
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn effective strategy to produce highly amenable cellulose and enhance lignin upgrading to aromatic and olefinic hydrocarbons†
Daniel
Vincent Sahayaraj‡
 ab,
Lusi
A‡
c,
Andrew J.
Kohler
ab,
Hamed
Bateni
ab,
Harish
Radhakrishnan
c,
Alireza
Saraeian
ab,
Brent H.
Shanks
ab,
Xianglan
Bai
c and
Jean-Philippe
Tessonnier
*ab
ab,
Lusi
A‡
c,
Andrew J.
Kohler
ab,
Hamed
Bateni
ab,
Harish
Radhakrishnan
c,
Alireza
Saraeian
ab,
Brent H.
Shanks
ab,
Xianglan
Bai
c and
Jean-Philippe
Tessonnier
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: tesso@iastate.edu
bCenter for Biorenewable Chemicals (CBiRC), Ames, Iowa 50011, USA
cDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA
First published on 24th October 2022
Abstract
Lignin is a promising renewable feedstock for the production of valuable phenolic and hydrocarbon building blocks. However, the economic viability of lignin upgrading strategies has so far been hampered by the recondensation of primary products and the preferential formation of char over bio-oil. Here, we demonstrate that lignin pretreatments effectively lower char formation and enhance carbon volatilization during pyrolysis. Various solvolytic and catalytic pretreatments were investigated using sub- and supercritical ethanol and supported metal catalysts. By combining GPC, GC-FID/MS, elemental analysis, HSQC NMR, and TGA, we decoupled the effects of solvolytic and catalytic steps and identified their independent roles on chemical modifications relative to the parent lignin. The pretreatment step enhanced the production of volatiles and concurrently reduced char formation during fast pyrolysis, from 58 C% to 9 C% for lignin deconstructed at 250 °C using Pd/C. The strategy was then extended directly to lignocellulosic biomass (corn stover, switchgrass, red oak) to fractionate and pretreat lignin in a one-pot approach. The lignin oil obtained from this process exhibited an excellent potential to be converted into platform chemicals. Upon catalytic fast pyrolysis of the lignin oil, 11–14 C% aromatic hydrocarbons were produced, while hydrodeoxygenation yielded 34–40 C% of aromatic hydrocarbons (50–56 C% total hydrocarbons). Similarly, the recovered carbohydrates-rich water-soluble fraction was subjected to hydrodeoxygenation and yielded 10–15 C% of aromatic hydrocarbons and 15–29 C% of C2–C6 alkenes (32–74 C% total hydrocarbons). Furthermore, the residual pulp recovered from this method was enriched in sugars and was three times more amenable to enzymatic hydrolysis than the parent biomass. This approach provides new opportunities for the selective and effective conversion of lignin into value-added chemicals and, thereby, enhanced carbon recovery, which is vital for implementing biomass as a feedstock for chemical manufacturing.
Broader contextTo achieve economic viability and strive, the bioeconomy needs to build on the existing bioethanol infrastructure and broaden the portfolio of products manufactured from biomass. To this end, substantial work has been done to convert cellulose to biobased chemicals through innovations in biocatalysis and thermocatalysis. In contrast, lignin, an aromatic polymer that accounts for 10–25% of lignocellulosic biomass, remains underutilized as a feedstock although its deconstruction could readily supply the chemical industry with platform aromatic hydrocarbons. The challenge comes for lignin's building blocks propensity to repolymerize and form a more condensed network. Reductive catalytic fractionation (RCF), a strategy that combines solvents and metal catalysts to deconstruct lignin and stabilize its building blocks, was recently proposed to directly convert the lignin component in whole biomass. Technologic and economic analyses have highlighted the promise of this strategy but also its present limitations. In particular, new valorization strategies must be explored and developed to also upgrade dimers and oligomers that account for up to 50% of the deconstructed lignin. This work identifies conversion pathways to enhance lignin deconstruction, mitigate carbon loss due to char formation, and facilitate the complete upgrading of lignin to valuable aromatic and olefinic hydrocarbons. When applied to whole biomass, the proposed process also enhances the enzymatic digestibility of cellulose, hence its potential for biofuel and biochemical production. Results obtained for the upgrading of corn stover, an abundant (>75 million tons per year) and underutilized byproduct of corn production, showed a 65% increase in lignin-derived aromatics along with a 200% increase in cellulose digestibility. |
1. Introduction
Lignocellulose comprises about 10–25% lignin, 20–30% hemicellulose, and 40–50% cellulose.1 Considering its structure, lignin is the largest renewable source of aromatic building blocks in nature and has significant potential to serve as a feedstock for the production of bulk phenolics and aromatic hydrocarbons such as benzene, toluene, and xylene (BTX) that are currently derived from petroleum.2 However, lignin valorization has proven to be challenging owing to its complex nature and recalcitrant structure, which is why nearly 95% of the extracted lignin is burnt for its energy value instead of being upgraded.3 When used only as an energy source, lignin is worth only about $150 per ton; in contrast, its full conversion to aromatics would increase its value to about $1200 per ton.4 Therefore, it is critical to design new conversion strategies that overcome present technical hurdles.The upgrading of lignocellulosic biomass using thermochemical methods typically starts with its fast pyrolysis.5–7 In this process, biomass or its components are heated to 450–600 °C, in the absence of oxygen, and using fast heating rates to maximize the production of bio-oil over solid products (biochar). Unfortunately, raw bio-oil presents adverse properties due to its high oxygen content (about 40%), namely poor thermal and chemical stability, corrosivity from high acidity, low heating value, and high viscosity compared to petroleum-derived oils.6,8,9 Several strategies were proposed to lower the bio-oils’ oxygen content, overcome its adverse properties, and improve its compatibility with existing infrastructure in the chemical industry. Most of these strategies focus on the production of aromatic hydrocarbons because of their high energy density and important role in chemical manufacturing. Catalytic fast pyrolysis (CFP) is a thermal upgrading strategy that enables the production of BTX, polycyclic aromatic hydrocarbons, alkanes, and olefins.5,10,11 The best results were obtained with ZSM-5 zeolite due to the unique size and shape selectivity provided by the crystallographic structure of this solid acid catalyst.12–14 Hydrodeoxygenation (HDO) presents another avenue for upgrading bio-oil by adding hydrogen gas as a reactant. Recently, low-pressure HDO reactions over transition metal oxides, particularly MoO3, were shown to completely remove oxygen from biomass/lignin pyrolytic vapors and produce valuable aliphatic and aromatic hydrocarbons.15,16
Although studies have shown that it is possible to convert lignin bio-oil into hydrocarbons, the yields are much lower than for whole biomass- and cellulose-derived bio-oils. For instance, 14 C% of aromatics were produced through CFP of pinewood while only 6 C% of aromatics were obtained from its lignin fraction using ZSM-5 at 600 °C.17,18 Similarly, HDO of cellulose using MoO3 in a tandem micropyrolyzer system at 400 °C generated 84 C% of hydrocarbons while only 30–32 C% were obtained with lignin as the starting material.19,20 Compared to whole biomass and cellulose, pyrolysis of lignin generates a significantly higher amount of char, which halved the bio-oil yields across all reaction temperatures.21–23 But, interestingly, when the aromatic hydrocarbon yields generated using a ZSM-5 catalyst at 500 °C were normalized on a pyrolysis vapor basis (in the absence of catalyst), the yields were comparable at 16% for all three components of biomass.24 Thus, the low hydrocarbon yields obtained for lignin CFP are not due to the nature of the species in the vapor phase but to the efficiency of the volatilization process during the initial pyrolysis step. Therefore, lignin pretreatments that would enhance the volatilization step are vital to increase the production of valuable products and lower carbon losses through char formation.
Several research groups have explored pretreatment strategies that could improve the carbon efficiency during catalytic deoxygenation. Feng et al. investigated a microwave-assisted formic acid pretreatment as a possible way to improve the production of aromatics for the CFP of lignocellulosic biomass. The pretreated beech wood produced a higher yield of valuable aromatic products than the untreated control (29.8 C% vs. 23.2 C%) along with a 10% reduction in char formation.25 The effects of HCl, NaOH, and H2O2 were also investigated for the pretreatment of kraft lignin prior to its pyrolysis, and upgrading at 650 °C.26 HCl was found to depolymerize the lignin matrix effectively, increasing the liquid yield, and, at optimized conditions, the selectivity to phenols reached greater than 50% using a large pore zeolite catalyst.26 More recently, Saraeian et al. demonstrated that partial depolymerization of lignin using a copper-doped porous metal oxide catalyst in methanol at 300 °C prior to pyrolysis significantly improved the yield of volatile products and decreased the yield of pyrolytic char.19 Furthermore, the yield of aromatics obtained from HDO using MoO3 increased by 90% for the depolymerized samples compared to the parent lignin.19 Although there is a plethora of studies on the pretreatment of biomass/lignin, the exact effect of this additional step remains poorly understood, and the key parameters that would unlock the selective upgrading of lignin to chemicals remain to be identified. Furthermore, accurate carbon tracking to account for losses encountered during the pretreatment step is necessary to draw quantitative conclusions about the beneficial effects of different pretreatments.
Here, we studied the independent and synergistic roles of solvolysis and catalysis during the pretreatment of technical lignin and raw biomass. We focused in particular on their role in enhancing (i) the deconstruction/volatilization of lignin during fast pyrolysis and (ii) the aromatic hydrocarbon yields achieved through subsequent CFP and HDO of the pyrolysis vapors. By combining various complementary characterization techniques, we first gained insights into the chemical transformations occurring during these pretreatments and the parameters that govern the size distribution of the lignin fragments. The product distributions from fast pyrolysis of the parent and pretreated lignin samples were compared to evaluate the effect of the pretreatment on bio-oil vapors and char yields. Next, the pretreated lignin obtained using the optimized reaction condition was upgraded to aromatic hydrocarbons through ex-situ CFP and low-pressure HDO, and the obtained yields were compared to the parent lignin.
Using the insights gained from the first part of our study on technical lignin pretreatments, we demonstrate the one-pot fractionation and pretreatment of lignin from raw biomass (Scheme 1). The pretreated lignin oil generated through this one-pot approach was readily converted to bio-oil through fast pyrolysis and was funneled to platform chemicals such as aromatics and alkenes at higher yields compared to lignin extracted using conventional fractionation methods. In addition, the residual carbohydrate pulp was preferred for enzymatic hydrolysis and, therefore, should be highly amenable to bioethanol/biochemical production. We further demonstrate the robustness of this ‘one-pot’ approach for various lignocellulosic feedstocks to elucidate the impact of structural variability using compositional analysis and carbon balances. The results provide valuable insights that further advance reductive catalytic fractionation (RCF) and lignin-first fractionation strategies, realizing an ‘ideal lignin stream’ through pretreatment and fractionation that could facilitate complete biomass utilization.
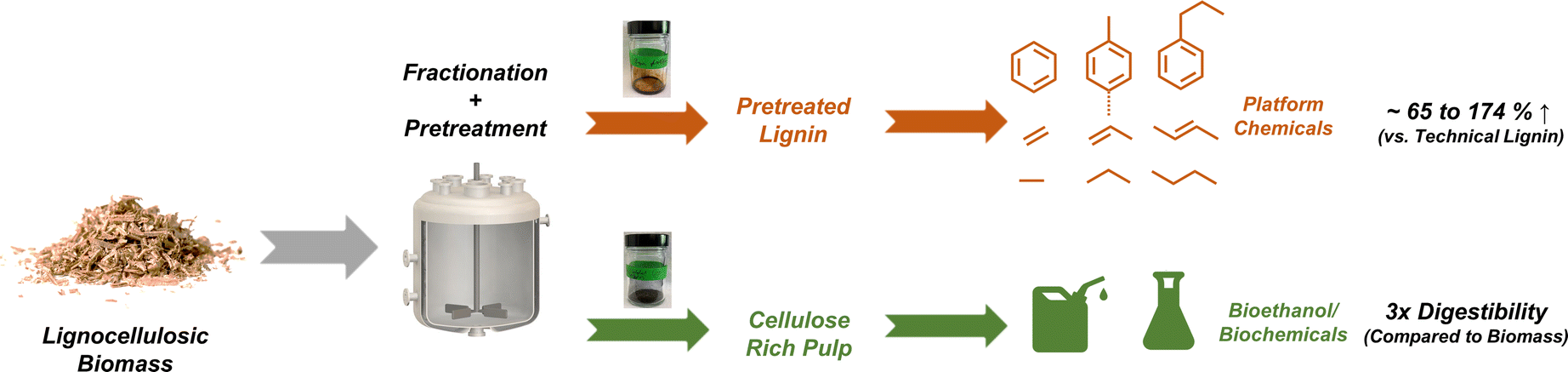 | ||
| Scheme 1 Illustration of the whole biomass upgrading strategy described in this study. The lignin oil recovered after one-pot fractionation and pretreatment was successfully upgraded to bulk platform chemicals at higher yields than conventional pulping approaches. The EtOH-insoluble pulp obtained in this study was enriched in sugars and showed improved digestibility, a desired property for bioethanol production. | ||
2. Results and discussion
2.1. Lignin pretreatment
As shown in Fig. 1, at 200 °C, 61–65 wt% of the pretreated sample relative to the initial lignin was isolated. When the reaction was performed under subcritical conditions, significant differences in product yields were not observed across the tested catalysts. Under supercritical conditions at 250 °C, the yield of the pretreated sample dropped to 50 wt%, and char was observed on the reactor walls in the absence of a catalyst. The benefits of adding a reductive catalyst at 250 °C were evident, as the yield of the products obtained increased by about 10 wt%. In contrast, the addition of HY zeolite, a solid acid catalyst, reduced the product recovery to 46 wt%.
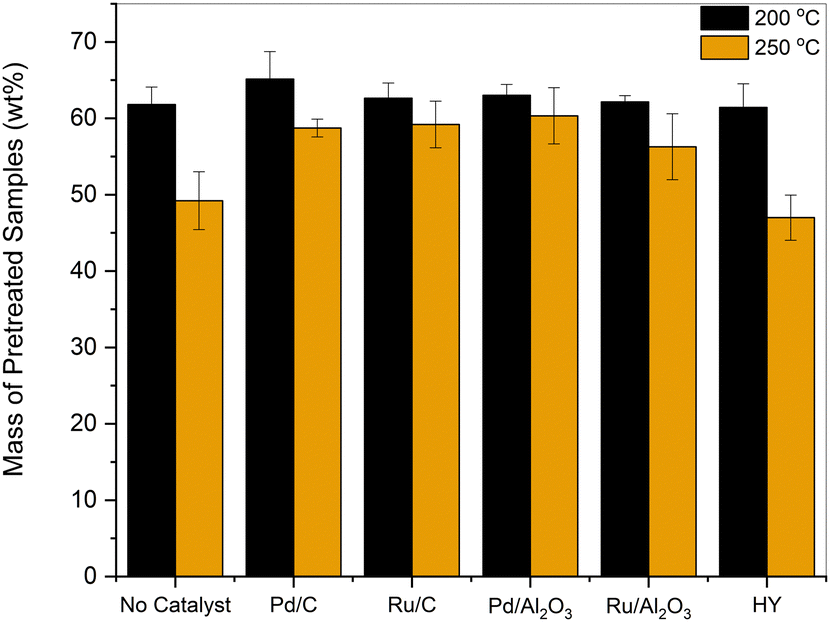 | ||
| Fig. 1 Amount of sample (wt%) recovered after treating organosolv lignin in EtOH at 200 and 250 °C for 3 h in the presence of various catalysts. | ||
The molecular weight distributions of the pretreated samples were measured to qualitatively assess the extent of deconstruction as a function of reaction conditions. The average molecular weight, Mw, of the products generated at 200 °C ranged from 1000 to 1386 Da, corresponding to approximately 18–25% of the parent lignin's molecular weight (Fig. S1a, ESI†). An apparent decrease in Mw to about 520–800 Da with a sharp peak corresponding to monomers around 200 Da was observed for the samples pretreated at 250 °C (Fig. S1b, ESI†).
Interestingly, the samples pretreated with and without catalyst showed almost identical molecular weight distributions, meaning the cleavage of the linkages in lignin occurred primarily through solvolysis. However, in the absence of catalyst, a 10 C% loss was observed along with noticeable char deposition on the reactor walls. These observations, when taken together, indicate that depolymerization and secondary recondensation reactions co-exist and compete with each other. Depolymerization reactions lead to smaller species such as monomers, while repolymerization reactions generate higher molecular weight products, including char, which are not recovered as pretreated lignin oil and are absent in the GPC data.33,35,36 At higher temperatures, the latter reactions are promoted, and thus the reductive passivation of the reactive intermediates becomes all the more necessary to prevent their repolymerization and enable a higher product recovery.33,37–39
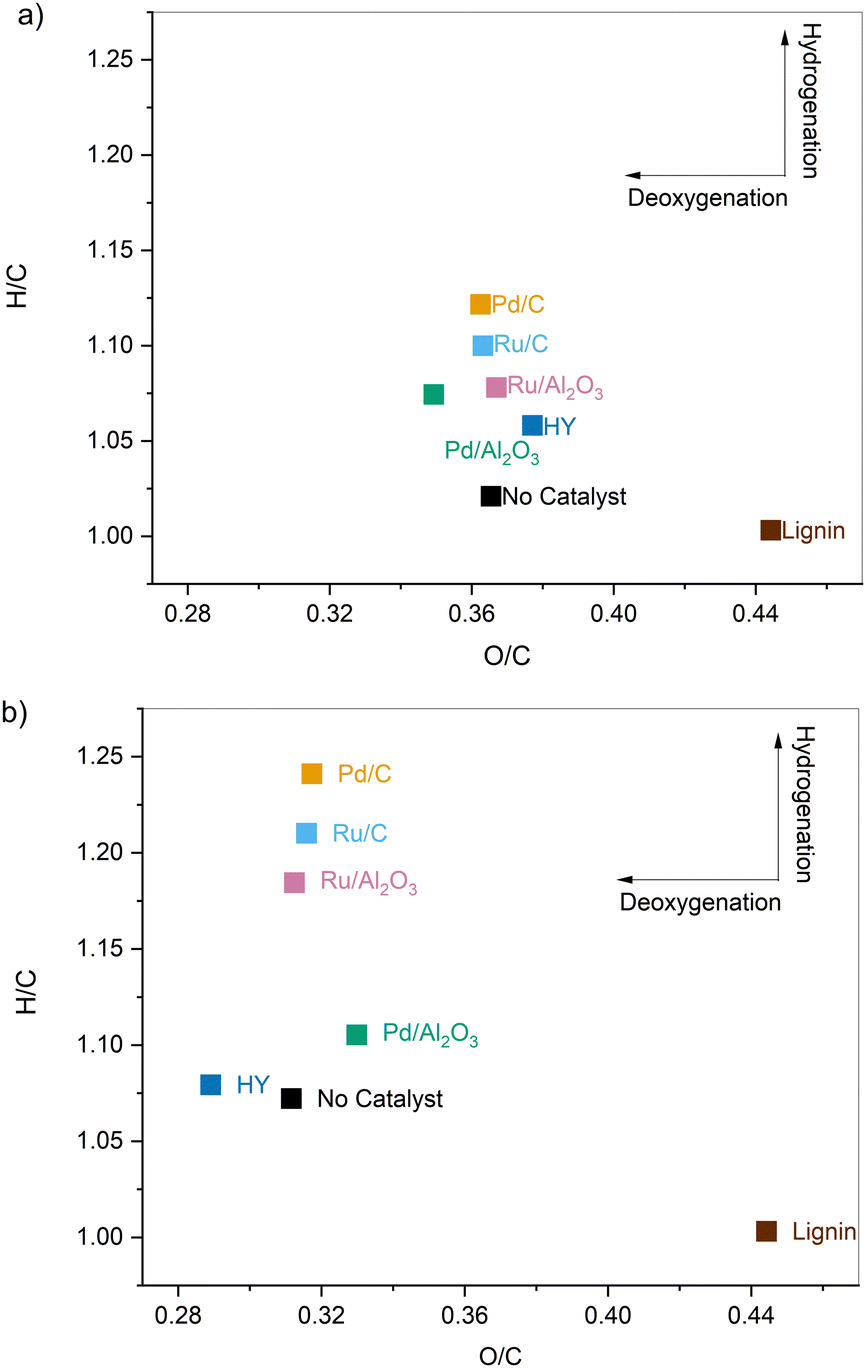 | ||
| Fig. 2 Van Krevelen diagrams depicting the H/C ratio versus O/C ratio for organosolv lignin and the samples pretreated in EtOH at (a) 200 °C and (b) 250 °C in the presence of various catalysts. | ||
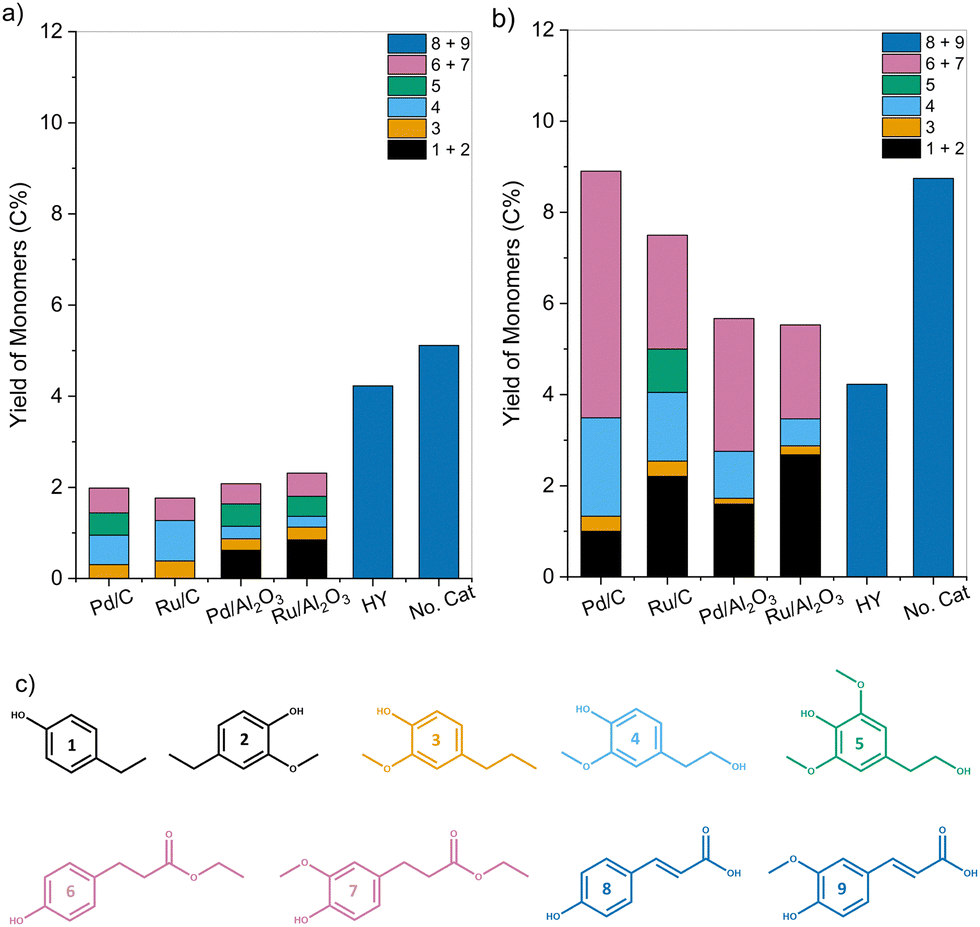 | ||
| Fig. 3 Yield of phenolic monomers (C%) obtained after subjecting organosolv corn stover lignin to a 3 hour pretreatment in EtOH in the presence of various catalysts at (a) 200 °C and (b) 250 °C. (c) The monomeric products consist of (1) ethyl phenol, (2) ethyl guaiacol, (3) 4-propyl guaiacol, (4) propanol guaiacol, (5) 4-(3-hydroxypropyl)-2,6-dimethoxyphenol, (6) ethyl 3-(4-hydroxyphenyl) propanoate, (7) ethyl 3-(4-hydroxy-3-methoxyphenyl) propanoate, (8) p-coumaric acid, (9) ferulic acid. | ||
Corn stover lignin is composed of up to ∼20% p-coumaric acid and ∼10% ferulic acid pendant units, which are linked through both ester and ether linkages.48 These linkages are cleaved under reaction conditions primarily through solvolysis even in the absence of a catalyst, as evidenced by the yields of monomers 8 and 9. In the presence of a transition metal catalyst, these hydroxycinnamic acids undergo esterification with EtOH followed by their subsequent hydrogenation to give monomers 6 and 7.34,49 Monomers 1 and 2 are formed through decarboxylation followed by hydrogenation of the unsaturated functionalities of formed hydroxycinnamic acids.50,51 Monomers 3, 4, and 5 are formed through the cleavage of β-O-4 linkages in lignin mediated by the supported metal catalyst.52 Only p-coumaric (8) and ferulic acid (9) were observed for lignin pretreated in the presence of HY zeolite catalyst. Moreover, the yield was lower than for samples deconstructed through solvolysis.
Together, these results indicate that lignin is first solvolytically deconstructed in a polar solvent at elevated temperatures to yield unsaturated products. These products, including monomers, undergo passivation over a hydrogenation catalyst producing saturated alkyl-phenols, guaiacols, ethyl ferulates, and coumarates.
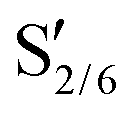 , G2 subunits), was found to be 30% for the parent lignin, which was in agreement with previous studies.53 Upon pretreatment, the signals of Aα and Aβ corresponding to benzylic alcohols and secondary alkyl protons of β-O-4 linkages decreased due to the scission of C–O–C linkages. The temperature was found to be the main driver for the cleavage of β-O-4 linkages as the β-O-4 concentration dropped to 6% when solvolysis was performed at 250 °C in the absence of any catalyst. Catalyst offered only modest gains, and further lowered the β-O-4 concentration to 2.3% when the pretreatment was performed at 250 °C in the presence of Pd/C.
, G2 subunits), was found to be 30% for the parent lignin, which was in agreement with previous studies.53 Upon pretreatment, the signals of Aα and Aβ corresponding to benzylic alcohols and secondary alkyl protons of β-O-4 linkages decreased due to the scission of C–O–C linkages. The temperature was found to be the main driver for the cleavage of β-O-4 linkages as the β-O-4 concentration dropped to 6% when solvolysis was performed at 250 °C in the absence of any catalyst. Catalyst offered only modest gains, and further lowered the β-O-4 concentration to 2.3% when the pretreatment was performed at 250 °C in the presence of Pd/C.
The increase in the presence of ethyl 3-(4-hydroxyphenyl) propanoate (dihydro pCA ethyl ester) and ethyl 3-(4-hydroxy-3-methoxyphenyl) propanoate (dihydro FA ethyl ester) C–H cross signals along with the disappearance of pCA and FA signals were observed for samples pretreated using a metal catalyst. These results are consistent with the major lignin monomer products detected by GC-FID/MS. These insights further corroborate that lignin is deconstructed solvolytically, and the heterogeneous metal catalyst is responsible for the stabilization of the extracted monomers through reductive chemistry. The mechanistic insights gained from elemental analysis (Van Krevelen diagrams), GC-FID/MS analysis of the monomers, and HSQC NMR analysis of the pretreated samples are further summarized in Fig. S3 (ESI†).
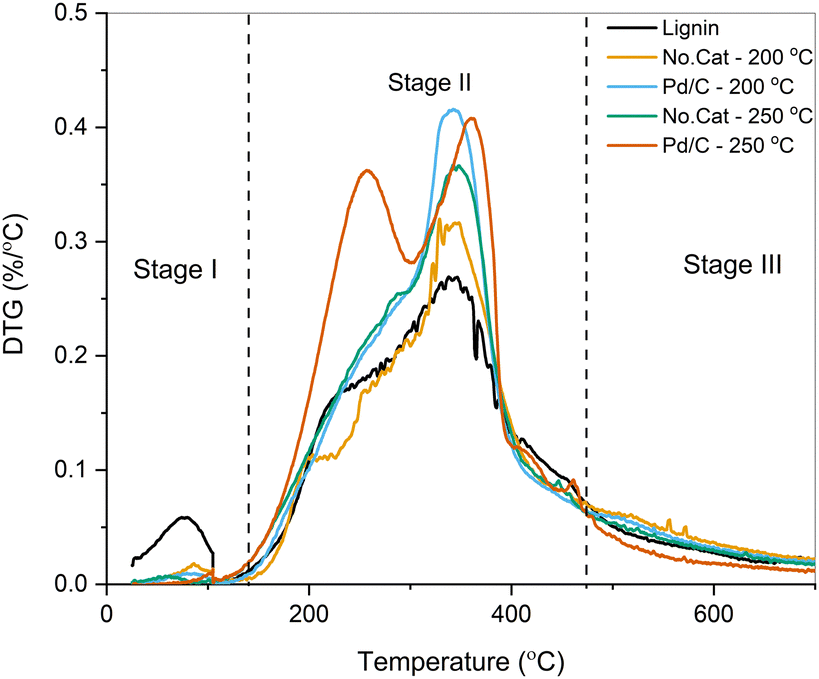 | ||
| Fig. 4 DTG curves of lignin and representative pretreated samples. | ||
As seen from the DTG curves, a larger fraction of the pretreated samples was volatilized compared to the parent lignin. The first contribution at ∼250 °C is related to the devolution of small phenolic compounds upon β-ether cleavage, the corresponding release of H2O, and the release of light gases from the cleavage of lateral chains in the lignin polymer.57 This contribution was prominent for pretreated samples, especially for the sample treated using Pd/C at 250 °C, possibly due to the volatilization of monomeric species. The peak at 340–350 °C corresponds to the decomposition of higher molecular weight phenolics, and the rate of volatilization was higher for the pretreated samples due to their partial deconstruction through solvolysis (vide supra).58 Proximate analysis for all the samples is reported in Fig. S5 (ESI†). The weight percentage of the volatiles fraction in the parent lignin was 63 wt%, while the pretreated samples showed a notable increase. Higher fractions of volatiles are preferred for lignin upgrading through pyrolysis, CFP, and vapor-phase HDO. By combining solvolytic deconstruction with Pd/C at 250 °C, the volatile content increased to 83 wt% along with a concomitant decrease in fixed, non-volatile, carbon (14 wt% vs. 26 wt%).
2.2. Upgrading of pretreated lignin through fast pyrolysis, catalytic fast pyrolysis, and hydrodeoxygenation
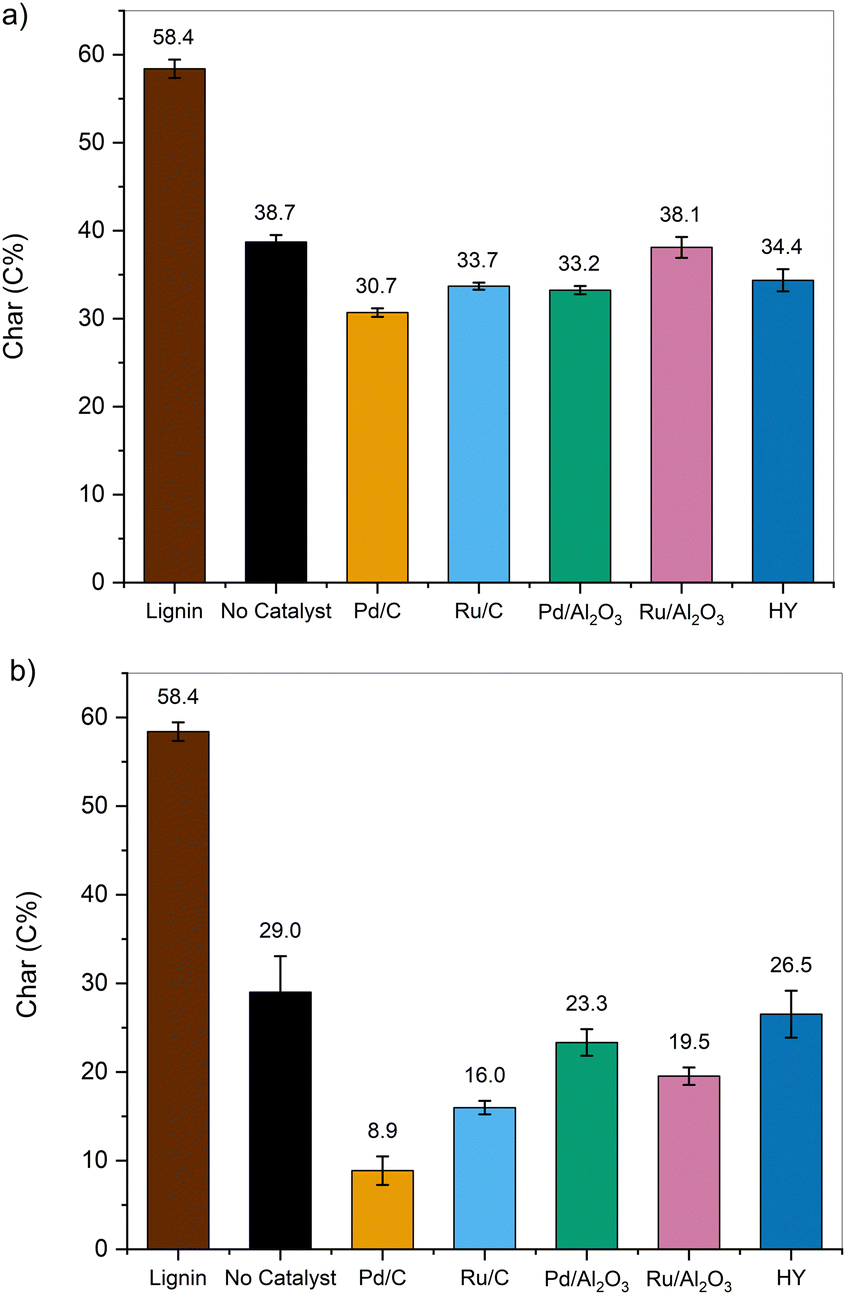 | ||
| Fig. 5 Char yields (in C%) measured after fast pyrolysis at 500 °C of the parent lignin and lignin samples pretreated in EtOH at (a) 200 °C and (b) 250 °C in the presence of various catalysts. | ||
It has been previously reported that the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond located in the phenolics side chains has a strong propensity towards (re)polymerization reactions.61,62 In a follow-up study, lignin-derived monomers were pyrolyzed to identify the mechanisms leading to char formation.63 It was observed that the extent of secondary char formation was strongly related to the types of functional groups on aromatic side chains, decreasing in the order of (CC) > (CO) > (O–CH3).63 Here, the hydrogenation of unsaturated bonds attenuated the formation of char through secondary reactions. Moreover, the combined yields of CO and CO2 were lower for the pretreated samples, potentially due to the lower number of C–O bonds susceptible to decarbonylation or decarboxylation reactions. Overall, the pretreatment step successfully reduced the yield of pyrolytic char from 58.4 C% for lignin to 8.8 C% for the sample deconstructed at 250 °C using Pd/C and enhanced the formation of volatile species.
C bond located in the phenolics side chains has a strong propensity towards (re)polymerization reactions.61,62 In a follow-up study, lignin-derived monomers were pyrolyzed to identify the mechanisms leading to char formation.63 It was observed that the extent of secondary char formation was strongly related to the types of functional groups on aromatic side chains, decreasing in the order of (CC) > (CO) > (O–CH3).63 Here, the hydrogenation of unsaturated bonds attenuated the formation of char through secondary reactions. Moreover, the combined yields of CO and CO2 were lower for the pretreated samples, potentially due to the lower number of C–O bonds susceptible to decarbonylation or decarboxylation reactions. Overall, the pretreatment step successfully reduced the yield of pyrolytic char from 58.4 C% for lignin to 8.8 C% for the sample deconstructed at 250 °C using Pd/C and enhanced the formation of volatile species.
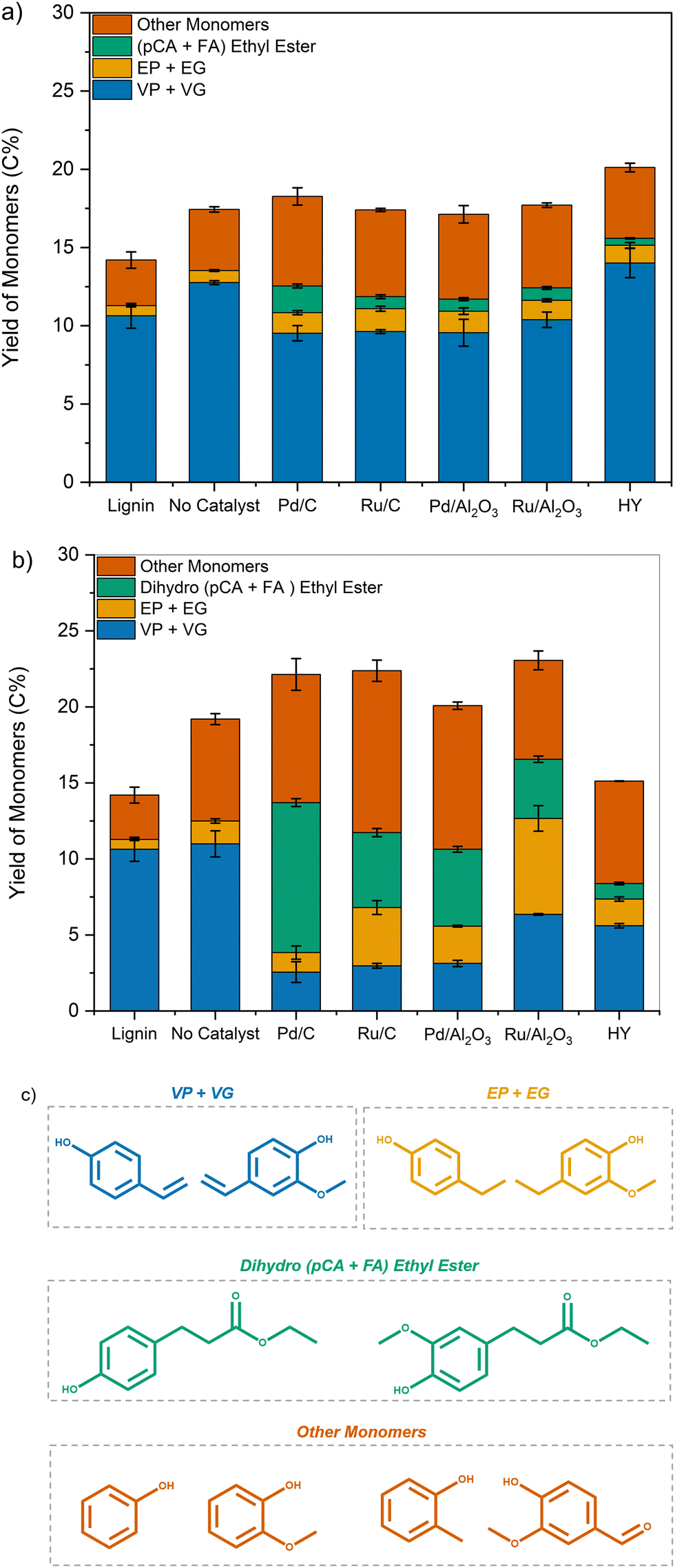 | ||
| Fig. 6 Yields of phenolic monomers (C%) obtained for the fast pyrolysis at 500 °C of lignin and samples pretreated in EtOH at (a) 200 °C and (b) 250 °C in the presence of various catalysts. (c) Molecular structures of the corresponding monomeric products. | ||
In contrast, samples pretreated using transition metal catalysts showed a decrease in VP and VG formation with a concomitant increase in EP and EG yields compared to that of lignin. Additionally, the formation of ethyl 3-(4-hydroxyphenyl) propanoate and ethyl 3-(4-hydroxy-3-methoxyphenyl) propanoate (dihydro (pCA + FA) ethyl ester) was also observed. For samples depolymerized using Pd/C at 250 °C, the yield of dihydro p-coumaric acid ethyl esters increased to 9.8 C%, while the yield of VP + VG dropped to 2.5 C%.
These results corroborate our previous observation that unsaturated functionalities present in lignin were reduced during pretreatment, leading to the formation of ethyl phenolics and dihydro p-coumaric acid ethyl esters instead of vinyl phenolics.
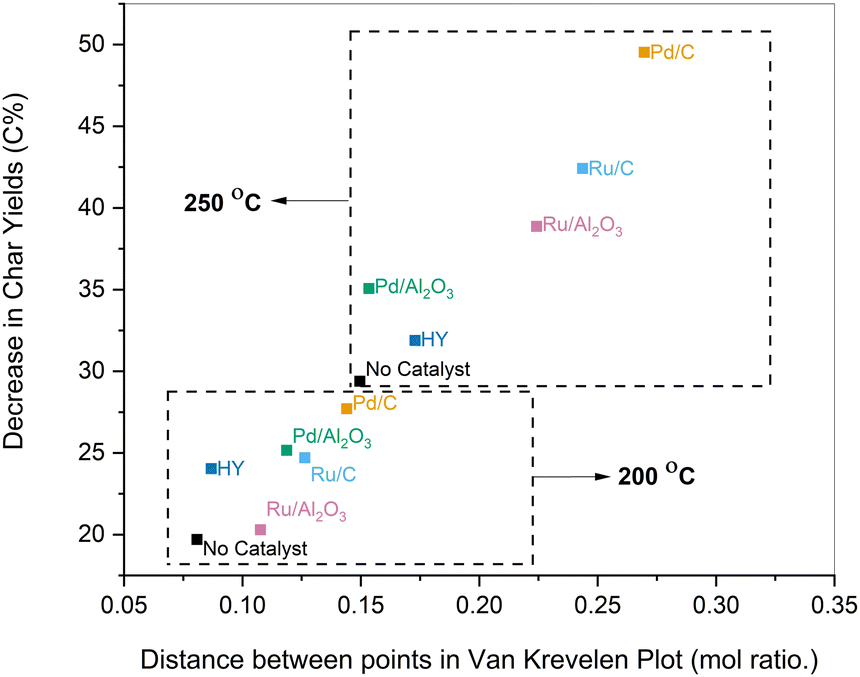 | ||
| Fig. 7 Reduction in char yields relative to lignin (C%) plotted against the distance between coordinates of lignin and pretreated samples in the Van Krevelen diagram for various catalysts and temperatures. | ||
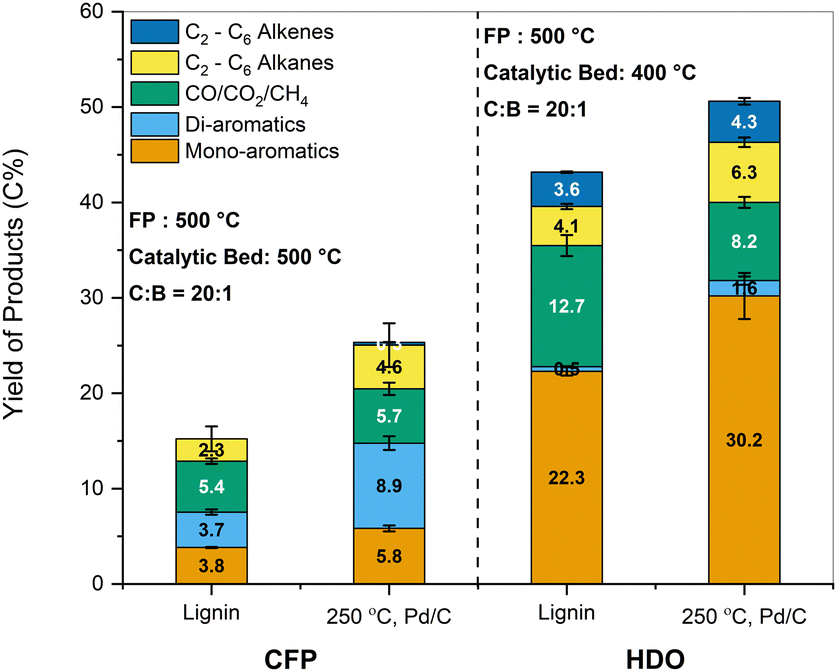 | ||
Fig. 8 Carbon yields of products obtained from CFP and HDO of lignin and sample pretreated in EtOH at 250 °C over Pd/C catalyst. Reaction conditions: CFP (ZSM-5 (Si/Al = 15) catalyst, fast pyrolysis temperature (FP): 500 °C, catalytic bed temperature: 500 °C, catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biomass ratio = 20:1) and HDO (MoO3 catalyst, fast pyrolysis temperature (FP): 500 °C, catalytic bed temperature: 500 °C, catalyst:biomass ratio = 20:1). :biomass ratio = 20:1) and HDO (MoO3 catalyst, fast pyrolysis temperature (FP): 500 °C, catalytic bed temperature: 500 °C, catalyst:biomass ratio = 20:1). | ||
The same samples were also subjected to HDO for comparison. The combined yields of mono-aromatics increased for the pretreated samples to 30.2 C% compared to 22.3 C% for the parent lignin. The yield of compounds such as benzene and ethylbenzene decreased slightly for the pretreated samples, while the yield of propyl benzene increased substantially from 0.95 C% to 6.10 C%. The change in selectivity could be explained by the presence of propyl guaiacol and propanol guaiacol as monomers in the pretreated sample. Saraeian et al. reported the formation of propyl benzene as a primary product during the HDO of propyl phenol using MoO3 at 400 °C.68 Similarly, Guan et al. reported a 98% yield of propyl benzene over Rh/Nb2O5 catalyst under 0.5 MPa H2 using 4-propyl guaiacol as the starting compound.69 In contrast to CFP, the formation of di-aromatics such as naphthalenes accounted for less than 2 C%. The yield of alkenes such as ethylene, propylene, and butene improved moderately upon pretreatment. Thus, the beneficial impact of the solvolytic and catalytic pretreatment was also reflected in enhanced aromatic and aliphatic hydrocarbon yields after CFP and HDO.
2.3. One-pot fractionation and pretreatment of lignin from whole biomass and upgrading to hydrocarbons
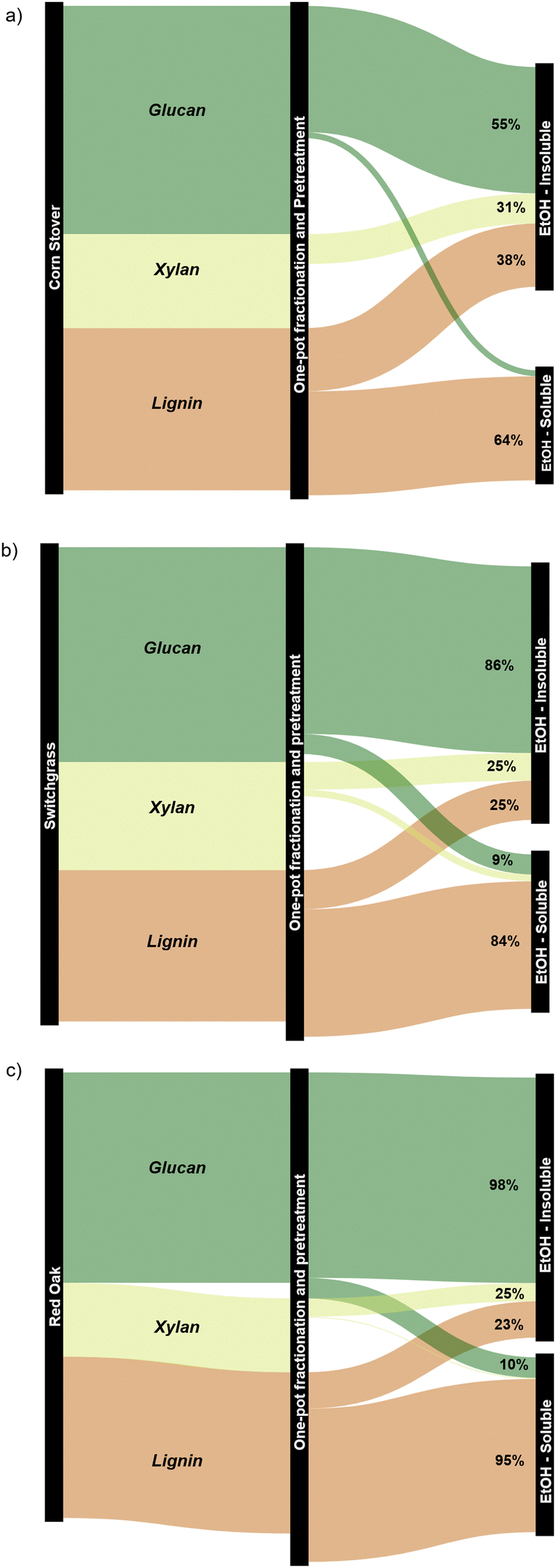 | ||
| Fig. 9 Component balance (upper bound values) for the products obtained from the one-pot fractionation and pretreatment of whole biomass in EtOH at 250 °C using Pd/C for (a) corn stover, (b) switchgrass, and (c) red oak. The corresponding component balances with the lower bound values are presented in Fig. S8 (ESI†). | ||
The ethanol-soluble fraction was rich in lignin extracted from the lignocellulose matrix. The extent of extraction varied between 64 and 95 wt% and was heavily dependent on the nature of the biomass sample. In some cases, the lignin component balance was greater than 100%, likely due to the incorporation of EtOH into the depolymerized lignin or the overestimation of the weights on the EtOH-insoluble pulp caused by the Pd/C catalyst, particularly for the upper bound measurements.
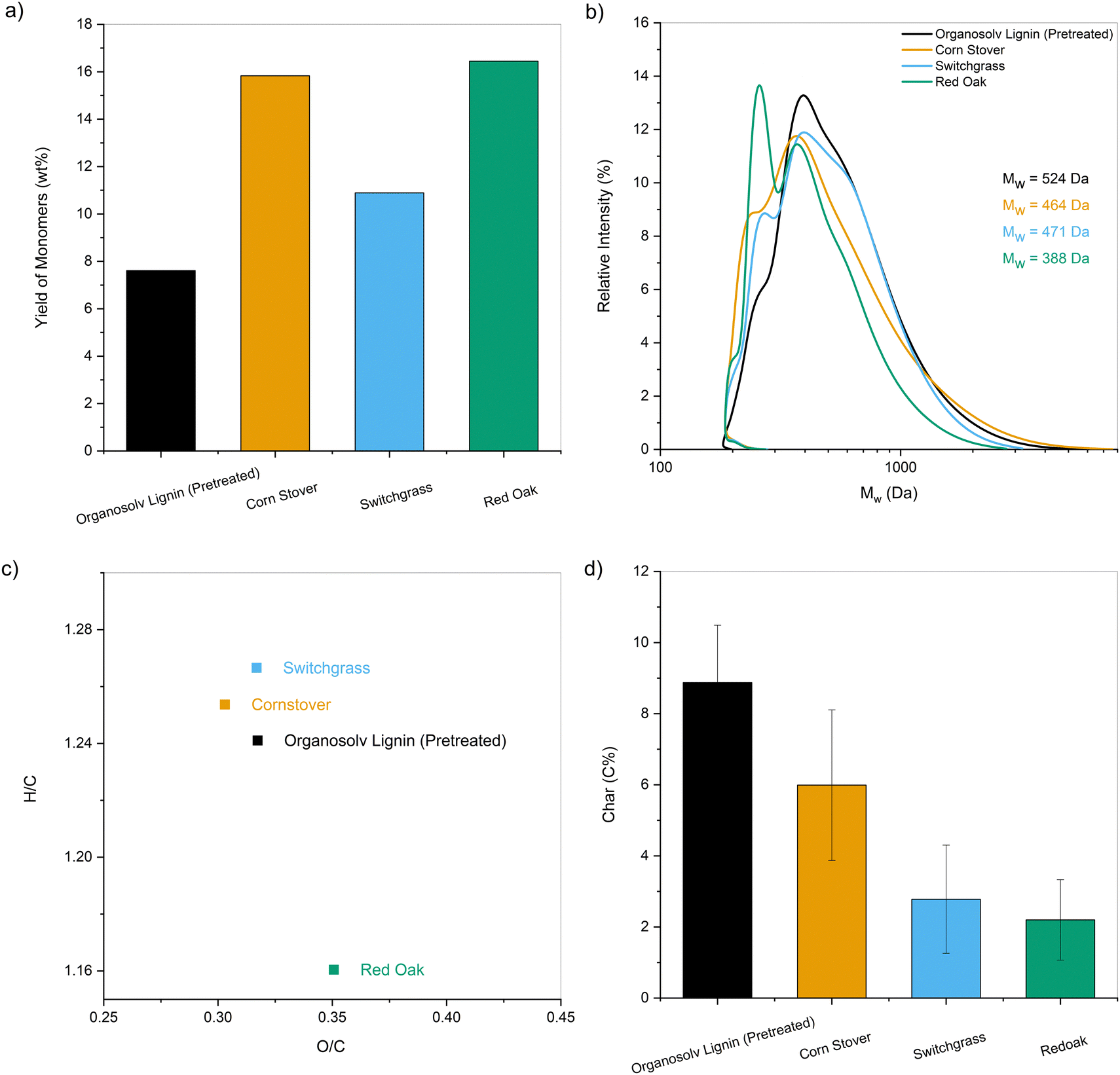 | ||
| Fig. 10 Characterization of the pretreated lignin oil obtained from the one-pot fractionation and pretreatment of whole biomass at 250 °C in EtOH over Pd/C. (a) Yield of monomers (wt%) relative to lignin content present in the starting biomass; (b) molecular weight distributions along with their average Mw; (c) Van Krevelen diagram; (d) char yields (C%) obtained after fast pyrolysis at 500 °C. The results for pretreated organosolv lignin are also provided for comparison. | ||
Similarly, alkene and aromatic hydrocarbon yields obtained for HDO of the lignin pyrolytic vapors using MoO3 catalyst range between 16 C% and 29 C%.16,19,68,75 As observed for CFP, an increase in yields of hydrocarbons was observed up to 33–36 C% for pretreated lignin oils obtained from biomass (Fig. S9b, ESI†). Further HDO optimization was performed by increasing the MoO3 catalyst to 20 mg for the lignin oils obtained from the one-pot fractionation and pretreatment of biomass. Compared to technical lignins, nearly twice the amount of carbon was volatilized during pyrolysis for the pretreated samples. Thus, the proportional increase in MoO3 kept the mass ratio of pyrolysis vapors to catalyst loaded constant for all samples. Fig. 11 presents the carbon yields obtained for the HDO of the one-pot pretreated lignin oils along with other technical lignins (the corresponding product distributions are provided in Table S2, ESI†). The yield of hydrocarbon-range products was significantly higher for the one-pot fractionated and pretreated samples than for the technical lignins (49–56 C% vs. 20–30 C%). It was apparent that lignin oil obtained after pretreatment could reduce the overall yield of char and increase the amount of volatilized carbon to increase the yield of valuable products upon upgrading.
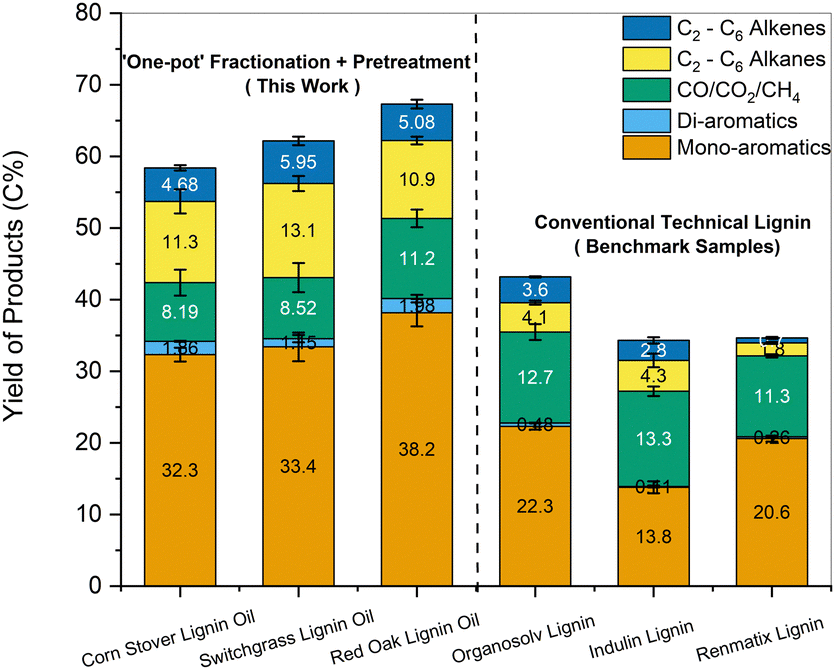 | ||
| Fig. 11 Carbon yields of products obtained from HDO of the lignin samples after ‘one-pot’ fractionation and pretreatment of whole biomass. Results obtained with conventional technical lignin samples are also provided for comparison. Conditions: MoO3 catalyst, fast pyrolysis: 500 °C, catalytic upgrading: 400 °C, catalyst:volatile = 40:1. | ||
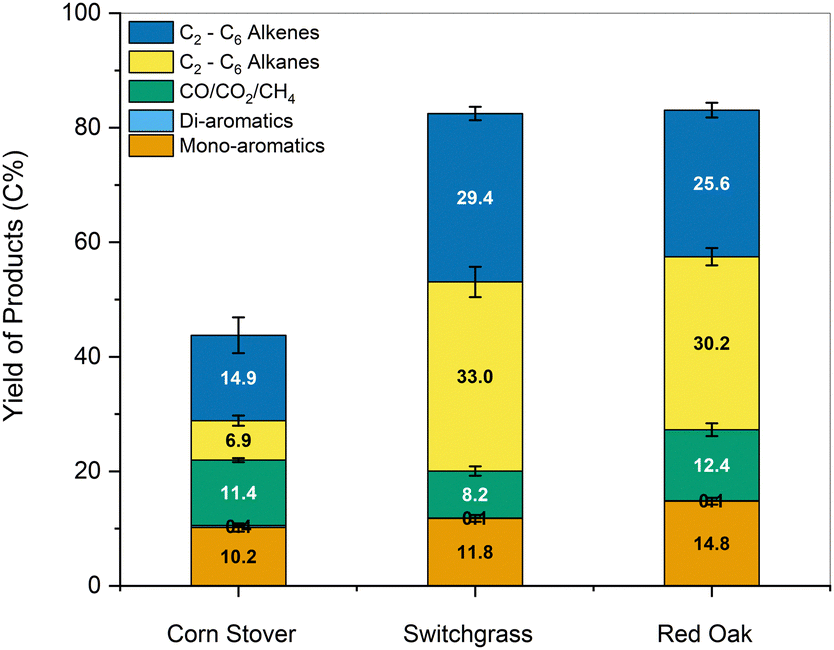 | ||
| Fig. 12 Carbon yields of products obtained from HDO of the water-soluble fraction recovered after ‘one-pot’ fractionation and pretreatment of whole biomass. Conditions: MoO3 catalyst, fast pyrolysis: 500 °C, catalytic upgrading: 400 °C, catalyst:volatile = 40:1. | ||
2.4. Enzymatic hydrolysis of EtOH-insoluble pulp
We further performed the enzymatic hydrolysis on the EtOH insoluble pulp obtained from corn stover to investigate whether the cocktail enzymes could digest the sugars present upon pretreatment. This analysis also helped rule out detrimental reactions between the reagents and sugars to which enzymes are sometimes sensitive. The results of the 72 h digestion are reported in Fig. 13 and Table S4 (ESI†). The residual solids showed a three-fold improvement in glucan and a four-fold improvement in xylan conversions compared to untreated biomass. Ferrini et al. stated that the quality of pulp obtained from RCF of biomass possessed superior quality compared to residual pulps obtained from organosolv fractionation. This was associated with the passivation of the lignin stream, which decreased the likelihood of lignin redeposition onto the fibers through recondensation processes.39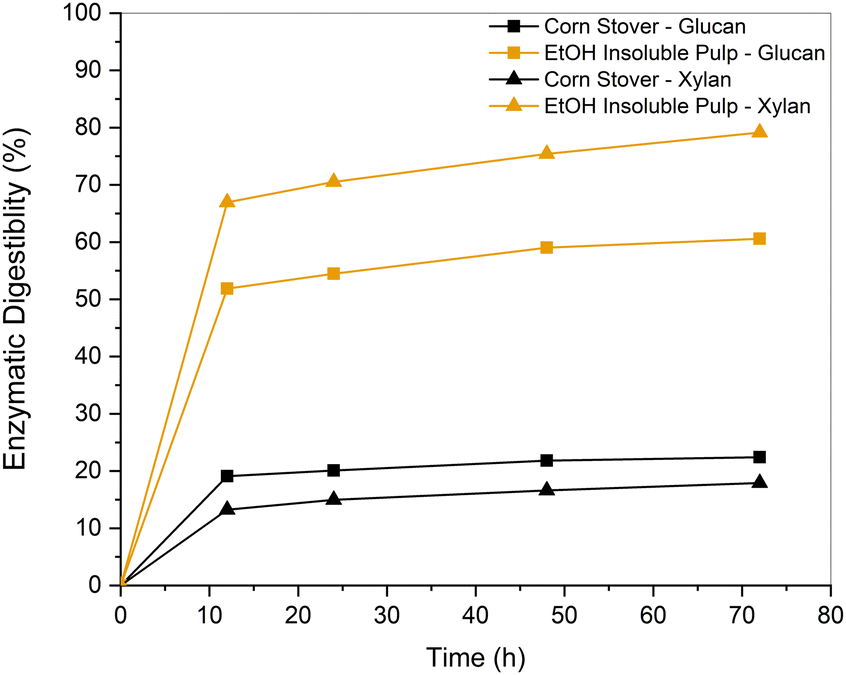 | ||
| Fig. 13 Enzymatic digestion performed on EtOH-insoluble pulp and untreated corn stover as a control experiment at a digestion time of 72 h. | ||
2.5. Remarks on the performance of the proposed approach for whole biomass upgrading in biorefineries
TEA/LCA studies highlighted the vital role of lignin valorization for biorefineries to become economically viable.19,32 To this end, various strategies were explored to deconstruct lignocellulosic biomass into separate carbohydrate-rich and lignin-rich streams that could be individually upgraded to value-added products. “Lignin-first” strategies that release lignin from plant biomass were shown to be the most effective to provide a processable lignin along with a cellulosic pulp amenable to bioethanol production.32,39,76 Significant work focused on the release of lignin monomers and small oligomers that could find applications in inks, adhesives, and other high-value products.76–78 However, there is a discrepancy between the size of these niche markets and the large volumes of bioethanol produced from the sugar-rich pulp. Therefore, pathways to commodity chemicals like mono-aromatics (benzene, toluene, xylene) and alkenes (ethylene, propylene, butene, butadiene) are significantly more attractive.19,77 Recent TEA revealed that the coproduction of olefins and aromatics could lower the minimum selling price of ethanol from $2 per gal to $0.75 per gal.19Several strategies were explored to convert “lignin-first” monomers to commodity aromatic hydrocarbons and oxygenates (phenol, cresol). However, very few studies tracked carbon atoms throughout the upgrading process, from the lignocellulosic feedstock to the final chemicals. The most comprehensive investigations to date are compiled in Table 1 and Table S5 (ESI†) and compared with the results obtained in the present work (our method for carbon tracking is provided in the ESI†). In their seminal Science article, the Sels group reported on the “lignin first” deconstruction and upgrading of birch and pine wood lignin to phenol, cresols, and propylene. They achieved a cumulative product yield of 33.3 wt% relative to the feedstock's lignin fraction (12.8 wt% relative to whole biomass).76 The Rinaldi group performed the hydrodeoxygenation of fractionated lignin-oil using a phosphidated Ni/SiO2 catalyst. Aromatic and aliphatic hydrocarbons were produced with excellent yields under their reaction conditions, reaching 33.8 and 11.3 wt%, respectively, relative to lignin (10 wt% total relative to whole biomass).79 However, these exceptional yields came at the expense of a broad product distribution ranging from C6 to C20, with a large number of branched and di-aromatic compounds, which would preferentially find applications as biofuels.
| Feedstock (reference) | Fractionation conditions | Upgrading conditions | Yield (wt% reference to lignin content) | Yield (wt% reference to mass of biomass) |
|---|---|---|---|---|
| Birchwood (Liao et al.)76 | 235 °C, methanol, 5 wt% Ru/C, 30 bar H2, 3 h | Hydroprocessing | Phenol: 19.8 wt% | Phenol: 7.1 wt% |
| 305 °C, 64 wt% Ni/SiO2 | Propylene: 9.3 wt% | Propylene: 3.7 wt% | ||
| Dealkylation | Cresols, benzenes, others:4.2 wt% | Cresols, benzenes, others:2.0 wt% | ||
| 410 °C, Z140-H | ||||
| Poplar (Cao et al.)79 | 180 °C, 2-propanol:water (7:3 v/v), RANEY® Ni, 3 h |
HDO | Aromatic C6–C10 and C14–C20:33.8 wt% |
Aromatic C6–C10 and C14–C20:7.5 wt% |
| 300 °C, phosphidated | Aliphatic C6–C10 and C14–C20:11.3 wt% |
Aliphatic C6–C10 and C14–C20:2.5 wt% |
||
| Ni/SiO2 | ||||
| Red Oak (this work) | 250 °C, ethanol, 5 wt% Pd/C, 30 bar H2, 3 h | Fast pyrolysis | Aromatics: 28.8 wt% | Aromatics: 9.5 wt% |
| 500 °C | Alkenes: 4.0 wt% | Alkenes: 3.3 wt% | ||
| HDO | Alkanes: 8.7 wt% | Alkanes: 5.2 wt% | ||
| 400 °C, MoO3 |
In comparison, the present work produced 28.8 wt% of mono-aromatics, 4.0 wt% of C2–C6 alkenes, and 8.7 wt% of C2–C6 alkanes when processing red oak. Benzene, toluene, xylenes, ethylbenzene, and propylbenzene accounted for 79% and 85% of the mono-aromatics produced from the lignin and water-soluble fractions, respectively (Tables S2 and S3, ESI†). Similarly, ethylene, propylene, and butenes represented 45–47% of all the olefins produced from the two fractions. In addition to generating drop-in commodity chemicals, our approach achieved a cumulative hydrocarbon yield of 18.0 wt% relative to the initial biomass in the case of red oak, along with 98% glucan retention in the pulp (see Section 2.3 and Fig. 9). For completeness, we also tracked carbon atoms from raw red oak biomass to final products. As can be seen in Fig. 14, 29.4 C% of red oak was converted to hydrocarbons while 43.5 C% were recovered as highly amenable pulp, giving a total carbon recovery of 72.9%. Our approach also presents technical advantages as the hydrodeoxygenation was performed under ambient pressure conditions, overcoming the need for costly high-pressure reactors for this step.
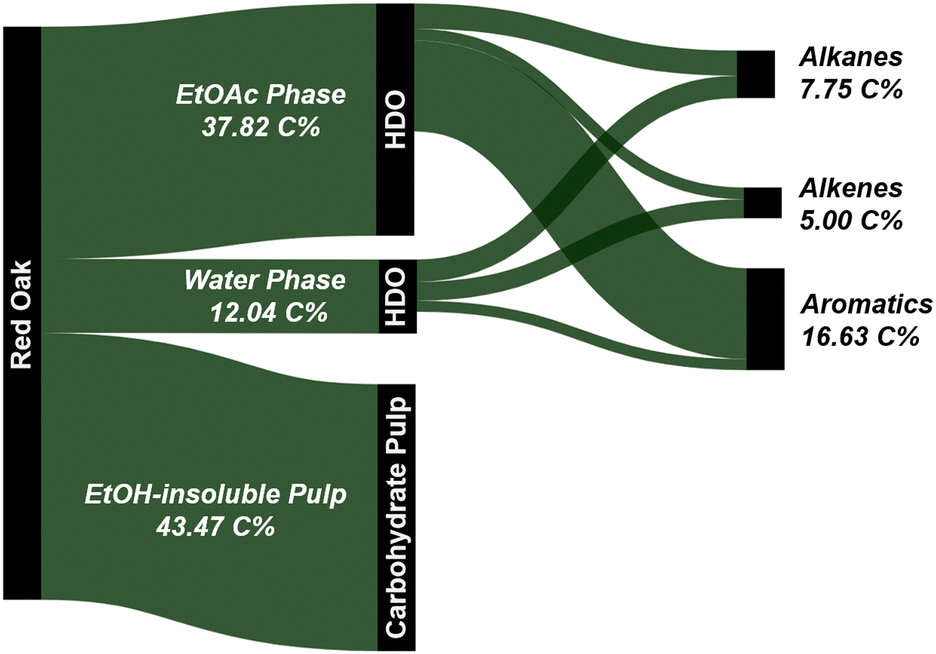 | ||
| Fig. 14 Carbon flow (upper bound values) for the one-pot fractionation and pretreatment of whole biomass in EtOH at 250 °C using Pd/C followed by HDO of both the ethyl acetate and water fractions. | ||
Global warming potential (GWP) calculations are particularly challenging for emerging technologies due to the many parameters involved during technology optimization and scale up. Nevertheless, the TEA-LCA study by Bartling et al. for an ethanol biorefinery incorporating RCF is encouraging as it revealed that the valorization of the lignin fraction yields a GWP of −0.175 kg CO2 eq. per kg when ethanol is used as a solvent, hence a net decrease in CO2 emissions.32 Moreover, the calculated GWP did not consider the GWP of the petrochemicals that would be displaced by the corresponding bioproducts. The manufacture of fossil-derived platform alkenes and aromatics (ethylene, propylene, and benzene/toluene/xylene) is responsible for nearly 250 megatons of carbon dioxide equivalent per year.80 Although a detailed TEA-LCA remains to be conducted for our approach, we anticipate that the advances presented here will further improve the GWP of lignin-first biorefineries and contribute to the decarbonization of the chemical industry.
3. Conclusion
The pretreatment of lignin was investigated using solvolytic and catalytic pretreatments to partially depolymerize it and enhance its conversion to aromatic hydrocarbons through CFP and HDO reactions. EtOH was chosen as the reaction solvent due to its ability to solubilize and depolymerize lignin along with Pd/C, Ru/C, Pd/Al2O3, Ru/Al2O3, and Y zeolite catalysts at sub- and supercritical conditions. At 200 °C, nearly 60 wt% of the loaded corn stover organosolv lignin was isolated after 3 hours irrespective of the catalyst added. At supercritical conditions (250 °C), a reductive catalyst was required to suppress detrimental recondensation reactions. Elemental analysis showed that a greater extent of deoxygenation took place at 250 °C compared to 200 °C. It was also inferred that deoxygenation was carried out mainly through solvolytic deconstruction. In contrast, the transition metal catalysts are primarily involved in the hydrogenation of the products, as evidenced by the similar O/C ratios and varying H/C ratios.The partially deconstructed samples’ molecular weight distributions ranged between 1000 and 1386 Da using subcritical EtOH and 520–800 Da with supercritical EtOH. The volatile monomeric products obtained upon pretreatment of corn stover lignin as a function of various reaction conditions and catalysts were analyzed by GC-FID/MS. Higher monomeric product yields were obtained with supercritical EtOH than with subcritical EtOH (4.0–7.5 wt% vs. 1.5–4.0 wt%), and the product distribution greatly varied upon the catalyst used in the system.
The thermal characteristics of the pretreated samples and parent lignin samples were studied using TGA and proximate analysis. In general, a larger portion of the products was volatilized from the pretreated samples than from the parent lignin. These results were corroborated by the proximate analysis. Specifically, 83 wt% of the volatile fraction was observed for the sample pretreated using Pd/C catalyst in EtOH at 250 °C compared to 63 wt% for the parent lignin. During fast pyrolysis of the pretreated samples, the yield of pyrolytic char dropped from 58.4 C% for lignin down to 8.8 C% for the sample deconstructed at 250 °C using Pd/C, and it subsequently enhanced the formation of volatile species.
The lignin sample that was deconstructed using Pd/C and supercritical EtOH was subjected to upgrading to aromatic hydrocarbons through CFP and HDO routes, and the yields were significantly higher than that of the parent lignin (96% increase for CFP and 38% increase for HDO).
We extended this approach to the one-pot fractionation and pretreatment of lignin from whole biomass for low-carbon-footprint chemical production. The lignin oil generated through this pretreatment was funneled to platform hydrocarbons, while the carbohydrate pulp was amenable to bioethanol production. The yield of mono-aromatics, di-aromatics, and alkenes improved significantly upon one-pot pretreatment and fractionation of lignin from biomass using Pd/C catalyst at 250 °C (39–45 C%) compared to other technical lignin (16–26 C%) generated using conventional fractionation techniques. The same HDO conditions were subsequently applied to the carbohydrates-rich water-soluble fraction to reduce waste and improve the overall carbon efficiency of the studied process. This step generated primarily C2–C4 alkenes and aromatic hydrocarbons. It also showed that all EtOH-soluble products could be upgraded using HDO, circumventing the need for a costly extraction/separation step. Overall, the production of platform alkenes and aromatics increased by up to 174%, while the residual pulp was rich in sugars and 200% more digestible, making it more amenable to bioethanol production. The total carbon efficiency of the complete process, from raw biomass to final products, was 72.9%.
4. Experimental
Chemicals and materials
Corn stover-derived organosolv lignin was obtained from Archer Daniels Midland (ADM). Renmatix lignin (RL) extracted from mixed hardwood using supercritical water extraction was provided by Renmatix and used without any further treatment. Indulin lignin (IL) extracted from pinewood is an unsulfonated kraft lignin obtained from Ingevity. Corn stover, switchgrass, and red oak biomass samples were provided by Iowa State University's BioCentury Research Farm. HPLC grade ethanol (EtOH, Decon Labs) was used as the reaction solvent for all experiments. Commercial 5 wt% Pd/C, 5 wt% Ru/C, 5 wt% Pd/Al2O3, 5 wt% Ru/Al2O3 were purchased from Sigma-Aldrich and were used as received. HY zeolite (CBV 720 with SiO2/Al2O3 = 30) and ZSM-5 (CBV 3024E with SiO2/Al2O3 = 30 in its ammonium form) were purchased from Zeolyst International. The purchased NH4-ZSM-5 was calcined in air at 550 °C for 10 hours (ramp: 5 °C min−1) to obtain the acidic form HZSM-5 of the zeolite before catalytic testing. MoO3 (Sigma-Aldrich, reagent grade) was calcined in a muffle furnace at 550 °C prior to catalytic testing. Polystyrene standards purchased from Agilent (Agilent Technologies, Inc., Santa Clara, CA) were used to calibrate molecular weights as a function of retention times for the GPC analysis. Tetrahydrofuran (Certified), which contains about 0.025% butylated hydroxytoluene as a preservative, was purchased from Fisher Scientific. For a complete list of commercial standards used for gas chromatography calibration, the reader is kindly referred to the ESI† (Table S6).Lignin pretreatment
Approximately 200 mg of lignin was added to 40 mL EtOH in a 75 mL Parr 4590 reactor along with 40 mg of the catalyst. After the reactor was sealed, it was purged three times with nitrogen. Next, the reactor was charged with 30 bar of hydrogen at room temperature. The reactor was then heated to either 200 or 250 °C (ramp: 10 °C min−1) and held at the target temperature for 3 h while stirring at 400 rpm. The pressure in the reaction vessel reached 70 bar at 200 °C and 110 bar at 250 °C. After the reaction, the reactor was cooled and depressurized at room temperature.Approximately 1.5 mL of the product dissolved in the solvent was collected, filtered using a 0.22 μm nylon syringe filter, and subjected to GC-FID/MS analysis. The remaining reactor content was recovered, the reactor was washed thoroughly with excess solvent, and the two solutions were combined in the same beaker. The mixture was then filtered using a 0.2 μm poly(ether sulfone) filter to separate the catalyst, and the obtained filtrate was dried. It is important to note that char and very high molecular weight oligomers are insoluble in EtOH at room temperature and were thus not recovered after pretreatment. The mass of the sample obtained after pretreatment was measured after drying it in a vacuum oven overnight at 40 °C. The obtained pretreated samples were further characterized through GPC, elemental analysis, HSQC NMR, and TGA analysis.
Fast pyrolysis of the parent lignin and pretreated samples
Untreated lignin and the pretreated samples were pyrolyzed in a two-stage Frontier tandem micropyrolyzer system (Frontier laboratory, Japan) equipped with an auto-shot sampler (Rx-3050 TR, Frontier Laboratories, Japan) connected to an Agilent 7890B GC-FID/TCD/MS (Fig. S10, ESI†). A deactivated stainless-steel cup containing approximately 500 μg of the sample was dropped into a first-stage reactor preheated at 500 °C, where the sample was pyrolyzed. Prior investigations have shown that the heating rate for this system is 500 °C s−1.81 Helium gas was used as both the sweep and carrier gas (Fig. S11, ESI†). The second-stage reactor was maintained at 300 °C to avoid condensation of the pyrolytic products. The vapors exiting the pyrolyzer were directly carried to the online GC for analysis. Char yields were measured gravimetrically by weighing the sample cups before and after pyrolysis. For the purpose of reporting on carbon yields, the carbon content of char was assumed to be approximately 90% based on previous reports.82CFP and HDO
CFP and HDO reactions were carried out in the same micropyrolyzer system as the one used for fast pyrolysis. For CFP, a quartz tube containing approximately 10 mg of the zeolite catalyst (210–297 μm) was inserted in the second-stage reactor to act as a packed bed reactor (Fig. S12, ESI†). The pyrolysis reactor and the catalyst bed temperatures were both set at 500 °C.For HDO, hydrogen gas was used as the sweep and carrier gas. The pyrolyzer and the second stage reactor were kept at 500 and 400 °C, respectively. Approximately 10 mg of MoO3 (37–105 μm) catalyst was mixed with ∼70 mg acid-washed glass beads (149–210 μm), fixed between two layers of quartz wool, and placed in the second reactor. The catalyst was reduced at 400 °C for 1 h in situ before reaction. The product yields were determined based on the following formula:
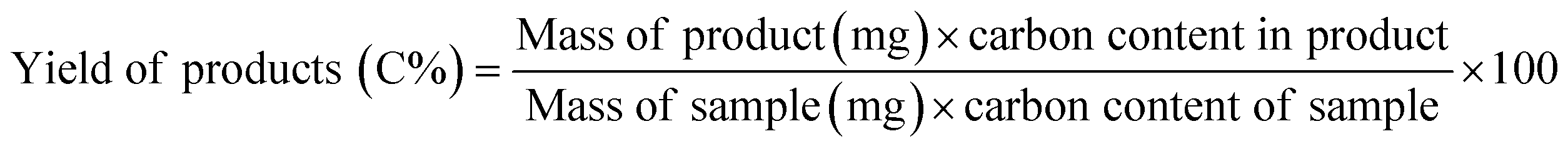
All interfaces, including the GC inlet, were kept at 300 °C to minimize condensation and repolymerization of generated vapors. The glass beads and quartz wool were previously tested and found to be inactive under reaction conditions.19
One-pot fractionation and pretreatment of lignin from biomass
Approximately 1 g of biomass with a particle size ≤2 mm, 100 mg of 5 wt% Pd/C, and 40 mL EtOH were added to a 75 mL Parr 4590 reactor. The reactor was sealed and purged with nitrogen three times. Next, the reactor was pressurized with 30 bar of hydrogen, heated to 250 °C (at a heating rate of 10 °C min−1), and held for 3 h while stirring at 400 rpm. To obtain an adequate amount of sample for further characterization, all experiments were repeated for a minimum of 5 times.After the reaction, the reactor was cooled down to room temperature and depressurized. The solid residue (Pd/C and EtOH-insoluble pulp) was separated by filtering the reactor contents using a 0.2 μm poly(ether sulfone) filter and washed with 30 mL EtOH. The catalyst was separated from the EtOH-insoluble pulp using a 75 μm sieve. EtOH was evaporated from the EtOH-soluble products by drying the filtrate overnight in a vacuum oven. The starting biomass, the EtOH-insoluble pulp, and the EtOH-soluble products were subjected to composition analysis to perform an overall mass and component balance. The EtOH-insoluble pulp obtained from corn stover was also subjected to enzymatic hydrolysis to determine the digestibility of pulp compared to the parent corn stover.
To separate the soluble lignin- and carbohydrates-derived products present in the EtOH-soluble fraction, liquid–liquid extractions were performed three times with ethyl acetate and water. The lignin oil was mainly concentrated in the ethyl acetate phase, while the aqueous phase contained dissolved sugars.69,83 The pretreated lignin oil was recovered by evaporating the ethyl acetate from the sample in a vacuum oven at 40 °C. The obtained lignin oil was characterized by GC-FID/MS, GPC, elemental analysis and further upgraded using CFP and HDO. Similarly, the sugar rich aqueous fraction was evaporated to remove water using a vacuum oven at 40 °C for two days. The dried sugar fraction obtained was also subjected to catalytic upgrading through HDO.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under the grant number CBET-1706046 and JPT's Richard C. Seagrave Professorship. The authors thank Iowa State University's BioCentury research farm for the biomass samples. We also acknowledge Dr Patrick Johnston for help with GPC and other analyses.References
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- M. Lievonen, J. J. Valle-Delgado, M.-L. Mattinen, E.-L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- C. C. Xu, L. Dessbesell, Y. Zhang and Z. Yuan, Biofuels, Bioprod. Biorefin., 2020, 15, 32–36 CrossRef.
- A. Saraeian, M. W. Nolte and B. H. Shanks, Renewable Sustainable Energy Rev., 2019, 104, 262–280 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Oasmaa, D. C. Elliott and J. Korhonen, Energy Fuels, 2010, 24, 6548–6554 CrossRef CAS.
- A. Eschenbacher, P. Fennell and A. D. Jensen, Energy Fuels, 2021, 35, 18333–18369 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Cheng, M. Addy, Q. Lu, M. M. Omar, Y. Liu, Y. Wang, L. Dai, E. Anderson, P. Peng, H. Lei and R. Ruan, Bioresour. Technol., 2017, 241, 1118–1126 CrossRef CAS PubMed.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 RSC.
- D. J. Mihalcik, C. A. Mullen and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2011, 92, 224–232 CrossRef CAS.
- Y. Yu, X. Li, L. Su, Y. Zhang, Y. Wang and H. Zhang, Appl. Catal., A, 2012, 447–448, 115–123 CrossRef CAS.
- S. Kelkar, C. M. Saffron, K. Andreassi, Z. Li, A. Murkute, D. J. Miller, T. J. Pinnavaia and R. M. Kriegel, Appl. Catal., B, 2015, 174, 85–95 CrossRef.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- M. W. Nolte, J. Zhang and B. H. Shanks, Green Chem., 2016, 18, 134–138 RSC.
- G. Zhou, P. A. Jensen, D. M. Le, N. O. Knudsen and A. D. Jensen, Green Chem., 2016, 18, 1965–1975 RSC.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98–110 RSC.
- A. Saraeian, A. Aui, Y. Gao, M. M. Wright, M. Foston and B. H. Shanks, Green Chem., 2020, 22, 2513–2525 RSC.
- M. W. Nolte, A. Saraeian and B. H. Shanks, Green Chem., 2017, 19, 3654–3664 RSC.
- K. B. Ansari, J. S. Arora, J. W. Chew, P. J. Dauenhauer and S. H. Mushrif, Ind. Eng. Chem. Res., 2019, 58, 15838–15852 CrossRef CAS.
- M. Carrier, M. Windt, B. Ziegler, J. Appelt, B. Saake, D. Meier and A. Bridgwater, ChemSusChem, 2017, 10, 3212–3224 CrossRef CAS PubMed.
- P. D. Muley, C. Henkel, K. K. Abdollahi, C. Marculescu and D. Boldor, Energy Convers. Manage., 2016, 117, 273–280 CrossRef CAS.
- A. R. Stanton, K. Iisa, C. Mukarakate and M. R. Nimlos, ACS Sustainable Chem. Eng., 2018, 6, 10030–10038 CrossRef CAS.
- Y. Feng, G. Li, X. Li, N. Zhu, B. Xiao, J. Li and Y. Wang, Bioresour. Technol., 2016, 214, 520–527 CrossRef CAS.
- Z. Ma, A. Ghosh, N. Asthana and J. van Bokhoven, ChemCatChem, 2017, 9, 954–961 CrossRef CAS.
- J. Akhtar and N. A. S. Amin, Renewable Sustainable Energy Rev., 2011, 15, 1615–1624 CrossRef CAS.
- H.-j Huang and X.-z Yuan, Prog. Energy Combust. Sci., 2015, 49, 59–80 CrossRef.
- M. Y. Lui, B. Chan, A. K. L. Yuen, A. F. Masters, A. Montoya and T. Maschmeyer, ChemSusChem, 2017, 10, 2140–2144 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- D. Vincent Sahayaraj, L. A, E. M. Mitchell, X. Bai and J.-P. Tessonnier, Green Chem., 2021, 23, 7731–7742 RSC.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- J.-Y. Kim, S. Oh, H. Hwang, T.-S. Cho, I.-G. Choi and J. W. Choi, Chemosphere, 2013, 93, 1755–1764 CrossRef CAS PubMed.
- R. J. A. Gosselink, W. Teunissen, J. E. G. van Dam, E. de Jong, G. Gellerstedt, E. L. Scott and J. P. M. Sanders, Bioresour. Technol., 2012, 106, 173–177 CrossRef CAS PubMed.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- O. E. Ebikade, N. Samulewicz, S. Xuan, J. D. Sheehan, C. Wu and D. G. Vlachos, Green Chem., 2020, 22, 7435–7447 RSC.
- P. Ferrini, C. A. Rezende and R. Rinaldi, ChemSusChem, 2016, 9, 3171–3180 CrossRef CAS PubMed.
- S. Ghoreishi, T. Barth and D. H. Hermundsgard, ACS Omega, 2019, 4, 19265–19278 CrossRef CAS PubMed.
- L. Chen, A. P. van Muyden, X. Cui, Z. Fei, N. Yan, G. Laurenczy and P. J. Dyson, JACS Au, 2021, 1, 729–733 CrossRef CAS PubMed.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- S. Qiu, M. Wang, Y. Fang and T. Tan, Sustainable Energy Fuels, 2020, 4, 5588–5594 RSC.
- T. Vangeel, T. Renders, K. Van Aelst, E. Cooreman, S. Van den Bosch, G. Van den Bossche, S. F. Koelewijn, C. M. Courtin and B. F. Sels, Green Chem., 2019, 21, 5841–5851 RSC.
- J. Park, H. S. Cahyadi, U. Mushtaq, D. Verma, D. Han, K.-W. Nam, S. K. Kwak and J. Kim, ACS Catal., 2020, 10, 12487–12506 CrossRef CAS.
- A. Bjelić, M. Grilc, M. Huš and B. Likozar, Chem. Eng. J., 2019, 359, 305–320 CrossRef.
- R. Shu, Q. Zhang, L. Ma, Y. Xu, P. Chen, C. Wang and T. Wang, Bioresour. Technol., 2016, 221, 568–575 CrossRef CAS PubMed.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1999, 47, 1988–1992 CrossRef CAS PubMed.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Liu, H. I. Kenttämaa and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 2316–2322 CrossRef CAS.
- Y. Ye, Y. Zhang, J. Fan and J. Chang, Bioresour. Technol., 2012, 118, 648–651 CrossRef CAS PubMed.
- Y. Ye, Y. Zhang, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2011, 51, 103–110 CrossRef.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- R. M. Kalinoski, W. Li, J. K. Mobley, S. O. Asare, M. Dorrani, B. C. Lynn, X. Chen and J. Shi, ACS Sustainable Chem. Eng., 2020, 8, 18455–18467 CrossRef CAS.
- P. Halder, S. Kundu, S. Patel, R. Parthasarathy, B. Pramanik, J. Paz-Ferreiro and K. Shah, Energy Convers. Manage., 2019, 200, 112067 CrossRef CAS.
- N. Muhammad, Z. Man, M. A. Bustam, M. I. A. Mutalib and S. Rafiq, J. Ind. Eng. Chem., 2013, 19, 207–214 CrossRef CAS.
- M. Wądrzyk, R. Janus, M. Lewandowski and A. Magdziarz, Renewable Energy, 2021, 177, 942–952 CrossRef.
- S. Wang, K. Wang, Q. Liu, Y. Gu, Z. Luo, K. Cen and T. Fransson, Biotechnol. Adv., 2009, 27, 562–567 CrossRef CAS PubMed.
- S. Zhou, Y. Xue, A. Sharma and X. Bai, ACS Sustainable Chem. Eng., 2016, 4, 6608–6617 CrossRef CAS.
- D. Duan, H. Lei, Y. Wang, R. Ruan, Y. Liu, L. Ding, Y. Zhang and L. Liu, J. Cleaner Prod., 2019, 231, 331–340 CrossRef CAS.
- Z.-P. Lei, Z.-Q. Hu, H.-F. Shui, S.-B. Ren, Z.-C. Wang, S.-G. Kang and C.-X. Pan, Fuel Process. Technol., 2015, 138, 612–615 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2009, 85, 237–246 CrossRef CAS.
- X. Bai, K. H. Kim, R. C. Brown, E. Dalluge, C. Hutchinson, Y. J. Lee and D. Dalluge, Fuel, 2014, 128, 170–179 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2009, 84, 79–83 CrossRef CAS.
- Y.-C. Qu, Z. Wang, Q. Lu and Y. Zhang, Ind. Eng. Chem. Res., 2013, 52, 12771–12776 CrossRef CAS.
- C. Liu, X. Wang, F. Lin, H. Zhang and R. Xiao, Fuel Process. Technol., 2018, 169, 50–57 CrossRef CAS.
- P. S. Marathe, R. J. M. Westerhof and S. R. A. Kersten, Appl. Energy, 2019, 236, 1125–1137 CrossRef CAS.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727–735 RSC.
- A. Saraeian, S. J. Burkhow, D. Jing, E. A. Smith and B. H. Shanks, ACS Sustainable Chem. Eng., 2021, 9, 6685–6696 CrossRef CAS.
- W. Guan, X. Chen, C. W. Tsang, H. Hu and C. Liang, ACS Sustainable Chem. Eng., 2021, 9, 3529–3541 CrossRef CAS.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. S. Doherty and I. M. O’Hara, Green Chem., 2016, 18, 360–381 RSC.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, Energy Fuels, 2010, 24, 683–689 CrossRef CAS.
- T. Phongpreecha, N. C. Hool, R. J. Stoklosa, A. S. Klett, C. E. Foster, A. Bhalla, D. Holmes, M. C. Thies and D. B. Hodge, Green Chem., 2017, 19, 5131–5143 RSC.
- Z. Luo, K. Lu, Y. Yang, S. Li and G. Li, RSC Adv., 2019, 9, 31960–31968 RSC.
- G. Zhou, P. A. Jensen, D. M. Le, N. O. Knudsen and A. D. Jensen, ACS Sustainable Chem. Eng., 2016, 4, 5432–5440 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. V. D. Bossche, J. V. Aelst, S. V. D. Bosch, T. Renders, K. Navare, T. Nicolaï, K. V. Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. V. Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- Z. Sun, J. Cheng, D. Wang, T.-Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, Joule, 2018, 2, 1118–1133 CrossRef CAS PubMed.
- IEA, Technology Roadmap - Energy and GHG Reductions in the Chemical Industry via Catalytic Processes, IEA, Paris, 2013, https://www.iea.org/reports/technology-roadmap-energy-and-ghg-reductions-in-the-chemical-industry-via-catalytic-processes Search PubMed.
- J. Zhang, M. W. Nolte and B. H. Shanks, ACS Sustainable Chem. Eng., 2014, 2, 2820–2830 CrossRef CAS.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. Geoffrey Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef CAS.
- T. Guo, X. Li, X. Liu, Y. Guo and Y. Wang, ChemSusChem, 2018, 11, 2758–2765 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental section; GPC, TGA, HSQC NMR and proximate analysis of the pretreated lignin samples; composition analysis and component balance for the one-pot fractionation and pretreatment of whole biomass; additional HDO results; pictures of the reactor setup used for FP, CFP, and HDO tests. See DOI: https://doi.org/10.1039/d2ee02304k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |