much less ORR performance in 0.1 M HClO4 than in 0.1 M KOH.10,11,23,34 As-DC1-1050 exhibits an E1/2 of 0.77 V in 0.1 M HClO4, further validating the utility of As-DC1-1050 in different electrolytes (Fig. S14, ESI†). Therefore, NaAsO2 is promisingly recycled for the synthesis of efficient ORR catalysts.
H2/O2 fuel cell and Zn–air battery performance
As-DC1-1050 was further configured into the cathodes of hydrogen–oxygen fuel cells (HOFCs) and zinc-air batteries (ZABs) to exhaustively demonstrate its application prospects. As shown in Fig. 5a, the As-DC1-1050 cathode exhibits a maximum power density of 701.9 mW cm−2 at 0.482 V, comparable to the maximum power density of 689.0 mW cm−2 at 0.452 V displayed by the 20% Pt/C cathode in HOFCs. Although As-DC1-1050 exhibits a much higher half-wave potential than 20% Pt/C, the maximum power density of the two is very close. This can be attributed to the fact that factors such as the spraying process of the cathode ink, the thickness of the catalyst, etc. may lead to fewer effective active sites for the As-DC1-1050 SAC to work in HOFCs.47 Additionally, the cathode catalyst thickness also affects the mass transfer process of O2 and intermediates, which in turn affects the performance of the catalyst in HOFCs.48 The As-DC1-1050 cathode was also operated at a practical fuel cell voltage of 0.67 V to investigate its durability.29 Strikingly, the As-DC1-1050 cathode displays a current density attenuation of only 9.38% after 290 h of operation (denoted as As-DC1-1050-290, Fig. 5b). Furthermore, the effectiveness of re-calcination in suppressing the performance degradation of As-DC1-1050 was further verified. The same constant voltage test was performed three times to obtain a sufficient amount of As-DC1-1050-290 after the stability test, and then As-DC1-1050-290 was heated to 400 °C for 30 min in a H2/Ar atmosphere (denoted as As-DC1-1050-290-R). Next, the As-DC1-1050-290-R cathode continued to be operated at a constant voltage of 0.67 V. As expected, the As-DC1-1050-290-R cathode demonstrates a current density of up to 96.37% of the initial current density of the As-DC1-1050 cathode. The As-DC1-1050-290-R cathode still maintains a current density of 90.14% after 300 h of consecutive operation, surpassing the state-of-the-art catalysts (Table S5, ESI†). Conclusively, re-calcination inhibits the performance degradation of As-DC1-1050 and the current density decays by only 9.86% after 590 h of stable operation in the HOFCs.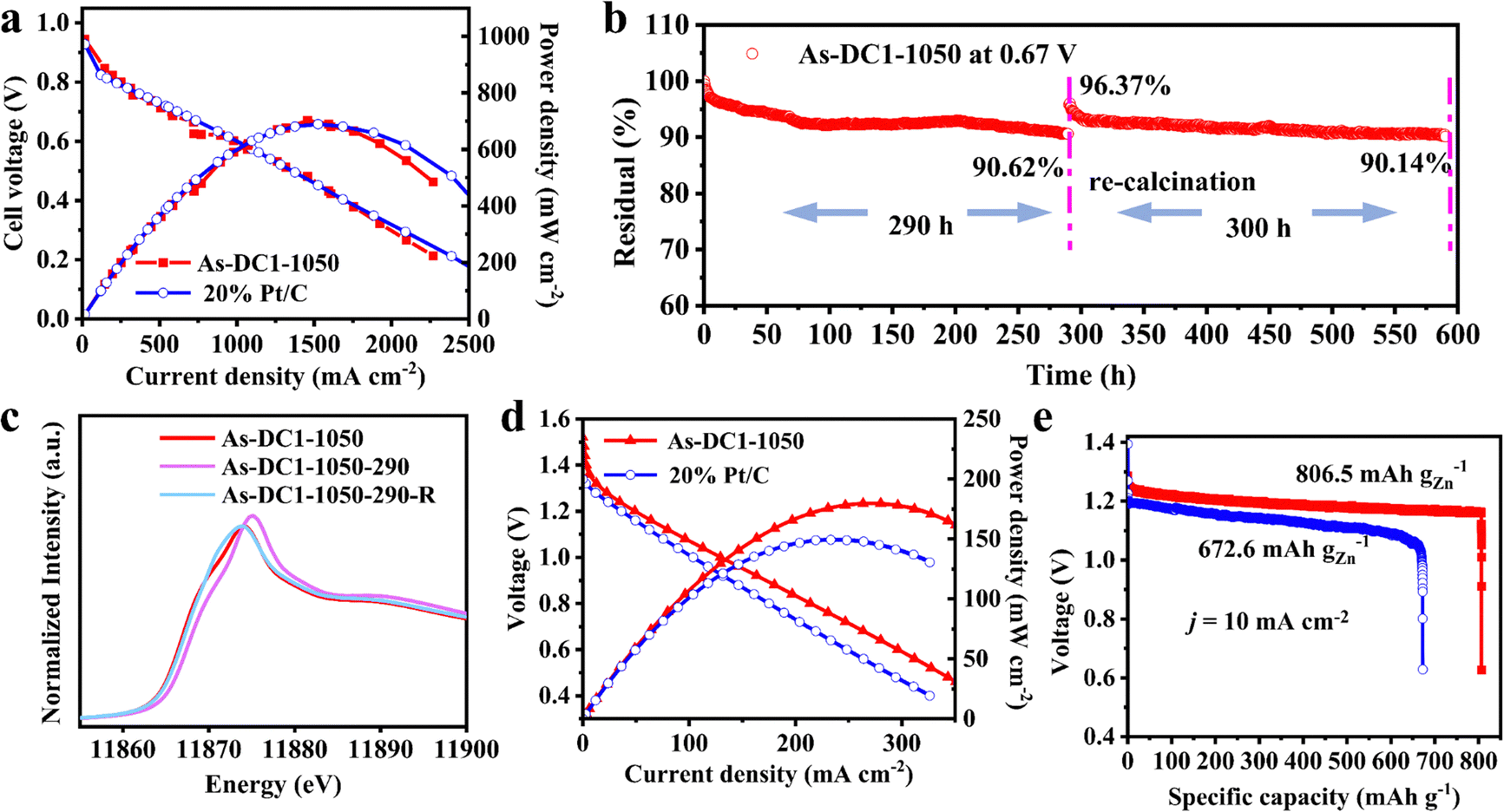 | ||
| Fig. 5 HOFC and ZAB performance with As-DC1-1050 and 20 wt% Pt/C cathode catalysts. (a) Polarization and power density curves in the HOFC. (b) The As-DC1-1050 cathode operated at 0.67 V for 290 h and for another 300 h after re-calcination in the HOFC. (c) As K-edge XANES spectra of As-DC1-1050, As-DC1-1050-290 and As-DC1-1050-290-R. (d) Polarization and power density curves and (e) specific capacities of the ZABs. | ||
XAS was further implemented to analyze the changes of As sites before and after the operation in HOFCs. The XANES results show a slightly higher intensity for As-DC1-1050-290 than for As-DC1-1050, indicating that As was partially oxidized after 290 h of operation (Fig. 5c). It is also consistent with the results shown in Fig. S9 and S10 (ESI†), demonstrating that oxidation of the carbon substrate and As site dominates the As-DC1-1050 degradation. As expected, the intensity of As-DC1-1050-290-R returns to approximately the same as that of As-DC1-1050. Furthermore, FT-EXAFS spectra indicate that As-DC1-1050-290-R shows a very similar profile to that of As-DC1-1050, which proves that the coordinative geometry of As is not significantly altered by re-calcination at 400 °C for only 30 min (Fig. S15, ESI†). TEM and EDS images further demonstrate that As-DC1-1050-290 does not show significant structural changes or clustering of As atoms before and after re-calcination (Fig. S16, ESI†). This further illuminates that re-calcination can effectively address the oxidation of As-DC1-1050 caused by prolonged operation, thereby inhibiting the reversible recession.
Additionally, As-DC1-1050 exhibits a maximum power density of 179.8 mW cm−2 at 0.663 V, even outperforming the maximum power density of 149.7 mW cm−2 at 0.620 V for 20% Pt/C in ZABs (Fig. 5d). As-DC1-1050 also displays an open-circuit voltage of 1.44 V comparable to that of Pt/C (1.43 V), and a higher specific capacity of 806.5 mA h gZn−1 at a current density of 10 mA cm−2 (Fig. 5e and Fig. S17, ESI†). Rate capability tests show that the output voltage of As-DC1-1050 even exceeds that of 20% Pt/C at high current densities greater than 50 mA cm−2. Galvanostatic cycling tests demonstrate that As-DC1-1050 can be operated reliably for more than 95 h at a current density of 5 mA cm−2, outperforming the stability of Pt/C. The attractive catalytic activity and stability at a lower cost suggest that As-DC1-1050 shows significant potential to replace commercial Pt/C in alkaline electrolytes (Table S6, ESI†).
Insights into the ORR mechanism
The above experimental results indicate that defects can significantly modulate the ORR activity of the As site on nitrogen-free carbon substrates. The binding energies of As in As-DC1-1050 at different potentials were further analyzed to investigate the oxygen reduction mechanism (Fig. 6a). The binding energy of As in As-DC1-1050 shifts positively as the applied voltage decreases. Compared to no bias, As-DC1-1050 works at 1.1 V with a positive shift in the binding energy of As, which is due to the adsorption of O2 in oxygen-saturated 0.1 M HClO4 at the As active site.47 The gradual formation of oxygen-containing intermediates (such as O* and OH*) at the As site contributes to a gradual increase in the binding energy of As when the applied voltage ranges from 0.9 to 0.5 V.3,32,36 This demonstrates that the high activity of As-DC1-1050 arises from the incorporation of single-atom As. In addition, As-DC1-1050-290, which was operated in a fuel cell after 290 h, was also subjected to XPS in situ strategies to analyze its oxygen reduction recession mechanism. Compared to As-DC1-1050, the binding energy of As in As-DC1-1050-290 shifts positively when the state is at no bias. This is attributed to the partial oxidation of As in As-DC1-1050-290 after the stability test, which is also consistent with the XANES results in Fig. 5c. Strikingly, compared to As-DC1-1050, the binding energy of As becomes smaller when As-DC1-1050-290 is operated at 0.5 V. The magnitude of the positive shift in the binding energy of As in As-DC1-1050-290 is significantly smaller than that of As in As-DC1-1050 when the applied voltage ranges from 0.9 to 0.5 V (Fig. 6a and Fig. S18, ESI†). This reveals that the formation of oxygen-containing intermediates at the As site in As-DC1-1050-290 is more difficult, resulting in a decrease in ORR activity.32,47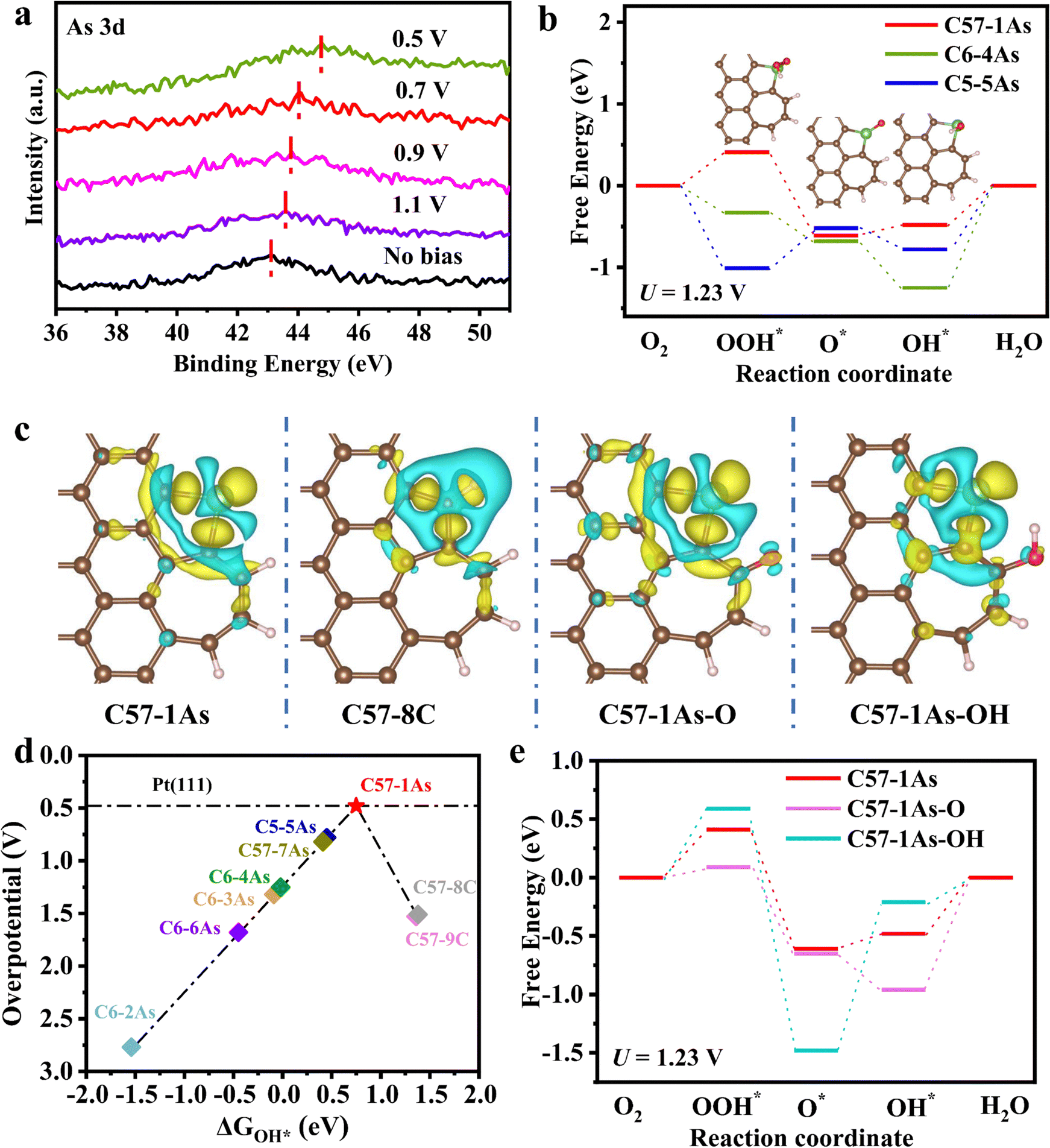 | ||
| Fig. 6 Mechanism analysis of As-based catalysts in ORR. (a) XPS of As-DC1-1050 at different potentials in 0.1 M HClO4. (b) Free-energy diagrams and key reaction intermediates of C57–1As, C6–4As and C5–5As at U = 1.23 V. The inset shows the key reaction intermediates on the C57–1As model. The elements carbon, hydrogen, oxygen and arsenic are denoted in brown, pink, red and green balls, respectively. (c) Differential charge density of C57–1As, C57–8C, C57–1As–O and C57–1As–OH. Cyan and yellow represent electron depletion and accumulation; the iso-surface value is 0.003eÅ−3. (d) ORR volcano plot of overpotential versus OH* adsorption energy for As-based catalysts and defective carbon. (e) Free-energy diagrams and key reaction intermediates of C57–1As, C57–1As–O and C57–1As–OH at U = 1.23 V. | ||
Here, density-functional theory (DFT) calculations were implemented to further elucidate the influence mechanism of the coordination ligation and local defects on the As site. Based on the experimentally observed images (Fig. S1, ESI†) and FT-EXAFS fitting results (Table S2, ESI†), structural models for As-DC1-1050 (C57–1As) and As-RC1-1050 (C6–4As) were constructed (Fig. S19a and d, ESI†).6 To clarify the influence of topological defects and edge effects on As active sites,12 five other As monatomic models were systematically proposed for comparison (Fig. S19b, c and e–g, ESI†). Additionally, given the possible enhanced activity of topological defects on carbon, two typical carbon defect models were also discussed9–11(Fig. S19h, ESI†). C57–8C (5.22 eV) and C57–9C (5.2 eV) exhibit a poor affinity for OOH*, indicating that OOH* is not effectively adsorbed at the carbon site,12,49 with overpotentials up to 1.53 V and 1.51 V, respectively (Table S7, ESI†). This demonstrates that the activity of topological defects on the carbon of C57–8C and C57–9C is essentially negligible. Adsorption free energies show that the As site on C57–1As exhibits a better affinity for oxygen intermediates compared to carbon (Table S7, ESI†). The charge distribution indicates that the single-atom As triggers the electron delocalization of C57–1As, which may significantly regulate the adsorption and hybridization of intermediates.6 For comparison, C57–8C and C57–9C show more charge depletion on the surface of the C site (Fig. 6c and Fig. S20, ESI†). Consequently, both XPS and theoretical analyses verify that single-atom As actually serves as a highly active centre for oxygen adsorption and activation.
The free energy diagrams of three typical models (C57–1As, C6–4As and C5–5As) were plotted to confirm the catalytic activity of As sites towards the four-electron associative ORR pathway (Fig. 6b and Fig. S21, S22, ESI†).6,35 To elucidate the effect of topological defects on activity, the overpotentials of the different active sites were also calculated and correlated with the adsorption energy of OH* to construct a typical volcano diagram (Fig. 6d). The As-based catalysts located on the left side bind strongly to OH*, thus eqn (1) is the potential-determining step.24,50
| OH* + H+ + e− → * + H2O | (1) |
The overpotential increases as the affinity of the As site for OH* increases. In particular, the strong binding of OH* to As may poison the active site As, which leads to an overpotential of As located in the bulk phase (C6–2As) as high as 2.77 V (Table S7, ESI†).50 C6–3As and C6–4As show smaller overpotential when As is located at the edge of the six-carbon ring, suggesting that edge effects can reduce the affinity of OH* for As. Similarly, As located at the edges of different defects also exhibits lower overpotential. Notably, C57–1As located at the top of the volcano exhibits the most desirable affinity for OH* (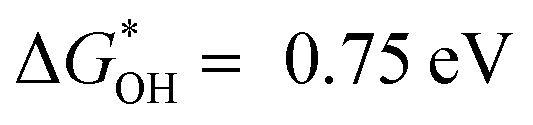 ), leading to the minimal ORR overpotential (0.48 V), comparable to that of the state-of-the-art Pt(111).27,50,51 Badel charge analysis shows a loss of 0.576e and 0.676e of As in C57–1As and C6–4As, respectively (Fig. S23, ESI†). This is also consistent with the results in Fig. 3a, where the valence state of As-DC1-1050 is slightly lower than that of As-RC1-1050. It indicates that the defect can inhibit the electron transfer from As to the neighbouring carbon atom, which may improve the activity of the catalyst.35 In order to investigate the adsorption behavior of the intermediates on the non-metal single-atom catalysts, the projected density of states (PDOS) of C57–1As, C6–4As, and C5–5As were calculated for comparison. Fig. S24 (ESI†) shows that the O-p states display an apparent hybridization with As-p states near the Fermi level, indicating the strongest interaction of As–OOH* in C5–5As–OOH* systems than other SACs.52 This phenomenon agrees well with the free energy level of OOH*, which suggests that the stronger the interaction between As and O, the easier the formation of OOH*. Conclusively, edge effects and topological defect types can modulate the affinity of As for OH* to achieve efficient adsorption–desorption behavior of oxygen intermediates.
), leading to the minimal ORR overpotential (0.48 V), comparable to that of the state-of-the-art Pt(111).27,50,51 Badel charge analysis shows a loss of 0.576e and 0.676e of As in C57–1As and C6–4As, respectively (Fig. S23, ESI†). This is also consistent with the results in Fig. 3a, where the valence state of As-DC1-1050 is slightly lower than that of As-RC1-1050. It indicates that the defect can inhibit the electron transfer from As to the neighbouring carbon atom, which may improve the activity of the catalyst.35 In order to investigate the adsorption behavior of the intermediates on the non-metal single-atom catalysts, the projected density of states (PDOS) of C57–1As, C6–4As, and C5–5As were calculated for comparison. Fig. S24 (ESI†) shows that the O-p states display an apparent hybridization with As-p states near the Fermi level, indicating the strongest interaction of As–OOH* in C5–5As–OOH* systems than other SACs.52 This phenomenon agrees well with the free energy level of OOH*, which suggests that the stronger the interaction between As and O, the easier the formation of OOH*. Conclusively, edge effects and topological defect types can modulate the affinity of As for OH* to achieve efficient adsorption–desorption behavior of oxygen intermediates.
To elucidate the decay mechanism of As-DC1-1050 after oxidation, the effect of oxygen-containing functional groups on the activity of As sites was further investigated. As shown in Fig. S9 and S10 (ESI†), oxygen and hydroxyl groups lead to a decline in the performance of As-DC1-1050. Previous research studies have shown that oxygen-containing functional groups are prone to arise around defects, and their effects on the active site are distance-limited.24,53 Therefore, the corresponding models of oxygen and hydroxyl groups adsorbed near the As site were proposed (Fig. S25, ESI†). The desorption and adsorption of OH* are the potential-determining steps for C57–1As–O and C57–1As–OH, respectively (Fig. 6e and Table S8, ESI†). The overpotential of C57–1As–O (0.96 V) and C57–1As–OH (1.27 V) increases significantly compared to that of C57–1As (0.48 V). The charge distribution clarifies that the introduction of oxygen and hydroxyl groups increases the charge depletion around the As site (Fig. 6c). In particular, the charge depletion caused by the hydroxyl group is particularly pronounced, resulting in a larger overpotential for C57–1As–OH.35 Consequently, the introduction of C–OH and C–O leads to an increased charge depletion around the As site, which dominates the decrease in As activity29 (Table S9, ESI†).
3. Conclusions
We have proposed a general strategy to synthesize high-loading (up to 13.78 wt%) non-metal monoatoms on carbon and further construct topological defects to modulate the activity of non-metal monoatoms. Impressively, As-DC1-1050 exhibited an E1/2 of 0.901 V and a degradation of 23 mV after 35000 cycles. As-DC1-1050 also exhibited a maximum power density of 701.9 mW cm−2 in HOFCs and 179.8 mW cm−2 in ZABs. Theoretical analysis revealed that the topological defects inhibit the electron transfer from As to the adjacent carbon atoms, diminishing the affinity of OH* over the As site for a lower overpotential (0.48 V). The results demonstrated that oxidation of the carbon substrate (oxygen and hydroxyl groups) associated with long-term operations dominated the As-DC1-1050 degradation. We further explored the possibility of utilizing re-calcination to inhibit the performance degradation of As SACs. The re-calcination process refreshed the carbon defect environment of As-DC1-1050, with a degradation of just 4 mV in half-wave potential after 35000 cycles and only 9.86% deterioration in current density after 590 h of operation in fuel cells. Our work provides attractive perspectives for the modulation and sustainability of efficient metal-free SACs.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Y. W., Y. J. L. and Y. Y. W. conceived the research. Y. W., Y. J. L. and Y. Y. W. wrote the paper. Y. W. W. contributed to the theoretical model construction and DFT calculations. Y. W., Y. Y. W. and J. L. contributed to the experimental planning. H. J. Z. and L. M. L. provided experimental measurements and STEM images. Y. J. L. synthesized the catalysts and performed the catalysis experiments. Z. P. G., J. W. and C. Z. F. discussed the results and polished the manuscript. All authors reviewed and edited the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (0301005202017, 2018CDQYFXCS0017, 106112017CDJXSYY0001), Thousand Young Talents Program of the Chinese Central Government (Grant No. 0220002102003), National Natural Science Foundation of China (NSFC, Grant No. 22371022, 22271029, U19A20100, 21971027, 21373280, 21403019), Beijing National Laboratory for Molecular Sciences (BNLMS) and Hundred Talents Program at Chongqing University (Grant No. 0903005203205), The State Key Laboratory of Mechanical Transmissions Project (SKLMT-ZZKT-2017M11), Natural Science Foundation of Chongqing (Grant No. cstc2019jcyj-msxmX0426), and Science and Technology Research Project of Education Agency in Chongqing (Grant No. KJZD-K201800102).References
- L. C. Kao, Y. Ha, W. J. Chang, X. F. Feng, Y. F. Ye, J. L. Chen, C. W. Pao, F. P. Yang, C. Zhu, W. L. Yang, J. H. Guo and S. Y. H. Liou, J. Am. Chem. Soc., 2021, 143, 16538–16548 CrossRef CAS PubMed.
- X. Y. Lou, R. Boada, L. Simonelli and M. Valiente, J. Colloid Interface Sci., 2022, 614, 460–467 CrossRef CAS PubMed.
- J. H. Park, S. J. Kim, I. H. Nam, J. Ryu, G. Y. Jung and Y. S. Han, Water Res., 2022, 222, 118873 CrossRef CAS.
- C. M. van Genuchten, Ind. Eng. Chem. Res., 2022, 61, 13154–13167 CrossRef CAS.
- Z. W. Liu, M. Li, F. Wang and Q. D. Wang, J. Power Sources, 2016, 306, 535–540 CrossRef CAS.
- H. Hu, J. J. Wang, B. F. Cui, X. R. Zheng, J. G. Lin, Y. D. Deng and X. P. Han, Angew. Chem., Int. Ed., 2022, 61, e202114441 CrossRef CAS PubMed.
- W. W. Fu, Y. W. Wang, W. Tian, H. J. Zhang, J. Li, S. Y. Wang and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 23791–23799 CrossRef CAS PubMed.
- Y. Q. Zhao, T. Ling, S. M. Chen, B. Jin, A. Vasileff, Y. Jiao, L. Song, J. Luo and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 12252–12257 CrossRef CAS PubMed.
- L. Tao, Q. Wang, S. Dou, Z. L. Ma, J. Huo, S. Y. Wang and L. M. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. S. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845–6851 CrossRef CAS.
- Y. Jia, L. Z. Zhang, A. J. Du, G. P. Gao, J. Chen, X. C. Yan, C. L. Brown and X. D. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- X. C. Yan, Y. Jia and X. D. Yao, Chem. Soc. Rev., 2018, 47, 7628–7658 RSC.
- S. H. Ye, F. Y. Luo, Q. L. Zhang, P. Y. Zhang, T. T. Xu, Q. Wang, D. S. He, L. C. Guo, Y. Zhang, C. X. He, X. P. Ouyang, M. Gu, J. H. Liu and X. L. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- S. H. Ye, W. Xiong, P. Liao, L. R. Zheng, X. Z. Ren, C. X. He, Q. L. Zhang and J. H. Liu, J. Mater. Chem. A, 2020, 8, 11246–11254 RSC.
- Z. D. Chen, W. D. Chen, L. R. Zheng, T. Huang, J. Hu, Y. Q. Lei, Q. Yuan, X. Z. Ren, Y. L. Li, L. Zhang, S. L. Huang, S. H. Ye, Q. L. Zhang, X. P. Ouyang, X. L. Sun and J. H. Liu, Sci. China: Chem., 2022, 65, 521–531 CrossRef CAS.
- S. H. Ye, S. H. Xie, Y. Q. Lei, X. Y. Yang, J. Hu, L. R. Zheng, Z. D. Chen, Y. H. Fu, X. Z. Ren, Y. L. Li, X. P. Ouyang, Q. L. Zhang, J. H. Liu and X. L. Sun, Nano Res., 2023, 16, 1869–1877 CrossRef CAS.
- M. T. Zhang, H. Li, J. X. Chen, F. X. Ma, L. Zhen, Z. H. Wen and C. Y. Xu, Adv. Funct. Mater., 2023, 33, 2209726 CrossRef CAS.
- H. Tian, A. L. Song, P. Zhang, K. A. Sun, J. J. Wang, B. Sun, Q. H. Fan, G. J. Shao, C. Chen, H. Liu, Y. D. Li and G. X. Wang, Adv. Mater., 2023, 35, 2210714 CrossRef CAS.
- S. Q. Huang, Z. L. Qiao, P. P. Sun, K. W. Qiao, K. Pei, L. Yang, H. X. Xu, S. T. Wang, Y. Huang, Y. S. Yan and D. P. Cao, Appl. Catal., B, 2022, 317, 121770 CrossRef CAS.
- C. X. Zhao, B. Q. Li, J. N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS.
- C. L. Yang, L. N. Wang, P. Yin, J. Y. Liu, M. X. Chen, Q. Q. Yan, Z. S. Wang, S. L. Xu, S. Q. Chu, C. H. Cui, H. X. Ju, J. F. Zhu, Y. Lin, J. L. Shui and H. W. Liang, Science, 2021, 374, 459–464 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- Y. Y. Shao, J. P. Dodelet, G. Wu and P. Zelenay, Adv. Mater., 2019, 31, 1807615 CrossRef.
- Z. Y. Lu, G. X. Chen, S. Siahrostami, Z. H. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. C. Lin, Y. Y. Liu, T. F. Jaramillo, J. K. Norskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- W. Wei, H. W. Liang, K. Parvez, X. D. Zhuang, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- X. G. Li, B. Y. Guan, S. Y. Gao and X. W. Lou, Energy Environ. Sci., 2019, 12, 648–655 RSC.
- Q. M. Deng, J. Zhao, T. T. Wu, G. B. Chen, H. A. Hansen and T. Vegge, J. Catal., 2019, 370, 378–384 CrossRef CAS.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS.
- S. W. Liu, C. Z. Li, M. J. Zachman, Y. C. Zeng, H. R. Yu, B. Y. Li, M. Y. Wang, J. Braaten, J. W. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. R. Shi, Z. X. Feng, H. Xu, G. F. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- X. Y. Xie, L. Shang, X. Y. Xiong, R. Shi and T. R. Zhang, Adv. Energy Mater., 2022, 12, 2102688 CrossRef CAS.
- Y. N. Shang, X. Xu, B. Y. Gao, S. B. Wang and X. G. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 RSC.
- L. C. Kao, Y. Ha, W. J. Chang, X. F. Feng, Y. F. Ye, J. L. Chen, C. W. Pao, F. P. Yang, C. Zhu, W. L. Yang, J. H. Guo and S. Y. H. Liou, J. Am. Chem. Soc., 2021, 143, 16538–16548 CrossRef CAS.
- Y. S. Han, J. H. Park, Y. Min and D. H. Lim, Chem. Eng. J., 2020, 397, 125426 CrossRef CAS.
- Z. Li, H. Cheng, Y. Lu, T. Wang, Y. F. Li, W. Zhang, G. J. He and Z. L. Tian, Adv. Energy. Mater, 2023, 13, 2203963 CrossRef CAS.
- J. J. Li, W. Xia, J. Tang, Y. Gao, C. Jiang, Y. N. Jia, T. Chen, Z. F. Hou, R. J. Qi, D. Jiang, T. Asahi, X. T. Xu, T. Wang, J. P. He and Y. Yamauchi, J. Am. Chem. Soc., 2022, 144, 9280–9291 CrossRef CAS PubMed.
- F. B. Biswas, S. Das, T. Nishimura, M. Endo, M. Fukuda, F. Morita, A. S. Mashio, T. Taniguchi, K. Maeda and H. Hasegawa, Chem. Eng. J., 2022, 450, 138232 CrossRef CAS.
- S. W. Li, J. J. Liu, Z. Yin, P. J. Ren, L. L. Lin, Y. Gong, C. Yang, X. S. Zheng, R. C. Cao, S. Y. Yao, Y. C. Deng, X. Liu, L. Gu, W. Zhou, J. F. Zhu, X. D. Wen, B. J. Xu and D. Ma, ACS Catal., 2020, 10, 907–913 CrossRef CAS.
- L. L. Li, X. Chang, X. Y. Lin, Z. J. Zhao and J. L. Gong, Chem. Soc. Rev., 2020, 49, 8156–8178 RSC.
- Y. Jia, L. Z. Zhang, L. Z. Zhuang, H. L. Liu, X. C. Yan, X. Wang, J. D. Liu, J. C. Wang, Y. R. Zheng, Z. H. Xiao, E. Taran, J. Chen, D. J. Yang, Z. H. Zhu, S. Y. Wang, L. M. Dai and X. D. Yao, Nat. Catal., 2019, 2, 688–695 CrossRef CAS.
- Q. Z. An, S. W. Bo, J. J. Jiang, C. Gong, H. Su, W. R. Cheng and Q. H. Liu, Adv. Sci., 2023, 10, 2205031 CrossRef CAS.
- Y. J. Luo, Y. W. Wang, Y. Y. Wang, H. M. Huang, L. Zhang, H. J. Zhang and Y. Wang, Appl. Catal., B, 2022, 317, 121797 CrossRef CAS.
- F. T. Kong, Y. F. Huang, M. X. Chen, G. Meng, H. Tian, Y. F. Chen, Z. W. Chang, C. Chen, W. P. Sun, X. Z. Cui and J. L. Shi, Appl. Catal., B, 2022, 317, 121768 CrossRef CAS.
- S. C. Ding, J. A. Barr, Q. R. Shi, Y. C. Zeng, P. Tieu, Z. Lyu, L. Z. Fang, T. Li, X. Q. Pan, S. P. Beckman, D. Du, H. F. Lin, J. C. Li, G. Wu and Y. H. Lin, Acs Nano, 2022, 16, 15165–15174 CrossRef CAS.
- L. S. Peng, L. Shang, T. R. Zhang and G. I. N. Waterhouse, Adv. Energy Mater., 2020, 10, 2003018 CrossRef CAS.
- F. Luo, A. R. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS.
- Q. Q. Zhang and J. Q. Guan, Energy Environ. Mater., 2021, 4, 307–335 CrossRef CAS.
- R. J. Gao, J. Wang, Z. F. Huang, R. R. Zhang, W. Wang, L. Pan, J. F. Zhang, W. K. Zhu, X. W. Zhang, C. X. Shi, J. Lim and J. J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- K. L. Wang and Y. Ding, Prog. Nat. Sci.: Mater. Int., 2020, 30, 775–786 CrossRef CAS.
- P. Zhang, J. S. Lian and Q. Jiang, Phys. Chem. Chem. Phys., 2012, 14, 11715–11723 RSC.
- H. X. Xu, D. J. Cheng, D. P. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- Y. J. Cheng, H. Q. Song, J. K. Yu, J. W. Chang, G. I. N. Waterhouse, Z. Y. Tang, B. Yang and S. Y. Lu, Chin. J. Catal., 2022, 43, 2443–2452 CrossRef CAS.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. H. Guo, P. D. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03274d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |