
Tailoring hydroxyl groups of organic phenazine anodes for high-performance and stable alkaline batteries†
Huilin
Cui
ab,
Dechao
Zhang
ab,
Zhuoxi
Wu
a,
Jiaxiong
Zhu
a,
Pei
Li
ab,
Chuan
Li
a,
Yue
Hou
ab,
Rong
Zhang
ab,
Xiaoqi
Wang
c,
Xu
Jin
c,
Shengchi
Bai
c and
Chunyi
Zhi
 *abd
*abd
aDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China
bHong Kong Center for Cerebro-Cardiovascular Health Engineering (COCHE), Shatin, NT, HKSAR, China. E-mail: cy.zhi@cityu.edu.hk
cResearch Institute of Petroleum Exploration & Development (RIPED), PetroChina Research Center of New Energy, No. 20 Xueyuan Road Haidian District, Beijing, 100083, P. R. China
dHong Kong Institute for Clean Energy, City University of Hong Kong, Kowloon 999077, Hong Kong
First published on 21st November 2023
Abstract
Alkaline-based aqueous batteries have attracted intensive research interests due to their high voltage, low cost, and high safety. However, metal anodes in alkaline electrolytes usually possess poor stability and severe side reactions. Organics are potential alternatives to address these problems, but they are typically not negative enough as anodes. Herein, a group of organic phenazine derivatives including phenazine (PZ), 2-hydroxyphenazine (PZ-OH), and 1,2-dihydroxyphenazine (PZ-2OH) are developed as anode materials for alkaline-based batteries. It is revealed that introducing hydroxyl groups can lower the redox potential by 0.4 V, and fast ion transport channels formed by intramolecular hydrogen bonds can remarkably improve redox kinetics. The optimized PZ-2OH‖Ni (OH)2 batteries deliver a high capacity of 208 mA h g−1anode, a high energy density of 247 W h kg−1anode, and ultra-stable cyclability up to 9000 cycles with a low-capacity decay rate (approximately 0.075‰ capacity decay rate per cycle). Meanwhile, we also demonstrate an alkaline PZ-2OH‖air cell, further proving the applicability of PZ-2OH under alkaline conditions. This work not only explores the effect of hydroxyl substituents on the electrochemical potential and reaction kinetics but also opens up the door to stable anodes for alkaline-based batteries.
Broader contextThe inherent safety and low cost make alkaline-based aqueous batteries ideal candidates for stationary energy storage. However, to date, the electrochemical performance of alkaline-based aqueous batteries has been limited, for example, in nickel-cadmium batteries due to their toxicity and stagnating energy density, in nickel metal batteries due to their low voltage and high cost, and in nickel-zinc batteries due to their relatively short cycle life. Therefore, an alternative anode material that can combine the advantages of low toxicity, cost-effectiveness, low potential, and super-stability needs to be developed urgently to solve these problems. Herein, we developed three phenazine derivatives as the candidate anodes with different numbers of hydroxyl group linking patterns. The results demonstrate the significant influence of hydroxyl groups, which could lower the redox potential by 0.4 V and that fast ion transport channels formed by intramolecular hydrogen bonds can remarkably improve redox kinetics. The optimized phenazine derivative anode with two hydroxyl groups shows both low potential and high rate performance and it can be successfully applied as the anode of alkaline nickel batteries and air batteries. The revealed profound hydroxyl group effect offers an in-depth insight into tailoring the electrochemical performance of organic-based electrodes and provides an efficient structure design strategy for alkaline-based batteries. |
Introduction
Towards solving the critical issue of the depletion of fossil fuels and the deterioration of the environment, great efforts have been devoted to the development of renewable energy sources. Electrochemical energy storage technologies are regarded as the high-efficiency energy storage devices for renewable energy, including organic lithium-ion batteries (LIBs) and nickel (Ni)-metal hydride (MH) and lead (Pb)-acid batteries.1,2 Among these, high energy density organic LIBs have revolutionized portable electronics and electric vehicles.3 However, fires and explosion accidents that occur in LIBs limit their application and promote the development of safe aqueous batteries according to some specific requirements.4 Alkaline aqueous batteries (ANABs) are intrinsically safe and possess potential high voltages.5–7 However, corrosion caused by the side reactions under alkaline conditions is unavoidable for anode materials, which leads to the poor cycling stability of ANABs.8Cadmium metal, the first-generation anode material for alkaline aqueous batteries, has been developed for over 100 years for its high energy density.9 However, it has been gradually phased out due to its toxicity and strong “memory” effect. Subsequently, super-stable alloys were explored and used to replace cadmium metals in ANABs.10 Nevertheless, a low nominal battery voltage (1.2 V) and the high cost of the alloy are still unsatisfactory. In recent years, zinc (Zn) metal has been extensively studied as an ideal anode material for alkaline-based batteries because of its low redox potential (−1.24 V versus standard hydrogen electrode (SHE)) and an impressively high theoretical specific capacity (820 mA h g−1).4,11,12 However, the relatively short cycle life caused by the zinc oxidation and the formation of an undesirable Zn(OH)4 passivation layer stalled the development of Zn anodes under alkaline conditions.2,8,13 To date, few metal materials can operate steadily in alkaline systems. In order to overcome these obstacles and achieve sustainable applications of alkaline-based batteries, it is crucial to design high-performance, low-cost, and environmentally friendly anode materials for ABANs.
In addition to metals, the efforts to design promising ABAN anode materials have been extended to organic molecule materials due to their low toxicity, cost effectiveness, and structural diversity. Meanwhile, replacing metal anodes with organic electrode materials can also potentially address the issues of dendrites, passivation, and corrosion of the metal anodes. Currently, the extensively studied organic electrode materials for the ABANs are conjugated quinone polymers, which store charge by means of an “ion coordination” mechanism and demonstrate broad applicability in various electrolyte systems (including Al3+, Zn2+, Li+, H+, and NH4+).14–18 However, the potentials of these materials are much higher than that of a zinc anode, resulting in low battery voltage and energy density. Among varieties of organic electrodes, phenazine (PZ) derivatives appear to be very promising electrode materials due to their potential high cycling stability.19,20 The nitrogen atom in phenazine compounds is highly electrically negative, which greatly influences the density distribution of electron clouds in the ring, causing the electron cloud of π transfer to the nitrogen atom. Thus, compared with quinone compounds, it is more difficult for the phenazine derivatives with higher electron cloud density to obtain electrons and thus have a relatively low redox potential.21–23 However, even though the redox potentials of phenazine derivatives are lower than those of most organic compounds, they are still much more positive than that of the zinc metal.14 In addition, the influences of grafted functional groups on the electrochemical redox properties of phenazine derivatives need to be explored.
Herein, we investigated the electrochemical behaviors of three kinds of phenazine derivatives, including phenazine (PZ), 2-hydroxyphenazine (PZ-OH), and 1,2-dihydroxyphenazine (PZ-2OH), which contain different numbers of hydroxyl groups. Comprehensive studies demonstrate that the hydroxyl substituted groups adjacent to phenazine can lower the redox potential by reducing the ability to lose electrons and improving the rate performance by forming hydrogen bonds with water molecules in the electrolyte. Owing to the high electrochemical performance of the optimized PZ-2OH anode, the designed alkaline-based aqueous batteries deliver a combination of high capacity and a high rate with robust cycling stability.
Results and discussion
Three kinds of phenazine derivatives (PZ, PZ-OH, and PZ-2OH) were successfully synthesized by a facile and environmentally benign condensation method, and the low-cost diaminobenzene and quinone were used as the precursors. The chemical structures of these phenazine derivatives are shown in Fig. 1a and confirmed by 1H nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FT-IR). The 1H NMR spectra show that PZ-OH and PZ-2OH exhibit obvious chemical shifts at 10.95, 8.19, 8.13, 7.90, 7.83, 7.60, 7.35 ppm and 10.99, 8.07, 7.74, 7.30 ppm, respectively (Fig. S1 and S2, ESI†),24,25 indicating that PZ-OH and PZ-2OH were successfully synthesized. The synthesized structure was further characterized by FT-IR (Fig. 1b). The emerging absorption peaks at 1627 cm−1, 1518 cm−1, 1147 cm−1, and 827 cm−1 in the FTIR spectra of phenazine derivatives indicate the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, and C–H linkages,18,26 while a broad peak in the FTIR spectra of PZ-OH and PZ-2OH at approximately 3300 cm−1 evidences the existence of a hydroxyl group.27 In addition, the powder X-ray diffraction (XRD) patterns demonstrated that phenazine-based organics have relatively ordered-aggregated structures due to the π–π stacking of individual phenazine derivative-based molecules (Fig. S3, ESI†), which can be verified by a peak at 291.3 eV in the high-resolution C 1s XPS of PZ-2OH (Fig. S4, ESI†). Meanwhile, the prepared phenazine derivatives show different colors ranging from yellow (PZ) to brown (PZ-OH), then to black (PZ-2OH) (Fig. S5, ESI†). The scanning electron microscopy (SEM) images showed that PZ has a rod morphology with a length of ∼300 μm, PZ-OH has a cluster morphology with a size of ∼10 μm, and PZ-2OH has a rod morphology with a size of ∼3 μm (Fig. S6, ESI†). Furthermore, compared with PZ powder, no significant sublimation behavior was noted in the PZ-OH and PZ-2OH powders due to the effect of strong interactions between hydroxyl groups (Fig. 2c). The TGA and DSC results show that the decomposition temperature gradually increased with increasing number of hydroxyl groups (Fig. S7, ESI†), suggesting that introduction of hydroxyl groups contributes to thermal stability of organic materials. These results show that the substituent groups significantly influence the physical properties (color, morphology, and surface area) and chemical properties (thermal stability) of these phenazine derivatives. Most importantly, as shown for the BET surface area of PZ, PZ-OH and PZ-2OH (Fig. S8, ESI†), a significant increase of the pore diameter can be observed from 2.90 m2 g−1 to 14.84 m2 g−1 and then to 19.50 m2 g−1, which may have some impacts on the electrochemical performance.
N, CC, and C–H linkages,18,26 while a broad peak in the FTIR spectra of PZ-OH and PZ-2OH at approximately 3300 cm−1 evidences the existence of a hydroxyl group.27 In addition, the powder X-ray diffraction (XRD) patterns demonstrated that phenazine-based organics have relatively ordered-aggregated structures due to the π–π stacking of individual phenazine derivative-based molecules (Fig. S3, ESI†), which can be verified by a peak at 291.3 eV in the high-resolution C 1s XPS of PZ-2OH (Fig. S4, ESI†). Meanwhile, the prepared phenazine derivatives show different colors ranging from yellow (PZ) to brown (PZ-OH), then to black (PZ-2OH) (Fig. S5, ESI†). The scanning electron microscopy (SEM) images showed that PZ has a rod morphology with a length of ∼300 μm, PZ-OH has a cluster morphology with a size of ∼10 μm, and PZ-2OH has a rod morphology with a size of ∼3 μm (Fig. S6, ESI†). Furthermore, compared with PZ powder, no significant sublimation behavior was noted in the PZ-OH and PZ-2OH powders due to the effect of strong interactions between hydroxyl groups (Fig. 2c). The TGA and DSC results show that the decomposition temperature gradually increased with increasing number of hydroxyl groups (Fig. S7, ESI†), suggesting that introduction of hydroxyl groups contributes to thermal stability of organic materials. These results show that the substituent groups significantly influence the physical properties (color, morphology, and surface area) and chemical properties (thermal stability) of these phenazine derivatives. Most importantly, as shown for the BET surface area of PZ, PZ-OH and PZ-2OH (Fig. S8, ESI†), a significant increase of the pore diameter can be observed from 2.90 m2 g−1 to 14.84 m2 g−1 and then to 19.50 m2 g−1, which may have some impacts on the electrochemical performance.
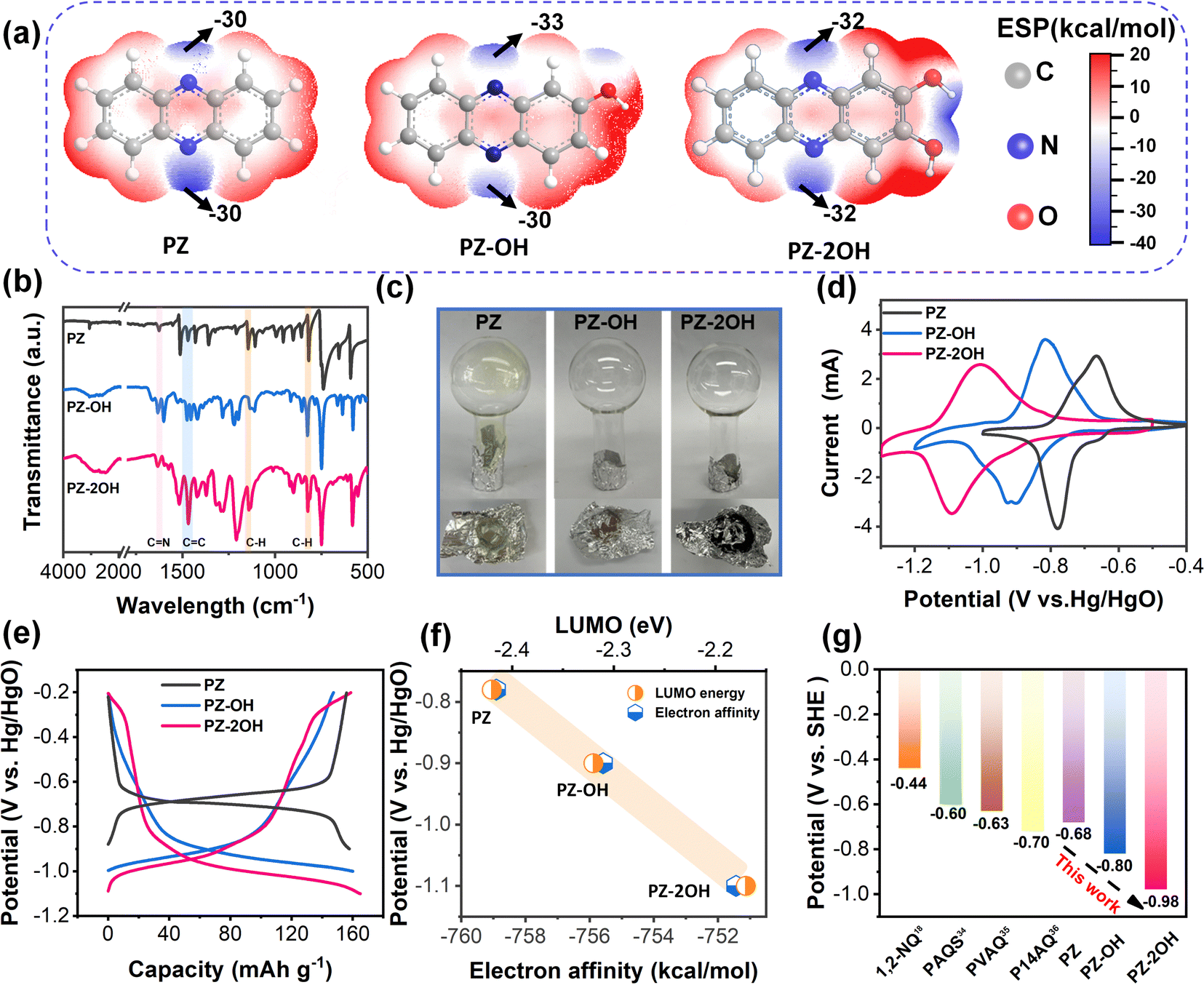 | ||
| Fig. 1 The characterization of phenazine derivatives. (a) Molecule structure and the calculated corresponding ESP distribution on the van der Waals surface of PZ, PZ-OH, and PZ-2OH molecules, respectively. (b) The FT-IR spectra. (c) Sublimation test of PZ, PZ-OH, and PZ-2OH at 180 °C. (d) CV curves of PZ, PZ-OH, and PZ-2OH. (e) Galvanostatic discharge/charge curves of PZ, PZ-OH, and PZ-2OH at a current density of 1 A g−1. (f) Calculated frontier molecular orbital energy and electron affinity of the three molecules (blue dots represent electron affinity and orange dots represent LUMO energy). (g) Comparison of the discharge potential between PZ-2OH and reported organic anodes in an alkaline-based system. | ||
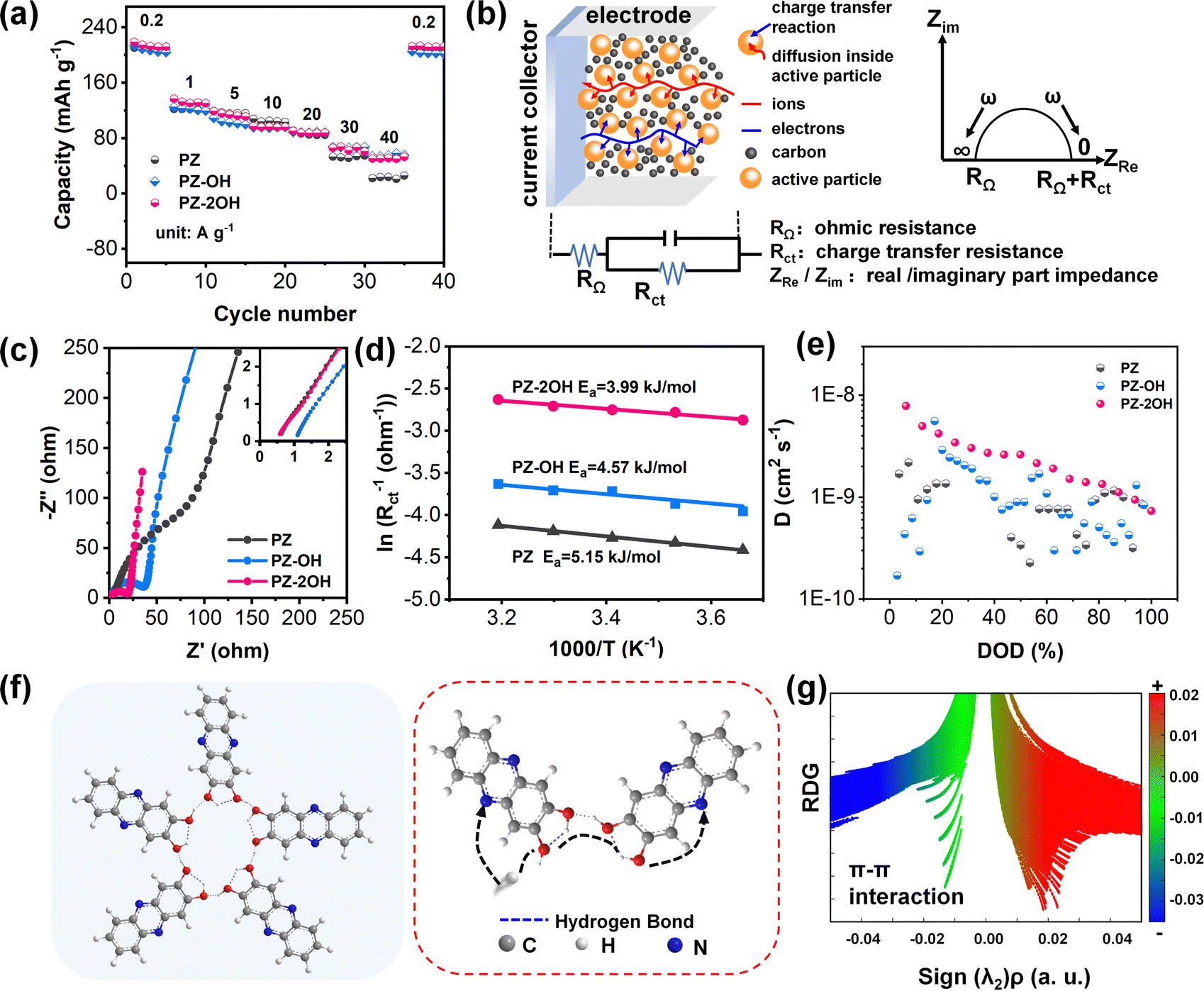 | ||
| Fig. 2 Electrochemical kinetic difference of PZ, PZ-OH, and PZ-2OH molecules. (a) Rate capabilities of PZ, PZ-OH, and PZ-2OH at various current densities. (b) Schematic diagram of the battery impedance spectrum. (c) EIS curves of PZ, PZ-OH, and PZ-2OH molecules. (d) The Arrhenius plots of ln(Rct) vs. 1000/T for PZ, PZ-OH, and PZ-2OH molecules. (e) The ion diffusion coefficient (D) for PZ, PZ-OH, and PZ-2OH molecules. (f) Schematic illustration of the ion conduction in the hydrogen bonding network in PZ-2OH. (g) Plots of the calculated RDG isosurface vs. sign(λ2) ρ for PZ-2OH. | ||
For further analysis of the structure and functional sites, theoretical calculations based on density functional theory (DFT), used in organic chemistry for the derivation of electrophilicity or nucleophilicity,28 were used for the simulation of the molecular electrostatic potential (ESP).29 At the van der Waals surface of the phenazine derivative molecules (Fig. 1a), the blue area indicates the molecules with more negative ESP, which have a tendency to undergo the electrophilic reaction.30 From the ESP mapping results, the CN bond regions of three phenazine derivatives with negative ESP values are identified as the highly reactive sites. The electrochemical performances of the as-synthesized PZ, PZ-OH and PZ-2OH were evaluated in 6 M KOH aqueous alkaline electrolyte based on the hydrogen evolution potentials’ negative shift as the KOH concentration increases (Fig. S9 and S10, ESI†).31 Cyclic voltammetry (CV) curves (Fig. 1d) of the three phenazine derivatives at a 5 mV s−1 scan rate show that there is a significant decrease in the oxidation and reduction potential upon introduction of hydroxy groups. The reduction potential drops dramatically from −0.78 V to −0.9 V and then down to −1.07 V for PZ, PZ-OH, and PZ-2OH, respectively. These results are consistent with the galvanostatic charge/discharge (GCD) curves, which show the average discharge voltage decreased from PZ, PZ-OH to PZ-2OH, respectively (Fig. 1e). However, the capacities of these three phenazine derivatives were not significantly changed, all being approximately 160 mA h g−1 at a current density of 0.5 A g−1.
To understand the potential characteristics, the electron affinities of three phenazine derivatives were analyzed by the first principles DFT method utilizing B3LYP. Herein, electron affinity represents the free energy variation between the neutral system and the negatively charged system. Fig. 1f shows that increasing the number of hydroxyl groups would generally reduce its electron affinity and decrease redox potential (Table S1, ESI†). Furthermore, the different redox potentials of three phenazine derivatives were interpreted using the LUMO energy levels (Fig. 1f and Fig. S11, ESI†). The redox potentials show a linear correlation with the electron affinity and the LUMO energy levels (Fig. 1f), that is, the phenazine derivatives with a lower LUMO energy and a more negative electron affinity value would have higher redox potentials. These results suggest that the high potential of molecular electrode materials is predictable by their more negative electron affinity and LUMO energy levels.32,33 More importantly, the discharge potential of PZ-2OH is lower than those of the reported cases of organic anodes, including 1,2-naphthoquinone (1,2-NQ),18 poly(anthraquinonyl sulfide) (PAQS),34 poly(2-vinylanthraquinone) (PVAQ),35 poly(1,4-anthraquinone) (P14AQ),36 in an aqueous alkaline electrolyte (Fig. 1g). Furthermore, carrying out theoretical simulations to predict the reduction potentials is helpful in finding suitable redox-active materials. The predicted redox potential values suggest that the redox potential of phenazine derivatives gradually shifts negatively from −0.976 V to −0.995 V, to −1.017 V, further to −1.037 V with the increase of the number of grafted hydroxyl groups, which is consistent with the calculated electron affinity results (Table S2, ESI†). However, the negative shift of the redox potential will inevitably lead to a more severe water decomposition problem, which is not conducive to the improvement of the performance of a phenazine derivative full battery. Therefore, considering the advantages of increasing the potential reduction of hydroxyl groups and the disadvantages of water decomposition, PZ-2OH shows excellent application potential in alkaline battery systems and is finally selected as the anode material.
To explore the impact of hydroxide groups on electrochemical performance, the rate capability of three phenazine derivatives was tested at current densities ranging from 0.2 A g−1 to 40 A g−1 (Fig. 2a). Hydroxide-enriched PZ-OH and PZ-2OH exhibited better rate capability than PZ, particularly with high current densities. The capacity of PZ-2OH and PZ-OH remains at 50 mA h g−1 when the current density reaches 40 mA g−1, retaining 20% of the initial capacity, whereas PZ only has a capacity of 20 mA h g−1. To further understand the effect of hydroxy-substituted phenazine on rate performance, electrochemical impedance spectroscopy (EIS) is employed for the analysis of the electrochemical kinetics mechanism. Since all the three electrodes are investigated in the same electrolytes, the impedance caused by the diffusion process is ignored. The effect on rate capability is mainly attributed to the difference in the charge transfer process, which is controlled by the electrochemical reaction steps. As shown in Fig. 2b, the electrochemical kinetics mechanism involves the combined controlled ohmic impedance and electrochemical transfer, which can be intuitively measured by the EIS of different electrodes. As demonstrated by the Nyquist plots, the ohmic impedance of PZ-2OH is smaller than that of PZ-OH and PZ (Fig. 2c), which is consistent with the Highest Occupied Molecular Orbital (HOMO)–LUMO energy level difference (PZ-2OH (ΔE = 3.52 eV) < PZ-OH (ΔE = 3.57 eV) < PZ (ΔE = 3.66 eV)) (Fig. S11, ESI†). The low HOMO–LUMO energy level gap indicates that electrons can easily transition from the highest to lowest occupied orbital.31 Furthermore, the PDOS analysis is shown in Fig. S12 (ESI†). PZ, PZ-OH, and PZ-2OH exhibit bandgaps of 1.893 and 1.244 and 0.059 eV, respectively. Among which, PZ-2OH has significantly smaller band gaps compared with PZ and PZ-OH, indicating the excellent electrical conductivity of PZ-2OH. Thus, the relatively high conductivity (smaller ohmic impedance) of PZ-2OH contributes to its high-rate capability. In addition, the ion diffusion resistance (σ) can be obtained by quantitatively analyzing the real part of impedance (Z′), which is linear with the reciprocal of the square root of frequency (ω−0.5).32 The smaller slope of the linear tendency implies faster ion transport. The PZ-2OH electrode delivered the lowest σ value of 11.11 Ω s−0.5, followed by PZ-OH (36.08 Ω s−0.5) and PZ (57.88 Ω s−0.5), indicating that the PZ-2OH anode possesses a more rapid ion transport kinetics (lower charge-transfer impedance) (Fig. S13, ESI†).
Moreover, the charge transfer behavior was further evaluated by the activation energy (Ea) (Fig. 2d). In accordance with the Arrhenius equation, the Ea value of PZ-2OH is 3.99 kJ mol−1 (Fig. 2d and Fig. S14, ESI†), which is smaller than those of PZ-OH (4.57 kJ mol−1) and PZ (5.15 kJ mol−1), demonstrating that the introduction of hydroxyl groups guaranteed fast ion diffusion and efficient charge transport. Besides, reaction kinetics, investigated using CV tests with scan rates from 1 to 10 mV s−1, also reflect the excellent rate performance of the PZ-2OH anode. The b values calculated from the oxidation and reduction peak currents for PZ, PZ-OH and PZ-2OH were (0.68, 0.54), (0.60, 0.57) and (0.84, 0.71), respectively, according to the relationship between the scan rate (v) and peak current (i) (Fig. S15 and S16, ESI†). The results demonstrate that in the redox processes the three phenazine derivatives exhibit ionic diffusion and capacitive behaviors simultaneously. The capacitive behavior increases gradually from PZ to PZ-OH and then to PZ-2OH (Fig. S16, ESI†). GITT testing further confirms that PZ-2OH has a higher ion diffusion coefficient compared to PZ-OH and PZ (Fig. 2e and Fig. S17, ESI†). The symmetric cell performances of PZ-derivatives in the non-faradaic region were investigated to estimate the double-layer capacitance (Cdl). The plots of current density ((ianodic − icathodic)/2/S) against the scan rate show that PZ-2OH exhibits much higher Cdl than PZ-OH and PZ, implying its higher electrochemically active surface area (Fig. S18, ESI†). From the above analysis, we could find that the ion diffusion kinetics of the phenazine derivative electrodes is affected by the hydroxyl groups, which are available to increase the probability of hydration by forming intramolecular/intermolecular hydrogen bonds, as shown in Fig. 2f. Compared to PZ-OH and PZ-2OH, it is not easy for PZ to hydrate because of the steric hindrances of adjacent H atoms.20 The formed hydrogen bonds contributed to the charge transfer. Therefore, the low ohmic impedance caused by high conductivity and the low charge transfer impedance caused by the ion channel constructed by the hydrogen bond network jointly facilitate the high-rate performance. The interaction of the hydrogen bond is also confirmed by the diagram of the calculated reduced density gradient (RDG) and the corresponding gradient isosurface (Fig. 2g). Specifically, the detected green spikes in the sign(λ2)ρ from −0.02 to 0.00 a.u. proved the existence of weak interactions between PZ-2OH molecules.
Considering the excellent electrochemical performance of the PZ-2OH anode in an alkaline electrolyte, a PZ-2OH‖Ni (OH)2 full cell was further assembled and investigated. Furthermore, the electrochemical performances of the PZ-2OH‖Ni (OH)2 full cell were evaluated in 6M KOH + 1 M LiOH aqueous electrolyte, and the addition of Li+ can suppress the O2 evolution in ANABs according to the previous reports.1 It is worth noting that a 5 μm thickness graphite oxide (GO) membrane was inserted between the anode and the separator, which acted as a strong shield to inhibit anode dissolution (Fig. S19, ESI†). In addition, a GO|| Ni(OH)2 full cell was also assembled to verify the capacity contribution of the inserted GO membrane. Both CV and GCD images confirmed that the GO membrane exhibits a negligible contribution to the full cell capacity (Fig. S20, ESI†). The typical CV curves of the PZ-2OH‖Ni (OH)2 full cell (Fig. 3a) show an oxidation peak at approximately 1.61 V and a reduction peak at 1.47 V, which arise from the redox reactions of the Ni(OH)2 cathode and the PZ-2OH anode. Such high discharge potential of PZ-2OH‖Ni (OH)2 batteries is superior to that of the previously reported Cd‖Ni and MH‖Ni batteries.2,9 Meanwhile, the green and cheap preparation method makes PZ-2OH demonstrate an obvious price advantage compared to other anodes for alkaline aqueous batteries (Fig. S21, ESI†). Fig. 3b and Fig. S22 (ESI†) show the charge/discharge profiles at different current densities and rate characteristics of the PZ-2OH‖Ni(OH)2 batteries. The full cell demonstrated a high discharge capacity (based on the mass of PZ-2OH) of 178 mA h g−1 at a current density of 0.2 A g−1 and a high-rate performance of 64 mA h g−1 at 10 A g−1. In addition, when the current density is restored to 0.2 A g−1, the discharge capacity is restored to 170 mA h g−1, demonstrating an excellent reversion capability. The high capacity and fast diffusion rate of the PZ-2OH anode material provide the PZ-2OH‖Ni (OH)2 full battery excellent power density (26.2 KW kg−1 at 10 A g−1) and energy density (247 W h kg−1 at 0.2 A g−1) (Fig. 3c). The high-power density exhibited by PZ-2OH‖Ni(OH)2 full battery makes it close to supercapacitors with fast charge behavior, while the high energy density it exhibits makes it also superior to those of the previously studied lead-acid and nickel-based batteries. Moreover, in various aqueous electrolytes, the discharge voltage of the full cell is higher than that reported for organic-based batteries, including acid electrolytes (organic pyrene-4,5,9,10-tetraone‖MnO2 (PTO‖MnO2),37 Pb‖p-chloranil/reduced graphene oxide (Pb‖PCHL-rGO),38 and all organic protons39), mild electrolytes (Zn‖tetraamino-p-benzoquinone (Zn‖TABQ),40 KMnHCF‖3,4,9,10-perylenetetracarboxylic diimide (KMnHCF ||PTCDI),41 and Mn‖tetrachloro-1,4-benzoquinone (Mn‖4-Cl-BQ)42), and alkaline electrolytes ((P14AQ‖Ni(OH)2),43 (PAQS‖Ni(OH)2)34) (Fig. 3d and Table S3, ESI†). The excellent rate performance of PZ-2OH‖Ni(OH)2 batteries was largely attributed to their fast kinetics, which was also explored by CV tests at different scan rates (Fig. S23 and S24, ESI†), and the PZ-2OH‖Ni(OH)2 cell in the reduction process and oxidation process shown slopes b of 0.84 and 0.91 respectively, suggesting pseudocapacitive dominance. In addition, due to the high concentration of alkaline electrolytes and the high electrochemical kinetics of the organic electrode, the PZ-2OH‖Ni(OH)2 batteries can work at −30 °C and maintain 73% capacity (Fig. 3e). Although the PZ-2OH‖Ni(OH)2 battery shows a certain degree of capacity decline at a circuit density of 1C (Fig. S25, ESI†), its cycling stability can improve as the current density increases to 0.5 A g−1, which has a durable cycling performance for more than 9000 cycles, and the average rate of the capacity decay rate per cycle is approximately 0.075‰ during the entire cycle process (Fig. 3f). To demonstrate the availability of the PZ-2OH‖Ni(OH)2 cell, we tested the battery performance based on the area capacity. As illustrated in Fig. S26 (ESI†), a high areal capacity of 14 mA h cm−2 can be obtained using a 14.8 mg cm−2 anode at an areal current density of 2 A cm−2, and a high-rate performance (20 A cm−2) can be achieved at various active current densities (Fig. S27, ESI†). Furthermore, the PZ-2OH‖Ni(OH)2 cell delivered stable cycle performances under 10 mg mass-loadings (Fig. S28, ESI†). Controlling the n/p ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5), the energy density of the PZ-2OH‖Ni(OH)2 cell reached 57.2 W h kg−1 (based on the total mass of the positive and negative electrodes) (Fig. S29, ESI†), which was comparable to those of lead-acid batteries. Therefore, PZ-2OH‖Ni(OH)2 has significant advantages over conventional Ni-based batteries (Fig. 3g). PZ-OH is cheaper than alloys, has lower toxicity than the cadmium metal, and has better stability than the Zn metal (Table S4, ESI†), demonstrating comprehensive performance advantages in price, toxicity, stability, and energy density.44
:1.5), the energy density of the PZ-2OH‖Ni(OH)2 cell reached 57.2 W h kg−1 (based on the total mass of the positive and negative electrodes) (Fig. S29, ESI†), which was comparable to those of lead-acid batteries. Therefore, PZ-2OH‖Ni(OH)2 has significant advantages over conventional Ni-based batteries (Fig. 3g). PZ-OH is cheaper than alloys, has lower toxicity than the cadmium metal, and has better stability than the Zn metal (Table S4, ESI†), demonstrating comprehensive performance advantages in price, toxicity, stability, and energy density.44
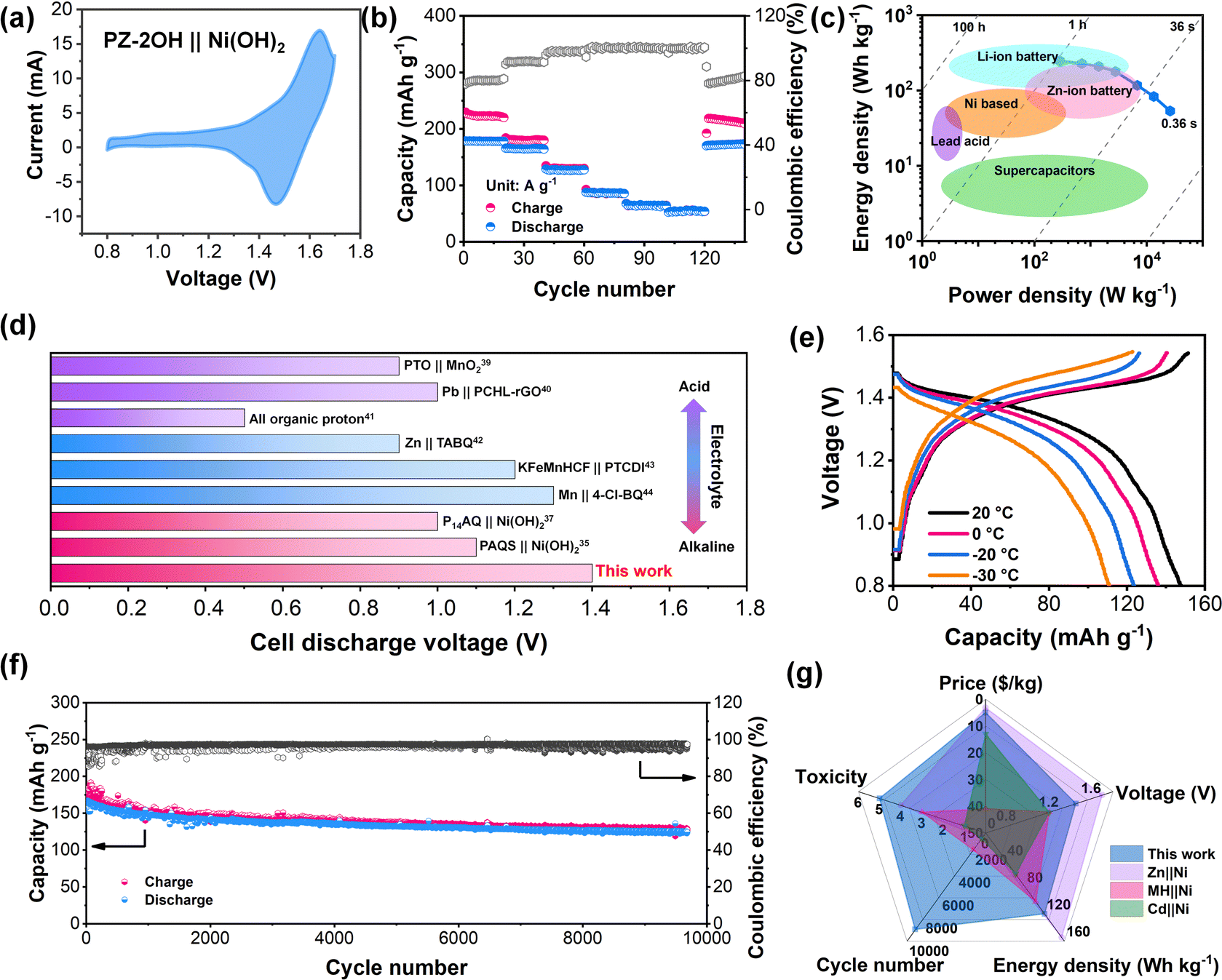 | ||
| Fig. 3 Electrochemical performance of the PZ-2OH‖Ni (OH)2 cells. (a) CV curves at 5 mV s−1. (b) Rate capability and Coulombic efficiency at current densities of 0.2 A g−1, 0.5 A g−1, 1 A g−1, 2 A g−1, 5 A g−1 and 10 A g−1. (c) Comparison of the energy density and power density of the PZ-2OH‖Ni (OH)2 cells with those of other energy storage systems, including Ni-based batteries, lead acid batteries, zinc-ion batteries, lithium-ion batteries, and supercapacitors. (d) Comparison of the cell discharge voltages of organic electrode materials in acid electrolytes, mild electrolytes, and alkaline electrolytes. (e) The GCD curves of the PZ-2OH‖Ni(OH)2 full cell in the temperature range of −30 °C to 20 °C and a current density of 1 A g−1. (f) Cycle life at a current density of 0.5 A g−1. (g) Comprehensive performance evaluation, including the price, energy density, voltage, toxicity and stability, between the conventional Ni-based batteries (Zn‖Ni, MH‖Ni, Cd‖Ni) and PZ-2OH‖Ni (OH)2. (The price is calculated according to the anode material; nontoxic indicators are graded from 1 to 5, and smaller non-toxic data mean more toxic.) | ||
To explore the charge storage mechanism, ex situ XPS and in situ Raman tests were used to analyze the structural changes of the PZ-2OH electrode in the labeled states during charge–discharge (Fig. 4a). Ex situ XPS spectra revealed the ion coordination/de-coordination mechanisms of the PZ-2OH anode at the pristine, full charge, and full discharge states. We fit the peak of the high-resolution N 1s spectra and the results show that the high-resolution N 1s spectrum of the pristine electrode could be divided into two peaks at 398.5 and 399.8 eV corresponding to conjugated (sp2) –N and non-conjugated (sp3) –NH– groups, respectively (Fig. 4c). During the charging process, the peaks of –N weakened gradually. In contrast, a new peak emerged at 397.84 eV ascribed to a newly formed K–N single bond when charged to 1.6 V, implying the formation of a C–N–K group via the interaction between K+ and C–N−. During the discharging process, the peak intensity of the CN double bond gradually enhanced and recovered to its pristine state, and simultaneously the peak of the K–N single bond gradually reduced. Therefore, potassium ions in the electrolyte are more likely coordinating ions, which will inevitably coordinate with C–N−. The reaction progress is monitored using the in situ Raman spectra (Fig. 4d), and the typical Raman peak shift of the CN bond is further shown in Fig. 4e. The absorption peak at 1678 cm−1 is attributed to CN stretching,19 which greatly weakens and disappears after the battery is charged to 1.6 V. A reverse evolution occurs when discharged to 0.8 V, demonstrating that the coordination/non-coordination behaviors are highly reversible. Combining ex situ XPS and the in situ Raman test characterization, it is found that the charge/discharge storage behavior of PZ-2OH is based on the n-type doping reaction (Fig. S18, ESI†). During the initial charging process, the PZ-2OH anode was reduced by accepting the delocalized electron and forming a negative radical. Meanwhile, K+ was inserted into PZ-2OH to ensure the electroneutrality. During the subsequent charging process, the negative PZ-2OH anode was reduced to the neutral state by losing the electron. In addition, the reduced states of the PZ-2OH anode were also monitored by ex situ FTIR (Fig. 4f) after different cycles. No obvious chemical decomposition was detected except the slight changes for the initial few cycles, indicating good chemical stability in strong alkaline solutions, which ensured the low capacity-fade rates during long cycling.
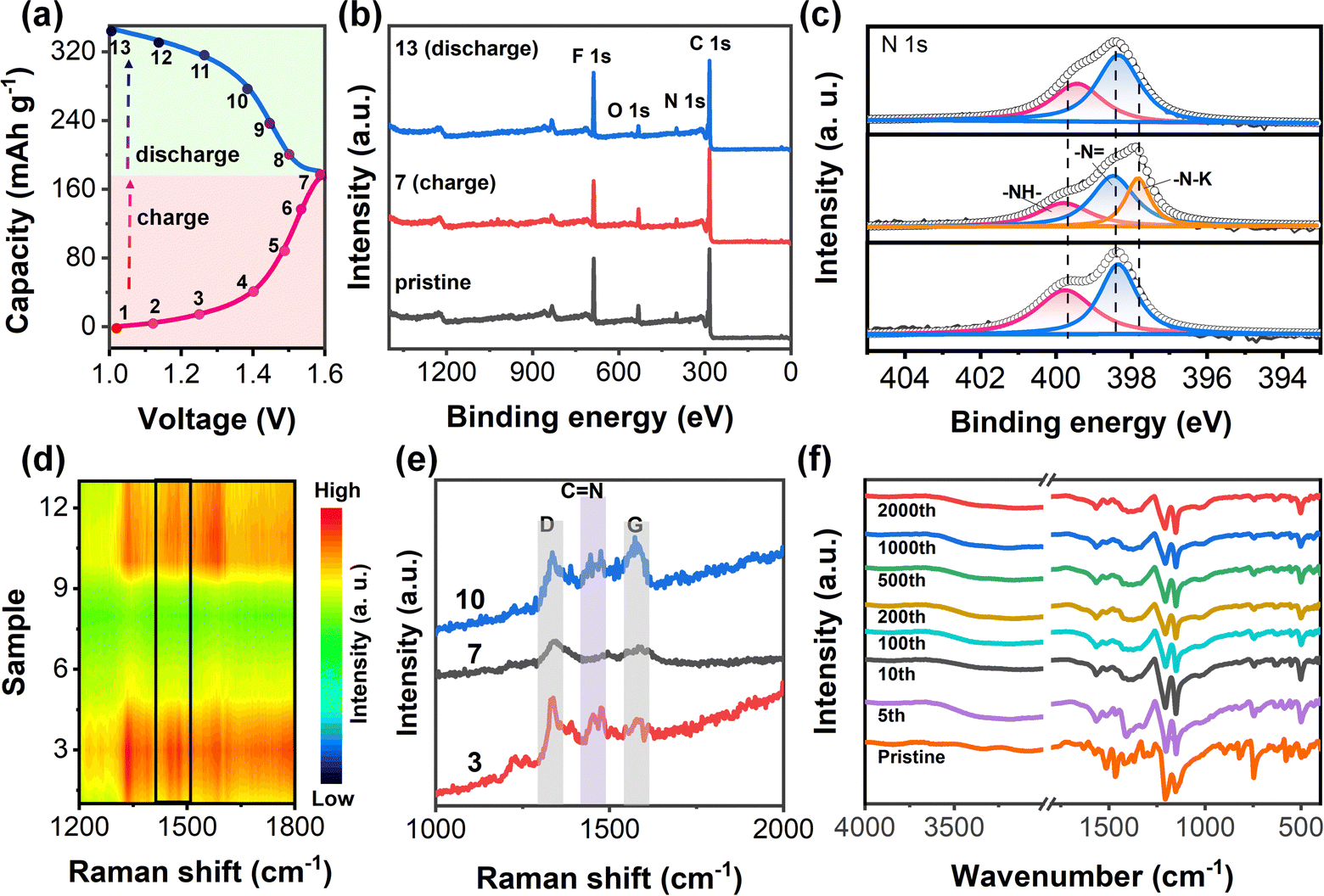 | ||
| Fig. 4 Ion coordination mechanism of PZ-2OH electrodes. (a) The charge/discharge curve of the PZ-2OH anode at a current density of 0.5 A g−1. Ex situ XPS (b) full spectra and (c) High resolution N 1s spectra at selected points. (d) In situ Raman spectra of PZ-2OH during the process of charge and discharge. (e) Typical Raman spectra of PZ-2OH at annotated points. (f) Ex situ FTIR of PZ-2OH after different cycles. | ||
To further verify the adaptability of PZ-2OH under alkaline conditions, the PZ-2OH‖air full battery was assembled (Fig. 5). The CV curves of the PZ-2OH‖air full cell showed typical oxidation and reduction peaks at 0.8 and 1.5 V (Fig. S30, ESI†), which confirms the possibility of PZ-2OH being used as an anode for air cells under alkaline conditions. The GCD curves and rate capabilities of the PZ-2OH‖air battery are shown in Fig. 5a and b, respectively. The cell delivered specific capacities of 181.3, 158.9, 136.7, 104.2, 72.9 and 52.3 mA h g−1 at 0.2, 0.5, 1, 2, 5 and 10 A g−1, respectively, based on the PZ-2OH mass, indicating the excellent rate performance. It is worth noting that the rate performance is poorer than that of the PZ-2OH half battery (Fig. 2a), which is caused by the limited catalytic rate of the air cathode at a high current. Moreover, the cycle performance of the PZ-2OH‖air full cell shows a high capacity retention of ∼65.8% in the initial 400 cycles, as demonstrated in Fig. 5c. A noticeable drop in capacity occurred as the cycle continued, which was caused by electrolyte loss in the open system. After the electrolyte was refilled into the battery, the capacity was immediately restored, suggesting that the reduction in capacity was mainly related to the loss of the electrolyte and decrease of ion transportation. The capacity of the full cell remains 80.4 mA h g−1 after 1000 cycles. Our results evidence that the developed PZ-2OH can work effectively in alkaline electrolytes and support various alkaline batteries to deliver superior energy/power density and rate capacity.
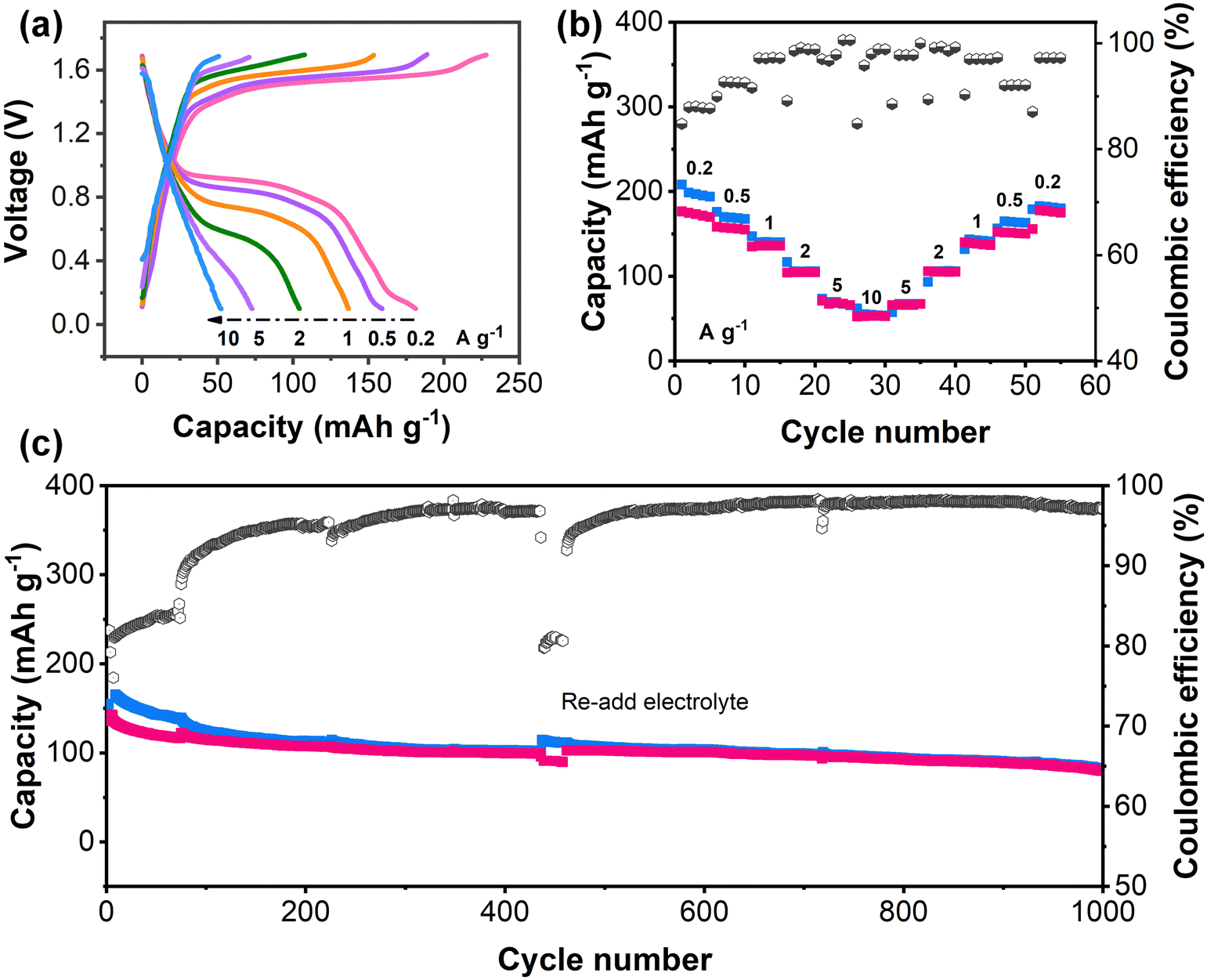 | ||
| Fig. 5 Electrochemical performance of the PZ-2OH‖air battery. (a) Typical GCD curves and (b) rate performance at a range of current densities. (c) Cycling stability at 0.5 A g−1. | ||
Conclusion
In this study, three kinds of phenazine derivatives (PZ, PZ-OH, and PZ-2OH) with different numbers of hydroxyl groups were synthesized as the anode materials for ANABs. The effects of hydroxyl groups on phenazine electrochemical properties were explored, which discloses a strong structure–electrochemical activity correlation. The introduced hydroxyl groups to phenazine acting as electron donor groups could remarkably lower the discharge potential from −0.78 V (PZ) to −1.07 V (PZ-2OH). Meanwhile, the phenazine derivatives with hydroxyl groups provided a fast charge transport channel through intermolecular interactions of hydroxyl functional groups, which reduced the charge-transfer impedance and delivered superior rate performance compared with phenazine. As a result, the PZ-2OH‖Ni(OH)2 battery shows excellent electrochemical performances, including a high reversible capacity of 178 mA h g−1 at 0.2 A g−1, a high power density of 26.2 KW kg−1 at 10 A g−1, and superior stable cyclability over 9000 cycles. In addition, the successful preparation of a PZ-2OH‖Air full cell further demonstrated the universal applicability of PZ-2OH materials in alkaline batteries. The present work demonstrates a wealth of opportunities for designing phenazine derivative anode materials to facilitate the exploration of high-performance ANABs.Author contributions
Conceptualization: H. C., D. Z., and C. Y. Z.; methodology: H. C., D. Z., Z. W., J. Z., and C. Y. Z.; investigation: H. C., D. Z., P. L., C. L.,Y. H., and R. Z.; computation: H. C.; writing – original draft: H. C.; writing – review and editing: H. C., D. Z., X. W., X. J., S. B. and C. Y. Z.; and D. Z. and C. Y. Z. jointly supervised this work.Conflicts of interest
The author declares no competing interests.Acknowledgements
This research was supported by the National Key R&D Program of China under Project 2019YFA0705104 and was supported in part by InnoHK Project on [Project 1.4 – Flexible and Stretchable Technologies (FAST) for monitoring of CVD risk factors: Soft Battery and self-powered, flexible medical devices] at Hong Kong Centre for Cerebro-cardiovascular Health Engineering (COCHE).References
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler, M. F. Burz, J. W. Long and D. R. Rolison, Science, 2017, 356, 415–418 CrossRef CAS.
- T. Sakai, I. Uehara and H. Ishikawa, J. Alloys Compd., 1999, 293, 762–769 CrossRef.
- T. J. Sun, C. Liu, J. Y. Wang, Q. S. Nian, Y. Z. Feng, Y. Zhang, Z. L. Tao and J. Chen, Nano Res., 2020, 13, 676–683 CrossRef CAS.
- W. Zhou, D. Zhu, J. He, J. Li, H. Chen, Y. Chen and D. Chao, Energy Environ. Sci., 2020, 13, 4157–4167 RSC.
- F. Wang, J. Xie, D. Zheng, F. Yang, H. Zhang and X. Lu, Adv. Mater., 2022, 34, 2200085 CrossRef CAS PubMed.
- L. Jiang, D. Dong and Y.-C. Lu, Nano Res. Energy, 2022, 1, e9120003 CrossRef.
- S. Chen, T. Wang, L. Ma, B. Zhou, J. Wu, D. Zhu, Y. Y. Li, J. Fan and C. Zhi, Chem, 2022, 9, 497–510 Search PubMed.
- M. J. D’Ambrose, D. E. Turney, G. G. Yadav, M. Nyce and S. Banerjee, ACS Appl. Energy Mater., 2021, 4, 3381–3392 CrossRef.
- M. Freitas and S. Rosalém, J. Power Sources, 2005, 139, 366–370 CrossRef CAS.
- F. Feng, M. Geng and D. Northwood, Int. J. Hydrogen Energy, 2001, 26, 725–734 CrossRef CAS.
- S. Chen, C. Peng, D. Xue, L. Ma and C. Zhi, Angew. Chem., 2022, 134, e202212767 CrossRef.
- J. S. Ko, A. B. Geltmacher, B. J. Hopkins, D. R. Rolison, J. W. Long and J. F. Parker, ACS Appl. Energy Mater., 2018, 2, 212–216 CrossRef.
- S. Yang, H. Lv, Y. Wang, X. Guo, L. Zhao, H. Li and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202209794 CrossRef CAS PubMed.
- J. Kim, S. Ko, C. Noh, H. Kim, S. Lee, D. Kim, H. Park, G. Kwon, G. Son and J. W. Ko, Angew. Chem., 2019, 131, 16920–16925 CrossRef.
- G. Liang, X. Li, Y. Wang, S. Yang, Z. Huang, Q. Yang, D. Wang, B. Dong, M. Zhu and C. Zhi, Nano Res. Energy, 2022, 1, e9120002 CrossRef.
- C. Luo, O. Borodin, X. Ji, S. Hou, K. J. Gaskell, X. Fan, J. Chen, T. Deng, R. Wang and J. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2004–2009 CrossRef CAS.
- H. Cui, L. Ma, Z. Huang, Z. Chen and C. Zhi, SmartMat, 2022, 3, 565–581 CrossRef CAS.
- Y. Li, Y. Lu, Y. Ni, S. Zheng, Z. Yan, K. Zhang, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 8066–8072 CrossRef CAS PubMed.
- S. Zhang, S. Long, H. Li and Q. Xu, Chem. Eng. J., 2020, 400, 125898 CrossRef CAS.
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401 CrossRef CAS.
- G. Dai, Y. Liu, Z. Niu, P. He, Y. Zhao, X. Zhang and H. Zhou, Matter, 2019, 1, 945–958 CrossRef.
- L. Miao, L. Liu, K. Zhang and J. Chen, ChemSusChem, 2020, 13, 2337–2344 CrossRef CAS PubMed.
- Z. Chen, H. Cui, Y. Hou, X. Wang, X. Jin, A. Chen, Q. Yang, D. Wang, Z. Huang and C. Zhi, Chem, 2022, 8, 2204–2216 CAS.
- A. Hollas, X. Wei, V. Murugesan, Z. Nie, B. Li, D. Reed, J. Liu, V. Sprenkle and W. Wang, Nat. Energy, 2018, 3, 508–514 CrossRef CAS.
- C. Wang, X. Li, B. Yu, Y. Wang, Z. Yang, H. Wang, H. Lin, J. Ma, G. Li and Z. Jin, ACS Energy Lett., 2020, 5, 411–417 CrossRef CAS.
- C. Zhang, W. Ma, C. Han, L.-W. Luo, A. Daniyar, S. Xiang, X. Wu, X. Ji and J.-X. Jiang, Energy Environ. Sci., 2021, 14, 462–472 RSC.
- M. Marques, A. Mamede, A. Vassalo, C. Makhoul, E. Cunha, D. Gonçalves, S. Parker and L. Batista de Carvalho, Sci. Rep., 2018, 8, 1–13 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong and J. Fan, Angew. Chem., 2022, 134, e202203453 CrossRef.
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., 2020, 132, 4950–4954 CrossRef.
- L. Ma, S. Chen, D. Wang, Q. Yang, F. Mo, G. Liang, N. Li, H. Zhang, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1803046 CrossRef.
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS PubMed.
- T. Liu, K. C. Kim, B. Lee, Z. Chen, S. Noda, S. S. Jang and S. W. Lee, Energy Environ. Sci., 2017, 10, 205–215 RSC.
- Y. L. Liang, Y. Jing, S. Gheytani, K. Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS PubMed.
- Y. Li, L. Liu, C. Liu, Y. Lu, R. Shi, F. Li and J. Chen, Chem, 2019, 5, 2159–2170 CAS.
- Z. Guo, J. Huang, X. Dong, Y. Xia, L. Yan, Z. Wang and Y. Wang, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- F. Yue, Z. Tie, S. Deng, S. Wang, M. Yang and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 13882–13886 CrossRef CAS PubMed.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- Z. Lin, H.-Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu and H. Li, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- S. Bi, S. Wang, F. Yue, Z. Tie and Z. Niu, Nat. Commun., 2021, 12, 6991 CrossRef CAS PubMed.
- C. Liu, T. Ma, K. Xia, X. Hou, Q. Nian, Y. Cai and J. Liang, Sustainable Energy Fuels, 2020, 4, 132–137 RSC.
- X. Jia, C. Liu, Z. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01212c |
| This journal is © The Royal Society of Chemistry 2024 |