
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of relative humidity, NOx, and ammonia on the physical properties of naphthalene secondary organic aerosols†
Alexandra L.
Klodt
 a,
Paige K.
Aiona‡
a,
Amanda C.
MacMillan‡
a,
Hyun
Ji (Julie) Lee§
a,
Xiaolu
Zhang¶
b,
Taylor
Helgestad||
b,
Gordon A.
Novak**
c,
Peng
Lin§
d,
Julia
Laskin
d,
Alexander
Laskin
de,
Timothy H.
Bertram
c,
Christopher D.
Cappa
b and
Sergey A.
Nizkorodov
*a
a,
Paige K.
Aiona‡
a,
Amanda C.
MacMillan‡
a,
Hyun
Ji (Julie) Lee§
a,
Xiaolu
Zhang¶
b,
Taylor
Helgestad||
b,
Gordon A.
Novak**
c,
Peng
Lin§
d,
Julia
Laskin
d,
Alexander
Laskin
de,
Timothy H.
Bertram
c,
Christopher D.
Cappa
b and
Sergey A.
Nizkorodov
*a
aDepartment of Chemistry, University of California, Irvine, California 92617, USA. E-mail: nizkorod@uci.edu
bDepartment of Civil and Environmental Engineering, University of California, Davis, California 95616, USA
cDepartment of Chemistry, University of Wisconsin, Madison, Wisconsin 53706, USA
dDepartment of Chemistry, USA
eDepartment of Earth, Atmospheric and Planetary Sciences, Purdue University, West Lafayette, Indiana 47906, USA
First published on 13th April 2023
Abstract
Light-absorbing organic aerosols, commonly referred to as “brown carbon” (BrC), have important climate effects. The primary source of BrC is biomass burning, but additional BrC can be formed by less constrained secondary atmospheric reactions, such as photooxidation of aromatic volatile organic compounds (VOCs) and reactions of carbonyl compounds in secondary organic aerosols (SOAs) with ammonia (NH3). As NH3 and VOC emissions increase globally, these insufficiently understood BrC production pathways may gain in importance relative to the primary BrC sources. This study examines the effects of NH3, NOx, and relative humidity (RH) on the optical and molecular properties of naphthalene (NAP) photooxidation SOAs. NAP SOAs were prepared by OH-initiated oxidation in a smog chamber at high and low levels of NOx, RH, and NH3. SOA hygroscopicity and optical properties were monitored online, and SOA samples were collected on filters for offline analysis. UV-visible spectroscopy and high-resolution mass spectrometry were used to determine mass absorption coefficients and molecular composition, respectively. Of the three parameters studied, RH exhibited the largest effects on both composition and optical properties – with an increase in the 〈O/C〉 ratio, relative dimer abundance, and absorption at wavelengths longer than 350 nm. Elevated NOx and NH3 concentrations increased the amount of nitrogen incorporated into the SOA, and NOx increased the amount of absorption at wavelengths longer than 350 nm while NH3 had little effect. The combination of elevated humidity and NOx led to the formation of 6-nitro-2-naphthol, a strong chromophore with a distinct peak at 400 nm in the absorption data. We find that RH, NOx, and NH3 all affect the characteristics of NAP SOAs in different ways: NOx and RH control both molecular composition and optical properties, while NH3 has a stronger effect on composition and a minor effect on the absorption coefficient.
Environmental significanceLight-absorbing aerosols formed by the photooxidation of aromatic volatile organic compounds have important climate effects, but the degree of light absorption depends on the chemical mechanism of photooxidation, environmental conditions and concentrations of various air pollutants. This study focuses on the effect of three parameters – relative humidity, concentration of nitrogen oxides, and concentration of ammonia – on the chemical composition and important physical properties of naphthalene-derived secondary organic aerosols. We observe that certain combinations of parameters during SOA formation lead to effects on the composition and optical properties that cannot be anticipated from varying the two parameters separately. This work demonstrates the importance of accurate representations of conditions in the atmosphere for simulating atmospheric processes. |
1 Introduction
The majority of atmospheric aerosols produce negative radiative forcing on climate, resulting in net cooling of the lower atmosphere, by scattering the incoming solar radiation and modifying cloud properties. However, certain aerosols, including black carbon (BC), brown carbon (BrC), and mineral dust, absorb visible radiation and convert it into heat thus reducing this cooling effect.1 Of these light-absorbing aerosols, BrC has the most uncertain effects on climate. BrC is composed of organic molecules that are capable of absorbing both visible and near-ultraviolet radiation, such as nitrophenols, which give BrC a characteristic yellow-brown color.2–4 BrC may be emitted from primary sources, such as biomass-burning and fossil fuel combustion,2,4–8 or produced by secondary processes.9,10 Examples of secondary BrC formation processes include high-NOx (=NO + NO2) photooxidation of aromatic volatile organic compounds (VOCs) resulting in secondary organic aerosols (SOAs),11,12,21,22,13–20 reactive uptake of oxygenated VOCs on highly acidic particles,23–28 aqueous-phase OH oxidation of aromatic compounds,29–32 reactions of triplet states with dissolved VOCs,33,34 and reactions of ammonia (NH3) and other nitrogen-containing species with gas-phase and particle phase carbonyl compounds.35–38The interest in BrC has resulted in multiple measurements of their optical properties, including extinction and absorption coefficients, single scattering albedo, absorption spectra and fluorescence spectra.10 Different mass spectrometry methods have also been used to investigate the chemical composition of BrC in order to identify the primary chromophores responsible for the brown color of BrC.10 From these measurements, nitroaromatics, or more specifically nitrophenols, have been identified as primary contributors to the light absorption by BrC.39 These compounds are readily produced during photooxidation of aromatics. For example, Romonosky et al.40 observed an elevated mass absorption coefficient of bulk SOA samples (MACbulk) under high-NOx conditions for SOAs generated from several aromatic precursors. Nakayama et al.15 found that increasing NOx concentrations during toluene photooxidation increases the SOA's imaginary and real refractive indices at 405 and 532 nm, using photoacoustic and cavity ring-down spectroscopy. Liu et al.41 also measured the real and imaginary refractive indices of toluene and m-xylene secondary organic materials, using spectroscopic ellipsometry and UV-vis spectroscopy. Both indices increased as a function of increasing NOx concentration, while decreasing with increasing wavelength. Using infrared spectroscopy, they also showed that the presence of NOx resulted in the production of organonitrogen species. Dingle et al.19 observed that the imaginary refractive index of SOAs prepared from toluene and 1-methylnaphthalene measured at 375 nm and 632 nm increase in the presence of NOx, and also increase with the extent of OH exposure. Lin et al.11 also found that elevated NOx conditions drastically increased the MAC of toluene photooxidation SOAs, while simultaneously altering the chemical composition by producing more nitrogen-containing compounds. Finally, Liu et al.12 worked with a SOA formed by photooxidation of toluene and trimethylbenzene in order to determine how light absorption of SOAs is affected by the precursor type, NOx levels, photolysis time, and RH. The presence of NOx increased SOA light absorption, with moderate humidity and short photolysis times resulting in the highest MAC.
The composition and optical properties of the naphthalene-derived secondary organic aerosol (NAP SOA), which is the main subject of this work, were previously examined under various SOA preparation conditions, particularly varied NOx concentrations. He et al.21 found that the imaginary part of the refractive index increased at higher NOx to VOC ratios for a NAP SOA prepared in an oxidation flow reactor due to greater formation of nitroaromatics. Xie et al.39 also found nitroaromatics to contribute strongly to the absorptivity of a NAP SOA prepared under high-NOx conditions, although less so than for SOAs prepared from benzene and m-cresol. Gemayel et al.42 identified several chromophoric compounds in a NAP SOA likely responsible for its photosensitizing properties. Finally, Siemens et al.22 characterized the composition of a NAP SOA prepared in an oxidation flow reactor under varying NOx levels to provide molecular-level information on the NAP SOA composition and its relationship with its optical properties.
Reduced nitrogen compounds, most prominently NH3 and amines, have been found to alter the optical properties of BrC by converting carbonyl compounds into strongly absorbing imines and nitrogen-containing heterocyclic compounds. Updyke et al.35 observed an increase in MACbulk during exposure of various types of absorbing and non-absorbing SOAs to gaseous NH3. For example, a white low-NOx trimethylbenzene SOA gained a beige color upon exposure, and the NAP SOA characteristic yellow color became more saturated. Liu et al.43 observed the formation of nitrogen-containing organics and an increase in the absorption coefficient during exposure of a low-NOx toluene SOA to ammonia. Cui et al.20 observed an enhancement in the absorption coefficient of a 1-methylnaphthalene SOA in the presence of NH3, especially under humidified conditions. Extensive studies have been conducted exploring the mechanism of NH3-driven browning reactions. The general mechanism appears to involve reactions of dicarbonyls with NH3 resulting in nitrogen-containing heterocyclic compounds, but the exact details depend on the specific system. For example, 1,2-dicarbonyls, such as glyoxal and methylglyoxal, form oligomeric compounds derived from imidazole44–47 and pyrazine,48 whereas 1,4-dicarbonyls such as ketolimonoaldehyde (a secondary limonene ozonolysis product) and 4-oxopentanal produce light absorbers based on pyrroles.49,50
Relative humidity (RH) has been found to affect the chemical composition of some SOA systems, and it may therefore influence optical properties. Song et al.27 found that high RH caused a decrease in light absorption in high-NOx α-pinene ozonolysis SOAs formed on seed particles. In contrast, Liu et al.12 found that toluene and trimethylbenzene photooxidation SOAs formed under dry conditions had a significantly lower MAC than those formed under humid conditions, with 30, 50, and 80% RH all resulting in comparable absorption. Moderately humid conditions (30%) resulted in more nitrogen-containing products than dry conditions, implicating the nitrogen-containing species as contributors to the increased MAC, as seen with the toluene SOA by Lin et al.11
In terms of synergetic effects between RH and NH3, Liu et al.43 observed that the extent of browning of low-NOx toluene SOAs by NH3 is reduced under dry conditions, where the high viscosity of SOA particles imposes a diffusion limitation on the rate of reaction between SOA carbonyls and ammonia. Smith et al.51 found that dry conditions favor more extensive reactions between NH3 and SOA compounds that form nitrogen-containing chromophores.
Most of the previous optical absorption measurements relied on experiments with filter samples or solution extracts of SOAs. In this study, we use more direct aerosol absorption measurements to study the effect of RH and the presence of NH3 and nitrogen oxides (NOx) on the production, optical properties, and molecular composition of BrC formed by the photooxidation of NAP. High-NOx NAP SOAs can be regarded as prototypical secondary BrC with a high MACbulk,17 approaching that for primary BrC produced by burning of biomass. SOAs are formed under either humid or dry and high- or low-NOx conditions, in the presence and absence of NH3 to examine possible synergistic effects between different sets of conditions. UV-visible, cavity ring-down, and photoacoustic spectroscopy are used to investigate differences in optical properties based on reaction conditions. Two methods of high-resolution mass spectrometry (HRMS) are used to find how these conditions affect the SOA chemical composition. Finally, Size-Resolved Cloud Condensation Nuclei (CCN) Analysis is used to probe the hygroscopic properties of the SOA. We find that RH, NOx, and NH3 all affect the characteristics of the NAP SOA in different ways: NOx and RH control both molecular composition and optical properties, while NH3 has a stronger effect on composition and a minor effect on the absorption coefficient.
2 Experimental
2.1 NAP SOA formation
SOAs were produced by the photooxidation of NAP in a 5 m3 Teflon aerosol smog chamber operated in a batch mode. The environmental variables included high- and low-NOx levels (likely below ∼1 ppb, near the detection limit of the NOy analyzer described in Section 2.2), high (at least 80%) and low (<2%) RH, and the absence and presence of ammonia (NH3). Prior to each study the chamber was flushed with purge air overnight. For elevated RH studies, humidification was accomplished by flushing the chamber with air passing through a Nafion multichannel humidifier. Approximately 40 μL of H2O2 (Aldrich; 30% by volume in water), which served as the OH precursor, was evaporated into the chamber to achieve a H2O2 mixing ratio of 2 ppm. A proton transfer time-of-flight mass spectrometer (PTR-ToF-MS; IONICON model 8000, Innsbruck, Austria) was used to monitor the rate of NAP depletion and estimate the OH steady-state concentration as described in the ESI (see Fig. S1).† The OH steady-state concentration was representative of peak atmospheric concentrations,52 about 4 × 106 molecules cm−3, similar to our previous studies.53,54 For high-NOx studies, NO was added to the chamber from a premixed gas cylinder (Praxair, 5000 ppm NO in N2) until a mixing ratio of 200–700 ppb of NO was achieved, depending on the experiment as described in Table 1. This is higher than ambient levels and definitively sufficient to suppress RO2 + RO2/HO2 reactions compared to RO2 + NO reactions. About 11–23 μL of a solution of 0.5 g mL−1 of NAP in dichloromethane was evaporated into the chamber to reach a mixing ratio of 100–400 ppb. No seed aerosol was used as it would interfere with the interpretation of optical measurements. All precursors were mixed by a fan inside the chamber, which was turned off before the start of photooxidation. Photooxidation was initiated by UV-B lamps (Solar Tec Systems model FS40T12/UVB) with emissions centered at 310 nm and ranged for 1.5–3 h.| Sample # | NAP (ppm) | H2O2 (ppm) | NOx (ppm) | RH (%) | NH3 (ppm)a | Reaction time (h) | Data sets presentedb,c |
|---|---|---|---|---|---|---|---|
| a The actual concentration of NH3 in the chamber was lower because of rapid loss of ammonia to the chamber walls. b Runs 1–4 involved both online and offline characterization of SOAs. Runs 5–23 were only done with offline methods. c Abbreviations are defined in the text. | |||||||
| 1 | 0.2 | 2.0 | 0.0 | 1 | 1.0 | 2.0 | SR-CCN, SEMS, CRD/PAS, nano-DESI-HRMS |
| 2 | 0.2 | 2.0 | 0.0 | 85 | 1.0 | 2.5 | SR-CCN, SEMS, CRD/PAS, nano-DESI-HRMS |
| 3 | 0.2 | 2.0 | 0.2 | 2 | 1.0 | 1.5 | SR-CCN, SEMS, CRD/PAS, nano-DESI-HRMS |
| 4 | 0.2 | 2.0 | 0.2 | 84 | 1.0 | 2.0 | SR-CCN, SEMS, CRD/PAS, nano-DESI-HRMS |
| 5 | 0.4 | 2.0 | 0.0 | 0 | — | 2.0 | Nano-DESI-HRMS |
| 6 | 0.4 | 2.0 | 0.0 | 99 | — | 2.0 | Nano-DESI-HRMS |
| 7 | 0.4 | 2.0 | 0.4 | 0 | — | 2.0 | Nano-DESI-HRMS |
| 8 | 0.4 | 2.0 | 0.4 | 86 | — | 2.0 | Nano-DESI-HRMS |
| 9 | 0.4 | 2.0 | 0.0 | 0 | — | 2.0 | UV-vis |
| 10 | 0.4 | 2.0 | 0.0 | 89 | — | 2.0 | UV-vis |
| 11 | 0.4 | 2.0 | 0.0 | 0 | 1.0 | 2.0 | UV-vis |
| 12 | 0.4 | 2.0 | 0.0 | 87 | 1.0 | 2.0 | UV-vis |
| 13 | 0.4 | 2.0 | 0.4 | 0 | — | 2.0 | UV-vis |
| 14 | 0.4 | 2.0 | 0.4 | 94 | — | 2.0 | UV-vis |
| 15 | 0.4 | 2.0 | 0.4 | 0 | 1.0 | 2.0 | UV-vis |
| 16 | 0.4 | 2.0 | 0.4 | 87 | 1.0 | 2.5 | UV-vis |
| 17 | 0.2 | 2.0 | 0.7 | 0 | — | 3.0 | UV-vis |
| 18 | 0.2 | 2.0 | 0.7 | 0 | — | 1.5 | UV-vis |
| 19 | 0.2 | 2.0 | 0.7 | 87 | — | 2.7 | UV-vis |
| 20 | 0.2 | 2.0 | 0.7 | 80 | — | 3.0 | UV-vis |
| 21 | 0.2 | 2.0 | 0.7 | 0 | — | 2.5 | HPLC-PDA-HRMS |
| 22 | 0.2 | 2.0 | 0.7 | 0 | — | 2.5 | HPLC-PDA-HRMS |
| 23 | 0.2 | 2.0 | 0.7 | 90 | — | 2.5 | HPLC-PDA-HRMS |
Table 1 summarizes the conditions in different experiments of this work. For samples 1–4, after photooxidation, NH3 was added to the chamber by evaporation of a known volume of NH4OH solution chosen to produce a NH3 mixing ratio of 1 ppm. It was later determined using an NH3/NOx analyzer (Ecotech 9842, Warren, RI, USA), which was installed after the first set of experiments had already completed, that most of the injected NH3 must have been quickly removed by the chamber walls. Based on these NH3/NOx analyzer measurements, the actual mixing ratio of NH3 in the chamber was considerably lower than the target value, possibly by as much as an order of magnitude, and hard to quantify as it also appeared to depend on the amount injected and history of prior experiments in the chamber. Samples 5–23 aimed to determine the effects of NH3 on the composition or optical properties using offline methods. In samples 11–12 and 15–16, NH4OH was evaporated into the chamber with a target mixing ratio of 1 ppm NH3 in a single injection, prior to photooxidation. Samples 5–9, 13–14, and 17–23 in Table 1 were prepared without addition of NH3. In all cases, the SOA was collected after each experiment by passing particles through an activated charcoal denuder and onto a PTFE filter at 15–20 SLM for 3–4.5 h.
2.2 Online analysis of the NAP SOA
Several instruments probed and sampled the SOA formed in the chamber simultaneously. Real time temperature and RH were measured with a Vaisala HMT330 probe and the concentrations of ozone (model 49i) and NO/NOy (model 42i-Y) were measured with Thermo Scientific monitors. Particle size distribution was monitored using a scanning mobility particle sizer (SMPS, TSI Model 3936), which was then used to estimate the mass of the NAP SOA collected on the filters used for offline analysis. SMPS data for all samples and ozone and NOy data for samples 17–23 are available online at the Index of Chamber Atmospheric Research in the United States (ICARUS).55 A Size-Resolved Cloud Condensation Nuclei (SR-CCN) system was used to directly measure aerosol hygroscopicity. Real-time optical measurements of absorption and extinction coefficients were taken at 405 and 532 nm using a system containing a scanning electrical mobility spectrometer (SEMS) and cavity ring-down-photoacoustic spectrometer (CRD-PAS) equipped with thermodenuders and humidifiers/driers at their inlets.2.3 Size-resolved cloud condensation nuclei instrument
The SR-CCN provided a direct measurement of aerosol hygroscopicity in the supersaturated water vapor regime. Dry aerosol particles were size selected using a differential mobility analyzer, and the monodisperse sample was sent to a condensation particle counter to count the total number of particles and a cloud condensation nuclei counter (DMT CCN-100) to count the cloud active fraction. A dry initial particle diameter of 100 nm was used in all cases. Cloud active particles are those that grow spontaneously at a given supersaturation. The ratio of CCN to total particles (CN) was measured as a function of supersaturation to give an activation efficiency spectrum: ε = f (supersaturation, CCN/CN). The supersaturation where the CCN/CN ratio is 0.5 is referred to as the critical supersaturation (Scrit). Activation at lower Scrit values for a given size indicates more hygroscopic particles. A single term hygroscopicity parameter, κ, was then calculated from the measured Scrit and dry particle diameter.562.4 Cavity ring-down photoacoustic spectroscopy instrument
A UC Davis cavity ring-down photoacoustic spectrometer (CRD-PAS) was used to measure light extinction and light absorption by the particles at 405 and 532 nm.57 The CRD has three pairs of sample and reference cells, with two of the pairs operating at 532 nm and one at 405 nm.58 The reference cells sampled the aerosol though filters to provide a quantitative measure of the contribution of gas-phase absorption to the observed signal (which was found to be negligible at both wavelengths). The sample flow was first dried to <30% RH using a diffusion drier and sent into the first pair of 532 nm sample/reference cells. For the second pair of the 532 nm sample/reference cells, the flow was re-humidified to ∼75–85% RH using Nafion flow-through humidifiers. At 405 nm, only dried particle extinction was measured. The flows exiting the two dry CRD channels were directed to two PAS channels, one at 405 nm and one at 532 nm. The PAS used in this work relies on an astigmatic multi-pass configuration in order to increase the sensitivity over single-pass instruments.59 The PAS was calibrated using gas-phase O3 produced from a mercury Pen-ray lamp by referencing the observed absorption signal to the simultaneously made CRDS extinction.60 The PAS calibration was further tested in the lab using gas-phase NO2 absorption, which yielded calibration coefficients within 5% of the O3-based calibrations. MACaerosol was then determined at 405 and 532 nm by dividing the absorption coefficients measured by the PAS by the mass concentration determined by the SEMS.61 The imaginary part of the refractive index was determined from the MACaerosol values using Mie theory as described previously.622.5 Nano-DESI HRMS measurements
Filters 1–8 were analyzed using a nanospray-desorption electrospray ionization source (nano-DESI) coupled to a LTQ-Orbitrap high-resolution mass spectrometer (HRMS) operated in positive ion mode.63,64 (Negative ion mode data were not collected for the nano-DESI analysis, but were collected for the chromatographic analysis of filters 21–23, as described below). Before analysis, the filters were taken out of the freezer, brought to room temperature, and cut in half. One half was taped to a microscope slide for the nano-DESI-HRMS analysis.63 A mixture of 70% acetonitrile and 30% deionized (DI) water was added dropwise onto the surface of the filter through a primary silica capillary to dissolve the NAP SOA, which was then transferred into the mass spectrometer via a nanospray capillary. The spray voltage for these runs was 3–5 kV, the MS heated inlet capillary temperature was 250 °C, and the mass resolving power was 105m/Δm at m/z 400. To obtain background spectra, the probe was placed over the area of the filter that did not contain any sample for 2–3 min, and then was moved to the center of the filter for 3 to 6 min to collect sample data. The sample was moved underneath the capillary droplet bridge to expose fresh spots on the filter to the droplet, with about 10 mm distance travelled by the droplet at the end of scan. The flow rate ranged from 600 to 800 nL min−1.HRMS peaks in the sample and background spectra with a peak to background ratio greater than 5 were extracted using Decon2LS software (https://omics.pnl.gov/software/decontools-decon2ls). Peaks were then aligned and assigned using a LabView program with a tolerance of ±0.001 m/z units, with constraints applied on the number of specified elements (C: 1–40, H: 2–80, O: 0–35, N: 0–2, and Na: 0–1) and elemental ratios (H/C: 0.3 to 1.5 and O/C: 0.0 to 2.30). All solvent peaks were removed, as well as peaks containing 13C atoms. Using the remaining peaks an internal calibration was done to achieve better mass precision and refine the assignments. Observed peaks were assumed as either protonated [M + H]+ or sodiated [M + Na]+ species; the elemental composition of their corresponding neutral species will be discussed throughout this paper.
2.6 UPLC-PDA-ESI-HRMS experiments
Filters 21–23 were analyzed by ultrahigh-performance liquid chromatography photodiode array high resolution mass spectrometry (UPLC-PDA-HRMS) to examine the compounds contributing to changes in the light absorption by the NAP SOA produced with and without humidity as described previously.54,65 Briefly, the instrument was a Thermo Q-Exactive Plus mass spectrometer (different from the one described above) with a heated electrospray ionization inlet. The resolving power at m/z 400 was 1.4 × 105. The separation was performed using a Phenomenex Luna Omega Polar C18, 150 × 2.1 mm column with 1.6 μm particles and 100 Å pores and the PDA scanned between 190 and 680 nm. The solvents for the UPLC consisted of water and acetonitrile both acidified with 0.1% formic acid as solvents A and B, respectively. The gradient was 95% solvent A for the first 3 min, and then a linear ramp to 95% solvent B for 9 min, a hold at this ratio for 2 min, and finally a linear ramp back to 95% solvent A in preparation for the next run. For mass spectrometry, the instrument was operated in both modes, with +3.5 kV as the spray voltage for ESI positive mode and −2.5 kV as the spray voltage for ESI negative mode. Samples were submitted dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:acetonitrile at about 200 μg mL−1 concentration.
:1 water:acetonitrile at about 200 μg mL−1 concentration.
The PDA data were analyzed using FreeStyle 1.6 from Thermo Scientific. PDA data were integrated from 280 to 680 nm (wavelengths that are able to penetrate into the troposphere). Then peaks in the PDA chromatogram were manually correlated to individual ions based on the time delay between the PDA and total ion chromatogram (TIC) of 0.06 min. FreeStyle was then used to determine the most abundant ions at these time points, thereby assigning likely formulae to the chromophores showing up in the PDA data. FreeStyle was also used to obtain the full MS spectra by integrating the TIC data (1–18 min) and generating a raw file. Subsequently, the analysis of the data was performed similarly to the nano-DESI data except for the alignment and assigning tolerance, which were set to 0.0005 m/z due to the increased m/z accuracy of this instrument. After neutral formulae were determined from the assignments, the positive and negative ion modes were combined, and the mass spectra below represent the reconstructed peak abundance of the unionized SOA components detected in either one or both ionization modes.
2.7 Spectrophotometry experiments
Samples 10–20 of Table 1 were used for offline UV-vis absorption spectroscopy measurements. For filters 9–16 NAP SOA samples collected on filters were extracted three times in succession, using 4 mL of DI water for each extraction. Filters were shaken for 10 min during each extraction. We then used the procedure described in Section S2 and Fig. S2 in the ESI† to estimate the completeness of the extraction at each step using absorbance data from each of the three extractions. This procedure made it possible to correct for the incomplete dissolution of the SOA material during the first extraction.A volume of 3 mL of the dissolved NAP SOA was transferred to a 1.0 cm quartz cuvette and analyzed using a dual-beam spectrometer (Shimadzu UV-2450) with a DI water blank. Absorption spectra were taken for each of the extracts for all 8 samples. The bulk mass absorption coefficient (MACbulk) of each sample was calculated using eqn (1):
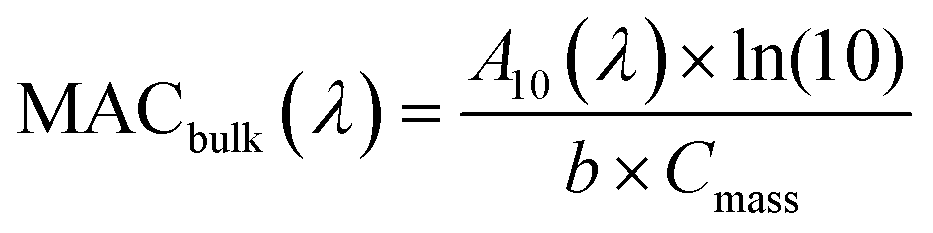 | (1) |
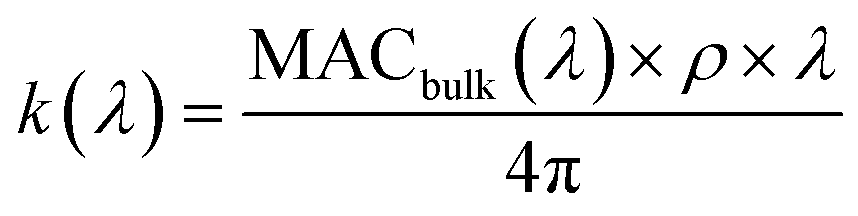 | (2) |
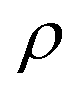 represents the density of the aerosol particles, which was assumed to be 1.5 g cm−3 based on previous studies.66,67
represents the density of the aerosol particles, which was assumed to be 1.5 g cm−3 based on previous studies.66,67
For filters 17–20, NAP SOA samples were extracted with methanol and for filters 21–23, which were used for mass spectrometry experiments, NAP SOA samples were extracted with acetonitrile (to prevent reactions between methanol and the SOA compounds that would interfere with the HRMS results).68 Extraction in these solvents was complete, as verified by taking the UV-vis spectra of secondary extracts, and no correction was necessary for the MACbulk value (see Fig. S3†). We expect MACbulk values in water and the organic solvents to be comparable based on previous work,35,54,69 although small solvatochromic effects cannot be ruled out.
3 Results and discussion
3.1 Molecular composition
Nano-DESI mass spectra for the samples 1–8 of Table 1 are shown in Fig. S4,† along with Van Krevelen Maximum Carbonyl Ratio (MCR)70 diagrams in Fig. S5† and double bond equivalent plots in Fig. S6.† In all cases, the mass spectra featured well-defined monomeric and dimeric regions, with the molecular weights of SOA compounds ranging from 100–250 and 250–500 Da, respectively. This is consistent with the mass spectra of naphthalene SOAs reported previously.17,22,71 While the overall shapes of the mass spectra were similar, the distribution of peak intensities changed from one mass spectrum to another depending on the conditions. Since the SOA composition and the corresponding mass spectra were found to depend on all three variables (concentrations of NOx, concentrations of NH3, and relative humidity) varied in this work, we will focus on comparisons of the samples in which only one parameter is changed at a time.Average elemental ratios (〈H/C〉, 〈O/C〉, and 〈N/C〉) were calculated for each compared subset. Specifically, averages were taken across all experiments with the variable of interest: RH, NOx, or NH3, with each experiment being used in multiple calculated averages. These average elemental ratios are plotted in Fig. 1. Additionally, p values were calculated using two-tailed t-tests to assess the significance of differences in the ratios observed under each condition, with the null hypothesis being that the elemental ratios are the same when the SOA is prepared at high and low RH, high and low NOx, or high and low ammonia. p values were set such that p < 0.10 represents a moderately significant effect, p < 0.05 represents a significant effect, and p < 0.01 represents a highly significant effect. Changes in elemental ratios were largely not significant across different SOA formation conditions. However, three exceptions to this were observed: the 〈O/C〉 ratio increased at low RH (p = 0.005) and 〈N/C〉 increased at high NOx concentrations (p = 0.096) and with added ammonia (p = 0.03). The marginal significance of the increase in the 〈N/C〉 ratio under high-NOx conditions was unexpected. This may be because nitroaromatic compounds, which are expected to be the majority of nitrogen-containing compounds under high-NOx conditions, are not detected efficiently in positive mode electrospray techniques.72 It may further be due to the influence of ammonia in some of the low-NOx experiments (i.e. samples 11 and 12 which were low-NOx and high-NH3) as well as the relatively low values of the measured 〈N/C〉 ratios.
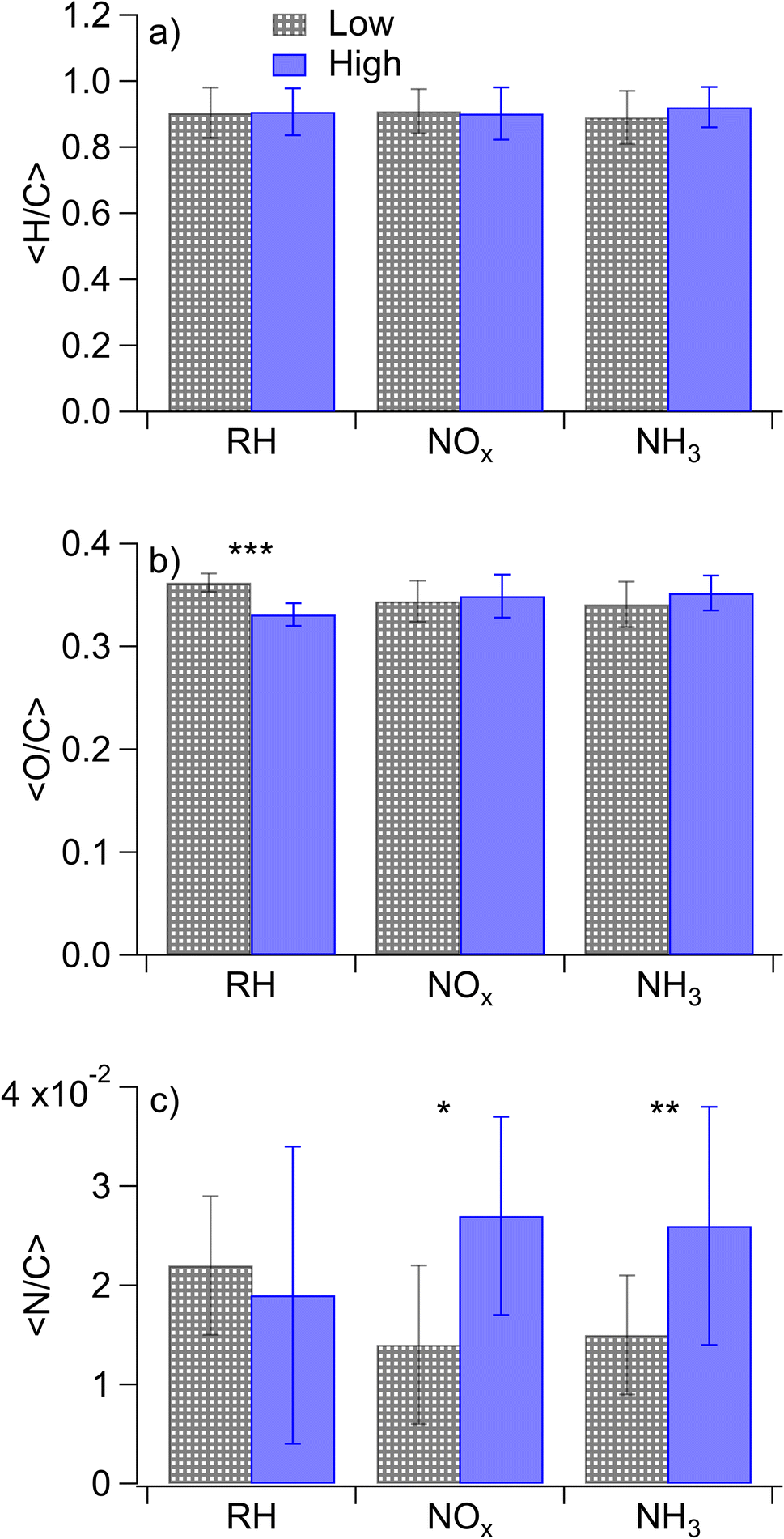 | ||
| Fig. 1 Summary of average elemental ratios averaged across all experiments with the corresponding parameters: high and low RH, high and low NOx, and with and without added NH3. Panel (a) shows average H/C ratios, panel (b) shows average O/C ratios, and panel (c) shows average N/C ratios. Error bars represent one standard deviation. * Designates a marginally significant difference (p = 0.096), ** designates a significant difference (p = 0.03), and *** designates a highly significant difference (p = 0.005) between the two conditions. | ||
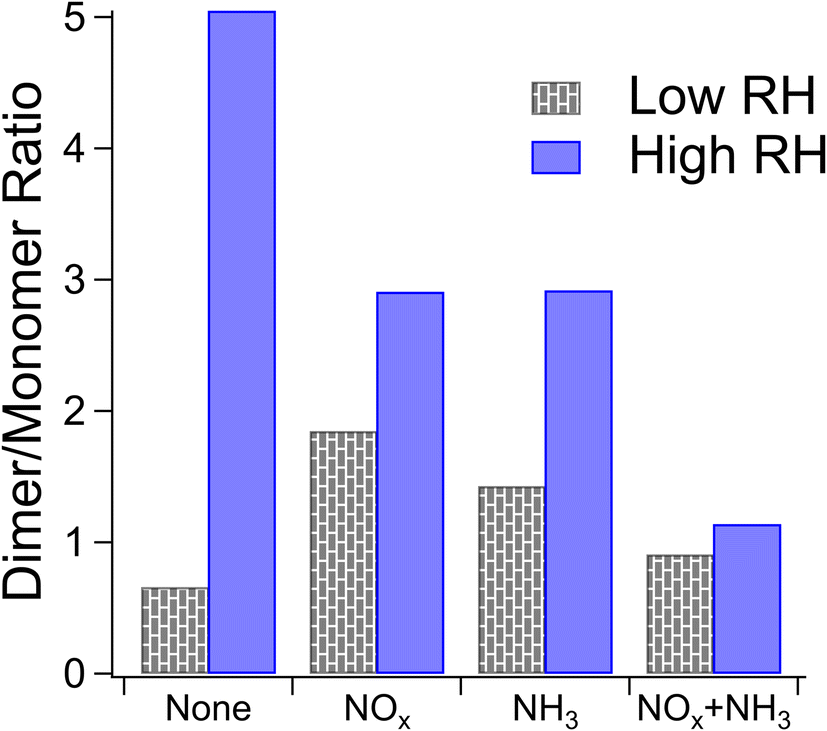 | ||
| Fig. 2 Dimer to monomer ratios under dry and humid conditions determined from nano-DESI HRMS data. Dry data are shown in patterned gray and humid data are shown in solid blue. Labels on the X-axis correspond to parameters (either NOx or NH3), which are elevated during SOA formation. | ||
The loss of high O/C ratios under humid conditions could be the result of wall loss effects as hydrophilic (i.e., more polar with higher O/C ratios) compounds partition to the walls of Teflon chambers more quickly under humid conditions.74 However, if this were the reason for our lower O/C ratios under humid conditions, the addition of NOx and ammonia should also increase this wall loss effect as they should lead to more polar molecules, but this is not observed. Rather, the effect of humidity on the Van Krevelen MCR diagram is reduced in the presence of NOx and NH3 with the percentage of the signal in the “highly unsaturated” region being nearly identical between dry and humid conditions when NOx or NH3 is elevated, although the diagonal lines of peaks representing condensation reactions are still evident. We therefore do not believe that wall loss effects sufficiently explain the differences in composition between the dry and humid conditions.
Previous work has shown increased oligomer formation at elevated RH for cyclohexene photooxidation SOAs.75 Other work has shown that in the case of SOAs from aromatic precursors, gas-phase carbonyls may hydrate on particles with adsorbed water and oligomerize without acidification.76 Carboxylic acids have been observed to form in the gas phase at faster rates at elevated RH for the OH oxidation of aromatic precursors,77 and photooxidation of aromatic compounds at elevated relative humidity produces higher levels of carboxylic acids and alcohols in the particle phase.78 As both carboxylic acids and alcohols are precursors to condensation reactions, higher concentrations of these species may lead to more condensation reactions as we observe in the Van Krevelen MCR diagrams in Fig. S5.† The increased rate of condensation reactions under humid conditions may therefore be due to a shift in reaction precursor concentrations. Other studies have shown suppressed oligomer formation at elevated RH for single-ring aromatic precursors, which was likely from the suppression of condensation reactions by water in the SOA particles.53,77,79 The NAP SOA studied here is less hygroscopic and water-soluble than SOAs from single-ring aromatic precursors in previous work – a result of its lower vapor pressure, which allows oxidation products from earlier generations with relatively low polarity to condense into particles. We observe a lower O/C ratio for our SOA (0.2 to 0.4, falling into the “highly unsaturated” MCR region) than for other SOA types where oligomer formation was suppressed (for instance 0.8 to 0.9 for toluene SOA and 0.45 to 0.6 for m-xylene SOA, falling into the “oxidized unsaturated” or “moderately oxidized” MCR regions).70,77 The O/C ratio controls water uptake,80 so less incorporation of water into the NAP SOA particles is expected than other types of SOA particles. Combining these effects, we suggest that in our system, elevated RH leads to higher concentrations of condensation reaction precursors but not enough extra water to suppress condensation reactions as observed for more hygroscopic SOA types.
NH3 + R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ⇌ R1R2C(OH)NH2 ⇌ R1R2CNH + H2O O ⇌ R1R2C(OH)NH2 ⇌ R1R2CNH + H2O | (3) |
| NH3 + HNO3 ⇌ NH4NO3 | (4) |
Finally, in our mass spectrometry analysis, we may observe compounds ionized by NH4+ rather than H+ when ammonia is present. This ionization mechanism will additionally be more prevalent when Reaction (4) occurs.
We estimate the relative prevalence of Reaction (3) by evaluating how often the mass difference of 0.9840 Da (corresponding to the combined effect of NH3 addition and loss of H2O) occurs in the nano-DESI mass spectrum, shown in Fig. 3. The addition of ammonia appears to increase the frequency of this mass difference, and the effect is smaller at high humidity under low-NOx conditions. This is consistent with Reaction (3). Previous studies have also observed a suppression of the formation of nitrogen-containing compounds from ammonia and amines at elevated RH (the addition of ammonia increased the 〈N/C〉 ratio to a lesser extent with increasing RH) and attributed it to the suppression of condensation reactions between carbonyls and ammonia as this reaction produces water as a product.51 With the addition of ammonia, we also observe an increase in the number of peaks with H/C ratios greater than 1 (Fig. S5†), presumably from the additional H atoms on NH3 reaction products. An alternative explanation for this observation may be more rapid partitioning to the walls under humid conditions; however this is not observed when NOx is also added. Instead, the addition of NOx appears to enhance the occurrence of a mass difference of 0.9840 Da, and the effect is greater under humid conditions. Both of these observations agree well with Aerosol Mass Spectrometry data reported by Cui et al.,20 who also concluded that NOx and the combination of NOx and water vapor promote the formation imine compounds with the addition of NH3. Under high-NOx conditions, the reaction of peroxy radicals (RO2) with NO produces more carbonyl compounds than the RO2/HO2 chemistry occurring in low-NOx samples.52 These carbonyls likely react with NH3, resulting in increased importance of carbonyl to imine transformations. The MCR analysis in Fig. S5† supports this theory, as the MCR is higher under high-NOx conditions than under low-NOx conditions.
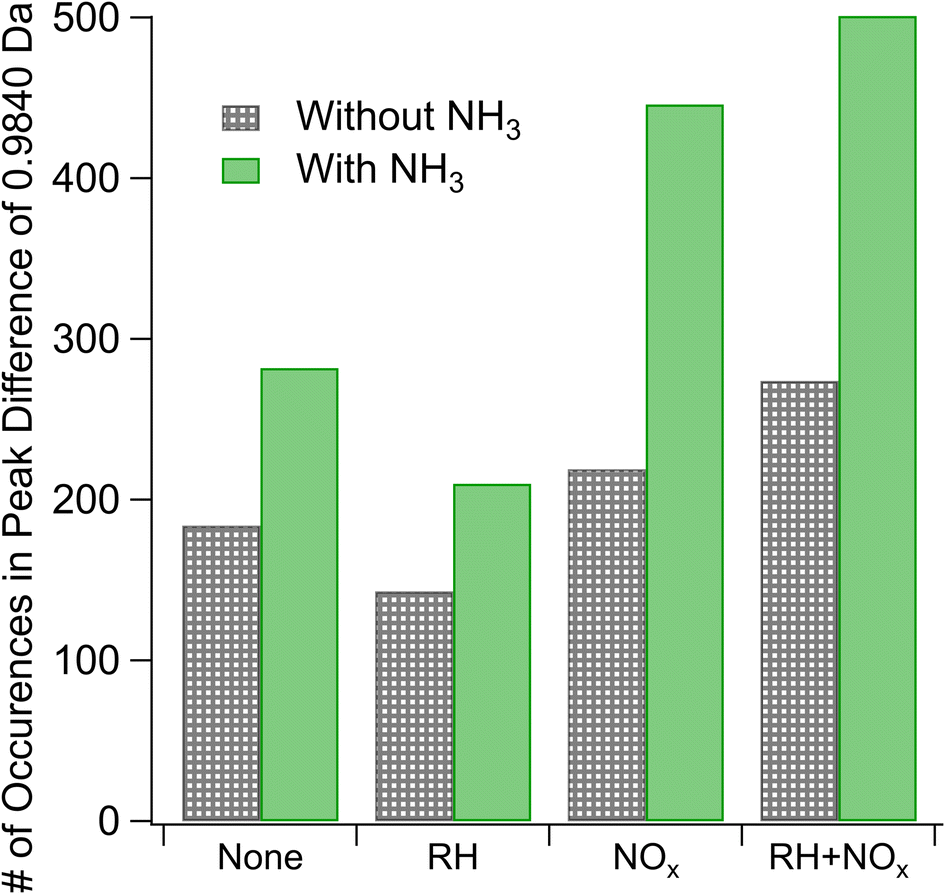 | ||
| Fig. 3 Differences in the occurrence of 0.9840 Da (corresponding to the mass difference when losing H2O and gaining NH3) for trials with and without ammonia added as measured by nano-DESI HRMS. Results without ammonia are shown in patterned gray and with ammonia are shown in solid green. Labels on the X-axis correspond to parameters (either RH or NOx), which are elevated during SOA formation. | ||
Reaction (4) may interfere with these results if, for instance, the compounds undergo a dimerization reaction which loses a water molecule as mentioned in Section 3.1.1 and then the resultant compound is ionized by particle-phase NH4+ formed through Reaction (4). The end difference in mass for this set of circumstances would be indistinguishable from that produced by Reaction (3). However, the dimer/monomer ratios in Fig. 2 and the occurrence of a mass difference of 0.9840 Da in Fig. 3 are anticorrelated, with the lowest relative abundance of dimers and the highest number of occurrences of our mass difference being for high-NOx and high-NH3 samples, suggesting that Reaction (4) does not explain the trends observed in Fig. 3.
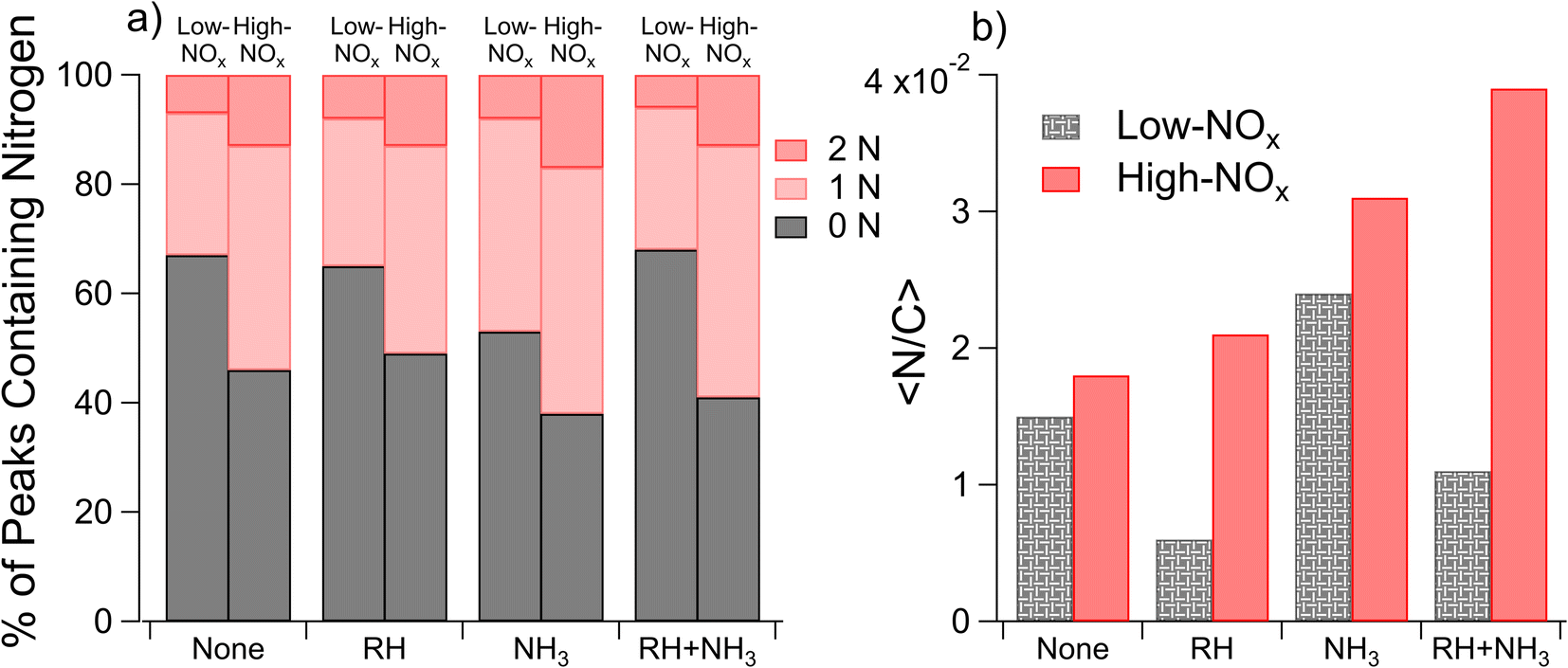 | ||
| Fig. 4 Comparison of nitrogen incorporation for high- and low-NOx samples as determined by nano-DESI. Panel (a) shows the percentage of the number of peaks assigned to formulae containing 0, 1, or 2 nitrogen atoms, with gray representing 0 nitrogen atoms, light pink representing 1 nitrogen atom, and dark pink representing 2 nitrogen atoms. Panel (b) shows the intensity-normalized 〈N/C〉 ratio for each set of samples under low- (in patterned gray) and high-NOx (in solid red) conditions. Labels on the X-axis correspond to parameters (either RH or NH3), which are elevated during SOA formation. | ||
3.2 Optical properties
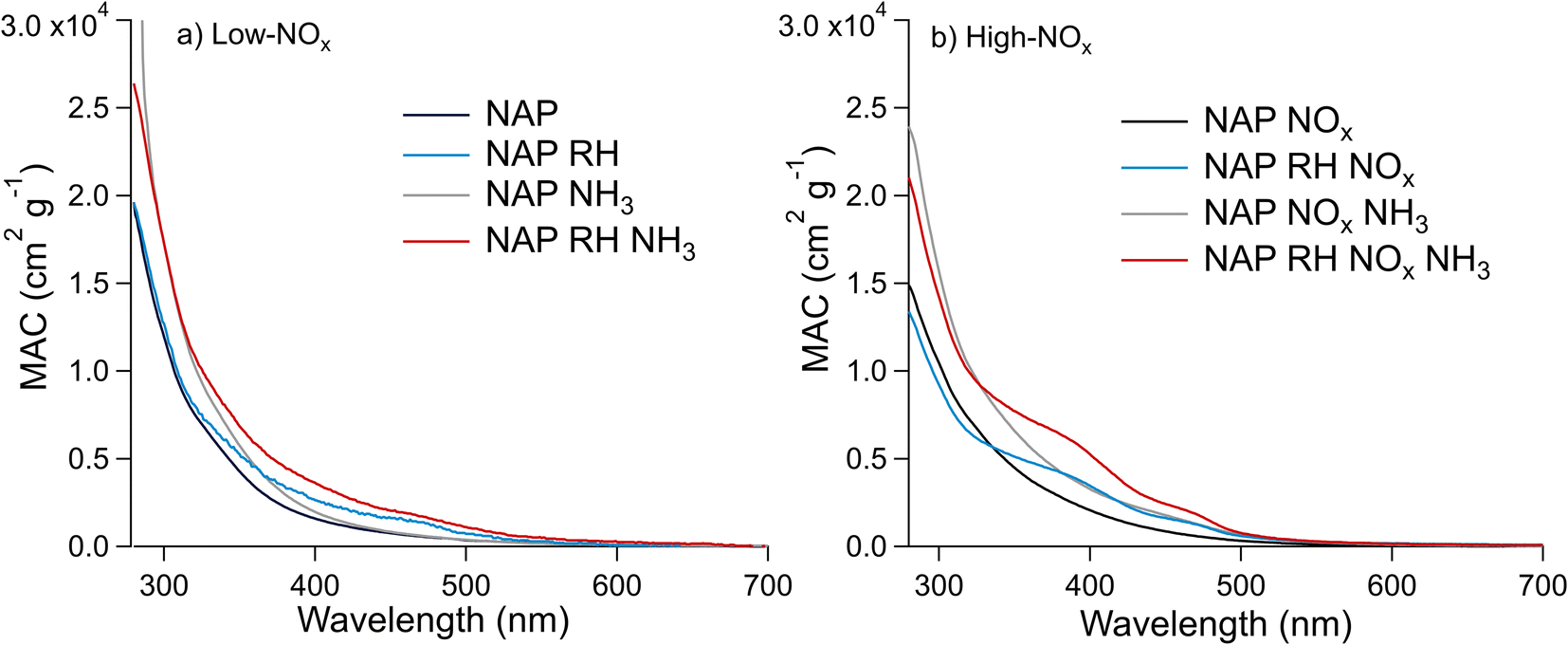 | ||
| Fig. 5 Effect of relative humidity and the presence of NH3 on the MACbulk of the NAP SOA under (a) low-NOx and (b) high-NOx conditions. These UV-vis spectra are for samples 9–16, which were extracted in water (see Section S2†). Trace labels denote parameters, which are elevated during SOA formation such that “NAP” represents low RH, low NOx, and low NH3, “NAP RH” represents high RH, low NOx, and low NH3, etc. | ||
The MACbulk values increase slightly in the presence of ammonia in Fig. 5a (low-NOx conditions), and to a greater extent in Fig. 5b (high-NOx conditions). This correlates with the extent of N-incorporation in the SOA as shown in Fig. 3, where the number of times the difference corresponding to the loss of H2O and gain of NH3 (0.9840 Da) occurred between peaks in the mass spectra was smaller under low-NOx conditions than under high-NOx conditions. These results are in agreement with Updyke et al.,35 where high-NOx conditions showed more browning with the addition of ammonia than low-NOx conditions, but the shape of the absorption spectrum did not change drastically for the NAP SOA under either condition. This result is likely because of greater formation of carbonyl compounds under high-NOx conditions as per Reaction (3).10,35,38 The effect of NH3 appears to be more significant in the presence of humidity, in agreement with similar observations of the enhanced effect of NH3 on the absorption coefficient of related SOAs from 1-methylnaphthalene.20
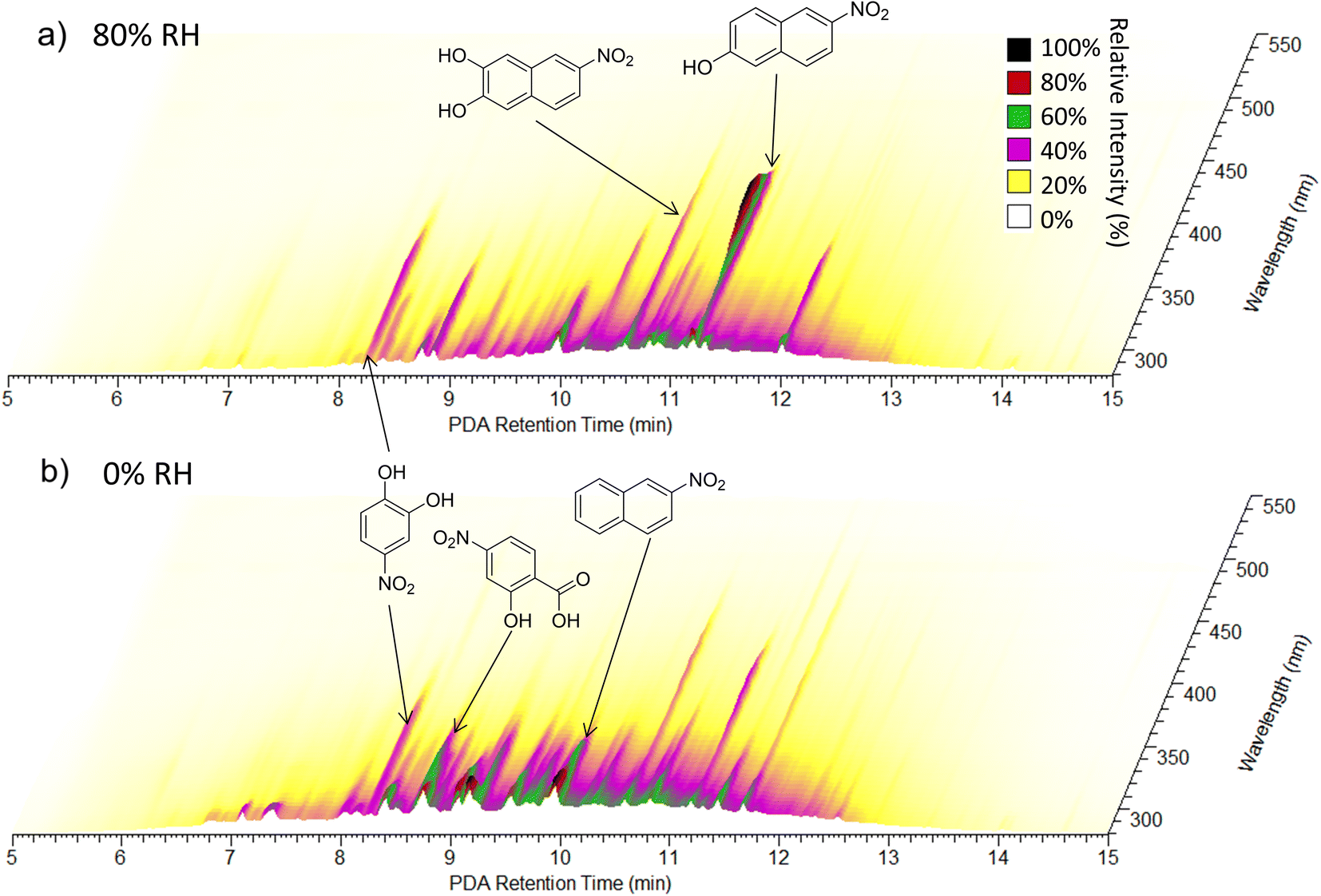 | ||
| Fig. 6 UPLC-PDA chromatograms for (a) the NAP SOA prepared under high-NOx at 80% RH and (b) the NAP SOA prepared under high-NOx at 0% RH. The retention time of the UPLC column is on the X-axis, the wavelength of absorption is on the Y-axis, and the absorbance is on the Z-axis. Spectra are from samples 23 (panel a) and 21 (panel b). Possible structures for the three most absorbing peaks in each panel are provided in the figure, and further individual peak assignments are provided in Fig. S11 and Table S1.† | ||
To further analyze the effect of relative humidity on the abundance of structural isomers of nitro-naphthol, we plotted the negative mode extracted ion chromatogram (EIC) trace of mass 188.035 (nitro-naphthol) for 0 and 80% RH high-NOx samples as shown in Fig. S9.† There are four major structural isomers of nitro-naphthol observed by this method in the NAP SOA samples, and the relative humidity during SOA formation changes the relative abundance of each of them. To determine if the differences in composition observed with high humidity could be replicated by exposing the SOA previously formed under dry conditions to liquid water, two samples of high-NOx SOAs were prepared under dry conditions (samples 21 and 22), and one was prepared under humid conditions (sample 23). One of the samples (sample 22) was extracted using acetonitrile, an equal volume of water was added, and then the SOA was allowed to age for three hours. This time frame was chosen to be slightly longer than the SOA formation experiments, which were 2.5 h under both humid and dry conditions. Note that it was necessary to extract using acetonitrile because the NAP SOA is only partially soluble in pure water. Exposure to water did not appear to change the relative abundance of structural isomers of nitro-naphthol (Fig. S9†) or the overall mass spectrum (Fig. S10†). Therefore, in addition to the effect on the dimer abundance and 〈O/C〉 ratio, RH appears to also influence the specific structural isomers that form during photooxidation of naphthalene (and not when the NAP SOA is dissolved in water during analysis).
Further analysis of Fig. 6 shows that under humid conditions, compounds elute on average later than under dry conditions (8 to 13 min as compared to 7 to 12.5 min), suggesting an increased abundance of less polar compounds under humid conditions, consistent with lower O/C ratios as measured using nano-DESI. A 2D representation of these data integrated from 280 to 680 nm is provided in Fig. S11† and assignments for major absorbers are provided in Table S1.† The major absorbers are generally C7 to C10 compounds, and the majority of them contain nitrogen.
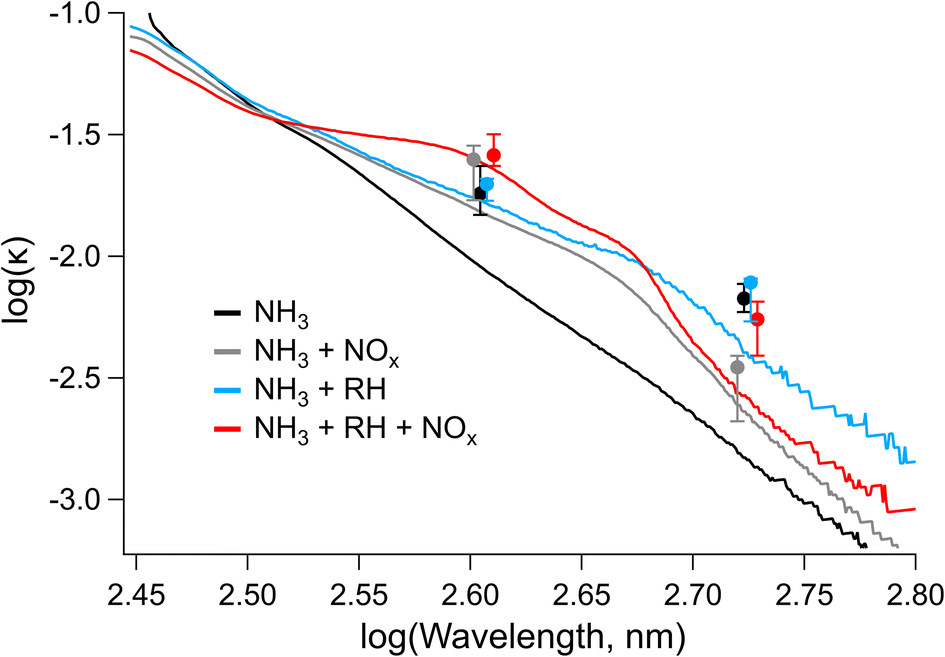 | ||
| Fig. 7 Imaginary refractive index (k) calculated from UV-vis spectra and from CRD-PAS data at 405 and 532 nm for samples 1–4. UV-vis data are shown as continuous lines, while CRD-PAS data are shown in filled circles. Error bars on the CRD-PAS data represent the range of measured values. CRD-PAS data points are taken at the same wavelengths but are offset in the x-direction for clarity. Note that all trails for which CRD-PAS data were collected correspond to conditions with NH3 present in the chamber. Daily data sets are included in Fig. S12–S15.† Labels on the legend correspond to parameters (NH3 and either RH or NOx), which were elevated during SOA formation. | ||
The values for the imaginary refractive index measured by CRD-PAS were consistently higher than the values measured offline by UV-vis, with the online (CRD-PAS) and offline (UV-vis) measurements agreeing within error for the high-RH samples but not the low-RH samples. This is likely due in part to the differing density of the aerosol particles between the high- and low-RH samples; the density we assumed for the offline conversion of MACbulk to k (1.5 g cm−3) may be more representative of high-RH samples than low-RH samples if the low-RH samples are of higher density. Previous studies have shown that elemental ratios can be used to predict the density of organic aerosol particles, with higher O/C ratios corresponding to higher densities.83,84 Our low-RH samples are therefore likely to be denser than our high-RH samples as the low-RH samples had significantly higher O/C ratios. The PAS values at 405 nm are largely within the error of each other. The only exception to this is the high-NOx + high-RH sample, which is higher than the two low-NOx samples, consistent with the formation of the peak at 400 nm corresponding the structural isomer of nitro-naphthol observed in Fig. 5 and 6. At 532 nm, the high-NOx + low-RH sample is lower than the other samples and the other samples are not different according to the CRD-PAS data. This trend is not reflected in the offline data, likely because of a larger error for these values: there was little signal in this region of UV-vis, and the wavelength is longer so errors will be amplified as per eqn (2).
By applying linear fits to the traces Fig. 7, the wavelength dependence of the absorption coefficient (w) can be determined and thereby the Absorption Ångström Exponent (AAE) can be estimated as described by Saleh et al.,85 where AAE ≈ w + 1. The results from this analysis are shown in Fig. 8 where they are placed in the context of the optically based BrC absorption strength classifications suggested by Saleh et al.85 Values are also included for the low-NH3 samples from Fig. 5, although they are not shown in Fig. 7. Based on the wavelength dependence of absorption and the value of k at 550 nm (the maximum of the solar spectrum), the NAP SOA falls into the classification of weakly absorbing BrC (W-BrC) under all preparation conditions. The AAE values for the samples range from 5.5 to 7.6. It can be seen in Fig. 8 that the best predictor of the AAE is relative humidity followed by NOx concentration. The presence of ammonia does not have a discernible effect on the AAE across the different preparation conditions.
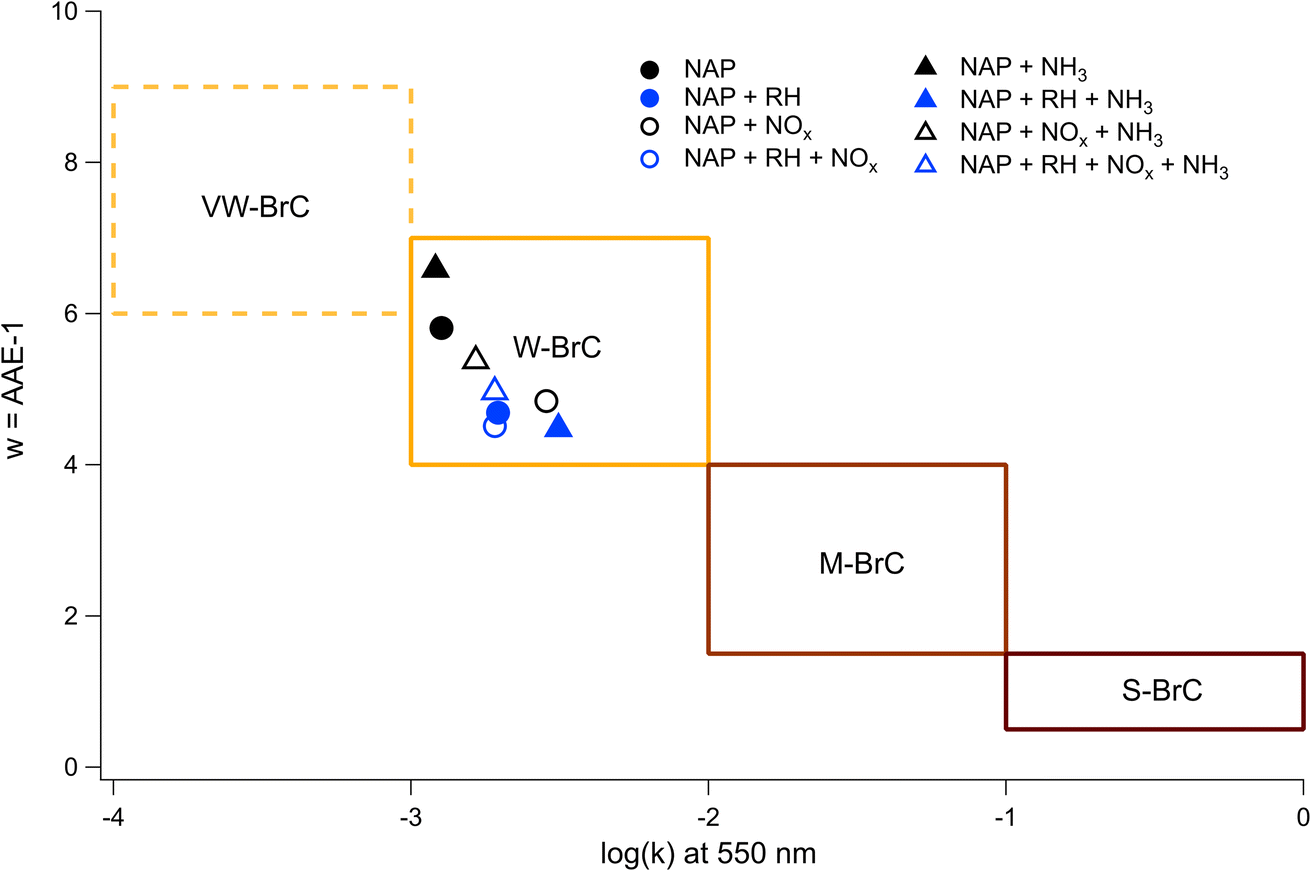 | ||
| Fig. 8 Values of w = AAE-1 and log(k) at 550 nm for the NAP SOA on the Saleh (2020) BrC classification plot. The legend specifies only parameters that were elevated during SOA formation. The way to read the markers is as follows: black represents low-RH and blue represents high-RH, circles represent low-NH3 and triangles represent high-NH3, and filled shapes represent low-NOx and empty shapes represent high-NOx. | ||
3.3 Hygroscopic properties
Fig. 9 summarizes the results of measurements of the hygroscopic properties of the NAP SOA (prepared with ammonia in the chamber) at varying RH and NOx concentrations. In both high- and low-RH samples, the addition of NOx increased the aerosol hygroscopicity. Additionally, under high-RH conditions in panel b, two activation events are observed, allowing the calculation of two values of κ. This is typical behavior for an externally mixed aerosol population, leading to two different activation events.86 In all samples, the measured κ values are representative of hygroscopic organic species (0.01 < κ < 0.5), while κ values typical of non-hygroscopic species (κ = 0) and inorganic species (0.5 < κ < 1.4) are not observed.56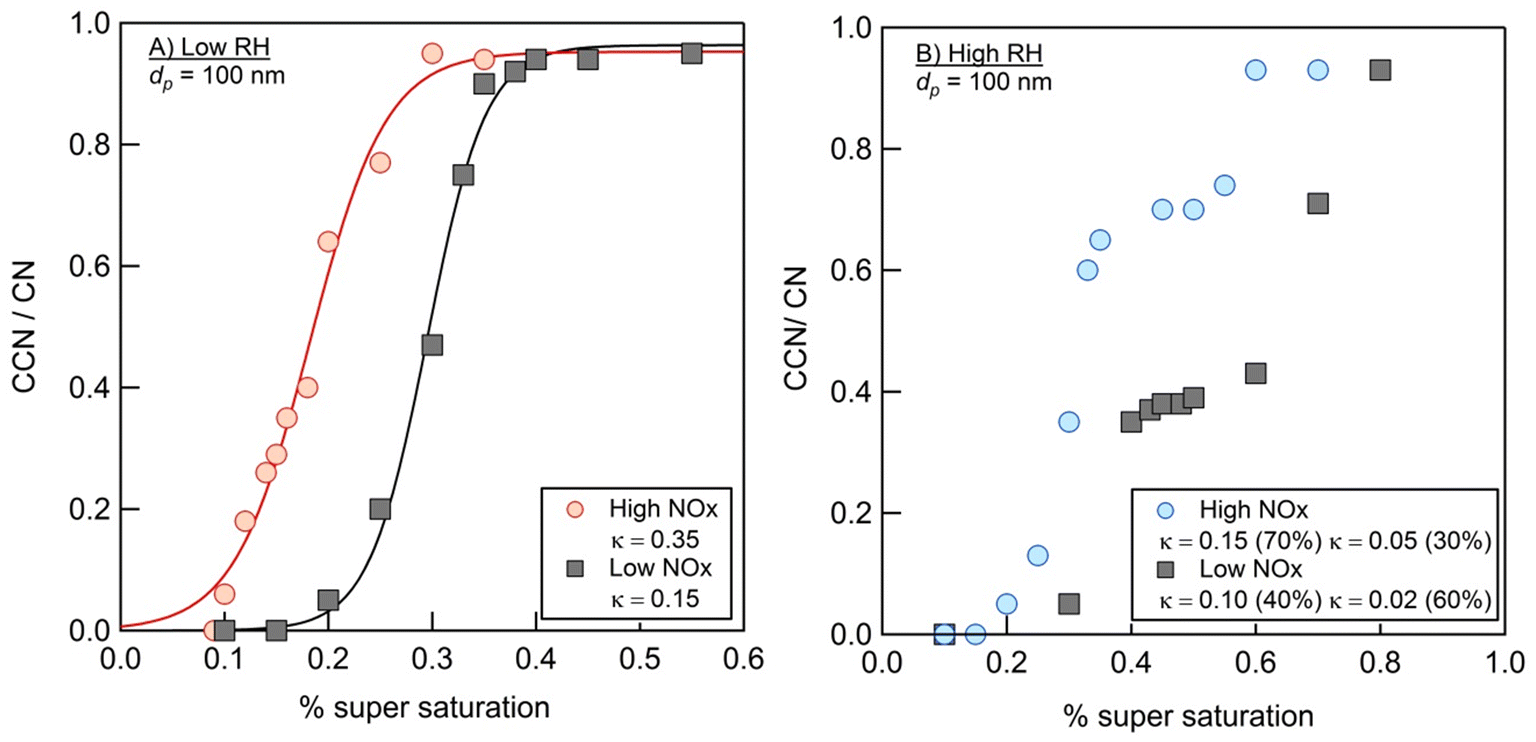 | ||
| Fig. 9 Fraction of particles that activate to CCN (CCN/CN) as a function of the % super saturation for samples 1 through 4. Panel A shows low RH studies, with the high-NOx data shown in red and the low-NOx data shown in black, and panel B shows high RH studies with the high-NOx data shown in blue and the low-NOx data shown in black. Note that all trails for which hygroscopicity data were collected contain ammonia. | ||
Previous work has measured κ for SOAs from other precursors to be near 0.1.87 The majority of the data presented here are consistent with this, with κ values ranging from 0.05 to 0.15. The two exceptions to this are the low-RH, high-NOx data, which result in a much greater κ = 0.35, and 60% component of the high-RH, low-NOx data, which result in a lower κ = 0.02. In general, the hygroscopicity data are consistent with the composition data. First, the incorporation of nitrogen, as observed for high-NOx samples in Fig. 4, likely increases hygroscopicity based on increased polarity with the incorporation of heteroatoms,56 as observed for high-NOx samples in Fig. 9. This could also be due to a small contribution of NH4NO3, which has a κ value of 0.67 and is likely to form when both NH3 and NOx are present.56,88 Second, a decrease in the 〈O/C〉 ratio and increase in dimer abundance as observed for high RH samples in Fig. 1 and 2 should decrease hygroscopicity, as observed for high RH samples in Fig. 9.89,90
4 Conclusions and future work
This work demonstrates that SOA formation conditions have complex effects on SOA composition and optical properties, and, importantly, in some cases the combination of two parameters (NH3, NOx, or RH) has different effects than only one of these parameters on its own. For instance, we observed much greater incorporation of NH3 when NOx was also present, but less incorporation of NH3 when RH was elevated and NOx was not. Or more dramatically, only the combination of high RH and high NOx produced a new absorption band at 400 nm attributable to 6-nitro-2-naphthol, a dominant contributor to the absorption coefficient of this aerosol. These examples suggest that, for a given set of experiments, some effects will not be independent of the control parameters (i.e., the parameters, such as RH, which are held constant while another parameter, like NOx level, is changed). Separate studies on the effects of one of these parameters may therefore show contradictory results depending on differences in the chosen control parameters. As a result, it will be imperative to simulate atmospheric conditions as accurately as possible for a robust representation of chemical processes in the atmosphere. Furthermore, additional studies should be conducted on the influence of parameter combinations (i.e., RH + NOx or NOx + NH3) on the properties of SOAs.Author contributions
The experiments and data analysis were conceived by SAN, THB and CC. ALK, PKA, ACM, JL and LMW carried out the smog chamber experiments; XZ and TH did the CRD/PAS measurements; GN did the SR-CCN measurements; PL, JL, and AL carried out the nano-DESI measurements; PKA and ALK did additional MAC measurements; ALK did LC measurements. ALK wrote the manuscript and all authors participated in its editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The UCI team acknowledges financial support from NSF grant AGS-1853639. The Purdue group acknowledges additional support from NOAA grant NA16OAR4310101. The LC instrument used in this work was purchased with the U.S. National Science Foundation grant CHE-1920242. The nano-DESI-HRMS measurements were performed at the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility located at PNNL, and sponsored by the Office of Biological and Environmental Research of the U.S. DOE.References
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Pean, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekci, R. Yu and B. Zhou, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York NY, USA, 2021, DOI:10.1017/9781009157896.
- M. O. Andreae and A. Gelencsér, Black Carbon or Brown Carbon? The Nature of Light-Absorbing Carbonaceous Aerosols, Atmos. Chem. Phys., 2006, 6(10), 3131–3148, DOI:10.5194/ACP-6-3131-2006.
- T. C. Bond, R. J. Charlson and L. Heintzenberg, Quantifying the Emission of Light-Absorbing Particles: Measurements Tailored to Climate Studies, Geophys. Res. Lett., 1998, 25(3), 337–340, DOI:10.1029/98GL00039.
- T. C. Bond, Spectral Dependence of Visible Light Absorption by Carbonaceous Particles Emitted from Coal Combustion, Geophys. Res. Lett., 2001, 28(21), 4075–4078, DOI:10.1029/2001GL013652.
- R. J. Hopkins, K. Lewis, Y. Desyaterik, Z. Wang, A. V. Tivanski, W. P. Arnott, A. Laskin and M. K. Gilles, Correlations between Optical, Chemical and Physical Properties of Biomass Burn Aerosols, Geophys. Res. Lett., 2007, 34, L18806, DOI:10.1029/2007GL030502.
- R. Saleh, E. S. Robinson, D. S. Tkacik, A. T. Ahern, S. Liu, A. C. Aiken, R. C. Sullivan, A. A. Presto, M. K. Dubey, R. J. Yokelson, N. M. Donahue and A. L. Robinson, Brownness of Organics in Aerosols from Biomass Burning Linked to Their Black Carbon Content, Nat. Geosci., 2014, 7(9), 647–650, DOI:10.1038/ngeo2220.
- R. A. Washenfelder, A. R. Attwood, C. A. Brock, H. Guo, L. Xu, R. J. Weber, N. L. Ng, H. M. Allen, B. R. Ayres, K. Baumann, R. C. Cohen, D. C. Draper, K. C. Duffey, E. Edgerton, J. L. Fry, W. W. Hu, J. L. Jimenez, B. B. Palm, P. Romer, E. A. Stone, P. J. Wooldridge and S. S. Brown, Biomass Burning Dominates Brown Carbon Absorption in the Rural Southeastern United States, Geophys. Res. Lett., 2015, 42(2), 653–664, DOI:10.1002/2014GL062444.
- A. P. S. Hettiyadura, V. Garcia, C. Li, C. P. West, J. Tomlin, Q. He, Y. Rudich and A. Laskin, Chemical Composition and Molecular-Specific Optical Properties of Atmospheric Brown Carbon Associated with Biomass Burning, Environ. Sci. Technol., 2021, 55, 2511–2521, DOI:10.1021/acs.est.0c05883.
- T. Moise, J. M. Flores and Y. Rudich, Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes, Chem. Rev., 2015, 115(10), 4400–4439, DOI:10.1021/CR5005259.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115(10), 4335–4382, DOI:10.1021/cr5006167.
- P. Lin, J. Liu, J. E. Shilling, S. M. Kathmann, J. Laskin and A. Laskin, Molecular Characterization of Brown Carbon (BrC) Chromophores in Secondary Organic Aerosol Generated from Photo-Oxidation of Toluene, Phys. Chem. Chem. Phys., 2015, 17(36), 23312–23325, 10.1039/C5CP02563J.
- J. Liu, P. Lin, A. Laskin, J. Laskin, S. M. Kathmann, M. Wise, R. Caylor, F. Imholt, V. Selimovic and J. E. Shilling, Optical Properties and Aging of Light-Absorbing Secondary Organic Aerosol, Atmos. Chem. Phys., 2016, 16(19), 12815–12827, DOI:10.5194/acp-16-12815-2016.
- A. T. Lambe, C. D. Cappa, P. Massoli, T. B. Onasch, S. D. Forestieri, A. T. Martin, M. J. Cummings, D. R. Croasdale, W. H. Brune, D. R. Worsnop and P. Davidovits, Relationship between Oxidation Level and Optical Properties of Secondary Organic Aerosol, Environ. Sci. Technol., 2013, 47(12), 6349–6357, DOI:10.1021/es401043j.
- T. Nakayama, Y. Matsumi, K. Sato, T. Imamura, A. Yamazaki and A. Uchiyama, Laboratory Studies on Optical Properties of Secondary Organic Aerosols Generated during the Photooxidation of Toluene and the Ozonolysis of α-Pinene, J. Geophys. Res. Atmos., 2010, 115(D24), 24204, DOI:10.1029/2010JD014387.
- T. Nakayama, K. Sato, Y. Matsumi, T. Imamura, A. Yamazaki and A. Uchiyama, Wavelength and NOx Dependent Complex Refractive Index of SOAs Generated from the Photooxidation of Toluene, Atmos. Chem. Phys., 2013, 13(2), 531–545, DOI:10.5194/ACP-13-531-2013.
- H. Kim and S. E. Paulson, Real Refractive Indices and Volatility of Secondary Organic Aerosol Generated from Photooxidation and Ozonolysis of Limonene, α-Pinene and Toluene, Atmos. Chem. Phys., 2013, 13(15), 7711–7723, DOI:10.5194/ACP-13-7711-2013.
- H. J. Lee, P. K. Aiona, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 2014, 48(17), 10217–10226, DOI:10.1021/es502515r.
- P. K. Aiona, J. L. Luek, S. A. Timko, L. C. Powers, M. Gonsior and S. A. Nizkorodov, Effect of Photolysis on Absorption and Fluorescence Spectra of Light-Absorbing Secondary Organic Aerosols, ACS Earth Space Chem., 2018, 2(3), 235–245, DOI:10.1021/acsearthspacechem.7b00153.
- J. H. Dingle, S. Zimmerman, A. L. Frie, J. Min, H. Jung and R. Bahreini, Complex Refractive Index, Single Scattering Albedo, and Mass Absorption Coefficient of Secondary Organic Aerosols Generated from Oxidation of Biogenic and Anthropogenic Precursors, Aerosol Sci. Technol., 2019, 53(4), 449–463, DOI:10.1080/02786826.2019.1571680.
- Y. Cui, A. L. Frie, J. H. Dingle, S. Zimmerman, I. Frausto-Vicencio, F. Hopkins and R. Bahreini, Influence of Ammonia and Relative Humidity on the Formation and Composition of Secondary Brown Carbon from Oxidation of 1-Methylnaphthalene and Longifolene, ACS Earth Space Chem., 2021, 5(4), 858–869, DOI:10.1021/ACSEARTHSPACECHEM.0C00353/ASSET/IMAGES/LARGE/SP0C00353_0005.JPEG.
- Q. He, C. Li, K. Siemens, A. C. Morales, A. P. S. Hettiyadura, A. Laskin and Y. Rudich, Optical Properties of Secondary Organic Aerosol Produced by Photooxidation of Naphthalene under NOx Condition, Environ. Sci. Technol., 2022, 56(8), 4816–4827, DOI:10.1021/ACS.EST.1C07328.
- K. Siemens, A. Morales, Q. He, C. Li, A. P. S. Hettiyadura, Y. Rudich and A. Laskin, Molecular Analysis of Secondary Brown Carbon Produced from the Photooxidation of Naphthalene, Environ. Sci. Technol., 2022, 56(6), 3340–3353, DOI:10.1021/ACS.EST.1C03135.
- A. Limbeck, M. Kulmala and H. Puxbaum, Secondary Organic Aerosol Formation in the Atmosphere via Heterogeneous Reaction of Gaseous Isoprene on Acidic Particles, Geophys. Res. Lett., 2003, 30(19), 1996, DOI:10.1029/2003GL017738.
- B. Nozière and W. Esteve, Organic Reactions Increasing the Absorption Index of Atmospheric Sulfuric Acid Aerosols, Geophys. Res. Lett., 2005, 32(3), 1–5, DOI:10.1029/2004GL021942.
- B. Nozière, D. Voisin, C. A. Longfellow, H. Friedli, B. E. Henry and D. R. Hanson, The Uptake of Methyl Vinyl Ketone, Methacrolein, and 2-Methyl-3-Butene-2-Ol onto Sulfuric Acid Solutions, J. Phys. Chem. A, 2006, 110(7), 2387–2395, DOI:10.1021/jp0555899.
- B. Nozière and W. Esteve, Light-Absorbing Aldol Condensation Products in Acidic Aerosols: Spectra, Kinetics, and Contribution to the Absorption Index, Atmos. Environ., 2007, 41(6), 1150–1163, DOI:10.1016/J.ATMOSENV.2006.10.001.
- C. Song, M. Gyawali, R. A. Zaveri, J. E. Shilling and W. P. Arnott, Light Absorption by Secondary Organic Aerosol from α-Pinene: Effects of Oxidants, Seed Aerosol Acidity, and Relative Humidity, J. Geophys. Res. Atmos., 2013, 118(20), 11741–11749, DOI:10.1002/JGRD.50767.
- Y. H. Lin, S. H. Budisulistiorini, K. Chu, R. A. Siejack, H. Zhang, M. Riva, Z. Zhang, A. Gold, K. E. Kautzman and J. D. Surratt, Light-Absorbing Oligomer Formation in Secondary Organic Aerosol from Reactive Uptake of Isoprene Epoxydiols, Environ. Sci. Technol., 2014, 48(20), 12012–12021, DOI:10.1021/es503142b.
- A. Gelencsér, A. Hoffer, G. Kiss, E. Tombácz, R. Kurdi and L. Bencze, In-Situ Formation of Light-Absorbing Organic Matter in Cloud Water, J. Atmos. Chem., 2003, 45(1), 25–33, DOI:10.1023/A:1024060428172.
- J. L. Chang and J. E. Thompson, Characterization of Colored Products Formed during Irradiation of Aqueous Solutions Containing H2O2 and Phenolic Compounds, Atmos. Environ., 2010, 44(4), 541–551, DOI:10.1016/J.ATMOSENV.2009.10.042.
- Y. L. Sun, Q. Zhang, C. Anastasio and J. Sun, Insights into Secondary Organic Aerosol Formed via Aqueous-Phase Reactions of Phenolic Compounds Based on High Resolution Mass Spectrometry, Atmos. Chem. Phys., 2010, 10(10), 4809–4822, DOI:10.5194/ACP-10-4809-2010.
- Y. J. Li, D. D. Huang, H. Y. Cheung, A. K. Y. Lee and C. K. Chan, Atmospheric Chemistry and Physics Aqueous-Phase Photochemical Oxidation and Direct Photolysis of Vanillin-a Model Compound of Methoxy Phenols from Biomass Burning, Atmos. Chem. Phys., 2014, 14, 2871–2885, DOI:10.5194/acp-14-2871-2014.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Chemical Characterization of SOA Formed from Aqueous-Phase Reactions of Phenols with the Triplet Excited State of Carbonyl and Hydroxyl Radical, Atmos. Chem. Phys., 2014, 14(24), 13801–13816, DOI:10.5194/ACP-14-13801-2014.
- W. Jiang, M. V. Misovich, A. P. S. Hettiyadura, A. Laskin, A. S. McFall, C. Anastasio and Q. Zhang, Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase - Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 2021, 55(8), 5199–5211, DOI:10.1021/ACS.EST.0C07581.
- K. M. Updyke, T. B. Nguyen and S. A. Nizkorodov, Formation of Brown Carbon via Reactions of Ammonia with Secondary Organic Aerosols from Biogenic and Anthropogenic Precursors, Atmos. Environ., 2012, 63, 22–31, DOI:10.1016/J.ATMOSENV.2012.09.012.
- H. J. Lee, A. Laskin, J. Laskin and S. A. Nizkorodov, Excitation-Emission Spectra and Fluorescence Quantum Yields for Fresh and Aged Biogenic Secondary Organic Aerosols, Environ. Sci. Technol., 2013, 47(11), 5763–5770, DOI:10.1021/es400644c.
- J. M. Flores, R. A. Washenfelder, G. Adler, H. J. Lee, L. Segev, J. Laskin, A. Laskin, S. A. Nizkorodov, S. S. Brown and Y. Rudich, Complex Refractive Indices in the Near-Ultraviolet Spectral Region of Biogenic Secondary Organic Aerosol Aged with Ammonia, Phys. Chem. Chem. Phys., 2014, 16(22), 10629–10642, 10.1039/C4CP01009D.
- D. L. Bones, D. K. Henricksen, S. A. Mang, M. Gonsior, A. P. Bateman, T. B. Nguyen, W. J. Cooper and S. A. Nizkorodov, Appearance of Strong Absorbers and Fluorophores in Limonene-O3 Secondary Organic Aerosol Due to NH4+-Mediated Chemical Aging over Long Time Scales, J. Geophys. Res. Atmos., 2010, 115(5), D05203, DOI:10.1029/2009JD012864.
- M. Xie, X. Chen, M. D. Hays, M. Lewandowski, J. Offenberg, T. E. Kleindienst and A. L. Holder, Light Absorption of Secondary Organic Aerosol: Composition and Contribution of Nitroaromatic Compounds, Environ. Sci. Technol., 2017, 51(20), 11607–11616, DOI:10.1021/ACS.EST.7B03263.
- D. E. Romonosky, N. N. Ali, M. N. Saiduddin, M. Wu, H. J. Lee, P. K. Aiona and S. A. Nizkorodov, Effective Absorption Cross Sections and Photolysis Rates of Anthropogenic and Biogenic Secondary Organic Aerosols, Atmos. Environ., 2016, 130, 172–179, DOI:10.1016/J.ATMOSENV.2015.10.019.
- P. F. Liu, N. Abdelmalki, H. M. Hung, Y. Wang, W. H. Brune and S. T. Martin, Ultraviolet and Visible Complex Refractive Indices of Secondary Organic Material Produced by Photooxidation of the Aromatic Compounds Toluene and M-Xylene, Atmos. Chem. Phys., 2015, 15(3), 1435–1446, DOI:10.5194/ACP-15-1435-2015.
- R. Gemayel, C. Emmelin, S. Perrier, S. Tomaz, V. J. Baboomian, D. A. Fishman, S. A. Nizkorodov, S. Dumas and C. George, Quenching of Ketone Triplet Excited States by Atmospheric Halides, Atmos. Environ., 2021, 1(1), 31–44, 10.1039/D0EA00011F.
- P. Liu, Y. J. Li, Y. Wang, A. P. Bateman, Y. Zhang, Z. Gong, A. K. Bertram and S. T. Martin, Highly Viscous States Affect the Browning of Atmospheric Organic Particulate Matter, ACS Cent. Sci., 2018, 4(2), 207–215, DOI:10.1021/ACSCENTSCI.7B00452.
- N. Sareen, A. N. Schwier, E. L. Shapiro, D. Mitroo and V. F. McNeill, Secondary Organic Material Formed by Methylglyoxal in Aqueous Aerosol Mimics, Atmos. Chem. Phys., 2010, 10(3), 997–1016, DOI:10.5194/ACP-10-997-2010.
- D. O. De Haan, A. L. Corrigan, M. A. Tolbert, J. L. Jimenez, S. E. Wood and J. J. Turley, Secondary Organic Aerosol Formation by Self-Reactions of Methylglyoxal and Glyoxal in Evaporating Droplets, Environ. Sci. Technol., 2009, 43(21), 8184–8190, DOI:10.1021/es902152t.
- D. O. De Haan, L. N. Hawkins, J. A. Kononenko, J. J. Turley, A. L. Corrigan, M. A. Tolbert and J. L. Jimenez, Formation of Nitrogen-Containing Oligomers by Methylglyoxal and Amines in Simulated Evaporating Cloud Droplets, Environ. Sci. Technol., 2011, 45(3), 984–991, DOI:10.1021/ES102933X.
- P. K. Aiona, H. J. Lee, R. Leslie, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Photochemistry of Products of the Aqueous Reaction of Methylglyoxal with Ammonium Sulfate, ACS Earth Space Chem., 2017, 1(8), 522–532, DOI:10.1021/ACSEARTHSPACECHEM.7B00075.
- D. N. Grace, J. R. Sharp, R. E. Holappa, E. N. Lugos, M. B. Sebold, D. R. Griffith, H. P. Hendrickson and M. M. Galloway, Heterocyclic Product Formation in Aqueous Brown Carbon Systems, ACS Earth Space Chem., 2019, 3(11), 2472–2481, DOI:10.1021/acsearthspacechem.9b00235.
- P. K. Aiona, H. J. Lee, P. Lin, F. Heller, A. Laskin, J. Laskin and S. A. Nizkorodov, A Role for 2-Methyl Pyrrole in the Browning of 4-Oxopentanal and Limonene Secondary Organic Aerosol, Environ. Sci. Technol., 2017, 51(19), 11048–11056, DOI:10.1021/ACS.EST.7B02293.
- T. B. Nguyen, A. Laskin, J. Laskin and S. A. Nizkorodov, Brown Carbon Formation from Ketoaldehydes of Biogenic Monoterpenes, Faraday Discuss., 2013, 165, 473–494, 10.1039/C3FD00036B.
- N. R. Smith, J. Montoya-Aguilera, D. Dabdub and S. A. Nizkorodov, Effect of Humidity on the Reactive Uptake of Ammonia and Dimethylamine by Nitrogen-Containing Secondary Organic Aerosol, Atmosphere, 2021, 12(11), 1502, DOI:10.3390/ATMOS12111502/S1.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere : Theory, Experiments, and Applications, Academic Press, 2000 Search PubMed.
- M. L. Hinks, J. Montoya-Aguilera, L. Ellison, P. Lin, A. Laskin, J. Laskin, M. Shiraiwa, D. Dabdub and S. A. Nizkorodov, Effect of Relative Humidity on the Composition of Secondary Organic Aerosol from the Oxidation of Toluene, Atmos. Chem. Phys., 2018, 18(3), 1643–1652, DOI:10.5194/ACP-18-1643-201.
- A. L. Klodt, M. Adamek, M. Dibley, S. A. Nizkorodov and R. E. O'Brien, Effects of the Sample Matrix on the Photobleaching and Photodegradation of Toluene-Derived Secondary Organic Aerosol Compounds, Atmos. Chem. Phys., 2022, 22(15), 10155–10171, DOI:10.5194/ACP-22-10155-2022.
- A. L. Klodt, Experiment Set: Naphthalene SOA for Humidity, NOx, and NH3 Effects, 2022, https://icarus.ucdavis.edu/chambers/531/experimentSets/255, accessed 2022-12-20 Search PubMed.
- M. D. Petters and S. M. Kreidenweis, A Single Parameter Representation of Hygroscopic Growth and Cloud Condensation Nucleus Activity, Atmos. Chem. Phys., 2007, 7(8), 1961–1971, DOI:10.5194/ACP-7-1961-2007.
- C. D. Cappa, T. B. Onasch, P. Massoli, D. R. Worsnop, T. S. Bates, E. S. Cross, P. Davidovits, J. Hakala, K. L. Hayden, B. T. Jobson, K. R. Kolesar, D. A. Lack, B. M. Lerner, S. M. Li, D. Mellon, I. Nuaaman, J. S. Olfert, T. Petäjä, P. K. Quinn, C. Song, R. Subramanian, E. J. Williams and R. A. Zaveri, Radiative Absorption Enhancements Due to the Mixing State of Atmospheric Black Carbon, Science, 2012, 337(6098), 1078–1081, DOI:10.1126/SCIENCE.1223447.
- J. M. Langridge, M. S. Richardson, D. Lack, D. Law and D. M. Murphy, Aircraft Instrument for Comprehensive Characterization of Aerosol Optical Properties, Part I: Wavelength-Dependent Optical Extinction and Its Relative Humidity Dependence Measured Using Cavity Ringdown Spectroscopy, Aerosol Sci. Technol., 2011, 45(11), 1305–1318, DOI:10.1080/02786826.2011.592745.
- D. A. Lack, M. S. Richardson, D. Law, J. M. Langridge, C. D. Cappa, R. J. McLaughlin and D. M. Murphy, Aircraft Instrument for Comprehensive Characterization of Aerosol Optical Properties, Part 2: Black and Brown Carbon Absorption and Absorption Enhancement Measured with Photo Acoustic Spectroscopy, Aerosol Sci. Technol., 2012, 46(5), 555–568, DOI:10.1080/02786826.2011.645955.
- N. W. Davies, M. I. Cotterell, C. Fox, K. Szpek, J. M. Haywood and J. M. Langridge, On the Accuracy of Aerosol Photoacoustic Spectrometer Calibrations Using Absorption by Ozone, Atmos. Meas. Tech., 2018, 11(4), 2313–2324, DOI:10.5194/AMT-11-2313-2018.
- D. A. Lack, J. M. Langridge, R. Bahreini, C. D. Cappa, A. M. Middlebrook and J. P. Schwarz, Brown Carbon and Internal Mixing in Biomass Burning Particles, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(37), 14802–14807, DOI:10.1073/pnas.1206575109.
- C. D. Cappa, X. Zhang, L. M. Russell, S. Collier, A. K. Y. Lee, C. L. Chen, R. Betha, S. Chen, J. Liu, D. J. Price, K. J. Sanchez, G. R. McMeeking, L. R. Williams, T. B. Onasch, D. R. Worsnop, J. Abbatt and Q. Zhang, Light Absorption by Ambient Black and Brown Carbon and Its Dependence on Black Carbon Coating State for Two California, USA, Cities in Winter and Summer, J. Geophys. Res. Atmos., 2019, 124(3), 1550–1577, DOI:10.1029/2018JD029501.
- P. J. Roach, J. Laskin and A. Laskin, Molecular Characterization of Organic Aerosols Using Nanospray-Desorption/Electrospray Ionization-Mass Spectrometry, Anal. Chem., 2010, 82(19), 7979–7986, DOI:10.1021/AC101449P.
- P. J. Roach, J. Laskin and A. Laskin, Nanospray Desorption Electrospray Ionization: An Ambient Method for Liquid-Extraction Surface Sampling in Mass Spectrometry, Analyst, 2010, 135(9), 2233–2236, 10.1039/C0AN00312C.
- H. Chin, K. S. Hopstock, L. T. Fleming, S. A. Nizkorodov and H. A. Al-Abadleh, Effect of Aromatic Ring Substituents on the Ability of Catechol to Produce Brown Carbon in Iron(III)-Catalyzed Reactions, Atmos. Environ., 2021, 1(2), 64–78, 10.1039/D0EA00007H.
- A. W. H. Chan, K. E. Kautzman, P. S. Chhabra, J. D. Surratt, M. N. Chan, J. D. Crounse, A. Kürten, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary Organic Aerosol Formation from Photooxidation of Naphthalene and Alkylnaphthalenes: Implications for Oxidation of Intermediate Volatility Organic Compounds (IVOCs), Atmos. Chem. Phys., 2009, 9(9), 3049–3060, DOI:10.5194/ACP-9-3049-2009.
- C. L. Chen, M. Kacarab, P. Tang and D. R. Cocker, SOA Formation from Naphthalene, 1-Methylnaphthalene, and 2-Methylnaphthalene Photooxidation, Atmos. Environ., 2016, 131, 424–433, DOI:10.1016/J.ATMOSENV.2016.02.007.
- A. P. Bateman, M. L. Walser, Y. Desyaterik, J. Laskin, A. Laskin and S. A. Nizkorodov, The Effect of Solvent on the Analysis of Secondary Organic Aerosol Using Electrospray Ionization Mass Spectrometry, Environ. Sci. Technol., 2008, 42(19), 7341–7346, DOI:10.1021/ES801226W.
- Y. Chen and T. C. Bond, Light Absorption by Organic Carbon from Wood Combustion, Atmos. Chem. Phys., 2010, 10(4), 1773–1787, DOI:10.5194/ACP-10-1773-2010.
- Y. Zhang, K. Wang, H. Tong, R. J. Huang and T. Hoffmann, The Maximum Carbonyl Ratio (MCR) as a New Index for the Structural Classification of Secondary Organic Aerosol Components, Rapid Commun. Mass Spectrom., 2021, 35(14), e9113, DOI:10.1002/RCM.9113.
- K. C. Edwards, A. L. Klodt, T. Galeazzo, M. Schervish, J. Wei, T. Fang, N. M. Donahue, B. Aumont, S. A. Nizkorodov and M. Shiraiwa, Effects of Nitrogen Oxides on the Production of Reactive Oxygen Species and Environmentally Persistent Free Radicals from α-Pinene and Naphthalene Secondary Organic Aerosols, J. Phys. Chem. A, 2022, 2022, 7361–7372, DOI:10.1021/acs.jpca.2c05532.
- P. Lin, L. T. Fleming, S. A. Nizkorodov, J. Laskin and A. Laskin, Comprehensive Molecular Characterization of Atmospheric Brown Carbon by High Resolution Mass Spectrometry with Electrospray and Atmospheric Pressure Photoionization, Anal. Chem., 2018, 90(21), 12493–12502, DOI:10.1021/acs.analchem.8b02177.
- C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. Decarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer and P. Artaxo, A Simplified Description of the Evolution of Organic Aerosol Composition in the Atmosphere, Geophys. Res. Lett., 2010, 37(8) DOI:10.1029/2010GL042737.
- Y. Huang, R. Zhao, S. M. Charan, C. M. Kenseth, X. Zhang and J. H. Seinfeld, Unified Theory of Vapor-Wall Mass Transport in Teflon-Walled Environmental Chambers, Environ. Sci. Technol., 2018, 52(4), 2134–2142, DOI:10.1021/ACS.EST.7B05575.
- S. Liu, N. T. Tsona, Q. Zhang, L. Jia, Y. Xu and L. Du, Influence of Relative Humidity on Cyclohexene SOA Formation from OH Photooxidation, Chemosphere, 2019, 231, 478–486, DOI:10.1016/J.CHEMOSPHERE.2019.05.131.
- M. Jang, B. Carroll, B. Chandramouli and R. M. Kamens, Particle Growth by Acid-Catalyzed Heterogeneous Reactions of Organic Carbonyls on Preexisting Aerosols, Environ. Sci. Technol., 2003, 37(17), 3828–3837, DOI:10.1021/ES021005U.
- T. Chen, B. Chu, Q. Ma, P. Zhang, J. Liu and H. He, Effect of Relative Humidity on SOA Formation from Aromatic Hydrocarbons: Implications from the Evolution of Gas- and Particle-Phase Species, Sci. Total Environ., 2021, 773, 145015, DOI:10.1016/J.SCITOTENV.2021.145015.
- L. Jia and Y. Xu, Effects of Relative Humidity on Ozone and Secondary Organic Aerosol Formation from the Photooxidation of Benzene and Ethylbenzene, Aerosol Sci. Technol., 2014, 48(1), 1–12, DOI:10.1080/02786826.2013.847269.
- L. Jia and Y. Xu, Different Roles of Water in Secondary Organic Aerosol Formation from Toluene and Isoprene, Atmos. Chem. Phys., 2018, 18, 8137–8154, DOI:10.5194/acp-18-8137-2018.
- P. Massoli, A. T. Lambe, A. T. Ahern, L. R. Williams, M. Ehn, J. Mikkilä, M. R. Canagaratna, W. H. Brune, T. B. Onasch, J. T. Jayne, T. Petäjä, M. Kulmala, A. Laaksonen, C. E. Kolb, P. Davidovits and D. R. Worsnop, Relationship between Aerosol Oxidation Level and Hygroscopic Properties of Laboratory Generated Secondary Organic Aerosol (SOA) Particles, Geophys. Res. Lett., 2010, 37(24) DOI:10.1029/2010GL045258.
- K. Mitra, H. R. Mishra, X. Pei and R. K. Pathak, Secondary Organic Aerosol (SOA) from Photo-Oxidation of Toluene: 1 Influence of Reactive Nitrogen, Acidity and Water Vapours on Optical Properties, Atmosphere, 2022, 13(7), 1099, DOI:10.3390/atmos13071099.
- G. Rusek and L. Prajer-Janczewska, Studies of the Electronic Structures in Mononitronaphthol-2 Derivatives by the UV and Vis Spectroscopic Methods, Spectrosc. Lett., 1978, 11(6), 381–387, DOI:10.1080/00387017808067759.
- M. Kuwata, S. R. Zorn and S. T. Martin, Using Elemental Ratios to Predict the Density of Organic Material Composed of Carbon, Hydrogen, and Oxygen, Environ. Sci. Technol., 2012, 46, 787–794, DOI:10.1021/es202525q.
- S. Nakao, P. Tang, X. Tang, C. H. Clark, L. Qi, E. Seo, A. Asa-Awuku and D. Cocker, Density and Elemental Ratios of Secondary Organic Aerosol: Application of a Density Prediction Method, Atmos. Environ., 2013, 68, 273–277, DOI:10.1016/J.ATMOSENV.2012.11.006.
- R. Saleh, From Measurements to Models: Toward Accurate Representation of Brown Carbon in Climate Calculations, Curr. Pollut. Rep., 2020, 6(2), 90–104, DOI:10.1007/S40726-020-00139-3.
- S. R. Schill, D. B. Collins, C. Lee, H. S. Morris, G. A. Novak, K. A. Prather, P. K. Quinn, C. M. Sultana, A. V. Tivanski, K. Zimmermann, C. D. Cappa and T. H. Bertram, The Impact of Aerosol Particle Mixing State on the Hygroscopicity of Sea Spray Aerosol, ACS Cent. Sci., 2015, 1(3), 132–141, DOI:10.1021/ACSCENTSCI.5B00174.
- A. J. Prenni, M. D. Petters, S. M. Kreidenweis, P. J. DeMott and P. J. Ziemann, Cloud Droplet Activation of Secondary Organic Aerosol, J. Geophys. Res. Atmos., 2007, 112(10) DOI:10.1029/2006JD007963.
- B. Svenningsson, J. Rissler, E. Swietlicki, M. Mircea, M. Bilde, M. C. Facchini, S. Decesari, S. Fuzzi, J. Zhou, J. Mønster and T. Rosenørn, Hygroscopic Growth and Critical Supersaturations for Mixed Aerosol Particles of Inorganic and Organic Compounds of Atmospheric Relevance, Atmos. Chem. Phys., 2006, 6(7), 1937–1952, DOI:10.5194/ACP-6-1937-2006.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326(5959), 1525–1529, DOI:10.1126/science.1180353.
- J. Wang, J. E. Shilling, J. Liu, A. Zelenyuk, D. M. Bell, M. D. Petters, R. Thalman, F. Mei, R. A. Zaveri and G. Zheng, Cloud Droplet Activation of Secondary Organic Aerosol Is Mainly Controlled by Molecular Weight, Not Water Solubility, Atmos. Chem. Phys., 2019, 19(2), 941–954, DOI:10.5194/ACP-19-941-2019.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00033h |
| ‡ Now at South Coast Air Quality Management District, Diamond Bar, California 91765. |
| § Now at California Air Resources Board, Riverside, California 92507. |
| ¶ Now at Air Quality Research Center, University of California, Davis, California 95616. |
| || Now at California Air Resources Board, Sacramento, CA 95814. |
| ** Now at Cooperative Institute for Research in Environmental Sciences, University of Colorado Boulder, Boulder, CO 80305 and NOAA Chemical Sciences Laboratory (CSL), Boulder, CO 80305. |
| This journal is © The Royal Society of Chemistry 2023 |