
A closely packed Pt1.5Ni1−x/Ni–N–C hybrid for relay catalysis towards oxygen reduction†
Wenxin
Guo‡
ab,
Xiaoping
Gao‡
a,
Mengzhao
Zhu‡
a,
Chenxi
Xu
c,
Xiaorong
Zhu
d,
Xuyan
Zhao
a,
Rongbo
Sun
a,
Zhenggang
Xue
e,
Jia
Song
a,
Lin
Tian
a,
Jie
Xu
f,
Wenxing
Chen
 g,
Yue
Lin
h,
Yafei
Li
i,
Huang
Zhou
*a and
Yuen
Wu
*ab
g,
Yue
Lin
h,
Yafei
Li
i,
Huang
Zhou
*a and
Yuen
Wu
*ab
aDepartment of Chemistry, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, Hefei 230026, China. E-mail: yuenwu@ustc.edu.cn
bDalian National Laboratory for Clean Energy, Dalian 116023, China
cSchool of Materials Science and Engineering, Hefei University of Technology, Hefei 230009, China
dSchool of Chemistry and Chemical Engineering, Nantong University, Nantong 226019, China
eNEST Laboratory, Department of Physics, Department of Chemistry, College of Science, Shanghai University, Shanghai 200444, China
fInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou 215123, China
gBeijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China
hHefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, China
iJiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China
First published on 28th November 2022
Abstract
Diminishing the usage of Pt without sacrificing its activity still remains a challenge in proton-exchange membrane fuel cells (PEMFCs). Here, we report a gas-promoted dealloying process to prepare a closely packed hybrid electrocatalyst containing Pt-based alloy nanocrystals (NCs) and dense isolated Ni sites. Driven by ammonia and heat, the initial Pt1.5Ni NC undergoes a dealloying process to form a stable Pt-skin Pt1.5Ni1−x alloy due to the continuous detachment of Ni atoms from it. Subsequently, these Ni atoms would be trapped by the adjacent defects on the carbon substrates, resulting in abundant Ni sites distributed closely around the dealloyed Pt1.5Ni1−x NC. For a multielectron transferred oxygen reduction reaction (ORR), the hybrid ensures the reduction of the two electrons at Ni single sites, and the corresponding intermediate (OOH*) rapidly migrates to the neighboring Pt-based NC to finish the subsequent electron transfer. This efficient relay catalytic process could greatly reduce the usage of Pt. The resulting catalyst exhibits excellent ORR activity with a mass activity (MA) of 4.10 A mgPt−1, exceeding that of commercial Pt/C by a factor of ∼15. More importantly, in practical H2/O2 fuel cell tests, a peak power density of 1.72 W cm−2 and a current density of 0.55 A cm−2 at 0.80 V can be achieved, both of which exceed DOE 2025 targets.
Broader contextAs a promising clean and efficient energy carrier, hydrogen is one of the best options to achieve energy technology revolution and large-scale decarbonization. Fuel cell electric vehicles (FCVs), a common terminal application of hydrogen energy, are expected to be an important electric transportation solution by converting the chemical energy of hydrogen into electrical energy through proton exchange membrane fuel cells (PEMFCs). The U.S. Department of Energy has set long-term goals for heavy-duty FCVs with fuel cell system lifetimes and costs of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours and $60 kW−1, respectively, which urgently requires the development of low-Pt loaded and highly performance catalysts to catalyze the kinetically sluggish cathodic oxygen reduction reaction (ORR), thereby reducing costs and further improving the economics of FCVs. Here, we integrated PtNi nanocrystals and dense isolated Ni sites on N–C to construct a closely packed hybrid for efficient relay catalysis, which greatly improved membrane electrode assembly (MEA) activity and stability while maintaining a low Pt loading. The rational design of high-efficiency relay catalysts not only offers the possibility of further promoting the development of FCV as automotive power sources, but also provides ideas to explore more electrocatalytic processes. 000 hours and $60 kW−1, respectively, which urgently requires the development of low-Pt loaded and highly performance catalysts to catalyze the kinetically sluggish cathodic oxygen reduction reaction (ORR), thereby reducing costs and further improving the economics of FCVs. Here, we integrated PtNi nanocrystals and dense isolated Ni sites on N–C to construct a closely packed hybrid for efficient relay catalysis, which greatly improved membrane electrode assembly (MEA) activity and stability while maintaining a low Pt loading. The rational design of high-efficiency relay catalysts not only offers the possibility of further promoting the development of FCV as automotive power sources, but also provides ideas to explore more electrocatalytic processes.
|
Introduction
Single-atom metal sites on nitrogen-doped carbon (M–N–C) have emerged as highly sought-after oxygen reduction reaction (ORR) catalysts for proton-exchange membrane fuel cells (PEMFCs) due to their potential to largely reduce the usage of Pt in platinum-group-metal (PGM)-based catalysts.1–6 However, most of the reported M–N–C electrocatalysts have difficulty in catalyzing the complex intermediates generated in multielectron processes, especially in acidic ORR, because of the obstacles in stabilizing intermediates at a single site in terms of the changing d-state energy and orbital symmetry.7–9 Specifically, for a 4e− ORR reduction process, isolated metal sites often face a barrier to adsorb the as-reduced intermediates, resulting in the difficulty in further reduction to H2O.10 For instance, the isolated Pt atom specifically exhibits the 2e− pathway for the ORR to H2O2 rather than 4e− to H2O, significantly limiting their applications in PEMFCs.11,12 This differs from conventional nanocrystal (NC) catalysts in that their broader continuous d orbitals are more likely to trap the reduction products generated during multielectron processes.13–15To better use M–N–C in the 4e− ORR to reduce the usage of Pt, an efficient approach is to replace only 1-2e− of the reduction process rather than the whole reduction process. Meanwhile, another site is also required to complete the transfer of other electrons by relay catalysis. In such a catalytic process, the intermediate species generated on M–N–C need to migrate promptly to the adjacent site, such as a NC or a cluster, while avoiding direct desorption without engaging in the subsequent reaction. This requires an effective coupling of the two active sites to shorten the diffusion distance of the intermediates during the ORR and carry out a cascade of multistep reactions. The complex sites of adjacent M–N–C and NC/cluster might ensure the complete and efficient reduction of continuously migrating intermediates in the 4e− process and might help to further reduce the effects of mass transfer and concentration polarization during the kinetic process.16,17 Thus, the full utilization of M–N–C by relay catalysis is a promising method to reduce the dosage of Pt as well as the catalyst cost.
Herein, we develop a gas-promoted dealloying process to directly construct a closely packed Pt–Ni alloy NC surrounded by single Ni sites, in which an efficient electron transfer relay can be achieved between these two types of sites to realize superior ORR performance. The obtained Pt–Ni NC possesses a stable platinum-rich surface due to the dealloying effect caused by heat and NH3 corrosion, in which abundant Ni atoms migrated from the initial Pt–Ni alloy and were trapped by adjacent nitrogen defect sites. This evolution process was well traced by aberration corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) and X-ray absorption fine structure (XAFS) spectroscopy. Compared with the untreated sample, the closely packed hybrid displays an enhanced ORR performance in acidic media with a mass activity (MA)/specific activity (SA) of 4.1 A mgPt−1/4.6 mA cm−2, which is 15/7 times that of commercial Pt/C and exceeds that of many noble metal catalysts. More importantly, the catalyst with only half of the Pt loading of Pt/C exhibits a much higher activity and durability in practical fuel cell tests. Finally, theoretical results reveal that the closely packed hybrid can ensure that the key intermediate species OOH* continuously migrates from the Ni single sites to the neighboring Pt-based NC, contributing to a relay reaction to enhance the catalytic performance.
Results and discussion
The detailed preparation route of relay catalysis for a closely packed hybrid electrocatalyst is illustrated in Fig. 1(a). First, the representative Pt-based NC of the octahedral Pt1.5Ni alloy with an average diameter of 7.66 nm was synthesized by a solvothermal method and subsequently loaded uniformly on nitrogen-doped carbon (N–C) derived from zeolitic imidazolium framework-8 (ZIF-8) (denoted as Pt1.5Ni/N–C, Fig. S1–S3, ESI†).18 The XRD pattern indicates that Zif-8-derived N–C is completely amorphous with no characteristic peaks. HAADF-STEM images proved that there are no metal species on the blank N–C (Fig. S2e–f, ESI†). Then, the obtained Pt1.5Ni/N–C was heated in a tube furnace (50–300 °C) under an NH3 atmosphere. With increasing temperature, abundant Ni atoms were hauled out of the Pt–Ni alloy particles to generate volatile species, leading to the gradual evolution of octahedral Pt1.5Ni to spherical-like Pt1.5Ni1−x nanoparticles (NPs) as the dealloying process proceeded (Fig. 1(b)).19,20 Meanwhile, the migrated Ni atoms were captured by the adjacent nitrogen defects on N–C. Afterward, a hybrid architecture consisting of the Pt–Ni alloy with a Pt-rich surface (∼7.38 nm) and closely isolated Ni sites was formed (denoted as Pt1.5Ni1−x/Ni–N–C, Fig. S4 and S5, ESI†). High-resolution TEM (HRTEM) images and the corresponding fast Fourier transform (FFT) pattern reveal that the obtained Pt1.5Ni1−x NPs have a spherical-like shape and good crystallinity, with lattice spacings of 0.230 nm and 0.196 nm corresponding to the mainly exposed stable Pt (111) and (200) planes, respectively (Fig. 1(c) and (d)).21 The Pt loading of 8 wt% for Pt1.5Ni1−x/Ni–N–C was identified by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The line scan profile taken from Fig. 1(f) depicts a discrete elemental distribution of Pt and Ni in a representative Pt1.5Ni1−x alloy and a surface layer with a Pt-rich shell (Fig. 1(e)). Energy dispersive X-ray spectroscopy (EDS) mappings in Fig. 1(f) (judged by the purple and blue circles) and Fig. S6, ESI† also show that the Ni species are closely and homogeneously distributed around the Pt–Ni alloy structure over the N–C substrate, suggesting the possibility of Ni atom migration by our strategy. To prove this, atomic resolution aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) was used. Fig. 1(g), (h) and Fig. S7, ESI† demonstrate the atomically dispersed Ni sites (marked by dark salmon circles) distributed uniformly adjacent to Pt1.5Ni1−x, indicating the formation of a closely packed hybrid (Pt1.5Ni1−x/Ni–N–C). | ||
| Fig. 1 Schematic illustrations and structural characterizations for the preparation of Pt1.5Ni1−x/Ni–N–C and reference materials. (a) Schematic illustration of the preparation of Pt1.5Ni1−x/Ni–N–C. Structural characterizations of Pt1.5Ni1−x/Ni–N–C: (b) TEM image, (c) HRTEM image, (d) FFT and (e) Line scan plot taken from (f), (f) EDS mappings, (g), (h) HADDF-STEM images. | ||
To investigate the intrinsic evolution mechanism from Pt1.5Ni/N–C to Pt1.5Ni1−x/Ni–N–C, representative atomic resolution HAADF-STEM images were acquired under NH3 with 50 °C as the reaction temperature interval. Fig. 2(a) reflects that the synthesized Pt1.5Ni NPs possess an octahedral morphology at room temperature with a lattice stripe of 0.23 nm, consistent with the (111) plane (Fig. S8, ESI†). The elemental mappings further reveal that the Pt1.5Ni NPs possess Pt-rich axes and corners and Ni-rich facet compositions (Fig. 2(a) and Fig. S9, ESI†). This can also be demonstrated by the line scan oriented along the 〈100〉 zone axes, which confirms the start signal with Ni enrichment in facets and Pt enrichment in central axes (Fig. 2(b)). At 50 °C, the initial octahedral morphology is almost unchanged, reflecting good structural stability (Fig. 2(c)). As the temperature increased to 100 °C and 200 °C, the particles gradually eroded into a concave shape, indicating that a dealloying process occurred on the surface. This phenomenon is due to the effect of NH3 molecules, resulting in the leaching of Ni atoms from the alloy particles and their eventual capture by N defects (Fig. 2(d) and (e)). Meanwhile, abundant Pt atoms migrated from Pt-rich edges to the vacant (111) facets under the sustained chaotic thermal motion of molecules. As the temperature rose further, the dealloying process intensified, resulting in the transformation of a truncated octahedron shape with a newly formed (100) plane, accompanied by abundant Ni single sites on the adjacent N–C (Fig. 2(f) and Fig. S10–S12, ESI†). At 300 °C, the truncated octahedron was further converted into a spherical-like shape, which was unchanged even at 350 °C, revealing good thermal stability (Fig. 2(g) and Fig. S13, ESI†).22,23 Density functional theory (DFT) calculations were performed to further probe the evolution mechanism. The results show the migration of a single Ni atom by NH3 coordination, and the subsequent capture by N4 defects is exothermic and requires a diffusion potential barrier of 2.85 eV to reach stable states, which is much smaller than the counterpart of Pt (3.78 eV, Fig. 2(h) and Fig. S14, ESI†). This further demonstrates that Ni atoms migrate more easily than Pt atoms in the bulk phase, agreeing well with the previous STEM-EDX observations (Fig. 1(f)–(h)).
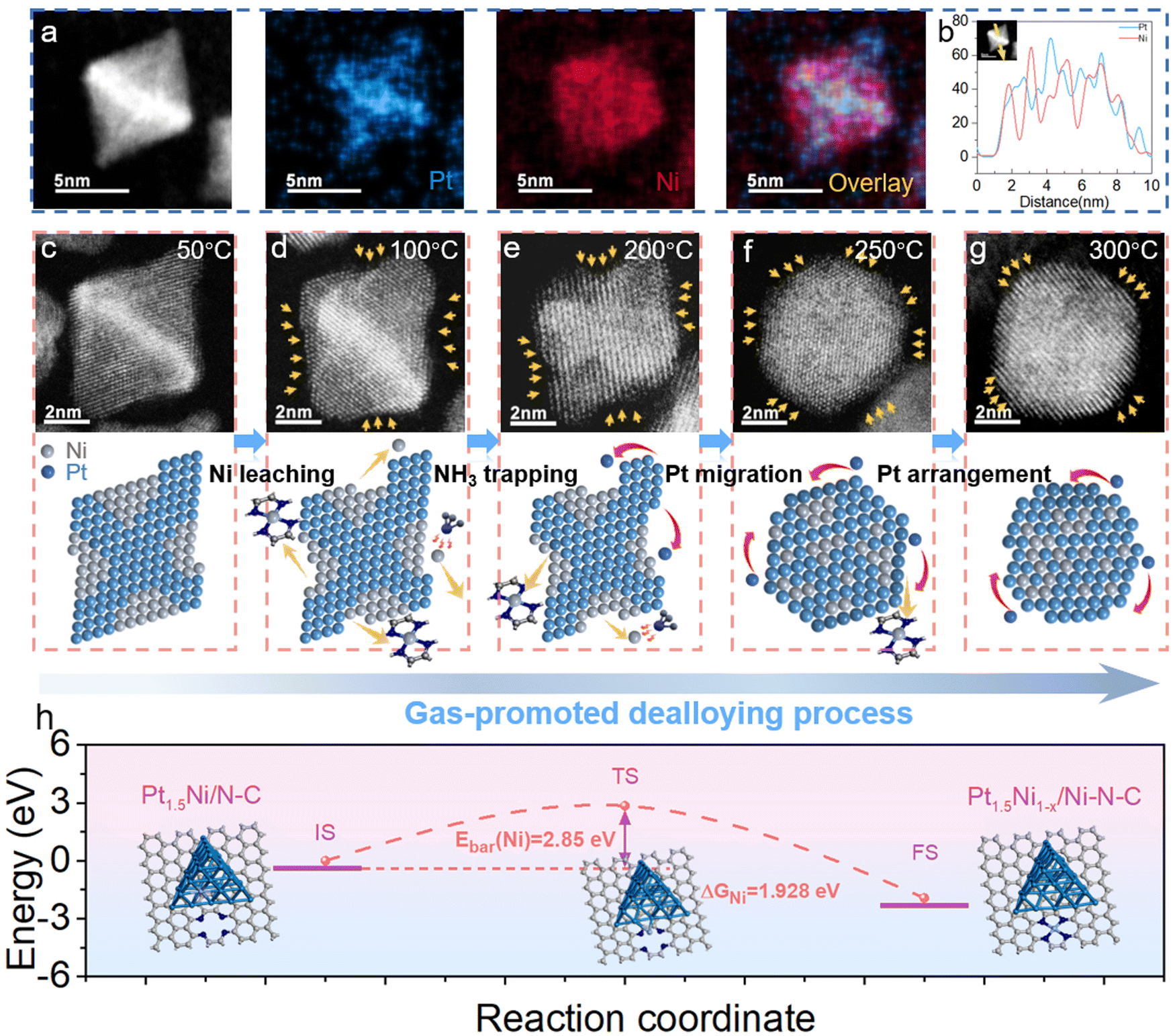 | ||
| Fig. 2 HADDF-STEM images and DFT calculations for the evolution of Pt1.5Ni NPs under NH3 and heat treatment. (a), (b) HADDF-STEM image, EDS mappings and line scan of Pt1.5Ni NPs, (c)–(g) HADDF-STEM images and geometric structure models for the evolution process of Pt1.5Ni NPs, (h) Calculated energies along the stretching pathway of the Ni atom from the Pt1.5Ni cluster to Ni–N4 sites by NH3 and the corresponding configurations. | ||
Powder X-ray diffraction (XRD) patterns were adopted to investigate the physical phase of Pt1.5Ni/N–C before and after treatment (Fig. 3(a)). The peak position corresponding to the (111) plane of the face-centered cubic (fcc) Pt–Ni alloy shifts slightly towards the lower 2θ degree of the Pt position, which could be attributed to the lattice spacing expansion caused by Ni leaching from the bulk phase of Pt1.5Ni.24 For Ni X-ray photoelectron spectroscopy (XPS) spectra (Fig. 3(b)), the relative peak ratio of Nix+(0 < x < 3) 2p3/2 in Pt1.5Ni1−x/Ni–N–C is significantly increased compared to Pt1.5Ni/N–C, suggesting that the partial Ni was oxidized in Pt1.5Ni1−x/Ni–N–C, which can also be confirmed by the near edge X-ray absorption fine structure (NEXAFS) spectra of Ni L-edge (Fig. S15, ESI†). The Pt1.5Ni1−x/Ni–N–C shows a weakened intensity due to the separation of the metallic phase. Alternatively, the collected Pt 4f XPS spectra in both Pt1.5Ni/N–C and Pt1.5Ni1−x/Ni–N–C indicate that the surface Pt is mainly in the metallic state (Fig. S16, ESI†).25 The high-resolution N 1s XPS spectra reveal that both Pt1.5Ni1−x/Ni–N–C and Pt1.5Ni/N–C possess pyridyl-N (398.5 eV), pyrrole-N (400.5 eV), metal-N (399.4 eV) and graphitic-N (401.4 eV),26 while the enhanced peak is detected at 399.4 eV in the former due to the generation of the Ni–N species (Fig. S17, ESI†). The N K-edge NEXAFS further shows the existence of π* excitation of pyridyl-N (398.9 eV) and graphitic-N (401.9 eV) species and σ* excitation of the C–N moieties (406.7 eV),27 illustrating that the samples were well graphitized (Fig. S18, ESI†). Moreover, the Raman spectra show an increased ID/IG value (the intensity ratio of the D-band and G-band, 0.93 vs. 0.95) after treatment, demonstrating the generation of carbon defects (Fig. S19, ESI†).
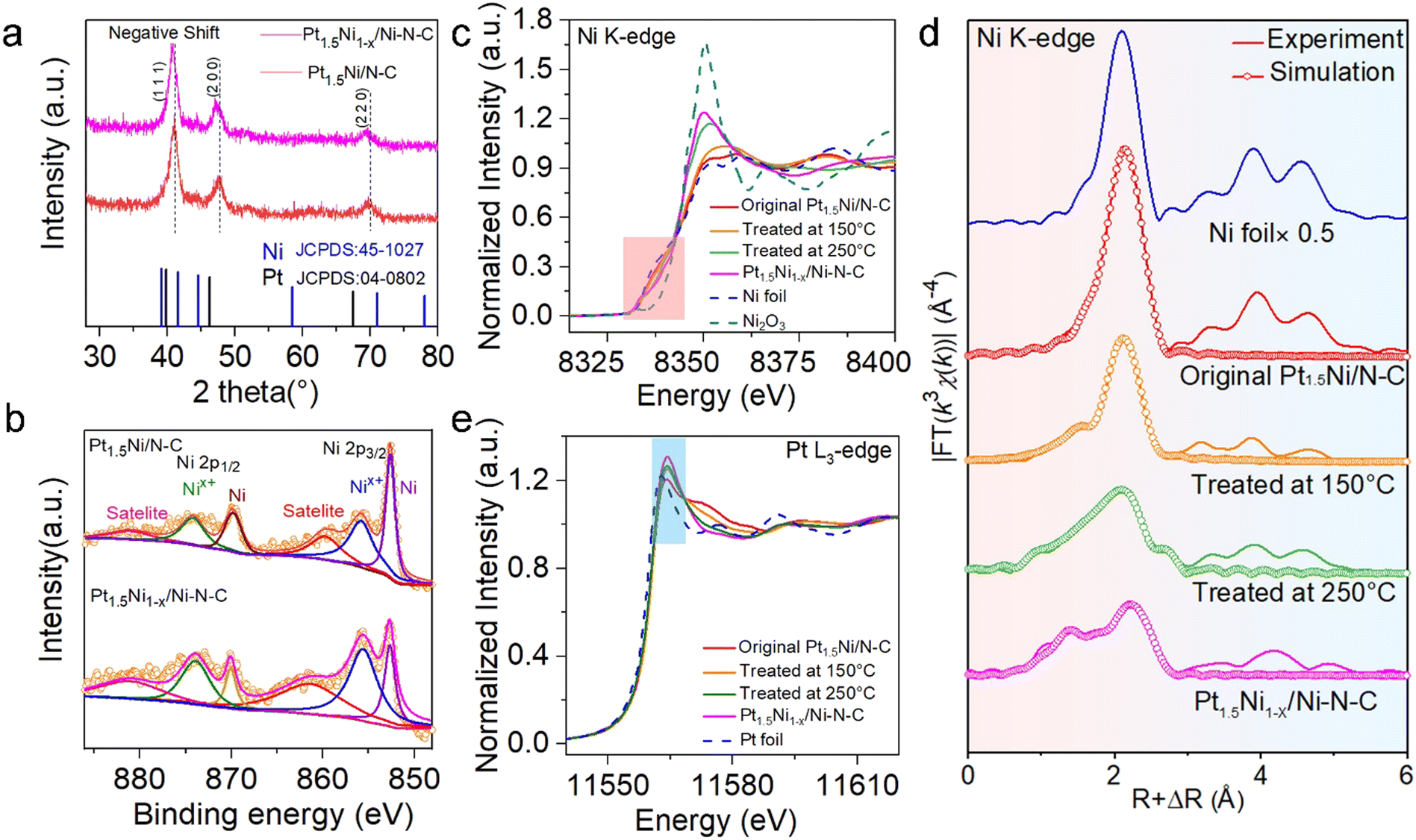 | ||
| Fig. 3 Structural characterizations of Pt1.5Ni1−x/Ni–N–C and reference materials. (a) XRD patterns and b) XPS of Ni 2p for Pt1.5Ni/N–C and Pt1.5Ni1−x/Ni–N–C, (c) Ni K-edge NEXAFS spectra, (d) the k3-weighted χ(k)-function of the EXAFS spectra and (e) Pt L3-edge NEXAFS spectra for Ni foil, Pt foil, Ni2O3, original Pt1.5Ni/N–C, Pt1.5Ni/N–C treated at 150 °C and 250 °C and Pt1.5Ni1−x/Ni–N–C (300 °C). | ||
NEXAFS and extended X-ray absorption fine structure (EXAFS) spectroscopy was performed to trace the changes in electronic states and coordination environment during the treatment. With the annealing temperature increasing, the Ni K-edge NEXAFS (Fig. 3(c)) spectra show that the white line (WL) intensity gradually increased and the absorption threshold position shifted toward Ni3+, indicating that Ni was further oxidized. Moreover, the EXAFS spectra of the R space show that the Ni–Ni bond had a certain degree of positive shift with respect to the Ni foil, and a new peak gradually emerged at ∼1.5 Å with lower intensity (Fig. 3(d)), revealing the gradual decrease in surface metallicity and the formation of Ni single sites. The fitting results of Pt1.5Ni1−x/Ni–N–C reveal that the peak located at 1.49 Å can be assigned to Ni–N bonds, and the corresponding coordinated number is 4. For Pt L3-edge XANES spectra, WL strength was enhanced gradually during annealing, which is related to the electron transfer within the alloy, further evidencing the decrease in alloying (Fig. 3(e)). More detailed fitting curves and parameters are displayed in the ESI,† Table S1.
To verify the catalytic performance of the hybrid (Pt1.5Ni1−x/Ni–N–C) obtained by the gas-promoted dealloying process, an ORR experiment was performed. The linear sweep voltammetry (LSV) curve indicates that the half-wave potential (E1/2) is 0.967 V for Pt1.5Ni1−x/Ni–N–C in 0.1 M HClO4 solution, which is superior to the untreated Pt1.5Ni/N–C (0.926 V), the commercial Pt/C (0.887 V) and most of the reported Pt-based catalysts (Fig. 4(a) and Table S2, ESI†). However, the single Ni site catalyst (Ni–N–C) prepared via an impregnation method (ESI†) shows a much poorer activity (E1/2 = 0.38 V). The fast ORR reaction kinetics and excellent activity of Pt1.5Ni1−x/Ni–N–C can be further observed by the small Tafel slope value Pt1.5Ni1−x/Ni–N–C (∼55.44 mV dec−1) < Pt1.5Ni/N–C (∼64.83 mV dec−1) < Pt/C (∼70.46 mV dec−1) < Ni–N–C (∼132.50 mV dec−1) (Fig. S20, ESI†).28 These results illustrate that the design of closely packed hybrids to obtain adjacent active sites is critical for acquiring a high ORR performance. The cyclic voltammetry (CV) curves were measured in a N2-saturated electrolyte (Fig. 4(b)) to obtain the electrochemically active surface area (ECSA) value of Pt1.5Ni1−x/Ni–N–C, which was estimated to be 89.88 m2 gPt−1, greater than that of Pt1.5Ni/N–C (76.07 m2 gPt−1), revealing that the former has a higher utilization of platinum.17,29 Combined with the almost unchanged surface area before and after treatment (766.9 m2 g−1 for Pt1.5Ni/N–C and 785.828 m2 g−1 for Pt1.5Ni1−x/Ni–N–C), it is reasonable to infer that Pt1.5Ni1−x/Ni–N–C possesses an increased number of Pt active sites (Fig. S21, ESI†). Furthermore, the rotating ring-disk electrode (RRDE) test shows an electron transfer number of ∼3.95 and a hydrogen peroxide yield of ∼4% for Pt1.5Ni1−x/Ni–N–C, suggesting a four-electron mechanism similar to that of commercial Pt/C (Fig. S22, ESI†). However, Ni–N–C displays an over 15% hydrogen peroxide yield with a much lower electron transfer number (∼3.4). This result suggests that the closely packed hybrid of Pt1.5Ni1−x/Ni–N–C can effectively inhibit the low electron transfer reaction on the Ni–N–C support, resulting in a superior catalytic performance. Additionally, Pt1.5Ni1−x/Ni–N–C realizes the optimal mass activities (MA) and specific activities (SA) under different potentials, especially for 4.1 AmgPt−1/4.6 mA cm−2 at 0.9 V, which exceeds that of Pt1.5Ni/N–C by a factor of ∼2.9/2.5 and Pt/C by a factor of ∼15.2/7.0 (Fig. 4(c) and (d)). The accelerated deterioration test (ADT) demonstrates that Pt1.5Ni1−x/Ni–N–C retains an outstanding stability after cycling with no observable decay of E1/2. The MA and SA of Pt1.5Ni1−x/Ni–N–C showed no obvious attenuation after 10000 cycles, while the catalyst showed a certain degree of attenuation at high potential and 20000 cycles. (Fig. 4(e) and Fig. S23–S25, ESI†). TEM images of Pt1.5Ni1−x/Ni–N–C after ADT manifest negligible changes in particle morphology and size (Fig. S26, ESI†), further indicating the good structural stability of the catalyst. In contrast, Pt/C shows a severe decrease in E1/2 after only 5000 cycles (Fig. S27 and S28, ESI†).
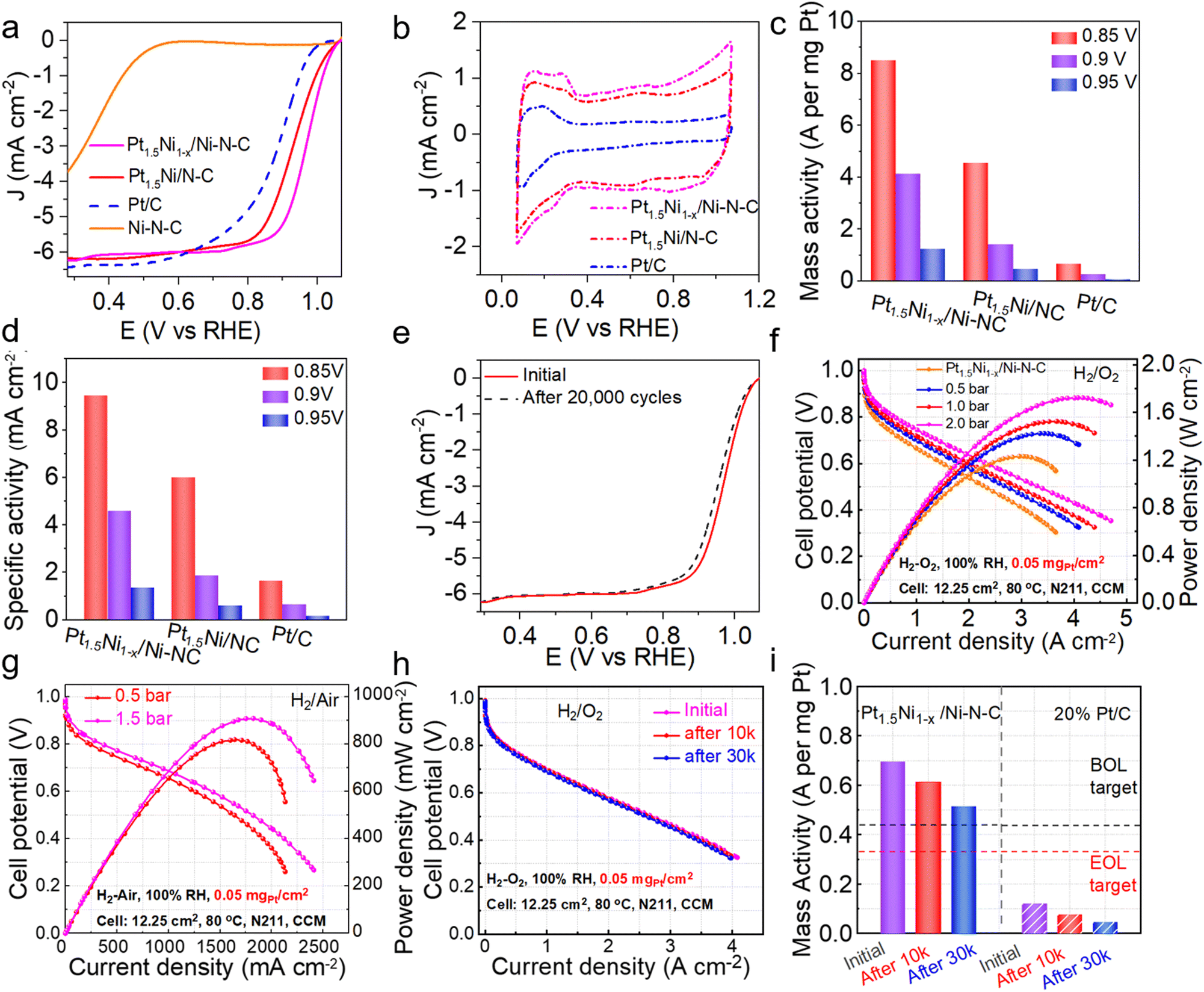 | ||
| Fig. 4 Catalytic performances of Pt1.5Ni1−x/Ni–N–C and reference materials. (a) ORR polarization curves and (b) cyclic voltammetry curves of Pt1.5Ni1−x/Ni–N–C, Pt1.5Ni/N–C, commercial Pt/C and Ni–N–C, (c) and (d) comparison of mass activity and specific activity at 0.85 V, 0.9 V, 0.95 V vs. RHE, (e) ORR polarization curves of Pt1.5Ni1−x/Ni–N–C before and after ADT, (f) and (g) H2–O2 and H2–air fuel cell i–V polarization and power density plots recorded under different O2/air pressures of Pt1.5Ni1−x/Ni–N–C, and (h) ADT of Pt1.5Ni1−x/Ni–N–C under H2/O2 at beginning-of-life (BOL), 10000 and 30000 cycles, respectively. (i) Mass activity of Pt1.5Ni1−x/Ni–N–C and commercial 20% Pt/C at 0.9 V iR-free before and after ADT (black and red dashed lines indicate the DOE BOL and end-of-life (EOL) targets, respectively). | ||
Moreover, to testify the practical performance of the prepared catalyst Pt1.5Ni1−x/Ni–N–C, we incorporated it into a membrane electrode assembly (MEA) as the cathode catalyst with a Pt loading of 0.05 mgPt cm−2 and assembled it into a PEMFC single cell. The current–voltage (i–V) polarization curves and power density distribution plots of Pt1.5Ni1−x/Ni–N–C demonstrate maximum power densities of 1.23, 1.42, 1.52 and 1.72 Wcm−2 in the H2/O2 cell under partial pressures of 0.0, 0.5, 1.0 and 2.0 bar, respectively (Fig. 4(f)), superior to Pt1.5Ni/N–C and commercial 20 wt% Pt/C MEA tested under the same conditions (Fig. S29 and S30, ESI†). Meanwhile, the through entire polarization scanning current densities of Pt1.5Ni1−x/Ni–N–CNi1−x/Ni–N–C MEA are higher than those of Pt1.5Ni/N–C and Pt/C MEA. For example, the current density for Pt1.5Ni1−x/Ni–N–C at the high voltage region (0.8 V) can reach 0.55 Acm−2, outperforming that for Pt1.5Ni/N–C (0.37 Acm−2) and Pt/C (0.34 Acm−2) at 2.0 bar. Pt1.5Ni1−x/Ni–N–C also exhibits maximum power densities of 0.91 and 0.82 Wcm−2 under different partial pressures and higher current densities compared with Pt1.5Ni/N–C and Pt/C across the entire polarization scan in H2–air cell tests (Fig. 4(g) and Fig. S31, S32, ESI†). In addition, Pt1.5Ni1−x/Ni–N–C MEA and Pt/C MEA were subjected to ADT experiments according to the U.S. DOE (Department of Energy) protocols.30 The i–V polarization curves of Pt1.5Ni1−x/Ni–N–C MEA show no significant decay in current density after 30000 continuous cycles and exhibit a low voltage drop of only 12 mV at a current density of 0.8 A cm−2, indicating a better MEA stability than Pt1.5Ni/N–C and Pt/C (18 mV and 130 mV decay at 0.8 A cm−2, respectively) (Fig. 4(h) and Fig. S33, S34, ESI†). Additionally, Fig. 4(i) reveals that Pt1.5Ni1−x/Ni–N–C MEA delivers a higher initial MA of 0.70 AmgPt−1 than Pt1.5Ni/N–C and only 26.3% decay after 30000 cycles, achieving the DOE 2025 target (0.44 AmgPt−1 for MA and <40% attenuation after 30000 cycles) (Fig. 4(i) and Fig. S35, ESI†). After 30000 cycles, AC HAADF-STEM and EDS mapping showed that the Pt1.5Ni1−x particles retained the original morphology and composition of the alloy, and the Ni sites were still isolated and dispersed on the NC substrate around the particles (Fig. S36 and S37, ESI†). To recap, the results gleaned above demonstrate the superior activity of Pt1.5Ni1−x/Ni–N–C in both RDE measurements and single fuel cell tests and perfectly cater to envision well-designed efficient PEMFC catalysts. This may be attributed to the relay transfer of reaction intermediates between Pt–Ni NC and Ni single sites, which can be further elucidated by the next theoretical calculations (Fig. 5).
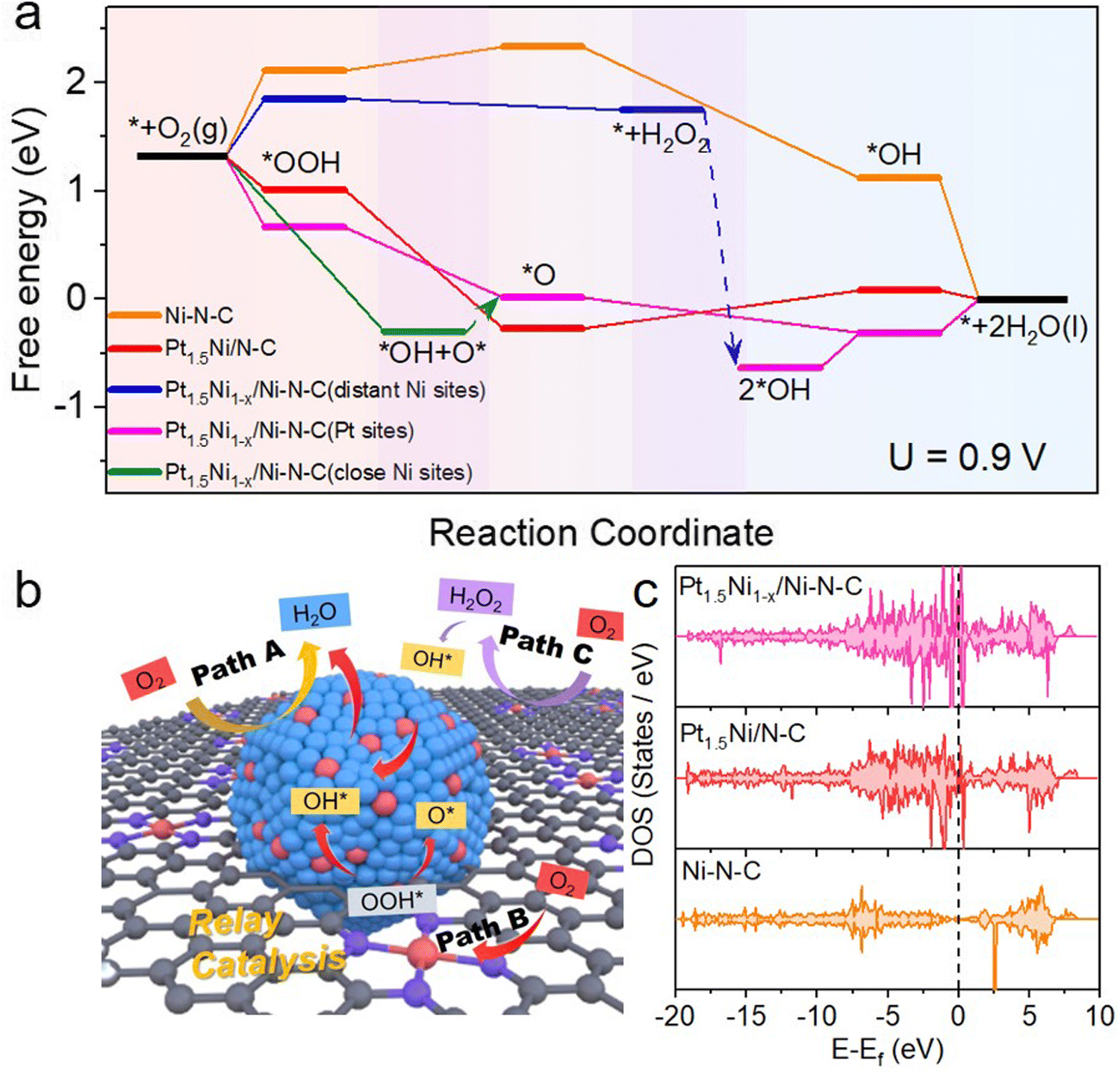 | ||
| Fig. 5 DFT calculations and schematic illustrations of Pt1.5Ni1−x/Ni–N–C and reference materials. (a) The ORR free energy diagram of Ni–N–C, Pt1.5Ni/N–C, Pt1.5Ni1−x/Ni–N–C with distant Ni sites, close Ni sites and Pt sites, (b) the schematics of Pt1.5Ni1−x/Ni–N–C (blue, red, purple, and gray spheres present Pt, Ni, N and C atoms, respectively), (c) the total density of states and the corresponding d band centers of Pt1.5Ni1−x/Ni–N–C, Pt1.5Ni/N–C and Ni–N–C. The Fermi-level is marked with a black dashed line. The d band center is marked with a purple line. | ||
To figure out the mechanism of the outstanding ORR activity observed on Pt1.5Ni1−x/Ni–N–C, comprehensive density functional theory (DFT) calculations were performed. According to the above physical characterization results and the consideration of the computational cost, the Pt1.5Ni1−x/Ni–N–C model consisting chiefly of a small PtNi cluster and its neighboring Ni–N–C sites was established to explicitly consider their roles in improving the ORR activities. The models of the Pt1.5Ni/N–C and Ni–N–C samples were also constructed for comparison (Fig. S38, ESI†). Based on the calculated free energy profile of the reaction path, the rate determining steps (RDS) of Pt1.5Ni1−x/Ni–N–C, Pt1.5Ni/N–C, and Ni–N–C are *OH + H+ + e− → * + H2O, *O + H+ + e− → *OH, and * + O2 + H+ + e− → *OOH, respectively. Their corresponding largest free energy changes are 0.31, 0.35, and 0.81 eV at 0.9 V, indicating that Pt1.5Ni1−x/Ni–N–C and Pt1.5Ni/N–C are more active in the four-electron ORR path than Ni–N–C. Meanwhile, the PtNi cluster sites show a much lower free energy change than that of the Ni–N–C sites, which is indicative of more inclination to the four-electron ORR path on the Pt1.5Ni1−x/Ni–N–C catalyst. The path shown by the yellow arrow in Fig. 5(b) and the pink line in Fig. 5(a) is path A, which occurs at the Pt NC site (Pt site) in Pt1.5Ni1−x/Ni–N–C, where O2 completes the reduction of 4 electrons directly. Due to the trace H2O2 products that have been proven in the RRDE evaluation (Fig. S22, ESI†), the pathway to produce H2O2 is further taken into consideration. The produced H2O2 does not bind to the distant Ni–N–C site (PGM-free site) and can be released and migrate to the PtNi cluster sites in the vicinity, as marked by the blue arrow in Fig. 5(a) and purple arrow in Fig. 5(b) (path C). Then, the H2O2 will be rapidly reduced to H2O on the PtNi cluster sites, which would effectively alleviate the damage of free radicals to carbon components and proton membranes. Interestingly, when the ORR process occurs on the close Ni–N–C site (neighboring the PtNi cluster site) of Pt1.5Ni1−x/Ni–N–C, the *OOH intermediate cannot be stably bound to the close Ni–N–C site and will migrate to the neighboring PtNi site and decompose into O* and *OH (Fig. 5(b) and Fig. S39, ESI†), as shown by the green line in Fig. 5(a) and red arrow route in Fig. 5(b) (path B). This mutual assistance of the PtNi cluster and Ni–N–C together completes a four-electron transfer path. The multisite synergy between the Ni–N–C sites and the PtNi cluster sites effectively activates oxygen molecules, which is beneficial to the further reduction process. Therefore, the catalytic efficiency of the multisite synergistic relay reaction path of Pt1.5Ni1−x/Ni–N–C is beyond those of Pt1.5Ni/N–C and Ni–N–C.
The density of states (DOS) of Pt1.5Ni1−x/Ni–N–C, Pt1.5Ni/N–C, and Ni–N–C as well as their corresponding d-band centers of the active atoms are subsequently calculated to understand the origin of the enhanced ORR activity of Pt1.5Ni1−x/Ni–N–C (Fig. 5(c) and Fig. S40, ESI†). The DOS in Fig. 5(c) shows that the Pt1.5Ni1−x/Ni–N–C and Pt1.5Ni/N–C catalysts have abundant electronic states near the Fermi-level in comparison with that of Ni–N–C, implying a greater charge transfer ability. Moreover, more abundant electronic states near the Fermi-level over Pt1.5Ni1−x/Ni–N–C than over Pt1.5Ni/N–C indicate the possible electron interaction between the PtNi clusters and the Ni–N–C sites, which helps to achieve an enhanced catalytic performance. In addition, the d band center of Pt and Ni in Pt1.5Ni1−x/Ni–N–C is closer to the Fermi-level than that of Ni in Ni–N–C (Fig. S40, ESI†), indicating the existence of electron interactions between the PtNi clusters and the Ni–N–C sites. This result also means stronger adsorption of intermediates on Pt1.5Ni1−x/Ni–N–C than on Ni–N–C, which is consistent with the calculated free energy profile.
Conclusions
In conclusion, we have developed an efficient gas-promoted dealloying process to synthesize a closely packed hybrid Pt1.5Ni1−x/Ni–N–C for high-efficiency PEMFCs. The enhancement of activity is due to the design of compactly adjacent active sites, allowing relay catalysis of the key intermediate OOH* on single atom sites and Pt alloy. This finding not only enriches the catalog of Pt-based ORR catalysts but also provides the idea for the design of effective relay catalysis.Author contributions
W. G. planned the synthesis, performed most of the reactions, collected and analyzed the data, and wrote the paper. W. G., M. Z. and X. Z. performed catalytic reactions. Y. W. and H. Z. conceived the experiments, designed the study and wrote the paper. J. X and Y. L. performed the electron microscopy characterization. W. C. carried out the X-ray structure characterization and proposed the structural model for the active sites. DFT calculations were performed by X. G., X. Z. and Y. L. The other authors performed some of the experiments and revised the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFE0191700), the National Natural Science Foundation of China (21522107), the Anhui Provincial Natural Science Foundation (2108085QB70 and 2108085UD06), the Key Technologies R & D Program of Anhui Province (2022a05020053), the Natural Science Foundation of Hefei, China (Grant No. 2021044), the China Postdoctoral Science Foundation funded project (BX20200317 and 2020M682030), the Fundamental Research Funds for the Central Universities (WK2060000021 and WK2060000025) and the Joint Funds from Hefei National Synchrotron Radiation Laboratory (KY2060000180 and KY2060000195). This work was partially carried out at the USTC Center for Micro and Nanoscale Research and Fabrication. The authors thank the CAS Fujian Institute of Innovation for funding support. The authors acknowledge the Experimental Center of Engineering and Materials Science at the University of Science and Technology of China. We thank the photoemission endstations BL1W1B in Beijing Synchrotron Radiation Facility (BSRF), BL14W1 in Shanghai Synchrotron Radiation Facility (SSRF), BL10B and BL11U in National Synchrotron Radiation Laboratory (NSRL) for the help in characterizations.Notes and references
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- H. Su, W. Zhou, H. Zhang, W. Zhou, X. Zhao, Y. Li, M. Liu, W. Cheng and Q. Liu, J. Am. Chem. Soc., 2020, 142, 12306–12313 CrossRef CAS PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbruster, A. Knop-Gericke and R. Schlogl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- J. Gao, H. b Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- S. Siahrostami, M. E. Bjorketun, P. Strasser, J. Greeley and J. Rossmeisl, Phys. Chem. Chem. Phys., 2013, 15, 9326–9334 RSC.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. T. Kim, K. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- T. Y. Yoo, J. M. Yoo, A. K. Sinha, M. S. Bootharaju, E. Jung, H. S. Lee, B. H. Lee, J. Kim, W. H. Antink, Y. M. Kim, J. Lee, E. Lee, D. W. Lee, S. P. Cho, S. J. Yoo, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 14190–14200 CrossRef CAS PubMed.
- E. Zhu, Y. Li, C.-Y. Chiu, X. Huang, M. Li, Z. Zhao, Y. Liu, X. Duan and Y. Huang, Nano Res., 2015, 9, 149–157 CrossRef.
- L. Chong, J. G. Wen, J. Kubal, F. G. Sen, J. X. Zou, J. Greeley, M. Chan, H. Barkholtz, W. J. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS.
- Z. Qiao, C. Wang, C. Li, Y. Zeng, S. Hwang, B. Li, S. Karakalos, J. Park, A. J. Kropf, E. C. Wegener, Q. Gong, H. Xu, G. Wang, D. J. Myers, J. Xie, J. S. Spendelow and G. Wu, Energy Environ. Sci., 2021, 14, 4948–4960 RSC.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef CAS.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- Z. Yang, B. Chen, W. Chen, Y. Qu, F. Zhou, C. Zhao, Q. Xu, Q. Zhang, X. Duan and Y. Wu, Nat. Commun., 2019, 10, 3734 CrossRef PubMed.
- X. Zhao, S. Chen, Z. Fang, J. Ding, W. Sang, Y. Wang, J. Zhao, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2015, 137, 2804–2807 CrossRef CAS PubMed.
- M. Gocyla, S. Kuehl, M. Shviro, H. Heyen, S. Selve, R. E. Dunin-Borkowski, M. Heggen and P. Strasser, ACS Nano, 2018, 12, 5306–5311 CrossRef CAS PubMed.
- V. Beermann, M. Gocyla, S. Kuhl, E. Padgett, H. Schmies, M. Goerlin, N. Erini, M. Shviro, M. Heggen, R. E. Dunin-Borkowski, D. A. Muller and P. Strasser, J. Am. Chem. Soc., 2017, 139, 16536–16547 CrossRef CAS.
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald, L. Gan, R. O'Malley, P. Strasser, F. T. Wagner and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 258–266 RSC.
- F. Kong, Z. Ren, M. Norouzi Banis, L. Du, X. Zhou, G. Chen, L. Zhang, J. Li, S. Wang, M. Li, K. Doyle-Davis, Y. Ma, R. Li, A. Young, L. Yang, M. Markiewicz, Y. Tong, G. Yin, C. Du, J. Luo and X. Sun, ACS Catal., 2020, 10, 4205–4214 CrossRef CAS.
- X. Ao, W. Zhang, B. Zhao, Y. Ding, G. Nam, L. Soule, A. Abdelhafiz, C. Wang and M. Liu, Energy Environ. Sci., 2020, 13, 3032–3040 RSC.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- X. L. Tian, X. Zhao, Y. Q. Su, L. J. Wang, H. M. Wang, D. Dang, B. Chi, H. F. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- The U.S. Department of Energy (DOE) Fuel Cell Technologies Office, (FCTO) Fuel Cells Multi-Year Research Development and Demonstration Plan (2016). https://www.energy.gov/sites/prod/files/2016/06/f32/fctomyrddfuelcells0.pdf.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02381d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |