
A semi-conductive organic cathode material enabled by extended conjugation for rechargeable aqueous zinc batteries†
Lu
Lin
,
Zirui
Lin
,
Jiaqi
Zhu
,
Kuo
Wang
,
Wanlong
Wu
,
Tong
Qiu
and
Xiaoqi
Sun
 *
*
Department of Chemistry, Northeastern University, Shenyang, 110819, China. E-mail: sunxiaoqi@mail.neu.edu.cn
First published on 16th December 2022
Abstract
Quinone cathode materials show great promise for aqueous zinc batteries. However, the poor electrical conductivity of organic compounds calls for the addition of high percentages of conductive agents during electrode preparation, usually ≥30%. Herein, we synthesize a semi-conductive piperazine-linked quinone of 2,3,7,8-tetraamino-5,10-dihydrophenazine-1,4,6,9-tetraone (TDT) for zinc batteries. Theoretical calculations suggest the extended conjugation among the π-electrons on the quinone rings and p-electrons on nitrogen sites in the structure. This allows electron delocalization throughout the entire molecule, and a small band gap of 1.5 eV is revealed by theoretical calculations and solid-state UV-vis spectroscopy. Besides, electron paramagnetic resonance demonstrates the formation of cationic radicals on the nitrogen of the middle piperazine ring. The radical is also stabilized by the extended conjugated system and further enhances the electrical conductivity. Thanks to the above factors, TDT exhibits a good electrical conductivity on the order of 0.1–1 mS cm−1. In aqueous zinc batteries, the TDT cathode with 10% carbon delivers a high capacity of 369 mA h g−1 at 0.2 A g−1 and retains 182 mA h g−1 at 10 A g−1. In situ UV-vis analysis further reveals the insolubility of TDT at all charged/discharged states, which ensures stable cycling for 3000 cycles.
Broader contextWith the requirements to minimize fossil fuel consumption, advanced rechargeable battery systems are in urgent demand. Aqueous zinc batteries, with high safety and low cost, have attracted extensive attentions as one of the most promising “beyond Li-ion” technologies for grid-level energy storage. Electrochemically active organic compounds are desirable cathode candidates thanks to their resource abundance, environmental friendliness and structural diversity. Nevertheless, their electrochemical performance is seriously limited by their poor electrical conductivities. A large amount of carbon additives, typically ≥30%, is required for electrode preparation, in order to provide satisfactory electron transport. This sacrifices the practical capacity and energy density of the system. Herein, we develop a semi-conductive small molecule quinone compound of 2,3,7,8-tetraamino-5,10-dihydrophenazine-1,4,6,9-tetraone (TDT) for aqueous zinc batteries. The molecule exhibits extended conjugation in the structure, which ensures electron delocalization and reduces the energy gap. A cation radical also forms on the nitrogen site. It is stabilized in the conjugated system and further enhances conductivity. The above factors endow the TDT compound with a semi-conductive nature with the electrical conductivity on the order of 0.1–1 mS cm−1. By applying much reduced carbon content of 10%, the TDT cathode delivers a high capacity of 369 mA h g−1 at 0.2 A g−1 and retains 182 mA h g−1 at 10 A g−1. Stable cycling is also realized for 3000 cycles. Our work proposes a potential path to fundamentally provide fast electron transport in organic electrode materials without relying on a high amount of carbon additives, which would encourage their applications in high-performance aqueous zinc batteries. |
Introduction
The “beyond Li-ion” technologies have gained great interest to break through the limitations of Li-ion batteries.1–4 Rechargeable aqueous zinc batteries present important advantages of low cost, high safety and high ionic conductivity with the application of water-based electrolytes. In addition, the zinc metal anode delivers high specific capacity (820 mA h g−1 and 5855 mA h cm−3), low redox potential (−0.76 V vs. standard hydrogen electrode) and good stability in water.5–8 Great efforts have been focused on developing cathode materials for aqueous zinc batteries.9–14 Organic compounds with carbonyl functional groups show promising electrochemical activities.15–17 The molecular engineering of substituent groups and tunnel sizes in organic structures provides feasibilities to further adjust the electrochemical behaviors.18,19 For instance, Chen et al. reported a calix[4]quinone (C4Q) molecule with open bowl structures as the cathode for zinc batteries, delivering a high capacity of 335 mA h g−1 at 20 mA g−1.20 Stoddart and co-workers proposed a triangular phenanthrenequinone-based macrocycle (PQ-Δ) with a layered structure, which allows the insertion of hydrated Zn2+ and displays a high capacity of 225 mA h g−1 at 30 mA g−1.21 Xia and co-workers proposed a pyrene-4,5,9,10-tetraone (PTO) cathode. It shows 336 mA h g−1 capacity at 40 mA g−1 with fast kinetics.22An important limitation of organic compounds, however, is the poor electrical conductivity. As a result, organic electrode preparation requires the addition of a large amount of carbon agents, typically ≥30%.20–28 This limits the practical capacity and energy density of the system. Studies have been carried out on the composite design of organic compounds with conductive materials, such as reduced graphene oxide.29 On the other hand, the enhancement of the conductivity of organic compounds themselves would fundamentally ensure the high electrochemical activity of the cathode materials. In theory, structures with extended π-electron conjugation would realize the delocalization of electrons and reduction of the energy gap between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO), providing a path to enhance the electrical conductivity.30,31 Besides, the introduction of radicals would also improve the conductivity.32,33
Following these rules, we herein synthesize a piperazine-linked quinone compound of 2,3,7,8-tetraamino-5,10-dihydrophenazine-1,4,6,9-tetraone (TDT) for aqueous zinc batteries. It contains two quinone rings linked by a piperazine ring in the middle. Theoretical calculations reveal the extended conjugation among π-electrons on quinone rings and p-electrons on nitrogen sites, so that electrons are delocalized throughout the entire molecule. The molecule possesses a small energy gap of 1.5 eV. Besides, electron paramagnetic resonance (EPR) analysis demonstrates the formation of a cationic radical on the nitrogen of the piperazine ring. The radical is stabilized by the conjugated structure and further enhances the electrical conductivity. As a result, TDT exhibits a semi-conductive nature with the electrical conductivity on the order of 0.1–1 mS cm−1. This enables the cathode material containing low carbon additive of 10% to show excellent electrochemical activity in aqueous zinc batteries. The TDT cathode delivers a high capacity of 369 mA h g−1 at 0.2 A g−1 and retains 182 mA h g−1 with the increase of the current density to 10 A g−1. TDT is also insoluble at all charged/discharged states as revealed by in situ UV-vis measurements. This ensures stable cycling for 3000 cycles.
Results and discussion
TDT is synthesized by the condensation reaction between two tetraamino-p-benzoquinone (TABQ) molecules. Fig. 1a shows the solid-state 13C nuclear magnetic resonance (NMR) of the obtained material. The chemical shifts (δ) at 174 ppm, 138 ppm, and 111 ppm correspond to the carbons on –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –C–NH2, and –C–NH– of the TDT structure, respectively. The intensity of the carbon on the carbonyl is usually low, as has also been experienced in previously reported compounds.34 The above carbon environments are also identified in the C 1s X-ray photoelectron spectroscopy (XPS, Fig. 1b). Besides, the N 1s XPS is fitted with –NH2 and –NH– at 399.1 eV and 400.7 eV, respectively (Fig. 1c). The O 1s XPS shows the dominant contribution from oxygen in carbonyls at 532.1 eV (Fig. 1d).35,36Fig. 1e shows the Fourier transform infrared (FT-IR) spectrum. The band between 3400 cm−1 and 3000 cm−1 results from N–H stretching vibration, and the peaks between 1400 cm−1 and 1170 cm−1 are attributed to the N–H bending vibration and C–N stretching vibration. The peaks between 1700 cm−1 and 1450 cm−1 correspond to the C–C and CC vibrations of quinone rings as well as the carbonyl groups. Table S1 (ESI†) lists the elemental composition of the as-prepared material measured by elemental analysis (EA), which agrees well with the theoretical composition of TDT. The above results confirm the successful synthesis of the TDT compound. Fig. 1f shows the scanning electron microscopy (SEM) image, presenting a short rod morphology. Thermogravimetric analysis (TGA) demonstrates the main weight loss above 300 °C (4% at 300 °C, 11% at 400 °C, 20% at 500 °C, Fig. S1, ESI†).
O, –C–NH2, and –C–NH– of the TDT structure, respectively. The intensity of the carbon on the carbonyl is usually low, as has also been experienced in previously reported compounds.34 The above carbon environments are also identified in the C 1s X-ray photoelectron spectroscopy (XPS, Fig. 1b). Besides, the N 1s XPS is fitted with –NH2 and –NH– at 399.1 eV and 400.7 eV, respectively (Fig. 1c). The O 1s XPS shows the dominant contribution from oxygen in carbonyls at 532.1 eV (Fig. 1d).35,36Fig. 1e shows the Fourier transform infrared (FT-IR) spectrum. The band between 3400 cm−1 and 3000 cm−1 results from N–H stretching vibration, and the peaks between 1400 cm−1 and 1170 cm−1 are attributed to the N–H bending vibration and C–N stretching vibration. The peaks between 1700 cm−1 and 1450 cm−1 correspond to the C–C and CC vibrations of quinone rings as well as the carbonyl groups. Table S1 (ESI†) lists the elemental composition of the as-prepared material measured by elemental analysis (EA), which agrees well with the theoretical composition of TDT. The above results confirm the successful synthesis of the TDT compound. Fig. 1f shows the scanning electron microscopy (SEM) image, presenting a short rod morphology. Thermogravimetric analysis (TGA) demonstrates the main weight loss above 300 °C (4% at 300 °C, 11% at 400 °C, 20% at 500 °C, Fig. S1, ESI†).
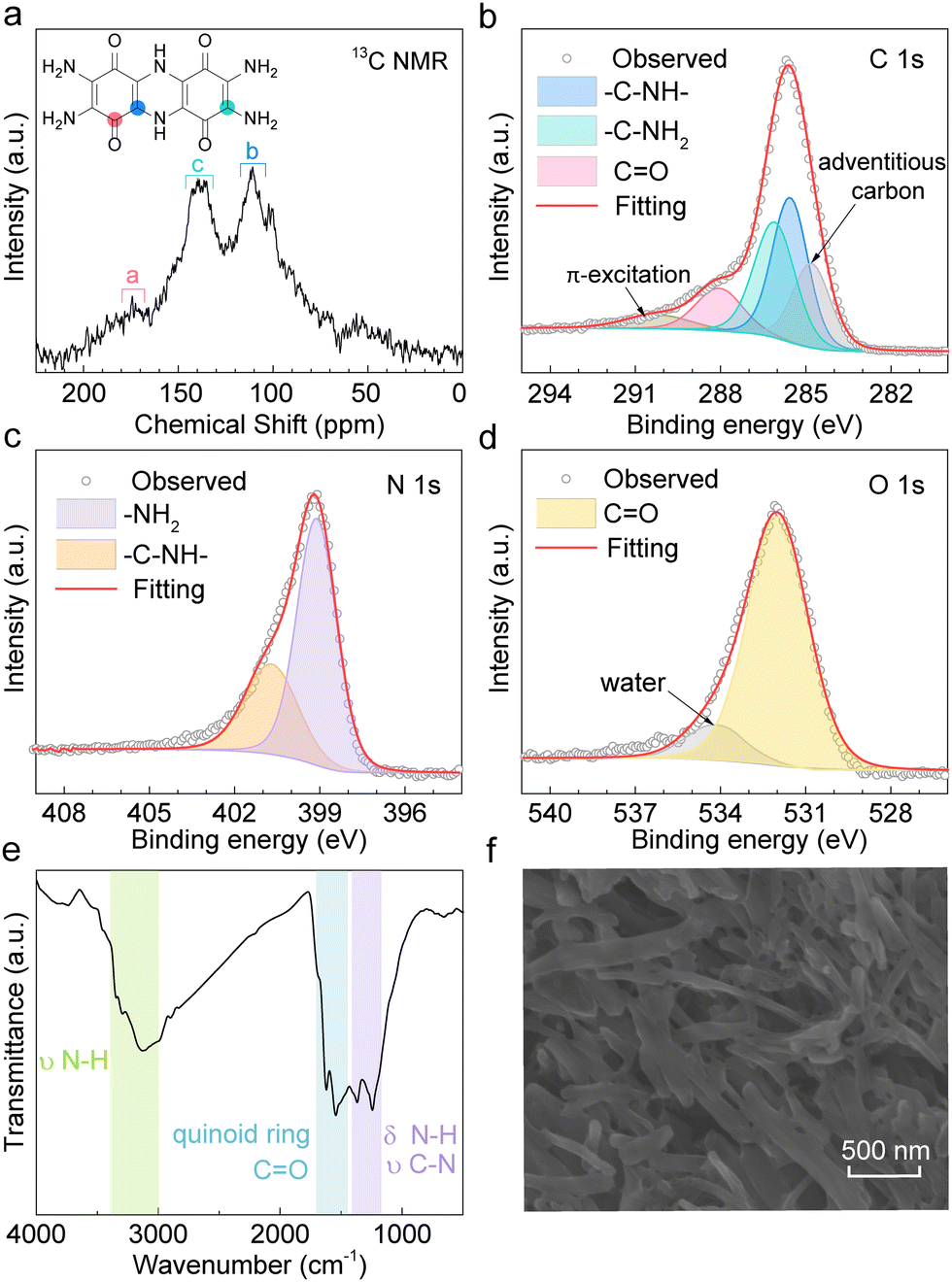 | ||
| Fig. 1 Characterizations of TDT. (a) Solid-state 13C NMR. (b) C 1s, (c) N 1s, and (d) O 1s XPS. (e) FT-IR. (f) SEM image. | ||
Density functional theory (DFT) calculations37 are carried out on the TDT molecule. The optimized structure demonstrates a highly conjugated system (Fig. 2a). The p-electrons on the nitrogen sites form extended conjugation with the π-electrons of the quinone rings, and the piperazine ring between the two quinone rings allows the delocalization of electrons through the entire molecule. Fig. 2b shows the localized orbital locator-π (LOL-π) map of TDT,38,39 which further confirms the high degree of conjugation in the structure and electron delocalization throughout the entire molecule. In theory, extended conjugations lead to the decrease of the HOMO and LUMO energy levels, with larger influence on the latter.30,31 As a result, the energy gap in the extended conjugated structures would be reduced. The detailed influence on the frontier molecular orbitals in TDT is calculated. The TABQ molecule, which does not contain the piperazine linkage and exhibits lower degree of conjugation, is applied as the comparing standard. The HOMO energy level decreases a little from −4.11 eV in TABQ to −4.65 eV in TDT, whereas a larger decrease from −2.13 eV to −3.15 eV is shown in the LUMO energy levels (Fig. 2c). This results in a reduced energy gap from 1.98 eV in TABQ to 1.50 eV in TDT. The direct band gaps are further calculated from solid-state UV-vis spectra (Fig. 2d).40 They are estimated to be 1.5 eV and 2.0 eV for TDT and TABQ, respectively, in good agreement with theoretical calculations.
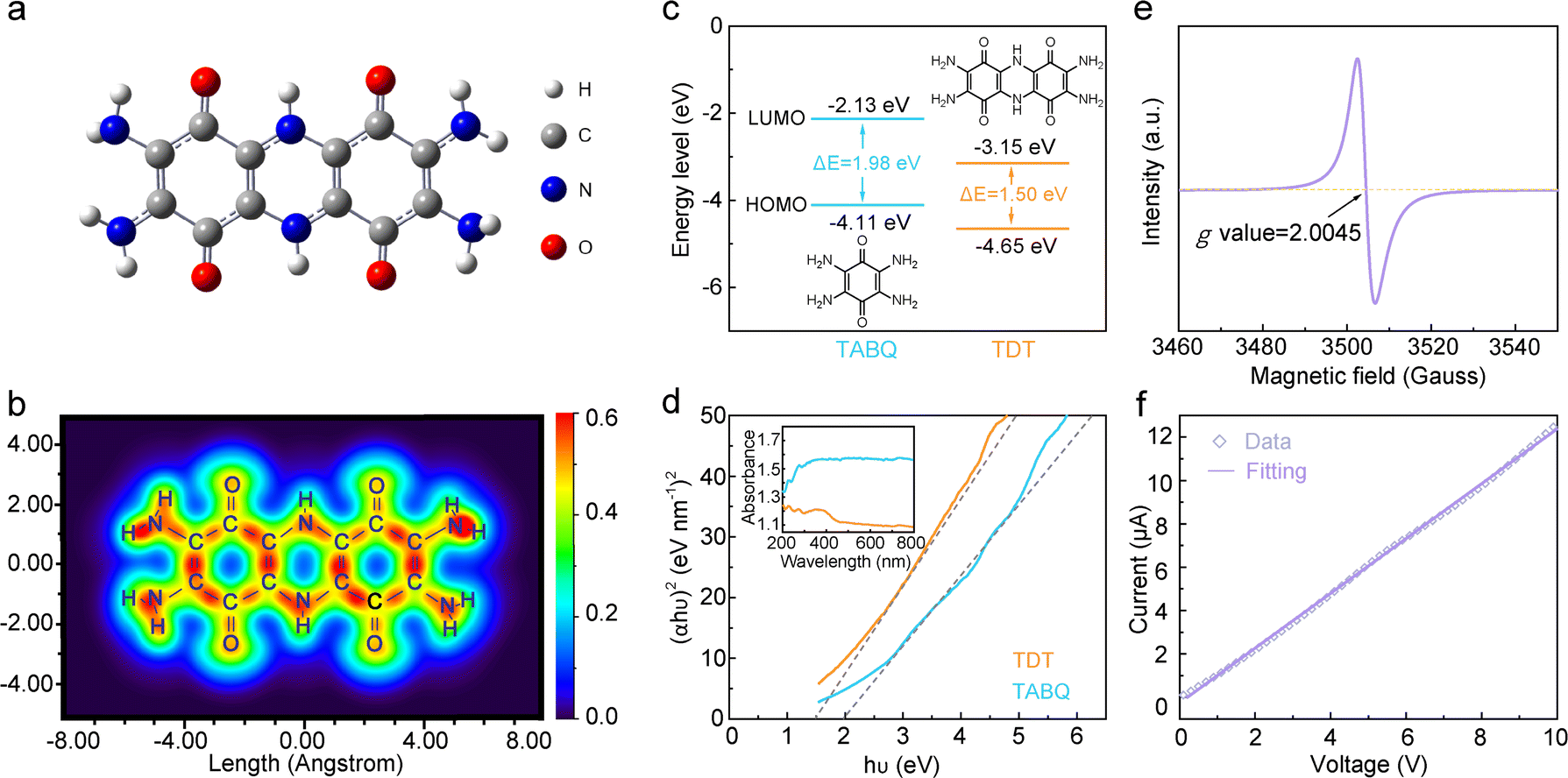 | ||
| Fig. 2 (a) The optimized structure and (b) LOL-π color-filled map of the TDT molecule. (c) Calculated HOMO and LUMO energy levels and (d) Tauc plots of TDT and TABQ (inset shows the UV-vis spectra). (e) EPR spectrum of TDT. (f) I–V plot of a TDT pellet. | ||
Fig. 2e shows the EPR spectrum of TDT, presenting an obvious signal at 3504 Gauss with the g value of 2.0045. This is typical for cationic radicals formed on nitrogen sites.32,41,42 The formation of a cationic radical generates an electron hole. The electron hole would realize high mobility in the conjugated structure under an external electric field, which enhances the electrical conductivity of the material. Meanwhile, the spin density of the cationic radical is delocalized in the conjugated system, so that the radical exhibits good stability.32,43–46 Overall, the nitrogen cation radical is stabilized by the extended conjugated structure and helps to enhance the electrical conductivity. Together with the electron delocalization and small band gap in TDT shown earlier, a good electrical conductivity can be expected for the compound.31,36
The electrical conductivity of TDT is firstly estimated by a two-probe system.47Fig. 2f shows the I–V plot of a TDT pellet. The electrical conductivity (σ) is determined to be 0.9 mS cm−1 according to the equation of σ = L/(RS), where R, S and L represent the resistance, surface area and thickness of the pellet. The electrical conductivity is further measured by a four-probe system at different pressures (Table S2, ESI†). TDT exhibits the conductivity of 0.56 mS cm−1 at 1 MPa, which is on the same order of magnitude as the two-probe measurement. With the increase of the pressure to 3 MPa, the conductivity increases to 12.3 mS cm−1. Notably, the electrical conductivity of TDT is in the range of semi-conductors (10−5–106 mS cm−1).48 This suggests the potential facile electron transport in the material when applied as an electrode.
With the semi-conductive nature of TDT, the cathode material is prepared by mixing TDT with carbon and binder at the weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. This carbon content is much lower than previous studies of organic electrodes where ≥30% carbon was applied.20–28 The electrochemical performance of the TDT cathode is tested with a Zn anode and 1 M ZnSO4 aqueous electrolyte. Fig. 3a shows the galvanostatic charge–discharge (GCD) curves at various current densities. At 0.2 A g−1, TDT delivers a high capacity of 369 mA h g−1. The capacity contribution from the carbon, binder and substrate is only around 15 mA h gTDT−1 (calculated based on the loading of TDT, Fig. S2, ESI†). The electrode also presents excellent rate capability. With 50 times increase of current density to 10 A g−1, it still achieves a high capacity of 182 mA h g−1. In order to take more benefit of the good conductivity of the compound, we further reduce the carbon content to 5% in the TDT cathode (9:0.5:0.5 ratio). It still delivers good capacities of 291, 241 and 172 mA h g−1 at 0.2 A g−1, 0.5 A g−1 and 1 A g−1, respectively (Fig. S3a, ESI†). The electrochemical performance is compared with the previously reported organic cathodes containing ≥30% carbon additives (Fig. 3b).20–28 The TDT cathode with a low carbon content of 10% provides superior capacities, and the performance is still among the best upon further reduction of the carbon content to 5%. It also presents a good advantage when considering the percentage of active material utilization (Fig. S4, ESI†). We further increase the active material loading from 1.2 mg cm−2 discussed above to 10 mg cm−2 (8:1:1 ratio). It retains high capacities of 345 mA h g−1, 307 mA h g−1 and 256 mA h g−1 at 0.2 A g−1, 0.5 A g−1 and 1 A g−1, respectively (Fig. S3b, ESI†). The excellent performance of the TDT electrode with low carbon content is attributed to its good electrical conductivity. For example, the electrical conductivities of commercially available PTO and tetrachloro-p-benzoquinone (TCBQ) are 3–6 orders of magnitude lower (Table S2, ESI†), together with larger calculated HOMO–LUMO gaps of 3.50 eV and 3.47 eV, respectively. Meanwhile, TDT delivers an average redox voltage of 0.78 V, which is among the range of reported n-type organic cathodes.20–28Fig. 3c shows the electrochemical impedance spectroscopy (EIS) at different potentials along discharge and charge. The intercepts at the x-axis of the Nyquist plots, representing the series resistance (Rs), are below 2 Ω. The small values again verify the good conductivity of TDT, which ensures its high capacity and excellent rate capability. The radii of the semi-circles in the medium-high frequency region, which correspond to the charge-transfer resistance (Rct), are similar at different states.
:1:1. This carbon content is much lower than previous studies of organic electrodes where ≥30% carbon was applied.20–28 The electrochemical performance of the TDT cathode is tested with a Zn anode and 1 M ZnSO4 aqueous electrolyte. Fig. 3a shows the galvanostatic charge–discharge (GCD) curves at various current densities. At 0.2 A g−1, TDT delivers a high capacity of 369 mA h g−1. The capacity contribution from the carbon, binder and substrate is only around 15 mA h gTDT−1 (calculated based on the loading of TDT, Fig. S2, ESI†). The electrode also presents excellent rate capability. With 50 times increase of current density to 10 A g−1, it still achieves a high capacity of 182 mA h g−1. In order to take more benefit of the good conductivity of the compound, we further reduce the carbon content to 5% in the TDT cathode (9:0.5:0.5 ratio). It still delivers good capacities of 291, 241 and 172 mA h g−1 at 0.2 A g−1, 0.5 A g−1 and 1 A g−1, respectively (Fig. S3a, ESI†). The electrochemical performance is compared with the previously reported organic cathodes containing ≥30% carbon additives (Fig. 3b).20–28 The TDT cathode with a low carbon content of 10% provides superior capacities, and the performance is still among the best upon further reduction of the carbon content to 5%. It also presents a good advantage when considering the percentage of active material utilization (Fig. S4, ESI†). We further increase the active material loading from 1.2 mg cm−2 discussed above to 10 mg cm−2 (8:1:1 ratio). It retains high capacities of 345 mA h g−1, 307 mA h g−1 and 256 mA h g−1 at 0.2 A g−1, 0.5 A g−1 and 1 A g−1, respectively (Fig. S3b, ESI†). The excellent performance of the TDT electrode with low carbon content is attributed to its good electrical conductivity. For example, the electrical conductivities of commercially available PTO and tetrachloro-p-benzoquinone (TCBQ) are 3–6 orders of magnitude lower (Table S2, ESI†), together with larger calculated HOMO–LUMO gaps of 3.50 eV and 3.47 eV, respectively. Meanwhile, TDT delivers an average redox voltage of 0.78 V, which is among the range of reported n-type organic cathodes.20–28Fig. 3c shows the electrochemical impedance spectroscopy (EIS) at different potentials along discharge and charge. The intercepts at the x-axis of the Nyquist plots, representing the series resistance (Rs), are below 2 Ω. The small values again verify the good conductivity of TDT, which ensures its high capacity and excellent rate capability. The radii of the semi-circles in the medium-high frequency region, which correspond to the charge-transfer resistance (Rct), are similar at different states.
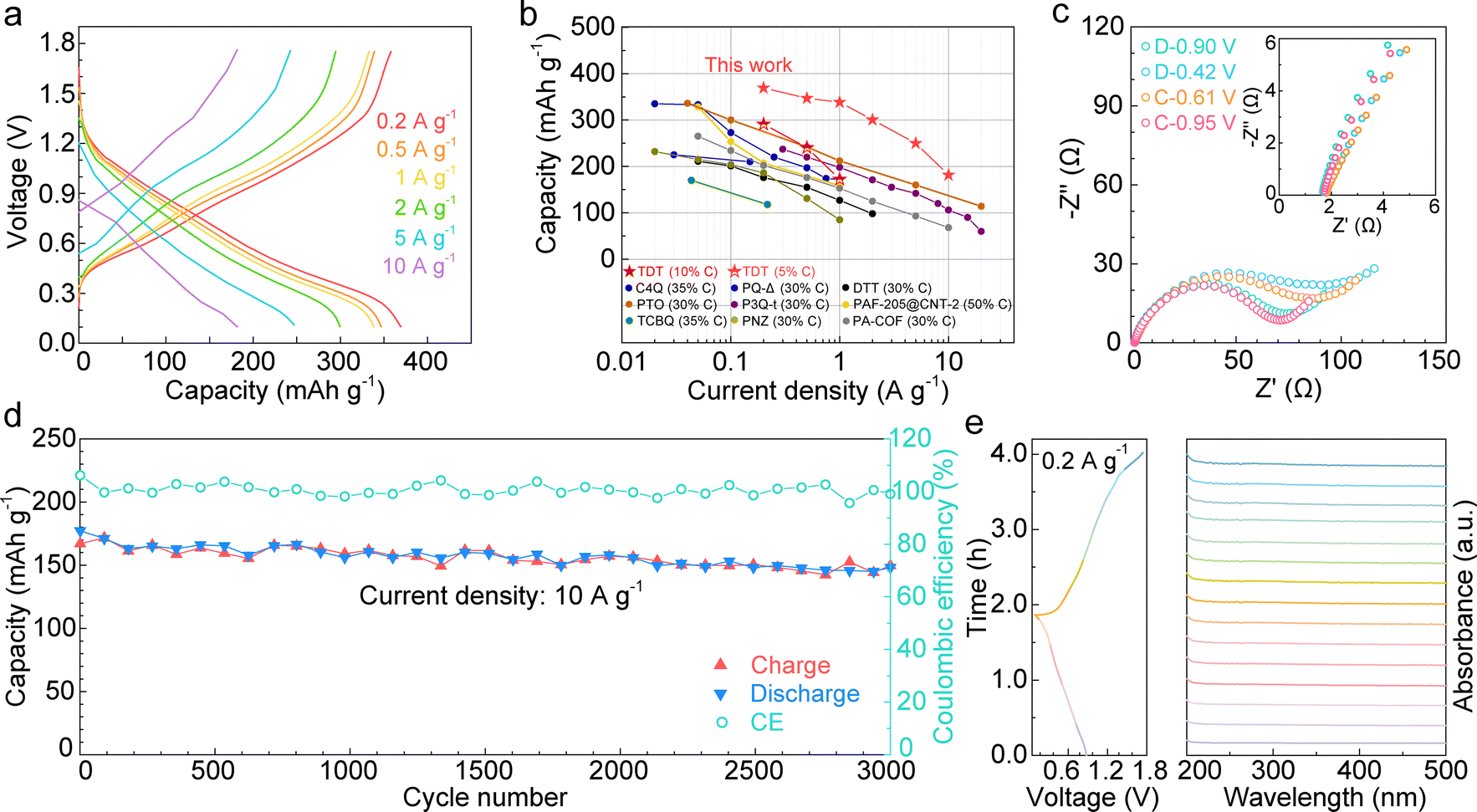 | ||
| Fig. 3 Electrochemical performance of the TDT cathode with 10% carbon in aqueous zinc batteries. (a) GCD curves under various current densities. (b) Comparison with previously reported organic electrodes containing ≥30% carbon additives.20–28 (c) Nyquist plots at different potentials along discharge and charge (inset shows the high frequency region). (d) Long-term cycling stability at 10 A g−1 for 3000 cycles. (e) In situ UV-vis analysis of the electrolyte during discharge and charge showing the insolubility of TDT. | ||
Long-term cycling of TDT is carried out at 10 A g−1 (Fig. 3d). The cathode exhibits excellent stability, with a high capacity of 148 mA h g−1 retained after 3000 cycles. The potential dissolution of the TDT active material is monitored by in situ UV-vis spectroscopy tests on the electrolyte during cycling (Fig. 3e). Negligible n–π* or π–π* absorption peaks from TDT are observed along the entire cycle, demonstrating the insolubility of TDT at all charged/discharged states. This ensures the excellent stability of the cathode over long-term cycling. FT-IR and SEM are carried out on the TDT cathode after 3000 cycles (Fig. S5, ESI†). The characteristic FT-IR peaks from the TDT material are retained. The peak at 1157 cm−1 is attributed to C–F vibration from the polytetrafluoroethylene (PTFE) binder, which is also noted in the un-cycled electrode. The rod morphology of the active material is preserved. This confirms the good stability of TDT upon electrochemical cycling.
The reaction kinetics of TDT were studied by cyclic voltammetry (CV) at various scan rates from 0.2 to 1.0 mV s−1.49–51 As shown in Fig. 4a, the overpotential of the two redox pairs in TDT only increases slightly with the increase of the scan rates. According to the relationship between the peak current i and scan rate v of i = avb, the b values are calculated to be 0.79, 0.92, 0.84, and 0.99 for the four redox peaks (Fig. 4b). The values between 0.5 and 1 suggest the combined diffusion-controlled and non-diffusion-controlled processes for energy storage. Their contributions are further separated by the equation of i = k1v (diffusion-controlled) + k2v1/2 (non-diffusion-controlled).51 This results in the non-diffusion-controlled contribution of 71.8% at 0.2 mV s−1, which increases to 85.6% at 1 mV s−1 (Fig. 4c and d). These results suggest the domination of the non-diffusion-controlled process in TDT, which ensures its high capacity and excellent rate capability.49,50
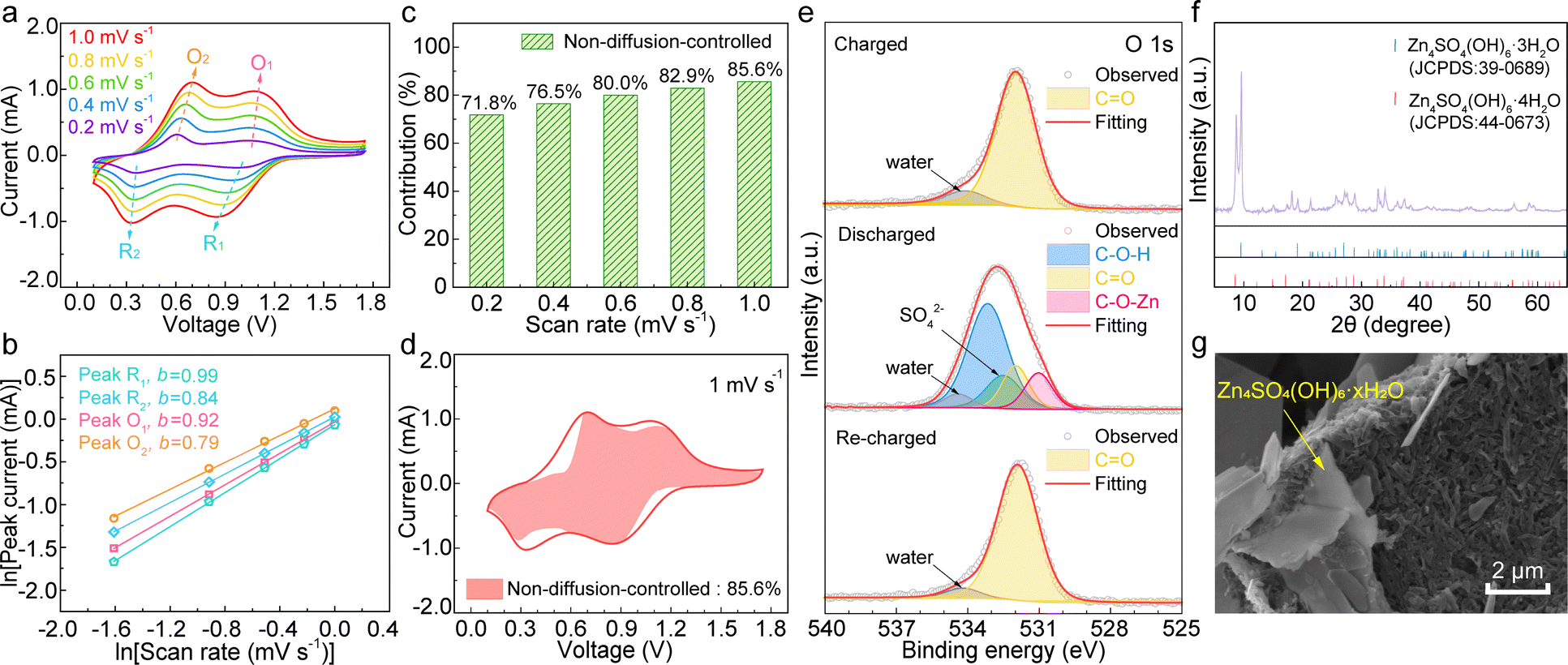 | ||
| Fig. 4 (a) CV curves of TDT at various scan rates. (b) The linear fits of ln(i) vs. ln(v) plots. (c) Non-diffusion-controlled capacity contributions at different scan rates and (d) the non-diffusion-controlled region in the CV curve at 1 mV s−1. (e) O 1s XPS fittings of TDT at different states. (f) XRD and (g) SEM image of the discharged TDT electrode. | ||
The energy storage mechanism of TDT is finally studied. Fig. 4e shows the fittings of the O 1s XPS spectra at different states. The charged spectrum is dominated by CO. Upon discharging, additional components of C–O–Zn and C–O–H appear at 531.0 eV and 533.2 eV, respectively.21 They are attributed to the reduction of the carbonyl groups on TDT together with the co-insertion of Zn2+ and H+. Besides, the SO42− signal results from Zn4SO4(OH)6·xH2O, which is also noted in the X-ray diffraction (XRD) pattern and SEM image (Fig. 4f and g). The signals in the Zn 2p XPS are from the inserted Zn2+ as well as Zn4SO4(OH)6·xH2O (Fig. S6, ESI†). The formation of zinc basic salts is associated with a local pH increase upon proton insertion into the cathode.52 Despite their insulating nature, they form as individual platelets rather than a uniform film blocking the active material. EIS at different potentials discussed earlier confirms small changes of Rs and Rct (Fig. 3c), suggesting that zinc basic salts do not hinder the reaction kinetics of the cathode. The re-charged O 1s XPS spectrum shows the reversed change. It demonstrates the fully reversible redox process of TDT in aqueous zinc cells. Galvanostatic intermittent titration technique (GITT) measurement was carried out for TDT, and the diffusion coefficients are calculated to be in the range of 10−9–10−11 cm2 s−1 (Fig. S7, ESI†). The diffusion coefficients keep decreasing upon discharge, which suggests more proton participation in the higher voltage region than in the lower one. The above analyses confirm the redox active site of the carbonyl together with the co-insertion of H+ and Zn2+ in TDT for energy storage.
Conclusion
In summary, we present a semi-conductive TDT compound as the cathode material for aqueous zinc batteries. Theoretical calculations and experimental analysis demonstrate the electron delocalization, the reduced energy gap and the formation of cation radicals in the material, which are realized by the extended conjugation within the entire molecule. The compound thus exhibits good electrical conductivity on the order of 0.1–1 mS cm−1. This enables the TDT cathode with a low carbon content of 10% to show excellent electrochemical performance. It delivers a high capacity of 369 mA h g−1 at 0.2 A g−1, and 182 mA h g−1 capacity is retained with the increase of the current density to 10 A g−1. Besides, in situ UV-vis measurements demonstrate the insolubility of TDT at all charged/discharged states, which ensures stable cycling for 3000 cycles. Mechanism studies suggest the redox processes on the carbonyl sites together with the co-storage of protons and zinc in TDT. Our work presents the synergistic approach of extended conjugation and cationic radicals to enhance the electrical conductivity of organic compounds. It fundamentally provides fast electron transport in electrode materials without relying on a high amount of carbon additives. This strategy will also be applicable for the design of other organic compounds as high-performance electrode materials in rechargeable batteries.Experimental section
Materials
TCBQ and potassium phthalimide were purchased from Aladdin Bio-Chem Technology (China). PTO was purchased from Bidepharm (China). Ketjen Black (KB) was purchased from Lion Specialty Chemicals (Japan). PTFE was purchased from Daikin Industries (Japan). Graphite foil was purchased from the SGL group (Germany). The other reagents were obtained from Sinopharm Chemical Reagent (China).Synthesis of TDT
The TABQ precursor was obtained by a previously reported method.52 Subsequently, 0.4 g of TABQ and 8.0 g of NaCl were added to an agate mortar, and 750 μL of hydrochloric acid (36–38%) was slowly added. The mixture was ground thoroughly until dry and annealed at 300 °C for 5 h under an argon atmosphere. The product was dispersed in 500 mL of 0.01 M sulfuric acid and boiled. The black precipitate was filtered and washed with deionized water and ethanol several times. [0.35 g, yield = 83%; solid-state 13C NMR (600 MHz, Magic Angle Spinning, Fig. 1a): δ (ppm): 174 (m); 138 (m); and 111 (m). EA: calculated (%) for C12H10N6O4: C 47.69, H 3.33, N 27.81, O 21.17; found (%): C 47.89, H 2.69, N 27.01, O 23.25].Characterizations
Solid-state 13C NMR was performed on JNM-ECZ600R 600M (Jeol, Japan). XPS was carried out on a K-Alpha+ X-ray Photoelectron Spectroscopy instrument (KRATOS Axis UltraDLD, England). The data were analyzed using CasaXPS software and calibrated by referencing the C 1s peak to 284.8 eV. FT-IR spectroscopy was carried out on a VERTEX70 (Bruker, Germany). XRD was carried out on an Empyrean diffractometer with Cu-Kα radiation (PANalytical B.V., Holland). EA was carried out by an Elementar Vario EL III (Elementar, Germany). The morphologies were obtained by a scanning electron microscope (HITACHI SU8010 SEM, Japan). TGA was carried out on a TGA/DSC3+ thermal analysis system (Mettler Toledo, Switzerland). Solid-state UV-vis spectroscopy was carried out by a UV3600IPPLUS (KRATOS, England). In situ UV-vis spectra were obtained by a Lambda XLS+ UV-vis spectrometer. EPR spectra were recorded on a Bruker EMXplus-10/12 spectrometer. The electrical conductivities were measured with pellets by a two-probe system using I–V plots or by a four-probe resistivity meter (YAOS FM100GH, China).Electrochemical measurements
The TDT cathodes were prepared by mixing the active material, KB conductive agent and PTFE binder at 8:1:1 weight ratio, except for the study of the cathode with 5% carbon where the ratio was 9:0.5:0.5, in deionized water and casted on a graphite foil substrate. The cathodes were dried at room temperature overnight. The mass loading of TDT was around 1.2 mg cm−2, except for the study of the cathode with a high mass loading of 10 mg cm−2. GCD measurements were carried out in CR2032-type coin cells with a zinc anode and 1 M ZnSO4 aqueous electrolyte. Filter paper and glass fiber separators were applied for low and high mass loading cathodes, respectively. EIS measurements were carried out in T-shaped three-electrode PFA Swagelok® type cells with a TDT working electrode, zinc counter electrode and SCE reference electrode. The potentials labeled in Fig. 3c in the main text were converted with respect to Zn. GITT measurement was carried out with a T-shaped three-electrode PFA Swagelok® type cell with TDT as the working electrode, and Zn as the counter and reference electrodes. Each pulse was applied at 50 mA g−1 for 300 s, followed by a 2 h rest. The voltage changes at the end of the relaxations were smaller than 8 mV h−1. The electrodes for ex situ characterizations were cycled at 0.2 A g−1. All electrochemical measurements were carried out on Bio-Logic VMP3 or LAND-CT2001A battery cycler.
Direct band gap calculation
The optical band gap energy (Eg) of TDT was calculated by the Tauc relation:53αhν = A(hν − Eg)n. In the equation, α was the absorption, hν was the incident photon energy and A was a constant. The coefficient ‘n’ was equal to 1/2 for direct band gap semiconductors. The Eg value corresponding to direct band gap transitions was given by: (αhν)2 = A(hν − Eg). Eg of TDT was calculated by plotting (αhν)2vs. hv and then extrapolating the straight portion of the curve to the hν axis at α = 0.Diffusion coefficient calculation
The diffusion coefficient (D) was calculated from GITT based on the following equation:52,54where τ was the relaxation time, ΔES was the steady-state potential change after a single pulse, and ΔEt was the potential change during a pulse after eliminating the iR drop. The diffusion length L was measured by the geometric thickness of the electrode.
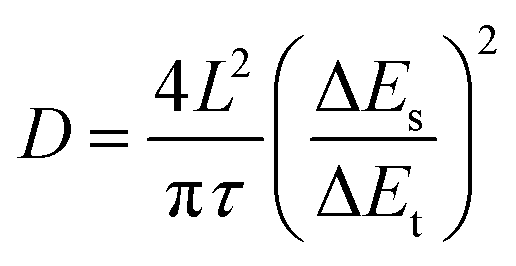
Computational details
DFT calculations were carried out with the Gaussian 16 software package.37 Geometrical optimizations were conducted with Becky's three-parameter exchange function combined with the Lee-Yang-Parr correlation functional (B3LYP) method and 6-31 G (d) basis set. Vibrational frequency calculations were performed on the optimized structures. An LOL-π map was obtained by the method proposed by Schmider and Becke using Multiwfn software.38,55Author contributions
L. L. and X. S. conceived and designed this work. L. L. carried out the synthesis, electrochemical measurements and computational calculations. Z. L and J. Z conducted part of the characterizations. L. L., Z. L., K. W., W. W. and T. Q. participated in the analysis of the data. All authors discussed and revised the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51974070, 52174276), the LiaoNing Revitalization Talents Program (XLYC1907069), and the Fundamental Research Funds for the Central Universities (N2105001). Special thanks are due to the instrumental analysis from the Analytical and Testing Center, Northeastern University and SDBSWeb: https://sdbs.db.aist.go.jp.References
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86, DOI:10.1038/s41586-019-1682-5
.
- J. B. Goodenough and Y. Kim, J. Am. Chem. Soc., 2009, 22, 587–603, DOI:10.1021/cm901452z
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, Energy Environ. Sci., 2020, 13, 2056, 10.1039/d0ee01017k
- J. Deng, C. Bae, J. Marcicki, A. Masias and T. Miller, Nat. Energy, 2018, 3, 261–266, DOI:10.1038/s41560-018-0122-3
- L. E. Blanc, D. Kundu and L. F. Linda, Joule, 2020, 4, 771–799, DOI:10.1016/j.joule.2020.03.002
- L. Miao, R. Wang, S. Di, Z. Qian, L. Zhang, W. Xin, M. Liu, Z. Zhu, S. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9978, DOI:10.1021/acsnano.2c02996
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2020, 15, 1638–1646, 10.1039/d1ee03882f
- H. Peng, C. Liu, N. Wang, C. Wang, D. Wang, Y. Li, B. Chen, J. Yang and Y. Qian, Energy Environ. Sci., 2022, 15, 1682–1693, 10.1039/d1ee03624f
- L. Yan, Y. Zhang, Z. Ni, Y. Zhang, J. Xu, T. Kong, J. Huang, W. Li, J. Ma and Y. Wang, J. Am. Chem. Soc., 2021, 143, 15369–15377, DOI:10.1021/jacs.1c06936
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170, DOI:10.1038/s41467-020-20331-9
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 16, 706–715, 10.1039/C8EE02892C
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119, DOI:10.1038/nenergy.2016.119
- S. Wei, S. Chen, X. Su, Z. Qi, C. Wang, B. Ganguli, P. Zhang, K. Zhu, Y. Cao, Q. He, D. Cao, X. Guo, W. Wen, X. Wu, P. M. Ajayan and L. Song, Energy Environ. Sci., 2021, 14, 3954–3964, 10.1039/d1ee00590a
- W. Sun, F. Wang, B. Zhang, M. Zhang and M. Winter, Science, 2021, 371, 46–51, DOI:10.1126/science.abb9554
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509, DOI:10.1002/aenm.201703509
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chemistry, 2018, 4, 2786–2813, DOI:10.1016/j.chempr.2018.09.005
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vald and D. Brandell, J. Power Sources, 2021, 482, 228814, DOI:10.1016/j.jpowsour.2020.228814
- H. Yang, J. Lee, J. Y. Cheong, Y. Wang, G. Duan, H. Hou, S. Jiang and I. Kim, Energy Environ. Sci., 2021, 14, 4228–4267, 10.1039/d1ee00419k
- Z. Tie and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303, DOI:10.1002/anie.202008960
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761, DOI:10.1126/sciadv.aao1761
- K. W. Nam, H. Kim, Y. Beldjoudi, T. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548, DOI:10.1021/jacs.9b12436
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741, DOI:10.1002/anie.201807121
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid and E. Tervoort, Chem. Mater., 2018, 30, 3874–3881, DOI:10.1021/acs.chemmater.8b01317
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, Z. Guo, Z. Wang, D. Bin and J. Ma, Adv. Mater., 2020, 32, 2000338, DOI:10.1002/adma.202000338
- X. Wang, J. Tang and W. Tang, Adv. Funct. Mater., 2022, 32, 2200517, DOI:10.1002/adfm.202200517
- J. Wang, Z. Liu, H. Wang, F. Cui and G. Zhu, Chem. Eng. J., 2022, 450, 138051, DOI:10.1016/j.cej.2022.138051
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401, DOI:10.1016/j.jpowsour.2020.228401
- W. Wang, V. S. Kale, Z. Cao, S. Kandambeth, W. Zhang, J. Ming, P. T. Parvatkar, E. Abou-Hamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2020, 5, 2256–2264, DOI:10.1021/acsenergylett.0c00903
- W. Ai, W. Zhou, Z. Du, C. Sun, J. Yang, Y. Chen, Z. Sun, S. Feng, J. Zhao, X. Dong, W. Huang and T. Yu, Adv. Funct. Mater., 2017, 27, 1603603, DOI:10.1002/adfm.201603603
- K. N. Houk, J. Am. Chem. Soc., 1973, 95, 4092–4094, DOI:10.1021/ja00793a069
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130, DOI:10.1021/jacs.5b00336
- Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878, DOI:10.1021/jacs.1c13012
- U. Geiser and J. A. Schlueter, Chem. Rev., 2004, 104, 203–5241, DOI:10.1021/cr0306844
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608, DOI:10.1002/ange.201607812
- H. Zhang, Y. Fang, F. Yang, X. Liu and X. Lu, Energy Environ. Sci., 2020, 13, 2515–2523, 10.1039/d0ee01723j
- J. Lee, H. Kim and M. J. Park, Chem. Mater., 2016, 28, 2408–2416, DOI:10.1021/acs.chemmater.6b00624
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- T. Lu and F. Chen, Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885
- T. Lu and Q. Chen, Theor. Chem. Acc., 2020, 139, 25, DOI:10.1007/s00214-019-2541-z
- J. Tong, Z. Song, D. Kim, X. Chen, C. Chen, A. F. Palmstrom, P. F. Ndione, M. O. Reese, S. P. Dunfield, O. G. Reid, J. Liu, F. Zhang, S. P. Harvey, J. Li, S. T. Christensen, J. Teeter, D. Zhao, M. M. Al-Jassim, M. F. A. M. J. M. Hest, C. Beard, S. E. Shaheen, J. J. Berry, Y. Yan and K. Zhu, Science, 2019, 364, 475–479, DOI:10.1126/science.aav7911
- Y. Miura, M. Momoki, T. Fuchikami, Y. Teki, K. Itoh and H. Mizutani, J. Org. Chem., 1996, 61, 4300–4308, DOI:10.1021/jo960347d
- S. T. Hauer, A. P. W. Schneeweis, S. D. Waniek, L. P. Sorge, K. Heinze and T. J. J. Müller, Org. Chem. Front., 2021, 8, 5744–5755, 10.1039/d1qo00867f
- P. P. Power, Chem. Rev., 2003, 103, 789–810, DOI:10.1021/cr020406p
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. Sci., 2020, 11, 1192, 10.1039/c9sc06143f
- Y. Tsujii, A. Tsuchida, Y. Onogi and M. Yamamoto, Macromolecules, 1990, 23, 4019–4023, DOI:10.1021/ma00219a023
- Y. Wang, Z. Chen, H. Huang, D. Wang, D. Liu and L. Wang, J. Mater. Chem. A, 2020, 8, 24675, 10.1039/d0ta08154j
- Z. Ye, S. Xie, Z. Cao, L. Wang, D. Xu, H. Zhang, J. Matz, P. Dong, H. Fang, J. Shen and M. Ye, Energy Storage Mater., 2021, 37, 378–386, DOI:10.1016/j.ensm.2021.02.022
- A. F. Lee, K. H. Jon, J. B. Mark and P. A. Steven, Chem. Commun., 2015, 51, 16886, 10.1039/C5CC07405C
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931, DOI:10.1021/jp074464w
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123–130, DOI:10.1038/s41560-018-0309-7
- J. Xie, F. Yu, J. Zhao, W. Guo, H. Zhang, G. Cui and Q. Zhang, Energy Storage Mater., 2020, 33, 283–289, DOI:10.1016/j.ensm.2020.08.027
- Z. Lin, H.-Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, Nat. Commun., 2021, 12, 4424, DOI:10.1038/s41467-021-24701-9
- V. M. Huxter, T. Mirkovic, P. S. Nair and G. D. Scholes, Adv. Mater., 2008, 20, 2439–2443, DOI:10.1002/adma.200702429
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569, DOI:10.1149/1.2133112
- H. L. Schmider and A. D. Becke, J. Mol. Struct.: THEOCHEM, 2000, 527, 51–61, DOI:10.1016/s0166-1280(00)00477-2
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02961h |
| This journal is © The Royal Society of Chemistry 2023 |