
Surface and lattice engineered ruthenium superstructures towards high-performance bifunctional hydrogen catalysis†
Leigang
Li
a,
Shangheng
Liu
a,
Changhong
Zhan
a,
Yan
Wen
a,
Zhefei
Sun
b,
Jiajia
Han
 *b,
Ting-Shan
Chan
c,
Qiaobao
Zhang
*b,
Zhiwei
Hu
d and
Xiaoqing
Huang
*a
*b,
Ting-Shan
Chan
c,
Qiaobao
Zhang
*b,
Zhiwei
Hu
d and
Xiaoqing
Huang
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: hxq006@xmu.edu.cn
bDepartment of Materials Science and Engineering, College of Materials, Xiamen University, Xiamen, 361005, China. E-mail: jiajiahan@xmu.edu.cn; zhangqiaobao@xmu.edu.cn
cNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu, 30076, Taiwan
dMax Planck Institute for Chemical Physics of Solids, Nothnitzer Strasse 40, Dresden, 01187, Germany
First published on 9th November 2022
Abstract
Developing high-performance bifunctional electrocatalysts towards the hydrogen evolution/oxidation reaction (HER/HOR) holds great significance for efficiently utilizing hydrogen energy. In this work, a unique class of Mo-modified Ru nanosheet assemblies (Mo–Ru NSAs) have been successfully prepared, where Mo possesses a unique configuration of both a metallic Mo atom and MoO3. Further structural optimization by density functional theory (DFT) calculations has revealed that the metallic Mo atom is embedded in the Ru lattice while MoO3 is adsorbed on a partially oxidized Mo atom. As a result, the surface electronic properties and lattice structures of Ru nanosheets have been dramatically altered, leading to optimized adsorption of intermediates and superior HER/HOR performance. In detail, 16 mV is sufficient to drive 10 mA cm−2 for the HER in 1 M KOH with a durable stability of 250 h. Furthermore, a high mass activity of 2.45 A mgRu−1 towards the HOR in 0.1 M KOH with high stability is also achieved. DFT calculations have further revealed that the coupling of a Mo atom and MoO3 can facilitate the rapid decomposition of H2O and generate highly active sites by steric hindrance, thereby enabling high bifunctional activity. It is anticipated that this work would enlighten the construction of more advanced bifunctional catalysts via surface and lattice engineering.
Broader contextDeveloping high-performance electrocatalysts towards the alkaline hydrogen evolution reaction (HER) and the hydrogen oxidation reaction (HOR) is crucial for hydrogen economy, where Ru-based materials are regarded as promising bifunctional HER/HOR electrocatalysts. However, the high potential barrier for water dissociation in alkaline media and the strong adsorption strength of intermediates over Ru have hindered the enhancement of the HER and HOR performance. Herein, for the first time, novel two-dimensional Ru nanosheet assemblies modified by Mo with a unique atomic configuration of the metallic Mo atom and MoO3 are prepared. The high-density coupling of the Mo atom and MoO3 has facilitated the rapid decomposition of H2O and generated highly active sites by steric hindrance, thereby giving rise to high bifunctional activity towards the HER and HOR, as evidenced by substantial experimental investigations and theoretical calculations. This work not only provides an approach to optimizing the surface electronic properties and lattice structures of electrocatalysts, but also would motivate the research on designing high-performance electrocatalysts for bifunctional hydrogen catalysis. |
Introduction
The increasingly urgent energy crisis and pollution problems have broken the carbon balance on the earth and the wide utilization of renewable energies is of paramount significance to solve these issues.1 Hydrogen (H2) energy has played a critical role in the development of renewable energy technology and the realization of a neutral carbon cycle.2–7 Water electrolyzers, producing high-purity H2 under mild conditions, and hydrogen fuel cells, converting H2 into high-density energy with zero emission of pollution, are two key devices to realize hydrogen economy.8–11 Compared to proton exchange membrane fuel cells, the anion exchange membrane fuel cells working under alkaline conditions are more attractive because of the mild working conditions and relatively low requirement of catalysts.12,13 However, the reaction kinetics of the hydrogen oxidation reaction (HOR) at the anode side is 2–3 orders of magnitude lower than that in acidic media, which has hindered the application of alkaline fuel cells.14–18 Platinum (Pt)-based nanostructures are still state-of-the-art electrocatalysts towards catalyzing the HOR but suffer from scarcity, high price and even poor stability.19,20 Regarding the hydrogen evolution reaction (HER) in alkaline electrolytes, the cathodic reaction of water electrolyzers faces the same situation.21–23 Therefore, it is imperative and significant to develop non-Pt electrocatalysts with high activity, durable stability and low price to catalyze the HER and HOR in alkaline electrolytes with satisfactory catalytic performance.In the pursuit of electrocatalysts toward promoting the HER and HOR, a wide range of efforts have been devoted to the design and fabrication of various metal-based nanostructures.24–26 Though diversified non-precious metal-based electrocatalysts have been investigated with significant progress, their activity is still far below that of precious metal-based catalysts. Among the precious metals, ruthenium (Ru) has been regarded as one of the most promising alternatives to substitute Pt for catalyzing the alkaline HER and HOR.27–31 Though it belonged to the platinum group, Ru is the cheapest platinum-group metal and its price is only 6–36% of that of Pt.29 Moreover, the abundance of Ru is much higher than that of Pt on the earth. Therefore, developing advanced Ru catalysts holds great significance for fulfilling the utilization of H2 energy. As a hydrogen catalysis electrocatalyst, however, the adsorption of reaction intermediates (H* and OH*) on Ru surfaces is not optimal enough to achieve satisfactory catalytic performance.32,33 Optimal adsorption of H* and OH* could facilitate H-related catalysis.34 In addition, enhanced H2O dissociation ability is beneficial for the HER in alkaline media.34 However, the relatively high potential barrier of H2O dissociation on Ru surfaces has restricted the enhancement of reaction kinetics of the HER.32 Therefore, substantial efforts are required for the construction of advanced Ru-based electrocatalysts with the optimized adsorption strength of intermediates and the enhanced H2O decomposition ability of alkaline electrolytes.
Herein, we have successfully fabricated Mo–Ru NSAs composed of two-dimensional (2D) nanosheets with a unique atomic configuration of Mo for the high-performance HER and HOR in alkaline electrolytes. In particular, an overpotential of as low as 16 mV is sufficient to deliver a current density of 10 mA cm−2 for the HER in 1 M KOH. More intriguingly, Mo–Ru NSAs can maintain a durable stability of as long as 250 h at 10 mA cm−2. Regarding the HOR, the catalytic activity is also significantly enhanced with a mass activity of 2.45 A mgRu−1 in 0.1 M KOH, which is 38.9 and 6.4 times that of Ru/C and Pt/C, respectively. The HOR stability of Mo–Ru NSAs is also much more durable than those of Ru/C and Pt/C. DFT calculations have revealed the weakened adsorption strength of H* and OH* near MoO3 on Mo–Ru NSAs. More attractively, the MoRu sites can enable spontaneous dissociation of H2O, facilitating the generation of H+ and OH−. With the increased coverage of H* and OH*, H* and OH* tend to move to MoO3, thereby further optimizing the adsorption strength of H* and OH*. The strong coupling of MoRu sites and MoO3 has enabled a more favorable reaction path and facilitated the energetically favorable progress of both the HER and HOR. This work with the demonstration of high-performance Mo-modified Ru nanosheet catalysts would benefit the preparation of more advanced Ru-based catalysts for bifunctional hydrogen catalysis and other catalytic applications.
Results and discussion
Mo–Ru NSAs were synthesized by a facile wet-chemical method, where metal precursors of ruthenium carbonyl (Ru3(CO)12) and molybdenum carbonyl (Mo(CO)6) were co-reduced in the solvent of oleylamine (OAm) with salicylic acid as the reducing agent (Fig. 1a). Transmission electron microscopy (TEM) and high-angle annular dark-field transmission electron microscopy (HAADF STEM) images were first obtained to investigate the overall morphology and microstructures of Mo–Ru NSAs. As demonstrated by the STEM and TEM images (Fig. 1b, c and Fig. S1, S2, ESI†), Mo–Ru NSAs are highly monodispersed. For each Mo–Ru NSA, it is formed by the self-assembly of abundant ultrathin nanosheets. Energy-dispersive X-ray spectroscopy (EDS) analysis indicates a chemical composition of 89.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.1 for Ru:Mo (molar ratio) (Fig. S3, ESI†). The phase information of the sample was investigated by powder X-ray diffraction (XRD) where only peaks related to hexagonal close-packed (hcp) Ru (PDF 06-0663) are observed (Fig. 1d). A high-magnification Cs-corrected STEM image was taken to analyze the microstructure and lattice spacings of 0.237 nm and 0.216 nm corresponding to the (100) facet and (002) facet of hcp Ru, respectively (Fig. 1e and f). The measured lattice spacings are both larger than those of bulk Ru, revealing the influence of Mo on the lattice structures of Ru nanosheets. EDS mapping (Fig. 1g) and line scan (Fig. S4, ESI†) were employed to analyze the elemental distribution in the sample, where Ru and Mo are observed to be uniformly distributed throughout the sample.
:10.1 for Ru:Mo (molar ratio) (Fig. S3, ESI†). The phase information of the sample was investigated by powder X-ray diffraction (XRD) where only peaks related to hexagonal close-packed (hcp) Ru (PDF 06-0663) are observed (Fig. 1d). A high-magnification Cs-corrected STEM image was taken to analyze the microstructure and lattice spacings of 0.237 nm and 0.216 nm corresponding to the (100) facet and (002) facet of hcp Ru, respectively (Fig. 1e and f). The measured lattice spacings are both larger than those of bulk Ru, revealing the influence of Mo on the lattice structures of Ru nanosheets. EDS mapping (Fig. 1g) and line scan (Fig. S4, ESI†) were employed to analyze the elemental distribution in the sample, where Ru and Mo are observed to be uniformly distributed throughout the sample.
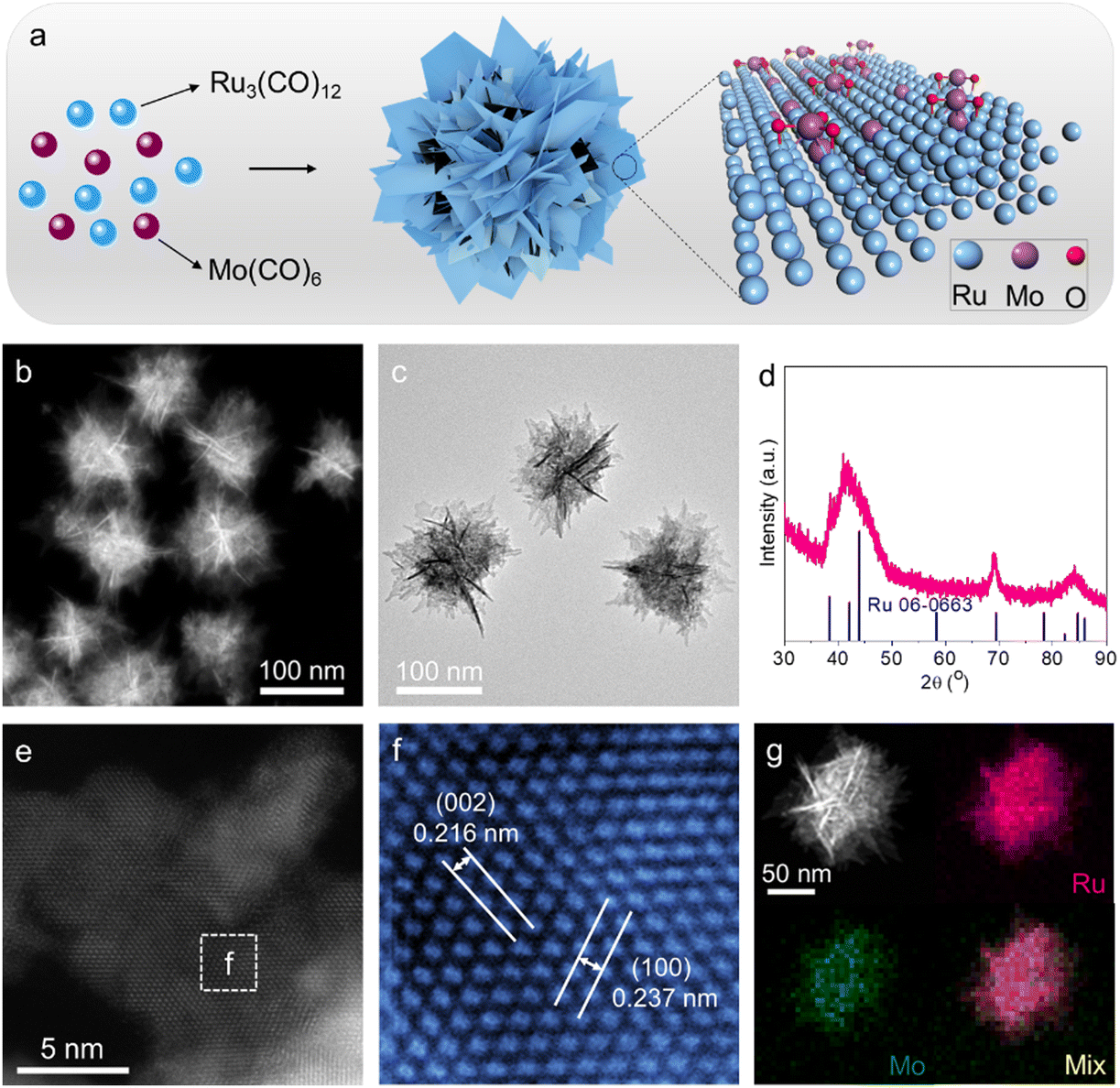 | ||
| Fig. 1 Structural characterization. (a) Schematic of the morphology and crystal atomic model of Mo–Ru NSAs. (b) HAADF STEM image, (c) TEM image, (d) XRD pattern, (e) and (f) Cs-corrected HAADF STEM images, and (g) STEM image and EDS mapping of Mo–Ru NSAs. | ||
More advanced characterization techniques were adopted to investigate the electronic and atomic structures of Mo–Ru NSAs. First, X-ray absorption spectroscopy (XAS) analysis was carried out for Mo–Ru NSAs with commercial Ru foil and RuO2 as references. Fig. 2a shows the Ru K-edge X-ray absorption near-edge structure (XANES) spectra of Mo–Ru NSAs, Ru foil and RuO2, where the absorption energy (E0) of Ru in Mo–Ru NSAs is close to that of Ru foil but far from that of RuO2. It indicates that Ru in Mo–Ru NSAs mainly exists in a metallic state. More interestingly, the absorption energy of Ru in Mo–Ru NSAs is lower than that of Ru foil (the inset in Fig. 2a), which indicates that electron transfer has occurred between Ru and Mo in Mo–Ru NSAs. The k2-weighted extended X-ray absorption fine structure (EXAFS) spectra of the Ru K-edge were obtained by Fourier transformation to analyze the local structures of Ru (Fig. 2b). The Ru foil presents a strong Ru–Ru peak at 2.36 Å while a Ru–O peak centered at 1.54 Å is observed for RuO2. For Mo–Ru NSAs, a stronger peak at 2.37 Å ascribed to the Ru–M (M = Ru/Mo) bond and a minor peak at 1.55 Å ascribed to the Ru–O bond are observed, indicating partially oxidized Ru. The small wavelet transformation for Ru has further vividly demonstrated the bonding information, where a strong Ru–Ru peak and a minor Ru–O peak are displayed for Mo–Ru NSAs (Fig. 2c–e).
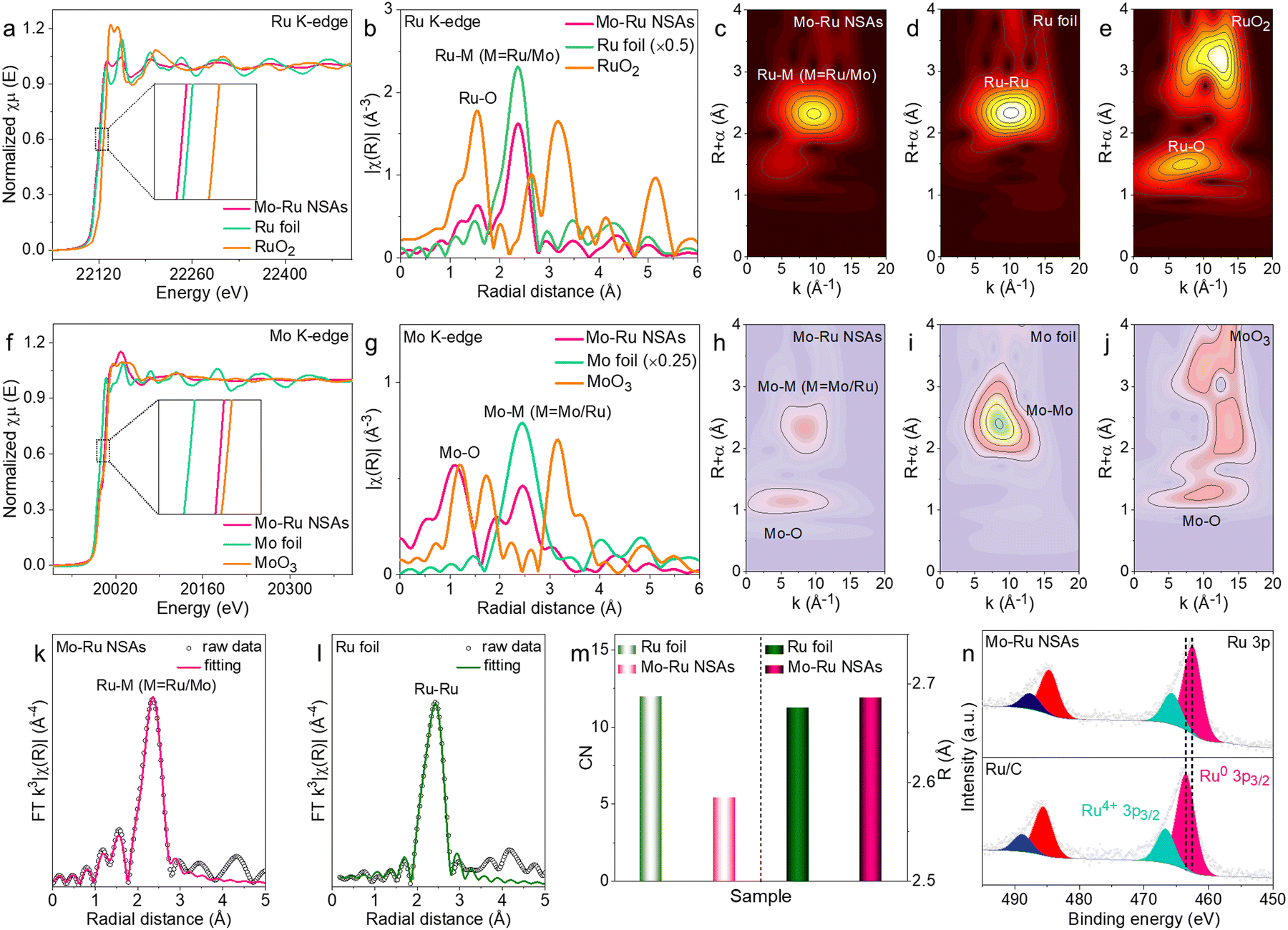 | ||
| Fig. 2 Electronic structure investigation. (a) Ru K-edge XANES and (b) Fourier transform EXAFS spectra of Mo–Ru NSAs, Ru foil and RuO2. Small wavelet transformation of (c) Mo–Ru NSAs, (d) Ru foil and (e) RuO2. (f) Mo K-edge XANES and (g) Fourier transform EXAFS spectra of Mo–Ru NSAs, Mo foil and MoO3. Small wavelet transformation of (h) Mo–Ru NSAs, (i) Mo foil and (j) MoO3. k3-weighted Fourier transformation and fitting of EXAFS spectra of (k) Mo–Ru NSAs and (l) Ru foil in R space. (m) Comparison of the coordination number of Ru and the Ru–Ru bond length of Mo–Ru NSAs and Ru foil. (n) Ru 3p XPS spectra of Mo–Ru NSAs and Ru foil. | ||
Besides, the Mo K-edge XANES and EXAFS spectra were also acquired for comprehensive analysis. Fig. 2f shows that the absorption energy for Mo in Mo–Ru NSAs is between that of Mo foil and MoO3, signifying that Mo atoms in Mo–Ru NSAs are in the oxidized state. However, the EXAFS spectra (Fig. 2g) and small wavelet transformation (Fig. 2h–j) of the Mo K-edge show the coexistence of the Mo–M (M = Mo/Ru) bond and the Mo–O bond for Mo–Ru NSAs, indicating that Mo atoms exist in both metallic and oxidized states in the sample. The coordination number (CN) of Ru in Mo–Ru NSAs was obtained by fitting the EXAFS spectra in R space with Ru foil as a reference (Fig. 2k–m) with the structural parameters listed in Table S1 (ESI†).35,36 Upon fitting, the CN of Ru in Mo–Ru NSAs is only 5.43, much smaller than that of Ru foil (12), which is consistent with the structural characteristics of Mo–Ru NSAs. The small coordination number of Ru indicates that Ru in Mo–Ru NSAs is highly unsaturated which may benefit the catalytic process by providing more active sites. Besides, the Ru–Ru length of Mo–Ru NSAs is slightly increased compared to Ru foil owing to the influence of Mo atoms. In the meantime, the EXAFS spectra of the Mo K-edge in R space were also fitted (Fig. S5, ESI†) with the structural parameters listed in Table S2 (ESI†). The surface electronic structures of Mo–Ru NSAs were analyzed and compared with that of Ru/C by X-ray photoelectron spectroscopy (XPS). As revealed by the Ru 3p XPS spectra of Mo–Ru NSAs and Ru/C, the surface of both samples is mainly in a metallic state with partial oxidation while the Mo 3d XPS spectrum shows the coexistence of metallic Mo atoms and oxidized Mo atoms (Fig. 2n and Fig. S6, ESI†). More interestingly, the binding energy of Ru 3p in Mo–Ru NSAs shows a negative shift of 0.98 eV. The negative shift indicates that Ru in Mo–Ru NSAs is electron-rich probably resulting from the electron transfer between Mo and Ru, which is in accordance with the XAS data. All the data above has demonstrated that Mo has significantly modified the lattice structures and surface electronic properties of Ru nanosheets.
According to the experimental results of XRD, we established the atomic model to explain the source of outstanding structural stability and potential electrocatalytic properties of Mo–Ru NSAs through DFT calculations. For the MoO3 decorated MoRu alloy, the surface of Ru containing one Mo atom was built and deposited by one MoO3. Based on the experimental characterization that indicates the coexistence of Ru–O, Mo–O and Mo–Mo bonds, we predict that MoO3 is extruded from the MoRu surface, which could change the electronic structures of the catalyst and affect the catalytic activity. Accordingly, MoO3 is placed on the MoRu(101) and MoRu(002) surfaces and then the structures are optimized (Fig. S7 and S8, ESI†). Fig. 3a and b presents the optimized region near MoO3, where the two-dimensional electron localization function (ELF) contours show the spatial electron localization of O and metal atoms, indicating the ionic bonding in the MoRu/MoO3 structure. The Bader charge analysis in Fig. 3c verifies the direction of interfacial electron transfer. It can be seen that the Mo atom in MoO3 loses about 2 electrons and transfers to the O atom, which is in line with the valence characteristics of metal oxides. Meanwhile, the Ru and Mo atoms on the surface of MoRu also contribute electrons to the O atoms, however, they were negatively charged on the surface of (002) without MoO3. Based on the experimental results that the Ru atoms gain electrons, there should be a large number of MoRu sites that are not covered by MoO3 on the surface of the catalyst. Returning to the case of MoRu/MoO3, such drastic charge transfer is beneficial for enhancing the binding between MoO3 and MoRu surfaces. As shown in Fig. 3d, the binding energies (Ebs) of MoO3 on MoRu(101) and MoRu(002) surfaces were calculated to be −5.32 and −5.08 eV, respectively. The large Eb values suggest the high stability of the catalyst under the studied reaction conditions. Consistent with the ELF results, differential charge densities in the inset of Fig. 3d also show that O atoms gain a large number of electrons while the surrounding Ru and Mo atoms contribute electrons during the formation of the MoRu/MoO3 structure. In addition, one can observe the electron cloud between Mo and Ru atoms, indicating the formation of a covalent-like bond between the metal atoms. The valence band spectra in Fig. 3e disclose that the characteristic 4d-band centers, which is a good descriptor for predicting the adsorbate-metal interactions,37,38 are considerably shifted upwards from −1.05 to −0.64 eV and −1.23 to −0.80 eV for MoRu(101) and MoRu(002) surfaces by the adsorption of MoO3. The upward shift of 4d-band centers affords a strong binding between surface Mo/Ru atoms and the adsorbed MoO3. It is evident that the electronic structure and bonding characteristics of our model are well consistent with the experimental results of XANES and EXAFS, accounting for their superior stability. Then, we plot the partial density of states (PDOS) of MoRu(101), MoRu(101)/MoO3, MoRu(002) and MoRu(002)/MoO3 surfaces, respectively (Fig. 3f). Since we focus on catalytic activity, only the PDOS near the Fermi level is shown. It can be seen that in the absence of MoO3, the MoRu surface conforms to the general characteristics of the electronic structure of the alloy, that is, the Ru-4d and Mo-4d orbitals have a major contribution to the Fermi level. When MoO3 is adsorbed on the MoRu surface, the O-p orbital in MoO3 is hybridized with the d-orbital of metal atoms, contributing to the Fermi level. Unlike the case of bulk MoO3, MoO3 displays a more metallic behaviour, which may exhibit excellent catalytic performance while improving its electron transportability.39
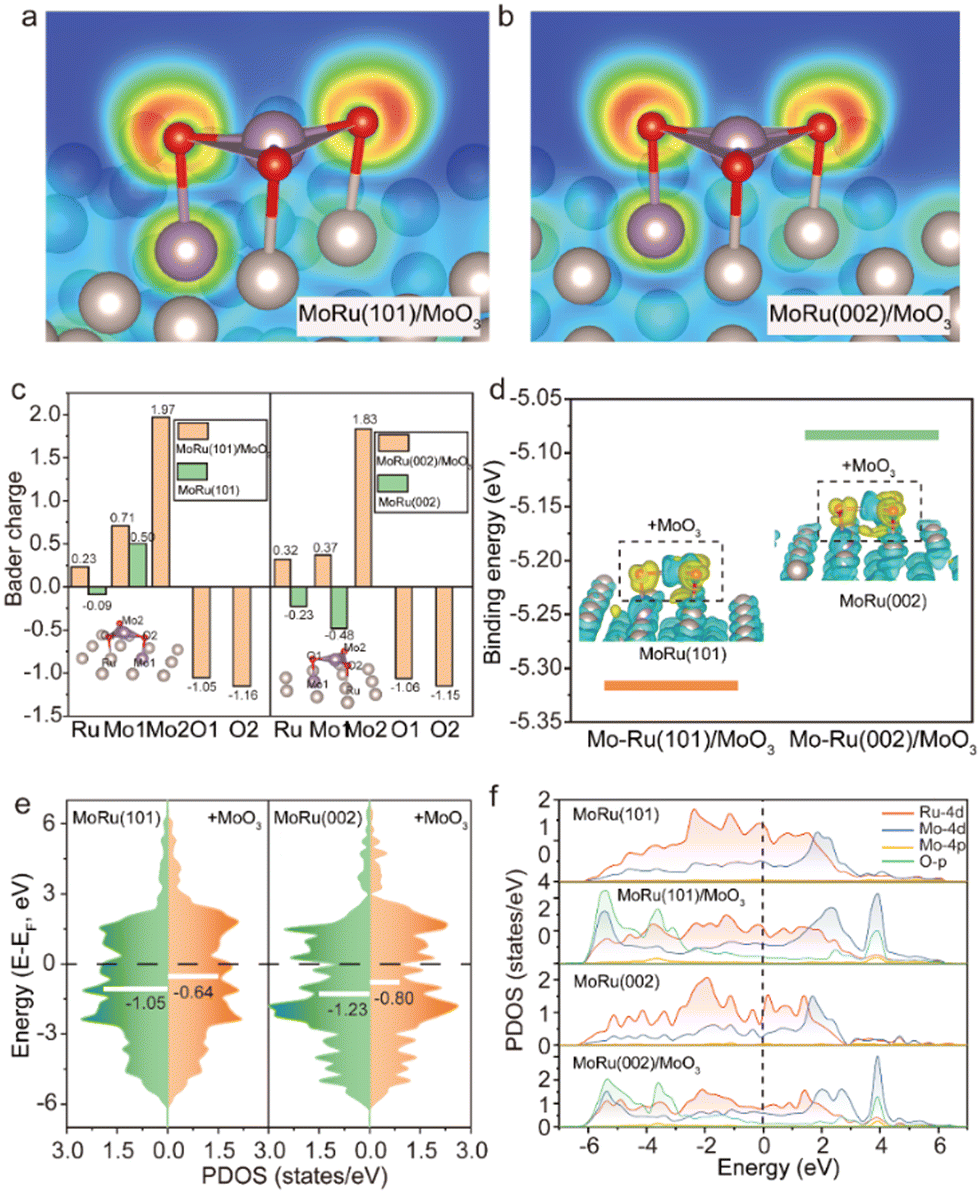 | ||
| Fig. 3 DFT calculation of MoRu(101)/MoO3 and MoRu(101)/MoO3 structures. Optimized configurations of (a) MoRu(101)/MoO3 and (b) MoRu(002)/MoO3 surfaces, rendered by the results of the electron localization function. (c) Bader charge for the atoms in MoRu and MoO3 of different surfaces. (d) Binding energy of MoO3 on MoRu(101) and MoRu(002) surfaces, respectively. The insets show the results of the corresponding charge density differences. Yellow and blue colors represent the accumulation and deficit of charge density, respectively. (e) Projected d-density of states of surface metal atoms in MoRu and MoO3 for different surfaces. The calculated d-band centers are marked with white lines. (f) Partial density of states for MoRu(101), MoRu(101)/MoO3, MoRu(002) and MoRu(101)/MoO3 surfaces, respectively. | ||
Possessing the intriguing morphology of 2D nanosheets where the lattice structures and surface electronic properties are drastically modified, it is attractive to investigate the electrochemical performance of Mo–Ru NSAs. In this work, the HER and HOR are selected as model reactions. Prior to electrochemical measurement, Mo–Ru NSAs were loaded on commercial carbon powder (Vulcan XC-72R) to improve the dispersibility and conductivity (Fig. S9, ESI†). The sample loaded on carbon powder was then annealed at 250 °C for 0.5 h in air to remove the adsorbed OAm (Fig. S10, ESI†). Combining the results of thermogravimetric analysis (TGA), XRD and chemical composition, the loading content of Ru is estimated to be about 34.0 wt% (Fig. S11, ESI†). Firstly, the HER performance of the annealed Mo–Ru NSAs was evaluated in 1 M KOH with commercial Ru/C and Pt/C measured for comparison. Fig. 4a displays the HER polarization curves of the catalysts in 1 M KOH. Apparently, the Mo–Ru NSAs show the highest HER activity with much faster reaction kinetics among the three catalysts. The overpotentials to reach 10, 30 and 50 mA cm−2 are all much lower than those of Ru/C and Pt/C (Fig. 4b). For example, the overpotential at 10 mA cm−2 is only 16 mV for Mo–Ru NSAs, which is much lower than those of Ru/C (61 mV) and Pt/C (40 mV). In addition, the specific current densities at different overpotentials (20, 40 and 60 mV) are all much higher than those of commercial Ru/C and Pt/C (Fig. 4c), signifying much faster reaction kinetics for the HER over Mo–Ru NSAs. In detail, the current density is 16.5 mA cm−2 for Mo–Ru NSAs at an overpotential of 20 mV, which is 13.8 and 9.2 times that of Ru/C (1.2 mA cm−2) and Pt/C (1.8 mA cm−2), respectively (Fig. 4c). To gain further insight into the reaction kinetics, the Tafel plots of the catalysts were obtained by plotting the overpotential against the current density in logarithm (Fig. 4d). Obviously, the Tafel slope value of Mo–Ru NSAs is much smaller than those of Ru/C and Pt/C, further confirming the much faster reaction kinetics of Mo–Ru NSAs.
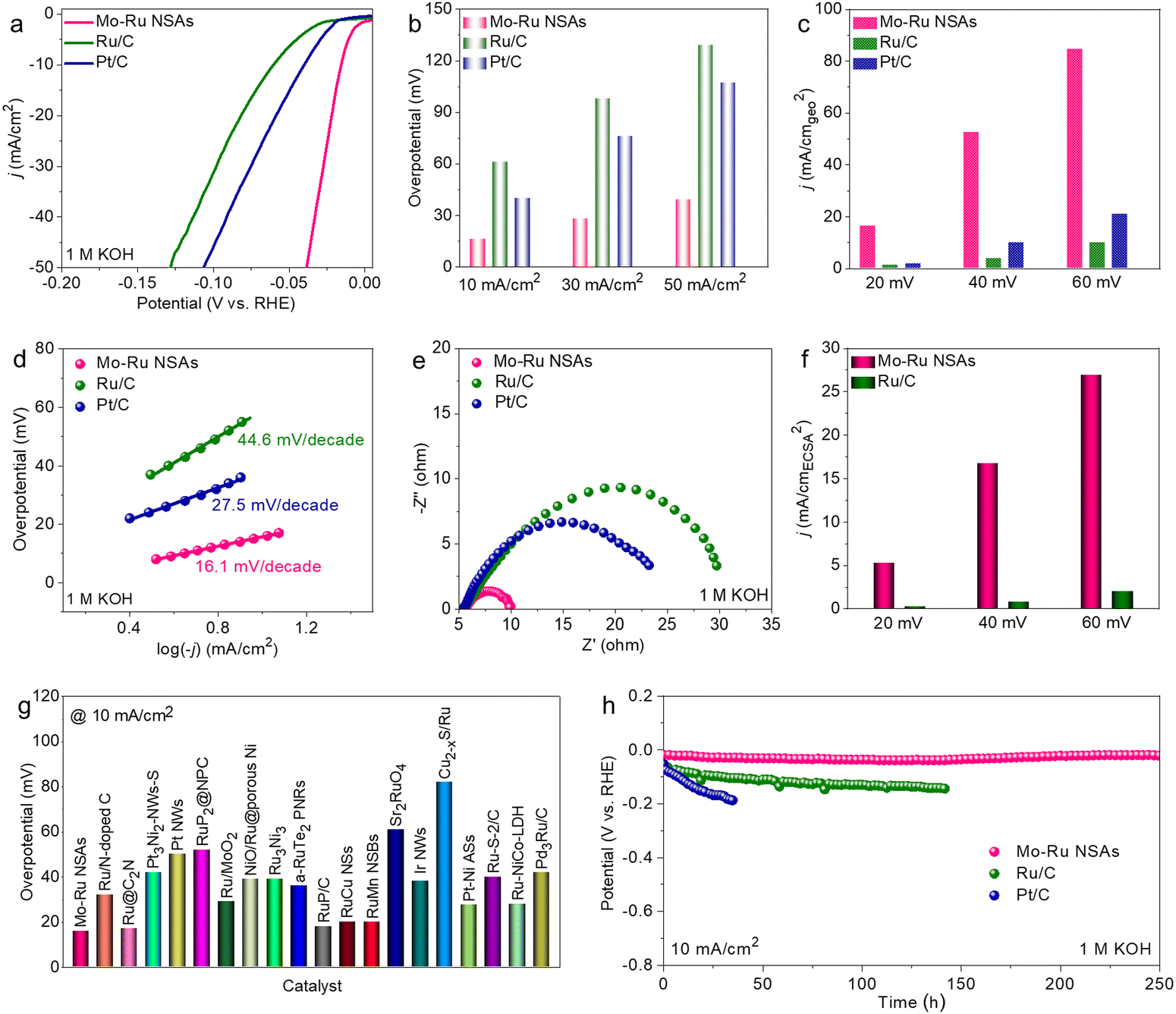 | ||
| Fig. 4 HER performance. (a) HER LSV curves of Mo–Ru NSAs, commercial Ru/C and Pt/C in 1 M KOH. (b) Overpotentials of reaching the current density of 10, 30 and 50 mA cm−2. (c) Current densities at overpotentials of 20, 40 and 60 mV. (d) Tafel plots of catalysts. (e) EIS Nyquist plots of the catalysts acquired in the frequency range of 0.1–106 Hz at an overpotential of 30 mV in 1 M KOH. (f) Comparison of the current densities between Mo–Ru NSAs and commercial Ru/C normalized to the ECSA at overpotentials of 20, 40 and 60 mV, respectively. (g) Comparison of the overpotentials reaching 10 mA cm−2. (h) Stability evaluation of the catalysts at a constant current density of 10 mA cm−2 by the chronopotentiometry method. | ||
To investigate the main factors that contribute to the superior HER activity of Mo–Ru NSAs, electrochemical impedance spectroscopy (EIS) spectra were acquired (Fig. 4e) in the frequency range of 0.1–106 Hz at an overpotential of 30 mV. The Mo–Ru NSAs possess much smaller resistance than Ru/C and Pt/C, indicating much faster electron transfer which is consistent with the ultrathin 2D nature of the nanosheets. Furthermore, the electrochemical surface areas (ECSAs) of Mo–Ru NSAs and Ru/C were evaluated by the double-layer capacitance (Cdl) of the catalysts which is proportional to the ECSAs (Fig. S12 and S13, ESI†). It is found that the Cdl value of Mo–Ru NSAs is smaller than that of commercial Ru/C, indicating a smaller ECSA value of Mo–Ru NSAs than Ru/C. The result is in agreement with the structural feature of commercial Ru/C where the Ru nanoparticles are ultrasmall (Fig. S14, ESI†). It can be therefore concluded that the difference in ECSA is not the factor that results in different HER activities. Under these conditions, it can also be deduced that Mo–Ru NSAs possess much higher intrinsic activity than commercial Ru/C considering its much smaller ECSA value. Fig. 4f presents the comparison of the current densities normalized to the relative ECSA of Mo–Ru NSAs and Ru/C at different overpotentials. The current density of Mo–Ru NSAs is 5.2 mA cmECSA−2 which is 21.7 times that of Ru/C (0.24 mA cmECSA−2). The activity difference is much larger than that normalized to the geometric area of the electrode. Mo–Ru NSAs represent a superior HER electrocatalyst in alkaline electrolytes with an ultralow overpotential of 16 mV at 10 mA cm−2 and surpass many other noble metal-based electrocatalysts reported in the literature (Fig. 4g and Table S3, ESI†). The HER stability of the catalysts was evaluated by the chronopotentiometry method at a constant current density of 10 mA cm−2. The catalytic activity of the commercial catalysts rapidly decayed with time especially for the benchmark HER catalyst of Pt/C (Fig. 4h). In sharp contrast, the Mo–Ru NSAs have exhibited significantly durable stability toward the HER. The Mo–Ru NSAs have maintained nearly constant HER activity during the 250 h stability test, representing an excellent electrocatalyst for the alkaline HER. In spite of the current superior catalytic performance, the activity and stability at large current densities would be explored in the future study.40 The spent Mo–Ru NSAs after the stability test were characterized by TEM and EDS (Fig. S15, ESI†). It was found that the morphology after the stability test can be well maintained and the composition was slightly changed.
With the superior HER performance, it is fascinating to investigate the catalytic properties of Mo–Ru NSAs towards the anodic HOR of fuel cells. The HOR polarization curves of Mo–Ru NSAs as well as Ru/C and Pt/C are acquired in H2-saturated 0.1 M KOH at a rotating speed of 1600 rpm on a rotating disk electrode (RDE). As shown in Fig. 5a, the Mo–Ru NSAs exhibit much higher HOR activity than Ru/C and Pt/C. Fig. 5b displays the kinetic current versus the potential in logarithm and the fitting results using the Butler–Volmer equation. In the observed potential range, the kinetic current of Mo–Ru NSAs is much higher than those of Ru/C and Pt/C, indicating much faster reaction kinetics over Mo–Ru NSAs towards the HOR. The kinetic current was normalized by the loading mass of Ru or Pt (denoted as ik,m) for quantitative comparison of the catalytic activity as shown in Fig. 5c. The mass activity of Mo–Ru NSAs (2.45 A mgRu−1) is 38.9 and 6.4 times those of Ru/C (0.063 A mg−1) and Pt/C (0.38 A mg−1) at 0.05 V vs. reversible hydrogen electrode (RHE), respectively. In the meantime, the exchange current normalized to the loading mass of noble metal (denoted as i0,m) is also obtained by the Butler–Volmer fitting (Fig. 5d). The i0,m value of Mo–Ru NSAs is 0.99 A mg−1, much higher than those of Ru/C (0.05 A mg−1) and Pt/C (0.18 A mg−1), indicating the much higher intrinsic activity of Mo–Ru NSAs. To analyze the electron transfer process, the HOR polarization curves at different rotating speeds of the catalysts were acquired (Fig. 5e and Fig. S16, ESI†) and Koutecky–Levich plots at 0.1 V vs. RHE were obtained (Fig. 5f). It is observed that the inverse of the current density is linear to the inverse of the square root of the rotating speed. The slopes of the Koutecky–Levich plots of Mo–Ru NSAs, Ru/C and Pt/C are fitted to be 12.24, 11.78 and 15.38 cm2 mA−1 rpm1/2, respectively, confirming the H2 mass-transport controlled process.16 Similar to the HER, the mass activity of Mo–Ru NSAs towards the HOR is compared with those reported in the literature where Mo–Ru NSAs outperform many noble metal-based electrocatalysts (Fig. 5g and Table S4, ESI†). The durability of the catalysts towards the HOR was investigated by monitoring the current-time response at 0.1 V vs. RHE in H2-saturated 0.1 M KOH (Fig. 5h). As shown, the activity of commercial Ru/C and Pt/C rapidly decayed with time especially at the initial stage with only 45.9% and 24.0% of the initial activity left after 10000 s. In contrast, the Mo–Ru NSAs present a highly stable catalytic process with only 22.8% activity loss after 10000 s. Characterization of the spent Mo–Ru NSAs after the HOR stability test also shows that there is no obvious change in the morphology but the chemical composition changed (Fig. S17, ESI†). More detailed microstructure analysis of catalysts after the catalytic stability test also demonstrates minor structural changes, indicating the structural stability of the catalysts (Fig. S18, ESI†).
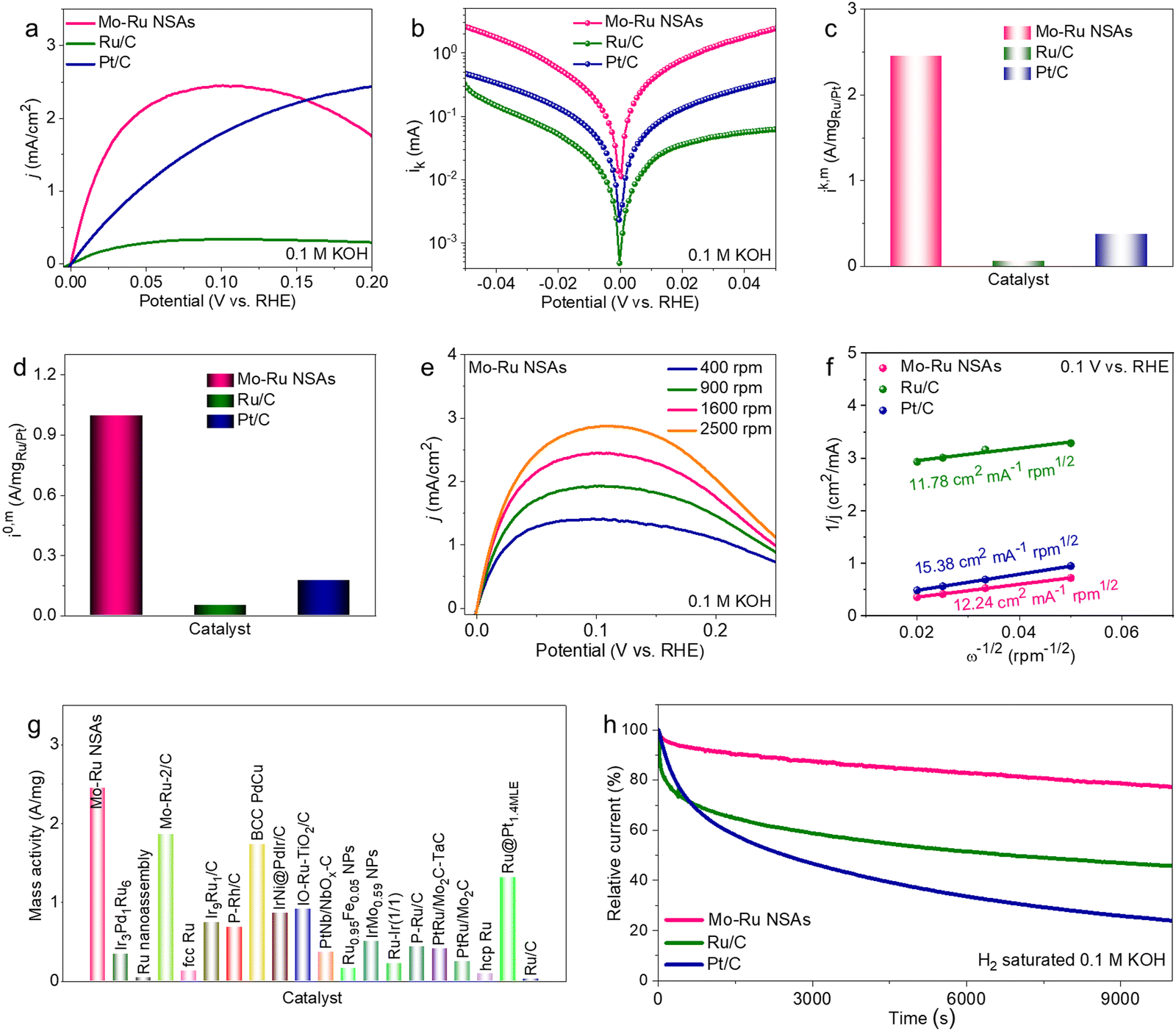 | ||
| Fig. 5 HOR performance. (a) HOR polarization curves of Mo–Ru NSAs, commercial Ru/C and Pt/C at a rotating speed of 1600 rpm in H2-saturated 0.1 M KOH. (b) Tafel plots of the kinetic current (ik) fitted by the Butler–Volmer equation. (c) Comparison of the kinetic current normalized to the mass of Ru/Pt at 0.05 V vs. RHE. (d) Comparison of the exchange current normalized to the mass of Ru/Pt. (e) HOR polarization curves of Mo–Ru NSAs at different rotating speeds. (f) Koutecky–Levich plots at 0.1 V vs. RHE. (g) Comparison of the mass activity of Mo–Ru NSAs with those reported in the literature. (h) Relative current-time chronopotentiometry measurement at 0.1 V vs. RHE in H2-saturated 0.1 M KOH. | ||
To understand the origin of the superior HER/HOR activity of Mo–Ru NSAs, DFT calculations were conducted to investigate the interaction of active sites on the surface of different catalysts and vital intermediates, that is, the adsorption energy (ΔE) and Gibbs free energy (ΔG) of adsorbed H*, 2H*, OH* and H2O* on MoRu(101), MoRu(002), MoRu(101)/MoO3 and MoRu(002)/MoO3 (MoO3 deposited on MoRu) surfaces. The cases of Ru(101) and Ru(002) were also considered for comparison. Fig. 6a shows the ΔE values of H* and OH*, which are pivotal intermediates in the HER and HOR, at different adsorption sites of different catalysts, where the corresponding adsorption site on both (101) and (002) surfaces for H* and OH* can be visualized in Fig. 6b and Fig. S19 (ESI†) (green five-pointed stars, blue hexagons and red squares represent weakly, moderately and strongly adsorbed sites, respectively). Compared with the strong binding of H* and OH* on Ru and Mo, both the H* and OH* binding to the sites near MoO3 can have a low ΔE that is very close to the best value (0 eV). In other words, as the coverage increases, H* and OH* tend to be close to MoO3, leading to a decrease in the ΔE and promoting the HER and HOR. Also, the calculated ΔG value of MoRu/MoO3 dramatically decreases to nearly 0 eV with the coverage increase of H* near MoO3 (Fig. 6c). Similar phenomena can be found in the adsorption of OH* and H2O* (Fig. S19, ESI†). We can contribute this to the enhanced electron density around the Mo and Ru atoms near MoO3.
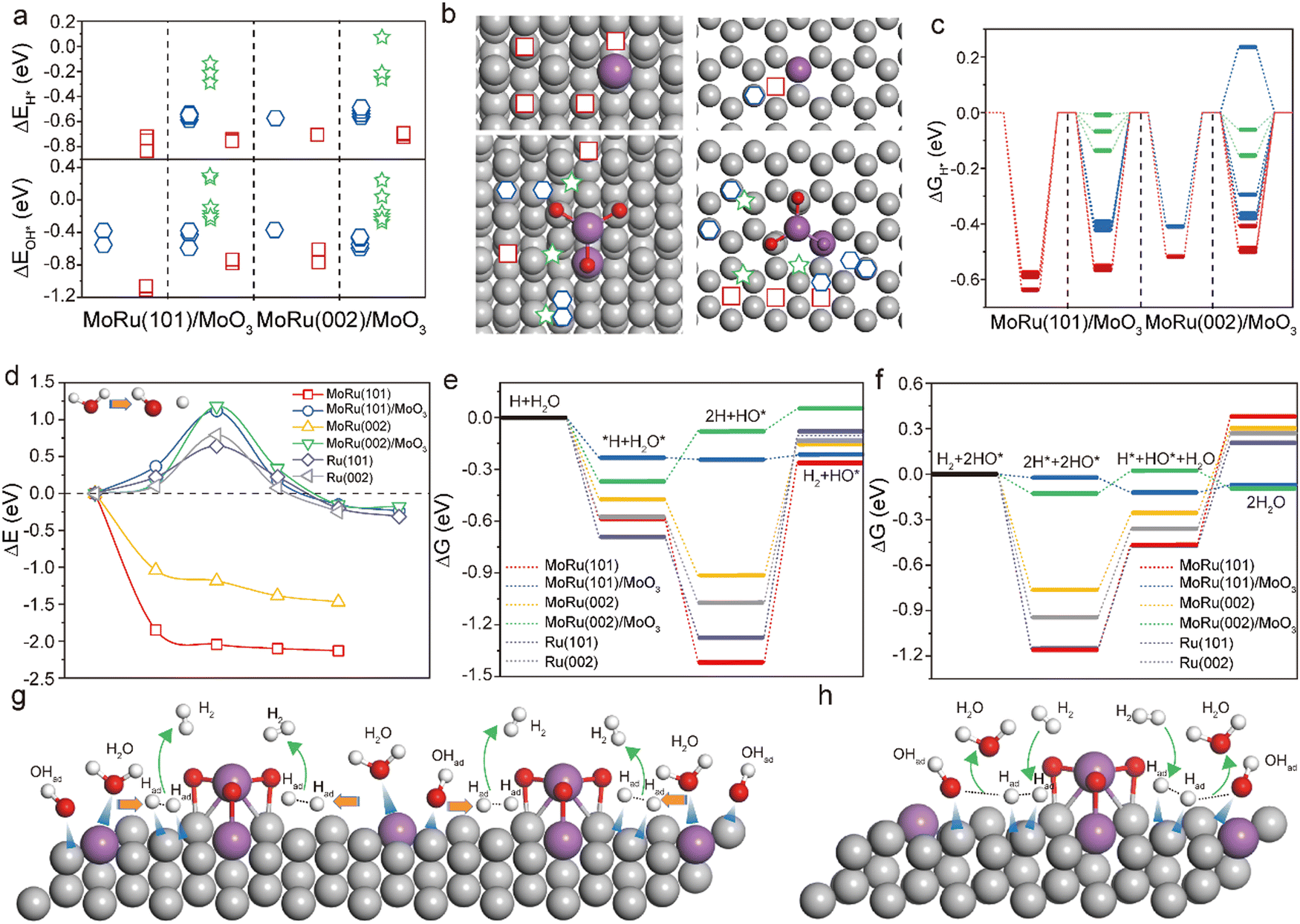 | ||
| Fig. 6 Theoretical investigation of the excellent electrocatalytic activity for the HER and HOR. (a) Adsorption energy ΔE of H* and OH* at different adsorption sites of different surfaces. (b) Diagram of the corresponding adsorption sites of H*, where green five-pointed stars, blue hexagons and red squares represent weakly, moderately and strongly adsorbed sites, respectively. (c) Gibbs free energy diagrams of absorbed H* on different surfaces. (d) Potential barrier for the decomposition of H2O on different surfaces. Free energy profile of (e) HOR and (f) HOR on different surfaces. Schematic of the proposed (g) HER and (h) HOR steps on the MoRu/MoO3 catalyst. | ||
For the HER process, the energetically easy decomposition of H2O is an important prerequisite for fast subsequent reactions.34 Our calculation results show that the surfaces of these different catalysts have strong adsorption capacity for H2O, with large absolute values of ΔE. However, at ambient temperature, due to the influence of vibrational entropy, the absolute value of ΔG at several sites approaches zero (Fig. S19, ESI†), showing a strong desorption tendency. From Fig. 6d, it can be seen that the decomposition of H2O near MoO3 needs to overcome a potential barrier of ∼1.2 eV, larger than that on the Ru surface. One interesting observation is that the MoRu sites enable the spontaneous decomposition of H2O*, which will greatly facilitate the generation of adsorbed H*. With the increasing coverage of adsorbed H*, a large number of weakly adsorbed states are formed near MoO3. In addition to H*, the MoRu/MoO3 surface can moderately adsorb H2O* and OH*. According to the calculation results of the Gibbs free energy of adsorption, the MoRu/MoO3 catalyst can obtain a more favorable reaction path, which enables the energetically favorable progress of both the HER and HOR (Fig. 6e and f), and achieves efficient bifunctional catalytic activity. This finding suggests that the combination of Mo and MoO3 does generate highly active sites over the Ru atoms. With an increase in product adsorption, these sites can serve as fast channels for the HER and HOR. Based on these results, a schematic of the proposed HER and HOR steps on the Mo–Ru NSA catalyst is shown in Fig. 6g and h, respectively. The schematic highlights the importance of the rapid decomposition of H2O on Mo sites and the MoO3 relying on steric hindrance to generate highly active sites to facilitate the reaction. The coupling of the Mo atom and MoO3 on the Ru surface enables efficient bifunctional catalytic activity.
Conclusions
To conclude, we have successfully fabricated Mo–Ru NSAs with unique atomic configurations of the metallic Mo atom and MoO3. The adsorption of the reaction intermediates is drastically optimized and the potential barrier of H2O dissociation is lowered over Mo–Ru NSAs. Compared with the state-of-the-art Pt/C and Ru/C, Mo–Ru NSAs have exhibited superior bifunctional HER and HOR performance in alkaline media. In particular, an overpotential of as low as 16 mV is sufficient to deliver a current density of 10 mA cm−2 for the HER in 1 M KOH and the mass activity of Mo–Ru NSAs for the HOR in 0.1 M KOH is 38.9 and 6.4 times that of Ru/C and Pt/C, respectively. DFT calculations reveal that the combination of MoO3 and MoRu can generate a large number of active adsorption sites. With the increasing coverage of the product, these sites can serve as fast channels for the HER and HOR, thereby enabling excellent bifunctional catalytic activity of Ru nanosheets. This work has paved a way toward the development of more advanced bifunctional electrocatalysts via surface and lattice engineering for hydrogen catalysis and beyond.Author contributions
X. H. and Q. Z. conceived and supervised the research. X. H. and L. L. designed the experiments. X. H., L. L., S. L., C. Z., Y. W. and Z. S. performed most of the experiments and data analysis. X. H., L. L., Q. Z. and J. H. participated in various aspects of the experiments and discussions. T. C. and Z. H. collected the XAS data. J. H. performed the DFT simulations. X. H., L. L. and J. H. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2020YFB1505802), the Ministry of Science and Technology (2017YFA0208200), the National Natural Science Foundation of China (22025108, U21A20327, 22121001, 52072323, 52122211), and the start-up supports from Xiamen University. The authors thank the BL01C1 beamline of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan).Notes and references
- W. Luo, Y. Wang and C. Cheng, Mater. Today Phys., 2020, 15, 100274 CrossRef.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- X. Zhang, Z. Luo, P. Yu, Y. Cai, Y. Du, D. Wu, S. Gao, C. Tan, Z. Li, M. Ren, T. Osipowicz, S. Chen, Z. Jiang, J. Li, Y. Huang, J. Yang, Y. Chen, C. Y. Ang, Y. Zhao, P. Wang, L. Song, X. Wu, Z. Liu, A. Borgna and H. Zhang, Nat. Catal., 2018, 1, 460–468 CrossRef CAS.
- L. Li, J. Huang, M. Li, Q. Li, L. Ouyang, M. Zhu and X. Yu, Int. J. Hydrogen Energy, 2013, 38, 16208–16214 CrossRef CAS.
- L. Li, Q. Gu, Z. Tang, X. Chen, Y. Tan, Q. Li and X. Yu, J. Mater. Chem. A, 2013, 1, 12263–12269 RSC.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- F. Xu, Y. Su and B. Lin, Front. Mater., 2020, 7, 4 CrossRef.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- J. Mao, C.-T. He, J. Pei, Y. Liu, J. Li, W. Chen, D. He, D. Wang and Y. Li, Nano Lett., 2020, 20, 3442–3448 CrossRef CAS PubMed.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- H. Wang, Y. Yang, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 4608–4616 CrossRef CAS.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
- H. Wang and H. D. Abruña, J. Phys. Chem. C, 2021, 125, 7188–7203 CrossRef CAS.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2019, 120, 851–918 CrossRef PubMed.
- P. Wang, K. Jiang, G. Wang, J. Yao and X. Huang, Angew. Chem. Int. Ed., 2016, 55, 12859–12863 CrossRef CAS PubMed.
- Z. Liu, J. Qi, M. Liu, S. Zhang, Q. Fan, H. Liu, K. Liu, H. Zheng, Y. Yin and C. Gao, Angew. Chem. Int. Ed., 2018, 57, 11678–11682 CrossRef CAS PubMed.
- X. Wang, Y. Zheng, W. Sheng, Z. J. Xu, M. Jaroniec and S.-Z. Qiao, Mater. Today, 2020, 36, 125–138 CrossRef CAS.
- J. Staszak-Jirkovský, Christos D. Malliakas, Pietro P. Lopes, N. Danilovic, Subrahmanyam S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, Vojislav R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- L. Li, L. Bu, B. Huang, P. Wang, C. Shen, S. Bai, T.-S. Chan, Q. Shao, Z. Hu and X. Huang, Adv. Mater., 2021, 33, 2105308 CrossRef CAS PubMed.
- Y. Zhao, X. Wang, G. Cheng and W. Luo, ACS Catal., 2020, 10, 11751–11757 CrossRef CAS.
- Y. Xue, L. Shi, X. Liu, J. Fang, X. Wang, B. P. Setzler, W. Zhu, Y. Yan and Z. Zhuang, Nat. Commun., 2020, 11, 5651 CrossRef CAS.
- T. Zhao, D. Xiao, Y. Chen, X. Tang, M. Gong, S. Deng, X. Liu, J. Ma, X. Zhao and D. Wang, J. Energy Chem., 2021, 61, 15–22 CrossRef CAS.
- Y. Zhou, Z. Xie, J. Jiang, J. Wang, X. Song, Q. He, W. Ding and Z. Wei, Nat. Catal., 2020, 3, 454–462 CrossRef CAS.
- J. Mao, C.-T. He, J. Pei, W. Chen, D. He, Y. He, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 4958 CrossRef.
- Y. Zhao, D. Wu and W. Luo, ACS Sustainable Chem. Eng., 2022, 10, 1616–1623 CrossRef CAS.
- X. Guo, X. Wan, Q. Liu, Y. Li, W. Li and J. Shui, eScience, 2022, 2, 304–310 CrossRef.
- C. Wang, P. Zhai, M. Xia, Y. Wu, B. Zhang, Z. Li, L. Ran, J. Gao, X. Zhang, Z. Fan, L. Sun and J. Hou, Angew. Chem. Int. Ed., 2021, 60, 27126–27134 CrossRef CAS PubMed.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef.
- H. Wang, J. Wang, Y. Pi, Q. Shao, Y. Tan and X. Huang, Angew. Chem. Int. Ed., 2019, 58, 2316–2320 CrossRef CAS PubMed.
- F.-Y. Yu, Z.-L. Lang, L.-Y. Yin, K. Feng, Y.-J. Xia, H.-Q. Tan, H.-T. Zhu, J. Zhong, Z.-H. Kang and Y.-G. Li, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- Q. Yang, Y. Jia, F. Wei, L. Zhuang, D. Yang, J. Liu, X. Wang, S. Lin, P. Yuan and X. Yao, Angew. Chem. Int. Ed., 2020, 59, 6122–6127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02076a |
| This journal is © The Royal Society of Chemistry 2023 |