
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu(IV)–Ru(IV) complexes having the doubly oxido-bridged core with a bridging carbonato or hydrogencarbonato ligand†
Tomoyo
Misawa-Suzuki
 * and
Hirotaka
Nagao
*
* and
Hirotaka
Nagao
*
Department of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioicho Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: t_misawa@sophia.ac.jp
First published on 10th February 2023
Abstract
Ru(IV)–Ru(IV) complexes having the doubly oxido-bridged diamond core with a bridging carbonato or hydrogencarbonato ligand, [{RuIV(ebpma)}2(μ-O)2(μ-O2CO(H)m)]Xn (ebpma; ethylbis(2-pyridylmethyl)amine, m = 0; [IV,IV]X2 (X = PF6, ClO4), m = 1; [IV,IV_1H](ClO4)3), were isolated via the oxidation of the corresponding carbonato-bridged Ru(III)–Ru(IV) complex ([III,IV]+), and “[IV,IV](ClO4)2 and [IV,IV_1H](ClO4)3” were structurally characterized. The electrochemical and spectroscopic properties of [IV,IV]2+ and [IV,IV_1H]3+ were investigated both in organic solvents and aqueous solutions. The reactivity toward organic solvents having (a) methyl group(s) and reactions with organic substrates were studied as well. This should be the first time when systematic comparisons of the Ru(IV)–Ru(IV) species and corresponding Ru(III)–Ru(IV) complexes in the same tridentate ligand system were made.
Introduction
Singly and doubly oxido-bridged multi-nuclear transition metal complexes have been of interest as the reaction centers of oxidation reactions. For the oxidation of methane, the active site of the reaction mediator, soluble methane monooxygenase (sMMO), shows a wide range of oxidation states from Fe(II)–Fe(II) to Fe(IV)–Fe(IV), with the elaborate changes of the frameworks during the reaction.1 Studies on the structural changes around the dinuclear oxido-bridged metal core accompanied by redox and protonation/deprotonation reactions are important for understanding the reaction mechanisms. A number of studies on the electronic structures, catalytic oxidation reactions and model reactions with sMMO analogues with diiron centers,2 or some with dinickel centers3 have been reported in homogeneous systems, which are not limited to the rich chemistry of mononuclear metal–oxido species by, e.g. the 8th group iron,4 ruthenium,5 and osmium6 centers. However, reports on those with diruthenium centers are limited although, only recently, a light-driven methane monooxygenation with the dinuclear Ru(IV)–Ru(IV) complex was reported.7In this sense, we have been investigating Ru(III)–Ru(IV) dimers having the {Ru2(μ-O(H))2(μ-O2Y–Z)}2+ (Y–Z = C–CH3 (acetato), N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(nitrato)) core.8c,d Instead of acetate, carbonate with a terminal oxygen, a potential site as a proton carrier site, was introduced as a bridging ligand.8e We have reported Ru(III)–Ru(IV) complexes with the doubly oxido- or hydroxido-bridged diamond core, M[{RuIII,IV(L)}2(μ-O)2(μ-O2CO)]2(PF6)3 (M[III,IV]2-(PF6)3; M+ = Na+, K+), [{RuIII,IV(L)}2(μ-O)2(μ-O2COH)](PF6)2 ([III,IV_1H](PF6)2), and M[{RuIII,IV(L)}2(μ-O)(μ-OH)(μ-O2COH)](ClO4)4 (M[III,IV_2H](ClO4)4; M+ = Na+, K+), in which L is the tridentate ethylbis(2-pyridylmethyl)amine (ebpma) ligand.8e The reversible protonation/deprotonation reactions, crystal structures, and electronic structures have been discussed.
O(nitrato)) core.8c,d Instead of acetate, carbonate with a terminal oxygen, a potential site as a proton carrier site, was introduced as a bridging ligand.8e We have reported Ru(III)–Ru(IV) complexes with the doubly oxido- or hydroxido-bridged diamond core, M[{RuIII,IV(L)}2(μ-O)2(μ-O2CO)]2(PF6)3 (M[III,IV]2-(PF6)3; M+ = Na+, K+), [{RuIII,IV(L)}2(μ-O)2(μ-O2COH)](PF6)2 ([III,IV_1H](PF6)2), and M[{RuIII,IV(L)}2(μ-O)(μ-OH)(μ-O2COH)](ClO4)4 (M[III,IV_2H](ClO4)4; M+ = Na+, K+), in which L is the tridentate ethylbis(2-pyridylmethyl)amine (ebpma) ligand.8e The reversible protonation/deprotonation reactions, crystal structures, and electronic structures have been discussed.
It is worth studying the fixation of hydrogencarbonate (HCO3−) on the transition metal center(s) as well as carbonate (CO32−) fixation for the evaluation of its activity using transition metal complexes. HCO3− exists in an equilibrium mixture of CO32− and H2CO3 (CO2 (aq.)) depending on pH. Thus, carbonate easily undergoes protonation, resulting in decarboxylation under acidic conditions (CO32− + 2H+ ⇄ CO2 + H2O). Isolation of hydrogencarbonato-bound transition metal complexes has been attempted in the monodentate coordination (e.g. {NiI–OC(O)OH},9 {ZnII–OC(O)OH}10) or in the bidentate coordination through its two oxygen atoms (e.g. {MoIV,IV2(μ-O2COH)},11 {RuI,I2(μ-O2COH)}12 or {RhIV,IV2(μ-O2COH)}13). However, no one has isolated HCO3−-bound transition metal complexes with both the Ru(IV)–Ru(IV) and Ru(III)–Ru(IV) centers, in which ancillary ligands are the same.
In this report, Ru(IV)–Ru(IV) complexes with the metal–oxido diamond core, [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2CO)](X)2·nH2O ([IV,IV](X)2·nH2O, X = PF6, ClO4) and [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2COH)](ClO4)3·HClO4·7H2O ([IV,IV_1H](ClO4)3·HClO4·7H2O), were isolated (Scheme 1) and structurally characterized by X-ray crystallography. Although there are a few studies on the Ru(IV)–Ru(IV) complexes with the singly,14 doubly15 or triply16 oxido-bridged core, the study on Ru(IV)–Ru(IV) complexes with the {Ru2(μ-O)2(μ-O2Y–Z)} framework should be the first report, to the best of our knowledge. The direct comparisons of the electronic structures and electrochemical and spectroscopic properties with those of the corresponding Ru(III)–Ru(IV) complexes in acetonitrile were achieved. The report on electrochemical and spectroscopic behaviors of [III,IV_1H]2+ or [III,IV_2H]3+ in acetonitrile is also the first report beyond those in aqueous solutions. The reactions of [IV,IV]2+ and [IV,IV_1H]3+ in solvents and in the presence of organic substrates were studied to gain mechanistic insights.
 | ||
| Scheme 1 Synthesis of Ru(IV)–Ru(IV) complexes having carbonato and hydrogencarbonato bidentate ligands. | ||
Results and discussion
Syntheses and electronic structures
The perchlorate salts of [IV,IV]2+ and [IV,IV_1H]3+ were isolated via the one-electron oxidation of Na[III,IV]2(ClO4)3·17H2O in water with an equimolar amount of (NH4)4[CeIV(SO4)4]·2H2O (CAS) at pH 2.8 and pH 1.2, respectively, at room temperature. The pH value was adjusted by using perchloric acid solutions for the formation of [IV,IV_1H]3+, in which one-protonation to the uncoordinated terminal oxygen of the carbonato ligand occurred after one-electron oxidation of the {RuIII,IV2(μ-O)2} core (Scheme 1). The pKa value of [IV,IV_1H]3+ was determined as 1.1 by spectroscopic studies in aqueous solution (Fig. S2, S3 and Table S1†). Since the cationic character was higher for the Ru(IV)–Ru(IV) core, the pKa value was lower than that of the corresponding Ru(III)–Ru(IV) complex, [III,IV_1H]2+ (pKa; 3.9).8e The spectroscopic studies in aqueous solutions revealed that a further one-protonated species, {[IV,IV_2H]4+}, could exist at pH lower than 0.6. Although the isolation of {[IV,IV_2H]4+} was attempted a couple of times, we had difficulties in reproducibility.[IV,IV](ClO4)2 and [IV,IV_1H](ClO4)3 showed the effective magnetic moments (μeff) of 1.04μB and 0.85μB at 298 K, indicating that although the antiferromagnetic coupling of the two paramagnetic d4 centers is observed, paramagnetic characters are maintained. The paramagnetic characters prevented us from fully understanding the 1H NMR signals although they were observed within the range of 0–12 ppm (Fig. S1†). In the IR spectra, the characteristic absorption band attributed to νas(CO) was observed at 1570(s) cm−1 for [IV,IV](ClO4)2·2H2O and 1591(s) cm−1 for [IV,IV](PF6)2·1.5H2O (Fig. S7†), which were 70–90 cm−1 higher than that of M[III,IV]2(PF6)3.8e The difference is originating from the more cationic character of the Ru(IV)–Ru(IV) centers, resulting in the stronger Ru(IV)–O(carbonato) bond, weaker bondings of the O–C–O moiety, and stronger CO bond. Also, the absence of M+ in the vicinity of [IV,IV]2+, although it was observed for [III,IV]+, might have some effects on the larger value for [IV,IV]2+. For [IV,IV_1H](ClO4)3, νas(CO) was observed at 1437 cm−1 (Fig. S7†), which is consistent with that of [III,IV_1H](PF6)2 (1437 cm−1),8e owing to the more delocalized electronic structure over the O–C(OH)–O moiety.
Single crystal X-ray crystallography
By X-ray crystallographic analyses of [IV,IV]2+ and [IV,IV_1H]3+, (Fig. 1, S18 and Tables 1, S4 and S5†), each ruthenium center is coordinated with tridentate ebpma in a facial manner, the bridging oxido or hydroxido ligands, and one of the oxygen atoms of the bridging carbonato or hydrogencarbonato ligand. Along with the complex cations, two or three ClO4− were co-crystalized with [IV,IV]2+ or [IV,IV_1H]3+, respectively, revealing that they surely have the Ru(IV)–Ru(IV) centers. The asymmetric unit of [IV,IV_1H]3+ (Fig. 1) contained one solvent water molecule in the vicinity of the terminal OH group of hydrogencarbonato, forming the hydrogen bond network with the perchlorate oxygen atoms. The perchlorate near [IV,IV_1H]3+ was neutral HClO4, revealed by one elongated Cl–O length. Thus, the whole chemical formula of [IV,IV_1H]3+ should be denoted as [IV,IV_1H](ClO4)3·HClO4·H2O. The Ru–O(oxido) lengths in [IV,IV]2+and [IV,IV_1H]3+ are closer to each other and comparable to those of the doubly oxido-bridged complexes in the Ru(IV)–Ru(IV) state, [{RuIV,IV(OHm)(L′)}2(μ-O)2]n+ (L′; [(η5-C5H5)Co(C2H5O)2PO3]−) (1.893(4)–1.926(4) Å),15a whereas shorter by 0.03–0.05 Å than the Ru3.5–O lengths in both [III,IV_1H]2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) and Na[III,IV_2H]3+,8e and by 0.1 Å shorter than the Ru3.5–OH lengths in Na[III,IV_2H]3+ (ave. 2.021 Å).8e The Ru⋯Ru distances (2.4261(3) and 2.4389(3) Å) are shorter than those in [{RuIV,IV(OHm)(L′)}2(μ-O)2]m+ (2.452(1)–2.505(4) Å)15a due to the difference in the framework. The Ru⋯Ru distances show that there is the dπ(Ru)–dπ(Ru) interaction in addition to the six-coordinate environment around the Ru centers. The shapes of the diamond core of [IV,IV]2+ and [IV,IV_1H]3+ are different from [{RuIV,IV(OHn)(L′)}2(μ-O)2]m+,15a because of the coordination of the bidentate (hydrogen)carbonato ligand on the {Ru2(μ-O)2} core. The O–O(H) distances are longer for the core with (i) the lower oxidation state of Ru, (ii) the hydroxido bridge rather than the oxido bridge, and (iii) HCO3− over CO32−; [III,IV_2H]3+ (3.058 Å) > [III,IV]+ (3.026 Å) > [III,IV_1H]3+ (2.994 Å) > [IV,IV]2+ (2.949 Å) > [IV,IV_1H]3+ (2.934 Å). The Ru⋯Ru distance (Å) is longer simply in the order of lower oxidation states of the Ru centers and with weaker σ-donating oxygen; [III,IV_2H]3+ (2.533 Å)8e > [III,IV_1H]2+ (2.454 Å)8e > [III,IV]+ (2.443 Å)8e > [IV,IV_1H]3+ (2.439 Å) > [IV,IV]2+ (2.426 Å).
and Na[III,IV_2H]3+,8e and by 0.1 Å shorter than the Ru3.5–OH lengths in Na[III,IV_2H]3+ (ave. 2.021 Å).8e The Ru⋯Ru distances (2.4261(3) and 2.4389(3) Å) are shorter than those in [{RuIV,IV(OHm)(L′)}2(μ-O)2]m+ (2.452(1)–2.505(4) Å)15a due to the difference in the framework. The Ru⋯Ru distances show that there is the dπ(Ru)–dπ(Ru) interaction in addition to the six-coordinate environment around the Ru centers. The shapes of the diamond core of [IV,IV]2+ and [IV,IV_1H]3+ are different from [{RuIV,IV(OHn)(L′)}2(μ-O)2]m+,15a because of the coordination of the bidentate (hydrogen)carbonato ligand on the {Ru2(μ-O)2} core. The O–O(H) distances are longer for the core with (i) the lower oxidation state of Ru, (ii) the hydroxido bridge rather than the oxido bridge, and (iii) HCO3− over CO32−; [III,IV_2H]3+ (3.058 Å) > [III,IV]+ (3.026 Å) > [III,IV_1H]3+ (2.994 Å) > [IV,IV]2+ (2.949 Å) > [IV,IV_1H]3+ (2.934 Å). The Ru⋯Ru distance (Å) is longer simply in the order of lower oxidation states of the Ru centers and with weaker σ-donating oxygen; [III,IV_2H]3+ (2.533 Å)8e > [III,IV_1H]2+ (2.454 Å)8e > [III,IV]+ (2.443 Å)8e > [IV,IV_1H]3+ (2.439 Å) > [IV,IV]2+ (2.426 Å).
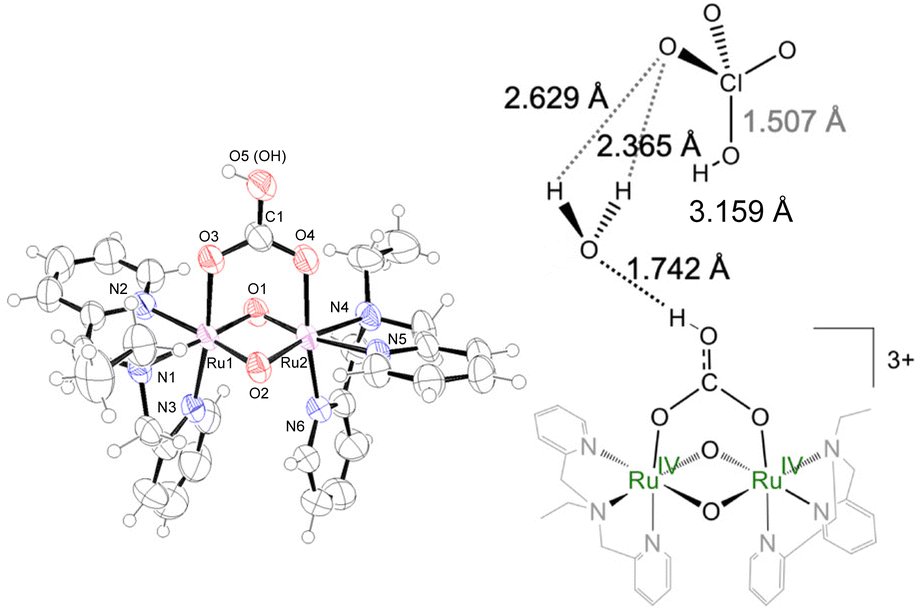 | ||
| Fig. 1 Crystal structure of [IV,IV_1H]3+ shown at the 50% probability level (left) and the hydrogen bonding network (right). | ||
| [IV,IV](ClO4)2·2H2O | [IV,IV_1H](ClO4)3·HClO4·H2O | K[III,IV]2(PF6)3·3CH3CN·4H2O8e | [III,IV_1H](ClO4)2·H2O8e | Na[III,IV_2H](ClO4)4·4H2O8e | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ru1,2–O1 | 1.905(3) | 1.914(3) | 1.918(2) | 1.899(2) | 1.940(5) | 1.940(4) | 1.9372(18) | 1.9329(17) | 1.954(4) | 1.953(4) |
| Ru1,2–O2 | 1.904(3) | 1.919(3) | 1.902(2) | 1.914(2) | 1.938(4) | 1.962(5) | 1.9360(18) | 1.9387(18) | 2.022(4) | 2.019(3) |
| Ru1⋯Ru2 | 2.4261(3) | 2.4389(3) | 2.4434(8) | 2.4547(2) | 2.5332(5) | |||||
| O1⋯O2 | 2.949 | 2.934 | 3.026 | 2.994 | 3.058 | |||||
| Ru–O3,4 | 2.031(3) | 2.022(3) | 2.069(2) | 2.065(2) | 2.055(4) | 2.063(4) | 2.0799(17) | 2.0968(18) | 2.078(4) | 2.083(4) |
| Ru–N1,4 | 2.147(3) | 2.146(3) | 2.130(3) | 2.134(3) | 2.195(5) | 2.168(5) | 2.158(2) | 2.156(2) | 2.131(4) | 2.123(4) |
| Ru–N2,5 | 2.091(3) | 2.091(3) | 2.078(3) | 2.086(3) | 2.113(7) | 2.100(6) | 2.087(2) | 2.093(2) | 2.072(3) | 2.088(4) |
| Ru–N3,6 | 2.076(3) | 2.072(3) | 2.047(3) | 2.056(3) | 2.061(5) | 2.057(5) | 2.039(2) | 2.042(2) | 2.042(5) | 2.045(5) |
| O3,4–C29 | 1.311(5) | 1.312(4) | 1.265(4) | 1.269(4) | 1.309(8) | 1.301(8) | 1.258(3) | 1.266(3) | 1.266(7) | 1.266(6) |
| C29–O5 | 1.228(5) | 1.297(4) | 1.252(7) | 1.315(3) | 1.313(7) | |||||
The Ru–N(amine) lengths were unambiguously influenced by the number of protons on the carbonato and oxido ligands, and the electronic state of the Ru centers; [III,IV]+ (ave. 2.182 Å)8e > [III,IV_1H]2+ (ave. 2.157 Å)8e > [IV,IV]2+ (ave. 2.147 Å) > [IV,IV_1H]3+ ≈ Na[III,IV_2H]3+ (ave. 2.131 Å).8e Carbonato is more σ-donating than hydrogencarbonato that makes the Ru–N(amine, σ-donor) lengths longer. Due to protonation to the oxido ligand, the basicity of oxygen became lower and, accordingly, the σ-donating N(amine) atom which located at the trans position to the oxido ligand was bound more strongly to the Ru centers. The CO length of [IV,IV]2+ is shorter by 0.025 Å than that of [III,IV]+, which is consistent with the higher wavenumber of νas(CO) for [IV,IV]2+. The metric parameters around hydrogencarbonato of [IV,IV_1H]3+ are in agreement with those in [III,IV_1H]2+8e and Na[III,IV_2H]3+8e (Table 1).
UV-vis-NIR spectroscopy in acetonitrile
The UV-vis-NIR absorption spectra of [IV,IV]2+ and [IV,IV_1H]3+ exhibited relatively weak and broad bands at 762 and 669 nm, and furthermore, much weaker and broader bands were also found at around 1000 and 1100 nm in CH3CN at 25 °C (Fig. S4†). Similar absorption bands at 632–694 nm and around 1070 nm have been reported for the diruthenium complexes having the {RuIV,IV2(μ-O)2} core.17 The spectroscopic parameters in CH3CN are listed in Tables 2 and S1† with the electrochemical information. In comparison with [III,IV]+ (1067 nm)8e and [III,IV_1H]2+ (972 nm) (Fig. S5†), or the corresponding acetato- or nitrato-bridged complexes ([{RuIII,IV(ebpma)}2(μ-O)2(μ-O2Y–Z)]2+; Y–Z = C–CH3, [III,IV(CH3CO2)]2+ or NO, [III,IV(NO3)]2+) at 977 or 762 nm (Fig. S6†),8c the characteristic broad bands were observed at shorter wavelengths, indicating that the energy difference of the MOs of the {RuIV,IV2(μ-O)2} core becomes higher than those of the {RuIII,IV2(μ-O)2} core. We could also conclude that the {Ru2(μ-O)2}–{Ru2(μ-O)2}* transition energies are higher and the intensities (ε/M−1 cm−1) are stronger for the HCO3−-bound complexes, [IV,IV_1H]3+ or [III,IV_1H]2+, than for the CO32−-bound ones, [IV,IV]2+ or [III,IV]+, respectively. Comparing the corresponding absorption bands of the Ru(IV)–Ru(IV) species prepared through electrochemical one-electron oxidation of [III,IV(CH3CO2)]2+ and [III,IV(NO3)]2+, namely, [IV,IV(CH3CO2)]3+ (677 nm) and [IV,IV(NO3)]3+ (586 nm),8c good correlations between the redox potentials and the absorption wavelengths are found (Table 2).
| λ (ε)/nm (M−1 cm−1) in acetonitrile {Ru2(μ-O)2}–{Ru2(μ-O)2}a | E 1/2/V (vs. Ag/AgNO3) Ru(IV)–Ru(IV)/Ru(III)–Ru(IV) | |||
|---|---|---|---|---|
| a {Ru2(μ-O)(μ-OH)} core. | ||||
| [IV,IV]2+ | 762 (1440) | [III,IV]+ | 1067 (660)8e | 0.15 |
| [IV,IV_1H]3+ | 669 (1970) | [III,IV_1H]2+ | 972 (1490) | 0.60 |
| — | [III,IV_2H]3+ | 964 (1330) | 1.32 | |
| [IV,IV(CH3CO2)]3+ | 677 (—) | [III,IV(CH3CO2)]2+ | 977 (1440)8c | 0.578c |
| — | [III,IV(CH3CO2)]3+a | 1187 (320)8d | 1.338d | |
| [IV,IV(NO3)]3+ | 586 (—) | [III,IV(NO3)]2+ | 762 (1300)8c | 0.938c |
Electrochemistry in acetonitrile
Cyclic voltammograms (CVs) show a reversible reduction wave at 0.15 V for [IV,IV]2+ or 0.60 V (E1/2vs. Ag|0.01 M AgNO3) for [IV,IV_1H]3+ in acetonitrile at 25 °C (Fig. S8†), which is assigned to the one-electron process between Ru(IV)–Ru(IV) and Ru(III)–Ru(IV) (Fig. S9, S13 and Tables S2, S3†). Hydrodynamic voltammetry (HDV) also supports the electronic state of Ru(IV)–Ru(IV) for [IV,IV]2+ and [IV,IV_1H]3+, by comparing the rest potentials with those of {Na+ + [III,IV]+} and [III,IV_1H]2+ (Fig. S10–S12 and S14–S16†). The one-electron oxidation process from [III,IV_2H]3+ to [IV,IV_2H]4+ was observed at 1.32 V (Epa) as an irreversible process. Through one-protonation to the uncoordinated oxygen of carbonato, which locates far from the {Ru2(μ-O)2} core, the redox potential shifted by +0.45 V and upon an additional one-protonation to the oxido ligand on the diamond core {Ru2(μ-O)2}, the potential shifted further by as much as +0.72 V. The differences in the effects of protonation to the {Ru2(μ-O)2} core or away from the core on the redox potentials were revealed. This must be the first discussion ever had, because even in our previous report on [III,IV(CH3CO2)]2+, and only the effect of one-protonation to one of the bridging oxido ligands of the {Ru2(μ-O)2} core (by +0.76 V) has been discussed.8d The one-electron redox reactions should occur at the {Ru2(μ-O)2} core, which was supported by theoretical calculations18 that revealed the SOMO delocalization mainly over the {Ru2(μ-O)2} core of both [IV,IV]2+ (Fig. S51 and Table S8†) and [IV,IV_1H]3+ (Fig. S52 and Table S9†), with a little contribution from the carbonato or hydrogencarbonato oxygen.As the one-electron redox potentials between Ru(IV)–Ru(IV) and Ru(III)–Ru(IV) become higher in the order of [III,IV(NO3)]2+ (0.93 V) > [III,IV_1H]2+ (0.60 V) ≈ [III,IV_(CH3CO2)]2+ (0.57 V) > [III,IV]+ (0.15 V), the {Ru2(μ-O)2}–{Ru2(μ-O)2}* transition energies become higher in the same order, owing to the greater stabilization of the {Ru2(μ-O)2} core (SOMO).
UV-vis-NIR spectroscopy and electrochemistry in water
In aqueous solutions containing HClO4(aq) and NaOH(aq), the spectroscopic properties were studied as a function of pH (Fig. S2 and Table S1†). Between pH 1.25 and 5.23, where [IV,IV]2+ should be present, λmax of the characteristic band, that was assigned to the transitions on the {Ru2(μ-O)2} core, was observed at 704 ± 4 nm within the apparatus resolution. The weaker band in the NIR region was observed as well at around 1090–1100 nm. The effects of water on [IV,IV]2+ seemed quite large since the differences in λmax were by as much as 25–70 nm compared to those in organic solvents (729–776 nm). Accordingly, the brown colored complex [IV,IV]2+ readily changed into emerald-green in aqueous solution. At pH 0.88, λmax was observed at 700 nm, corresponding to [IV,IV_1H]3+; however at pH 0.55, one more proton addition to [IV,IV_1H]3+ seemed to occur since the λmax shifted by 10 nm to 681 nm. We believe that {[IV,IV_2H]4+} could be present in solution without decarboxylation even under such harsh acidic conditions, although it was not isolated.The CPET processes were observed in the CVs in aqueous solutions at different pH values (Fig. S17,† shown vs. Ag|AgCl); at pH 2.7, 0.39 V (vs. Ag|Ag+) for a 1e−/1H+ process of [III,IV_1H]2+/[IV,IV]2+ (eqn (2)), at pH 1.8, 0.44 V for a 1e−/2H+ process of [III,IV_2H]3+/[IV,IV]2+ (eqn (3)) and at pH 1.1, 0.53 V for a 1e−/1H+ process of [III,IV_2H]3+/[IV,IV_1H]3+ (eqn (6)). Above pH 4, the redox potentials between Ru(IV)–Ru(IV) and Ru(III)–Ru(IV) were similar, and thus one-electron transfer without proton transfer occurred between [IV,IV]2+ and [III,IV]+ (eqn (1)).
| [IV,IV]2+ + e− → [III,IV]+ | (1) |
| [IV,IV]2+ + e− + H+ → [III,IV\_1H]2+ | (2) |
| [IV,IV]2+ + e− + 2H+ → [III,IV\_2H]3+ | (3) |
| [IV,IV\_1H]3+ → [IV,IV]2+ + H+ | (4) |
| [IV,IV\_1H]3+ + e− → [III,IV\_1H]2+ | (5) |
| [IV,IV\_1H]3+ + e− + H+ → [III,IV\_2H]3+ | (6) |
| [IV,IV\_1H]3+ + e− + 2H+ → {[III,IV\_3H]4+}* | (7) |
Spectroscopic studies on the reactions in various solvents and with substrates
The solvent dependency of UV-vis-NIR spectroscopic behaviors was also of interest. However, both [IV,IV]2+ and [IV,IV_1H]3+ were more reactive than to evaluate the spectroscopic properties at 25 °C. The spectroscopic data regarding the reactions are listed in Tables 3 and S6.† Although some of them showed a relationship between BDEC–H of the solvent and the kobs value (e.g. between acetone and acetonitrile), others did not (e.g. between methanol and DMSO), and thus some more factors for the reaction rates might be considered.| Solv.a | Before reaction | After reaction | k obs, h−1 at 0.13 mM, unless otherwise noted | ||
|---|---|---|---|---|---|
| Complex | λ max/nm | Product | λ max/nm | ||
| a BDEsC–H (kcal mol−1);19 DMSO 94, CH3OH 96.06, (CH3)2CO 96.0, and CH3CN 97.0. b Immediately deprotonated upon dissolution. c The spectra of authentic samples are shown in Fig. S4 (acetonitrile), S21 (methanol), S22 (DMSO), S23 (nitromethane) and S34 (acetone).† d The data of authentic samples of pH 6.3 and 1.0 aqueous solutions are from ref. 8e and also shown in Table S1.† | |||||
| DMSO (dehydrated) | [IV,IV_1H]3+ → [IV,IV]2+b | 756 | [III,IV_2H]3+ | 341, 965 (342, 966)c | 0.135 (initial) |
| 0.039 (following) | |||||
| Methanol (dehydrated) | [IV,IV_1H]3+ → [IV,IV]2+b | 729 | [III,IV_2H]3+ | 338, 966 (337, 970)c | 2.3 |
| Acetone (dehydrated) | [IV,IV]2+ | 776 | [III,IV_2H]3+ | 965 (967)c | 0.22 (0.14 mM) |
| Acetone | 773 | [III,IV_2H]3+ | 962–963 (967)c | 0.27 (0.20 mM) | |
| Acetonitrile | 762 | [III,IV_2H]3+ | 959 (964)c | 0.043 (0.20 mM) | |
| pH 4 water (HClO4–NaOH) | 705 | [III,IV]+ | 335, ∼1028 (337, 1035)d | — | |
| Acetone (dehydrated) | [IV,IV_1H]3+ | 666 | [III,IV_2H]3+ | 964 (967)c | 0.14 (initial) |
| 0.013 (following) | |||||
| Acetone | 665 | [III,IV_1H]2+; >0.2 mM | 974–977 (∼973)c | 0.40 (initial) | |
| [III,IV_2H]3+; <0.2 mM | 964–965 (967)c | 0.22 (following) | |||
| Acetonitrile | 670 | [III,IV_2H]3+ | 963 (964)c | 0.0019 (0.30 mM) | |
| Nitromethane | 674 | [III,IV_1H]2+ | 962 (966)c | 0.0039 | |
| pH 1.1 water (HClO4–NaOH) | 697 | [III,IV_2H]3+ | ∼293, ∼1180 (∼298, 1170)d | — | |
Upon dissolution of [IV,IV_1H]3+ in dehydrated DMSO (Fig. S19†) and methanol (Fig. 2), the proton at hydrogencarbonato of [IV,IV_1H]3+ was dissociated (eqn (4)), and then 1e−/2H+ transfer to [IV,IV]2+ proceeded showing isosbestic points, affording [III,IV_2H]3+ quantitatively (eqn (3)). In nitromethane, the 1e− transfer to [IV,IV_1H]3+ proceeded to change into [III,IV_1H]2+ (eqn (5), Fig. S20†). In acetone or acetonitrile, 1e− transfer (eqn 5) or CPET (1e/1H+ or 1e−/2H+ transfer) occurred to [IV,IV]2+ and [IV,IV_1H]3+ (eqn (2), (3), (6) and (7)).
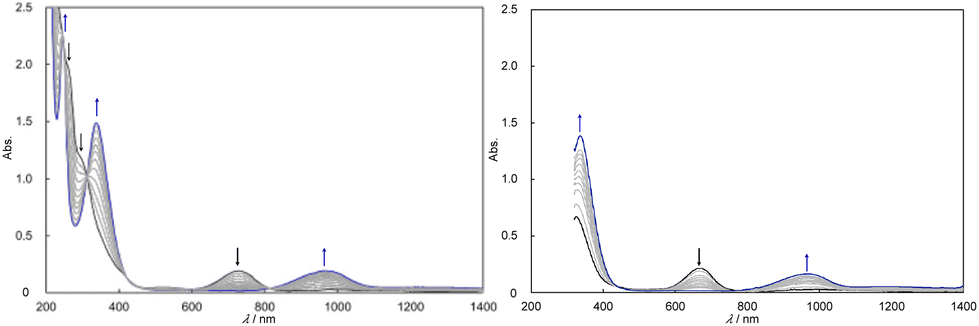 | ||
| Fig. 2 Spectral changes of [IV,IV_1H]3+ (0.13 mM) into [III,IV_1H]2+via the formation of [IV,IV]2+ in dehydrated methanol (left) and those directly into [III,IV_1H]2+ in acetone (right) under air at 25 °C. | ||
The reactions of [IV,IV]2+ in acetone proceeded with isosbestic points, forming only [III,IV_2H]3+ through the 1e−/2H+ transfer (eqn (3), Fig. S26†). The kobs values were analyzed as the pseudo first-order reaction and the rate constants (kobs, s−1) were investigated at different complex concentrations (Fig. S26–S28†). [IV,IV]2+ was quantitatively changed into [III,IV_2H]3+ by 1e−/2H+ transfer (eqn (3)) and kobs was not related to the [IV,IV]2+ concentration (Fig. S29†). In the case of [IV,IV_1H]3+, different reactions occurred depending on the complex concentration (lower/higher than 0.2 mM) (Fig. 2, S30 and S33†). The kobs value was analyzed by two-step first-order linear approximations and the reaction rates were in a linear and negative relationship with the concentration of [IV,IV_1H]3+ (Fig. S33†). The reaction rates were higher below 0.2 mM, where the reaction product was [III,IV_2H]3+via 1e−/1H+ transfer (eqn (6)), and slower above 0.2 mM, where only [III,IV_1H]2+ was formed via 1e− transfer (eqn (5)).
The kinetic isotopic effect (KIE, divided by the number of equivalent active hydrogen atoms) was observed in acetone-d6 for both [IV,IV]2+ (5.1) (Fig. S28†) and [IV,IV_1H]3+ (2.2–24) (Fig. S32†) as summarized in Table S6.† Hydrogen atom abstraction (HAA) of acetone should be involved. However, because they are stoichiometric reactions and the concentration of product(s) is rather low, the analysis of each reaction product was demanding.
The effects of water content in acetone on the reaction rates were observed as well. The reaction rates (kobs, h−1) of [IV,IV]2+ in 99.5% acetone were a little larger than those in dehydrated acetone (Fig. S27†). The reaction rates of [IV,IV_1H]3+ in 99.5% acetone were 0.50 (initial) and 0.29 (following) (Fig. S30vi†) and those in dehydrated acetone–D2O (v/v = 99.5/0.5) were 0.39 and 0.21, whereas 0.14 and 0.013 in dehydrated acetone (Fig. S31†). These results revealed that the KIE due to the D–O cleavage of D2O was hardly observed and the C–H activation of acetone could possibly occur. For comparisons, the reactivity was evaluated in water as well. In pH 4 water, [IV,IV]2+ showed a decay into [III,IV]+ by 1e− transfer (eqn (1), Fig. S24†), which was different from the reactions in organic solvents. In pH 1 water, [IV,IV_1H]3+ was changed into [III,IV_2H]2+ through the 1e−/1H+ transfer (eqn (6), Fig. S25†).
In acetonitrile, the reaction rates were much slower than those in acetone. [IV,IV]2+ underwent 1e−/2H+ transfer to quantitatively give [III,IV_2H]2+ (eqn (3), Fig. S35†). The reaction in acetonitrile-d3 was performed for the 0.82 mM solution of [IV,IV]2+ and the KIE was 1.5 for the initial reaction, which lasted only for a moment, and 1.2 for the following reaction (Fig. S35†). Therefore, HAA of acetonitrile might be involved in the reactions of [IV,IV]2+. [IV,IV_1H]3+ showed much lower reactivity than [IV,IV]2+, taking over a month to complete the reactions.
Then, in dehydrated acetonitrile, the oxidation reactions of cyclic organic compounds, benzyl alcohol, cyclohexene, cumene and ethylbenzene were studied. Based on the pKa values and redox potentials (Scheme 2) of [IV,IV]2+ and [IV,IV_1H]3+, the hydrogen accepting ability or BDEC–H was calculated to be 84.3 and 92.7 kcal mol−1, respectively, according to eqn (8).20
| BDE (kcal mol−1) = 1.37(pKa) + 23.06E1/2 + CH | (8) |
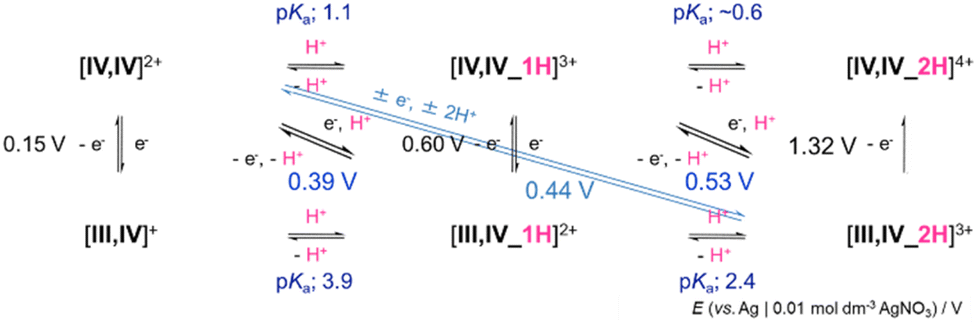 | ||
| Scheme 2 Summary of pKa values and redox potentials (vs. Ag|0.01 M AgNO3). | ||
The dependency of the reaction rate on the concentration of the substrate (0.05–0.2 M) was studied. All could be analysed as the first-order linear relationships (kobs, s−1) and the second-order rate constants (k, M−1 s−1) were evaluated (Fig. S37–S50, Table S7†). For the reactions with ethylbenzene, only those of [IV,IV]2+ were investigated since the reaction rates of [IV,IV_1H]3+ were too slow to discuss at 25 °C. Independent of the kind of substrate, [IV,IV]2+ underwent 1e−/2H+ transfer to form [III,IV_2H]3+ (eqn 3) and [IV,IV_1H]3+ changed solely into {[III,IV_3H]4+}* (eqn (7)) or into a mixture of {[III,IV_3H]4+}* and [III,IV_2H]3+ (eqn (6) and (7)), where {[III,IV_3H]4+}* seemed to be a transient intermediate that we have not identified. The reaction rates of 1e−/2H+ transfer to [IV,IV]2+ to form [III,IV_2H]3+ (eqn 3) (Fig. S38, S42, S46 and S50†) were greater than those of 1e−/1H+ or 1e−/2H+ transfer to [IV,IV_1H]3+ (Fig. S40, S44 and S48†) (Table S7†), although BDEC–H of [IV,IV]2+ is smaller than that of [IV,IV_1H]3+. These results indicated that the uncoordinated terminal oxygen of carbonato in [IV,IV]2+ would better function as a one-proton acceptor than the oxido ligand(s). Although the plot of log(k′, M−1 s−1) against BDE was drawn based on the Bell–Evans–Polanyi equation (eqn (9)),21 the relationships were not understandable, indicating that multiple reaction mechanisms are involved depending on the substrate.
| logk = αΔH° + C | (9) |
Reactions with deuterated reagents, benzyl alcohol-d2 (BA-d2; C6H5CD2OH, Fig. S37iv and S39iii†) and ethylbenzene-d10 (EB-d10, Fig. S49ii†), were performed. KIE values (kD, s−1/kH, s−1) were observed to be 2.6 and 6.1 for [IV,IV]2+ with BA (0.15 M) and EB (0.20 M) and 43 for [IV,IV_1H]3+ with BA (0.15 M)), which ensured that the HAA occurred and should probably be the rate determining step. The hydrogen tunnelling effect would contribute to the large KIE values.5t HAA would be achieved by a 1e− transfer to the Ru(IV) center and for [IV,IV]2+, by one proton transfer to the terminal oxygen carbonato ligand to form [III,IV_1H]2+. Then, one of the oxido ligands undergoes protonation that could originate from unavoidable water in solution to finally yield [III,IV_2H]3+. With respect to [IV,IV_1H]3+, one or both of the oxido ligand(s) function(s) as (a) proton acceptor(s) to form [III,IV_2H]3+ or {[III,IV_3H]4+}*, respectively. The higher oxidation potential of [IV,IV_1H]3+ compared to that of [IV,IV]2+ results in greater BDEC–H; however, the basicity of carbonato oxygen may have an advantage, which may lead to a higher reaction rate. The analyses of the reaction products and reaction mechanisms including oxygen sources and oxygenation processes or product selectivity are underway.
Experimental
Syntheses and characterization of the complexes
All of the chemical reagents were used as purchased without further purification.Ethylbis(2-pyridylmethyl)amine (ebpma) was prepared by a procedure given in ref. 8a. [{RuIII,IVCl2(ebpma)}2(μ-O)]PF6·(CH3)2CO was synthesized by a previously reported procedure8c from fac-[RuIIICl3(ebpma)]8avia a triply chlorido-bridged diruthenium(II) complex, [{RuII,II(ebpma)}2(μ-Cl)3]PF6.8b Na[{RuIII,IV(ebpma)}2(μ-O)2(μ-O2CO)]2(PF6)3·7H2O (Na[III,IV]2(PF6)3·7H2O) [{RuIII,IV(ebpma)}2(μ-O)2(μ-O2COH)](PF6)2·1.5H2O ([III,IV_1H](PF6)2·1.5H2O) were prepared according to the procedures reported in ref. 8e.
X = ClO4, n = 2. [IV,IV](ClO4)2·2H2O was synthesized by a reaction of Na[III,IV]2(ClO4)3·17H2O8e (100 mg, 0.094 mmol) in an aqueous solution containing (NH4)4[CeIV(SO4)4]·2H2O (67 mg, 0.106 mmol) at pH 2.8. The volume of the obtained dark blue–green solution was reduced to less than 1 cm3 by a slow evaporation under air, yielding brown single crystals. Yield; 50 mg, 52%. E. A./% calcd for C29H39N6O23Cl2Ru2 ([{RuIV(ebpma)}2(μ-O)2(μ-O2CO)](ClO4)2·2H2O); C, 34.78; H, 3.99; N, 8.39; found; C, 34.56; H, 3.76; N, 8.37%. FAB MS (+) (relative intensity, m/z ([M]2+ = [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2CO)]2+); 375.2 (33%, [M]2+/2), 850.4 (100%, [M]2+ + ClO4−). IR spectrum (KBr disk); νas(CO); 1570(sh), ν(ClO4); 1078, 1109, 1123 cm−1.
X = PF6, n = 1.5. The PF6–salt, [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2CO)](PF6)2·1.5H2O, was synthesized in a similar manner to that of [IV,IV](ClO4)2·2H2O by using Na[III,IV]2(PF6)3·7H2O8e as a starting material. E. A./% calcd for C29H37N6O6.5Ru2P2F12; calcd C, 32.67; H, 3.47; N, 7.88; found; C, 32.57; H, 3.27; N, 7.92%. IR spectrum (KBr disk); νas(CO); 1591(sh), 1612 (s), 1635 (sh) cm−1, νas(PF6); 839 cm−1. ΛM = 214 S cm2 mol−1 (1.0 mM).
Measurements
Elemental analyses were performed with a PerkinElmer Model 2400-II system or performed in the Institute of Physical and Chemical Research (RIKEN). Fast atom bombardment mass spectrometry (FAB MS) was performed with a JEOL Model JMS SX700 V system, using m-nitrobenzyl alcohol as a matrix. The effective magnetic moments, μeff/μB, were measured with a Sherwood Scientific Magway MSB MK1 balance at 298 K using Hg[CoII(SCN)4] as a calibrant. X-band electron spin resonance (ESR) spectra were recorded with a JEOL Model JESFA300 ESR spectrometer at 93 K. IR spectra were recorded on a Shimadzu IR Model Affinity-1 spectrophotometer with 2.0 cm−1 resolution using the samples prepared as KBr disks. UV-vis-NIR spectra were measured on a Shimadzu UV-3600 Plus or 1900i in an organic solvent or in aqueous solution using a quartz cell of 1 cm path length. Cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) were performed in CH3CN containing 0.1 mol dm−3 tetra-n-butylammonium perchlorate (TBAP, Nakarai Tesque Ltd) as a supporting electrolyte with a platinum or a glassy carbon disk working electrode (ϕ = 1.6 mm), an Ag|0.01 mol dm−3 AgNO3 reference electrode, and a platinum wire counter electrode using an ALS 630E electrochemical analyser. At the end of each measurement, ferrocene ([FeIII(Cp)2]+/[FeII(Cp)2], 0.074 V vs. Ag|0.01 mol dm−3 AgNO3 (CH3CN)) was added as an internal standard to tune the redox potentials. For the measurements in 0.1 mol dm−3 NaOH–HClO4 aqueous solutions, a glassy carbon working electrode (ϕ = 1.0 mm), an Ag|AgCl 3.0 mol dm−3 NaCl reference electrode, and a platinum wire counter electrode were used. Hydrodynamic voltammetry (HDV) was performed with a rotating glassy carbon disk working electrode (ϕ = 3.0 mm) by a BAS RDE-2 rotating electrode.X-ray crystallography
Single crystals suitable for X-ray diffraction were grown by slow evaporation of the pH 2.8 or 1.1 solution of [IV,IV](ClO4)2·2H2O and [IV,IV_1H](ClO4)3·HClO4·H2O, respectively, at around 50 °C. The intensity data were collected on a Rigaku Model Synergy-S system, using multilayer mirror monochromated CuKα radiation (1.54184 Å). All the calculations were performed using CrysAlisPro (Data Collection and Processing Software, Rigaku Corporation (2015)). Structures were solved by direct methods, expanded using Fourier techniques and refined using full-matrix least-squares techniques on F2 using SHELXT with Olex2 1.2 (CCDC 2115482 for [IV,IV](ClO4)2·2H2O and 2115484 for [IV,IV_1H](ClO4)3·HClO4·H2O).†Reactions in organic solvents and in the presence of organic compounds
Superdehydrated solvents are of 99.8% grade. Deuterated purity for acetone-d6 was 99.6%, for acetonitrile-d3 99.8%, for benzylalcohol-d2 99% and for ethylbenzene-d10 99%. C–H oxidation reactions of organic compounds were studied in superdehydrated CH3CN under air at 25 °C with complexes of 0.20 mM and substrates ranging from 0.05 M to 0.2 M.DFT calculations
Density functional theory (DFT) calculations were performed using the Gaussian 09 program.18 The geometrical optimizations were performed at the unrestricted B3LYP level of DFT, with Los Alamos effective core potential plus minimal basis (LANL2MB) or double-ζ (LANL2DZ) basis sets.Conclusions
The Ru(IV)–Ru(IV) complexes with the {Ru2(μ-O)2} core and a bridging carbonato or hydrogencarbonato ligand, which are structurally analogous to the reaction centers of sMMO, in particular, the plausible intermediate Q having the “closed-core”, were isolated and studied in detail. It should be emphasized that the carbonato or hydrogencarbonato ligand was stabilized in the Ru(IV)–Ru(IV) centers and they did not undergo decarboxylation even at as low as pH 0.5. The direct comparisons of spectroscopic properties and structural parameters with those of the Ru(III)–Ru(IV) species are worth noting since all the complexes are designed using the same tridentate ligand (ebpma). The electron transition energies on the {Ru2(μ-O)2} core were in relation to the electronic state of the Ru centers [IV,IV]2+ (λmax = 760 nm) and [IV,IV_1H]3+ (669 nm) vs. [III,IV]+ (1067 nm) and [III,IV_1H]2+ (972 nm), and to the electrostatic nature of the bridging ligand, (O2Y–Z). The structural parameters of the {Ru2(μ-O)2} diamond core changed due to the higher cationic character of the Ru(IV)–Ru(IV) centers compared to the Ru(III)–Ru(IV) ones. However, it was also true that the ebpma ligand seemed to play a nice role in minimizing the impact of the change in the electronic structure on the shape of the diamond core, {Ru2(μ-O)2}.Reactions in solvents and oxidation reactions of organic substrates using these Ru(IV)–Ru(IV) complexes were investigated in dehydrated acetonitrile, which was revealed to be the only solvent suitable for the reaction mediator for our system. The relationships between the reaction rate and the BDE of substrates, and the different reaction processes between [IV,IV]2+ and [IV,IV_1H]3+ were found. Further investigations on the reaction products will be performed by analysing the reaction conditions in detail. This work would contribute to the strctural model of metalloenzymes and would also give insights into some model reactions of C–H activation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate Prof. Dr Shinkoh Nanbu for the theoretical calculations. This work was supported by JSPS KAKENHI Grant Number 22K14696.References
- (a) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280 CrossRef CAS PubMed; (b) R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431 CrossRef CAS PubMed.
- (a) A. B. Jacobs, R. Banerjee, D. E. Deweese, A. Braun, J. T. Babicz Jr., L. B. Gee, K. D. Sutherlin, L. H. Böttger, Y. Yoda, M. Saito, S. Kitao, Y. Kobayashi, M. Seto, K. Tamasaku, J. D. Lipscomb, K. Park and E. I. Solomon, J. Am. Chem. Soc., 2021, 143, 16007 CrossRef CAS PubMed; (b) G. Xue, R. D. Hont, E. Münck and L. Que Jr., Nat. Chem., 2010, 2, 400 CrossRef CAS PubMed; (c) M. Martinho, G. Xue, A. T. Fiedler, L. Que Jr., E. L. Bominaar and E. Münck, J. Am. Chem. Soc., 2009, 131, 5823 CrossRef CAS PubMed; E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987 Search PubMed.
- Y. Morimoto, Y. Takagi, T. Saito, T. Ohta, T. Ogura, N. Tohnai, M. Nakano and S. Itoh, Angew. Chem., Int. Ed., 2018, 57, 7640 CrossRef CAS PubMed.
- (a) R. Singh, G. Ganguly, S. O. Malinkin, S. Demeshko, F. Meyer, E. Nordlander and T. K. Paine, Inorg. Chem., 2019, 54, 1862 CrossRef PubMed and the references therein; (b) S. T. Kleespies, W. N. Oloo, A. Mukherjee and L. Que Jr., Inorg. Chem., 2015, 54, 5053 CrossRef CAS PubMed and the references therein; (c) D. Wang, M. Zhang, P. Bühlmann and L. Que Jr., J. Am. Chem. Soc., 2010, 132, 7638 CrossRef CAS PubMed; (d) C. R. Goldsmith and T. D. P. Stack, Inorg. Chem., 2006, 45, 6048 CrossRef CAS PubMed; (e) J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que Jr., J. Am. Chem. Soc., 2004, 126, 472 CrossRef CAS PubMed; (f) C. R. Goldsmith, R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 2002, 124, 83 CrossRef CAS PubMed.
- (a) H. Kotani, H. Shimomura, K. Ikeda, T. Ishizuka, Y. Shiota, K. Yoshizawa and T. Kojima, J. Am. Chem. Soc., 2020, 142, 16982 CrossRef CAS PubMed; (b) H. Kotani, H. Shimomura, M. Horimoto, T. Ishizuka, Y. Shiota, K. Yoshizawa, S. Yanagisawa, Y. Kawahara-Nakagawa, M. Kubo and T. Kojima, Dalton Trans., 2019, 48, 13154 RSC; (c) C.-W. Tse, Y. Liu, T. W.-S. Chow, C. Ma, W.-P. Yip, X.-Y. Chang, K.-H. Low, J.-S. Huang and C.-M. Che, Chem. Sci., 2018, 9, 2803 RSC; (d) R. Ray, S. Chandra, D. Maiti and G. K. Lahiri, Chem. – Eur. J., 2016, 22, 8814 CrossRef CAS PubMed; (e) T. Ishizuka, H. Kotani and T. Kojima, Dalton Trans., 2016, 45, 16727 RSC; (f) S. N. Dhuri, Y.-M. Lee, M. S. Seo, J. Cho, D. D. Narulkar, S. Fukuzumi and W. Nam, Dalton Trans., 2015, 44, 7634 RSC; (g) S. N. Dhuri, K.-B. Cho, Y.-M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623 CrossRef CAS PubMed; (h) T. Ishizuka, S. Ohzu and T. Kojima, Synlett, 2014, 25, 1667 CrossRef; (i) T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692 CrossRef CAS PubMed; (j) Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772 CrossRef CAS PubMed; (k) C.-M. Che, J.-L. Zhang, R. Zhang, J.-S. Huang, T.-S. Lai, W.-M. Tsui, X.-G. Zhou, Z.-Y. Zhou, N. Zhu and C. K. Chang, Chem. – Eur. J., 2005, 11, 7040 CrossRef CAS PubMed; (l) R. Zhang, W.-Y. Yu and C.-M. Che, Tetrahedron: Asymmetry, 2005, 16, 3520 CrossRef CAS; (m) W. K. Seok and T. J. Meyer, Inorg. Chem., 2005, 44, 3931 CrossRef CAS PubMed; (n) T. J. Meyer, Inorg. Chem., 2005, 44, 2150 CrossRef PubMed; (o) J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351 CrossRef CAS PubMed; (p) W. W. Y. Lam, S.-M. Yiu, D. T. Y. Yiu, T.-C. Lau, W.-P. Yip and C.-M. Che, Inorg. Chem., 2003, 42, 8011 CrossRef CAS PubMed; (q) S. Funyu, T. Isobe, S. Takagi, D. A. Tryk and H. Inoue, J. Am. Chem. Soc., 2003, 125, 5734 CrossRef CAS PubMed; (r) D. T. Y. Yiu, M. F. W. Lee, W. W. Y. Lam and T.-C. Lau, Inorg. Chem., 2003, 42, 1225 CrossRef CAS PubMed; (s) C.-M. Che, K.-W. Cheng, M. C. W. Chan, T.-C. Lau and C.-K. Mak, J. Org. Chem., 2000, 65, 7996 CrossRef CAS PubMed; (t) W.-C. Cheng, W.-Y. Yu, C.-K. Li and C.-M. Che, J. Org. Chem., 1995, 60, 6840 CrossRef CAS; (u) C.-M. Che, V. Q.-Q. Yam and T. C. W. Mak, J. Am. Chem. Soc., 1990, 112, 2284 CrossRef CAS; (v) J. C. Dobson and T. J. Meyer, Inorg. Chem., 1988, 27, 3283 CrossRef CAS.
- (a) T. Fujimoto, Y. Hirata, H. Sugimoto, M. Miyanishi, Y. Shiota, K. Yoshizawa and S. Itoh, Dalton Trans., 2022, 51, 1123 RSC; (b) T. Fujimoto, Y. Hirata, H. Sugimoto, M. Miyanishi, Y. Shiota, K. Yoshizawa and S. Itoh, Bull. Chem. Soc. Jpn., 2022, 95, 64 CrossRef CAS; (c) D. M. E. van Niekerk, T. E. Geswindt and W. J. Gerber, Inorg. Chem., 2021, 60, 782 CrossRef CAS PubMed; (d) T.-C. Lau, Angew. Chem., Int. Ed., 2016, 55, 288 CrossRef PubMed; (e) Y. Liu, S.-M. Ng, W. W. Y. Lam, S.-M. Yiu and T.-C. Lau, J. Am. Chem. Soc., 2004, 126, 14921 CrossRef PubMed.
- T. Nakano, T. Abe, T. Matsumoto, K. Kimura, G. Nakamura, S. Hayami, Y. Shiota, K. Yoshizawa and S. Ogo, RSC Adv., 2022, 12, 1253 Search PubMed.
- (a) Y. Shimizu, S. Fukui, T. Oi and H. Nagao, Bull. Chem. Soc. Jpn., 2008, 81, 1285 CrossRef CAS; (b) T. Suzuki, K. Matsuya, T. Kawamoto and H. Nagao, Eur. J. Inorg. Chem., 2014, 722 CrossRef CAS; (c) T. Suzuki, Y. Suzuki, T. Kawamoto, R. Miyamoto, S. Nanbu and H. Nagao, Inorg. Chem., 2016, 55, 6830 CrossRef CAS PubMed; (d) T. Misawa-Suzuki, T. Watanabe, M. Okamura, S. Nanbu and H. Nagao, Inorg. Chem., 2020, 59, 612 CrossRef CAS PubMed; (e) T. Misawa-Suzuki, S. Mafune and H. Nagao, Inorg. Chem., 2021, 60, 9996 CrossRef CAS PubMed.
- K. J. Jonasson, A. H. Mousa and O. F. Wendt, Polyhedron, 2018, 143, 132 CrossRef CAS.
- A. Looney, R. Han, K. McNeill and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4690 CrossRef CAS.
- F. Bottomley and J. Chen, Organometallics, 1992, 11, 3404 CrossRef CAS.
- T. Ghatak, A. Sinha, S. M. W. Rahaman and J. K. Bera, Inorg. Chim. Acta, 2011, 372, 94 CrossRef CAS.
- N. J. Meanwell, A. J. Smith, H. Adams, S. Okeya and P. M. Maitlis, Organometallics, 1983, 2, 1705 CrossRef CAS.
- (a) S. K. Tyrlik, M. Kisielinska and J. C. Huffman, Transition Met. Chem., 1995, 20, 413 CrossRef CAS; (b) M. Orts-Arroyo, I. Castro, F. Llobet and J. Martínez-Lillo, Cryst. Growth Des., 2020, 20, 2044 CrossRef CAS.
- (a) J. M. Power, K. Evertz, L. Henling, R. Marsh, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1990, 29, 5058 CrossRef CAS; (b) E. P. Kelson, L. M. Healing, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1993, 32, 2863 CrossRef CAS.
- P. Neubold, S. P. C. D. Vedova, B. K. Wieghardt, B. Number and J. Weiss, Angew. Chem., Int. Ed. Engl., 1989, 28, 763 CrossRef.
- A. Geilenkirchen, P. Neubold, R. Schneider, K. Wieghardt, U. Florke, H.-J. Haupt and B. Nuber, J. Chem. Soc., Dalton Trans., 1994, 457 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, FL, 2007 Search PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed.
- (a) J. R. Murdoch, J. Am. Chem. Soc., 1972, 94, 4410 CrossRef CAS; (b) J. E. Leffler, Science, 1953, 117, 340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115482 and 2115484. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04080h |
| This journal is © The Royal Society of Chemistry 2023 |