
Active site imprinting on Ti oxocluster metal–organic frameworks for photocatalytic hydrogen release from formic acid†
Alberto
García-Baldoví
a,
Raquel
Del Angel
b,
Georges
Mouchaham
 *b,
Shanping
Liu
c,
Dong
Fan
c,
Guillaume
Maurin
c,
Sergio
Navalón
d,
Christian
Serre
*b and
Hermenegildo
Garcia
*a
*b,
Shanping
Liu
c,
Dong
Fan
c,
Guillaume
Maurin
c,
Sergio
Navalón
d,
Christian
Serre
*b and
Hermenegildo
Garcia
*a
aInstituto Universitario de Tecnología Química, Consejo Superior de Investigaciones Científicas-Universitat Politècnica de València, Universitat Politècnica de Valencia, Av. De los Naranjos s/n, 46023 Valencia, Spain. E-mail: hgarcia@qim.upv.es
bInstitut des Matériaux Poreux de Paris, Ecole Normale Supérieure, ESPCI Paris, CNRS, PSL University, Paris, France. E-mail: georges.mouchaham@ens.psl.eu; christian.serre@ens.psl.eu
cICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France
dDepartment of Chemistry, Universitat Politècnica de València, C/Camino de Vera, s/n, 46022, Valencia, Spain
First published on 9th November 2022
Abstract
On-board hydrogen release from liquid organic carriers is a process that can make feasible the use of H2 as a transportation fuel. Formic acid is considered as one of the most convenient liquid hydrogen organic carriers, and since it can be easily obtained from CO2, it is water soluble which makes it unnecessary to recover the H2-depleted byproducts. Compared to the more conventional thermal catalytic decomposition of formic acid, the use of light in combination with a photocatalyst has been much less explored. Herein, we report a new paradigm in MOF photocatalysis with the use of a microporous titanium oxocluster based metal–organic framework (Ti-MOF) endowed with formate-imprinted active sites, namely MIP-177-LT (MIP stands for Materials from Institute of Porous Materials of Paris, LT for Low Temperature), as a highly efficient photocatalyst for H2 release from formic acid without the need to neutralize acidity or use sacrificial agents or noble metals. Noteworthily, a quantum efficiency of 22% has been determined for the photocatalytic H2 release that is highly remarkable for a non-toxic noble metal-free photocatalyst.
Broader contextDue to the high mass energy density, green hydrogen is considered as the most adequate energy vector for transportation to replace fossil fuels. However, one of the major hurdles in hydrogen technology is the low volumetric energy density of compressed hydrogen. One way to overcome this limitation is the use of a liquid organic compound that can release on-board hydrogen upon demand. Among these liquid organic hydrogen carriers, formic acid is one of the most promising due to its easy preparation from CO2 and decomposition. In the present submission, it is reported that due to formate-imprinting a Ti-containing metal–organic framework, stable in formic acid reflux, is observed in the absence of any noble metal high photocatalytic H2 release with an apparent quantum efficiency of 22% in the UV region. Compared to conventional thermal catalysis, light has the advantage of an instantaneous on–off response to H2 release. The present photocatalytic material only contains abundant first-row transition metal (Ti) as the metal. |
Introduction
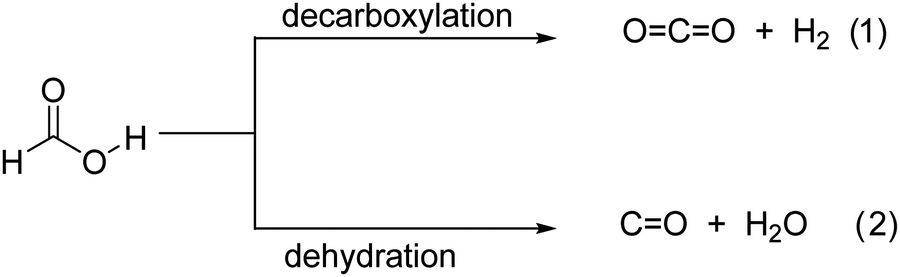
In the forthcoming decarbonized energy, hydrogen will play a key role as an energy vector.1 However, implementation of the hydrogen technology is hampered by several bottlenecks related to hydrogen generation from water using renewable energy, safe and adequate hydrogen storage and non-polluting hydrogen consumption. In the context of the use of hydrogen in transportation, one of the major problems is the low volumetric energy density of compressed hydrogen.2 One approach to overcome this issue is the use of liquid organic compounds (known as liquid organic hydrogen carriers, LOHCs) that can release onboard hydrogen on demand.3,4 Among the requirements for ideal LOHCs, the most important ones are easy synthesis, facile hydrogen release, lack of toxicity, water solubility and a high percentage of hydrogen release. Thus, formic acid (HCOOH, FA) is among the most investigated LOHCs not only owing to its hydrogen content (4.4 wt%),5 stability and non-toxic character, but also because of its availability from biomass as well as from CO2 hydrogenation. Besides, the HCOOH/CO2 system can be considered as an ideal environment-friendly system for hydrogen storage provided that CO2 is obtained from air, in which case the whole hydrogen storage/hydrogen release cycle is CO2 neutral. Indeed, dehydrogenation and re-hydrogenation reactions (HCOOH ↔ H2 + CO2; ΔG = −48.4 kJ mol−1)6 can be considered as a neutral CO2-footprint cycle.5Catalytic FA decomposition at room temperature has been reported using noble metals,6–10 making the process less applicable. Other transition metals can also promote hydrogen release from FA,11–15 but they require some heating, occur at a slower rate and are much less efficient. In addition, due to the known dissolution of metals in an acidic medium and the ability of FA to form soluble metal-coordination complexes, corrosion of the catalyst and/or metal leaching are some of the limitations generally encountered in such a process.16 To circumvent this issue, hydrogen release measurements have been reported for formate salts,17 but this makes again the process less attractive since no FA is directly used, but a derivative containing unwanted alkali metal ions or other charge compensating cations, such as NH4+. This makes necessary FA neutralization with bases.18 Moreover, another common problem of FA decomposition is the occurrence of some degree of FA dehydration (HCOOH ↔ H2O + CO; ΔG = − 28.5 kJ mol−1) taking place to some extent concomitantly with FA dehydrogenation.19 It appears that the monodentate binding mode of FA on the catalyst surface at the expense of bridging mode adsorption is more favorable for the unwanted dehydration reaction.6 The presence of CO from FA dehydration, even in minute proportions, is highly detrimental to the use of the released H2 in fuel cells, due to catalyst poisoning.20 Therefore, a suitable design of the active site to reach a highly selective dehydrogenation reaction is desirable.
One alternative to conventional thermal catalysis that has been considerably less explored is the photocatalytic FA decomposition. Photocatalysis can promote FA decarboxylation.21 In a general reaction mechanism, photogenerated holes abstract one electron from formic acid or formate giving a HCOO˙radical that decomposes spontaneously, giving a CO2 molecule and a hydrogen atom.21 Photogenerated electrons are consumed by H+ forming another hydrogen atom. Typical photocatalysts for FA decomposition are TiO2 materials in various crystalline phases, structural morphologies and dopants.22–25 However, even when using TiO2 as a photocatalyst, noble metal co-catalysts such as Pt,26,27 Au23,25 and Pd28 are generally employed to increase charge separation and to favor the occurrence of chemical reactions. Although much less studied, other photocatalysts besides TiO2, either independently or as heterojunctions, have also been tested for photocatalytic FA decomposition.21 The list includes toxic CdS29 or metal phosphides prepared using poisonous and hazardous phosphine,30 as well as BiVO4,31 reduced graphene oxide32–34 and carbon nitride.35 Also in these cases, these photocatalysts contain in most cases noble metals to enhance their catalytic activity.
Metal–organic frameworks (MOFs) are becoming very attractive candidates for catalysis in general and, for photocatalysis in particular.36–38 Thanks to the versatility of their hybrid (organic/inorganic) porous and highly ordered (crystalline) structure, their features can be finely tuned to deliver great catalytic performance with a minimal energetic penalty. MOFs have also been employed as passive hosts of active CdS, plasmonic Au@Pd or In2S3/Co(salen) photocatalysts for FA decomposition,39–41 observing in some cases the evolution of a significant CO proportion,39,41 whose presence is highly detrimental for the operation of hydrogen fuel cells. In these examples MOFs act as porous supports with negligible intrinsic activity. However, as far as we know, MOFs by themselves have not yet been considered as candidates for hydrogen release from FA in the absence of any sacrificial agents.
MOF synthesis is a very active area of research.42,43 As an example of the various synthetic approaches offered by MOFs, there is a current trend in MOF synthesis of using, besides the organic building units (multitopic ligands) required to connect the inorganic building units (IBUs), other organic additives known as modulators.42 These modulators are competing during the MOF synthesis with the structural ligands in terms of coordination with the metal ions of the IBUs, allowing a better control over the nucleation process and, hence, over the overall crystallinity of the resulting MOF sample. Beyond their impact on the crystallization process, these modulators can induce the formation of structural defects that could be beneficial for enhancing the MOF activity in various applications, particularly in catalysis and photocatalysis, by promoting accessible active metal sites and charge carrier trapping sites, respectively.44,45
Among the various modulators reported, monotopic carboxylic acids and particularly FA are some of the preferred agents.46,47 Depending on the MOF structure, the nature of the building units, and the synthetic conditions, the modulator can either (i) replace the structural linker, leading to a ligand defective structure, and/or (ii) occupy a free position inherently not affecting the structuring ligands. In both cases, the modulators can play a role in creating potential accessible active sites. It occurred to us that FA as a modulator can also be positively used to “imprint” in the resulting MOF structure an active site for photocatalytic FA decomposition in photoresponsive MOFs.
In this regard, Ti-MOFs are a particularly attractive sub-class of materials of most relevance as photocatalyst candidates, owing to their IBUs consisting of Ti metal ions are known for their photophysical response and redox photoactivity.48 While in general transition metals with incompletely filled d orbitals undergo a fast d ← d electronic relaxation of the excited states, d0 or d10 transition metals, like Ti4+ or Zn2+, not suffering from this deactivation pathway, are photocatalytically more efficient.48 Our hypothesis is that one of these Ti-MOFs, denoted MIP-177-LT,49 prepared in FA as a pure solvent under solvothermal conditions should be particularly efficient for FA decomposition, due to FA modulator templating during its synthesis.49 MIP-177-LT with a robust 3D microporous structure of formula Ti12O15(mdip)3(formate)6 (mdip: methylene di-isophthalate) is built up of Ti12O15 oxoclusters as IBUs, where the TiIV ions are connected by μ3-oxo bridges, carboxylate groups of different mdip ligands and formate groups as depicted in Fig. 1.49 As shown in this figure, there are two types of formate groups. Half of them (highlighted in green) are linking two adjacent equatorial Ti ions in each Ti12-oxocluster, and they are accessible through channels running along the c-axis. A second half (highlighted in yellow) are bridging two adjacent Ti12O15 clusters and they are not exposed to the micropores. MIP-177-LT exhibits also remarkable chemical stability, among the best MOFs, including stability against strong acids such as concentrated HCl, sulfuric and nitric acids, aqua regia and even 6 M H3PO4 which has been of interest for proton conductivity50 or release of nitric oxide under simulated body fluid conditions.51
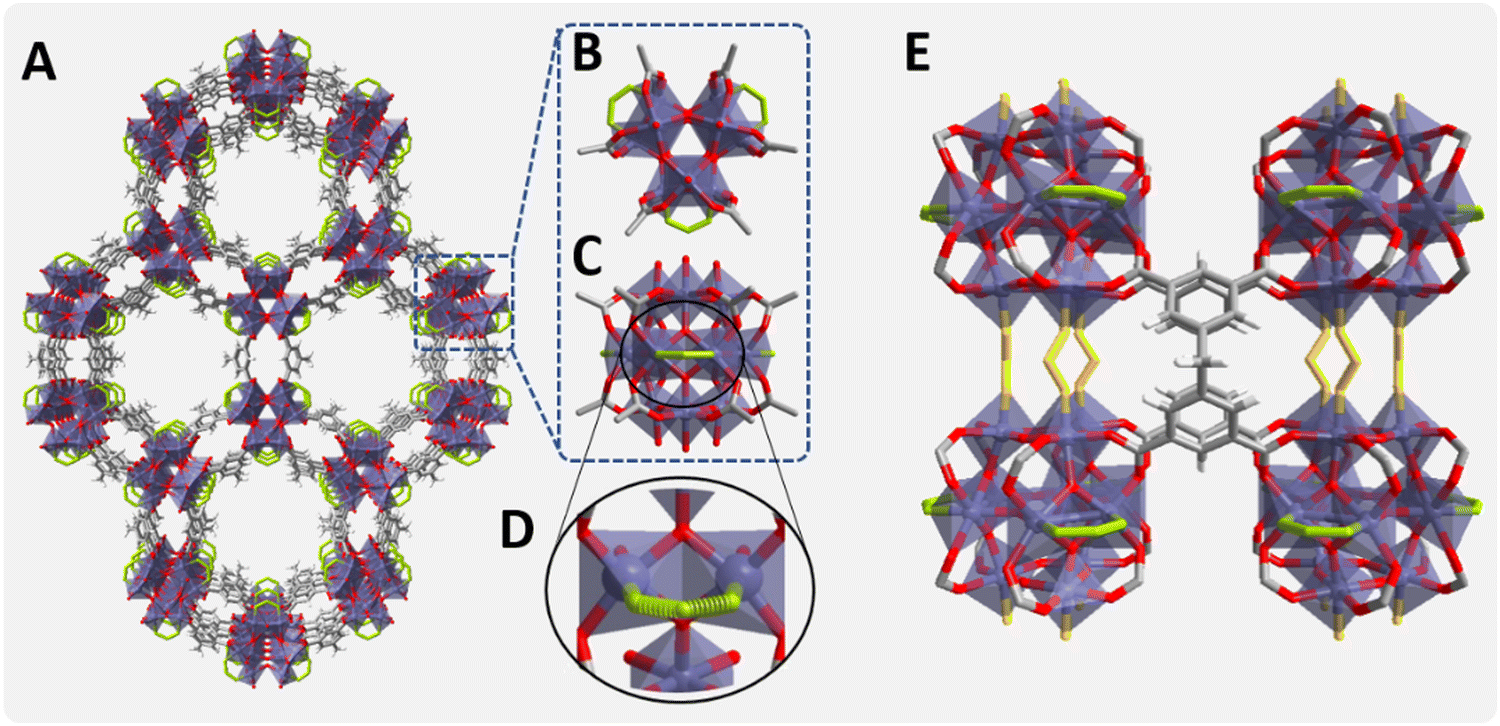 | ||
| Fig. 1 (A), Crystal structure of MIP-177-LT. (B and C) Ti12O15 cluster showing non-bridging formates (equatorial) in green, top and side views, respectively. (D) View of the equatorial formate-imprinted sites. (E) View showing in yellow the bridging formates between two adjacent Ti12 oxoclusters aligned along the c-axis. | ||
Considering the remarkable photocatalytic activity of Ti-MOFs, and the high chemical stability of MIP-177-LT to strong acids being prepared in FA at reflux temperature, it motivated us to consider this MOF as a formate-imprinted catalyst to promote the photocatalytic hydrogen release from FA. Formate imprinting makes the recognition of FA by active sites possible, fitting adequately in the structure. As will be shown below, containing exclusively Ti as the only metal in the absence of noble metals, MIL-177-LT is a very efficient and stable photocatalyst for hydrogen release from aqueous solutions of FA, comparing favorably with other Ti- and Zr-containing MOFs and with conventional TiO2 photocatalysts.
Results and discussion
The concept of the present study is summarized in Fig. 2 in which key aspects, like formate molecular imprinting, structural stability against strongly corrosive acids, the presence of Ti oxoclusters and other features of MIP-177-LT have been highlighted. The synthesis of MIP-177-LT was performed as reported49 from Ti(iPrO)4 as the molecular source of TiIV and di(isophthalyl)methane (H4mdip) as a linker in FA as a solvent under reflux conditions during 48 hours. MIP-177-LT synthesis is reliable and it can be scaled up to 100 g. The samples used for the present study exhibit the expected PXRD pattern, chemical composition and spectroscopic properties as reported before.49Fig. 3 shows the XRD characterization data of the MIP-177-LT sample under study. Besides the MIP-177-LT, another MIL-177-LT sample was subjected to acid treatment (MIP-177-LT-AT), to remove formate groups coordinated on the Ti sites, while only slightly decreasing somehow the BET area of the material that goes from 640 m2 g−1 for MIP-177-LT to 560 m2 g−1 for MIP-177-LT-AT. These two MOFs showed some differences in their optical properties, where a blue shift can be observed on the absorption onset wavelength in the diffuse reflectance UV-Vis absorption spectrum of MIP-177-LT-AT compared to MIP-177-LT (Fig. S1, ESI†), probably due to the removal of oligomeric TiOx impurities occupying some pore space. Additional characterization, including diffuse-reflectance optical spectra, diffraction patterns, N2 and CO2 adsorption/desorption isotherms and SEM images of the MIP-177-LT and MIP-177-LT-AT samples used in the present study, including particle morphology and XPS data are presented also in Fig. S1 (ESI†). For the sake of comparison, this study was also completed with MIL-125(Ti) and UiO-66(Zr) taken as reference MOF photocatalysts and commercial P25 TiO2. | ||
| Fig. 2 MIP-177 optimum features for effective photocatalytic H2 release from FA. | ||
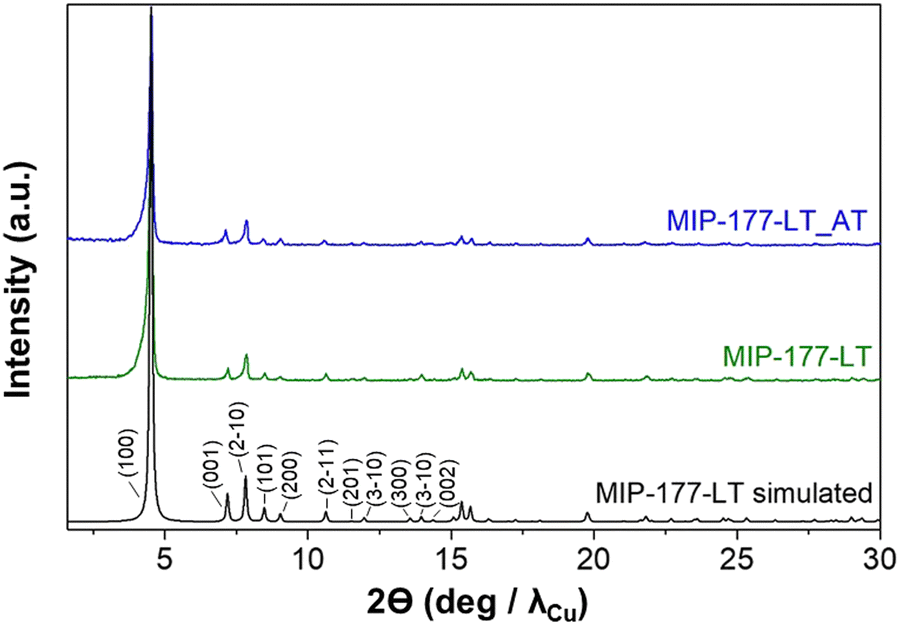 | ||
| Fig. 3 PXRD patterns of the pristine and the acid treated (AT) MIP-177-LT samples, in comparison with the simulated one for the crystal structure of MIP-177-LT in which the Miller indices are indicated for the first 10 reflections. | ||
Photocatalytic stability
Prior to the evaluation of the photocatalytic H2 release from FA, blank controls irradiating MIP-177-LT and MIP-177-LT-AT photocatalysts were performed. These initial screening experiments serve to determine the photostability of MIP-177-LT and MIP-177-LT-AT. Irradiations were carried out for 24 h periods as dry powders upon exposure to light of different wavelength regions. The results are summarized in Table S1 (ESI†). Previous reports in the literature have shown that carboxylate MOFs can undergo partial photodecarboxylation under UV light to a significant extent of up to 33% of the total carboxylate groups of the material during prolonged (months) irradiation times under UV-Vis light.52In the present case, it was found that irradiation through Pyrex with the full spectral output of a 300 W Xe lamp for 24 h of 10 mg of MIP-177-LT gives rise to the evolution of 8 μmol CO2 accompanied by two-orders of magnitude lesser amounts of H2. This CO2 evolution corresponds to about 8.8% of the carboxylate groups present in the material according to its theoretical Ti12O15(mdip)3(formate)6 formula. In comparison, CO2 evolution for the same period of time decreased by factors of 3.5 and 5.7 times less when the sample was irradiated with 360 and 400 nm cut-off filters, respectively. This corresponds to the photochemical decomposition in 24 h of 2.5 and 1.5% of the carboxylate groups present in the material according to the theoretical formula, meaning that short wavelengths from 300 to 360 nm are responsible for most (71%) of the MIP-177-LT self-decomposition and that irradiation with wavelengths longer than 400 nm diminishes MIP-177-LT damage in 91%.
Similar photostability studies were also performed on MIP-177-LT-AT. The same general trend, but with an even lesser CO2 evolution (maximum of 2.00 μmol at 24 h) was observed. Incidentally, for MIP-177-LT-AT generation of CH4 in a higher proportion than the H2 amount was detected.
These preliminary studies indicate that deep UV radiation of wavelengths shorter than 360 nm is responsible for most of the CO2 evolution and that part of these gases derive from species present inside the channels that can be removed by acid treatment, since MIP-177-LT-AT has 70% lesser CO2 and CH4 evolution. This less-damaging spectral range corresponds to the solar spectrum at the earth's surface.
To put into context these CO2 evolution values with those that will be reported below in FA decomposition, they are three orders of magnitude lower and in the case of H2 even lower. Therefore, the conclusion of these preliminary control studies is that self-decomposition is not relevant compared to the photocatalytic FA decomposition activity data presented herein below.
H2 release from photocatalytic FA decomposition
Photocatalytic H2 release from FA experiments was carried out at room temperature under continuous magnetic stirring of 10 mg of either MIP-177-LT or MIP-177-LT-AT in 15 mL of 10−3 M FA solution in water in a closed 51 mL Pyrex reactor upon illumination of a 300 W Xe lamp (see Fig. S2, ESI†). Gas evolution was periodically analyzed for 2 h irradiation runs by injecting the gases of the headspace in a micro-GC. According to the dehydrogenation and dehydration reactions (eqn (1) and (2)), photocatalytic decomposition of FA can afford stoichiometric H2 and CO2 or stoichiometric H2O and CO, respectively. In agreement with the preliminary photostability study, control experiments in the absence of FA under otherwise identical conditions or irradiation of FA in the absence of any MIP-177 photocatalysts show a negligible amount of H2 compared to the large values observed for the FA solutions.Noteworthily, the decomposition products observed in all the experiments with the presence of MIP-177 photocatalysts were predominantly H2 and CO2, accompanied by much lesser amounts of CH4. Importantly, the amount of CO was below the detection limit of our GC analysis, corresponding to CO concentrations lower than 0.1% in the gas composition, much smaller than the values of about 10% reported for TiO2.21,39 7.0 μmol H2, corresponding to over 46% of the theoretical stoichiometric amount for complete FA dehydrogenation (15 μmol), were quantified at 2 h of reaction time using fresh MIP-177-LT. Notably, while H2 evolution increased quite linearly with the irradiation time, the CO2 concentration profile slightly deviated from that of H2 at short irradiation times, but finally both reached a quasi-stoichiometric amount at 2 h of reaction time. Fig. 4A shows the temporal gas evolution measured for the fresh MIP-177-LT sample. Note that the continuous lines do not correspond to the fitting of the experimental points to a kinetic model.
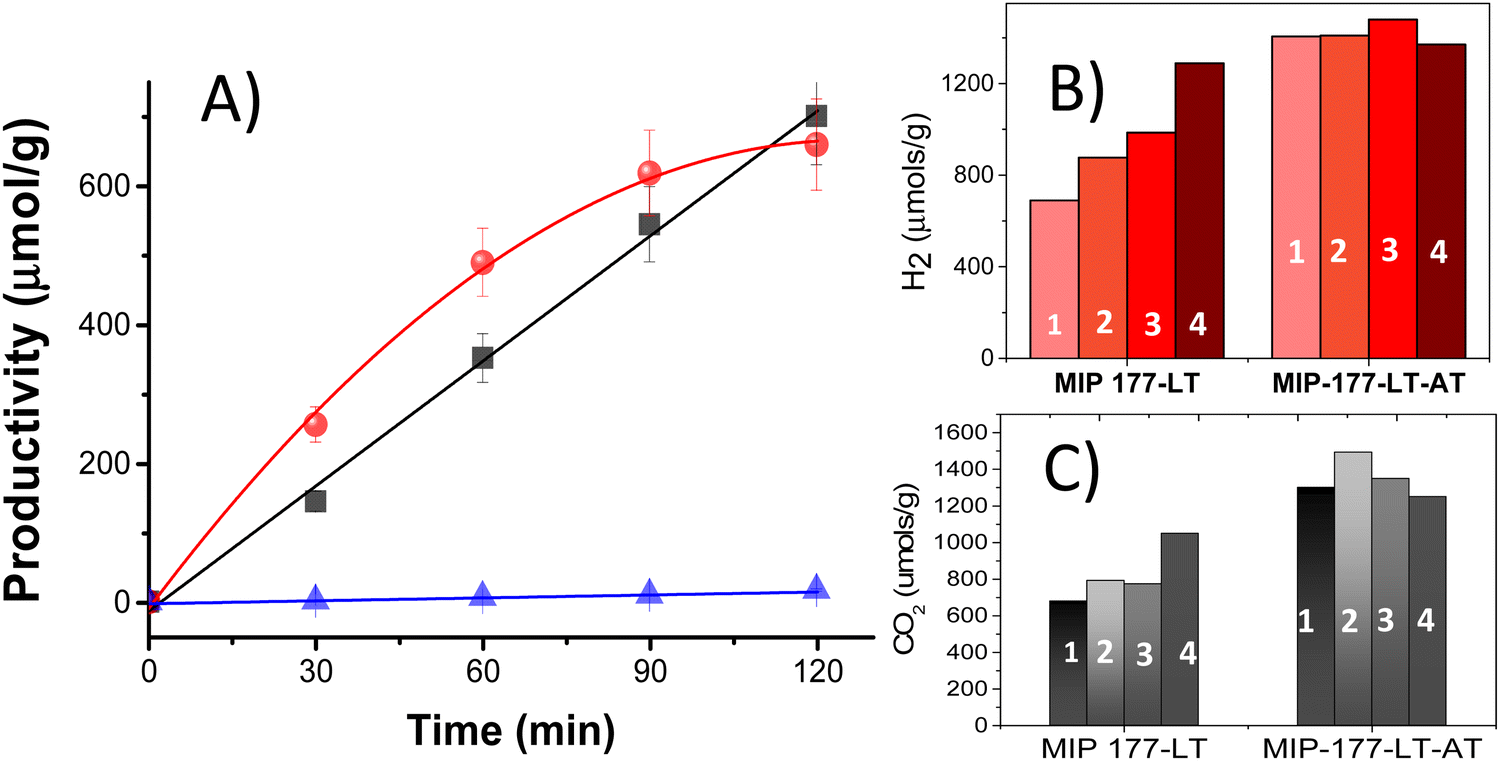 | ||
| Fig. 4 (A) Time dependent profile of H2 (black), CO2 (red) and CH4 (blue) upon UV-Vis irradiation of a 10−3 M FA aqueous solution in the presence of MIP-177-LT as a photocatalyst. The continuous lines do not correspond to a fitting using a kinetic model. (B) H2 or CO2 (C) production at 2 h of irradiation time in the presence of MIP-177-LT or MIP-177-LT-AT upon the reuse number as indicated in the drawing. Reaction conditions: photocatalyst 10 mg, solution volume 20 mL, FA concentration 10−3 M, and 300 W Xe lamp through Pyrex. | ||
In accordance with the expected influence of acid washings removing impurities, the photocatalytic activity of the acid treated MIP-177-LT-AT was even better and exhibit almost complete FA decomposition in 2 h with quasi-stoichiometric amounts of H2 (13.0 μmol g−1) and CO2 at 2 h of irradiation time (Fig. 4B and C). Also, some methane formation was observed for MIP-177-LT, while in comparison the presence of methane in the case of MIP-177-LT-AT as a photocatalyst was barely detectable.
Powder XRD and XPS data of the MIP-177-LT samples after the photocatalytic FA decomposition did not reveal any change (Fig. S3, ESI†), confirming the excellent stability of the materials under the reaction conditions, in good accordance with the reported stability of MIP-177-LT under strongly acidic conditions. In addition, surface area measurements of the MIP-177-LT sample used as a photocatalyst for H2 release and exhaustive washing with water indicated a BET area of 404 m2 g−1, that is somewhat lower than the value obtained for the fresh material, but still notably high confirming porosity stability. Similarly, SEM images of the used MIP-177-LT sample (Fig. S3, ESI†) show some abrasion of the particles that otherwise maintain the crystalline morphology of the fresh MIP-177-LT material.
In agreement with the catalyst stability in FA, reusability tests provided an important observation in the case of MIP-177-LT, i.e., H2 and CO2 evolution become faster and the final H2 amount evolved at 2 h reaction time continuously grows upon reuse as presented in Fig. 4B. Thus, the final H2 amount increased from 700 μmol g−1 to 877, 896 and 1289 μmol g−1 for the first, second, third and fourth use, respectively. The H2 amount released in the fourth use is about 86% of the maximum theoretical H2 amount according to the 10−3 M FA concentration. The apparent turnover frequency (TOF) obtained by dividing the evolved H2 mols by the mols of Ti atoms present in the photocatalyst also grows from 2.5 × 10−2 to 12.7 10−2 h−1 (see the ESI† for details of apparent TOF calculation). As commented earlier and seen in Fig. 4A, the use of fresh MIP-177-LT samples lead to the production of a higher CO2 concentration than the corresponding stoichiometric H2 amount at a short reaction time. In comparison, as the same MIP-177-LT sample is increasingly reused, CO2 evolution followed the same time-dependent profile as that of H2 (Fig. S4, ESI†) and the amounts of evolved CO2 and H2 in the fourth use were even significantly higher than for the fresh MIP-177-LT sample (Fig. 4B and C). Therefore, the enhanced photocatalytic CO2 evolution observed for the as-synthesized MIP-177-LT sample at initial reaction times is specific to this material and it tends to become corrected to the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2/H2 ratio upon extended irradiation. Furthermore, it is worth noting that the acid-treated MIP-177-LT-AT sample exhibits 1:1 CO2/H2 evolution since the start of the irradiation (Fig. S4, ESI†), meaning again that non-stoichiometric CO2/H2 evolution is exclusive of the fresh MIP-177-LT sample. Assuming that the charge separation state efficiency and other parameters related to the photocatalytic mechanisms dependent on the intrinsic electronic MIP-177-LT properties are constant upon reuse, TOF data indicate that about 5.1 more sites are becoming active upon MIP-177-LT reuse. This can be interpreted considering that only a fraction of the adjacent Ti atoms accessible from the pore space was initially available for the photocatalytic reaction in the fresh photocatalyst, and this percentage increases upon continuous irradiation, probably due to the decomposition of the structural formate groups bridging two equatorial Ti atoms of the same Ti12 cluster. The TOF enhancement by a factor of about 5 upon reuse would indicate that for the extensively used MIP-177-LT electrons and holes located at any of the six positions of equatorial bridging formates accessible from the micropores have become photocatalytically active upon extended use, increasing in this way the efficiency of charge separation in the generation of CO2 and H2.
:1 CO2/H2 ratio upon extended irradiation. Furthermore, it is worth noting that the acid-treated MIP-177-LT-AT sample exhibits 1:1 CO2/H2 evolution since the start of the irradiation (Fig. S4, ESI†), meaning again that non-stoichiometric CO2/H2 evolution is exclusive of the fresh MIP-177-LT sample. Assuming that the charge separation state efficiency and other parameters related to the photocatalytic mechanisms dependent on the intrinsic electronic MIP-177-LT properties are constant upon reuse, TOF data indicate that about 5.1 more sites are becoming active upon MIP-177-LT reuse. This can be interpreted considering that only a fraction of the adjacent Ti atoms accessible from the pore space was initially available for the photocatalytic reaction in the fresh photocatalyst, and this percentage increases upon continuous irradiation, probably due to the decomposition of the structural formate groups bridging two equatorial Ti atoms of the same Ti12 cluster. The TOF enhancement by a factor of about 5 upon reuse would indicate that for the extensively used MIP-177-LT electrons and holes located at any of the six positions of equatorial bridging formates accessible from the micropores have become photocatalytically active upon extended use, increasing in this way the efficiency of charge separation in the generation of CO2 and H2.
Notably, when considering the acid treated MIP-177-LT-AT that contains much less amounts of formates onto the Ti12O15 oxoclusters due to acid treatment, the photocatalytic activity from the first to the fourth use becomes rather constant, although some increase in the TOF value from 0.35 h−1 for the fresh sample to a TOF of 0.46 h−1 for the fourth times used material was also observed. Most likely 0.46 h−1 is about the maximum TOF value that can be achieved with the light power used in the present photocatalytic decomposition study. TOF values indicate that the performance of MIP-177-LT-AT is about four-fold higher than that of fresh MIP-177-LT, illustrating again the benefits of acid treatment to achieve optimal photocatalytic activity. Interestingly, according to the stoichiometry of eqn (1) one can easily verify that a complete decomposition of all the FA present in the aqueous solution was achieved at 2 h irradiation for the MIP-177-LT-AT photocatalyst.
In addition, PXRD analysis after the five times used MIP-177-LT-AT sample did not reveal an apparent change in the diffraction pattern compared to the fresh material (Fig. S5, ESI†) again in accordance with the stability of MIP-177-LT in acidic media.
Benchmarking
To put the photocatalytic activity of MIP-177-LT and MIP-177-LT-AT into context, similar experiments on the photocatalytic H2 release by FA decomposition were performed using the same mass of other benchmark MOF photocatalysts, namely, MIL-125(Ti) and UiO-66(Zr). As stated earlier, to our knowledge, there are no precedents in the literature on the use of bare MOFs as active photocatalysts for the photocatalytic FA decomposition, while a recent study from El-Roz et al. showed high potential for the UiO-66(Zr)-(COOH)2-Cu, where Cu-anhydride complex is at the origin of the activity.53 As summarized in Table 1, H2 evolution was also observed for these MOFs with a final H2 production at 2 h of 260 and 360 μmol gphotocatalyst−1 for the bare MIL-125(Ti) and UiO-66(Zr), respectively. This represents one half or less of the photocatalytic activity of fresh MIP-177-LT and about seven times lower than that of the highest performing MIP-177-LT-AT. Note that out of a few disordered defects, MIL-125(Ti) does not contain any structural sites with formate imprinting. UiO-66(Zr) exhibits ordered or disordered sites, depending on the synthesis conditions, that are likely to be accessible to formate anchoring, but it is based on a different coordination mode of the metal (Zr4+ is 8 coordinated against 6-fold for Ti4+ ions). Considering the unique structure of MIP-177-LT, we attribute this strong catalytic enhancement to the effect of formate imprinting in the structure due to the specific arrangement of Ti atoms in the oxocluster. In comparison, the photocatalytic activity of MIL-125(Ti) and UiO-66(Zr) must correspond to the general electron/hole activity of the photogenerated charge separate state to decompose FA.54To further support that formate imprinting of Ti12O15 metal nodes in MIP-177-LT is responsible for this excellent performance in H2 release from FA, photocurrent measurements using the same mass of the three MOFs were conducted. The results are presented in Fig. S6 (ESI†). As can be seen, under the same conditions the photocurrent density of UiO-66(Zr) and MIL-125(Ti) was six and three times higher than that of MIP-177-LT, respectively, meaning that the photogenerated charge carriers that can be extracted from these MOFs are higher than in MIP-177-LT. Note that the current intensity measuring the number of electrons extracted from the material gives a quantitative determination of the number of active sites in each material. Now, from the results of the photocurrent measurements, it can be concluded that in spite of this lower charge extraction in MIP-177-LT, its photocatalytic activity for FA decomposition is much higher, indicating that the Ti12O15 oxocluster organization is optimum to favor FA decomposition in spite of the lesser number of holes.
To consider a broader context, commercial P25 TiO2 was finally evaluated as a photocatalyst under the same conditions. Note that most of the studies with TiO2 as a photocatalyst use noble metal co-catalysts and herein the purpose was to evaluate the activity of plain TiO2 that could be compared with the Ti12O15 clusters of MIP-177-LT. The results presented in Table 1 show that P25 exhibits H2 production at 2 h of irradiation time similar to that measured for UiO-66(Zr), however, it is much lower than those of the formate imprinted MIP-177-LT that upon reuse can be almost an order of magnitude more active than P25 TiO2. Furthermore, apparent activation energy (Ea) values for the photocatalytic FA decomposition at temperatures ranging from 25 °C to 40 °C were determined for both MIP-177-LT and TiO2 as photocatalysts. The results are presented in Fig. S7 in the ESI.† The corresponding Ea values of MIP-177-LT and TiO2 are 57.5 kJ mol−1 and 107.0 kJ mol−1, respectively. This trend clearly states that the FA decomposition is much more favorable in MIP-177-LT vs. TiO2.
| Photocatalysts | H2 production (μmol gphotocatalyst−1) | TOF (h−1) |
|---|---|---|
| MIL-125(Ti) | 266 | 0.021 |
| UiO-66(Zr) | 360 | 0.031 |
| P-25 TiO2 | 340 | 0.009 |
| MIP-177-LT-AT (fresh) | 1300 | 0.35 |
Influence of FA concentration
Taking advantage of the exceptional chemical stability of MIP-177-LT in strong acid media, additional photocatalytic measurements were carried out with FA concentrations of 10−2 and 1 M using MIP-177-LT as a reference photocatalyst. The results summarized in Table S2 (ESI†) show that the H2 evolution rate does not increase upon increasing the FA concentration from 10−3 to 10−2 M. The highest concentration of 1 M, makes H2 evolution even slower than when the concentration is 10−2 M. PXRD of the MIP-177-LT samples after their use as photocatalysts showed no appreciable changes in the PXRD patterns. Chemical analysis of the FA solution revealed an increase in the Ti content as a function of FA concentration that is <0.001, 0.012 and 4.86% of the total Ti content present in the fresh MIP-177-LT used as a photocatalyst for 10−3, 10−2 and 1 M FA solutions, respectively.To understand the negative effect of excessive FA concentration, analogous studies on the influence of the concentration were carried out for aqueous solutions of sodium formate (NaHCO2). The pH value of NaHCO2 at 10−2 and 1 M was almost neutral in both cases. An increase in the temporal H2 evolution plot upon increasing the NaHCO2 concentration from 10−2 to 1 M was observed (Table 2). Accordingly, it is proposed that the MIP-177-LT structure is suitable to adsorb and recognize formate species, rather than FA. Adsorption of HCO2− on the imprinting active sites results in enhanced photocatalytic H2 evolution.
| Time | H2 release (μmol gphotocatalyst−1) | |
|---|---|---|
| NaHCO2 concentration 10−2 M | NaHCO2 concentration 1 M | |
| 30 | 120 | 280 |
| 60 | 324 | 645 |
| 90 | 430 | 916 |
| 120 | 870 | 1250 |
Now, when using aqueous FA solution and according to the equilibrium constant, formate should also be present when the concentration of FA in aqueous solution is 10−2 M and it is able to compete for the MIP-177 sites at this concentration. In contrast, the formate concentration becomes negligible compared to that of FA as the concentration of FA increases to 1 M. In addition, FA undergoes a strong association with the formation of FA dimers (eqn (3)) at a high concentration as observed by 1H NMR spectroscopy.55 These two facts, the decrease in HCO2− concentration relative to the FA concentration and FA dimer association, are proposed to be responsible for the unexpectedly lower observed photocatalytic activity of MIP-177-LT as the FA concentration increases. Therefore, in agreement with the structure of MIP-177-LT these measurements clearly reveal that it is formate the real species that is undergoing decomposition by the imprinted adjacent bi-Ti sites. The bidentate bridging of formate according to Fig. 1 justifies also the absence of CO formation that occurs for monodentate FA bridging.6
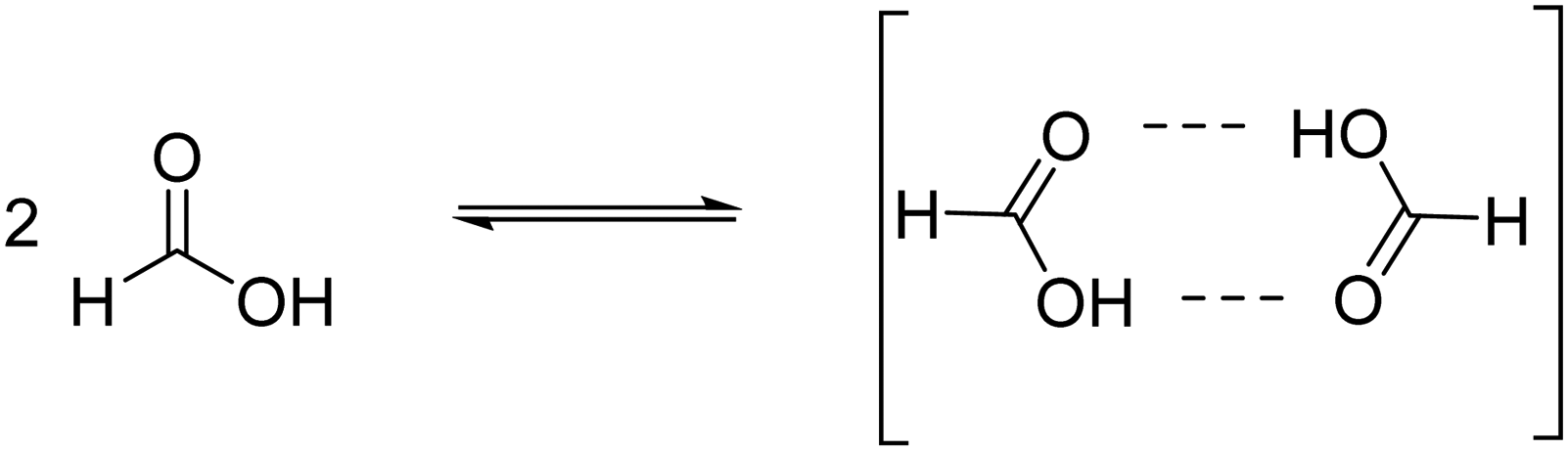 | (3) |
Reaction mechanism
It is proposed that light absorption generates a transient state of charge separation resulting in electrons located at TiIV and holes in oxygen atoms of the clusters and/or the mdip carboxylates. These holes at TiIII then get one electron from formate promoting decarboxylation of the attached HCO2˙ radical intermediate, then evolving CO2 and forming a surface H atom that will associate with a H atom appearing in the reduction of H+ by photogenerated electrons resulting in a H2 molecule. Scheme 1 summarizes the mechanistic proposal along with the experimental energy of the lowest unoccupied and highest occupied crystal orbitals (LUCO and HOCO) of MIP-177-LT vs. the H+/H2 and HCO2H/CO2 redox potentials. The main peculiarity of MIP-177-LT compared to the other two MOFs included in Table 1 is the presence of formate-imprinted sites in the structure that should favor the adsorption of formates at the active site where holes are formed, thus increasing the efficiency of the process.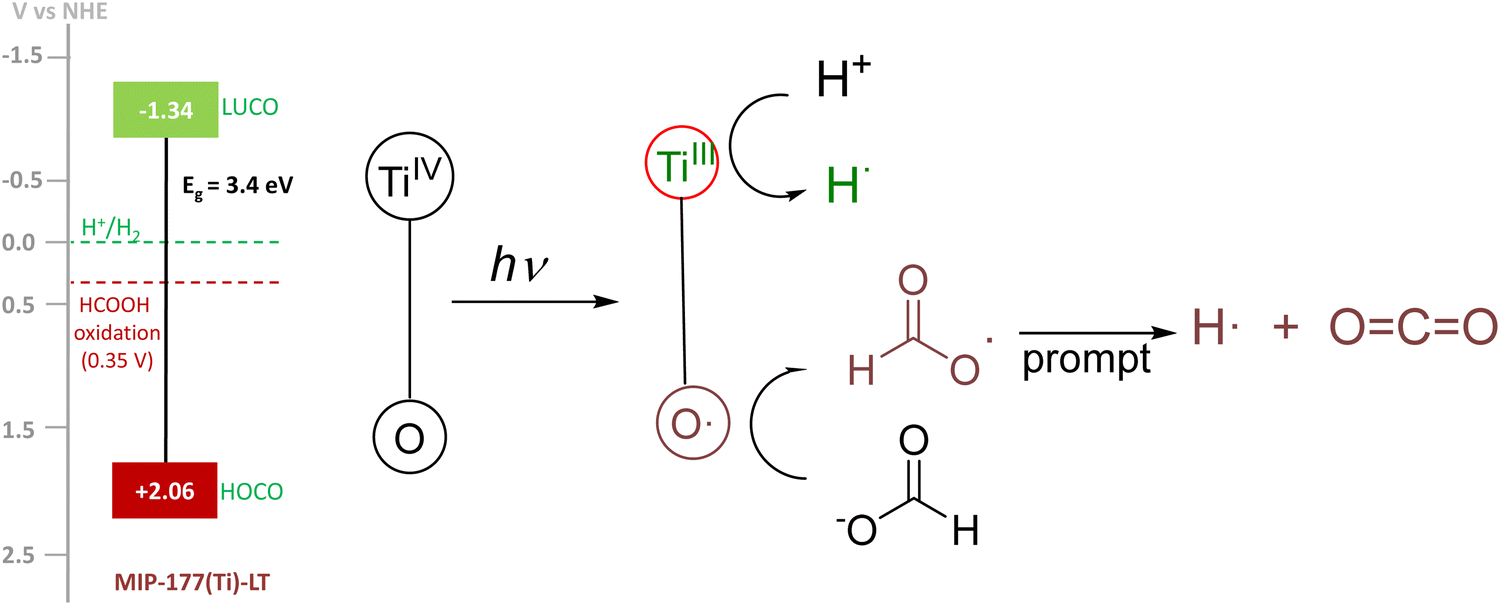 | ||
| Scheme 1 Proposed reaction mechanism. Upon light absorption, an electron migrates by ligand-to-metal photoinduced charge transfer to TiIV. Photogenerated electrons on TiIII produce H atoms and photogenerated holes produce formate radicals that decompose into CO2 and H atoms. | ||
To support this possible mechanism, photocatalytic formate decomposition was performed in the presence of quenchers. Table S3 (ESI†) provides a summary of the quenching study. In this way, performing the photocatalytic formic acid decomposition in the presence of 10−3 M cerium ammonium nitrate results in a significant diminution of the amount of evolved H2 to about one third of the volume in the absence of Ce(NH4)2(NO3)6, while CO2 evolution was enhanced somewhat from 701 to 794 μmol gphotocatalyst−1 at 2 h of reaction time. This observation is compatible with the mechanistic proposal, holes being consumed by formates evolving CO2, while photogenerated electrons convert CeIV into CeIII, thwarting in part H2 evolution that becomes unbalanced.
In contrast, all the attempts to stop or diminish CO2 evolution in MIP-177-LT as a reference photocatalyst by using hole quenchers such as hydrogen sulfite or hydrogen sulfide were unsuccessful (Table S4, ESI†), probably reflecting the preferential selective formate adsorption on the Ti atoms with respect to the quenchers.
Molecular insights into the reaction mechanism
Density functional theory (DFT) calculations implementing D3 dispersion correction (see the ESI†) were further performed to shed light on the catalytic reaction mechanism and to understand the electronic and structural origins of the outstanding performance exhibited by MIP-177(Ti)-LT. Herein, a representative Ti12O15 cluster model was cleaved from the periodic MOF structure integrating an unsaturated TiIV/TiIII pair to account for the light-induced switching of one active site from TiIV to TiIII as revealed experimentally and in line with previously reported Ti-MOFs.56,57 The first step of the reaction, i.e. chemisorption of HCOOH throughout this Ti active site, is associated with an adsorption energy of −0.79 eV and Ti–O(HCOOH) distances of 2.65 Å (TiIII) and 2.61 Å (TiIV) (Table S5, ESI†), which suggest the favorable formation of the initial state, although without leading to a highly stable state that would be detrimental to proceed the following steps of the mechanism. The calculated Gibbs-free energy profile for the dehydrogenation of *HCOOH through the *HCOO (formate) pathway was further explored (Fig. 5a) and was found to be more thermodynamically favorable as compared to the dehydration process examined in Fig. S7 (ESI†). The adsorbed *HCOOH molecule undergoes O–H bond cleavage to form *HCOO* and *H first. At this step, the released H is coordinated to the μO site, while the O of *HCOO* still bonds to the Ti site. Then, *HCOO undergoes a reorientation and the C–H bond breaks to form CO2. Note that these calculations do not refer to the macroscopic kinetics of the reaction and they cannot capture a different time-dependent evolution profile for CO2 and H2 production at a short reaction time.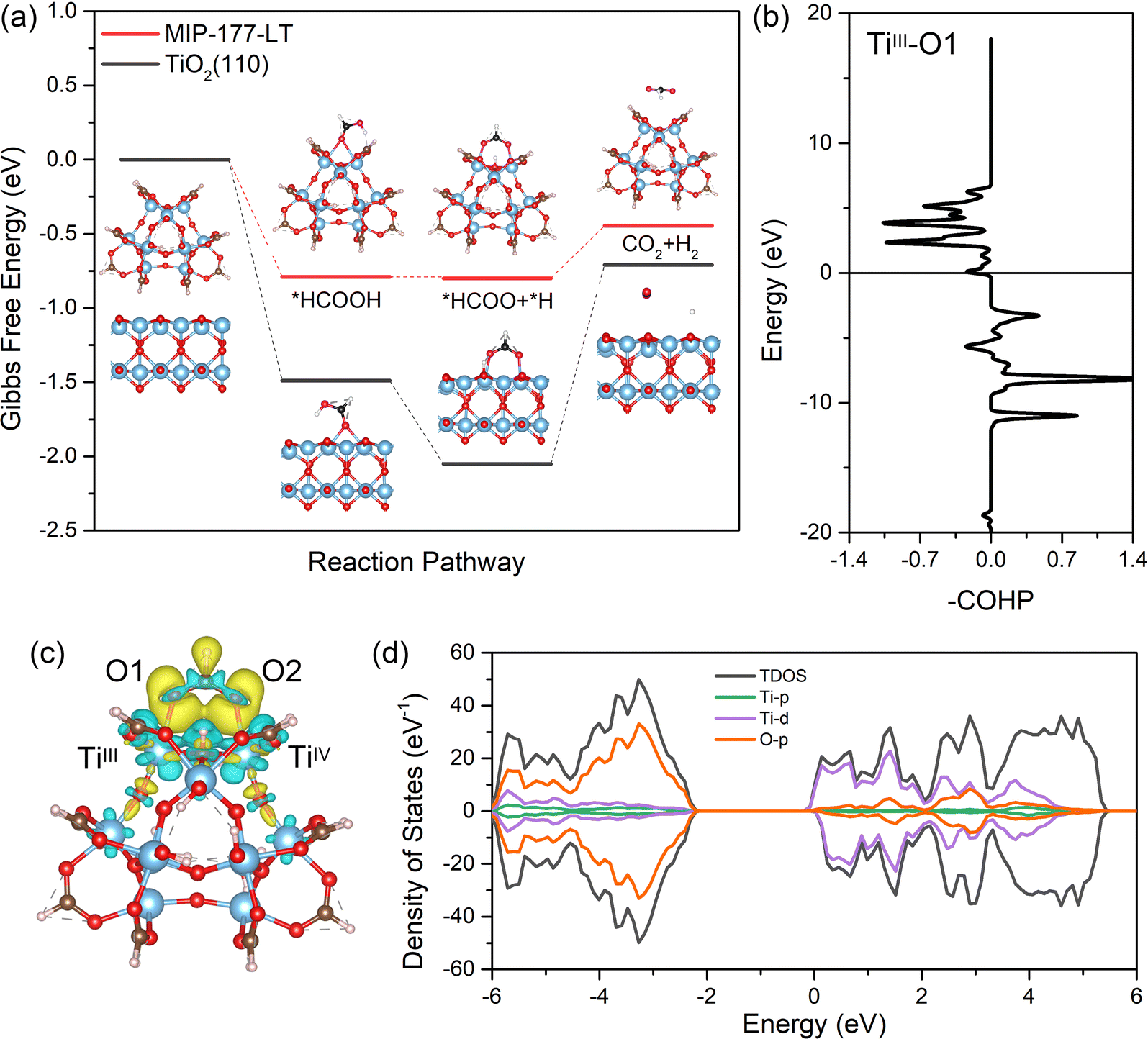 | ||
| Fig. 5 (a) Gibbs free energy profile for the conversion of HCOOH into H2 and CO2 simulated for MIP-177(Ti)-LT and TiO2(110). The total free energy of MIP-177(Ti)-LT and TiO2 models with a gas-phase HCOOH molecule was set as the zero point in the energy profile. The color scheme for FA: O, pink; H, white; C, black; The color scheme for catalysts: Ti, blue; O, red; and H: light pink. (b) COHP analysis of *HCOO on MIP-177-LT for the TiIII adsorption site. Positive and negative values of –COHP correspond to the bonding and antibonding interactions. (c) Charge density difference of *HCOO on the MIP-177(Ti)-LT cluster model. Yellow and blue lobes refer to electron accumulation and depletion regions. (d) Total and projected Density of States (DOS) of *HCOO on the MIP-177(Ti)-LT cluster model. | ||
For comparison, the TiO2(110) as a benchmark photocatalyst was also computationally explored. We first calculated that the adsorption energy of HCOOH (−1.49 eV) on TiO2(110) is much higher than that calculated for MIP-177(Ti)-LT (−0.79 eV) in line with substantially shorter Ti–O(HCOOH) distances (2.10 Å and 2.18 Å vs. 2.65 Å and 2.61 Å for MIP-177(Ti)-LT) (details shown in Table S5, ESI†). The adsorption of *HCOO has also more energetics on the TiO2(110) surface than that of MIP-177(Ti)-LT (energy difference: 0.76 eV), which makes the cleavage of the C–H bond from *HCOO much less thermodynamically preferable on TiO2 (ΔG = 1.34 eV) compared to MIP-177(Ti)-LT (ΔG = 0.35 eV) as revealed in Fig. 5a. This predicted trend is in line with the much lower activation energy value measured for the photocatalytic FA decomposition in MIP-177-LT (57.5 kJ mol−1) vs. TiO2 (107.0 kJ mol−1). The local environment of the Ti12O15 inorganic node of MIP-177(Ti)-LT combined with its structural adaptability towards guest adsorption (see the guest-induced structural relaxation in Fig. S8, S9 and Table S5, ESI†) is a key to offer optimum energetics for the coordination of *HCOOH and *HCOO to promote effective catalytic activity according to the Sabatier principle.58
According to the Gibbs-free energy profile, the C–H cleavage of *HCOO is the key step for the H2 release reaction. We analyzed the crystal orbital Hamilton populations (COHP) for *HCOO adsorption configuration and we found that the interaction between *HCOO and MIP-177(Ti)-LT comes from the anti-bonding between O of *HCOO and Ti of MIP-177(Ti)-LT (Fig. 5b). In addition, for MIP-177(Ti)-LT, the charge density difference analysis clearly showed that there is an accumulation of electrons on the Ti atom and depletion on the O atom (Fig. 5c). This electron transfer is at the origin of the relatively strong interaction between the Ti and O atoms, beneficial for the following transformation of electrons to the C atom, promoting the cleavage of C–H. The density of states (DOS) analysis (Fig. 5d) evidenced that the O-p states contribute to the lowest unoccupied molecular orbital (LUMO) which accepts electrons, while Ti-d states contribute to the highest occupied molecular orbital (HOMO) which provides electrons. The same conclusion can be drawn from the analysis of the *HCOOH adsorption configuration (Fig. S10, ESI†). This whole analysis supports that the light-induced TiIII plays a critical role in the whole photocatalytic FA decomposition of MIP-177(Ti)-LT.
Photo response of MIP-177-LT
Further studies were aimed at determining the chromophores responsible for photodecarboxylation by studying the influence of the irradiation wavelength in H2 release from FA using MIP-177-LT as a photocatalyst. The results are presented in Fig. 6. It can be seen that irradiation in the deep UV at 238 nm exhibits a high apparent quantum yield of 22%. This remarkable quantum yield decreases as the irradiation wavelength becomes longer. At 300 nm, the apparent quantum yield is 0.5%. This dependence of the photocatalytic activity with the wavelength indicates that direct excitation of the Ti ← O ligand-to-metal charge transfer band of Ti12O15 clusters that should have an absorption band at 240 nm is the most efficient photophysical process to promote FA decarboxylation and H2 release.59,60 Excitation at the localized organic mdip linker with absorption extending to 274 and 300 nm can also promote H2 evolution through a Ti12O15 ← mdip ligand to metal charge transfer,57 but with much less efficiency. This lesser efficiency indicates the incomplete quenching of localized mdip excitons by the Ti12O15 cluster, probably because other deactivation pathways are available to the ligand excited state including radiationless decay through conformational phenyl ring rotation or emissive photoluminescence.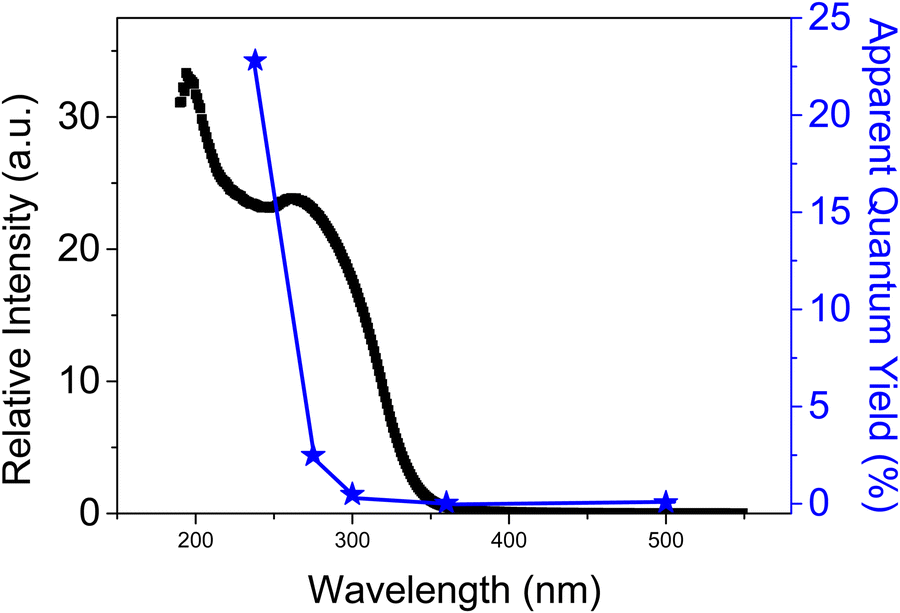 | ||
| Fig. 6 Photoresponse of MIP-177-LT for FA decarboxylation at different wavelengths (left axis) compared with the diffuse reflectance UV-Vis absorption spectrum of MIP-177-LT. | ||
The photoresponse in the visible region, although existing in MIP-177-LT, probably due to the action of structural defects or impurities, is orders of magnitude lower than that in the UV region. In fact, one of the improvements to increase the photocatalytic activity of MIP-177-LT could be extending its photoresponse towards longer wavelengths by appropriate design of mixed metal or mixed ligand MIP-177 following reported strategies.
Scope
Photocatalytic decarboxylation is a general reaction that has gained renewed interest as a way to obtain hydrocarbon fuels from biomass-derived aliphatic carboxylic acids.61,62 As FA, other carboxylic acids can undergo photocatalytic decarboxylation to form the corresponding alkyl radicals that can abstract one hydrogen from the solvent, dimerize or undergo trapping after consecutive reactions.61,63,64 It was of interest to determine whether or not the formate-imprinted photocatalytic site of MIP-177-LT can also promote photodecarboxylation of other carboxylic acids with similar high activity. Therefore, besides FA decomposition, the photocatalytic activity of MIP-177-LT was also assessed in the photocatalytic decarboxylation of acetic, propionic and methacrylic acids.As expected, all the tested aliphatic carboxylic acids undergo photodecarboxylation promoted by MIP-177-LT. The main product was CO2, accompanied by much lesser amounts of H2 and light alkanes. The results are presented in Table 3. As can be seen, acetic acid at 10−3 M concentration undergoes a complete photocatalytic decarboxylation after 16 h of irradiation time. The initial evolution of H2 during the irradiation indicates that formate groups present in MIP-177-LT are undergoing photodecarboxylation in the first stages, before observing acetic acid decarboxylation. Interestingly, a second use of MIP-177-LT shows a considerable deactivation degree of about 90% that further decreases to a residual 5% of the initial activity in the third use. This behavior contrasts with the previously commented growth in MIP-177-LT catalytic activity using FA and indicates the poisoning of the active sites by some blocking by-products. The fact that methane is not evolved in the expected stoichiometric amounts with respect to CO2 suggests that some byproducts such as methoxy groups could be blocking the sites.
| Carboxylic acid | H2 (μmol gphotocatalyst−1) | CO2 (μmol gphotocatalyst−1) | Other |
|---|---|---|---|
| Acetic acid (16 h) | 1350 | 1405 | CH4 |
| Propionic acid (2 h) | 1487 | 922 | CH4, CH3CH3 |
| Methacrylic acid | 1410 | 1490 | CH4, CH3–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
Propionic acid also undergoes photodecarboxylation to a considerable extent after 2 h of 300 W Xe lamp irradiation (Table 3) and undergoes deactivation in the second use to a value corresponding to only about 20% of the activity of the fresh MIP-177-LT. Although the origin of deactivation deserves a deeper study, the PXRD pattern of the deactivated MIP-177-LT after its use as a photocatalyst shows that the crystallinity of the samples has been preserved, indicating that MIP-177-LT decomposition is not the reason for the loss of photocatalytic activity.
Similar trends were also observed for the photocatalytic decomposition of methacrylic acid promoted by MIP-177-LT, i.e., the evolution of CO2 without being accompanied by similar proportions of other organic hydrocarbons and considerable deactivation upon reuse.
Although further studies are necessary to understand the photocatalytic data and performance of MIL-177-LT for acetic, propionic and methacrylic acid, the behavior with these carboxylic acids clearly contrasts with that of FA, thus reinforcing the importance of formate imprinting on the Ti12O15 nodes in the photocatalytic H2 release activity and stability.
Conclusions
Ti-MOF MIP-177-LT was demonstrated to combine several properties that make this material highly active for photocatalytic H2 release from FA, especially the presence of accessible formate-imprinted active sites in equatorial Ti atoms and their structural robustness in strong acidic media. These unique features led to FA decomposition activity over one order of magnitude superior to the ones of the benchmark MOF photocatalysts MIL-125(Ti) and UiO-66(Zr) or over 30 times that of TiO2 P25, reaching an apparent quantum yield as high as 22% in the UV region. Noteworthily, the FA photocatalytic decomposition rate of the fresh MIP-177-LT increases upon irradiation and is disfavored at high FA concentrations, facts that are related to the preference of the material for formate with respect to its acidic form. Acid washing has a beneficial influence on the photocatalytic activity of MIP-177-LT-AT by removing impurities and releasing the active centers, reaching a maximum TOF value of 0.46 h−1. Quenching studies and DFT calculations support that H2 consumes photogenerated electrons, but CO2 evolution could not be quenched by strong electron donors indicating the high selectivity of the active sites for formate coordination. Considering the advantage of light over heat for instantaneous on–off control of H2 release, the present results open up the way for efficient MOF photocatalysts by design based on earth abundant and affordable elements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Spanish Ministerio de Ciencia e Innovación (PID2021-126071OB-C21 and Severo Ochoa) and Generalitat Valenciana (Prometeo 2017/083) is gratefully acknowledged. R. D. A. is thankful to CONACYT for the PhD grant 2018-000003-01EXTF-00075 (CVU: 697332). S. N. is gracious for the financial support from Grant PID2021-123856OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. The computational work was performed using HPC resources from GENCI-CINES (Grant A0120907613).References
- Z. Abdin, A. Zafaranloo, A. Rafiee, W. Mérida, W. Lipiński and K. R. Khalilpour, Renewable Sustainable Energy Rev., 2020, 120, 109620 CrossRef CAS.
- S. Sharma and S. K. Ghoshal, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS.
- T. He, Q. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- S.-M. Lu, Z. Wang, J. Wang, J. Li and C. Li, Green Chem., 2018, 20, 1835–1840 RSC.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., 2013, 125, 4502–4505 CrossRef.
- Q. Zhang, Q. Mao, Y. Zhou, L. Zou, D. Zhu, Y. Huang, H. Gao, X. Luo, Y. Mao and Z. Liang, ACS Sustainable Chem. Eng., 2022, 10, 4599–4609 CrossRef CAS.
- D. Mellmann, E. Barsch, M. Bauer, K. Grabow, A. Boddien, A. Kammer, P. Sponholz, U. Bentrup, R. Jackstell and H. Junge, Chem. – Eur. J., 2014, 20, 13589–13602 CrossRef CAS PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- A. Léval, A. Agapova, C. Steinlechner, E. Alberico, H. Junge and M. Beller, Green Chem., 2020, 22, 913–920 RSC.
- N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Coord. Chem. Rev., 2018, 373, 317–332 CrossRef CAS.
- V. Singh and R. Singh, Corros. Sci., 1995, 37, 1399–1410 CrossRef CAS.
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef.
- K. Sordakis, A. F. Dalebrook and G. Laurenczy, ChemCatChem, 2015, 7, 2332–2339 CrossRef CAS.
- J. R. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- B. M. Besancon, V. Hasanov, R. Imbault-Lastapis, R. Benesch, M. Barrio and M. J. Mølnvik, Int. J. Hydrogen Energy, 2009, 34, 2350–2360 CrossRef CAS.
- M. Navlani-García, D. Salinas-Torres, K. Mori, Y. Kuwahara and H. Yamashita, Heterog. Photocatal., 2020, 193–223 Search PubMed.
- H. J. Yun, H. Lee, J. B. Joo, W. Kim and J. Yi, J. Phys. Chem. C, 2009, 113, 3050–3055 CrossRef CAS.
- Z. Zhang, S.-W. Cao, Y. Liao and C. Xue, Appl. Catal., B, 2015, 162, 204–209 CrossRef CAS.
- Y. Ji and Y. Luo, J. Power Sources, 2016, 306, 208–212 CrossRef CAS.
- A. Gazsi, G. Schubert, P. Pusztai and F. Solymosi, Int. J. Hydrogen Energy, 2013, 38, 7756–7766 CrossRef CAS.
- C. Tao, W. Guopeng, F. Zhaochi, H. Gengshen, S. Weiguang, Y. Pinliang and L. Can, Chin. J. Catal., 2008, 29, 105–107 CrossRef.
- K. L. Miller, C. W. Lee, J. L. Falconer and J. W. Medlin, J. Catal., 2010, 275, 294–299 CrossRef CAS.
- B. Wu, J. Lee, S. Mubeen, Y. S. Jun, G. D. Stucky and M. Moskovits, Adv. Opt. Mater., 2016, 4, 1041–1046 CrossRef CAS.
- I. Willner and Z. Goren, J. Chem. Soc., Chem. Commun., 1986, 172–173 RSC.
- S. Cao, Y. Chen, H. Wang, J. Chen, X. Shi, H. Li, P. Cheng, X. Liu, M. Liu and L. Piao, Joule, 2018, 2, 549–557 CrossRef CAS.
- R. Zhu, F. Tian, R. Yang, J. He, J. Zhong and B. Chen, Renewable Energy, 2019, 139, 22–27 CrossRef.
- M. Hamandi, G. Berhault, C. Guillard and H. Kochkar, Appl. Catal., B, 2017, 209, 203–213 CrossRef CAS.
- Q. Zhang, C.-F. Lin, Y. H. Jing and C.-T. Chang, J. Air Waste Manage. Assoc., 2014, 64, 578–585 CrossRef CAS.
- M. Hamandi, G. Berhault, C. Guillard and H. Kochkar, Mol. Catal., 2017, 432, 125–130 CrossRef CAS.
- C. Wan, L. Zhou, L. Sun, L. Xu, D.-G. Cheng, F. Chen, X. Zhan and Y. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef CAS.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- X. Deng, Z. Li and H. García, Chem. – Eur. J., 2017, 23, 11189–11209 CrossRef CAS PubMed.
- M. Zeng, Z. Chai, X. Deng, Q. Li, S. Feng, J. Wang and D. Xu, Nano Res., 2016, 9, 2729–2734 CrossRef CAS.
- M. Wen, K. Mori, Y. Kuwahara and H. Yamashita, ACS Energy Lett., 2017, 2, 1–7 CrossRef CAS.
- M. Zhang, W. Lin, L. Ma, Y. Pi and T. Wang, Chem. Commun., 2022, 58, 7140–7143 RSC.
- D. Jiang, C. Huang, J. Zhu, P. Wang, Z. Liu and D. Fang, Coord. Chem. Rev., 2021, 444, 214064 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- S. A. Younis, E. E. Kwon, M. Qasim, K.-H. Kim, T. Kim, D. Kukkar, X. Dou and I. Ali, Prog. Energy Combust. Sci., 2020, 81, 100870 CrossRef.
- M. Ghorbanloo, V. Safarifard and A. Morsali, New J. Chem., 2017, 41, 3957–3965 RSC.
- J. Sun, J. Wan, Y. Wang, Z. Yan, Y. Ma, S. Ding, M. Tang and Y. Xie, J. Hazard. Mater., 2022, 429, 128299 CrossRef CAS.
- G. Wißmann, A. Schaate, S. Lilienthal, I. Bremer, A. M. Schneider and P. Behrens, Microporous Mesoporous Mater., 2012, 152, 64–70 CrossRef.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
- S. Wang, T. Kitao, N. Guillou, M. Wahiduzzaman, C. Martineau-Corcos, F. Nouar, A. Tissot, L. Binet, N. Ramsahye, S. Devautour-Vinot, S. Kitagawa, S. Seki, Y. Tsutsui, V. Briois, N. Steunou, G. Maurin, T. Uemura and C. Serre, Nat. Commun., 2018, 9, 1660 CrossRef PubMed.
- S. Nandi, S. Wang, M. Wahiduzzaman, V. Yadav, K. Taksande, G. Maurin, C. Serre and S. Devautour-Vinot, ACS Appl. Mater. Interfaces, 2021, 13, 20194–20200 CrossRef CAS PubMed.
- R. V. Pinto, S. Wang, S. R. Tavares, J. Pires, F. Antunes, A. Vimont, G. Clet, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2020, 59, 5135–5143 CrossRef CAS PubMed.
- D. Mateo, A. Santiago-Portillo, J. Albero, S. Navalón, M. Alvaro and H. García, Angew. Chem., 2019, 131, 18007–18012 CrossRef.
- H. Issa Hamoud, P. Damacet, D. Fan, N. Assaad, O. I. Lebedev, A. Krystianiak, A. Gouda, O. Heintz, M. Daturi, G. Maurin, M. Hmadeh and M. El-Roz, J. Am. Chem. Soc., 2022, 144(36), 16433–16446 CrossRef CAS PubMed.
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121(21), 13051–13085 CrossRef CAS PubMed.
- W. Reutemann and H. Kieczka, Ullmann's encyclopedia of industrial chemistry, 2011, vol. 1 Search PubMed.
- M. de Miguel, F. Ragon, T. Devic, C. Serre, P. Horcajada and H. García, Chem. Phys. Chem., 2012, 13, 3651–3654 CrossRef CAS PubMed.
- A. Santiago Portillo, H. G. Baldoví, M. T. Garcia Fernandez, S. Navalon, P. Atienzar, B. Ferrer, M. Alvaro, H. Garcia and Z. Li, J. Phys. Chem. C, 2017, 121, 7015–7024 CrossRef CAS.
- A. Medford, A. Vojvodic, J. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- C. Aprile, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2008, 10, 769–783 RSC.
- D. Budac and P. Wan, J. Photochem. Photobiol., A, 1992, 67, 135–166 CrossRef CAS.
- X. Du, Y. Peng, J. Albero, D. Li, C. Hu and H. García, ChemSusChem, 2022, 15, e202102107 CAS.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- J. Shi, T. Yuan, M. Zheng and X. Wang, ACS Catal., 2021, 11, 3040–3047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02258c |
| This journal is © The Royal Society of Chemistry 2023 |