
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand and metal-centred reactivity in 2,6-bis(imino)-1,4-dihydropyridinate Zn(II) alkyls: the dual behaviour of an intriguing type of complex†
J. M.
Delgado-Collado
,
M.
Gallardo-Villagrán
,
E.
Álvarez
 ,
J.
Cámpora
* and
A.
Rodríguez-Delgado
*
,
J.
Cámpora
* and
A.
Rodríguez-Delgado
*
Instituto de Investigaciones Químicas, CSIC-Universidad de Sevilla, c/Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: campora@iiq.csic.es; arodríguez@us.es
First published on 3rd July 2023
Abstract
Dihydropyridines, either free or metal-coordinated, are organic hydride transfer reductants that perform on the same premises as the natural redox cofactor NAD(P)+/NAD(P)H. 1-Bn and 1-Me are alkylzinc complexes containing dihydropyridinate-based pincer ligands that have been synthesized through different routes involving the addition of ZnR2 (R = Bn, Me) to the 2,6-bis(imino)-pyridine and 2,6-bis(imino)-4-Bn-dihydropyridine (iPrBIP and 4-BniPrBIPH2) ligands, respectively. The alkyls complexes 1-R react with fluorinated alcohols RFOH (RF = C6F5 or t-C4F9) yielding isolable fluoroalkoxides 2-F5 and 2-F9, in which the reactive 1,4-dihydropyridinate ligand remains unchanged. The crystal structure of 2-F5 shows the shortest Zn⋯F–C interaction reported so far, involving one of the o-F atoms of the C6F5 group. However, the mechanism of the alcoholysis reactions is not straightforward, as NMR monitoring revealed that acidic RFOH first protonates the dihydropyridine nitrogen, releasing the dihydropyridine base 4-BniPrBIPH2 and a highly reactive Zn(R)(ORF) species that re-captures the dihydropyridine in a subsequent step, eliminating the corresponding alkane (R–H). Depending on the mixing conditions, the pincer dihydropyridinate ligand may undergo aromatization to produce the new Zn(II) dialkoxides 3-F5 and 3-F9 stabilised by a neutral iPrBIP ligand [(4R-iPrBIP)Zn(ORF)2]. These protonation and hydride transfer reactions illustrate the dual reactivity of the pincer 1,4-dihydropyridinate zinc entity.
Introduction
2,6-Bis(imino)pyridines (BIP) are a versatile family of pincer-type ligands with many applications in coordination chemistry and catalysis.1 The extended conjugated π-orbitals of these molecules give rise to a characteristic reactivity that the coordinate metal fragment can modulate.2 The ability of BIP ligands to reversibly accept electrons (up to four)3 results in their chemical “non-innocence”,4 a potent feature that enhances the redox properties of abundant “base” metals, allowing these to emulate such noble elements in their capacity to catalyse many types of reactions.5 Furthermore, the BIP ligands may also experience valuable transformations that lead to unusual ligand architectures.6 Among these, the de-aromatization of the central pyridine ring by alkyl transfer from the metal is particularly relevant. This type of transformation was accidentally discovered in the context of olefin polymerisation catalysis, in attempts to synthesise well-defined organotransition metal BIP complexes using conventional transmetallation procedures.7 One of the most frequent processes is the alkyl transfer to the remote C4 site in the pyridine ring, which affords 2,6-diimino-4-alkyl-dihydropyridinate(−1) complexes, [(4-R-HBIP)M(R)n], often with complete regioselectivity. These compounds (Scheme 1, inset) are very reactive and undergo further transformations. Characteristic examples of this reactivity for different organometallic units containing first-row transition (Mn(II),8–10 Cr(III)7b), post-transition (Zn(II)11) and main-group metals (Al(III),12 Mg(II)13 and heavier alkali-earth dications14) are displayed in Scheme 1.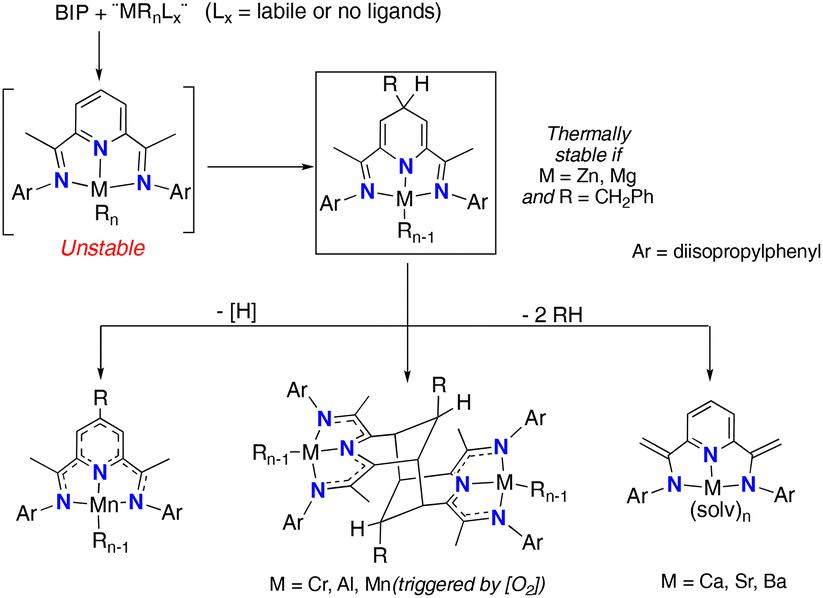 | ||
| Scheme 1 Selective complexation of organometallic fragments, alkyl migration to the pyridine C4 position and further transformations. | ||
Our group has been interested in BIP chemistry since these early findings were first made,8 as a means to develop synthetically useful transformations.8b We have shown that ligand exchange reactions in suitable transition or main-group metal alkyl precursors [MRnLn] (L is a labile coligand) provide a mild and efficient pathway to BIP-based complexes.8 In general, polyalkyl metal complexes [(BIP)MRn] (with n > 1) are unstable.2a,9 However, generating these compounds under controlled conditions allows harnessing their complicated reactivity.
In this context, we have been particularly interested in the chemistry of 2,6-bis(imino)-1,4-dihydropyridinate ligands. We,15 and others,16 have called attention to the analogy between these BIP-based systems and essential reversible biologic redox cofactors (e.g., NAD(P)H). Furthermore, the interaction between metals and organic hydrides is attracting interest as a sustainable approach to multielectron reduction in synthesis and catalysis.17,18 Among the different 2,6-diimino-4-R-pyridinate complexes, the benzyl Zn(II) derivative 1-Bn provides an attractive entry in this chemistry due to its diamagnetism, thermal stability, and easy accessibility from the reaction between ZnBn2 and iPrBIP.15
As shown in Scheme 2, 1-Bn exhibits dual reactivity with Lewis and Brønsted acids. B(C6F5)3 selectively accepts hydride from the C4 site of the dihydropyridine ring. On the contrary, weak protic acids react at the metal site. Alcohols (methanol, for example) cleanly release the free 1,4-dihydropyridine base, which can be recovered in a highly selective manner. This is a synthetically valuable reaction, as 4-R-H2iPrBIP can be transferred as a pincer-like tridentate ligand to other metal fragments.12b,15 However, despite our efforts, we have not been able to detect any intermediate Zn(II) dihydropyridine-alkoxide intermediates in this reaction. Dihydropyridinate Zn(II) alkoxides could have interesting applications, e.g., as biomimetic hydridic reductants, or ring opening polymerisation catalysts.19 Therefore, we decided to replace methanol with the more acidic fluorinated alcohols such as nonafluoro-tert-butanol (CF3)3COH (HF9) and pentafluorophenol C6F5OH (HF5), as we expected the corresponding fluoroalkoxide complexes to be more stable and amenable for isolation. As suspected, reactions with fluoroalcohols have provided relevant insights into the unusual reactivity of the dihydropyridinate-Zn(II) system, and a Zn(II) perfluorophenoxide has been isolated and structurally characterised, revealing a remarkable Zn⋯F interaction. Here, we describe the results of this investigation.
 | ||
| Scheme 2 Synthesis of 1-Bn and reactions with strong Lewis and weak protic acids (B(C6F5)3 and MeOH), respectively. | ||
Results and discussion
The starting point for this investigation was the synthesis of dihydropyridinate-zinc alkoxides by metathetical exchange of the Zn–C bond in 1-Bn with methanol and other alcohols. As mentioned in the Introduction, methanol releases free the 4-Bn-H2iPrBIP base (since then H2BnBIP for simplicity), but no intermediates have been detected. We then turned to the more acidic HF9 or HF5 to detect the target alkoxide species. In addition, we have expanded the scope of our studies to the methyl analogue of 1-Bn, a more reactive methyl complex 1-Me, which was synthesised from H2BnBIP and ZnMe2, as an extension of our general methodology for this type of dihydropyridinates (Scheme 3). | ||
| Scheme 3 Synthesis of 1-Me from ZnMe2. | ||
It is worth mentioning that the ligand transfer shown in Scheme 3 is the only possible route to access methylzinc dihydropyridinates because selective methyl migrations are virtually unknown in the BIP system. Furthermore, unlike other zinc dialkyls, such as Zn(Bn)2 or Zn(allyl)2, ZnMe2 does not react with the BIP ligands.20 The reaction of ZnMe2 with H2BnBIP occurs immediately, even at sub-ambient temperature, cleanly providing the target product with high yield. 1-Me, that has been unambiguously characterized by 1H, 13C NMR and elemental analysis. Like 1-Bn, 1-Me is a dark purple solid, highly soluble in nonpolar solvents and exceedingly sensitive to traces of moisture and air. In solution, both exhibit remarkable thermal stability, with their 1H NMR spectra showing no alteration after heating to 120 °C. Like methanol, an excess of fluoroalcohol (>2 equiv.) immediately cleaves 1-Bn or 1-Me in noncoordinating solvents (e.g., C6D6), releasing the H2BnBIP ligand. After filtration, the 19F NMR spectrum of the supernatant solution shows a number of signals, likely arising from oligomeric zinc fluoroalkoxides.21 However, if equimolar amounts of the corresponding fluoroalcohol are used, new complexes were formed, whose signals are observed in the 1H NMR spectra of the reaction mixtures, along with those of the corresponding R–H (toluene from 1-Bn or methane from 1-Me). For each fluoroalcohol, the NMR spectrum of the main product is the same, independently of the starting material (1-Bn or 1-Me). This suggests that two new fluoroalkoxide derivatives, 2-F9 and 2-F5, respectively, are formed in these reactions. This conclusion is supported by the 19F spectra of the main species, which show characteristic sets of signals (a singlet for 2-F9 at −75.5 ppm, and a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 set of multiplets for 2-F5 at −167.9; −168.6 and −180.8 ppm) that differ significantly from those of the fluoroalcohols HF9 or HF5, respectively. Reddish purple samples of 2-F9 and 2-F5 were isolated from the reaction mixtures, pure enough to perform their spectroscopic characterisation but crystallisation of pure samples was difficult. Compound 2-F9 proved to be more challenging due to its high solubility in hydrocarbon solvents, and quality single crystals could not be grown. However, this was finally achieved for 2-F5, whose X-ray diffraction structure is shown in Fig. 1. The structure of this compound is unusual in several aspects and is discussed later in detail.
:2:1 set of multiplets for 2-F5 at −167.9; −168.6 and −180.8 ppm) that differ significantly from those of the fluoroalcohols HF9 or HF5, respectively. Reddish purple samples of 2-F9 and 2-F5 were isolated from the reaction mixtures, pure enough to perform their spectroscopic characterisation but crystallisation of pure samples was difficult. Compound 2-F9 proved to be more challenging due to its high solubility in hydrocarbon solvents, and quality single crystals could not be grown. However, this was finally achieved for 2-F5, whose X-ray diffraction structure is shown in Fig. 1. The structure of this compound is unusual in several aspects and is discussed later in detail.
 | ||
| Fig. 1 ORTEP representation of the structure of compound 2-F5. Selected bonds (Å) and angles (°): Zn(1)–O(1), 1.989(5); Zn(1)–N(1), 1.904(5); Zn(1)–N(2), 2.176(5); Zn(1)–N(3), 2.178(5); C(1)–C(2), 1.370(9); N(2)–C(6), 1.300(8); Zn(1)–F(1), 2.406(5) F(1)–C(42), 1.349(8); F(5)–(C46), 1.348(9); O(1)–Zn(1)–N(1), 135.2(2); O(1)–Zn(1)–N(2), 105.9(2); O(1)–Zn(1)–N(3), 106.1(2); N(2)–Zn(1)–N(3), 147.98(19); N(1)–Zn(1)–N(2), 79.3(2); C(1)–N(1)–C(5), 118.8(5); N(1)–Zn(1)–F(1), 148.1(2); Zn(1)–F(1)–C(42), 104.6(4). | ||
The difficulties encountered for the isolation of pure samples of 2-F9 and 2-F5 are not exclusively due to their high solubilities. Although 2-F9 and 2-F5 are by large the main species generated in these alcoholyses, we never observed fully selective reactions between the starting materials and the fluoroalcohols. Complex 1-Me reacts faster than 1-Bn, and the outcomes of its reactions are in general cleaner than those performed with 1-Bn, but both reactions are in general very sensitive to the dosage of the fluoroalcohols. Even a slight excess of the latter leads to complex reaction mixtures, containing a number of side products, whereas deficiency of the alcohol in the reaction media obviously causes some starting material to remain, which is difficult to eliminate. We finally concluded that the best results are obtained when 1-Me is reacted with 0.95 equivalents of the corresponding fluoroalcohol. When the alcoholyses were monitored by 1H and 19F NMR (0.45 M in C6D6), we were surprised to observe that the initial action of the fluoroalcohols is not to cleave the Zn–C, but the Zn–N bond, releasing H2BnBIP, which is unambiguously identified in the 1H NMR spectra of the mixtures within minutes after reacting (Scheme 4). The initial protonation step is faster with pentafluorophenol than with nonafluoro-t-butanol. When C6D6 solutions of 1-Me and C6F5OH are mixed in 1:0.95 molar ratio (for a total [Zn] = 0.45 M), only a small amount of 1-Me survives after 5 min (<10%, compatible with the slight reagent deficiency), while ca. 90% of the ligand appears as free dihydropyridine. The fate of the organozinc fragment was unclear at this stage; likely, it is released as oligomeric methyl-fluorophenoxides [Zn(Me)(OC6F5)]x. Up to four Zn–Me signals of comparable intensity were observed in the high-field region of the 1H NMR spectrum (−0.28, −0.29, −0.38, −0.41 ppm), none of them corresponding to ZnMe2 (−0.69 ppm). As the reaction mixture settled, the intensity of these signals and those of H2BnBIP decay simultaneously as those corresponding to 2-F5 increase, indicating that the free base is captured by the alkyl–alkoxide zinc species. Note that the formation of methane is irreversible and drives the whole process; even if the methylzinc fluoroalkoxide species disproportionate giving ZnMe2 (and the dialkoxide), this would react with the free H2BnBIP, regenerating some 1-Me, that would eventually evolve into 2-F5 or 2-F9. A small amount of 1-Me (<10%) remains in quasi-stationary concentration for 6–7 h, disappearing after 24 h to leave a relatively clean spectrum corresponding mainly to 2-F5, with low-intensity signals of unknown by-products (approx. 10%); see below. Similar observations were made in a version of this experiment using nonafluoro-t-butanol instead of pentafluorophenol. In this case, however, the initial protonation occurs at a slower pace, and a significant fraction of surviving 1-Me remains more than 20% for up to 3 days under the above-stated conditions. Free H2BnBIP was also detected, but in a lower proportion, with the maximum concentration (30%) being attained after 35 min, as the rate of the protonation step is now comparable to that of its recapture rate by the alkyl–alkoxide zinc products. Very likely, the different reactivity of the two fluoroalcohols stems from steric effects, as C6F5OH is only slightly more acidic than t-C4F9OH (their pKa's in MeCN are 20.1 and 20.5, respectively).22 As mentioned above, under the same experimental conditions, the reactivity of 1-Bn is similar to that of 1-Me. However, because the former compound reacts slower, the initial step, namely, the formation of H2BnBIP and [Zn(Bn)(ORF)], is less clearly appreciated because it enters in competition with subsequent side processes.
 | ||
| Scheme 4 Two-step mechanism for the reaction of alkylzinc dihydropyridinates with fluoroalkoxides. | ||
We mentioned before that the reactions of the alkylzinc dihydropyridinate complexes with the fluorinated alcohols HF5 and HF9 are not fully selective, as unknown by-products are invariably detected at the end of the processes. In a further effort to solve these problems and in order to gain more knowledge about the nature of these minor species, we conducted two-step neutralisation experiments, first by adding a substoichiometric amounts of the HORF to 1-Me in toluene to take the conversion to 50–70% of the starting material, and allowing enough time to fully consume the fluoroalcohol. The precise amount of reagent needed to complete the neutralisation was then estimated from the 1H NMR spectrum of the crude mixture, and it was subsequently added to complete the reaction. The result of this experiment was completely unexpected. Whereas control NMR spectra of the initial reaction mixtures showed the usual mixture of 1-Me and the corresponding monoalkoxide species, the second addition caused extensive decomposition, revealed in complex 1H and 19F spectra where the signals of the same by-products detected before were clearly seen. A study consisting of evaporation and recrystallisation of the red residues from a mixture led to the isolation of crystals of, 3-F5 and 3-F9, which turned out to be suitable for X-ray diffraction studies. Their molecular structures are shown in Fig. 2. The reproducibility of these experiments was checked for 3-F9, and the same structure was resolved from crystals formed in a second reaction. Unfortunately, the small amount of crystalline material did not allow completing the characterization of these products.
 | ||
| Fig. 2 ORTEP representation and significant bond distances and angles for 3-F5 (left) and 3-F9 (right). 3-F5: Zn(1)–O(1), 1.926(3); Zn(1)–O(2), 1.926(3); Zn(1)–N(1), 2.043(6); Zn(1)–N(2), 2.408(6); Zn(1)–N(3), 2.308(6); C(1)–C(2), 1.369(10); C(2)–C(3), 1.374(11); N(2)–C(6), 1.254(9); O(1)–Zn(1)–N(1), 130.39(12); O(1)–Zn(1)–N(2), 98.76(14); O(1)–Zn(1)–N(3), 101.68(15); O(2)–Zn(1)–N(1), 130.39 (12); O(2)–Zn(1)–N(2), 98.76(14); O(2)–Zn(1)–N(3), 101.68(15); N(2)–Zn(1)–N(3), 148.24(19); N(1)–Zn(1)–N(2), 73.4(2); N(1)–C(5)–C(13) 116.5(6); O(1)–Zn(1)–O(2) 99.1(2) 3–F9: Zn(1)–O(1), 1.861(5); Zn(1)–O(2), 1.905(5); Zn(1)–N(1), 2.131(5); Zn(1)–N(2), 2.047(6); C(1)–C(2), 1.376(10); C(2)–C(3), 1.409(10) N(2)–C(6), 1.291(9); O(1)–Zn(1)–N(1), 119.1(2); O(1)–Zn(1)–N(2), 122.5(3); O(2)–Zn(1)–N(1), 106.2(2); O(2)–Zn(1)–N(2), 99.7 (3); N(1)–Zn(1)–N(2), 80.2(2); C(1)–N(1)–C(5) and 118.7(6). | ||
As can be seen, 3-F5 and 3-F9 are bis(aryloxo)zinc(II) adducts showing the aromatised 4-Bn-iPrBIP ligand. The main difference between both structures is the tridentate vs. bidentate coordination of the BIP ligand in the fluorophenoxide and fluoroalkoxide, respectively. In 3-F9, the simultaneous coordination of the three nitrogen atoms is probably hindered by the bulkiness of the t-C4F9O fragment, leaving one of the imine functionalities pending uncoordinated, but 3-F5 reveals that the imine N(2) atom is more weakly bonded to the metal, (2.403(6) Å) than the other (Zn–N(3), 2.308(6) Å). In addition, the main BIP ligand plane in 3-F5 is on a crystallographic mirror plane, which results in two perfectly symmetrical Zn–O bonds (1.926(3) Å) and coordination environment close to a trigonal bipyramid. In contrast, the zinc centre in 3-F9 could be described as a severely distorted tetrahedron, with one of the Zn–O bonds significantly shorter than the other (1.861(5) vs. 1.904(5) Å). Differently from 2-F5 (see below), none of these two structures has Zn⋯F distances shorter than 2.9 Å.
The most relevant feature associated with the isolation of compounds 3 is the aromatization of the central heterocycle, which becomes a regular pyridine ring. Although at this stage of our research we have no direct evidence on how the formation of these compounds occurs, we believe that transient species Zn(R)(ORF) or Zn(ORF)2 released at the initial phase of the reaction may play the role of a Lewis acids, in the same way as B(C6F5)3 (see Scheme 2). Specifically, the addition of HORF to a solution enriched in 2-F5 or 2-F9 might favour the removal of hydrides from these compounds; redistribution of the fluoroalkoxide leads to the observed bis-aryloxides along with highly reactive hydrido-zinc species23 that continue to react, eventually causing the complicated 19F spectra observed in both reactions (see Scheme 5).
 | ||
| Scheme 5 Proposed mechanism for the formation of 3-F5 and 3-F9. | ||
These results illustrate the extreme sensitivity of seemingly simple alkyl/alcohol neutralisation to reaction conditions. Although they can be optimised to favour the isolation of the dihydropyridinate-fluoroalkoxide complexes 2, competitive reactions are also at play and formation of side products like 3 and other species is probably unavoidable.
As commented above, the crystal structure of 2-F5 is remarkable in several aspects. Only a handful of structurally characterized complexes containing pincer-type ligands built on a 4-dihydropyridinate central ring,24 and just a few of them are 2,6-bis(imino)-dihydropyridinates related to BIP chemistry.11–13,25 Among the latter, the only direct zinc precedent for 2-F5 is its organozinc precursor 1-Bn,11 reported by us.
Despite the known ability of alkoxide ligands to bridge Lewis acidic metal centres, 2-F5 is monomeric. The Zn⋯F1 distance, 2.406(5) Å, is short for a nonbonding contact and this suggests the existence of a chemical interaction along the Zn⋯F vector, implying that the Zn(II) centre has some degree coordinative unsaturation. Therefore, the central Zn atom is to be considered as pentacoordinate. Atom F1 sits almost on the very same plane as defined by the three nitrogen atoms of the dihydropyridinate ligand. The four nearly coplanar atoms configure the basis of a square pyramid (SQP), with the oxygen atom placed at the apex, as represented in Fig. 3. The alternative representation as a trigonal bipyramid (TBP), with N2, O1, and F1 in the equatorial plane and N1 and N3 in the axis, gives a poorer description of the coordination polyhedron. Taking the two wider angles N2–Zn–N3 = 147.98(19)° and N1–Zn–F1 = 148.1(2)° as β and α, respectively, the geometry index τ5, defined as shown in Fig. 2,26 is nearly zero, as expected for SQP, while it would take a value close to 1 for a TBP. Moreover, as the Zn atom tends to occupy the geometric centre of the square pyramid, it is lifted 0.54 Å over the basal plane. This is not far from the centre of the SQP (1/4 of the height of the pyramid) since the distance from O1 to the basal plane is 2.27 Å.
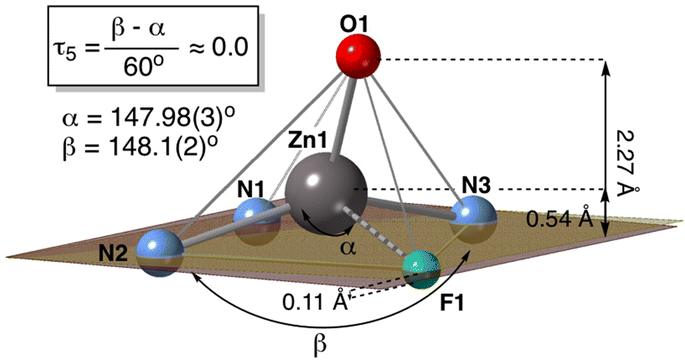 | ||
| Fig. 3 Simplified representation of the SQP coordination environment of complex 2-F5. The plane defined by the three nitrogen atoms is represented in clear colour, and the nearby basal plane in a darker hue, with the distance between both planes, indicated at the F1 atom. For other indications on the figure, see the main text. | ||
The dihydropyridinate and pentafluorophenoxide ligands are almost exactly orthogonal: the angle formed by the average planes that contain the ZnOC6F5 and Zn(N(C2)N(C2)N) fragments is 89.2°. In addition to the Zn⋯F interaction, the symmetric configuration of its coordination sphere points to a tightly coordinated Zn centre, possibly as a consequence of the Lewis acidity imparted by the electron-withdrawing pentafluorophenoxide. In many Zn(II) complexes with BIP, or BIP-based ligands, there is one N(imine) → Zn bond significantly longer than the other,15 as plainly illustrated for the neutral bis(alkoxides) 3. This effect is absent from 2-F5. Consequently, the dative Zn–N(imine) bonds in the latter (Zn–N2, 2.176(5) and Zn–N3 2.178(5) Å) are essentially identical, and significantly shorter than in 1-Bn, 2.3006(16) and 2.3545(17) Å.11 Still, the most revealing feature in 2-F5 is the short Zn⋯F1 interaction mentioned above.
Short Zn⋯F contacts involving organic C–F bonds have been reported in the literature. For example, Darensbourg discussed similar Zn⋯F interactions in the range 2.73–2.91 Å in the crystal structure of the binuclear 2,6-difluorophenoxide [Zn(μ-OC6F2H3)(OC6F2H3)(THF)]2.27 These distances are comparable to, or marginally below the sum of Bondi's van der Waals radii for F and Zn (2.86 Å). As a consequence, these were classified as non-bonding contacts. Furthermore, the pentafluorophenolate solvate [Zn(μ-OC6F5)(OC6F5)(THF)2]2, featuring the same C6F5O ligand as 2-F5 but with a coordinatively saturated, five coordinated metal center shows no Zn⋯F contacts,28 and the same can be said for compounds 3-F5 or 3-F9. A search in the Cambridge Structural Database29 for structures with Zn⋯F–C distances of less than 3 Å retrieved 116 hits and 222 contacts,29 27 of them intramolecular and 195 intermolecular. Our search was limited to fluoro-organic fragments to exclude special bonding situations. For example, attractive electrostatic forces would dominate intermolecular interactions between cationic Zn species and inorganic anions (e.g. BF4− or SbF6−). However, the shortest Zn⋯F–C were found in four ionic complexes with organic fluorinated anions,30 with the hitherto closest approach, 2.471 Å, corresponding to the interaction of a linear cation [(NHC)Zn(C6F5)]+ with [B(C6F5)4]− as its counteranion.30a Most of the remaining Zn⋯F–C contacts (192 = 86%) fall within the range of 2.7–3.0 Å. Only 10 contacts (5%) are shorter than 2.7 Å, and none of these is under 2.5 Å. Thus, to our knowledge, the 2.406(5) Å Zn⋯F distance of 2-F5 is the closest Zn⋯F–C contact observed so far.
The van der Waals radii concept was recently revisited by Álvarez,31 who concluded that distances shorter than the radii sum by more than 1.3 Å correspond to a chemical bond, and those 0.7 to 1.3 Å shorter fall within the van der Waals gap, suggesting a special bonding situation that requires a deeper analysis. The sum of Álvarez’ van der Waals radii of Zn and F, is 3.85 Å. In consequence, the “van der Waals gap” for weak Zn⋯F interactions lies between 3.15 and 2.55 Å, which fits very well the conclusions of our search in the CSD database. As mentioned, just a few structures involving organic fluorides contain Zn⋯F–C separations under 2.6 Å. Thus, the 2.406 Å Zn⋯F(1) distance observed in 2-F5 is shorter than expected for a weak dipolar or a dispersive interaction, as it is in the upper edge for a formal Zn–F bond. Indeed, features such as the shift of the Zn atom outside of the flat dihydropyridinate and the narrowing of the N1–Zn–O angle (compared to the analogous N–Zn–C in 1-Bn) suggest that a chemical bond between F1 and Zn completes the SQP coordination environment of Zn. Therefore, we decided to investigate this molecule in more detail to verify the existence of a bond between the Zn atom and F1.
To determine whether the short Zn⋯F contact corresponds to a genuine chemical interaction, we first studied the topology of the electron density wave function in the experimental X-ray geometry of the 2-F5 molecule with Bader's AIM theory (Fig. 4). For this purpose, we first allowed H atoms in their original positions to relax into realistic positions with molecular mechanics (Merck Molecular Force Field), while keeping the heavier atoms. Then, we performed a single-point calculation using an advanced DFT method (ωB97M-V/def2-TZVPPD) to build a high-quality wavefunction. Fig. 3 shows a molecular diagram of 2-F5 showing a contour plot of the Laplacian of the electron density, sliced along the plane defined by the atoms Zn, F1, and C42. As can be seen, there is a Bond Critical Point (BCP) between F(1) and Zn, confirming the existence of a chemical interaction, and a Ring Critical Point (RCP) characterises the OC6F5 as a chelate ligand. Furthermore, the Dispersion Index (DI) coefficients (the AIM analogue of bond orders) indicate that the F(1) and Zn atoms share 0.11 electron pairs; for comparison, other DI values for Zn–L bonds are 0.32 for the almost symmetrical dative bonds from the imine N(2) or N(3) to Zn, and 0.53 and 0.44 electron pairs for the covalent Zn–N(1) and Zn–O bonds, respectively. These DI coefficients show that, although weak, the Zn⋯F interaction is not negligible. A similar conclusion is drawn by comparing the electron densities at the corresponding BCPs. The electron accumulation on the F⋯Zn bond path (electron density on the F1–Zn BCP = 0.0256 e au−3) is taken from the σ-C–F bond, which shows a slightly decreased electron density on the C(43)–F(1) BCP (0.2479 e au−3) in comparison with the average in the five C–F BCPs of the pentafluorophenoxide ligand (0.2624 e au−3). The X-ray diffraction structure confirms that the F(1)–C(42) bond is, indeed, virtually identical than its non-coordinated congener F(5)–C(46) (1.349(8) vs. 1.348(9) Å), although the difference is in the limit of the accuracy of the data.
 | ||
| Fig. 4 Molecular diagram of 2-F5 generated by the AIMALL software showing a slice of the Laplacian of the electron density (−∇2ρ, contour plot) along the plane defined by atoms Zn, F1, and C42. Critical bond (BCP) and critical ring points (RCP) are represented with small green and red spheres, respectively. Hydrogen atoms and their associated BCPs and RCPs have been omitted for clarity. | ||
To gain a more direct indication of the strength of the Zn⋯F1 interaction, we conducted a variable temperature 19F NMR study of 2-F5 in CD2Cl2 between +25 and −80 °C (Fig. S6, ESI†). The Zn⋯F1 interaction should cause differentiation of the signals of the perfluoroaryl ring. However, the 19F spectrum of this complex recorded in CD2Cl2 at room temperature or slightly below shows sharp multiplets in a 2:2:1 intensity ratio, consistent with the free rotation of the C6F5 ring. On cooling, the signals of o- and m-F become broader, while the p-F multiplet remains sharp, pointing to the influence of the Zn⋯F1 interaction as the most likely cause. This effect becomes evident at 0 °C. De-coalescence of the signal for the ortho-19F nuclei was observed at ca. −60 °C (at 376.5 MHz). At this temperature, the broad o,o′-F signal, at −170.5 ppm, merges with the baseline. Further cooling causes the apparent symmetry of C6F5 to break down. At −70 °C, one of the ortho-F signals appears at −172.45 ppm, whereas the other is obscured by that of the meta-F, giving a broad feature for 3 F at −168.5 ppm.
Unfortunately, the limit for a slow exchange rate was not reached and the spectrum remains partially unresolved at −85 °C, the lowest temperature reachable in CD2Cl2. The line broadening is not due to technical reasons but to fluxional behaviour, as the signal of a small amount of C6F6 added as an internal reference remains sharp over the entire temperature range. These difficulties prevented us from executing a full line-shaped analysis of the spectra. However, a DFT calculation (ωB97M-V/def2-TZVPP/CPCM, see ESI† for details) of the 19F chemical shifts, based on the molecular geometry of the solid-state XRD data, confirmed that the signal of the coordinated 19F should shift to a higher field (i.e., more negative shift), and that the resonance of the free o′-F atom (F5) would appear approximately in the same region as that of the meta-19F nuclei. The separation between the o- and o′-19F signals (Δδo,o′) predicted by the calculation, 7.9 ppm, is comparable to the experimental separation, ca. 4.0 ppm. The lower value of the experimental Δδo,o′ suggests some the weakening of the Zn⋯F interaction in solution.
Combining the experimental coalescence temperature (213 K) and the observed Δδo,o′ (4.0 ppm ≈ 1500 Hz), the energy barrier for the fluxional exchange between ortho-19F signals can be estimated as 8.9 kcal mol−1. Comparable energy barriers have been measured for the axial-equatorial flip in five-coordinated iPrBIP complexes, for example 10.5 kcal mol−1 (at −55 °C) for the thermally unstable zinc complex [(iPrBIP)Zn(Bn)2].11 However, this motion cannot be responsible for the fluxionality of 2-F5 since the o,o′-F exchange implies breaking the F⋯Zn interaction. Indeed, the 8.9 kcal mol−1 barrier could be taken as a measure the strength of the interaction, but only if the process is intramolecular and the exchange rate is not limited by a restricted rotation of the pentafluorophenoxide ligand. To clarify this point further, we attempted to model the o-F exchange between the coordinated and free sites using DFT methods. We found that the functional ωB97X-D with a double-zeta quality basis set reproduces the geometry of 2-F5 reasonably well, with a somewhat longer Zn⋯F distance, 2.427 Å. To limit the conformational freedom of the original molecule and simplify the calculation, we used a slightly modified model, 2′-F5, with a Me group instead of the 4-Bn at the C4 site of the dihydropyridinate ring. However, we were unable to locate stable stationary points (either and intermediate or a transition state) to exchange both o-F atoms through a simple rotation of the C6F5 ring. The difficulty lies in the steric interactions between the pentafluorophenoxide ligand and the bulky N-aryl substituents during this rotation. Instead, we found that a flip movement of the O-C6F5 fragment allows shifting the Zn⋯F1 to a Zn⋯F5 interaction. This process exchanges two diastereoisomers, 2′-F5 and 2′′-F5 (Scheme 6), and not merely the ortho-F sites within a single molecule. In line with the X-ray structural data, 2′-F5 is marginally more stable than 2′′-F5. A stationary point was found in the pathway from 2′-F5 to 2′′-F5, which corresponds to a transition state (TS) in a very flat region of the potential energy surface, with a single but minimal imaginary frequency (26 cm−1). Given the remoteness of the 4-R substituent from the C6F5 group, the 19F NMR spectra of the diastereomeric forms of 2-F5 are probably very similar. Resolving the signals of the isomer is not possible within the temperature range accessible in CD2Cl2, and therefore this mechanism is still compatible with our NMR data. The free energy computed at the same level of the theory used for geometry optimisation (approx. 5 kcal mol−1) is somewhat low but not too far from our expectations for a preliminary calculation and could be justified by the weaker Zn⋯F interaction found at this level of the theory when a solvent model is applied (Zn–F(1) = 2.48 Å). Disappointingly, a finer single-point evaluation of the electronic energy using triple-Z quality basis functions and the same functional applied for chemical shift calculation further reduced the energy barrier to ca. 2 kcal mol−1. The DFT method may be underestimating the energy barrier because it does not capture the full strength of the Zn⋯F interaction. Thus, attempts to improve the accuracy of the DFT calculations running geometry optimisations on 2′-F5 with improved basis functions (6-31+G* or 6-311G*) produced significantly longer Zn⋯F bond lengths. In addition, it might also be that the corrections for dispersive forces are overestimating the intensity of the aryl–aryl π-stacking interactions, visible in the molecular drawing shown in Scheme 6, which contribute to stabilising the transition state TS.
 | ||
| Scheme 6 Intramolecular mechanism proposed for the o,o′-F exchange between free and bound sites in the simplified model 2′-F5 (R = Me instead of Bn to reduce conformational freedom in the calculations). | ||
Harder and co-workers have recently reported the syntheses of magnesium and zinc complex cations featuring coordinated halobenzenes (X-Ph) of composition [(diketiminate)M(X-Ph)]+.32,33 They show that the uneven distribution of negative charge on the halogen atom (with a “nucleophilic belt” and a “sigma hole”)34 causes the M⋯X–C interaction to be angular for heavier halogens. In the case of Mg, the fluorobenzene complex displays a Mg⋯F–C interaction that is the weakest and the least directional, this being consistent with its prevailingly electrostatic nature.32 However, although isostructural complexes with angular M⋯X–C interactions were observed in the Zn analogues with bromobenzene or chlorobenzene, the metal–halogen interaction is disfavoured by the corresponding fluorobenzene derivative and the complex adopts a π-bonded (η1) Zn⋯C interaction, instead.33 This was attributed to the more covalent character of the interactions for M = Zn. The intramolecular configuration of the halogen–Zn interaction in 2-F5 forces an angular disposition of the Zn⋯F–C interaction (104.6(4)°), which is close to the angles found in Harder's chloro- and bromobenzene diketiminate-Zn complexes (107.6 and 104.1, respectively). Thus, the angular configuration imposed by the intramolecular coordination in 2-F5 may play a stabilizing role, by placing the fluorine atom in the appropriate configuration to increase the covalent contribution to the Zn⋯F interaction.
Conclusions
In this work, we report the syntheses of mononuclear aryloxy- and alkoxo-zinc compounds stabilised by pincer-type 2,6-diimino-4-alkyl-dihydropyridinate ligands via protolytic alkyl/alcohol reactions. The availability of the new zinc alkyl precursor, 1-Me, synthesized through a ligand-transfer methodology, and the use of fluoroalcohols HO-RF (RF = C6F5 or C(CF3)3) were instrumental in the isolation of stable products, and for the clarification of the mechanism that, surprisingly, does not involve a classic σ-bond exchange. We showed that the initial attack of the fluoroalcohol does not cleave the Zn–C bond in 1-Me or 1-Bn, but the alcohol proton is initially delivered to the basic dihydropyridinate nitrogen, releasing a transient methyl- or benzylzinc species, [Zn(R)(ORF)]n. The latter reacts with the newly formed free 1,4-dihydropyridine base H2BnBIP in a subsequent step, giving rise to the final alkoxo-product 2-F5 or 2-F9, together with the corresponding alkane H–R. Moreover, we observed that the outcome of the reaction is very sensitive to the mixing conditions. Competitive aromatisation of the dihydropyridine ring can also occur, producing new alkoxides [(iPrBIP)Zn(ORF)2] (3-F5 or 3-F9). We are still investigating the origin of these compounds, but we suspect that the aromatisation of the dihydropyridinate ring is induced by transient Zn electrophiles when the product (2-F5 or 2-F9) overreacts with the corresponding fluoroalcohol.In addition, we discovered that the fluoroaryloxide derivative 2-F5 exhibits a remarkable coordination mode through a halogen bond interaction, with the shortest Zn⋯F–C distance so far recorded in the CSD database. This provides additional complex stabilisation, mitigating the coordinative unsaturation of the Zn centre. VT-NMR affords an energy barrier of ca. 8.9 kcal mol−1 for the reversible dissociation of the halogen bridge. Although attempts to model the fluxional exchange of 2-F5 by DFT calculations provided an underestimation of the strength of the Zn⋯F interaction, AIM analyses on the experimental geometry confirm the existence of a substantial interaction, probably due to the favourable disposition of the fluoroaryloxo fragment, which allows a weak dative donation from the weak “nucleophilic belt” of electron density of the fluorine atom.
As a final remark, we would like to stress that the presence of two distinct reactivity sites in the 2,6-diimino-4R-dihydropyridinate ligand dominates the reactivity of its Zn(II) complexes, leading to some unusual chemistry. The results presented in this work suggest that the dual reactivity of the dihydropyridine ring, combined with the labile coordination of the Zn centre to BIP and H2BIP ligands, could be exploited in biomimetic hydrogen transfer reactions similar to the NAD(P)+/NAD(P)H system.
Experimental section
General considerations
Most of the compounds presented in this work are highly sensitive to oxygen and moisture. Therefore, inert atmosphere Schlenk techniques or/and a N2-filled glove box were routinely used in manipulations and procedures. The solvents (toluene, hexane, pentane, and diethyl ether) were rigorously degassed, dried, and distilled immediately prior to use. NMR spectra were recorded on Bruker equipment, model DPX-300 and DPX-400. The chemical shifts of the 19F{1H} spectra were referenced to an internal C6F6 signals but are expressed with regard to CFCl3, while the 1H and 13C{1H} spectra are referenced to tetramethylsilane (TMS). Spectral assignations were routinely helped with bidimensional (2D) 1H–1H COSY, 1H–13C HMBC and HSQC heterocorrelation spectra. Prior use, the deuterated solvents used (CD2Cl2 and C6D6) were dried over the corresponding desiccant agent (e.g., C6D6 in sodium), filtered, and distilled under reduced pressure. Elemental analysis, and X-ray diffraction measurements were carried out at the Instituto de Investigaciones Químicas. ZnMe2 (0.2 M toluene solution), t-C4F9OH, C6F5OH and BnMgCl (1.0 M in diethyl ether) were purchased from Sigma-Aldrich and employed as received. The Grignard reagent (2 M in THF) was titrated prior to use. Compounds 2,6-[2,6-iPr2C6H3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)]2-C5H3N (iPrBIP), 2,6-[2,6-iPr2C6H3NC(Me)]2-4-Bn-C5H4N (H2BniPrBIP), and [ZnBn(4-BniPrBIPH)] (1-Bn) were synthesised according to experimental procedures described in the literature.13
C(Me)]2-C5H3N (iPrBIP), 2,6-[2,6-iPr2C6H3NC(Me)]2-4-Bn-C5H4N (H2BniPrBIP), and [ZnBn(4-BniPrBIPH)] (1-Bn) were synthesised according to experimental procedures described in the literature.13
Synthesis of [ZnMe(4-BniPrBIPH)] (1-Me)
In a nitrogen-filled glovebox, 1.6 mL of a solution of ZnMe2 (0.2 M in toluene, 0.32 mmol) are slowly added to a yellow solution of 4-BniPrBIPH2 (182 mg; 0.32 mmol), dissolved in 15 mL of toluene, cooled to −25 °C. The solution instantly changed to purple. After 16 hours stirring at room temperature, the solvent and volatiles were removed at reduced pressure, obtaining a purple solid, which was extracted with hexane (2 × 15 mL), filtered and dried under vacuum. The 1H-NMR spectrum of the resulting purple solid in C6D6 shows only signals of compound 1-Me (187 mg, 90%), which was recrystallised from cold hexane (−23 °C). 1H NMR (25 °C, 400 MHz, C6D6): δ −0.62 (s, 3H, ZnMe) 1.00 (d, 3JHH = 6.4 Hz, 6H, CHMeMe), 1.04 (d, 3JHH = 6.4 Hz, 6H, CHMeMe), 1.13–1.17 (overlapped doublets, 3JHH = 6.6 Hz, 12H, CHMeMe), 1.71 (s, 6H, Me(CN)), 2.74 (sept, 4H, 3JHH = 6.9 Hz, CHMeMe), 2.91 (d, 3JHH = 7.0 Hz, 2H, CH2, Py–Bn), 4.00 (m, 1H, 4-CHPy), 5.22 (d, 3JHH = 4.8 Hz, 2H, 3,5-CHPy), 7.06 (m, 6H, CHN–Ar), 7.20 (m, 5H, CHAr, Py–Bn). 13C{1H} RMN (C6D6, 25 °C, 100 MHz): δ −14.23 (ZnMe), 15.97 (Me(CN)), 23.64 (CHMeMe), 24.00 (CHMeMe), 28.81 (CHMeMe), 39.83 (4-CHpy), 47.27 (CH2, Py–Bn), 106.25 (3,5-CHPy), 123.68 (p-CHN–Ar), 125.38 (m-CHN–Ar), 126.37 (p-CHAr, Py–Bn), 128.68 (o-CHAr, Py–Bn), 129.96 (m-CHAr, Py–Bn), 138.08 (i-CAr, Py–Bn), 139.77 (2-CPy), 143.73 (o-CN–Ar), 144.11 (i-CN–Ar), 167.73 (Me(CN)). Anal. calcd for C41H53N3Zn: C, 75.38, H, 8.18, N, 6.43. Found: C, 75.29, H, 8.21, N, 6.16.Synthesis of [ZnOC6F5(4-BniPrBIPH)] (2-F5)
Procedure A. A colourless solution of F5C6OH (24.4 mg, 0.133 mmol) in toluene (5 mL) was added to a purple 5 mL solution of compound 1-Me (87.5 mg, 0.133 mmol) in the same solvent at room temperature. The resultant mixture immediately changed to red-purple. This was stirred during 24 h, and the solvent and volatiles were evaporated, leaving a red-purple solid. The 1H-NMR of the residue product confirms the formation of 2-F5 along with minor impurities. The solid was extracted in hexane (2 × 10 mL), filtered and dried under vacuum, leaving a dark purple solid, whose NMR spectra showed only the signals of compound 2-F5 (83.2 mg, 75%). X-ray quality crystals were grown from a cold hexane (−30 °C). 1H NMR (C6D6, 25 °C, 400 MHz): δ 0.86–0.90 (overlapped doublets, 3JHH = 6.4 Hz, 12H, CHMeMe), 1.28–1.32 (overlapped doublets, 3JHH = 6.6 Hz, 12H, CHMeMe), 1.65 (s, 6H, Me(CN)), 2.75 (sept, 3JHH = 6.7 Hz, 4H, CHMeMe), 2.90 (d, 3JHH = 7.0 Hz, 2H, CH2, Py–Bn), 3.84 (m, 1H, 4-CHPy), 5.18 (d, 3JHH = 4.8 Hz, 2H, 3,5-CHPy), 6.88 (m, 6H, CHN–Ar), 7.21 (m, 5H, CHAr, Py–Bn). 19F{1H} NMR (C6D6, 25 °C, 376 MHz): δ −167.90 (t, 3JFF = 22.6 Hz, 2F, m-F), −168.60 (dd, 3JFF = 10.0 Hz, 2F, o-F), −180.80 (tt, 3JFF = 9.9 Hz, 1F, p-F). 13C{1H} NMR (C6D6, 25 °C, 100 MHz): δ 13.21 (CHMeMe), 16.02 (Me(CN)), 22.36 (CHMeMe), 24.20 (CHMeMe), 28.49 (CH(iPr)), 39.55 (4-CHpy), 45.21 (CH2, Py–Bn), 106.55 (3,5-CHPy), 123.21 (p-CHN–Ar), 123.51 (m-CHN–Ar), 126.09 (p-CHAr, Py–Bn), 128.95 (o-CHAr, Py–Bn), 129.44 (m-CHAr, Py–Bn), 135.57 (i-CAr, Py–Bn), 138.43 (2-CPy), 141.15 (o-CN–Ar), 142.04 (i-CN–Ar), 171.07 (Me(CN)). Anal. calcd for C46H50F5N3OZn: C, 67.27, H, 6.14, N, 5.12. Found: C, 67.12, H, 6.39, N, 5.47.Procedure B. A 5 mL toluene solution of F5C6OH (24.1 mg, 0.131 mmol) was added by syringe to a dark blue solution of 1-Bn (95.4 mg, 0.131 mmol) in 15 mL of toluene at −20 °C, observing an instantaneous colour change to red-purple. The mixture was gradually warmed and kept under magnetic stirring for 40 h at room temperature, the time after which it was evaporated under vacuum, obtaining a sticky solid. This was dissolved in 10 mL of pentane, filtered and dried, isolating 71.2 mg of a dark purple microcrystalline solid that after NMR analysis corresponds to complex 2-F5 (71.2 mg, 65%). Then, this product was dissolved in 15 mL of dried pentane, filtered and evaporated until 1/3 of its original amount, prior storing at −30 °C. After 24 h, a significant amount of a red-purple microcrystalline solid suitable for X-ray diffraction studies was formed.
NMR monitoring of the reaction of 1-Me with F5C6OH
A colourless solution of F5C6OH (4.9 mg, 0.027 mmol) in C6D6 (0.2 mL) was added to a purple 0.4 mL solution of compound 1-Me (18.3 mg, 0.028 mmol) in the same solvent at approx. −25 °C. The resulting solution was then transferred to a J-Young capped type NMR tube, placed in the probe of the NMR spectrometer and analysed by 1H and 19F NMR within 10 min at room temperature. Successive spectra were recorded every 30 min during the first 90 min and each hour afterwards, during the following 4 hours. The last NMR spectra were registered within 24 hours, the time after which the reaction monitoring was concluded, since the relative amounts of the reaction products of the reaction were observed to remain constant with respect to the previous NMR recorded 8 h before and the signals of the starting material (1-Me) represented less than 10% of the amount used in the reaction.VT-NMR monitoring of the dynamic behaviour of 2-F5
A dark violet 0.02 M solution of [ZnOC6F5(4-BniPrBIPH)] (2-F5) (12 mg; 0.014 mmol) in 0.7 mL of CD2Cl2 was transferred to a J-Young capped type NMR tube at room temperature to which C6F6 was added as internal 19F-NMR reference. Then, the NMR tube was placed in the probe of the NMR spectrometer and both 1H- and 19F-NMR spectra were recorded at 298 K (25 °C). Thereafter, successive spectra were recorded at every 10 °C until reaching −85 °C (see ESI, Fig. S9†). At this point, the temperature of the NMR probe was increased stepwise and new spectra were recorded at 20 °C intervals. The spectra registered in the ascendant and descendent series were undistinguishable. 1H NMR (CD2Cl2, 25 °C, 400 MHz): δ 1.17 (d, 3J = 6.8 Hz, 6H, CHMeMe), 1.13 (d, 3J = 6.8 Hz, 12H, CHMeMe), 1.09–1.06 (d, 3J = 6.8 Hz 6H, CHMeMe), 2.07 (s, 6H, Me(CN)), 2.70 (dh, J = 27.1, 6.8 Hz, 4H, CHMeMe), 2.94 (d, J = 6.8 Hz, CH2 Py–Bn 2H), 3.86–3.65 (m, 1H, 4-CHPy), 5.43 (d, J = 5.0 Hz, 2H, 3,5-CHPy), 7.10–7.04 (m, 6H, CHN–Ar), 7.27–7.37 (m, 5H, CHAr, Py–Bn). 19F{1H} NMR (CD2Cl2, 25 °C, 376 MHz): δ −162.65 (s, 6F, C6F6as internal reference), −168.63 to −168.75 (m, 4F, m-F and o-F), −181.95 – −182.18 (m, 1F, p-F).Synthesis of [ZnOC4F9(4-BniPrBIPH)] (2-F9)
NMR tube scale reaction. A 0.7 mL deuterated benzene solution of 1-Bn (19.7 mg, 0.027 mmol) were added over 6.5 mg (0.027 mmol) of t-C4F9OH at −20 °C (approx.) via pipette. The color of the resultant solution turned instantly form dark blue to dark purple. This reaction mixture was transferred to a J-Young capped type NMR tube which was then placed in the probe of the NMR spectrometer and analysed by 1H and 19F NMR within 30 min at room temperature, observing predominantly the signals of metal free 4-BniPrBIPH, 2-F9 and the starting material 1-Bn in a relative ratio of 3
:1.5:1, respectively. A new 1H-NMR recorded with 120 min showed the same species but their relative ratio had changed to 2:2:1. Then, after 5 h the reaction mixture still showed relative proportions of 2:5:1, in which the major species corresponds to the alkoxide product 2-F9. Then, the mixture was left settling and a new NMR recorded within 3 days, displaying virtually only the signals of compound 2-F9 and toluene.
NMR monitoring of the reaction of 1-Me with t-C4F9OH
A colourless solution of t-C4F9OH (6.2 mg, 0.026 mmol) in C6D6 (0.2 mL) was added to a purple 0.4 mL solution of compound 1-Me (18.0 mg, 0.027 mmol) in the same solvent at approx. −20 °C. The resultant solution was then transferred to a J-Young capped type NMR tube, placed in the probe of the NMR spectrometer and analysed by 1H and 19F NMR within 10 min. Successive spectra were recorded at room temperature every 30 min during the first 90 min and each hour thereafter, during 5 hours. New NMR spectra were registered in 30 h. The relative ratios of reactants and products kept changing until 120 hours, time after which the ratios of the resultant species were observed to be unchanged with to respect of the previous NMR recorded after 96 h and the signals of the starting material (1-Me) represented less than 10% of the amount employed.X-ray structural analysis for 2-F5, 3-F5 and 3-F9
Crystals suitable for X-ray diffraction analysis were coated with dry perfluoropolyether, mounted on glass fibres, and fixed in a cold nitrogen stream to the goniometer head. Data collection35 were performed on a Bruker-AXS, D8 Quest ECO diffractometer equipped with a micro-focus IμS 3.0 source, using graphite monochromatized Mo radiation λ(Mo Kα) = 0.71073 Å and with an area detector Bruker Photon II 14 – CPAD. The data were reduced (SAINT) and corrected for absorption effects by the multiscan method (SADABS).36 The structures were solved by direct methods (SIR2002, SHELXS)37 and refined against all F2 data by full-matrix least-squares techniques (SHELXL-2018/3)38 minimizing w[Fo2 − Fc2]2. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in calculated positions and allowed to ride on their carrier atoms with the isotropic temperature factors Uiso fixed at 1.2 times (1.5 times for methyl groups) of the Ueq values of the respective carrier atoms. After numerous attempts using different crystallization conditions, the 2-F5 crystals could only be obtained as twins with at least 5 domains, although one of them represents 50% of the total, while the remaining domains had percentages lower than 10%. However, we were unable to obtain reliable refinement data by incorporating more than one domain and refining the BASF parameter and HKLF5 reflection file. Consequently, we only utilized the most intense and significant reflections from the primary or main domain. In this way we avoid the twinning problem, allowing us to obtain crystallographic data of somewhat more acceptable quality despite the very poor quality of the crystal. The crystal structure of 2-F5 crystallizes in the centrosymmetric space group P21/n. Several carbon atoms exhibit positional disorder, but modelling or applying ADP restraints was unnecessary.Similarly, to 2-F5, the 3-F5 crystals could only be acquired as twins with a minimum of 5 domains. One of the domains predominated at a 70% ratio, while the remaining domains constituted a very minor percentage. Additionally, obtaining satisfactory refinement data by incorporating more than one domain and refining the BASF parameter and HKLF5 reflection file was unsuccessful. Consequently, by treating the main domain with a high percentage as an untwined crystal, we circumvented the twinning issue, enabling us to obtain crystallographic data of acceptable quality despite the poor quality of crystal. The crystal structure of 3-F5 crystallizes in the centrosymmetric space group Pnma. The benzyl group and most of the fluorine atoms display some disorder, which did not require modelling but necessitated the application of ADP restraints to two carbon atoms in the benzyl group. In the asymmetric unit of the crystal, in addition to the structure of the zinc complex, a molecule of dichloromethane is observed as a crystallization solvent, wherein one of the chlorine atoms coincides with an inversion centre. The crystal structure of 3-F9 crystallizes in the non-centrosymmetric space group Pn and was refined as a 2-component inversion twin with a domain ratio of 0.97:0.03. In this crystal structure, an isopropyl group from an aniline, two carbon atoms from a nonafluoro tert-butyl alkoxide, and most of the fluorine atoms in both perfluoro alkoxides exhibit some disorder, necessitating the application of certain ADP restraints. CCDC 2256678 (2-F5), 2256679 (3-F5) and 2256680 (3-F9)† contain the supplementary crystallographic data for this paper.
565 reflections measured, 4708 independent reflections (Rint = 0.1018). The final R1 values were 0.0760 (I > 2σ(I)). The final wR(F2) values were 0.1922 (I > 2σ(I)). The final R1 values were 0.1172 (all data). The final wR(F2) values were 0.2109 (all data). The goodness of fit on F2 was 1.054.
883 reflections measured, 8705 independent reflections (Rint = 0.0830). The final R1 values were 0.0549 (I > 2σ(I)). The final wR(F2) values were 0.1237 (I > 2σ(I)). The final R1 values were 0.0716 (all data). The final wR(F2) values were 0.1321 (all data). The goodness of fit on F2 was 1.042.
Computational data
For computational details see ESI.†Author contributions
J. M. Delgado-Collado: investigation, validation, M. Gallardo-Villagrán: investigation, validation. E. Álvarez: investigation, formal analysis. J. Cámpora: conceptualization, writing – review and editing, visualization, supervision, funding acquisition and project administration. A. Rodríguez-Delgado: conceptualization, investigation, validation, writing – original draft – review and editing, visualization, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Research Agency (AEI), and Junta de Andalucía through the European Union (Feder Funds) projects PGC2018-095768-B-100 and PY20_0104, respectively.References
- (a) Z. Flisak and W.-H. Sun, ACS Catal., 2015, 5, 4713–4724 CrossRef CAS; (b) B. L. Small, Acc. Chem. Res., 2015, 48, 2599–2611 CrossRef CAS PubMed.
- (a) Q. Knijnenburg, S. Gambarotta and P. H. M. Budzelaar, Dalton Trans., 2006, 5442–5448 RSC; (b) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS PubMed.
- (a) C. Römelt, T. Weyhermüller and K. Wieghardt, Coord. Chem. Rev., 2019, 380, 287–317 CrossRef; (b) D. Sieh, M. Schlimm, L. Andernach, F. Angersbach, S. Nückel, J. Schöffel, N. Šušnjar and P. Burger, Eur. J. Inorg. Chem., 2012, 444–462 CrossRef CAS; (c) T. W. Myers, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Dalton Trans., 2016, 45, 5989–5998 RSC.
- S. Blanchard, E. Derat, M. Desage-El Murr, L. Fensterbank, M. Malacria and V. Mouriès-Mansuy, Eur. J. Inorg. Chem., 2012, 376–389 CrossRef CAS.
- (a) P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed; (b) P. J. Chirik, Angew. Chem., Int. Ed., 2017, 56, 5170–5181 CrossRef CAS PubMed; (c) L. A. Berben, Chem. – Eur. J., 2015, 21, 2734–2742 CrossRef CAS PubMed.
- J. Cámpora, A. Rodríguez-Delgado and P. Palma, in Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018, pp. 539–586, DOI:10.1016/B978-0-12-812931-9.00026-8.
- (a) D. Reardon, F. Conan, S. Gambarotta, G. Yap and Q. Wang, J. Am. Chem. Soc., 1999, 121, 9318–9325 CrossRef CAS; (b) H. Sugiyama, G. Aharonian, S. Gambarotta, G. P. A. Yap and P. H. M. Budzelaar, J. Am. Chem. Soc., 2002, 124, 12268–12274 CrossRef CAS PubMed; (c) J. Scott, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, J. Am. Chem. Soc., 2005, 127, 13019–13029 CrossRef CAS PubMed.
- (a) J. Cámpora, A. M. Naz, P. Palma and E. Álvarez, Organometallics, 2005, 24, 4878–4871 CrossRef; (b) J. Cámpora, C. M. Pérez, A. Rodríguez-Delgado, A. M. Naz, P. Palma and E. Álvarez, Organometallics, 2007, 26, 1104–1107 CrossRef.
- C. M. Pérez, A. Rodríguez-Delgado, P. Palma, E. Álvarez, E. Gutiérrez-Puebla and J. Cámpora, Chem. – Eur. J., 2010, 16, 13834–13842 CrossRef PubMed.
- J. J. Sandoval, C. Melero, P. Palma, E. Álvarez, A. Rodríguez-Delgado and J. Cámpora, Organometallics, 2016, 35, 3336–3343 CrossRef CAS.
- J. J. Sandoval, P. Palma, E. Álvarez, A. Rodríguez-Delgado and J. Cámpora, Chem. Commun., 2013, 49, 6791–6793 RSC.
- (a) Q. Knijnenburg, J. M. M. Smits and P. H. M. Budzelaar, Organometallics, 2006, 25, 1036–1046 CrossRef CAS; (b) M. Gallardo-Villagrán, F. Vidal, P. Palma, E. Álvarez, E. Y. X. Chen, J. Cámpora and A. Rodríguez-Delgado, Dalton Trans., 2019, 48, 9104–9116 RSC.
- J. J. Sandoval, P. Palma, E. Álvarez, J. Cámpora and A. Rodríguez-Delgado, Organometallics, 2016, 35, 3197–3204 CrossRef CAS.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2010, 29, 4203–4206 CrossRef CAS.
- J. J. Sandoval, E. Álvarez, P. Palma, A. Rodríguez-Delgado and J. Cámpora, Organometallics, 2018, 37, 1734–1744 CrossRef CAS.
- T. J. Sherbow, L. W. T. Parsons, N. A. Phan, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2020, 59, 17614–17619 CrossRef CAS PubMed.
- A. McSkimming and S. B. Colbran, Chem. Soc. Rev., 2013, 42, 5439–5488 RSC.
- (a) G. Hamasaka, H. Tsuji, M. Ehara and Y. Uozumi, RSC Adv., 2019, 9, 10201–10210 RSC; (b) P. T. Smith, S. Weng and C. J. Chang, Inorg. Chem., 2020, 59, 9270–9278 CrossRef CAS PubMed; (c) R. Arevalo, R. Lopez, L. R. Falvello, L. Riera and J. Perez, Chem. – Eur. J., 2021, 27, 379–389 CrossRef CAS PubMed; (d) D. Ghosh, G. R. Kumar, S. Subramanian and K. Tanaka, ChemSusChem, 2021, 14, 824–841 CrossRef CAS PubMed; (e) M. F. Pang, L. L. Shi, Y. F. Xie, T. Y. Geng, L. Liu, R. Z. Liao, C. H. Tung and W. G. Wang, ACS Catal., 2022, 12, 5013–5021 CrossRef CAS; (f) G. B. Shen, B. C. Qian, G. S. Zhang, G. Z. Luo, Y. H. Fu and X. Q. Zhu, Org. Chem. Front., 2022, 9, 6833–6848 RSC; (g) M. R. Schreier, B. Pfund, D. M. Steffen and O. S. Wenger, Inorg. Chem., 2023, 62(20), 7636–7643 CrossRef CAS PubMed.
- Leading references: (a) M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS; (b) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2001, 125, 11350–11359 CrossRef PubMed; (c) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (d) Ch. M. Thomas, O. L. Casagrande Jr., Ch. W. Lehmann, Th. Roisnel and J.-F. Carpentier, Inorg. Chem., 2007, 46, 329–340 CrossRef PubMed; (e) Y. Wang, W. Zhao, D. Liu, Sh. Li, X. Liu, D. Cui and X. Chen, Organometallics, 2012, 31, 4182–4190 CrossRef CAS; (f) A. B. Biernesser, Bo Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed; (g) Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, Sh. Li and D. Cui, Chem. Commun., 2014, 50, 11411–11414 RSC; (h) C. M. Manna, A. Kaur, L. M. Yablon, F. Haeffner, Bo Li and J. A. Byers, J. Am. Chem. Soc., 2015, 137, 14235–14235 Search PubMed; (i) A. Kronast, M. Reiter, P. T. Altenbuchner, Ch. Jandl, A. Pȯthig and B. Rieger, Organometallics, 2016, 35, 681–685 CrossRef CAS; (j) D. E. Stasiw, A. L. Luke, T. Rossen, A. Bleague, M. Mandal, B. D. Neisen, Ch. J. Cramer, M. Kol and W. B. Tolman, Inorg. Chem., 2017, 56, 14366–14372 CrossRef CAS PubMed; (k) M. A. Ortuño, B. Dereli, K. R. D. Chiaie, A. B. Biernesser, M. Qi, J. A. Byers and C. J. Cramer, Inorg. Chem., 2018, 57, 2064–2071 CrossRef PubMed; (l) M. Qi, Qi Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed; (m) M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS; (n) J. E. Chellali, A. K. Alverson and J. R. Robinson, ACS Catal., 2022, 12, 5585–5594 CrossRef CAS; (o) F. d Vries and E. Otten, ACS Catal., 2022, 12, 4125–4130 CrossRef PubMed.
- I. J. Blackmore, V. C. Gibson, P. B. Hitchcock, C. W. Rees, D. J. Williams and A. J. P. White, J. Am. Chem. Soc., 2005, 127, 6012–6020 CrossRef CAS PubMed.
- (a) Y. Sarazin, J. A. Wright, D. A. J. Harding, E. Martin, T. J. Woodman, D. L. Hughes and M. Bochmann, J. Organomet. Chem., 2008, 693, 1494–1501 CrossRef CAS; (b) T. O. Petersen, S. Weigel, D. Kratzert, A. Fischer and I. Krossing, ChemistrySelect, 2017, 2, 265–273 CrossRef CAS.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Eur. J. Org. Chem., 2021, 1407–1419 CrossRef.
- A.-K. Wiegand, A. Rit and J. Okuda, Coord. Chem. Rev., 2016, 314, 71–82 CrossRef CAS.
- For crystal structures featuring pincer ligands with a central 4-hydropyridinate rings, see: (a) S. Kundu, W. W. Brennessel and W. D. Jones, Inorg. Chem., 2011, 50, 9443–9453 CrossRef CAS PubMed; (b) S. Lapointe, E. Khaskin, R. R. Fayzullin and J. R. Khusnutdinova, Organometallics, 2019, 38, 4433–4447 CrossRef CAS; (c) T. Mehdoui, J.-C. Berthet, P. Thuéry, L. Salmon, E. Rivière and M. Ephritikhine, Chem. – Eur. J., 2005, 11, 6994–7006 CrossRef CAS PubMed; (d) T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed; (e) G. Zhang, J. Wu, S. Zheng, M. C. Neary, J. Mao, M. Flores, R. J. Trovitch and P. A. Dub, J. Am. Chem. Soc., 2019, 141, 15230–15239 CrossRef CAS PubMed; (f) R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, S. DeBeer and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 13168–13184 CrossRef CAS PubMed; (g) R. Fandos, A. Otero and A. M. Rodríguez, Polyhedron, 2018, 143, 171–175 CrossRef CAS.
- T. K. Mukhopadhyay, C. L. Rock, M. Hong, D. C. Ashley, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2017, 139, 4901–4915 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS.
- Y. Sarazin, J. A. Wright, D. A. J. Harding, E. Martin, T. J. Woodman, D. L. Hughes and M. Bochmann, J. Organomet. Chem., 2008, 693, 1494–1501 CrossRef CAS.
- CSD Database 5.43, last updated, Nov. 2023. Cambridge Crystallographic Data Centre (CCDC).
- (a) R. Guermazi, D. Specklin, C. Gourlaouen, P. de Frémont and S. Dagorne, Eur. J. Inorg. Chem., 2022, e202101002 CAS; (b) J.-C. Bruyere, D. Specklin, C. Gourlaouen, R. Lapenta, L. F. Veiros, A. Grassi, S. Milione, L. Ruhlmann, C. Boudon and S. Dagorne, Chem. – Eur. J., 2019, 25, 8061–8069 CrossRef CAS PubMed; (c) T. O. Petersen and I. Krossing, personal communication to CCDC, deposition number 1432546.
- S. Álvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- A. Friedrich, J. Pahl, J. Eyselein, J. Langer, N. van Eikema Hommes, A. Görling and S. Harder, Chem. Sci., 2021, 12, 2410–2418 RSC.
- A. Friedrich, J. Eyselein, J. Langer and S. Harder, Organometallics, 2021, 40, 448–457 CrossRef CAS.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, in Halogen Bonding I: Impact on Materials Chemistry and Life Sciences, ed. P. Metrangolo and G. Resnati, Springer International Publishing, Cham, 2015, pp. 1–17, DOI:10.1007/128_2014_573.
- Bruker APEX3 software suite, Bruker AXS, Inc.; Madison, WI, 53711, 2016.G Search PubMed.
- Bruker Advanced X-ray solutions, SAINT and SADABS programs, Bruker AXS Inc., Madison, WI53711, 2012 Search PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, SIR2002: the Program, J. Appl. Crystallogr., 2003, 36, 1103–1104 CrossRef CAS.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and computational details including XYZ-files. CCDC 2256678–2256680. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01492d |
| This journal is © The Royal Society of Chemistry 2023 |