
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, X-ray characterization and DFT analysis of dicyanidoaurate telluronium salts: on the importance of charge assisted chalcogen bonds†
Simone
Ghinato
a,
Alessia
Giordana
 *a,
Eliano
Diana
a,
Rosa M.
Gomila
b,
Emanuele
Priola
*a and
Antonio
Frontera
*b
*a,
Eliano
Diana
a,
Rosa M.
Gomila
b,
Emanuele
Priola
*a and
Antonio
Frontera
*b
aUniversità degli Studi di Torino, Department of Chemistry, Via Pietro Giuria 7, 10125 Torino, Italy. E-mail: alessia.giordana@unito.it; emanuele.priola@unito.it
bDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 11th October 2023
Abstract
In this manuscript we report the synthesis and X-ray characterization of two cyanidoaurate telluronium salts, namely (3-fluorophenyl)(methyl)(phenyl)telluronium dicyanidoaurate [(3-F-Ph)(Me)(Ph)Te][Au(CN)2] (1) and methyldiphenyltelluronium dicyanidoaurate [(Me)(Ph)2Te][Au(CN)2] (2). In the solid state, the tellurium atom establishes three concurrent and directional chalcogen bonds (ChBs) with the adjacent anions, in both compounds. These charge-assisted ChBs (CAChBs) have been analyzed using DFT calculations and several computational tools. The MEP surface analysis discloses the existence of three σ-holes at the Te-atoms capable of establishing strong CAChBs with the counter-ions. In addition, significant charge transfer from the lone pair orbital at the N-atom of the anion to the antibonding σ*(Te–C) orbital of the cation is observed in some cases.
Introduction
Organotelluronium salts are interesting compounds with applications as reactants in organic synthetic chemistry. In particular, they are precursors of telluronium ylides that can react with carbonyl compounds to yield oxiranes and secondary alcohols.1 These compounds have been known for over a century2 and their solid state architecture3–14 was described as being governed by tellurium–anion interactions.15 They were classified as secondary weak interactions that expanded the TeR3E (E = lone pair) trigonal pyramidal geometry into a five- or six-coordinate entity.16,17 Solid-state 125Te-NMR spectroscopy was also used as a sensitive tool for the identification of these weak secondary bonding interactions between the cations and anions.13More recently, noncovalent interactions involving elements of the p-block of the periodic table, in general behaving as electrophiles, have been attracting considerable attention.18–23 Several theoretical24 and experimental25 works have demonstrated that the electron density distribution in the covalently bonded atoms of p-block is anisotropic and shows regions of lower (positive) or higher (negative) electron density. The location and number of positive regions are related to the position and number of covalent bonds formed by the atoms.26 For instance, chalcogen atoms typically form two covalent bonds and, consequently, two regions of depleted electron density (named σ-holes) are usually located opposite these bonds.27 With electron-rich sites, these σ-holes form highly directional interactions which are named chalcogen bonds (ChBs).28 Similar names are used for interactions involving elements of groups 17 and 15: halogen bonds (HaBs),18 and pnictogen bonds (PnBs),20,29 respectively.
Interest in ChB has increased exponentially in the last ten years.19,30–32 It has been recently used in catalysis,33,34 molecular recognition, and crystal engineering.35 More recently, the utilization of trivalent sulfur cations as ChB donor sites (R3S+⋯A) has been studied both theoretically and experimentally.36–38 Trisubstituted sulfur atoms afforded charge-assisted chalcogen bonds (CAChBs), which are particularly strong thanks to the contribution of the cation–anion electrostatic attraction. Moreover, the existence of three σ-holes in the chalcogen atom enables the possibility of establishing up to three ChBs.36–38 In this regard, Resnati et al.36 have demonstrated the existence and relevance of CAChBs in crystal structures and enzyme inhibitors of α-glucosidase and analyzed the energetic features of supramolecular assemblies of sulfonium, selenonium and telluronium salts. In these systems, the Ch-atoms possess three σ-holes available to interact with electron rich atoms, mimicking the behavior of pnictogen bonds.
X-ray structures of telluronium cations where the counterion contains gold are very rare. To the best of our knowledge, only two examples are available in the literature: one corresponds to triphenyltelluronium with tetrachloridoaurate (refcode MIHSOL)39 and the other to trimethyltelluronium with bis(4,5-dimercapto-1,3-dithiole-2-thionato)-gold(III) (refcode LALFUZ),40 with both compounds incorporating Au(III) in a square planar environment, see Scheme 1. In this manuscript, we report for the first time asymmetric telluronium salts including Au(I) dicyanoaurate as a counterion. In particular, we analyze the structure and bonding of (3-fluorophenyl)(methyl)(phenyl)telluronium dicyanidoaurate [(3-F-Ph)(Me)(Ph)Te][Au(CN)2] 1 and methyldiphenyltelluronium dicyanidoaurate [(Me)(Ph)2Te][Au(CN)2] 2 and the effect of the geometry on the cation–anion interactions. The assemblies, constructed by chalcogen bonding interactions, have been described and studied using DFT calculations, molecular electrostatic potential (MEP) surface analysis and two computational tools based on the topology of the electron density, QTAIM41 and NCIPlot.42 Moreover, the donor–acceptor orbitals involved in the ChBs were analyzed using the natural bond orbital (NBO) methodology43 and the Kitaura–Morokuma44 energy decomposition analysis.
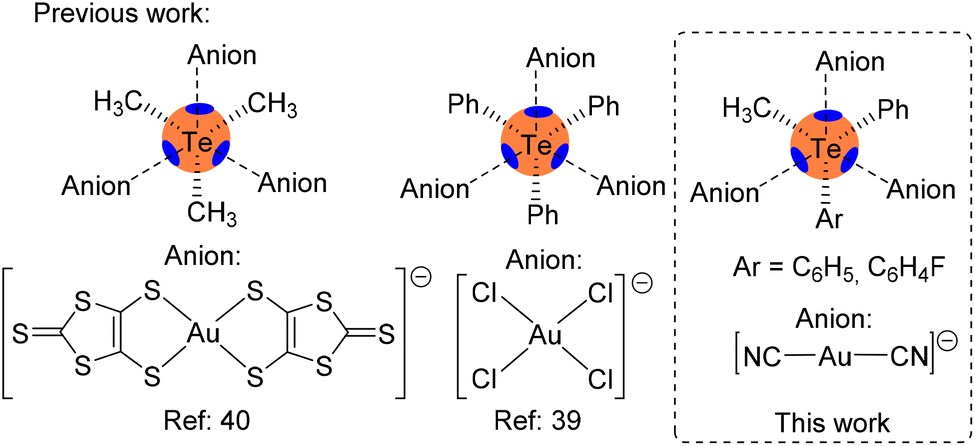 | ||
| Scheme 1 Left: previous examples of telluronium cations with Au-based anions. Right: the telluronium salts studied herein. | ||
Materials and methods
Synthesis of complexes 1 and 2
Compounds 1 and 2 have been obtained by metathesis. Ethanolic solutions of equimolar amounts of K[Au(CN)2] (Merck, used without any purification) and the corresponding tetrafluoroborate salts, 3a and 3b, respectively (synthesized and purified as described in Scheme 2 and the ESI†), were mixed and from slow evaporation, few prismatic colorless crystals started to appear after a few weeks. Crystals were separated from the mother solution before the formation of other by-products.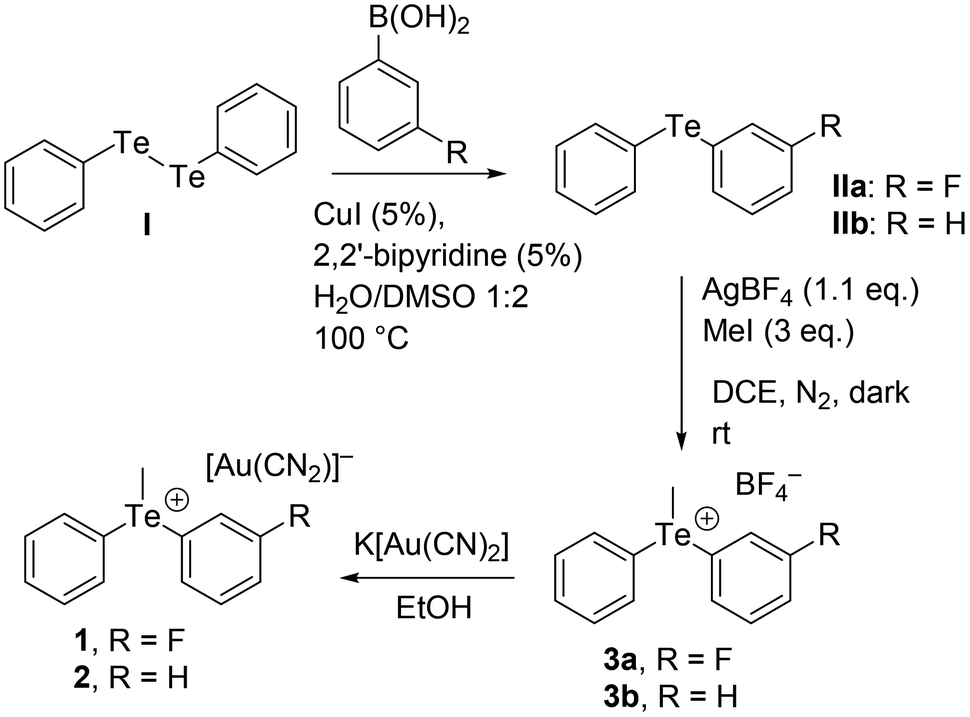 | ||
| Scheme 2 Synthetic route to compounds 1–3. | ||
Single-crystal X-ray diffraction
Single crystal data of crystallized compounds were collected on a Gemini R Ultra diffractometer (Agilent Technologies UK Ltd, Oxford, U.K.) using graphite-monochromatic Cu Kα radiation (λ = 1.5406 Å) with the ω-scan method. Copper derived radiation has been preferred for cases of very weakly diffracting crystals. CrysAlisPro software was used for retrieving cell parameters, performing data reduction and absorption correction (with the multi-scan technique). All structures were solved by direct methods using SHELXS-14 and refined with full-matrix least-squares on F2 using SHELXL-14 using the Olex2 program. All non-hydrogen atoms were anisotropically refined. Hydrogen atoms were calculated and refined as riding on the corresponding atom. Images of the structures were obtained using Mercury software. Crystal data and refinement, selected bond lengths and angle amplitudes and the asymmetric units of compounds are reported in the ESI.† The crystallographic data for the crystallized compounds have been deposited at the Cambridge Crystallographic Data Centre as supplementary publications under CCDC numbers 2284002 and 2284003.†Characterization
FT-ATR spectra were recorded in the 50–4000 cm−1 range using a Bruker Vertex 70 spectrophotometer equipped with a Harrick MVP2 ATR cell and DTGS detectors (either with Si or KBr beamsplitters). Micro Raman spectra of single crystals were recorded using a Horiba Jobin Yvon HR800 spectrometer, equipped with an Olympus BX41 microscope, by exciting with a 532 nm laser.DFT calculations
The energy and EDA calculations were carried out using the Turbomole 7.7 program45 and the PBE0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46-D447/def2-TZVP48 level of theory. For gold, the def2-TZVP basis set used in this work included effective core potentials (ECP),49 and relativistic effects were used for the inner electrons.48 The crystallographic coordinates were used to evaluate the interactions in the solid state of compounds 1 and 2, since we were interested in studying the interactions as they stand in the solid state. The dimers and trimers extracted from the solid-state structures were selected to study the chalcogen bonding interactions. The interaction energies were computed by subtracting the sum of the energies of the monomers from those of the assembly. Bader's “Atoms in molecules” theory (QTAIM)41 and the noncovalent interaction plot (NCIPlot)42 were used to study the interactions discussed herein using the Multiwfn program50 and represented using the VMD visualization software.51 The molecular electrostatic potential (MEP) surfaces were computed using the 0.001 a.u. isosurface as the best estimation of the van der Waals surface at the same level of theory and represented using the GaussView program.52 For the NCIPlot representations, the following settings were used: RDG isosurface = 0.4, density cut-off = 0.04 a.u., and color scale −0.03 ≤ (signλ2)ρ ≤ 0.03 a.u. Natural bond orbital (NBO)43 calculations were performed using the NBO7.0 program.53
46-D447/def2-TZVP48 level of theory. For gold, the def2-TZVP basis set used in this work included effective core potentials (ECP),49 and relativistic effects were used for the inner electrons.48 The crystallographic coordinates were used to evaluate the interactions in the solid state of compounds 1 and 2, since we were interested in studying the interactions as they stand in the solid state. The dimers and trimers extracted from the solid-state structures were selected to study the chalcogen bonding interactions. The interaction energies were computed by subtracting the sum of the energies of the monomers from those of the assembly. Bader's “Atoms in molecules” theory (QTAIM)41 and the noncovalent interaction plot (NCIPlot)42 were used to study the interactions discussed herein using the Multiwfn program50 and represented using the VMD visualization software.51 The molecular electrostatic potential (MEP) surfaces were computed using the 0.001 a.u. isosurface as the best estimation of the van der Waals surface at the same level of theory and represented using the GaussView program.52 For the NCIPlot representations, the following settings were used: RDG isosurface = 0.4, density cut-off = 0.04 a.u., and color scale −0.03 ≤ (signλ2)ρ ≤ 0.03 a.u. Natural bond orbital (NBO)43 calculations were performed using the NBO7.0 program.53
Results and discussion
Description of the structures
Both the dicyanidoaurate salts crystallize from ethanol in the monoclinic space group P21/n and are isomorphous. Both salts present a crystal packing similar to the dicyanoargentate derivative of the methyldiphenyltelluronium cation (refcode HUHBUH) (Fig. 1).54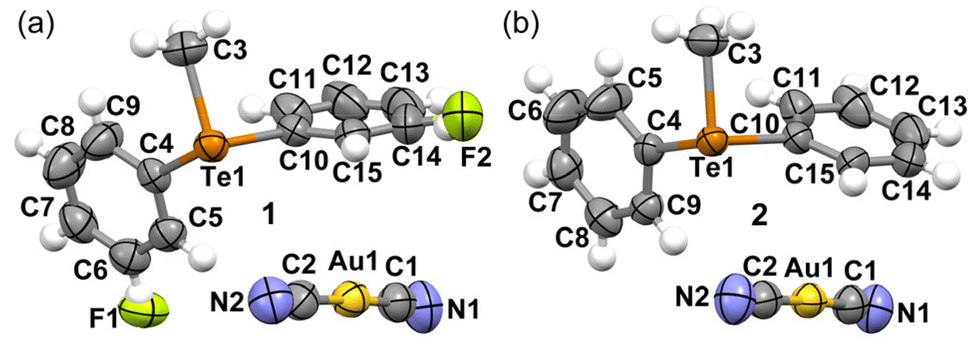 | ||
| Fig. 1 Ellipsoid plots (50% probability) of the asymmetric units of salts 1 (a) and 2 (b). | ||
In both crystal structures (Fig. 2), the telluronium cation forms dimeric nodal chains. These chains are constructed with two bridging dicyanoaurate chalcogen-bonded cyanides and two terminal dicyanidoaurate chalcogen-bonded cyanides, as shown in Fig. 2b for compound 2. These interactions constitute the secondary coordination sphere for the tellurium(IV) centre, as illustrated in Scheme 1 and detailed in Table 1.
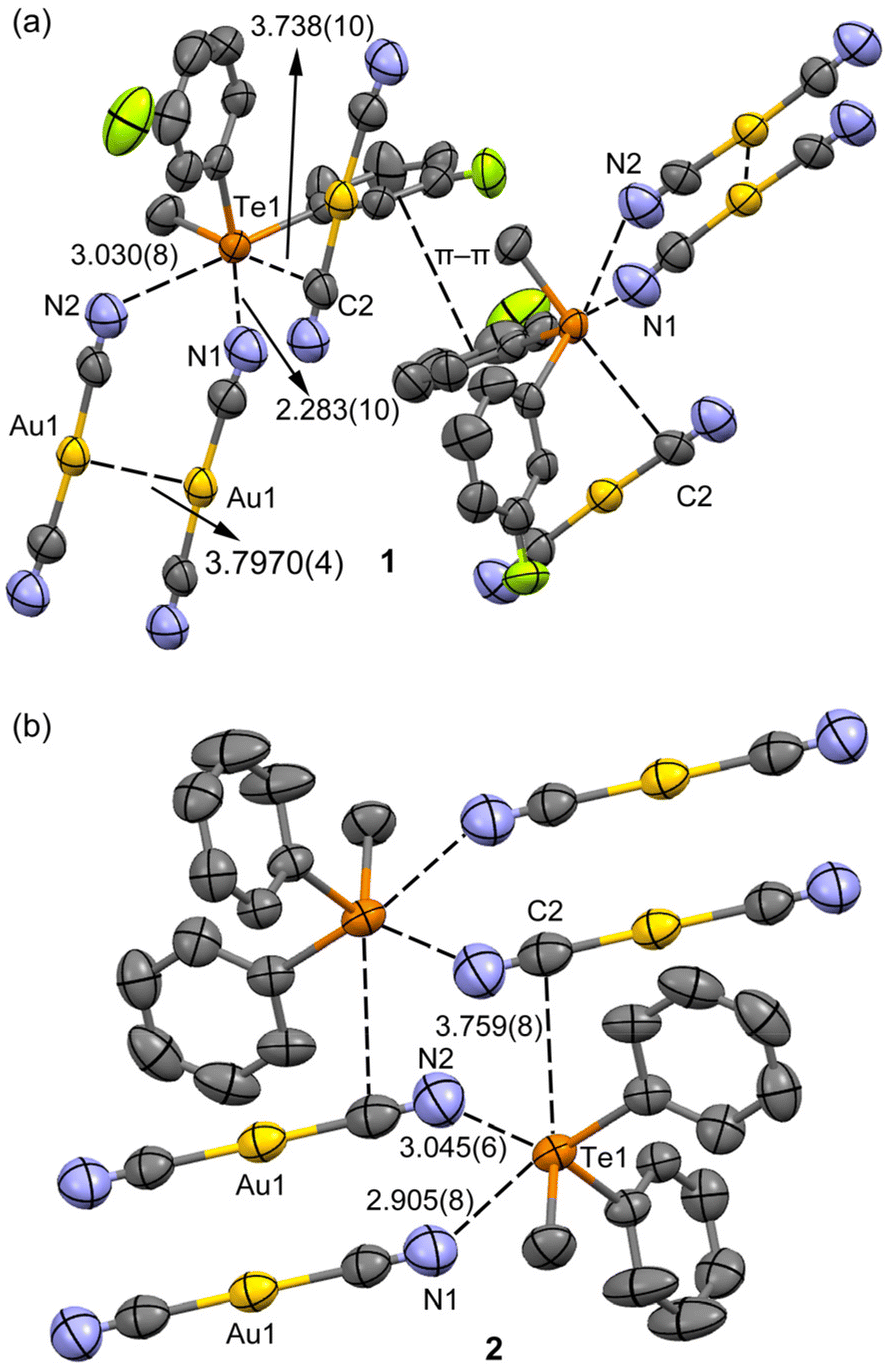 | ||
| Fig. 2 (a) Details of the ChBs and π⋯π stacking interactions in 1 and (b) the dimeric motif in 2. Distances in Å. Geometric features of the π-stacking: Cg⋯Cg distance: 4.026 Å, and angles between ring planes: 9.87°. | ||
| Cmpnd | ChBa | d Ch⋯A (Å) | Angle (°) | δ Ch⋯A | Strengthc |
|---|---|---|---|---|---|
| a i = −1 + x, y, and z; ii = 1 − x, −y, and 1 − z; iii = −x, 2 − y, and −z. b δ Ch⋯A = {[dCh⋯A−(rvdWCh + rvdWA)]/(rvdWCh + rvdWA)}·100. c m: medium; s: strong; w: weak. | |||||
| 1 | C10−Te1⋯N1i | 2.873(10) | 170.8(3) | –20.4 | m–s |
| 1 | C4−Te1⋯N2ii | 3.030(8) | 169.4(3) | –16.1 | w |
| 1 | C3−Te1⋯C2i | 3.738(10) | 164.5(3) | –0.6 | w |
| 2 | C10−Te1⋯N1i | 2.905(8) | 170.9(2) | –19.5 | m–s |
| 2 | C4−Te1⋯N2iii | 3.045(6) | 170.8(2) | –15.7 | w |
| 2 | C3−Te1⋯C2 | 3.759(8) | 164.3(2) | 0.0 | w |
Comparative analysis of the distances reveals a noticeable increase in the strength of chalcogen bonding (ChB) upon the introduction of a fluorine substituent, as presented in Table 1. Notably, no metallophilic interactions are discerned in structures 1 and 2. However, a robust argentophilic interaction is prominent in HUHBUH (with distances of d(Au1⋯Au1) = 3.7970(4) Å in 1, d(Au1⋯Au1) = 3.9272(4) Å in 2, and d(Ag1⋯Ag2) = 3.171(2) Å in HUHBUH). This observation suggests that while ChB is consistently present, it exhibits greater directionality within dicyanoaurate compared to dicyanoargentate. In the latter case, the transition towards argentophilicity is more feasible, albeit typically less energetically favourable than in the case of gold. This underscores the importance of interaction directionality and appropriate counterion geometry in establishing robust metallophilic interactions.55
The δCh⋯A values for the Te⋯N and Te⋯C chalcogen bonding interactions in compounds 1 and 2 were derived using the equation proposed by Aragoni et al.56 and are given in Table 1. These values shed light on the relative shortening of the ChB distances compared to the sum of the involved vdW radii and also offer a reference against average values obtained from the CSD. A ChB strength scale, based on these δCh⋯A values, has been suggested in ref. 56. Analysing the δCh⋯A values from Table 1 and referencing the literature scale,56 we deduce that both compounds presented feature one medium-strong ChB (Te1⋯N1 contact) and two weaker interactions (Te1⋯N2 and Te1⋯C2).
Upon examining the arrangement of aromatic rings, an intriguing observation emerged: the fluoro derivative displayed a shifted π⋯π stacking interaction (Fig. 2a) absent in the unsubstituted salt. This particular stacking interaction could be attributed to dipolar forces stemming from the asymmetrically charged phenyl groups of the telluronium cation. Furthermore, it is noteworthy that in both cases, the dicyanoaurate did not participate in the classical Au⋯π interactions. This characteristic is intriguing, considering that the coulombic component aligns with certain previously studied systems.57 These findings emphasize the pivotal role of ChBs in shaping the X-ray packing of both structures, complemented by directional anion⋯π interactions explored in the subsequent sections.
Table 2 shows a compilation of the geometric characteristics of the ChBs in other asymmetric telluronium salts sourced from the Cambridge Structural Database (CSD). Broadly speaking, these distances align well with those in Table 1 for compounds 1 and 2 discussed in this study. Additionally, the Te⋯μ-CN distances in compounds 1 and 2 show strong consistency with the Te⋯π(C6H5) distances in the CSD refcode LOXPOD, where the ChBs form between the Te atom and the π-system of the aromatic rings.
| CSD reference | Anion | Lengths (Å) |
|---|---|---|
| GUNVIX | BF4− | 3.138–3.287 |
| IXOMUG | BF4− | 2.871–3.263 |
| JOMQIL | Cl− | 3.237–3.317 |
| LOXPOD | B(C6H5)4− | 3.575–3.750 |
| OHIRON | RSO3− | 2.840–3.013 |
| POXGAK | Cl− | 3.151 |
| QOBXEK | Br− | 3.633 |
| REQNAY/REQNOM | CF3SO3− | 2.895–3.101 |
| REQPOO/REQPUU | CF3SO3− | 2.872–3.029 |
| TAJJAP | ClO4− | 3.331–3.558 |
| TIZVAZ | RSO3− | 2.877–3.051 |
| XUPWUB | p-MePhSO3− | 2.815–2.945 |
| YODVUK/YODWEB | I− | 3.347–3.655 |
| YODWAR/YODWIZ | I−/I3− | 3.502–3.991 |
Spectroscopic characterization
Infrared and Raman spectra of 1, 2 and their tetrafluoroborate salt precursors ([(3-F-Ph)(Me)(Ph)Te](BF4) (3a) and [(Me)(Ph)2Te](BF4) (3b)) were recorded on solid samples and the vibrational frequencies and assignment are listed in Table S7, ESI.†The infrared spectra of crystalline [(Ph)2(Me)Te]I have been reported,58 and are substantially similar to those of 3b. The vibrational modes of Te–C have been assigned by comparison with the vibrational spectra of Ph2TeAlg2.59 The vibrational mode of 3a has been assigned by comparison with that of 3b and the vibrational frequencies of fluorobenzene.60 The vibrational frequencies of telluronium cations 3a and 3b seem quite insensitive to the change of BF4− with [Au(CN)2]−. A similar behavior is found in [Au(CN)2]−, which has vibrational frequencies very similar to those of the crystalline K[Au(CN)2] salt.61 Only one vibrational mode seems sensible to the intermolecular interaction between Te and the cyanide group: the stretching of the Te–CH3 bond that undergoes a lowering of nearly 5 cm−1 by moving from BF4− to the [Au(CN)2]− anion, which suggests a lowering of the Te–C bond order in consequence of the interaction with a nitrogen lone pair donor.
DFT calculations
To begin, the computation of molecular electrostatic potential (MEP) surfaces for both salts was undertaken to investigate the presence and strength of σ-holes centred on the Te-atoms. To model the salts, the dicyanidoaurate anion closest to the cation, yet not interacting with the σ-holes, was employed. This choice ensured the use of neutral systems, facilitating an accurate analysis of the relative σ-hole intensities. The MEP surfaces are illustrated in Fig. 3 (left), revealing that the MEP minima for both compounds are situated at the N-atom of the cyanido ligand (−63.4 kcal mol−1 and −64.0 kcal mol−1 for 1 and 2, respectively).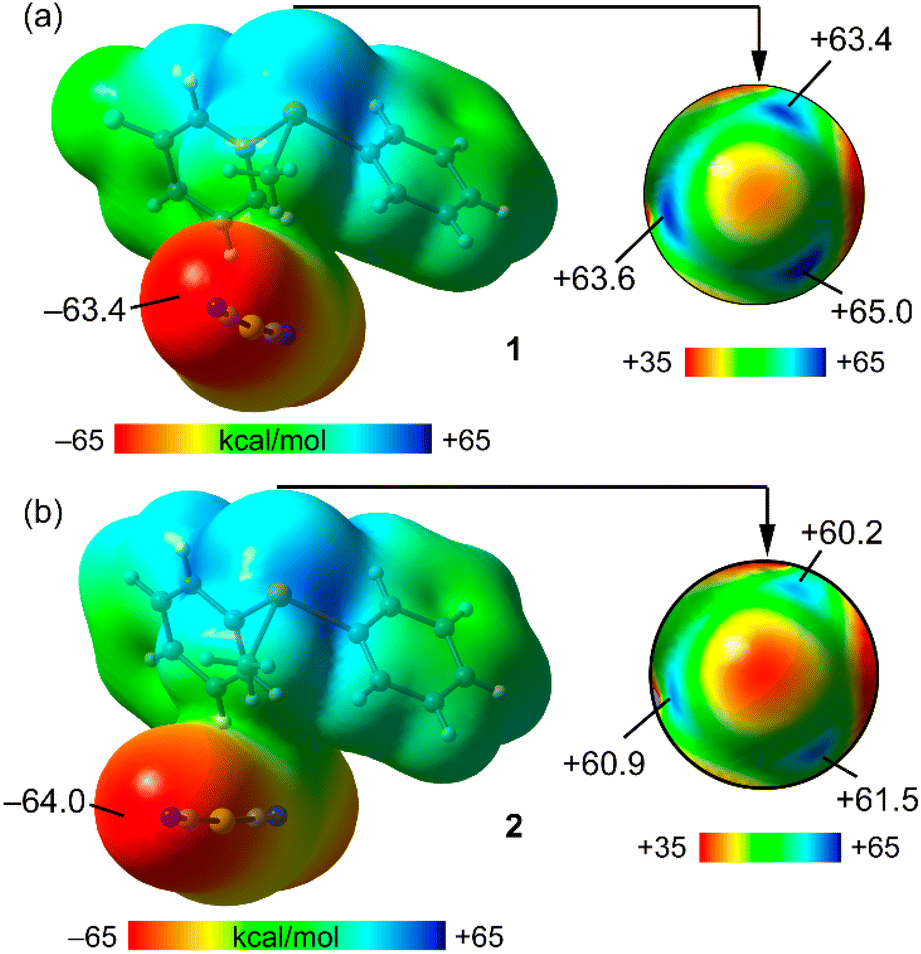 | ||
| Fig. 3 MEP surfaces (isovalue, 0.001 a.u.) of compounds 1 (a) and 2 (b). Values at selected points are indicated in kcal mol−1. | ||
For a detailed representation of the σ-holes and the electron density anisotropy at the Te-atom, a distinct scale was applied to portray a section of the MEP surface encompassing the Te-atom in Fig. 3 (right). Consequently, three distinct σ-holes emerged, positioned on the extension of the three Te–C covalent bonds. In compound 1, the MEP maximum was positioned at the σ-hole corresponding to the 3-fluorophenyl ring (+65.0 kcal mol−1), while the other two σ-holes exhibited closely similar and slightly lower MEP values (+63.4 kcal mol−1 and +63.6 kcal mol−1). This configuration establishes all three σ-holes as favourable sites for interactions with counterions.
In compound 2, the MEP values at the three σ-holes were comparable, ranging from +60.2 kcal mol−1 to +61.5 kcal mol−1, modestly lower when contrasted with those of compound 1.
As elucidated in the preceding sections, each telluronium cation forms three charge-assisted chalcogen bonds (CAChBs) with dicyanidoaurate anions. The energetic attributes of these CAChBs were individually investigated using DFT calculations and QTAIM/NCIPlot analysis. For energy calculations, the entire salt was treated as a unified entity, and its interaction with an additional anion was computed. For example, in compound 1, the association [(3-F-Ph)(Me)(Ph)Te][Au(CN)2] + [Au(CN)2]− was evaluated to yield [(Me)(Ph)2Te][Au(CN)2]⋯[Au(CN)2]− for each ChB (referred to as dimers A–C in Fig. 4, as discussed below).
Fig. 4 showcases comprehensive QTAIM/NCIPlot analyses for the three ChB assemblies within compound 1. Employing both methods in tandem allows for a spatial representation of interactions, discerning their attractive or repulsive nature. In our NCIPlot illustrations, blue signifies potent and attractive interactions, while green denotes weaker and attractive interactions. As depicted in Fig. 4, within all the three studied dimers (A–C), the ChB interaction is defined by a bond critical point (BCP, marked by a red sphere) and a bond path (highlighted by an orange line) linking the N-atom of the anion to the Te-atom of the cation, thus affirming the existence of these three ChBs.
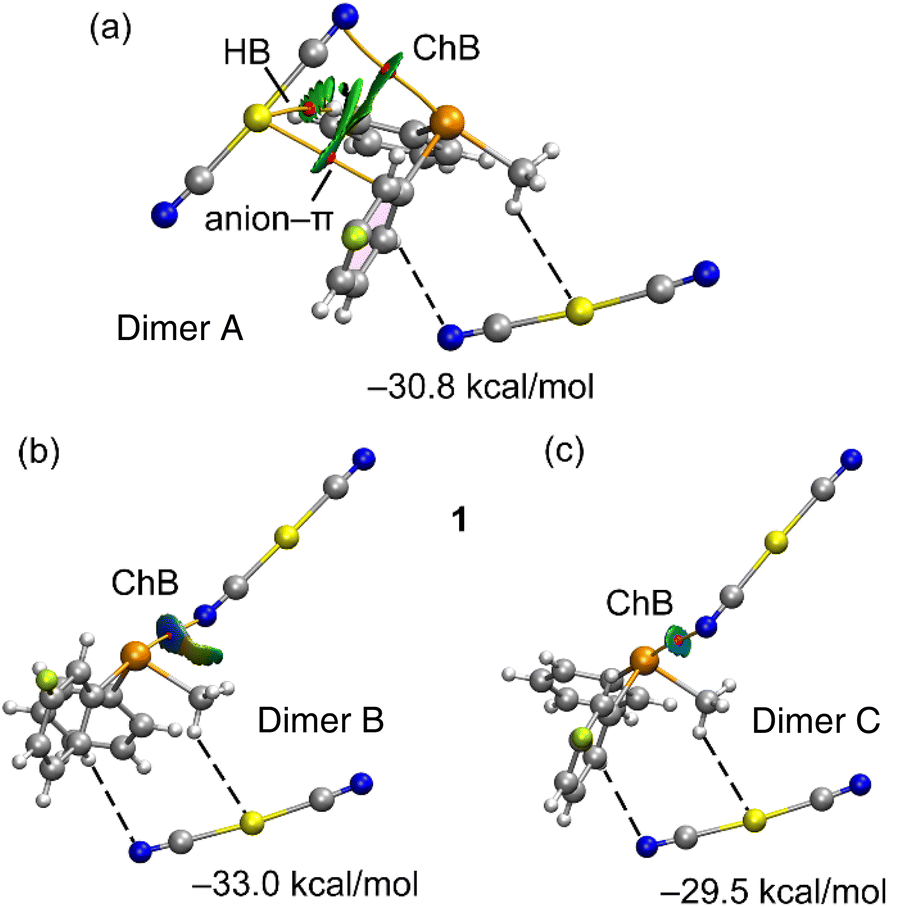 | ||
| Fig. 4 QTAIM/NCIPlot analysis of ChB dimer A (a) dimer B (b) and dimer C (c) of 1. Only intermolecular interactions are represented. See theoretical methods for the NCIPlot settings. The dimerization energies are indicated and have been computed considering the telluronium and one [Au(CN)2]− anion (the one connected with dashed lines) as a monomer. | ||
Furthermore, isosurfaces of the reduced density gradient (RDG) manifest coinciding with the BCP positions upon complexation. Notably, the coloration of the RDG isosurfaces differs: green (indicative of weak interactions) for dimer A, dark blue (signifying strong interactions) for dimer B, and a moderately strong bluish hue for dimer C, consistent with the experimental distances. In congruence with the intensity of the σ-hole opposite the 3-fluorophenyl ring, the binding energies incorporated in Fig. 4 reveal that dimer B possesses the most substantial dimerization energy. This outcome aligns with the RDG isosurface colour and the intensity of the aforementioned σ-hole. The other two dimers display closely similar interaction energies due to additional contacts compensating for the greater ChB strength in dimer C. Particularly noteworthy are a C–H⋯Au hydrogen bond and an anion–π interaction, each characterized by BCPs and green RDG isosurfaces.
The notable dimerization energies highlighted in Fig. 4 further underscore the pivotal role of the three ChBs in guiding the solid-state structure of compound 1.
A parallel analysis has been conducted for compound 2, which is elucidated comprehensively in Fig. 5. The arrangement of bond critical points (BCPs), bond paths, and reduced density gradient (RDG) isosurfaces closely mirrors the description provided earlier for compound 1. Remarkably, the dimerization energies are less negative (indicative of weaker interactions) in accordance with the MEP analysis revealing reduced MEP values at the σ-holes within compound 2. A prominent energy difference of 2.5 kcal mol−1 between the dimers underscores the influence of fluorine substitution on the strength of ChB.
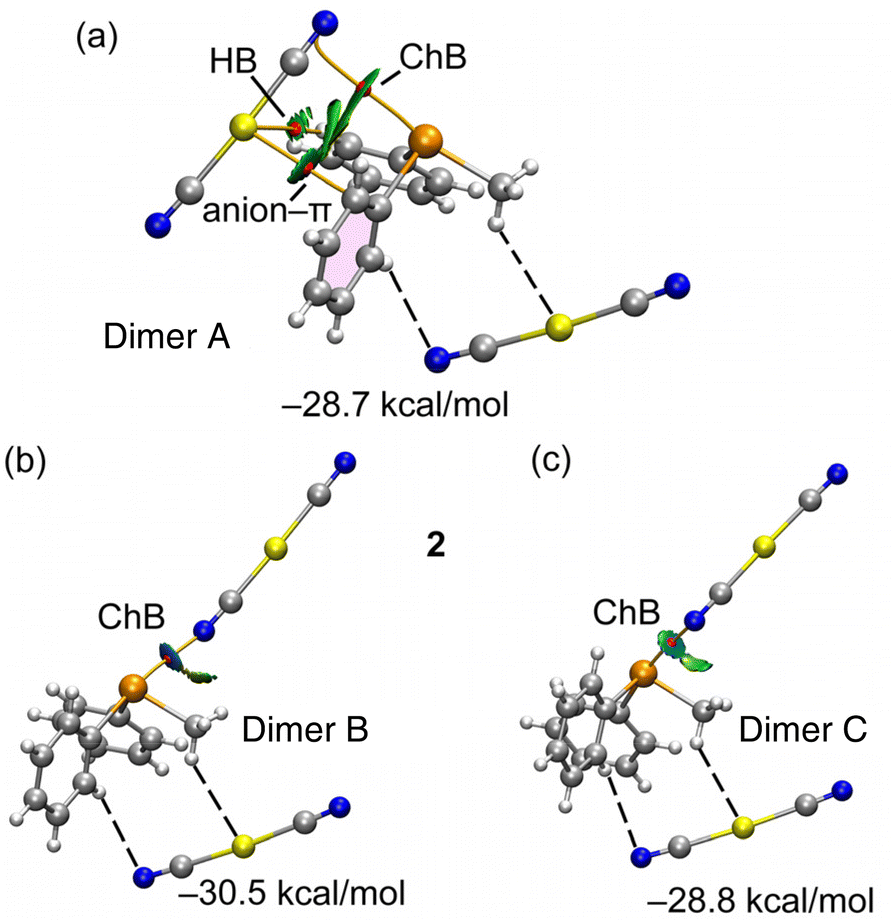 | ||
| Fig. 5 QTAIM/NCIPlot analysis of ChB dimer A (a) dimer B (b) and dimer C (c) of 2. Only intermolecular interactions are represented. See theoretical methods for the NCIPlot settings. The dimerization energies are indicated and have been computed considering the telluronium and one [Au(CN)2]− anion (the one connected with dashed lines) as a monomer. | ||
At this juncture, it is insightful to compare the strength of the CAChBs discussed in this study with those documented in the existing literature for neutral ChB donors.62 While the ChB distances for neutral Te compounds might be comparable or even shorter,62c the interaction energies for the CAChBs examined in this study are notably greater due to the supplementary ion-pair effect.
To discern charge transfer effects within the ChB interactions observed in compounds 1 and 2, NBO analysis was employed. This approach is adept at deciphering donor–acceptor interactions between orbitals and revealing their stabilization energies through second-order perturbation analysis. The summarized outcomes are presented in Table 3. A close review of the results reveals the existence of two distinct orbital donor–acceptor interactions. Both these contributions are shared across compounds 1 and 2, as well as within the three chalcogen bonding dimers. The primary contribution involves electron donation from a lone pair (LP) orbital of nitrogen, belonging to the Au-coordinated cyanido ligand, to the antibonding σ*(Te–C) orbital, a characteristic trait of σ-hole interactions. A secondary yet less significant contribution emerges from a π-orbital of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond to the antibonding σ*(Te–C) orbital.
N bond to the antibonding σ*(Te–C) orbital.
| Compound | Dimer | NBOs | E (2) |
|---|---|---|---|
| 1 | A | LP(N)→σ*(Te–C) | 0.08 |
| π(CN) → σ*(Te–C) | 0.74 | ||
| 1 | B | LP(N) → σ*(Te–C) | 7.76 |
| π(CN) → σ*(Te–C) | 0.76 | ||
| 1 | C | LP(N) → σ*(Te–C) | 3.16 |
| π(CN) → σ*(Te–C) | 1.20 | ||
| 2 | A | LP(N) → σ*(Te–C) | 0.05 |
| π(CN) → σ*(Te–C) | 0.61 | ||
| 2 | B | LP(N) → σ*(Te–C) | 6.82 |
| π(CN) → σ*(Te–C) | 0.58 | ||
| 2 | C | LP(N) → σ*(Te–C) | 2.73 |
| π(CN) → σ*(Te–C) | 1.45 |
By evaluating various dimers in this study and considering the second-order orbital energies highlighted in Table 3, it becomes evident that dimer B showcases the most substantial LP(N) → σ*(Te–C) contribution (7.76 kcal mol−1 in 1 and 6.82 kcal mol−1 in 2). This alignment effectively corresponds to the highest binding energy observed for dimer B and the shortest Te⋯N distances encountered. In addition, it agrees well with the δCh⋯A values mentioned before (see Table 1). The σ-hole character of the interaction is demonstrated through the analysis of the NBO plots presented in Fig. 6 for dimer B of both compounds. In these plots, the SP-hybridized lone pair originating from one N-atom of the dicyanidoaurate anion aligns with the σ*(Te–C) orbital of the telluronium cation in both compounds.
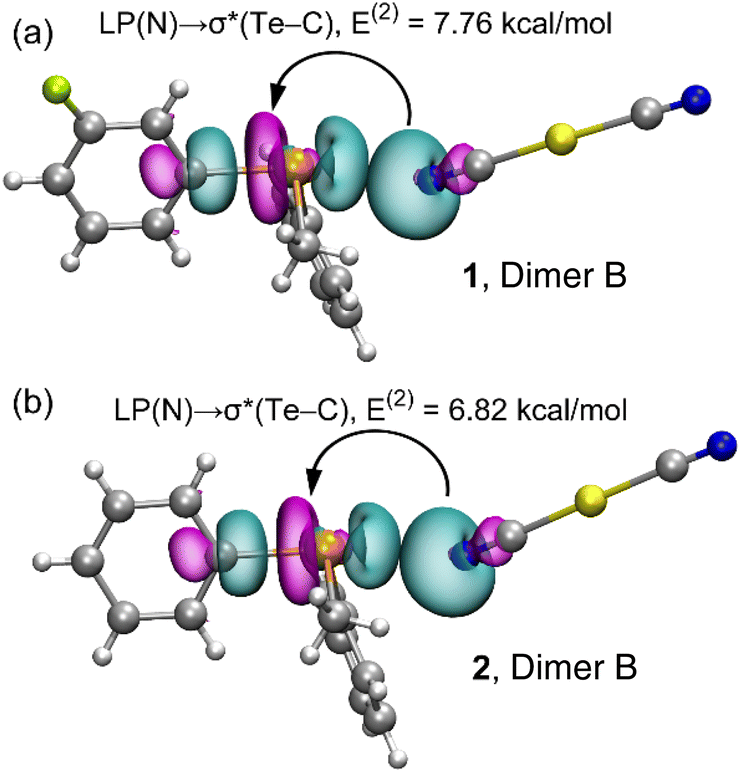 | ||
| Fig. 6 Plots of the NBOs involved in the electron charge transfer and the associated stabilization energy in the dimer B of compounds 1 (a) and 2 (b). | ||
To complement the insights gleaned from the MEP, NBO, and QTAIM/NCIPlot analyses, an energy decomposition analysis (EDA) was undertaken for the three dimers within compound 1, taken as an illustrative example. The summarized outcomes are presented in Fig. 7, revealing that in all the dimers, the electrostatic contribution takes precedence (indicated by fuchsia bars), followed by the orbital contribution.
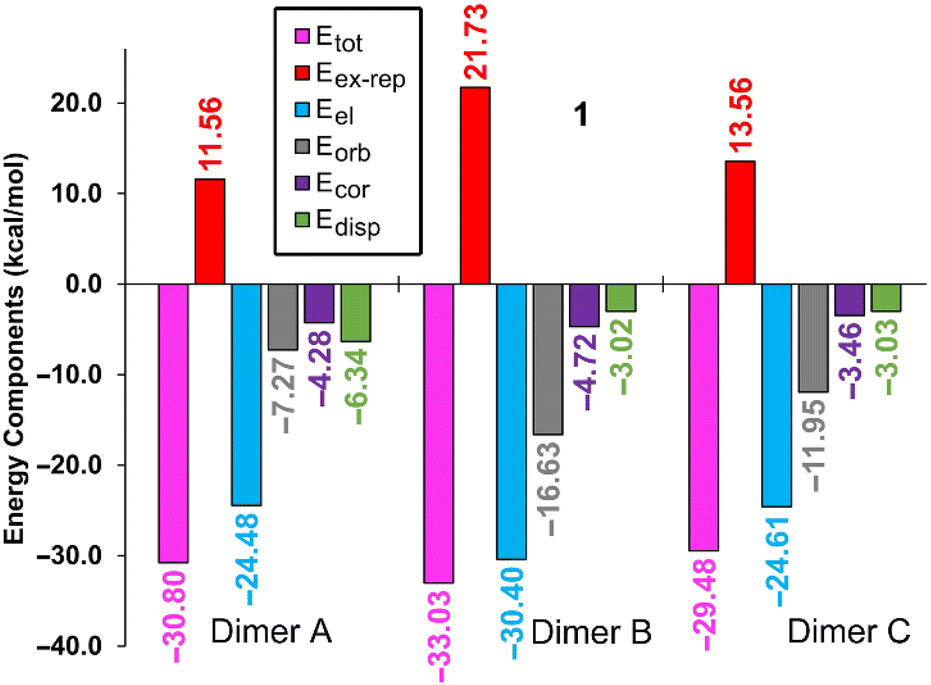 | ||
| Fig. 7 Bar plots of the total (Etot), electrostatic (Eel), exchange repulsion (Eex-rep), orbital (Eorb), correlation (Ecor) and dispersion (Edisp) components of the binding energies of the chalcogen bonding assemblies of compound 1. All values are in kcal mol−1. | ||
In both compounds (see Table 3), dimer C displays notable contributions from both LP(N) → σ*(Te–C) interactions (3.16 kcal mol−1 in 1 and 2.73 kcal mol−1 in 2) and π(CN) → σ*(Te–C) interactions (1.20 kcal mol−1 in 1 and 1.45 kcal mol−1 in 2). Notably, dimer A in both compounds exhibits a minimal orbital contribution, likely attributed to the elongated Te⋯N distances and the resulting diminished orbital overlap.
For the dimer exhibiting the largest contribution within both complexes (dimer B), the associated NBOs are plotted in Fig. 6. This contribution in the EDA analysis is represented by the grey bar. Interestingly, a discernible trend emerges wherein the orbital contribution is most prominent in dimer B, followed by dimer C, and is least evident in dimer A. This pattern harmonizes effectively with the findings from the NBO analysis (Table 3).
Furthermore, the EDA calculations reveal that the underlying nature of the CAChBs is predominantly governed by electrostatics and charge transfer. Within dimers B and C, both the correlation term (Ecor) and the dispersion term (Edisp) hold similar values. In contrast, dimer A displays a larger dispersion term (Edisp), aligning with the establishment of an anion–π interaction where dispersion effects play a pivotal role.63
Conclusions
Two new telluronium salts featuring linear dicyanidoaurate anion [Au(CN)2]− counterions have been successfully synthesized and subjected to detailed structural characterization. Given the ionic nature of these structures, the primary driving force shaping the resultant X-ray structures is the coulombic attraction. However, this work underscores the considerable influence of directional chalcogen bonds on the final assemblies.The molecular electrostatic potential (MEP) calculations have unequivocally identified three σ-holes of comparable intensity situated at the tellurium atom. Consequently, our scrutiny of the supramolecular assemblies has focused on the formation of three distinct and directional charge-assisted chalcogen bond (CAChB) interactions facilitated by the tellurium atom. These interactions bear significant energetic implications, with compound 1 manifesting slightly heightened favourability due to the presence of the electron-withdrawing fluorine atom.
Furthermore, a comprehensive investigation of the CAChB interactions has been conducted through the combined utilization of the QTAIM and NCIPlot analyses. The NBO analysis has unveiled pivotal donor–acceptor interactions, prominently featuring electron donation from a lone pair (LP) located at the N-atom of the dicyanidoaurate anion to the vacant σ*(Te–C) antibonding orbital. The aggregate stabilization energy attributable to these charge-transfer phenomena varies markedly across different CAChB interactions, intimately tied to the Te⋯N distance.
Employing energy decomposition analysis (EDA), we have substantiated that electrostatic and charge transfer effects are preeminent within the Te⋯N contacts. The findings detailed in this study hold potential to captivate the interests of the inorganic crystal engineering and supramolecular chemistry communities, offering insights that further our comprehension of chalcogen bonding interactions.
Author contributions
S. G., A. G., E. D., R. M. G., E. P., and A. F.: investigation and data curation, A. G., E. P. and A. F.: project administration, research direction, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the “Ministerio de Ciencia, Investigacion y Universidades/Agencia Estatal de Investigación” (MICIU/AEI/10.13039/501100011033) of Spain (project PID2020-115637GB-I00, FEDER funds). The authors thank the CTI (UIB) for computational facilities. A. F. is grateful to the Alexander von Humboldt Foundation for the J. C. Mutis Award.The micro-Raman measurements were obtained using the equipment acquired by the Interdepartmental Center “G. Scansetti” for Studies on Asbestos and Other Toxic Particulates with a grant from Compagnia di San Paolo, Torino, Italy.
The authors acknowledge support from Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001).
References
- Z.-L. Zhou, Y.-Z. Huang, Y. Tang, Z.-H. Chen, L.-P. Shi, X.-L. Jin and Q.-C. Yang, Organometallics, 1994, 13, 1575 CrossRef CAS.
- K. J. Irgolic, The Organic Chemistry of Tellurium, Gordon and Breach, New York, 1974, p. 199 Search PubMed.
- F. Einstein, J. Trotter and C. Williston, J. Chem. Soc. A, 1967, 2018 RSC.
- J.-S. Lee, D. D. Titus and R. F. Ziolo, J. Chem. Soc., Chem. Commun., 1976, 501 RSC.
- R. F. Ziolo and K. Pritchett, J. Organomet. Chem., 1976, 116, 211 CrossRef CAS.
- D. D. Titus, J.-S. Lee and R. F. Ziolo, J. Organomet. Chem., 1976, 120, 381 CrossRef CAS.
- (a) J.-S. Lee and D. D. Titus, J. Cryst. Mol. Struct., 1976, 6, 279 CrossRef CAS; (b) J.-S. Lee, D. D. Titus and R. F. Ziolo, Inorg. Chem., 1977, 16, 2487 CrossRef CAS.
- E. Denes, A. Beleagă and A. Silvestru, Inorg. Chim. Acta, 2020, 511, 119841 CrossRef CAS.
- P. A. Ash, J.-S. Lee, D. D. Titus, K. Bowman and R. F. Ziolo, J. Organomet. Chem., 1977, 135, 91 CrossRef CAS.
- R. V. Mitcham, B. Lee, K. B. Mertes and R. F. Ziolo, Inorg. Chem., 1979, 18, 3498 CrossRef CAS.
- R. F. Ziolo and M. Extine, Inorg. Chem., 1980, 19, 2964 CAS.
- M. N. Ponnuswamy and J. Trotter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1671 CrossRef.
- M. J. Collins, J. A. Ripmeester and J. F. Sawyer, J. Am. Chem. Soc., 1988, 110, 8583 CrossRef CAS.
- J. Jeske, W.-W. du Mont and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1996, 35, 2653 CrossRef CAS.
- (a) N. Sudha and H. B. Singh, Coord. Chem. Rev., 1994, 135, 469–515 CrossRef; (b) I. Haiduc, R. B. King and M. G. Newton, Chem. Rev., 1994, 94, 301–326 CAS; (c) E. R. T. Tiekink and J. Zuckerman-Schpector, Coord. Chem. Rev., 2010, 254, 46–76 CAS; (d) T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725–1739 CAS.
- R. F. Ziolo and J. M. Troup, Inorg. Chem., 1979, 18, 2271 CrossRef CAS.
- B. H. Christian, M. J. Collins, R. J. Gillespie and J. F. Sawyer, Inorg. Chem., 1986, 25, 777 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed.
- S. Scheiner, Acc. Chem. Res., 2013, 46, 280–288 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed.
- A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed.
- S. Grabowski, J. Comput. Chem., 2018, 39, 472–480 CrossRef CAS PubMed.
- (a) P. Politzer and J. S. Murray, Crystals, 2017, 7, 212–226 CrossRef; (b) I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS; (c) J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS PubMed.
- B. Mallada, A. Gallardo, M. Lamanec, B. de la Torre, V. Špirko, P. Hobza and P. Jelinek, Science, 2021, 374, 863–867 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697–2702 CrossRef CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- (a) J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CrossRef CAS; (b) P. Scilabra, G. Terraneo and G. Resnati, J. Fluorine Chem., 2017, 203, 62–74 CrossRef CAS.
- (a) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed; (b) S. Scheiner, Chem. – Eur. J., 2016, 22, 18850–18858 CrossRef CAS PubMed.
- E. Alikhani, F. Fuster, B. Madebene and S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 2430–2442 RSC.
- W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
- M. Macchione, A. Goujon, K. Strakova, H. V. Humeniuk, G. Licari, E. Tajkhorshid, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2019, 58, 15752–15756 CrossRef CAS PubMed.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- B. Galmés, A. Juan-Bals, A. Frontera and G. Resnati, Chem. – Eur. J., 2020, 26, 4599–4606 CrossRef PubMed.
- U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2014, 118, 3183–3192 CrossRef CAS PubMed.
- S. Scheiner, J. Phys. Chem. A, 2015, 119, 9189–9199 CrossRef CAS PubMed.
- R. Oilunkaniemi, J. Pietikainen, R. S. Laitinen and M. Ahlgren, J. Organomet. Chem., 2001, 640, 50 CrossRef CAS.
- K. Swaminathan, P. J. Carroll, Kochurani, H. B. Singh and H. D. Bhargava, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 1243 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Hacer, H. Horn and C. Kömel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, USA, 2018 Search PubMed.
- T. M. Klapotke, B. Krumm, P. Mayer, H. Piotrowski, I. Schwab and M. Vogt, Eur. J. Inorg. Chem., 2002, 2701–2709 CrossRef CAS.
- (a) E. Priola, G. Volpi, R. Rabezzana, E. Borfecchia, C. Garino, P. Benzi, A. Martini, L. Operti and E. Diana, Inorg. Chem., 2020, 59, 203–213 CrossRef CAS PubMed; (b) A. Giordana, R. M. Gomila, R. Rabezzana, E. Laurenti, E. Priola, B. Eftekhar-Sis, G. Mahmoudi and A. Frontera, ChemPlusChem, 2023, 88, e202300052 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, V. Lippolis, A. Pintus, Y. Torubaev and E. Podda, Molecules, 2023, 28, 3133 CrossRef CAS PubMed.
- (a) E. Priola, A. Giordana, P. P. Mazzeo, G. Mahmoudi, R. M. Gomila, F. I. Zubkov, K. M. Pokazeev, K. S. Valchuk, A. Bacchi, E. Zangrando and A. Frontera, Dalton Trans., 2021, 50, 16954–16960 RSC; (b) E. Priola, N. Curetti, D. Marabello, J. Andreo, A. Giordana, L. Andreo, P. Benna, P. T. C. Freire, P. Benzi, L. Operti and E. Diana, CrystEngComm, 2022, 24, 2336–2348 RSC.
- N. S. Dance, W. R. McWhinnie, J. Mallaki and Z. Monsef-Mirzai, J. Organomet. Chem., 1980, 198, 131–143 CrossRef CAS.
- N. S. Dance and W. R. McWhinnie, J. Chem. Soc., Dalton Trans., 1975, 43–45 RSC.
- D. H. Whiffen, J. Chem. Soc., 1956, 1350–1356 RSC.
- B. M. Chadwick and S. G. Frankiss, J. Mol. Struct., 1976, 31, 1–9 CrossRef CAS.
- (a) S. Kolb, G. A. Oliver and D. B. Werz, Chalcogen bonding in supramolecular structures, anion recognition, and catalysis, in Comprehensive Inorganic Chemistry III, ed. R. S. Laitinen, Elsevier, Oxford, 2023, vol. 1, pp. 602–651 Search PubMed; (b) S. Kolb, G. A. Oliver and D. B. Werz, Angew. Chem., Int. Ed., 2020, 59, 22306–22310 CrossRef CAS PubMed; (c) R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholde, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed; T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725–1739 Search PubMed; A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 Search PubMed.
- (a) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed; (b) D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3539–3542 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic tables, IR and NMR spectra. CCDC 2284002 and 2284003. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02787b |
| This journal is © The Royal Society of Chemistry 2023 |