
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal complexes of cyclam with two 2,2,2-trifluoroethylphosphinate pendant arms as probes for 19F magnetic resonance imaging†
Filip
Koucký
,
Jan
Kotek
 *,
Ivana
Císařová
,
Jana
Havlíčková
,
Vojtěch
Kubíček
and
Petr
Hermann
*,
Ivana
Císařová
,
Jana
Havlíčková
,
Vojtěch
Kubíček
and
Petr
Hermann
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 42 Prague 2, Czech Republic. E-mail: modrej@natur.cuni.cz
First published on 16th June 2023
Abstract
A new cyclam-based ligand bearing two methylene(2,2,2-trifluoroethyl)phosphinate pendant arms was synthesized and its coordination behaviour towards selected divalent transition metal ions [Co(II), Ni(II), Cu(II), Zn(II)] was studied. The ligand was found to be very selective for the Cu(II) ion according to the common Williams–Irving trend. Complexes with all the studied metal ions were structurally characterized. The Cu(II) ion forms two isomeric complexes; the pentacoordinated isomer pc-[Cu(L)] is the kinetic product and the octahedral trans-O,O′-[Cu(L)] isomer is the final (thermodynamic) product of the complexation reaction. Other studied metal ions form octahedral cis-O,O′-[M(L)] complexes. The complexes with paramagnetic metal ions showed a significant shortening of 19F NMR longitudinal relaxation times (T1) to the millisecond range [Ni(II) and Cu(II) complexes] or tens of milliseconds [Co(II) complex] at the temperature and magnetic field relevant for 19F magnetic resonance imaging (MRI). Such a short T1 results from a short distance between the paramagnetic metal ion and the fluorine atoms (∼6.1–6.4 Å). The complexes show high kinetic inertness towards acid-assisted dissociation; in particular, the trans-O,O′-[Cu(L)] complex was found to be extremely inert with a dissociation half-time of 2.8 h in 1 M HCl at 90 °C. Together with the short relaxation time, it potentially enables in vitro/in vivo utilization of the complexes as efficient contrast agents for 19F MRI.
Introduction
Among diagnostic techniques, magnetic resonance imaging (MRI) plays a crucial role in modern medicine. Its main advantage comes from the use of non-ionizing radiation, compared to classical radiomedicinal methods such as computed tomography (CT), single-photon emission computed tomography (SPECT) and positron emission tomography (PET) where ionizing radiation is used. However, MRI suffers from much lower sensitivity compared to SPECT and PET. Classical MRI detects an NMR effect of water 1H nuclei where sensitivity and image contrast can be improved by the application of so-called contrast agents (CAs). These compounds influence proton relaxation times (longitudinal, T1, and transversal, T2) of water molecules present in their vicinity, and the use of an appropriate pulse sequence increases or decreases the water 1H signals of the given tissue selectively.1 In addition, modern MRI techniques can also detect 1H signals of other compounds present in the organism (e.g. fat). Furthermore, the use of responsive (“smart”) contrast agents which modulate their response according to surrounding conditions can provide information on the physiological status of tissues.2 Alternatively, NMR spectra of parts of tissues could be acquired by a method called magnetic resonance spectroscopy imaging (MRSI).3 Also other NMR active nuclei such as 13C, 19F or 31P can be detected by MRI.4,5 Among them, 19F is of special interest as its gyromagnetic momentum is close to that of 1H which enables detection of 19F NMR signals on proton MRI scanners with only small hardware and software modifications. Furthermore, fluorine is a monoisotopic element and has almost no abundance in living organisms which predisposes it for use in “hot-spot” imaging. The need for “hot-spots” overlaid with anatomical images can be elegantly solved by 1H/19F MRI tandem imaging on the same hardware. Such an imaging method could be particularly useful e.g. in monitoring the fate of transplanted cells (cell tracking).6–8A number of compounds have been tested as 19F NMR contrast agents. They were usually perfluorinated hydrocarbons and their derivatives (for simplicity, they are called perfluorocarbons, PFCs), and some of them have been already studied for a long time as potential blood substitutes.9,10 The list of compounds includes e.g. hexafluorobenzene,11 perfluorodecalin,12 tris(perfluoroalkyl)amines,13 perfluorinated crown ethers,14–18 fluorinated polymers,19etc., which are used in the form of nanoemulsions. However, these organic compounds show very long T1 relaxation times. Therefore, a long delay between excitation pulses is needed, which prolongs the total acquisition time. To shorten the time of imaging, complexes of fluorine-containing ligands with paramagnetic metal ions were introduced.20–22 They are usually complexes of lanthanide(III) ions [with a generally large magnetic momentum, especially for Dy(III) and Ho(III)] with ligands based on a cyclen (1,4,7,10-tetraazacyclododecane) skeleton (e.g. H4dota derivatives, Fig. 1),23,24 which ensures kinetic inertness of these contrast agents.25,26 More recently, transition metal ion complexes have been also found to effectively shorten T1 relaxation times.27–30 Here, especially complexes of cyclam-based ligands (cyclam = 1,4,8,11-tetraazacyclotetradecane, Fig. 1) were found to be promising.31–36 In general, the cyclam derivatives are not suitable for complexation of lanthanide ions due to an inappropriately large macrocyclic cavity, but they are well known for effective binding of heavier first-row transition metal ions. In particular, Cu(II) and Ni(II) ions are bound very selectively into thermodynamically stable and kinetically inert complexes.37–41 However, some Ni(II) complexes of 1,8-bis(2,2,2-trifluoroethyl)-cyclam derivatives (1,8-tfe2cyclams, Fig. 1) showed decreased stability and kinetic inertness, and have in general very short relaxation times (in the millisecond range) due to a very short distance between the paramagnetic centre and fluorine atoms, which prevents simple measurement and places non-trivial demands on the hardware used for imaging.32 In this respect, analogous Co(II) complexes with T1 ∼ 10–15 ms seem to be more promising.34 In addition, we designed a series of ligands with larger separation of the fluorine atoms from the paramagnetic metal ion centre [1,8-(tfe-NHR)2cyclams, Fig. 1].42 It was shown that complexation of Cu(II) to these structures shortens significantly fluorine relaxation times to an optimal range (order of tenths of milliseconds).42 Some time ago, we introduced (2,2,2-trifluoroethyl)phosphinic acid as a suitable pendant arm in the cyclen-based ligand H4dotptfe (Fig. 1) and studied its lanthanide(III) complexes.43 To extend our work in this field, we report on the results of our study of a new cyclam derivative with two (2,2,2-trifluoroethyl)phosphinic acid pendant moieties in the 1,8-positions (H2L, Fig. 1).
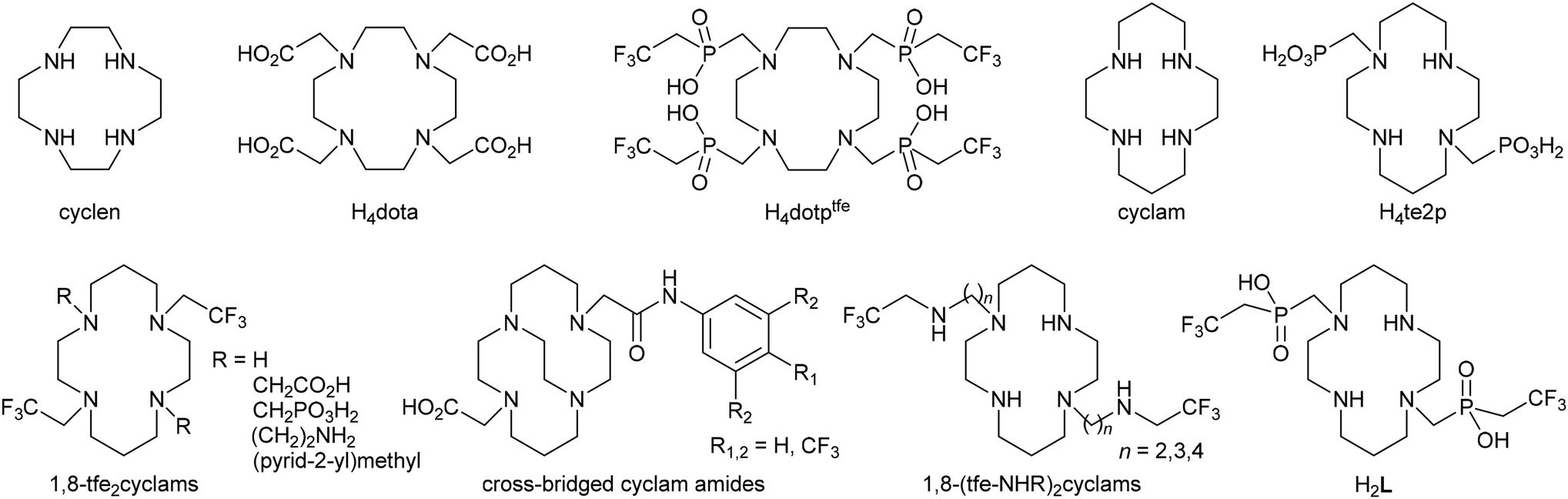 | ||
| Fig. 1 Ligands mentioned in the text. | ||
Experimental
General
Commercial chemicals (Fluka, Aldrich, CheMatech, Lachema, Fluorochem) were used as obtained. Anhydrous solvents were obtained by established procedures44 or purchased. The 1,8-dibenzyl-cyclam 1 was prepared by the reported procedure.45Thin-layer chromatography (TLC) was performed on silica-coated aluminium sheets, silica gel 60 F254 (Merck). Spots were visualized using UV light (254 and 366 nm), spraying with 0.5% ethanolic solution of ninhydrin, and dipping in 5% aq. solution of CuSO4, iodine vapour or in aq. solution of 1% KMnO4 and 2% Na2CO3.
NMR spectra were recorded on NMR spectrometers Varian VNMRS300 equipped with a pseudo-4-chanel probe (frequencies 299.9 MHz for 1H, 282.2 MHz for 19F and 121.4 MHz for 31P), Varian Inova 400 MHz (frequencies 400.0 MHz for 1H, 100.6 MHz for 13C, 376.3 MHz for 19F and 161.9 MHz for 31P) equipped with an ID-PFG probe for 19F relaxation experiments or with an ASW 4NUC probe, or Bruker Avance III 600 MHz (frequencies 600.2 MHz for 1H, 150.9 MHz for 13C and 564.7 MHz for 19F) equipped with a cryoprobe (1H, 13C, 15N) or with a BBO probe (19F). Representative NMR spectra of the prepared compounds are given in the ESI.† The spectra were acquired at 25 °C unless stated otherwise. Internal references for 1H and 13C NMR spectra were t-BuOH for D2O solutions and the CHD2OD residual peak for MeOH-d4 solutions, respectively. Aq. H3PO4 (3%) was used as the external reference for 31P NMR, and ca. 1% triflic acid for 19F NMR. These secondary references were referenced to 85% H3PO4 (0.0 ppm) and to Freon-11 (0.0 ppm) respectively. Chemical shifts are given in ppm and coupling constants at Hz. Chemical shifts of paramagnetic compounds were corrected for the bulk magnetic susceptibility effect: to the sample, a small amount of 2,2,2-trifluoroethanol was added, and the chemical shift was measured using an insert cuvette containing the secondary reference. The same insert cuvette was used to measure the chemical shift of 2,2,2-trifluoroethanol in a pure solvent, and the difference between the signals of trifluoroethanol was used for chemical shift correction of the paramagnetic compound. The T1 relaxation times of the 19F NMR signal were measured with the inversion recovery sequence. The T2 relaxation times of diamagnetic compounds were measured with the CPMG sequence on Varian Inova 400 MHz and Bruker III 600 MHz; the measurement is not accessible on VNMRS300 as the probe used does not provide accurate 180° pulses for 19F nuclei. The 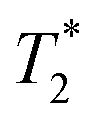 relaxation times were calculated for all paramagnetic complexes from half-widths of the NMR signals.
relaxation times were calculated for all paramagnetic complexes from half-widths of the NMR signals.
Mass spectra were recorded on a Waters ACQUITY QDa, which is a part of the Waters Arc HPLC system. Data were processed using Empower 3 software. Samples were dissolved in water, MeOH or MeCN. The HPLC was run on the same device using the Cortecs C18 2.7 μm, 4.6 × 50 mm column and H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN 100 to 0% gradient [0.1% trifluoroacetic acid (tfa) used as a modifier].
:MeCN 100 to 0% gradient [0.1% trifluoroacetic acid (tfa) used as a modifier].
The UV-Vis spectra were recorded on a spectrometer Specord 50 Plus (Analytic Jena) in a quartz–glass cell with an optical path of 1 cm in the range of 350–1100 nm.
Ligand synthesis
Synthesis of the studied ligand was performed according to Scheme 1.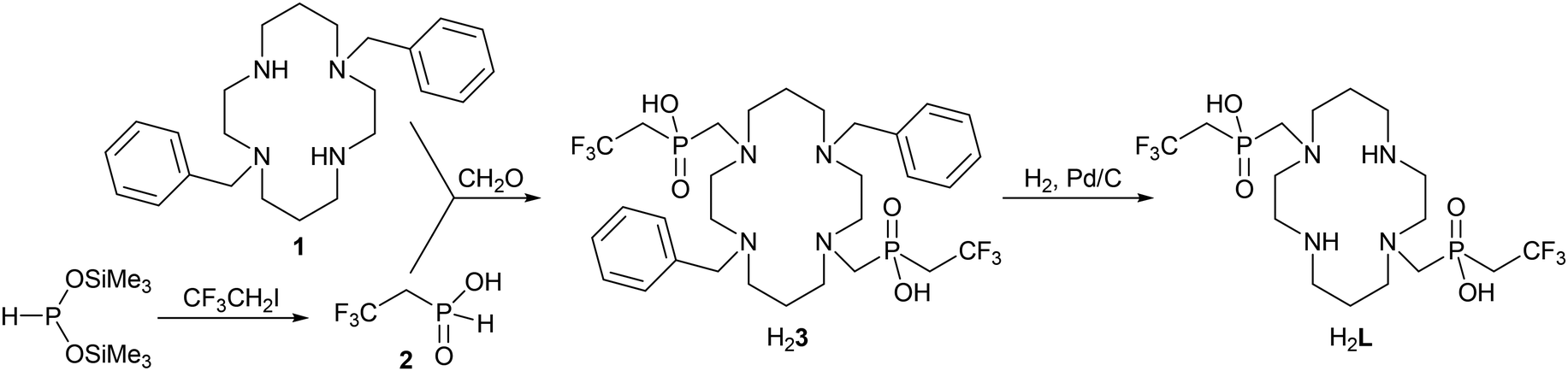 | ||
| Scheme 1 Synthesis of ligand H2L. | ||
The volatiles were evaporated on a rotary evaporator at a bath temperature of 50 °C. The remaining material was poured onto a column of a strong cation exchange resin in a H+-cycle (Dowex 50, 500 ml) and the mixture of acids was eluted with water (DIPEA was retained on the column). Acid fractions were concentrated on a rotary evaporator at 40 °C. The 1H NMR spectrum confirmed the absence of DIPEA in the eluted material. After this procedure, ethyl ester of 2 present in the initial mixture was completely hydrolysed.
A solution of Pb(OAc)2 in 20% acetic acid (1 M, 70 ml) was added into the evaporated eluate obtained above and diluted with water (50 ml). The mixture was stirred at room temperature for 3 h and the yellow precipitate of PbI2 was removed by filtration on a Büchner funnel through several layers of filtration paper. Excess Pb(II) in the filtrate was removed by bubbling hydrogen sulphide through the solution and the PbS precipitate was filtered off using a Büchner funnel with a paper mesh. Volatiles from the filtrate were removed at 50 °C using a rotary evaporator.
The product was purified on a silica column (300 ml) using conc. aq. NH3:EtOH 1:20 as a mobile phase [Rf(product) 0.65, Rf(H3PO3) 0.35]. Fractions containing the pure product were combined and evaporated giving the ammonium salt of (2,2,2-trifluoroethyl)phosphinic acid as a white solid. Free acid was obtained by passing a solution of the ammonium salt through a column of a strong cation exchange resin (Dowex 50, H+-form, 100 ml, elution with water) and evaporation of the eluate at 40 °C. Yield 9.0 g (57%) as a clear colourless oil.
NMR (ammonium salt, D2O, pD 4.6). 1H: 7.15 (d; 1H; HP; 1JHP 547), 2.63 (m; 2H; CH2). 13C{1H}: 37.5 (dq; CH2; 2JCF 27.3, 1JCP 81.3), 125.8 (qd; CF3; 1JCF 275; 2JCP 3.1). 19F: −57.7 (pseudo-q; 3JFH ≈ 3JFP ≈ 12.0). 31P{1H}: 13.3 (q, 3JPF 12.0). 31P: 13.3 (dm, 1JPH 547). MS-ESI, (+): 149.0 [M + H]+; (−): 146.7 [M − H]−. TLC (conc. aq. NH3:EtOH 1:20): Rf 0.65 (ninhydrin, yellow spot; KMnO4/Na2CO3, yellow spot).
EA (H23·6CF3CO2H·2H2O): found (calc. for C42H54F24N4O18P2, Mr 1420.81) C: 35.68 (35.51), H: 3.60 (3.83), N: 3.73 (3.94), F: 31.28 (32.09), P: 4.13 (4.36). NMR (H23·6H2O, MeOH-d4, atom numbering scheme is given in the ESI†). 1H: 2.11–2.30 (4H, m, H6); 2.33–2.40 (2H, m, H2); 2.43–2.58 (6H, m, H9 and H8); 2.60–2.67 (2H, m, H8); 2.78–2.86 (4H, m, H5 and H7); 2.91–3.01 (4H, m, H3 and H7); 3.26–3.34 (1H, m, H3); 3.34–3.41 (2H, m, H2); 3.68–3.74 (2H, m, H5); 4.21–4.27 (2H, m, H11); 4.39–4.46 (2H, m, H11); 7.47–7.53 (6H, m, H14 and H15); 7.56–7.61 (4H, m, H13). 13C{1H}: 24.56 (s, C6); 37.32 (dq, C9, 2JCP 79.6, 2JCF 28.2); 49.9 (s, C2); 50.3 (s, C5); 52.0 (s, C3); 54.9 (d, C8, 2JCP 119); 58.5 (s, C7); 58.8 (s, C11); 127.8 (q, C10, 1JCF 275.1 Hz); 130.8 (s, C12); 131.1 (s, C14); 131.9 (s, C15); 134.1 (s, C13). 19F: −58.06 (dt; 3JFH 12.3, 3JFP 6.8). 31P{1H}: 23.4 (q, 3JPF 6.9). 31P: 23.5 (m). MS-ESI, (+): 701.4 [M + H]+; (−): 699.4 [M − H]−. TLC (conc. aq. NH3:EtOH 1:10): Rf 0.80 (CuSO4, green spot).
EA [(H6L)Cl4·4H2O]: found (calc. for C16H44Cl4F6N4O8P2Mr 738.29) C: 27.07 (26.03), H: 5.30 (6.01), N: 7.94 (7.59), F: 15.63 (15.44), P: 8.16 (8.39), Cl: 19.36 (19.21). NMR [(H6L)Cl4·4H2O, D2O, pD 9.0 (NaOD), atom numbering scheme is given in the ESI†]. 1H: 1.97 (4H, bs, H6); 2.72 (4H, pseudo-p, 3JHF ≈ 3JHP ≈ 12.4, H9); 2.85 (4H, bs, H2 and H8); 2.92 (8H, bs, H2 and H7); 3.12 (4H, bs, H3); 3.21 (4H, t, 3JHH 5.9, H5). 13C{1H}: 24.1 (s, C6); 35.1 (dq, C9, 1JCP 79.5, 2JCF 28.6); 45.9 (s, C3); 48.2 (s, C5); 54.5 (d, C8, 1JCP 117.3); 55.4 (d, C2, 3JCP 6.6); 58.8 (d, C7, 3JCP 5.5); 125.7 (qd, C10, 1JCF 274.9, 2JCP 3.2). 19F: −57.34 (dt; 3JFH 12.0, 3JFP 7.2). 31P{1H}: 28.5 (q, 3JPF 7.3). 31P: 28.5 (m). MS-ESI, (+): 521.2 [M + H]+, 1041.5 [2M + H]+. TLC (conc. aq. NH3:EtOH 1:5): Rf 0.80 (CuSO4, violet spot).
Preparation of complexes of H23
Mixing H23·6CF3CO2H·2H2O (50 mg) with Cu(OAc)2·H2O (8 mg, 1.1 equiv.) in water:MeOH (1:1, 2 ml) and neutralization with 2% aq. LiOH afforded an emerald green precipitate of the pc-[Cu(3)] complex which is only sparingly soluble in alcohols. A few green single crystals were isolated after the concentration of the mother liquor and acetone vapour diffusion. The X-ray structural analysis of these crystals revealed the composition as pc-[Cu(3)]·Cu(AcO)1.8(tfa)0.2·4H2O·0.5acetone.
Attempts to prepare complexes of H23 with Co(II), Ni(II) and Zn(II) by mixing H23 with MCl2 in aq. MeOH solutions and addition of aq. ammonia as a base led to the formation of precipitates insoluble in all common solvents (H2O, MeOH, EtOH, DMSO, DMF, CHCl3).
Preparation of complexes of H2L
Solutions of complexes for 19F NMR characterization were prepared directly in the NMR tube by mixing 150 μl of 18.4 mM solution of (H6L)Cl4·4H2O used for potentiometry, HEPES buffer (300 μl, 0.5 M, pH 7.4) and an appropriate volume of the solution of the metal salt (ca. 50 mM). The Zn(II) complex was prepared by mixing the ligand and a slight excess (1.2 equiv.) of ZnCl2 to ensure the absence of free H2L as, due to diamagnetism of the sample, the signal of free H2L would be hardly distinguishable from the signal(s) of the complex isomers; it would complicate the interpretation of the NMR spectra. The Ni(II) and Co(II) complexes were prepared using a slightly sub-stoichiometric amount of Ni(NO3)2 or Co(NO3)2, respectively. In these cases, the signal of the diamagnetic H2L excess was used as an internal reference. For the Co(II) complex, the NMR tube was flushed with Ar and pH was increased to 8.0 with aq. NaOH to ensure the full complexation of Co(II) ions (according to the results of the potentiometric study, see below). The Co(II) complex showed one 19F NMR signal (−38.6 ppm, Fig. S1†) which remained unchanged with further standing (only some negligible oxidation to diamagnetic Co(III) was observed if the tube was opened to air; a very small diamagnetic signal appeared); further study revealed its cis-O,O′-[Co(L)] geometry (see below). However, solutions containing Ni(II) and Zn(II) complexes showed complicated spectra. Therefore, the samples were equilibrated for several days at 80 °C. The measured 19F NMR spectra of the fully equilibrated solutions showed one major signal of the Ni(II) complex at −47.1 ppm (∼80%) besides two minor signals at −43.7 and −47.7 ppm with equal intensity (each ∼10%), as shown in Fig. S2.† Based on a further 19F NMR study, the cis-O,O′-[Ni(L)] geometry was suggested with R/S isomerism of the phosphorus atom (see below). In the case of the Zn(II)–H2L system, an isomeric mixture of three complexes was observed (pseudo-quartets due to 19F–31P/1H couplings centred at −57.15 pm, ∼40%; −57.23 ppm, ∼55%; −57.30 ppm, ∼5%, Fig. S3†). In the case of the diamagnetic Zn(II)–H2L system, it was possible to acquire the 31P NMR spectra also, which were consistent with the 19F NMR data (three signals in 31P NMR spectra, 30.1 pm, ∼55%; 29.4 ppm, ∼5%; 28.7 ppm, ∼40%, Fig. S4†). The same 19F and 31P NMR spectra were observed for several independently prepared samples of the [Zn(L)] complex after the equilibration.
An alternative way was used to prepare complex solutions without additional salts and excess ligand: a solution of H2L (46.0 mM, 840 μl, ca. 20 mg of H2L) was mixed with 1.5 equiv. of CoCO3 (7 mg) or with freshly precipitated M(OH)2 excess [M = Ni, Zn prepared by the addition of aq. LiOH to aq. MCl2 (2.0 equiv.) and washing the precipitate with water by repeated centrifugation]. The mixtures were stirred in a closed vial at 80 °C for 5 d. After cooling, the excess of the solid phase was filtered off and the filtrate was concentrated. The measured 19F NMR spectra confirmed the absence of free H2L and showed the same signals of the complexes as found in the samples mentioned above, including the same ratio of the isomeric species.
The formation of the complexes was confirmed also by mass spectrometry. MS-ESI, (+): [Co(L)]: 578.2 [59CoL+ H]+; 577.2, [59CoL]+; [Ni(L)]: 577.1 [58NiL + H]+; [Zn(L)]: 583.2 [64ZnL + H]+.
Single crystals of cis-O,O′-[Co(L)]·LiCl·3H2O were prepared by acetone vapour diffusion into a solution of the complex prepared from a mixture of H2L and CoCl2 neutralized with LiOH.
Single crystals of cis-O,O′-[Ni(L)]·3.5H2O and cis-O,O′-[Zn(L)]·2H2O·0.5acetone were formed on acetone vapour diffusion into solutions of the corresponding complexes prepared from M(OH)2 as described above.
Alternatively, 46.0 mM solution of H2L (840 μl, ca. 20 mg of H2L) was mixed with freshly precipitated Cu(OH)2 prepared by addition of 2% aq. LiOH to aq. solution of CuCl2·2H2O (13 mg, 2.0 equiv.) and washing the precipitate with water by repeated centrifugation. The mixture was stirred in a closed vial at room temperature overnight. The excess of Cu(OH)2 was filtered off and the filtrate was concentrated. The TLC analysis showed exclusive formation of the pc-[Cu(L)] isomer. The measured 19F NMR spectra confirmed the absence of free H2L and presence of the signal belonging to the pc-[Cu(L)] complex as mentioned above. TLC (conc. aq. NH3:EtOH 2:3): Rf 0.80 (blue spot, Fig. S7†). MS-ESI: (+): 581.9 [M + H]+, 603.9 [M + Na]+.
Single crystals of pc-[Cu(L)]·3H2O were formed on acetone vapour diffusion into a concentrated aqueous solution of the complex.
Alternatively, 46.0 mM solution of H2L (1.25 ml, ca. 30 mg of H2L) and CuCl2·2H2O (10 mg, 1 equiv.) was neutralized to pH 7.4 with LiOH and heated at 90 °C for 5 d. The volatiles were evaporated and the complex was purified by chromatography on silica using conc. aq. NH3:EtOH 2:3 as a mobile phase. The violet-coloured fraction was evaporated to dryness. The measured 19F NMR spectra confirmed the absence of free ligand and presence of one signal of the trans-Cu(II)-complex. TLC (conc. aq. NH3:EtOH 2:3): Rf 0.95 (violet spot, Fig. S7†). MS-ESI: (+): 581.9 [M + H]+, 603.9 [M + Na]+.
Single crystals of trans-O,O′-[Cu(L)]·NH4(Cl0.54Br0.46) were formed on acetone vapour diffusion into concentrated chromatographic fractions containing the complex.
Single crystals of trans-O,O′-[Cu(L)]·(H3O)(ClO4)·H2O were crystallized by cooling of the saturated solution of the complex in boiling 1 M HClO4.
Single-crystal X-ray diffraction study
The selected crystals were mounted on a glass fibre in a random orientation and the diffraction data were collected by using a Nonius KappaCCD diffractometer equipped with a Bruker APEX-II CCD detector using Mo-Kα radiation (λ = 0.71073 Å) at 150 K (Cryostream Cooler, Oxford Cryosystem) {H23·6H2O, H2L·4AcOH and cis-O,O′-[Ni(L)]·3.5H2O}, or with a Bruker D8 VENTURE Duo diffractometer with an IμS micro-focus sealed tube using Mo-Kα radiation at 150 K {pc-[Cu(3)]·Cu(AcO)1.8(tfa)0.2·4H2O·0.5acetone, pc-[Cu(L)]·3H2O and trans-O,O′-[Cu(L)]·NH4(Cl0.54Br0.46)} or at 120 K {H2L·LiCl·6H2O, cis-O,O′-[Co(L)]·LiCl·3H2O, trans-O,O′-[Cu(L)]·(H3O)(ClO4)·H2O, cis-O,O′-[Zn(L)]·2H2O·0.5(C3H6O)}, or using Cu-Kα radiation (λ = 1.54178 Å) at 120 K [H23·6CF3CO2H·2H2O, (H4L)Cl2·4H2O].Data were analysed using the SAINT V8.34A–V8.40B software package (Bruker AXS Inc., 2015–2019). Data were corrected for absorption effects using the multi-scan method (SADABS).46 All structures were solved by the direct methods (SHELXT2014)47 and refined using full-matrix least-squares techniques (SHELXL2014).48 Details on structure refinement are given in the ESI.†
All the data for the structures reported here have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 2262037–2262048 (for an overview of experimental crystallographic data, see Table S1†).
Potentiometric study
The methodology of potentiometric titrations and processing of the experimental data were analogous to those previously reported.39,49 Titrations were carried out in a vessel tempered to 25 ± 0.1 °C at the ionic strength I = 0.1 M (NMe4)Cl. The water ion product, pKw = 13.81, and stability constants of M(II)–OH− systems were taken from the literature.50The ligand stock solution was prepared by the dissolution of (H6L)Cl4·4H2O (1.3217 g) in a 100 ml volumetric flask. The solid (H6L)Cl4·4H2O is slightly unstable due to the loss of HCl and water and, thus, the exact ligand concentration was determined by 19F qNMR (18.4 mM) using standardized aq. solution of tfa. This value agrees well with the value calculated during the fitting of the protonation constants (difference < 1%). The overall protonation constants βn are concentration constants and are defined by βn = [HnL]/([H]n·[L]) (stepwise protonation constants are defined as logK1 = logβ1; logKn = logβn − logβn−1). The overall stability constants are defined by the general equation βhml = [MmHhLl]/([M]m·[H]h·[L]l). Here, the formation of only M:L = 1:1 complexes (m = l = 1) was suggested. The constants (with their standard deviations) were calculated with the OPIUM program.51 The protonation constants were determined using standard titrations in the pH range 1.6–12.1 with cL = 0.004 M and starting volume approx. 5 ml (pH means “analytical” pH, –log[H+]). Equilibrium was established slowly in the metal(II)-containing systems. Therefore, stability constants of the complexes were obtained by the “out-of-cell” method. Each solution corresponding to one titration point of a common titration was prepared under an Ar stream in tubes with ground joints (pH 1.6–6.5, three titrations with 15 points) from the ligand, metal ion and HCl/(NMe4)Cl stock solutions and water (starting volume ca. 1 ml, M:L molar ratio 0.95:1, cL = 0.004 M). Then, a known amount of (NMe4)OH standard solution was added under Ar. The tubes were firmly closed with stoppers and the solutions were left to equilibrate at room temperature. Afterwards, pH was measured with a freshly calibrated combined glass electrode in each tube. One set of tubes containing the Cu(II)–H2L system was equilibrated for three weeks and the others for four weeks. Systems with Co(II), Ni(II) and Zn(II) were equilibrated for six weeks (one set of each metal ion) and eight weeks (the remaining sets). Identical results were obtained in both time points for each metal ion system proving a reaching the thermodynamic equilibrium after the shorter time period used.
Study of kinetic inertness of the complexes
The stock solution of the complex was mixed with water and stock solution of HCl to reach the final HCl concentration of 1.0 M in order to obtain dissociation data comparable with the related systems.32 The acid-assisted dissociation of Cu(II) complexes was followed by a decrease of their CT bands in the UV-Vis spectra (315 and 280 nm for the pc- and the trans-isomer, respectively); the temperature of the experiment was chosen to obtain data within a reasonable time (<∼24 h): 25 °C and 90 °C for the pc- and the trans-isomer, respectively. Acid-assisted dissociation of the Co(II) and Ni(II) complexes was followed by 19F NMR (a decrease in the intensity of the signal of the complex, an increase in the intensity of the signal of the free ligand, and integrated with respect to tfa used as an external reference). Temperatures 25 °C and 90 °C were used for the Co(II) and the Ni(II) complexes, respectively.Results and discussion
Synthesis of H2L
Synthesis of the studied ligand was performed as shown in the Experimental section (Scheme 1).The key starting compound, 2,2,2-trifluoroethylphosphinic acid 2, was prepared by an Arbuzov-type reaction between 2,2,2-trifluoroethyl iodide and bis(trimethylsilyl)hypophosphite which was generated in situ from hypophosphorous acid and trimethylsilyl chloride in the presence of N,N-diisopropylethylamine (DIPEA) as a base. The silyl ester intermediate was hydrolysed with aqueous ethanol leading to the product 2 with conversion in the range of 60–80% (according to the intensity of the 31P NMR signal (14.5 ppm, 1JPH = 576 Hz) and phosphorous acid as a dominant by-product (1.8 ppm, 1JPH = 660 Hz). Despite an excess of the alkylation agent used, no bis(2,2,2-trifluoroethyl)phosphinic acid was formed during the reaction, probably due to the low nucleophilicity of the monosubstituted derivative and volatility of the starting alkylation agent (Ar flow was used to ensure an inert atmosphere).
DIPEA was removed on a strong cation exchange resin in the H+-form and the acid eluate containing predominantly product 2, phosphorous acid and hydroiodic acid was evaporated at <40 °C (in bath) to avoid oxidation/disproportionation of product 2. However, under such conditions, it was impossible to quantitatively remove HI. Any HI present in the crude product mixture forms elemental iodine on standing which oxidizes the compound 2 to 2,2,2-trifluoroethylphosphonic acid. HI was removed with an excess of lead(II) which was then precipitated by gaseous H2S. The separation of product 2 and phosphorous acid was accomplished by chromatography on silica using aq. NH3:EtOH 1:20 as a mobile phase. After chromatography, the product was isolated in the form of a semi-solid ammonium salt. It was converted to free acid on a strong cation exchanger in the H+-form. Free acid 2 was isolated as a colourless oil in a moderate yield.
A Mannich-type reaction between 1,8-dibenzyl-cyclam 1, 2,2,2-trifluoroethylphosphinic acid 2 and paraformaldehyde was performed in a strong acid solution (water:trifluoroacetic acid 1:1) at 60 °C. Aq. HCl usually used as a solvent for this type of reaction cannot be used here due to the insolubility of the starting amine 1 in the solvent. In less acidic media (acetic acid), the reaction did not proceed even at 80 °C but the oxidation of the P–H bond was observed instead. The product H23 is insoluble in the reaction medium and was separated by filtration as an adduct with trifluoroacetic acid. Re-crystallization of the product from water:trifluoroacetic acid (1:1) afforded single crystals of H23·6CF3CO2H·2H2O suitable for X-ray diffraction analysis (see the ESI†). Despite the presence of several strong acid molecules in the solid-state structure, the macrocycle is only double-protonated and the protons are located on amino groups bearing the benzyl groups. Trifluoroacetic acid was removed by the re-crystallization of H23·6CF3CO2H·2H2O from water and H23·6H2O was obtained. Slow cooling of the saturated hot aq. solution afforded crystals suitable for the X-ray diffraction study (see the ESI†).
Debenzylation of the intermediate H23 to H2L was performed in 90% v/v aq. acetic acid under a H2 atmosphere using Pd/C as a catalyst. After catalyst removal, ligand H2L was isolated by the crystallization of the concentrated reaction mixture as a zwitterionic adduct with acetic acid H2L·4AcOH. The procedure afforded single crystals of sufficient quality for the determination of the crystal structure. It revealed deprotonation of the phosphinate arms and protonation of the secondary amino groups, i.e. H2L is present in a zwitterionic form (Fig. 2).
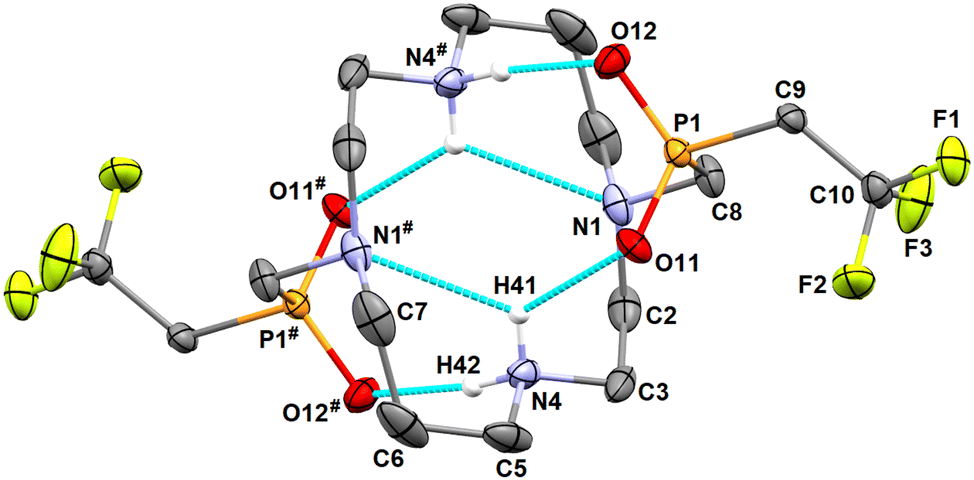 | ||
| Fig. 2 Molecular structure of H2L found in the crystal structure of H2L·4AcOH. Carbon-bound hydrogen atoms are omitted for clarity. Intramolecular hydrogen bonds are shown as turquoise dashed lines. Selected centrosymmetry-related atoms are labelled with #. | ||
The zwitterionic molecule of H2L possesses a centre of symmetry and adopts a “common” angular conformation similar to those found in the crystal structure of the cyclam itself and in its diprotonated forms.52,53 The phosphinate pendant arms are turned above/below the macrocyclic cavity and stabilize the molecular conformation by strong intramolecular hydrogen bonds [d(N4#⋯O11) = 2.71 Å, d(N4⋯O12) = 2.85 Å]. Both molecules of the co-crystallized acetic acid are protonated and are bound by very strong hydrogen bonds to the phosphinate oxygen atoms {d(O[AcOH]⋯O11/O12) = 2.58 Å, Fig. S21†}.
The ligand H2L can be easily converted into hydrochloride (H6L)Cl4·4H2O by the crystallization of H2L·4AcOH from aq. HCl (6 M). Further re-crystallization of this material from water afforded (H4L)Cl2·4H2O which was structurally characterized. In the presence of HCl, all macrocycle amino groups of the ligand molecule are protonated which leads to the conformation (3,4,3,4)-D52,53 of the cyclam ring. The nitrogen atoms bearing the phosphinate pendant arms form the corners of the (3,4,3,4)-D rectangle (Fig. 3). Such a conformation maximizes the separation of the positive charges but avoids the formation of intramolecular hydrogen bonds, and only a wide network of intermolecular contacts is formed.
 | ||
| Fig. 3 Molecular structure of one of independent (H4L)2+ cations found in the crystal structure of (H4L)Cl2·4H2O. Carbon-bound hydrogen atoms are omitted for clarity. Selected centrosymmetry-related atoms are labelled with #. | ||
Attempts to structurally characterize the ligand in its deprotonated form by the crystallization of H2L solution neutralised by LiOH to pH ca. 9 (a higher pH leads to a slow degradation of the ligand molecule) led to the formation of single crystals of zwitterionic H2L as a LiCl adduct, H2L·LiCl·6H2O. The protonation of the secondary amino groups found in this crystal structure points to their high basicity (see the ESI, Fig. S22†).
Syntheses and solid-state structures of the complexes
Compound H23 can also potentially serve as a ligand for transition metal ions. It was tested by the complexation of copper(II) ions. However, the compound H23 is insoluble in water, and only slightly soluble in aq. MeOH and EtOH mixtures. Therefore, the complexation reaction was performed in these media. The reaction of H23 with Cu(II) affords an emerald green complex somewhat soluble in aq. MeOH, EtOH or i-PrOH which can be precipitated by the addition of acetone. Despite a number of attempts, a few single crystals suitable for the determination of the molecular structure of the complex were isolated only in the case where a slight excess of copper(II) acetate was used as a metal source and the ligand was used in the form of H23·6CF3CO2H·2H2O. The X-ray structural analysis revealed the composition of such crystals to be pc-[Cu(3)]·Cu(AcO)1.8(tfa)0.2·4H2O·0.5acetone with a pentacoordinated (pc) geometry of the [Cu(3)] species, leaving one of the phosphinate pendant arms uncoordinated (Fig. 4). Similarly to other pc-complexes of cyclam-1,8-bis(methylphosphorus acids) and related N-alkylated compounds (1,8-H4te2p, Fig. 1),39 macrocycles adopt the conformation trans-I (according to commonly accepted nomenclature, although the term “trans” is misleading in this case as there is no steric possibility to occupy two trans positions above and below the plane of the macrocycle).54 The coordination sphere of the central Cu(II) ion is intermediate between a trigonal bipyramid (nitrogen atoms N1 and N8 bearing the phosphinate pendant arms placed in apical positions) and a tetragonal pyramid (a N4 base with an apical oxygen atom), as evidenced by the criterion τ = 0.505.55 Excess of Cu(II) and acetate and trifluoroacetate anions present in the solution form a centrosymmetric dimeric core of copper-acetate-like structural motif where the apical positions of the metal coordination spheres are occupied by an oxygen atom of the uncoordinated phosphinate pendant arm from the pc-[Cu(3)] species (Fig. S8†). Selected geometric parameters of the coordination sphere are outlined in Tables 1 and S2.† No changes in the UV-Vis spectra upon heating the solution indicate that pc-[Cu(3)] species with the conformation trans-I cannot be rearranged into other isomers with a different macrocycle conformation. The stability of the conformation trans-I is in agreement with previous observations for the complexes of 1,8-H4te2p ligands alkylated on N4/11 nitrogen atoms (Fig. 1).39 | ||
| Fig. 4 Molecular structure of pc-[Cu(3)] complex found in the crystal structure of pc-[Cu(3)]·Cu(AcO)1.8(tfa)0.2·4H2O·0.5acetone. Hydrogen atoms are omitted for clarity. | ||
| Bond (Å) | pc-[Cu(3)] | cis-O,O′-[Co(L)] | cis-O,O′-[Ni(L)] | pc-[Cu(L)] | trans-O,O′-[Cu(L)]a | trans-O,O′-[Cu(L)]b | cis-O,O′-[Zn(L)] | ||
|---|---|---|---|---|---|---|---|---|---|
| Mol. 1 | Mol. 2 | Mol. 1 | Mol. 2 | ||||||
| a trans-O,O′-[Cu(L)]·NH4(Cl0·54Br0.46). b trans-O,O′-[Cu(L)]·(H3O)(ClO4)·H2O. # Centrosymmetry-related atoms (N8 = N1#, N11 = N4#, O21 = O11#). | |||||||||
| M–N1 | 2.102(3) | 2.182(2) | 2.212(2) | 2.154(1) | 2.067(1) | 2.069(1) | 2.068(1) | 2.099(1) | 2.183(1) |
| M–N4 | 2.122(3) | 2.115(2) | 2.124(2) | 2.104(1) | 2.017(1) | 2.006(1) | 2.021(1) | 2.024(2) | 2.130(1) |
| M–N8 | 2.088(3) | 2.174(2) | 2.189(1) | 2.129(1) | 2.064(1) | 2.069(1)# | 2.068(1)# | 2.099(1)# | 2.217(1) |
| M–N11 | 2.119(3) | 2.134(2) | 2.121(2) | 2.083(1) | 2.022(1) | 2.006(1)# | 2.021(1)# | 2.024(2)# | 2.161(1) |
| M–O11 | 2.093(3) | 2.094(1) | 2.081(1) | 2.071(1) | 2.148(1) | 2.433(1) | 2.412(1) | 2.372(1) | 2.154(1) |
| M–O21 | — | 2.085(1) | 2.109(1) | 2.113(1) | — | 2.433(1)# | 2.412(1)# | 2.372(1)# | 2.080(1) |
Attempts to prepare complexes of H23 with other studied transition metal ions [Co(II), Ni(II) and Zn(II)] by mixing H23 with metal chlorides in aq. MeOH solutions and adding ammonia as a base led to the formation of precipitates insoluble in common solvents which cannot be further studied in solution and, therefore, other study of the complexes was impossible. Different behaviours of the Cu(II) complexes and precipitated phases formed with other transition metal ions come probably from the molecular structures – except for the Cu(II) ion, where a pentacoordinated species with the hydrophilic phosphinate arm pointing out of the hydrophobic coordination sphere is formed, other metal ions prefer an octahedral sphere (see below). In these cases, the metal ions are hexadentately wrapped with the (3)2− anion. Hydrophobic benzyl and trifluoroethyl side groups point away and give to the electroneutral complex species a non-polar character. The molecules are thus hydrophobically packed in the solid state and are intact to the solvent.
All studied transition metal ions, Co(II)–Zn(II), were successfully complexed with H2L. In a typical complexation experiment, an excess of freshly prepared metal(II) hydroxide was reacted with H2L. After several hours, the excess of M(OH)2 was filtered off. As Co(OH)2 could be possibly oxidized, the cobalt(II) complex was prepared by the neutralization of H2L and CoCl2 solution with LiOH or by the reaction of aq. solution of H2L with CoCO3. The absence of the free ligand in solutions of the complexes was confirmed by the 19F NMR spectra. In the spectra, one signal was present in the case of the Co(II)–H2L system but for the other complexes a number of isomeric complex species were obviously initially formed. Heating of the solutions led to spectra with one dominant signal [Ni(II) complex], one broad symmetric signal [Cu(II) complex] and several signals [Zn(II) complex]. The solutions were concentrated and a slow addition of acetone afforded single crystals suitable for structural characterization. For the Co(II), Ni(II) and Zn(II) complexes, crystals contain the cis-O,O′-[M(L)] complex species having a somewhat distorted octahedral sphere; particularly, bond angles related to small five-membered chelate rings are somewhat smaller than 90° (Tables 1 and S2†). In all these complexes, the cyclam conformation cis-V was found.54 All coordinated nitrogen atoms of the macrocycle (N1, N4, N8, N11) have the same stereochemical descriptors according to the Cahn–Ingold–Prelog rules, leading to enantiomeric R,R,R,R and S,S,S,S pairs. Due to crystallographic centrosymmetry, both enantiomers are present in the crystal structures. However, coordination of the phosphinate pendant arm leads to a new stereocentre on the phosphorus atoms. In the solid state, they have the same absolute configuration as the nitrogen atoms of the macrocycle, resulting in the presence of all-N-(R)-all-P-(R) and all-N-(S)-all-P-(S) species.
The cobalt(II) complex crystallizes in the form of cis-O,O′-[Co(L)]·LiCl·3H2O. In the crystal structure, two independent complex molecules are present in the independent unit. However, their geometry is very similar and, therefore, only one of them is shown in Fig. 5. Structural overlay of both independent complex molecules is shown in Fig. S9,† and selected geometric parameters are listed in Tables 1 and S2.†
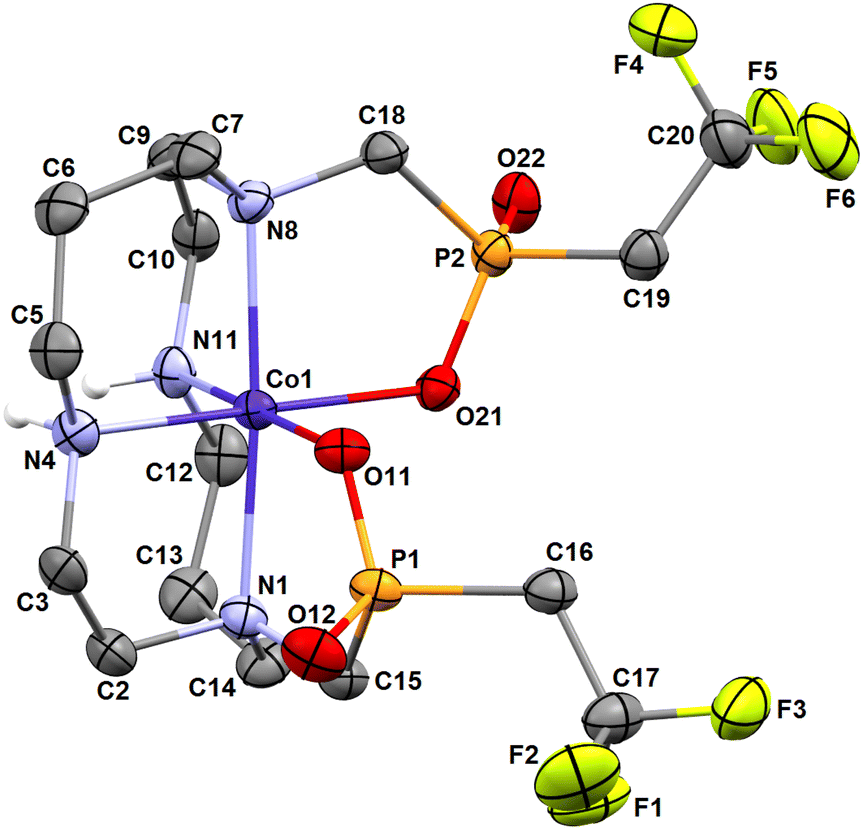 | ||
| Fig. 5 Molecular structure of cis-O,O′-[Co(L)] complex found in the crystal structure of cis-O,O′-[Co(L)]·LiCl·3H2O. Carbon-bound hydrogen atoms are omitted for clarity. | ||
In the case of the Ni(II)–H2L system, single crystals of the composition cis-O,O′-[Ni(L)]·3.5H2O were isolated. This isomer is stable and is not rearranged to the trans-O,O′ type even on prolonged reflux in an aq. solution, as evidenced by no changes in the 19F NMR spectra. Similar preferential formation of the cis isomer was previously observed for the Ni(II)–H4te2p system.56 The molecular structure of the cis-O,O′-[Ni(L)] complex is very similar to that of the Co(II) complex discussed above (see Tables 1 and S2†) and is shown in Fig. S10.†
From an isomeric mixture of Zn(II) complexes, crystals suitable for diffraction analysis had the composition cis-O,O′-[Zn(L)]·2H2O·0.5acetone. The geometry of the complex species is very similar to that of the Co(II) and Ni(II) complexes discussed above (Fig. S11, Tables 1 and S2†).
If the complexation of Cu(II) proceeds in a slightly acidic solution (pH 3–4) and at a low temperature, a blue isomer is formed exclusively, which was identified by X-ray diffraction as the pentacoordinated species pc-[Cu(L)] with the cyclam conformation trans-I.54 This blue isomer gradually rearranges to the violet one upon prolonged heating (90 °C, 5 d, Fig. S7†) which was identified as trans-O,O′-[Cu(L)] with the cyclam conformation trans-III.54 If the pH used for the complexation reaction was 7.4, a small amount of the trans isomer was formed besides the major pc-[Cu(L)] isomer. Both isomers can be separated by TLC/HPLC. From the mixture, the isomers can be isolated by column chromatography on silica using aq. NH3–EtOH mixture.
The pc-isomer is a kinetic product of the complexation reaction and, at room temperature, it rearranges only very slowly into the trans one, which is obviously the thermodynamic product; a spot of the trans-isomer detectable by TLC appears in a stock solution of the pc-isomer after standing at room temperature for several months. The rearrangement can be completed after refluxing an aq. solution of the pc-isomer for several days (rearrangement is somewhat faster in neutral than in the acidic solution). This isomerism is fully analogous to the behaviour of Cu(II) complexes of 1,8-H4te2p (Fig. 1).39
The molecular structures of both isomers are shown in Fig. 6 and 7. Similarly to the pc-[Cu(3)] species discussed above and to analogous complexes of the 1,8-H4te2p ligand family39 and the recently reported pc-complex with cyclam derivative bearing two [di(phenyl)phosphoryl]methyl pendant arms,57 the pc-[Cu(L)] species adopts an intermediate structure between a trigonal bipyramid and a square pyramid (τ = 0.520).55 The trans-O,O′-[Cu(L)] complex is a tetragonal bipyramid with an axial coordination of the pendant arms elongated by the Jahn–Teller phenomenon. Similar elongation was observed in the related structure of trans-O,O′-[Cu(1,8-H2te2p)].39 Selected geometric parameters of the reported structures are outlined in Tables 1 and S2.†
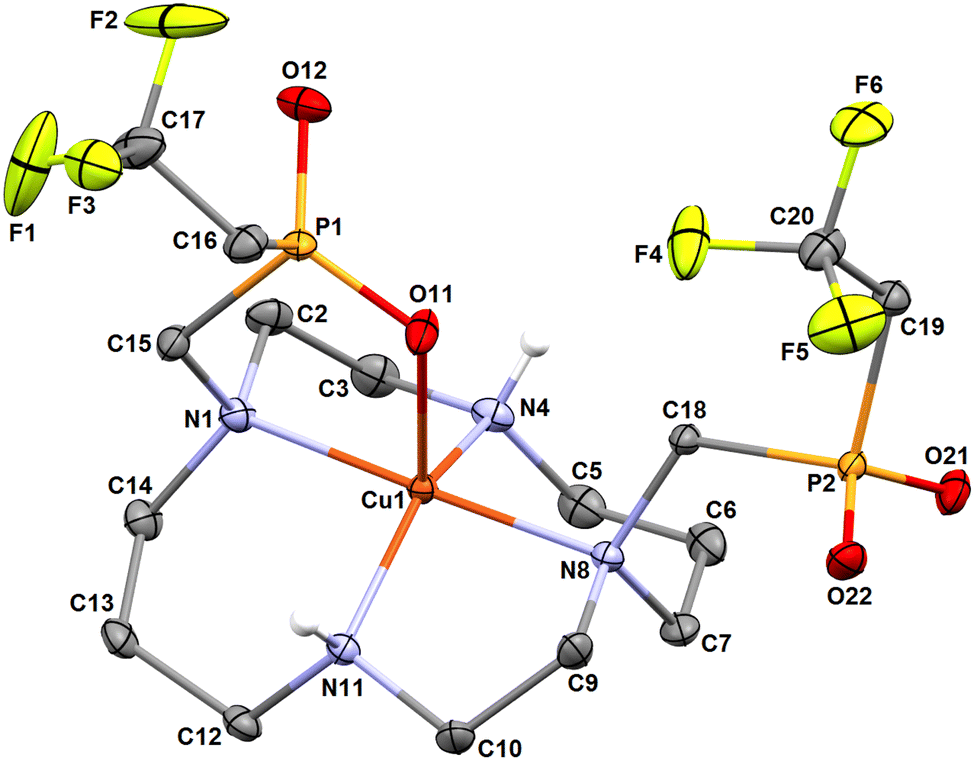 | ||
| Fig. 6 Molecular structure of pc-[Cu(L)] complex found in the crystal structure of pc-[Cu(L)]·3H2O. Carbon-bound hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 7 Molecular structure of trans-O,O′-[Cu(L)] complex found in the crystal structure of trans-O,O′-[Cu(L)]·NH4(Cl0.54Br0.46). Carbon-bound hydrogen atoms are omitted for clarity. Selected centrosymmetry-related atoms are labelled with #. | ||
During the preliminary test of kinetic inertness of trans-O,O′-[Cu(L)] (see below), single crystals with the composition trans-O,O′-[Cu(L)]·(H3O)(ClO4)·H2O were crystallized by cooling of a saturated solution of the complex in boiling 1 M HClO4. The formation of this phase clearly indicates the extreme kinetic inertness of the complex. In the crystal structure, the oxonium ion serves as a bridge between non-coordinated oxygen atoms of the pendant arms of two neighbouring trans-O,O′-[Cu(L)] complex molecules (Fig. S12†). However, geometries of the complex species is essentially the same as those observed in the above discussed crystal structure of trans-O,O′-[Cu(L)]·NH4(Cl0.54Br0.46) (Tables 1 and S2†).
Solution equilibrium studies
The acid–base behaviour of H2L and stability of its complexes were studied by potentiometric titrations. The calculated constants are listed in Table 2. Similarly to other cyclam-based ligands, H2L shows two protonation constants in the strongly alkaline region (logK1,2 > 10). They correspond to the protonation of the secondary amino groups of the macrocycle and it is consistent with the structure of the ligand's zwitterionic form found in the solid state. In the pH range used for titration (starting from pH 1.6), no further protonation was observed. It points to the low basicity of the amino groups bearing the phosphinate moieties and to the high acidity of the (2,2,2-trifluoroethyl)phosphinic acid pendant arms. It is consistent with the observation that the ligand H2L crystallizes from aq. acetic acid as the adduct with protonated AcOH (Fig. 2). It should be noted that the compound H23 crystallizes from aq. trifluoroacetic acid similarly to the zwitterionic adduct with a protonated trifluoroacetic acid molecule, H23·6CF3CO2H·2H2O. To obtain the (H4L)2+ species with a fully protonated macrocycle, crystallization from aq. HCl had to be used (Fig. 3). Consistent with the absence of further protonation in the pH range of potentiometric titration, no change of 1H, 13C, 19F and 31P chemical shifts was observed in the pH range 1–10, and third/more protonation(s) can be suggested to proceed on tertiary amino groups of the macrocycle with logKh < 1 as can be seen from the chemical shift change in strongly acidic solutions (Fig. S13†). The distribution diagram of the ligand is shown in Fig. S14.† Compared to the related 1,8-phosphinoxide derivative57 and the ligands of the H4te2p phosphonate family,58 the ring basicity of H2L (logK1 + logK2) is lower as a consequence of the electron-withdrawing character of the phosphinate moieties.59
Kh) of H2L and its stability constants (logβML) with selected transition metal ions, and comparison with related ligands. Charges of the species are omitted for clarity
| Equilibrium | H2Lb | 1,8-H4te2p | Cyclam | H4tetai |
|---|---|---|---|---|
|
a Mixture of isomers (see text).
b This work.
c Ref. 58.
d Overall constant logβ2 over two undistinguishable protonation steps.
e Ref. 40.
f Ref. 39.
g Ref. 60.
h Ref. 37.
i Ref. 38.
|
||||
| H + L = HL | 11.755(3) | —c,d | 11.29g | 10.52 |
| H + HL = H2L | 10.599(3) | 26.41c,d | 10.19g | 10.18 |
| Co + L = cis-[Co(L)] | 14.00(8) | 19.28e | 14.3h | 16.38 |
| Ni + L = cis-[Ni(L)] | 16.46(8)a | 21.99e | 22.2h | 19.83 |
| Cu + L = pc-[Cu(L)] | 22.76(5) | 25.40f | 28.1h | 20.49 |
| Zn + L = [Zn(L)] | 15.79(3)a | 20.35e | 15.2h | 16.40 |
Complexation reaction with transition metal ions is relatively slow and, therefore, an out-of-cell method had to be used, with equilibration times of 3 weeks for the Cu(II)–H2L system and 6 weeks for the other ions. Under these conditions, pentacoordinated isomer pc-[Cu(L)] and octahedral cis-isomers of [Co(L)] and [Ni(L)] complexes were formed exclusively, as identified by TLC/HPLC and 19F NMR spectroscopy (see below). However, for the Zn(II)–H2L system, a mixture of at least three isomers was formed according to the 19F NMR spectra. The same mixture of isomers was obtained in independent reactions (see above). According to the Williams–Irving trend, H2L shows a very high selectivity for Cu(II) over its neighbours Ni(II) and Zn(II). The selectivity is about 6–7 orders in magnitude, similarly to that found for related 1,8-disubstituted ligands.39,40,57 Although the stability constants of the complexes are in general somewhat lower than those reported for the cyclam itself, 1,8-H4te2p and H4teta, the stabilities are still sufficient for potential applications as the metal ions are quantitatively complexed under physiological conditions (and the formed complexes are kinetically inert, which is in general more important for possible applications, see below). The Cu(II) ion is fully complexed at pH ∼3, whereas full complexations of Ni(II) and Zn(II) proceed at pH ∼6, and that of Co(II) ion at pH ∼7. The distribution diagrams of the species are shown in Fig. 8.
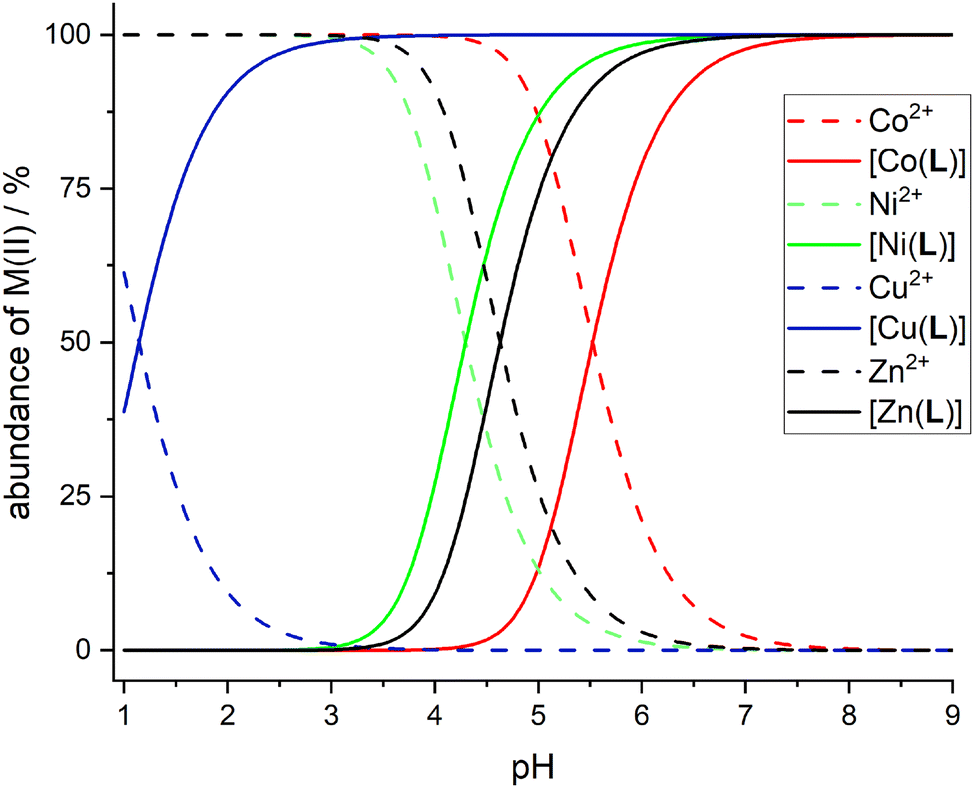 | ||
| Fig. 8 Distribution diagrams of the M(II)–H2L systems; c(M) = c(H2L) = 0.004 M. Co(II) – red lines, Ni(II) – green lines, Cu(II) – blue lines, Zn(II) – black lines. Full lines show distribution of the [M(L)] complex species, dashed lines show distribution of the free metal aqua ions. | ||
Solution structure of the complexes
The Co(II), Ni(II) and Zn(II) complexes isolated in the solid state have the cis-O,O′-geometry with the cyclam conformation cis-V.54 In the case of H2L, all nitrogen atoms have to have formally the same absolute configuration, “all-N-(R)” or “all-N-(S)”. Coordination of the phosphinate pendant arm results in four different substituents around the phosphorus atom and, thus, R/S absolute configurations of the phosphorus atom can be distinguished, leading to diastereoisomerism. In all structurally characterized octahedral complexes of H2L, the same absolute configuration on both phosphorus atoms and on macrocycle nitrogen atoms was found in the solid state, giving rise to “all-N-(R) + P-(R,R)” and “all-N-(S) + P-(S,S)” enantiomeric pairs. These enantiomers should show only one 19F NMR signal due to effective C2-symmetry.Overlaid 19F NMR spectra are shown in Fig. 9 for visual comparison (induced shift, linewidth) of the measured 19F NMR signals of the studied complexes.
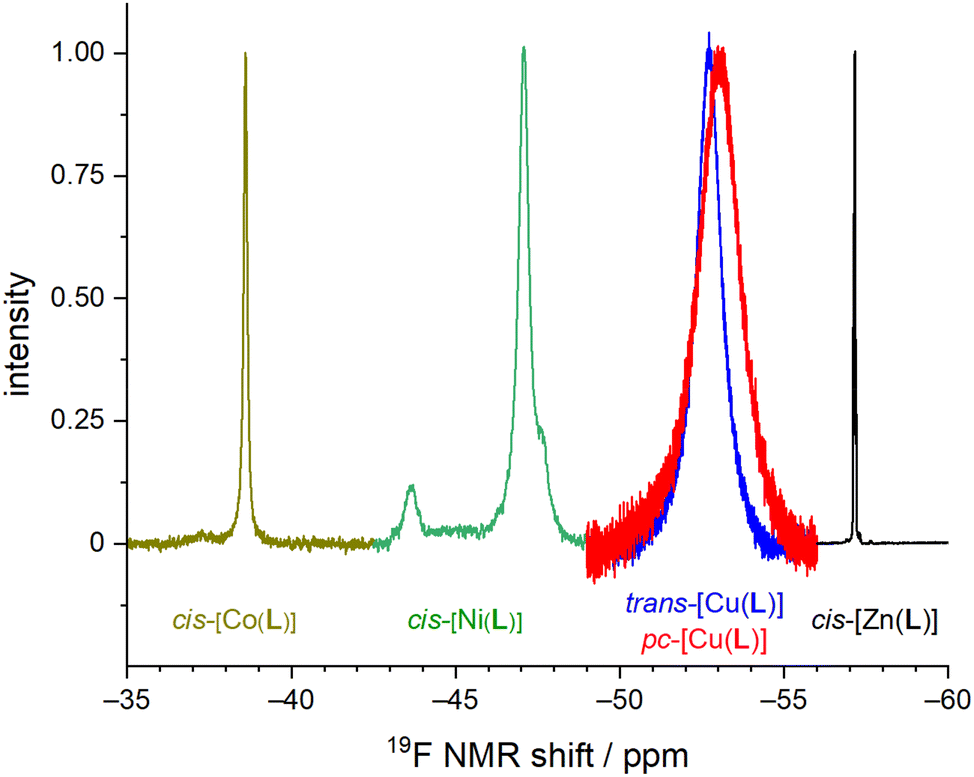 | ||
| Fig. 9 Representation of normalized 19F NMR spectra of studied M(II)–H2L complexes. | ||
The 19F NMR spectra of the equilibrated solution of the Co(II)–H2L complex show one broad symmetric signal at −38.6 ppm (Fig. S1†) which agrees with a presence of the cis-O,O′-species found in the solid state. In contrast, in a solution of the Ni(II) complex, three signals were found – a major signal at −47.1 ppm (∼80%) and two equal minor signals at −43.7 and −47.7 ppm (each ∼10%), as shown in Fig. S2.† The pc-[Cu(L)] isomer shows two very broad signals in the 19F NMR spectra as expected for the coordinated and the non-coordinated pendant arms (−52.6 ppm and −53.2 ppm, Fig. S5†), whereas the trans-O,O′-[Cu(L)] isomer shows one symmetric signal (−52.7 ppm, Fig. S6†). In the case of the pc-[Cu(L)] isomer, no coalescence of both signals was observed upon heating. It points that the pentacoordinated sphere is not fluxional. In a solution of the Zn(II) complex, pseudo-quartets (due to F–P,H couplings) centred at −57.15 pm (∼40%), −57.23 ppm (∼55%) and −57.30 ppm (∼5%) were observed (Fig. S3†). An analogous pattern was found also in the 31P NMR spectra (30.1 pm, ∼55%; 29.4 ppm, ∼5%; 28.7 ppm, ∼40%, Fig. S4†). The 31P NMR spectra of other complexes do not show any measurable signals due to the strong paramagnetic effect on the phosphorus atoms.
Based on the numbers of the signals in the 19F NMR spectra, one can conclude that a mixture of isomers is present in the solutions of the Ni(II) and Zn(II) complexes. The assignment of the signals was performed by the measurement of the 19F NMR spectra of freshly dissolved solid samples: batches of crystalline material were prepared from which unit cell parameters were determined for several selected crystals to confirm that they correspond to cis-O,O′-[Ni(L)]·3.5H2O and cis-O,O′-[Zn(L)]·2H2O·0.5(C3H6O), respectively. In the case of the Ni(II) complex, the 19F NMR spectra contains all three signals as described above immediately after dissolution. Thus, an equilibration is evidently very fast process (<3 min). As two minor signals have the same intensity, the pair of minor signals probably corresponds to “all-N-(R) + P-(R,S)” and “all-N-(S) + P-(S,R)” enantiomers formed by a mutual exchange of the coordinated and non-coordinated oxygen atoms of one pendant arm. It seems more accessible than a rearrangement of macrocycle conformation to e.g. conformation trans-III as Ni(II) complexes of cyclam-based ligands are rigid and such a rearrangement is usually energetically demanding and a very slow process.56 In the case of the Zn(II) complex, the situation is much more complicated. Freshly dissolved sample of cis-O,O′-[Zn(L)] with “all-N-(R) + P-(R,R)” and “all-N-(S) + P-(S,S)” configurations shows a multiplet centred at δF = −57.15 ppm (pseudo-quartet due to F–H,P coupling) and δP = 28.69 ppm (quartet due to P–F coupling in 31P{1H} spectra), so these signals can be assigned to the isomer found in the solid state. However, the other two signals increase gradually, and one of them becomes finally dominant. The final equilibrated mixture (5 d, 80 °C) shows the same spectra containing three multiplets as described above for the synthetic mixture and the signal of the original “all-N-(R) + P-(R,R)” and “all-N-(S) + P-(S,S)” enantiomers has finally only 40% abundance.
Obviously, at least two new isomeric complex species are present in the solution. They could be e.g. diastereoisomers “all-N-(R) + P-(R,S)” and “all-N-(R) + P-(S,S)” and their enantiomers as the phosphinate group oxygen atom exchange seems to be the most accessible process. However, some signal overlap has to be supposed as such a mixture should show four individual signals. Alternatively, a partial change of the macrocycle conformation cis-V to other possible conformations could occur; such a process has been documented well for Zn(II) complexes of cyclam derivatives.61
Kinetic inertness of the complexes
Kinetic inertness is suggested as a more important parameter for in vivo use than thermodynamic stability25,62 and, therefore, it was roughly determined by acid-assisted dissociation in 1 M HCl. pc-[Cu(L)] was found to be moderately inert with a half-life of 15.4 min at room temperature. However, the trans-O,O′-[Cu(L)] complex was found to be extremely inert; no change in the UV-Vis spectra was found at room temperature after 24 h. Therefore, dissociation reaction was followed at 90 °C. Under these conditions, dissociation half-time of 2.8 h was observed (Fig. S15†). The behaviour of both Cu(II)–H2L complexes is very similar to that of Cu(II) complexes of 1,8-H4te2p (Fig. 1).39As the complexes of Co(II) and Ni(II) ions show no significant absorption in the near UV region and d–d transition bands have a very low intensity, their kinetic inertness was followed by 19F NMR. Both cis-O,O′-[M(L)] were found to be very inert. The observed half-life of cis-O,O′-[Co(L)] in 1 M HCl at 25 °C was 3.0 h (Fig. S16†). In the case of cis-O,O′-[Ni(L)], no changes in the 19F NMR spectra were found after standing for 24 h under these conditions and, therefore, temperature was increased to 90 °C. At such an increased temperature, the dissociation half-life time was 36 min (Fig. S16†). The inertness of cis-O,O′-[Ni(L)] is thus comparable or somewhat lower than that reported for the trans-O,O′-[Ni(1,8-tfe2cyclam-R2)] family, but significantly higher compared to that of the cis-isomer of Ni(II) complex with 1,8-tfe2cyclam with no coordinating pendant arms (Fig. 1, R = H).32
19F NMR relaxation of the complexes
As the distance between the metal centre and the fluorine atoms is a crucial parameter influencing the relaxation rate of the 19F NMR signal, it is compiled in Table 3. It can be seen that the geometry of the coordination sphere influences the M⋯F distance only negligibly – in the cis-isomers, the distances are in the range of 5.6–6.5 Å, which is very similar to range of 5.8–6.8 Å found in the pc-Cu(II) complexes for the coordinated pendant arm and 5.8–7.0 Å found for the trans-Cu(II) species. The range of the distances of fluorine atoms belonging to the non-coordinated pendant arms in the pc-Cu(II) complexes is slightly larger, 7.2–7.7 Å and 5.1–7.0 Å in the pc-[Cu(3)] and pc-[Cu(L)] complexes, respectively. The observed distances are relevant for their significant influence on the relaxation times.30 As the trifluoroethyl group undergoes free rotation, mean distances (Table 3) are used in the following discussion, although effective distances in the solution can differ due to molecular flexibility and can be shorter.30 Solutions of the prepared complexes underwent detailed 19F NMR study. The T1 and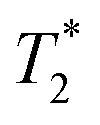 relaxation times were measured at available magnetic fields (7.05/9.40/14.1 T, 282/376/565 MHz) at 25 and 37 °C and the results are outlined in Table 4. The measurements confirmed significant shortening of the relaxation times in all studied paramagnetic complexes. A source of the T1 shortening lies in the dipolar mechanism as the number of bonds between the metal ion and fluorine atom is too high (5) to consider a significant contact contribution and Curie relaxation can be neglected for small molecules with small magnetic moments.30,34
relaxation times were measured at available magnetic fields (7.05/9.40/14.1 T, 282/376/565 MHz) at 25 and 37 °C and the results are outlined in Table 4. The measurements confirmed significant shortening of the relaxation times in all studied paramagnetic complexes. A source of the T1 shortening lies in the dipolar mechanism as the number of bonds between the metal ion and fluorine atom is too high (5) to consider a significant contact contribution and Curie relaxation can be neglected for small molecules with small magnetic moments.30,34
| Distances (Å) | pc-[Cu(3)]a | cis-O,O′-[Co(L)] | cis-O,O′-[Ni(L)] | pc-[Cu(L)]a | trans-O,O′-[Cu(L)]b | trans-O,O′-[Cu(L)]c | cis-O,O′-[Zn(L)] | ||
|---|---|---|---|---|---|---|---|---|---|
| Mol. 1 | Mol. 2 | Mol. 1 | Mol. 2 | ||||||
| a F1–F3 belong to the coordinated pendant arm, F4–F6 belong to the non-coordinated pendant arm. b trans-O,O′-[Cu(L)]·NH4(Cl0.54Br0.46). c trans-O,O′-[Cu(L)]·(H3O)(ClO4)·H2O. # Centrosymmetry-related atoms (F4 = F1#, F5 = F2#, F6 = F3#). $ Fluorine atoms of coordinated/non-coordinated pendant arms. | |||||||||
| M⋯F1 | 5.884(3) | 5.570(1) | 5.702(1) | 5.620(1) | 5.813(1) | 5.813(1) | 5.809(1) | 5.904(1) | 5.637(1) |
| M⋯F2 | 6.284(3) | 6.266(1) | 6.205(1) | 6.240(1) | 6.338(1) | 6.414(1) | 6.370(1) | 6.377(1) | 6.362(1) |
| M⋯F3 | 6.768(3) | 6.484(2) | 6.413(1) | 6.415(1) | 6.701(1) | 6.908(1) | 6.973(1) | 6.993(1) | 6.502(1) |
| M⋯F4 | 7.170(3) | 5.815(2) | 5.868(2) | 5.596(1) | 5.075(1) | 5.813(1)# | 5.809(1)# | 5.904(1)# | 5.668(1) |
| M⋯F5 | 7.647(3) | 6.330(1) | 6.322(1) | 6.319(1) | 6.590(1) | 6.414(1)# | 6.370(1)# | 6.377(1)# | 6.261(1) |
| M⋯F6 | 7.747(3) | 6.634(1) | 6.658(1) | 6.478(1) | 6.966(1) | 6.908(1)# | 6.973(1)# | 6.993(1)# | 6.439(1) |
| Average | 6.31/7.52$ | 6.18 | 6.19 | 6.11 | 6.28/6.21$ | 6.38 | 6.38 | 6.42 | 6.14 |
 for other complexes). Chemical shifts are corrected for bulk magnetic susceptibility effect
for other complexes). Chemical shifts are corrected for bulk magnetic susceptibility effect
| Parameter | H2L | cis-O,O′-[Co(L)] | cis-O,O′-[Ni(L)] | pc-[Cu(L)] | trans-O,O′-[Cu(L)] | cis-O,O′-[Zn(L)]d |
|---|---|---|---|---|---|---|
| a The pH 8.0 was used to assure the full complexation. b The pH 4.0 was used to avoid formation of the trans-O,O′-isomer. c The pendant arms are not equivalent in the pc-[Cu(L)] complex – one is coordinated, whereas the second is not. However, deconvolution of the measured signal into two peaks −52.6 and −53.2 ppm was possible only at 14.1 T. Both signals are very broad and strongly overlap (Fig. S5†). At lower fields, nearly symmetric signal was observed (Fig. 9). d Mixture of isomers is present in solution; the data are given for the isomer characterized by X-ray crystallography. | ||||||
| pH | 7.4 | 8.0a | 7.4 | 4.0b | 7.4 | 7.4 |
| 19F δ/ppm | −57.13 | −38.6 | −47.1 | −52.9c | −52.7 | −57.15 |
| T 1 25 °C (282 MHz)/ms | 1.38(7) × 103 | 35(2) | 3.6(2) | 6.5(3) | 5.1(3) | 1.11(6) × 103 |
| T 2 25 °C (282 MHz)/ms | — | 7.2(7) | 2.9(3) | 0.5(1) | 0.7(1) | — |
| (T2/T1)282 MHz, 25 °C | — | 0.21 | 0.81 | 0.08 | 0.14 | — |
| T 1 37 °C (282 MHz)/ms | 1.79(9) × 103 | 41(2) | 4.5(3) | 8.5(4) | 7.0(4) | 1.42(7) × 103 |
| T 2 37 °C (282 MHz)/ms | — | 3.5(3) | 3.1(3) | 0.7(1) | 0.8(1) | — |
| (T2/T1)282 MHz, 37 °C | — | 0.09 | 0.69 | 0.08 | 0.11 | — |
| T 1 25 °C (376 MHz)/ms | 1.11(5) × 103 | 33(2) | 3.3(2) | 5.9(3) | 5.4(3) | 0.87(4) × 103 |
| T 2 25 °C (376 MHz)/ms | 0.90(5) × 103 | 6.3(6) | 2.2(2) | 0.5(1) | 0.9(1) | 0.61(6) × 103 |
| (T2/T1)376 MHz, 25 °C | 0.81 | 0.19 | 0.67 | 0.08 | 0.17 | 0.70 |
| T 1 37 °C (376 MHz)/ms | 1.29(6) × 103 | 35(2) | 3.6(2) | 8.7(4) | 6.7(3) | 1.04(5) × 103 |
| T 2 37 °C (376 MHz)/ms | 0.92(5) × 103 | 4.7(5) | 2.5(2) | 0.6(1) | 1.1(1) | 0.87(4) × 103 |
| (T2/T1)376 MHz, 37 °C | 0.71 | 0.13 | 0.69 | 0.07 | 0.16 | 0.84 |
| T 1 25 °C (565 MHz)/ms | 0.74(4) × 103 | 28(2) | 3.2(2) | 7.5(4) | 6.1(3) | 0.60(3) × 103 |
| T 2 25 °C (565 MHz)/ms | 0.60(3) × 103 | 6.3(6) | 1.9(2) | 0.6(1)/0.8(1) c | 1.2(1) | 0.48(3) × 103 |
| (T2/T1)565 MHz, 25 °C | 0.81 | 0.23 | 0.59 | 0.10 | 0.20 | 0.80 |
| T 1 37 °C (565 MHz)/ms | 0.94(5) × 103 | 33(2) | 3.9(2) | 9.3(5) | 7.2(4) | 0.77(4) × 103 |
| T 2 37 °C (565 MHz)/ms | 0.74(4) × 103 | 3.4(3) | 2.4(2) | 0.7(1)/1.0(1)c | 1.5(1) | 0.62(3) × 103 |
| (T2/T1)565 MHz, 37 °C | 0.79 | 0.10 | 0.62 | 0.09 | 0.21 | 0.81 |
The cis-O,O′-[Co(L)] species showed longitudinal relaxation times in the range 30–40 ms which is very convenient for practical utilization. It is ca. 2–3 times longer (at the same magnetic fields and temperatures) compared to that of Co(II) complexes of 1,8-tfe2-cyclams (Fig. 1, R = CH2CO2H, CH2PO3H2) studied previously.34 The difference comes from longer distances between the central metal ion and fluorine atoms in cis-O,O′-[Co(L)] when compared to that in the complexes of 1,8-tfe2-cyclams (6.19 Å and 5 bonds for cis-O,O′-[Co(L)] compared to 5.25 Å and 4 bonds for Co(II)–1,8-tfe2-cyclams). Furthermore, the 19F NMR signal is relatively narrow (half-width 40–90 Hz depending on the temperature and magnetic field used) when compared to other studied paramagnetic complexes {Ni(II) 100–170 Hz, pc-[Cu(L)] 370–640 Hz, trans-O,O′-[Cu(L)] 200–450 Hz}, although in combination with a relatively long T1 it gives a rather low 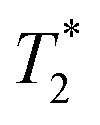 /T1 ratio (0.1–0.2).
/T1 ratio (0.1–0.2).
The 19F NMR signal of cis-O,O′-[Ni(L)] relaxes significantly faster than that of the Co(II) complex but it shows ca. 2-times slower relaxation (T1 ∼3–4 ms) in comparison with that of the Ni(II) complexes of 1,8-tfe2-cyclam derivatives (Fig. 1, R = H, CH2CO2H, CH2PO3H2, (CH2)2NH2, pyrid-2-ylmethyl), whose T1 was typically in range of 1–2 ms.31,32 It corresponds to the distance change (average M⋯F 6.11 Å for the cis-O,O′-[Ni(L)] complex compared to 5.16–5.34 for the Ni(II)–1,8-tfe2-cyclam complexes). The relaxation time is still too short for a comfortable standard imaging, but the high  /T1 ratio 0.6–0.8 of the signal can be potentially utilized in special imaging experiments.63,64 For the agent useful in standard imaging, a longer separation of the metal ion and fluorine atoms is obviously needed when using Ni(II) ion, as evidenced by T1 in the order of tens of milliseconds observed for Ni(II) complexes of cross-bridged cyclam containing [(trifluoromethyl)phenyl]acetamide pendant arms (Fig. 1, 8 bond separation, 7.28 Å, 10 ms; 9 bond separation, 8.72 Å, 29 ms).33
/T1 ratio 0.6–0.8 of the signal can be potentially utilized in special imaging experiments.63,64 For the agent useful in standard imaging, a longer separation of the metal ion and fluorine atoms is obviously needed when using Ni(II) ion, as evidenced by T1 in the order of tens of milliseconds observed for Ni(II) complexes of cross-bridged cyclam containing [(trifluoromethyl)phenyl]acetamide pendant arms (Fig. 1, 8 bond separation, 7.28 Å, 10 ms; 9 bond separation, 8.72 Å, 29 ms).33
Observed T1 for the Cu(II)–H2L complexes (ca. 5–9 ms) is slightly lower than values found for isomeric Cu(II) complexes of 1,8-(tfe-NHCH2CH2)2cyclam (7–12 ms)42 whereas the average M⋯F distances found in the Cu(II)–H2L complexes are somewhat longer (6.36 Å) than those observed for the Cu(II)–1,8-(tfe-NHCH2CH2)2cyclam (5.6–6.1 Å).42 However, the signals of the Cu(II)–H2L complexes are very broad. The 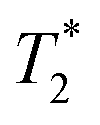 /T1 ratio is ca. 0.1 for the pentacoordinated and ca. 0.2 for the trans-O,O′-[Cu(L)] isomer, respectively.
/T1 ratio is ca. 0.1 for the pentacoordinated and ca. 0.2 for the trans-O,O′-[Cu(L)] isomer, respectively.
The T1 data acquired at 25 °C have been further assessed using the equation set of the Bloch–Redfield–Wangsness theory.30 Preliminary calculation confirmed that the contribution of Curie relaxation is for the presented set of complexes negligible and, thus, only the dipolar relaxation mechanism was considered. This phenomenon is quantified by eqn (1):
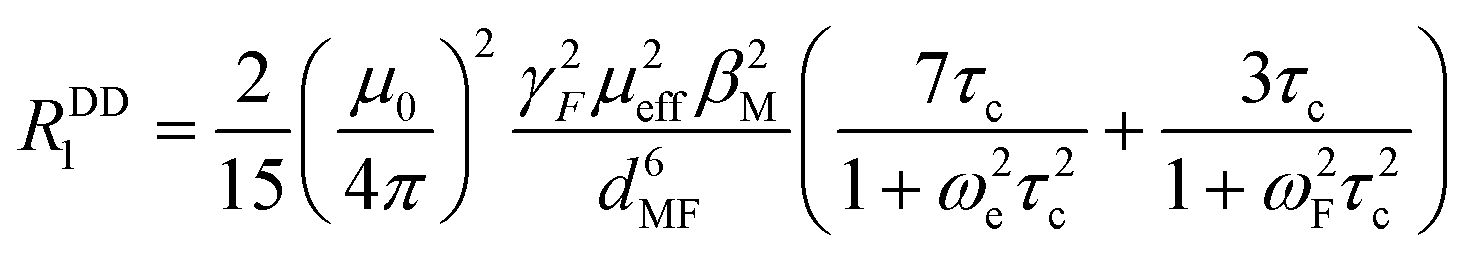 | (1) |
| τc−1 = τe−1 + τR−1 | (2) |
 | ||
| Fig. 10 Longitudinal relaxation rates of studied paramagnetic complexes. Parameters used for the fitting: τR = 170 ps (common for all complexes), dMF = 6.18 Å and μeff = 4.7 for cis-O,O′-[Co(L)], dMF = 6.11 Å and μeff = 3.5 for cis-O,O′-[Ni(L)], dMF = 6.25 Å and μeff = 1.9 for pc-[Cu(L)] and dMF = 6.30 Å and μeff = 1.9 for trans-O,O′-[Cu(L)]. Calculated values of τe were 1.0 × 10−12 s {cis-O,O′-[Co(L)]}, 8.5 × 10−11 s {cis-O,O′-[Ni(L)]}, 4 × 10−10 s {pc-[Cu(L)]} and 5 × 10−10 s {trans-O,O′-[Cu(L)]}, respectively. | ||
Of the used paramagnetic metal ions, the situation is the most straightforward for the Co2+ complex, as τe of this ion is significantly shorter than τR (by ca. 2 orders of magnitude) and, thus, τc is governed dominantly by τe. In contrast, for Ni2+ and Cu2+, contributions of both τe and τR to τc are significant, and thus, their values significantly correlate (i.e. a slight change in the suggested τR brings a significant change in τe, and vice versa).
Conclusions
A new cyclam-based ligand substituted in the 1,8-positions with two (2,2,2-trifluoroethyl)phosphinate pendant arms was found to bind divalent transition metal ions Co(II), Ni(II), Cu(II) and Zn(II) in stable complexes with a high selectivity for the Cu(II) ion. The Cu(II) ion forms two isomeric complexes: the pentacoordinated isomer pc-[Cu(L)] is formed as the kinetic product of the complexation reaction and rearranges to the thermodynamic product, the octahedral trans-O,O′-[Cu(L)] isomer. The Co(II), Ni(II) and Zn(II) ions form octahedral cis-O,O′-[M(L)] complexes. The complexes are kinetically inert with respect to acid-assisted dissociation. Paramagnetic metal ion complexes show very short relaxation times of the 19F NMR signal {cis-O,O′-[Co(L)] 30–40 ms, cis-O,O′-[Ni(L)] 3–4 ms, pc-[Cu(L)] ∼9 ms, trans-O,O′-[Cu(L)] ∼7 ms; 37 °C, 7–14 T}, which is a result of a short distance between the paramagnetic metal ion and the fluorine atoms (mean values found in the crystal structures are ∼6.1–6.4 Å). It makes these systems promising and potentially useful as contrast agents in 19F magnetic resonance imaging (19F MRI).Author contributions
FK: investigation, formal analysis, and writing. JK: conceptualization, investigation, formal analysis, supervision, and writing. IC: investigation and formal analysis. JH: investigation. VK: formal analysis. PH: conceptualization, funding acquisition, supervision, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the Czech Science Foundation (GAČR) 22-34083S is acknowledged. The project was performed in the frame of COST-action CA 18202 NECTAR.References
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach, L. Helm and É. Tóth, John Wiley & Sons, 2nd ed., 2013 Search PubMed.
- C. S. Bonnet and É. Tóth, Chimia, 2016, 70, 102–108 CrossRef CAS PubMed.
- B. Condon, EMPA J., 2011, 2, 403–410 Search PubMed.
- Y. A. Pirogov, Phys. Procedia, 2016, 82, 3–7 CrossRef CAS.
- D. Bartusik-Aebisher, Z. Bober, J. Zalejska-Fiolka, A. Kawczyk-Krupka and D. Aebisher, Molecules, 2022, 27, 6493 CrossRef CAS PubMed.
- E. T. Ahrens, R. Flores, H. Xu and P. A. Morel, Nat. Biotechnol., 2005, 23, 983–987 CrossRef CAS PubMed.
- Y. Mo, C. Huang, C. Liu, Z. Duan, J. Liu and D. Wu, Macromol. Rapid Commun., 2023, 2200744 CrossRef PubMed.
- B. L. Bona, O. Koshkina, C. Chirizzi, V. Dichiarante, P. Metrangolo and F. Baldelli Bombelli, Acc. Mater. Res., 2023, 4, 71–85 CrossRef CAS.
- K. Waxman, Ann. Emerg. Med., 1986, 15, 1423–1424 CrossRef CAS PubMed.
- K. C. Lowe, Blood Rev., 1999, 13, 171–184 CrossRef CAS PubMed.
- L. Mignion, J. Magat, O. Schakman, E. Marbaix, B. Gallez and B. F. Jordan, Magn. Reson. Med., 2013, 69, 248–254 CrossRef CAS PubMed.
- E. Maevsky, G. Ivanitsky, L. Bogdanova, O. Axenova, N. Karmen, E. Zhiburt, R. Senina, S. Pushkin, I. Maslennikov, A. Orlov and I. Marinicheva, Artif. Cells, Blood Substitutes, Biotechnol., 2005, 33, 37–46 CrossRef CAS PubMed.
- R. P. Mason, P. P. Antich, E. E. Babcock, J. L. Gerberich and R. L. Nunnally, Magn. Reson. Imaging, 1989, 7, 475–485 CrossRef CAS PubMed.
- B. J. Dardzinski and C. H. Sotak, Magn. Reson. Med., 1994, 32, 88–97 CrossRef CAS PubMed.
- G. M. Lanza, X. Yu, P. M. Winter, D. R. Abendschein, K. K. Karukstis, M. J. Scott, L. K. Chinen, R. W. Fuhrhop, D. E. Scherrer and S. A. Wickline, Circulation, 2002, 106, 2842–2847 CrossRef CAS PubMed.
- A. M. Morawski, P. M. Winter, X. Yu, R. W. Fuhrhop, M. J. Scott, F. Hockett, J. D. Robertson, P. J. Gaffney, G. M. Lanza and S. A. Wickline, Magn. Reson. Med., 2004, 52, 1255–1262 CrossRef CAS PubMed.
- S. D. Caruthers, A. M. Neubauer, F. D. Hockett, R. Lamerichs, P. M. Winter, M. J. Scott, P. J. Gaffney, S. A. Wickline and G. M. Lanza, Invest. Radiol., 2006, 41, 305–312 CrossRef PubMed.
- A. M. Neubauer, J. Myerson, D. Caruthers, F. D. Hockett, P. M. Winter, J. Chen, P. J. Gaffney, J. D. Robertson, G. M. Lanza and S. A. Wickline, Magn. Reson. Med., 2008, 60, 1066–1072 CrossRef CAS PubMed.
- Y. Li, J. Cui, C. Li, H. Zhou, J. Chang, O. Aras and F. An, ChemMedChem, 2022, 17, e202100701 CAS.
- P. Hermann, J. Blahut, J. Kotek and V. Herynek, Met. Ions Life Sci., 2021, 22, 239–270 Search PubMed.
- D. Janasik and T. Krawczyk, Chem. – Eur. J., 2022, 28, e202102556 CAS.
- A. Li, X. Luo, D. Chen, L. Li, H. Lin and J. Gao, Anal. Chem., 2023, 95, 70–82 CrossRef CAS PubMed.
- A. M. Kenwright, I. Kuprov, E. De Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC.
- R. Pujales-Paradela, T. Savić, P. Pérez-Lourido, D. Esteban-Gómez, G. Angelovski, M. Botta and C. Platas-Iglesias, Inorg. Chem., 2019, 58, 7571–7583 CrossRef CAS PubMed.
- E. Brücher, G. Tircsó, Z. Baranyai, Z. Kovács and A. D. Sherry, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach, L. Helm and É. Tóth, John Wiley & Sons, 2nd ed., 2013, ch. 4 Search PubMed.
- J. Neburkova, A. M. Rulseh, S. L. Y. Chang, H. Raabova, J. Vejpravova, M. Dracinsky, J. Tarabek, J. Kotek, M. Pingle, P. Majer, J. Vymazal and P. Cigler, Nanoscale Adv., 2020, 2, 5567–5571 RSC.
- K. Srivastava, E. A. Weitz, K. L. Peterson, M. Marjańska and V. C. Pierre, Inorg. Chem., 2017, 56, 1546–1557 CrossRef CAS PubMed.
- M. Yu, B. S. Bouley, D. Xie and E. L. Que, Dalton Trans., 2019, 48, 9337–9341 RSC.
- A. Gupta, P. Caravan, W. S. Price, C. Platas-Iglesias and E. M. Gale, Inorg. Chem., 2020, 59, 6648–6678 CrossRef CAS PubMed.
- M. Zalewski, D. Janasik, A. Wierzbicka and T. Krawczyk, Inorg. Chem., 2022, 61, 19524–19542 CrossRef CAS PubMed.
- J. Blahut, P. Hermann, A. Gálisová, V. Herynek, I. Císařová, Z. Tošner and J. Kotek, Dalton Trans., 2016, 45, 474–478 RSC.
- J. Blahut, K. Bernášek, A. Gálisová, V. Herynek, I. Císařová, J. Kotek, J. Lang, S. Matějková and P. Hermann, Inorg. Chem., 2017, 56, 13337–13348 CrossRef CAS PubMed.
- R. Pujales-Paradela, T. Savić, I. Brandariz, P. Pérez-Lourido, G. Angelovski, D. Esteban-Gómez and C. Platas-Iglesias, Chem. Commun., 2019, 55, 4115–4118 RSC.
- J. Blahut, L. Benda, J. Kotek, G. Pintacuda and P. Hermann, Inorg. Chem., 2020, 59, 10071–10082 CrossRef CAS PubMed.
- D. Xie, M. Yu, R. T. Kadakia and E. L. Que, Acc. Chem. Res., 2020, 53, 2–10 CrossRef CAS PubMed.
- J. S. Enriquez, M. Yu, B. S. Bouley, D. Xie and E. L. Que, Dalton Trans., 2018, 47, 15024–15030 RSC.
- NIST Standard Reference Database 46 (Critically Selected Stability Constants of Metal Complexes),Version 7.0, 2003, distributed by NIST standard Reference Data, Gaithersburg, MD 20899, USA Search PubMed.
- S. Chaves, R. Delgado and J. J. R. F. da Silva, Talanta, 1992, 39, 249–254 CrossRef CAS PubMed.
- J. Kotek, P. Lubal, P. Hermann, I. Císařová, I. Lukeš, T. Godula, I. Svobodová, P. Táborský and J. Havel, Chem. – Eur. J., 2003, 9, 233–248 CrossRef CAS PubMed.
- I. Svobodová, P. Lubal, J. Plutnar, J. Havlíčková, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2006, 5184–5197 RSC.
- J. Havlíčková, H. Medová, T. Vitha, J. Kotek, I. Císařová and P. Hermann, Dalton Trans., 2008, 5378–5386 RSC.
- Z. Kotková, F. Koucký, J. Kotek, I. Císařová, D. Parker and P. Hermann, Dalton Trans., 2023, 52, 1861–1875 RSC.
- V. Herynek, M. Martinisková, Y. Bobrova, A. Gálisová, J. Kotek, P. Hermann, F. Koucký, D. Jirák and M. Hájek, Magn. Reson. Mater. Phys., Biol. Med., 2019, 32, 115–122 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth Heinemann An imprint of Elsevier Science, 5th edn, 2003 Search PubMed.
- G. Royal, V. Dahaoui-Gindrey, S. Dahaoui, A. Tabard, R. Guilard, P. Pullumbi and C. Lecomte, Eur. J. Org. Chem., 1998, 1971–1975 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXT2014/5. Program for Crystal Structure Solution from Diffraction Data, University of Göttingen, Göttingen, 2014 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, University of Göttingen, Göttingen, 2014 Search PubMed; (b) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed; (c) G. M. Sheldrick, SHELXL-2014/7. Program for Crystal Structure Refinement from Diffraction Data, University of Göttingen, Göttingen, 2014 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- C. F. Baes, Jr. and R. E. Mesmer, The Hydrolysis of Cations, Wiley, New York, 1976 Search PubMed.
- M. Kývala and I. Lukeš, International Conference, Chemometrics ×95, Abstract book p. 63, Pardubice (Czech Republic), 1995; full version of ™OPIUM is available (free of charge) on https://www.natur.cuni.cz/~kyvala/opium.html.
- M. Meyer, V. Dahaoui-Ginderey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 178–180, 1313–1405 CrossRef CAS.
- P. Hermann, J. Kotek and V. Kubíček, Comprehensive Heterocyclic Chemistry, ed. D. StC. Black, J. Cossy and C. V. Stevens, Elsevier, 2022. Vol. 14, ch. 14.11, pp. 591–683 Search PubMed.
- B. Bosnich, C. K. Poon and M. L. Tobe, Inorg. Chem., 1965, 4, 1102–1108 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- J. Kotek, P. Vojtíšek, I. Císařová, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2001, 66, 363–381 CrossRef CAS.
- M. M. Le Roy, S. Héry, N. Saffon-Merceron, C. Platas-Iglesias, T. Troadec and R. Tripier, Inorg. Chem., 2023, 62, 8112–8122 CrossRef CAS PubMed.
- J. Kotek, P. Vojtíšek, I. Císařová, P. Hermann, P. Jurečka, J. Rohovec and I. Lukeš, Collect. Czech. Chem. Commun., 2000, 65, 1289–1316 CrossRef CAS.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef.
- R. D. Hancock, R. J. Motekaitis, J. Mashishi, I. Cukrowski, J. H. Reibenspies and A. E. Martell, J. Chem. Soc., Perkin Trans. 2, 1996, 1925–1929 RSC.
- X. Liang and P. J. Sadler, Chem. Soc. Rev., 2004, 33, 246–266 RSC.
- T. J. Clough, L. Jiang, K.-L. Wong and N. J. Long, Nat. Commun., 2019, 10, 1420 CrossRef PubMed.
- K. H. Chalmers, A. M. Kenwright, D. Parker and A. M. Blamire, Magn. Reson. Med., 2011, 66, 931–936 CrossRef CAS PubMed.
- F. Schmid, C. Höltke, D. Parker and C. Faber, Magn. Reson. Med., 2013, 69, 1056–1062 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of studied complexes, colour change of isomeric Cu(II) complexes, experimental data and details for the crystal structure refinement, ligand distribution diagram, details of the kinetic inertness study, NMR characterization of organic compounds, and discussion of the crystal structures of synthetic intermediates. CCDC 2262037–2262048. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01420g |
| This journal is © The Royal Society of Chemistry 2023 |