
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IR spectra and structures of saturated ruthenium cluster carbonyl cations Run(CO)m+ (n = 1–6)†
David
Yubero Valdivielso
,
Christian
Kerpal‡
,
Wieland
Schöllkopf
 ,
Gerard
Meijer
and
André
Fielicke
*
,
Gerard
Meijer
and
André
Fielicke
*
Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: fielicke@fhi-berlin.mpg.de
First published on 27th June 2023
Abstract
A series of saturated ruthenium cluster carbonyls (Ru(CO)5+, Ru2(CO)9+, Ru3(CO)12+, Ru4(CO)14+, Ru5(CO)16+ and Ru6(CO)18+) have been synthesized in the gas phase and subsequently characterized by infrared spectroscopy. Their size-specific IR spectra in the region of the carbonyl stretch vibration (1900–2150 cm−1) and in the region of the Ru–C–O bending modes (420–620 cm−1) are obtained by infrared multiple photon dissociation spectroscopy. The structures of these cluster carbonyls are assigned by comparison with results from density functional calculations. A multitude of differently activated CO ligands are identified in these cationic cluster carbonyls, reaching from terminal, over non-symmetrically bridging (semi-bridging) ligands with varying degrees of interaction to additional Ru atoms towards symmetrically bridging CO ligands.
Introduction
Metal cluster carbonyls have been an important class of substances for developing a profound understanding of the binding in polynuclear coordination compounds.1–5 Much of the knowledge on their structures is based on the characterization of compounds that are stable and synthesizable often in bulk quantities, e.g. via X-ray crystallography. Other approaches that can also be applied to less stable metal carbonyls rely on their synthesis in (cryogenic) inert matrices6–9 or molecular beams,10,11 where they are typically characterized by infrared spectroscopy. The latter makes use of the often very strong absorptions related to C–O stretch vibrations and the pronounced sensitivity of its vibrational frequency, ν(CO), towards the binding environment.12,13Gas-phase studies that can provide – via combination with mass spectrometry – direct information on the size and stoichiometry of a cluster carbonyl are usually limited to charged carbonyls, as the susceptibility to fragmentation during the ionization process can add severe complications to the studies of neutral metal cluster carbonyls.14 Only very recently the combination of IR excitation with ‘soft’ ionization using a tunable vacuum UV free electron laser (FEL) opened up new possibilities for gas-phase experiments on neutral metal carbonyls.15–17 So far, the majority of such studies in the gas phase, however, have focussed on charged mono and binuclear carbonyls. Recently, also the synthesis and characterization of certain polynuclear metal carbonyl cations (and dications) have been reported in the condensed phase, which become accessible, inter alia, in the presence of weakly coordinating anions.18–20 A pertinent example is the formation of dications M3(CO)142+ (M = Ru, Os) with a linear-chain arrangement of the metal skeleton.21
Solution studies of metal cluster carbonyls, and in particular also of ruthenium carbonyls, have intensively utilized IR spectroscopy in the range of ν(CO) to study the dynamics of such cluster compounds. Particular interest is, e.g., on their photochemical or thermal disintegration, vibrational interactions within the cluster complex or with the solvent, and the reorganization of their ligand shells.22,23 In these solution phase experiments, intermediates may be detected based on their spectral signature; however, their identification has not always been straightforward.24 Furthermore, in contrast to their fixed structure in the solid state, polynuclear carbonyls in solution show often a pronounced fluxional behaviour due to intramolecular CO exchange.25,26 For example, Ru3(CO)12 is well known to exhibit rapid scrambling between axial and equatorial CO ligands with a low barrier of about 20 kJ mol−1.27 The exact reorganization mechanism and whether it involves bridging CO ligands that have been found as structural units in the ground state structure of the lighter homologue Fe3(CO)12 are still under discussion. However, intermediates containing bridging CO ligands have been experimentally detected during the photoinduced dissociation of Ru3(CO)12,22,28,29 and their possible role in the scrambling process has been postulated based on quantum chemical calculations.30,31 As we will show later in this article, semi-bridging CO ligands are present in cationic Ru3(CO)12+ and other cationic Ru carbonyls isolated in the gas phase.
Ionic carbonyls in the gas phase have the advantage that their composition can be precisely determined by means of mass spectrometry. By applying infrared multiple photon dissociation (IR-MPD) spectroscopy, size-selective structural characterization becomes possible without the interference through possible fragmentation products.10,32–34 Already in the late 1980s, cationic polynuclear transition metal cluster carbonyls were synthesized in the gas phase via reactions of transition metal clusters with CO and their compositions were analysed by means of mass spectrometry.35 At comparably high CO partial pressure and sufficient reaction time, i.e. many reactive collisions between cluster and CO molecules, characteristic abundance patterns of metal cluster carbonyls are observed, relating the nuclearity of the metal cluster core, n, to a particularly stable stoichiometry of the corresponding carbonyl. In this way, cationic cluster carbonyls have also been synthesized, and they do not have known neutral complexes as stable counterparts. The experimentally observed stoichiometries of these gas-phase carbonyl clusters are well understood in the context of the Wade–Mingos rules,36,37 an extension of the 18 electron rule for transition metal complexes to polynuclear compounds. Within this polyhedral skeletal electron pair theory (PSEPT), the cluster's nuclearity, composition, and electronic structure are related to predict the geometry of the clusters.
The Wade–Mingos rules aim to describe the bonding and topology of the metal skeleton in polynuclear, usually closed-shell, complexes. They treat the ligand as a source of a given number of electrons shared over all the valence orbitals of the cluster complex and, therefore, cannot provide detailed structural information on the ligand arrangement in the complex. Instead, usually space-filling arguments are used to rationalize the arrangement of the ligand sphere. Infrared spectroscopy offers additional information as it is sensitive to structural details of a carbonyl complex, e.g., via the symmetry selection rule for IR activity. Furthermore, as ν(CO) is a measure of the activation of C–O through the metal–CO interaction, or more specifically through the π back-bonding, it is particularly useful to discern CO ligands in different metal coordination sites and, in particular, to differentiate between terminal and bridging CO ligands.
Cationic Rh cluster carbonyls, for instance, have been investigated by IR-MPD spectroscopy, which then allowed for a comparison between structures of the cations and those of the known stable neutral Rh carbonyls. As a result it was concluded that ionization leads to a destabilization of the μ2 bridging CO ligands in Rh2(CO)8 (2 μ2-CO) and Rh4(CO)12 (3 μ2-CO) such that the cations only contain terminal CO ligands, while the structure of Rh6(CO)16+ fully resembles that of the neutral octahedral species and contains 4 μ3 face-capping CO ligands.34
Gas-phase synthesis of cationic Ru carbonyls has been reported before for the tetra to hexanuclear carbonyls of Ru and their structures as derived via application of the Wade–Mingos rules have been compared to those predicted by means of density functional theory calculations.38 Here, we substantiate these structural assignments by comparing gas-phase IR-MPD spectra observed for the full series of saturated cationic Ru carbonyls Ru(CO)5+, Ru2(CO)9+, Ru3(CO)12+, Ru4(CO)14+, Ru5(CO)16+ and Ru6(CO)18 with IR spectra calculated using DFT for the putative ground-state isomers of these carbonyls.39
Experimental and theoretical methods
Experimental methods
Ruthenium carbonyl cluster cations are produced in the gas phase via laser vaporization of metallic Ru in the presence of He and CO. The initially formed ruthenium cluster cations react with CO gas at ∼30 °C in the high pressure region of the cluster source before the mixture expands into a vacuum forming a molecular beam. The distribution of cationic species embedded in the cluster beam is analysed by reflectron time-of-flight mass spectrometry. The clusters are produced at 10 Hz; on alternate shots, the beam is exposed to the focussed infrared beam of the FHI-FEL40 that acts as an intense and tunable IR source. Upon resonant excitation and absorption of multiple IR photons, a carbonyl complex can fragment, leading to a reduction of its detected ion intensity. This intensity change, normalized by the intensity in the ion pulses that are not exposed to IR, is converted to relative IR cross sections. Thereby, also the variation of IR fluence over the tuning range of the FEL is taken into account. Typical IR pulse energies in these experiments were 3 mJ in the region of ν(CO) and 8–16 mJ at longer wavelengths (here generally called far-IR) at a pulse duration of 5–8 μs. The accuracy of the evaluated band positions in the IR spectra is limited by the step-size of IR frequencies at which separate mass spectra have been recorded that is in the ν(CO) region 5 cm−1 and in the far-IR region 2 cm−1. Further details on the experiment and data evaluation have been reported elsewhere.41,42Theoretical methods
Structures of the Ru cluster carbonyls, their relative energies, and IR spectra were calculated using density functional theory (DFT) within TURBOMOLE V6.4.43 Initial guess structures were taken from the known structures of a variety of stable neutral carbonyl compounds and from structures generated via a modified basin hopping algorithm44 that treats CO as a pseudo-atom. Basin hopping was performed using RI-DFT with the PBE functional and def-SV(P) basis sets as well as the def-ecp effective core potential for Ru as implemented in TURBOMOLE. Further optimizations and frequency calculations made use of the PBE0 hybrid functional and the larger def2-TZVP/def2-ecp combination.45–48 Calculated harmonic vibrational frequencies were scaled by factors of 0.955 and 0.933 in the region of ν(CO) and in lower frequency modes, respectively, which have been optimized for the herein discussed carbonyls. This scaling also shows excellent agreement between calculated and experimental values8 for the most intense ν(CO) band in Ru3(CO)12 (ESI, Fig. S1†). For a better visual comparison between experimental and calculated spectra, the latter were folded with a Gaussian line shape function with a full-width at half maximum of 1% of the centre frequency.Initial structural optimizations were performed without any symmetry constraints; however, the optimized structures were carefully checked for the presence of any point group symmetry and, if appropriate, re-optimized within the resulting symmetry. All of the structures reported below are true minima, and only for the lowest here reported state of Ru6(CO)18+ (C2, 2B) a small imaginary frequency (8.1i cm−1) remained; due to the size of the system we refrained from further optimizations.
Results and discussion
Structure of Ru carbonyls
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 55 shows good agreement for both. Only the axial d(C–O) is slightly overestimated in the calculated results by ∼1% and the corresponding d(Ru–C) is too small by the same value, while the deviations for the other atomic distances are smaller (see the ESI, Table S1†).
55 shows good agreement for both. Only the axial d(C–O) is slightly overestimated in the calculated results by ∼1% and the corresponding d(Ru–C) is too small by the same value, while the deviations for the other atomic distances are smaller (see the ESI, Table S1†).
 | ||
| Fig. 1 Ball and stick representations of the putative global minimum structures for cationic ruthenium carbonyls and for neutral Ru3(CO)12 with point group symmetries and electronic states given. For Ru(CO)5+ and Ru6(CO)18+ also a higher energy isomer is shown. The colour scheme is as follows: Ru – teal, C – grey, O – red (terminal CO), C – light grey, O – orange (semi-bridging CO), C – lightgrey, and O – yellow (symmetrically bridging CO). For better clarity, some of the longer Ru–C bonds for semi-bridging ligands are depicted as dashed lines only. | ||
| VE | Rule | Structure class | Skeleton structure | |
|---|---|---|---|---|
| Ru(CO)5+ | 18 | — | Atom | |
| Ru2(CO)9+ | 34 | — | Dimer | |
| Ru3(CO)12+ | 48 | 16n | Ring | Triangle |
| Ru4(CO)14+ | 60 | 15n | Three-connected | Tetrahedron |
| Ru5(CO)16+ | 72 | 14n + 2 | Closo deltahedral | Trigonal bipyramid |
| Ru6(CO)18+ | 84 | 14n | Hypercloso deltahedral | Capped trigonal bipyramid |
The structural representations in Fig. 1 distinguish between terminal, symmetrically μ2-bridging, and non-symmetrically bridging or semi-bridging CO ligands. This differentiation between binding modes is based on the bridge asymmetry parameter α as introduced by Curtis.60

Clearly, this still remains an arbitrary definition and in fact there is a rather smooth structural transition from ‘true’ terminal to bridging CO ligands which goes along with a lowering of the Ru–C–O bond angle, a lengthening of the C–O bond, and a red-shift of the corresponding ν(CO), as discussed below.
Saturation composition and implications from PSEPT
The reaction of gas-phase Ru cluster cations with CO molecules in the cluster source leads, at sufficiently high CO partial pressures and under stabilizing multiple collision conditions with background He gas, to a distribution of cationic carbonyl complexes (Fig. 2). The relevant partial pressures of He and CO in the source are difficult to quantify as they are both introduced by separate pulsed valves, but by comparison with the conditions in similar cluster sources a total pressure of a few tens of mbar can be deduced, consisting mostly of He carrier gas.63–65 The reaction time can be approximated by the time the complexes spend in the cluster source, which is on the order of 100 μs. Under such conditions, for a given nuclearity of the metal cluster, a carbonyl with a specific stoichiometry (marked as n,m in Fig. 2) is formed as the major product. The mass spectrometric signals appear rather broad which is due to the natural isotopic distribution of Ru, but their position and overall width agree well with the distributions expected for the given carbonyl compositions. Furthermore, the resolution of the mass spectrometer of m/Δm ≈ 1000 is sufficient to resolve all mass signals for the lighter carbonyls showing no obvious additional signals other than those from the isotopes of Ru and C. | ||
| Fig. 2 Mass spectrum of saturated Ru cluster carbonyls Run(CO)m+. Compositions of particularly intense, i.e. stable, Ru carbonyls are indicated as (n,m). Additional minor signals are from less stable CO complexes. | ||
Similar mass spectrometric results have been reported before for mass selected Ru4+, Ru5+ and Ru6+ reacted with CO in a gas-filled radio frequency ion trap, leading to the same complex compositions.38 These experiments showed that complex formation proceeds via fast successive addition of CO molecules with the saturated complex being the only observable product. While Fig. 2 includes complexes up to Ru8(CO)21+, the following discussion will be limited to mono to hexanuclear carbonyls as for the larger complexes no insightful IR spectra have been obtained.
The composition of saturated cluster carbonyls as derived from the mass spectrum is listed in Table 1 together with the structural predictions according to PSEPT.1,2,5,36,66 As discussed by Lang et al., the saturated cationic carbonyls observed in their study follow the PSEPT concept as applied to the neutral species, i.e. a half-filled valence orbital can be treated as fully occupied.38 In this scheme, ruthenium with its [Kr]4d75s1 configuration of the neutral atom is assumed to contribute 8 valence electrons to the total valence electron counts, while a single CO ligand acts here as a 2 electron donor. We note that this may not be a general rule as shown for the bridging CO ligands in a recent, very detailed comparative bonding analysis for M2(CO)9 (M = Fe, Ru, Os).52 The total number of valence electrons for the respective neutral cluster carbonyls and the deduced assignments are shown in Table 1. The last column of this table contains the structure derived for the metal cluster skeletons of these carbonyl clusters.
IR-MPD spectra and structural assignments
 | ||
| Fig. 3 Overview of IR-MPD spectra of Run(CO)m+. The coloured areas highlight typical ranges for terminal (blue), symmetrically bridging (orange) and non-symmetric semi-bridging CO ligands. Bands observed at longer wavelengths are assigned to delocalized Ru–C–O bending modes. In the gap between 640 and 1900 cm−1 no vibrational fundamentals are expected for these complexes. The intensities in the high frequency range above 1900 cm−1 are multiplied by a factor of 0.1 for a better visualization on a common intensity scale. For penta and hexanuclear carbonyls IR-MPD spectra obtained with twice the IR intensity are included in the far-IR range (grey lines) which shows additional minor features below 500 cm−1. | ||
The IR spectra show clear signals in two ranges. The bands experimentally detected in the upper far-IR (460–570 cm−1) all can be assigned, by comparison with the results of DFT calculations, to strongly delocalized Ru–C–O bending modes, δ(Ru–C–O), each involving many or even all carbonyl groups. Other modes at longer wavelengths have much lower IR intensities and are not experimentally observed as they do not lead to enough energy absorption to induce CO losses. Calculations locate the Ru–CO stretch vibrations in the 370–450 cm−1 range. They are highly coupled with bending and other deformation modes of the carbonyl network. Vibrations involving the Ru cluster are found below 180 cm−1, which is also well below the region of skeleton vibrations of bare Ru clusters which reach up to ∼290 cm−1 for cationic Run+ (n = 7–9) clusters.59 For further comparison, ν(Ru–Ru) of the isolated dimer has been measured at 347 cm−1via Raman spectroscopy in a cryogenic argon matrix.68 The intense bands seen between 1920 and 2130 cm−1 correspond to ν(CO) modes. Note that in the region of ν(CO) the intensity in the IR spectra is scaled by a factor of 0.1. The average position of the ν(CO) bands shifts with increasing nuclearity to lower frequencies which reflects an increase of C–O bond activation in the carbonyls. This can be related to a more effective π back-donation in the larger cluster complexes due to the decreasing CO/Ru ratio (5 in Ru(CO)5+vs. 3 in Ru6(CO)18+) together with a better delocalization of the charge in the larger clusters.69 More detailed assignments can be based on a comparison with the results of DFT calculations. In short, bands above 2040 cm−1 are seen for all of these cationic carbonyls and can be related to terminal (μ1) CO ligands. The range between 1980 and 2040 cm−1 is characteristic for non-symmetrically bridging (semi-bridging) CO with clear signals present, e.g., for Ru3(CO)12+ and Ru6(CO)18+. At even lower frequencies (∼1920–1960 cm−1), bands related to symmetrically μ2-bridging CO are found, with Ru4(CO)14+ showing the most prominent signal. Ru5(CO)16+ shows weak signals for both symmetrical and non-symmetrically bridging CO ligands. No bands are observed at a lower frequency and therefore the presence of higher coordinated, i.e. μ3-face capping, CO ligands can be excluded for the carbonyls studied here. The range between 640 and 1900 cm−1 is not shown, but has been measured partially: the far-IR measurements extend up to 750 cm−1 and the higher frequency range has been studied from 1800 cm−1 upwards, but no additional signals are seen in these regions.
 | ||
| Fig. 4 Experimental IR-MPD spectra (black) of saturated cationic Ru cluster carbonyls compared to the predicted harmonic IR spectra (green) of the putative global minima as obtained using DFT. For Ru(CO)5+ and Ru6(CO)18+, calculated spectra of higher energy isomers (orange) are included for comparison. The experimental and calculated intensities in the frequency range above 1900 cm−1 are multiplied by a factor of 0.1 for a better visualization on a common intensity scale. For penta and hexanuclear carbonyls IR-MPD spectra obtained with twice the IR intensity are included in the far-IR range (grey lines). | ||

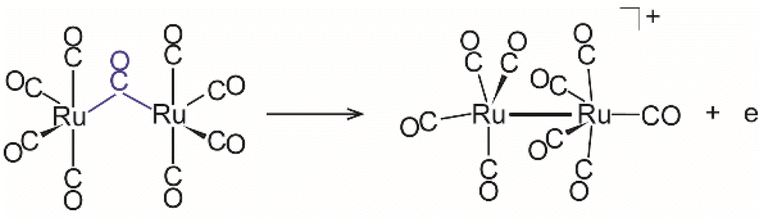

Terminal, semi-bridging and symmetrically bridging carbonyl ligands
The presented assignments of the IR spectra based on DFT calculations reveal that the cationic ruthenium cluster carbonyls provide diverse examples of coordinated CO ligands reaching from terminal, over semi-bridging to symmetrically μ2-bridging CO and, thereby, allow for a comparison of these structural elements with chemically very similar compounds. They are all homoleptic Ru carbonyls, while the former comparisons usually invoke complexes containing different transition metals and also other types of ligands which may account for additional steric and electronic constraints affecting the CO coordination. At this point, we do not intend to analyse the electronic structure of the cluster carbonyls further but rather discuss the resulting Ru–C–O binding geometries that can be related to the IR spectroscopic data. However, due to the open shell structure of the here discussed carbonyl cations, one may expect Jahn–Teller type distortions leading to overall symmetry reductions. This may provide the basis for a larger variation of CO coordination in the cations compared to closed shell structures of higher symmetry.More detailed discussions of the structural parameters of differently coordinated CO ligands and the consequences for their electronic structures, in particular the M–M interactions, have been given elsewhere.71,74–76 According to Crabtree et al.74 one can distinguish four coordination types for CO interacting with a pair of metal atoms, (i) terminal ligands which essentially interact only with one of the metal atoms in a nearly linear M–C–O arrangement and (ii) symmetrically bridging ligands with two equivalent M–C bonds and the CO axis standing (close to) perpendicular to the bridged M–M pair. Bent semi-bridging (iii) ligands with one short and one long M–C distance and an M–C–O angle significantly deviating from linearity are formed by π back-bonding into the CO's 2π* orbital from the more distant metal atom via interaction with an M–M antibonding orbital. They are seen as intermediates between the terminal and symmetrical-bridging CO ligands. A linear semi-bridging CO (iv) indicates, however, according to Hall76,77 π back-bonding from a filled π orbital along the M–M entity, i.e., it stabilizes the M–M bonding orbital, while the bent semi-bridging carbonyl stabilizes an antibonding orbital. In both cases the C–O bond is more weakened compared to the terminal CO ligands due to back-bonding from two metal centres which increases the electron density in the CO's 2π* orbital. Similar binding schemes can be discussed for CO interaction, e.g., with the three metal atoms of a triangular face, with the limiting cases of terminal CO binding and central face-capping (μ3-bridging).76 The structural difference between linear and bent semi-bridging CO ligands can be visualized according to Curtis by plotting ∠(Ru–C–O) vs. the bridge asymmetry parameter. In the case of the here studied Ru cluster carbonyls (see Fig. 5a), however, the consistent decrease of ∠(Ru–C–O) for α < 0.6 indicates that only bent semi-bridging ligands are present.
 | ||
| Fig. 5 (a) Curtis plot of the Ru–C–O angle vs. the bridge asymmetry parameter α. (b) Distribution of C–O bond lengths in cationic saturated Ru cluster carbonyls, Run(CO)m+ (n = 1–6), as a function of the Ru–C–O angle calculated using DFT. In both plots, the values for the calculated D3 structure of neutral Ru3(CO)12 are given for comparison (black crosses). | ||
Fig. 5b displays the gradual change in the CO coordination geometries for the cationic Ru cluster carbonyls by plotting the calculated C–O bond length as a function of the Ru–C–O bond angle. As discussed before in the assignments for the specific carbonyls, the presence of these ligands with different d(C–O) has been verified based on their characteristic absorptions in the ν(CO) region. Terminal CO, where the π system extending over the Ru–C–O unit stabilises a linear arrangement (∠(Ru–C–O) = 175.5–180°), has the shortest d(C–O) ranging from 112.6–113.7 pm. The closest to linear Ru–C–O units are found in Ru(CO)5+, where no interaction with further metal atoms can occur. In the carbonyls of higher nuclearity, however, such interactions can provide additional π back-donation leading to a further increase of d(C–O). Accordingly, the longest C–O bonds are found for the symmetrically μ2-bridging ligands in Ru4(CO)14+ and Ru5(CO)16+ with d(C–O) of 114.5 and 114.8 pm and ∠(Ru–C–O) of 139.7 and 141.0°, respectively. In between those are the bent semi-bridging ligands with d(C–O) ranging from 113.4 to 114.4 pm and ∠(Ru–C–O) = 172.5–156.4°.
Both plots in Fig. 5 also include the CO ligands in the predicted D3 ground state of neutral Ru3(CO)12 (black crosses). Structurally, the (close to) equatorial CO ligands (α = 0.96) clearly fall into the class of terminal CO ligands, while the axial ones are very similar to semi-bridging ligands, in particular to the ones identified in the structure of the corresponding cation Ru3(CO)12+. This is illustrated also by the values of their bridge asymmetry parameter of α = 0.63, which is slightly above the cut-off of 0.6 for semi-bridging carbonyls according to Curtis et al.,61 but still in the range suggested later by Parmelee and Mankad which reaches up to α = 0.7.62 In the cation, three semi-bridging CO ligands have a bridge asymmetry parameter α between 0.34 and 0.51, and two more ligands fall into the range of α between 0.6 and 0.7 (see Fig. 5a). The strong structural similarities of the D3 structure of Ru3(CO)12 and that of Ru3(CO)12+ can be realized from Fig. 1, despite the lower symmetry of the cation.
Conclusions
The IR spectra of saturated cationic ruthenium cluster carbonyls containing 1–6 Ru atoms have been obtained via infrared multiple photon dissociation spectroscopy and assigned by comparison with the spectra calculated using DFT for the putative global minima as obtained by a basin hopping global optimization algorithm. The metal skeletal arrangements of the assigned structures follow the predictions according to the polyhedral skeleton electron pair theory for the corresponding closed-shell neutral species. This supports the conclusions already drawn by Lang et al.38 that, at least for the Ru carbonyls under study, within PSEPT counting schemes a half-filled valence orbital (in the cation) can be treated as fully occupied.The smallest carbonyls, Ru(CO)5+ and Ru2(CO)9+, only contain terminal CO ligands. The larger ones contain a variety of differently coordinated carbonyl ligands including terminal CO ligands, semi-bridging CO ligands, and symmetrically bridging CO units. The corresponding C–O stretch vibrations follow the well-known trend of ν(CO) showing the highest values for terminal CO ligands and gradually decreasing with increasing interaction of the CO ligand with additional Ru atoms. This gradual change goes along with a lengthening of the C–O bonds and a bending of the Ru–C–O units which is in-line with the effect of the Ru → CO π back-donation mechanism for classical transition metal carbonyls.
The large variety of differently coordinated CO ligands in the Ru carbonyl clusters analysed in this study allows for a more systematic structural comparison of the ligand environment in these carbonyls including also the most stable structure of neutral Ru3(CO)12. As a result, we notice a strong similarity between the predicted D3 symmetric ground state of neutral Ru3(CO)12 and its cation in terms of the coordination geometry of the ‘axial’ CO ligands. One may speculate if the weakly semi-bridging character of these ligands is related to the dynamical properties of Ru3(CO)12.
More direct information on the binding interactions between transition metal atoms and CO ligands and the transition between terminal, semi- and symmetrically bridging configurations can be obtained by analysing the electron density distributions in related compounds, although the major focus of these studies often is on the metal–metal interaction. However, these experimental studies are typically performed via X-ray diffraction on crystalline samples, where, taking the example of neutral Ru3(CO)12, the molecular structure and dynamical behaviour may significantly differ from that of the isolated molecule due to substantial intermolecular interactions in the crystal. Similarly, the binding interactions may be further analysed theoretically, i.e. using the atoms-in-molecules approach,78 but such analysis would go well beyond the scope of this study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gert von Helden for fruitful discussions and Dan J. Harding for help with implementing the modified basin hopping routine. We gratefully acknowledge support by the Deutsche Forschungsgemeinschaft (FI 893/3 and FI 893/5). D. Y. V. thanks the International Max Planck Research School "Functional Interfaces in Physics and Chemistry" for support. Open Access funding provided by the Max Planck Society is acknowledged.References
- J. W. Lauher, J. Am. Chem. Soc., 1978, 100, 5305–5314 CrossRef CAS.
- D. M. P. Mingos and D. J. Wales, Introduction to Cluster Chemistry, Prentice-Hall, London, 1990 Search PubMed.
- P. J. Dyson and J. S. McIndoe, Transition Metal Carbonyl Cluster Chemistry, Gordon and Breach Science Publishers, Amsterdam, 2000 Search PubMed.
- B. K. Teo, G. Longoni and F. R. K. Chung, Inorg. Chem., 1984, 23, 1257–1266 CrossRef CAS.
- K. Wade, J. Chem. Soc., Dalton Trans., 1971, 792–793 CAS.
- M. Zhou, L. Andrews and C. W. Bauschlicher Jr., Chem. Rev., 2001, 101, 1931–1961 CrossRef CAS.
- H.-J. Himmel and O. Hübner, Mol. Catal., 2014, 25–52 CAS.
- J. G. Bentsen and M. S. Wrighton, J. Am. Chem. Soc., 1987, 109, 4530–4544 CrossRef CAS.
- S. Fedrigo, T. L. Haslett and M. Moskovits, Chem. Phys. Lett., 1999, 307, 333–338 CrossRef CAS.
- A. M. Ricks, Z. E. Reed and M. A. Duncan, J. Mol. Spectrosc., 2011, 266, 63–74 CrossRef CAS.
- G.-J. Wang and M.-F. Zhou, Chin. J. Chem. Phys., 2018, 31, 1–11 CrossRef CAS.
- N. Sheppard and T. T. Nguyen, in Advances in Infrared and Raman Spectroscopy, ed. R. E. Hester and R. J. H. Clark, Heyden, London, 1978, vol. 5, pp. 67–148 Search PubMed.
- A. Fielicke, P. Gruene, G. Meijer and D. M. Rayner, Surf. Sci., 2009, 603, 1427–1433 CrossRef CAS.
- O. Schalk, I. Josefsson, R. Richter, K. C. Prince, M. Odelius and M. Mucke, J. Chem. Phys., 2015, 143, 154305 CrossRef.
- C. Wang, Q. Li, X. Kong, H. Zheng, T. Wang, Y. Zhao, G. Li, H. Xie, J. Yang, G. Wu, W. Zhang, D. Dai, M. Zhou, X. Yang and L. Jiang, J. Phys. Chem. Lett., 2021, 12, 1012–1017 CrossRef CAS PubMed.
- G. Li, C. Wang, H.-J. Zheng, T.-T. Wang, H. Xie, X.-M. Yang and L. Jiang, Chin. J. Chem. Phys., 2021, 34, 51–60 CrossRef CAS.
- C. Wang, C.-Y. Tian, Y. Zhao, S. Jiang, T. Wang, H. Zheng, W. Yan, G. Li, H. Xie, J. Li, H.-S. Hu, X. Yang and L. Jiang, Angew. Chem., Int. Ed., 2023, e202305490 Search PubMed.
- M. Bodenbinder, G. Balzer-Jöllenbeck, H. Willner, R. J. Batchelor, F. W. B. Einstein, C. Wang and F. Aubke, Inorg. Chem., 1996, 35, 82–92 CrossRef CAS.
- Q. Xu, B. T. Heaton, C. Jacob, K. Mogi, Y. Ichihashi, Y. Souma, K. Kanamori and T. Eguchi, J. Am. Chem. Soc., 2000, 122, 6862–6870 CrossRef CAS.
- P. J. Malinowski and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 13460–13462 CrossRef CAS PubMed.
- M. Sellin, C. Friedmann, M. Mayländer, S. Richert and I. Krossing, Chem. Sci., 2022, 13, 9147–9158 RSC.
- M. R. Harpham, A. B. Stickrath, X. Zhang, J. Huang, M. W. Mara, L. X. Chen and D.-J. Liu, J. Phys. Chem. A, 2013, 117, 9807–9813 CrossRef CAS PubMed.
- X. Dong, F. Yang, J. Zhao and J. Wang, J. Phys. Chem. B, 2018, 122, 1296–1305 CrossRef CAS PubMed.
- Q. Kong, J. H. Lee, A. Plech, M. Wulff, H. Ihee and M. H. J. Koch, Angew. Chem., Int. Ed., 2008, 47, 5550–5553 CrossRef CAS PubMed.
- E. Band and E. L. Muetterties, Chem. Rev., 1978, 78, 639–658 CrossRef CAS.
- L. J. Farrugia, J. Chem. Soc., Dalton Trans., 1997, 1783–1792 RSC.
- S. Aime, W. Dastru, R. Gobetto, J. Krause and L. Milone, Organometallics, 1995, 14, 4435–4438 CrossRef CAS.
- Q. Y. Kong, M. G. Laursen, K. Haldrup, K. S. Kjær, D. Khakhulin, E. Biasin, T. B. van Driel, M. Wulff, V. Kabanova, R. Vuilleumier, S. Bratos, M. M. Nielsen, K. J. Gaffney, T. C. Weng and M. H. J. Koch, Photochem. Photobiol. Sci., 2019, 18, 319–327 CrossRef CAS PubMed.
- F. W. Vergeer, F. Hartl, P. Matousek, D. J. Stufkens and M. Towrie, Chem. Commun., 2002, 1220–1221 RSC.
- X. Feng, J. Gu, Y. Xie, R. B. King and H. F. Schaefer, J. Chem. Theory Comput., 2007, 3, 1580–1587 CrossRef CAS.
- B. Peng, Q.-S. Li, Y. Xie, R. B. King and H. F. Schaefer III, Dalton Trans., 2008, 6977–6986 RSC.
- D. T. Moore, J. Oomens, J. R. Eyler, G. Meijer, G. von Helden and D. P. Ridge, J. Am. Chem. Soc., 2004, 126, 14726–14727 CrossRef CAS.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Am. Chem. Soc., 2005, 127, 8416–8423 CrossRef CAS PubMed.
- I. Swart, F. M. F. de Groot, B. M. Weckhuysen, D. M. Rayner, G. Meijer and A. Fielicke, J. Am. Chem. Soc., 2008, 130, 2126–2127 CrossRef CAS PubMed.
- P. Fayet, M. J. McGlinchey and L. H. Wöste, J. Am. Chem. Soc., 1987, 109, 1733–1738 CrossRef CAS.
- D. M. P. Mingos, T. Slee and L. Zhenyang, Chem. Rev., 1990, 90, 383–402 CrossRef CAS.
- D. M. P. Mingos and D. J. Wales, J. Am. Chem. Soc., 1990, 112, 930–932 CrossRef CAS.
- S. M. Lang, S. U. Fortig, T. M. Bernhardt, M. Krstic and V. Bonacic-Koutecky, J. Phys. Chem. A, 2014, 118, 8356–8359 CrossRef CAS PubMed.
- D. Yubero Valdivielso, PhD thesis, Technische Universität Berlin, 2022.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. Todd, in Proc. SPIE 9512, Advances in X-ray Free-Electron Lasers Instrumentation III, SPIE, 2015, p. 95121L Search PubMed.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Phys. Chem. B, 2004, 108, 14591–14598 CrossRef CAS.
- A. Fielicke, G. von Helden and G. Meijer, Eur. Phys. J. D, 2005, 34, 83–88 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- D. J. Harding, C. Kerpal, G. Meijer and A. Fielicke, J. Phys. Chem. Lett., 2013, 4, 892–896 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- D. Andrae, U. Haeusermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. Rushman, G. N. Van Buuren, M. Shiralian and R. K. Pomeroy, Organometallics, 1983, 2, 693–694 CrossRef CAS.
- J. R. Moss and W. A. G. Graham, J. Chem. Soc., Dalton Trans., 1977, 95–99 RSC.
- F.-W. Grevels, W. E. Klotzbuecher, J. Schrickel and K. Schaffner, J. Am. Chem. Soc., 1994, 116, 6229–6237 CrossRef CAS.
- S. Pan, L. Zhao, H. V. R. Dias and G. Frenking, Inorg. Chem., 2018, 57, 7780–7791 CrossRef CAS PubMed.
- T. Bennett, R. H. Adnan, J. F. Alvino, V. Golovko, G. G. Andersson and G. F. Metha, Inorg. Chem., 2014, 53, 4340–4349 CrossRef CAS PubMed.
- R. White, T. Bennett, V. Golovko, G. G. Andersson and G. F. Metha, ChemistrySelect, 2016, 1, 1163–1167 CrossRef CAS.
- M. R. Churchill, F. J. Hollander and J. P. Hutchinson, Inorg. Chem., 1977, 16, 2655–2659 CrossRef CAS.
- L. L. Wang and D. D. Johnson, J. Phys. Chem. B, 2005, 109, 23113–23117 CrossRef CAS PubMed.
- J. Kim and J. Kim, Chem. Phys. Lett., 2014, 592, 24–29 CrossRef CAS.
- E. Waldt, A.-S. Hehn, R. Ahlrichs, M. M. Kappes and D. Schooss, J. Chem. Phys., 2015, 142, 024319 CrossRef PubMed.
- C. Kerpal, D. J. Harding, D. M. Rayner, J. T. Lyon and A. Fielicke, J. Phys. Chem. C, 2015, 119, 10869–10875 CrossRef CAS.
- R. J. Klingler, W. M. Butler and M. D. Curtis, J. Am. Chem. Soc., 1978, 100, 5034–5039 CrossRef CAS.
- M. D. Curtis, K. R. Han and W. M. Butler, Inorg. Chem., 1980, 19, 2096–2101 CrossRef CAS.
- S. R. Parmelee and N. P. Mankad, Dalton Trans., 2015, 44, 17007–17014 RSC.
- S. C. Richtsmeier, E. K. Parks, K. Liu, L. G. Pobo and S. J. Riley, J. Chem. Phys., 1985, 82, 3659–3665 CrossRef CAS.
- J. Woenckhaus and J. A. Becker, Rev. Sci. Instrum., 1994, 65, 2019–2022 CrossRef CAS.
- A. Fielicke and K. Rademann, J. Phys. Chem. A, 2000, 104, 6979–6982 CrossRef CAS.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- B. Peng, F. Gu, X. Zhang, Q. Luo and Q. Li, Sci. China, Ser. B: Chem., 2009, 52, 1938–1944 CrossRef CAS.
- H. M. Wang, Y. F. Liu, H. Haouari, R. Craig, J. R. Lombardi and D. M. Lindsay, J. Chem. Phys., 1997, 106, 6534–6537 CrossRef CAS.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Chem. Phys., 2006, 124, 194305 CrossRef PubMed.
- E. Bernhardt, C. Bach, B. Bley, R. Wartchow, U. Westphal, I. H. T. Sham, B. von Ahsen, C. Wang, H. Willner, R. C. Thompson and F. Aubke, Inorg. Chem., 2005, 44, 4189–4205 CrossRef CAS PubMed.
- F. A. Cotton, in Prog. Inorg. Chem., 1976, pp. 1–28 Search PubMed.
- Q. Kong, J. H. Lee, K. H. Kim, J. Kim, M. Wulff, H. Ihee and M. H. J. Koch, J. Am. Chem. Soc., 2010, 132, 2600–2607 CrossRef CAS PubMed.
- P. F. Jackson, B. F. G. Johnson, J. Lewis, M. McPartlin and W. J. H. Nelson, J. Chem. Soc., Chem. Commun., 1979, 735–736 RSC.
- R. H. Crabtree and M. Lavin, Inorg. Chem., 1986, 25, 805–812 CrossRef CAS.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238, 383–412 CrossRef.
- S. Ding and M. B. Hall, in The Chemical Bond I: 100 Years Old and Getting Stronger, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2016, pp. 199–248 Search PubMed.
- B. J. Morris-Sherwood, C. B. Powell and M. B. Hall, J. Am. Chem. Soc., 1984, 106, 5079–5083 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Comparison of calculated and experimental IR spectra for neutral Ru3(CO)12. Experimental and calculated bond lengths in Ru3(CO)12. Cartesian atomic coordinates for the optimized structures, vibrational frequencies and IR intensities. See DOI: https://doi.org/10.1039/d3dt01129a |
| ‡ Present address: Department of Physics and Astronomy, University of North Carolina at Asheville, One University Heights, Asheville, NC 28804, USA. |
| This journal is © The Royal Society of Chemistry 2023 |