DOI:
10.1039/D2DT03644D
(Paper)
Dalton Trans., 2023,
52, 2255-2261
White light and colour-tunable emission from a single component europium-1,8-naphthalimide thin film†
Received
13th November 2022
, Accepted 1st December 2022
First published on 9th February 2023
Abstract
The synthesis and fabrication of spin coated films of a new Eu3+ complex [Eu(1)3] derived from the 1,8-naphthalimide containing ligand 1H is presented. The complex is multi-emissive displaying blue emission from the 1,8-naphthalimide fluorophore and red emission from the Eu3+ centre in both solution-state and solid-state. This allows the overall emission to be tuned by changing the excitaton wavelength, where varing degrees of red and blue emission intensity alter the overall emission colour from blue, to red and including white-light emission. The complex was spin-coated onto quartz slides giving 134 nm thick coatings that retained the multi-emissive and colour tunable properties. Overall, resulting in a colour-tunable system which in solution, solid, and thin film states can alter the overall colour from deep red to dark blue.
Introduction
Multi-emissive materials continue to attract significant attention from the chemistry research community. Their propensity towards advanced applications (ratiometric sensors,1–3 molecular barcodes,4 logic gates,5 imaging agents,6 and white emitting materials)7–11 means they are high value targets. Various approaches to generating multi-emissive materials exist,12–15 including the formation of supramolecular materials from trivalent lanthanide ions (Ln3+).7,11,16 Advantages of the supramolecular approach include combining both Ln3+ and ligand functionalities easily into a single component (i.e. a luminescent metal and a luminescent ligand to give multi-emission), inherently soluble systems that allow for solution processibility,17 and modular design meaning the systems can be easily tuned or improved. Ln3+ are particularly attractive for the aforementioned applications owing to their excellent photophysical properties (long-lived excited states, solid-state emission, line-like emission bands and tunable quantum yields).7,18 Whilst many multi-emissive Ln3+ containing systems are Metal Organic Frameworks (MOFs),3 we are interested in discrete systems as these are typically more soluble and processable. Choice of ligand fluorophore plays an important role – it must be emissive in the correct wavelength range, easily tuned, and not reduce solubility. 1,8-Naphthalimides (Nap) are excellent candidates,19 as they emit across a broad range of wavelengths,20 their solubility is tunable, they are synthetically easy to include into ligands, and have proven layer forming abilities.21–23 Naphthalimides have previously been used to generate Ln3+ complexes21–30 including in white emissive lanthanide systems.8,10 Ward and co-workers showed that single component multi-emission was possible by combining Eu3+ and Nap in a macrocyclic complex, achieving white emission in solution (CIE coordinates of x,y = 0.27, 0.25) and blue emission in thin films prepared by evaporation.10 In comparison Yan and co-workers achieved white emission in the solid-state using Eu3+ and Nap carboxylate ligands (CIE coordinates x,y = 0.34, 0.31–pure white is considered x,y = 0.33).8,10,31 However, to the best of our knowledge the naphthalimide–lanthanide combination has not been used to generate white-emissive thin-films. Herein, we report our approach to generating multi-emissive lanthanide thin-films by combining 1,8-naphthalimides and pyridine-2-carboxylate-6-amide (PDC) Ln3+ binding groups (Fig. 1). PDC is a relatively under-utilised motif for lanthanide assemblies but has both benefits of improved coordination of the more common dipicolinic acid (DPA) from the free carboxylate, and easy modification of the 2,6-pyridinedicarboxylate amide (PDA) through the amide side arm. PDC systems have shown to be effective sensitizers for a range of visibly emitting lanthanides.32 With this in mind, we have utilised our previously developed 1,2,3-triazole “click” synthetic strategy32–34 to functionalise PDC with a blue emissive Nap and complex with the visibly emissive lanthanide ion Eu3+.
 |
| | Fig. 1 Two stage synthesis of Ligand 1H with CuACC “click” reaction between precursors A and B followed by deprotection of benzyl ester resulting in ligand 1H. | |
Results and discussion
Ligand synthesis and characterisation
Ligand 1H was synthesised in a converging seven-step synthesis starting from commercially available DPA and 1,8-naphthalic anhydride (Fig. 1).32,33,35,36 1,8-Naphthalic anhydride was converted into N-(3-azidopropyl)-1,8-naphthalimide (B) in a three step process following literature procedures35,36 and then underwent a CuAAC reaction with our previously reported mono-alkyne (A)32 at 80 °C in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:H2O forming the benzyl protected intermediate (1Bz).32 Removal of the benzyl protecting group was carried out using LiOH following reported procedures32 and resulted in 1H as a off-white solid in 59% yield. All compounds were characterised using 1H-NMR, 13C-NMR, FTIR and Mass Spectrometry. Successful formation of 1H was confirmed by LRMS with peaks at 485.10 m/z and 507.00 m/z corresponding to the [1H + H]+ species (calc. for C25H21N6O5+, 485.16) and [1H + Na]+ species (calc. for C25H20N6O5Na+, 507.14) respectively. 1H-NMR also indicated successful formation of 1H, particularly when comparing the spectra of 1H with intermediate 1Bz and precursors A and B (Fig. S21†). The appearance of a singlet resonance peak at ≈8.0 ppm assigned to the 1,2,3-triazole proton along with the corresponding disappearance of the terminal alkyne proton signal at 3.2 ppm of precursor A and downfield shifts observed in linking methylene groups bonded to the 1,2,3-triazole ring (t) (3.5 to 4.1 ppm for N3–CH2 to t–CH2 and 4.2 to 4.6 ppm for NH–CH2–C CH to NH–CH2–t) post CuAAC reaction is evidence of successful 1,2,3-triazole formation in intermediate 1Bz. Deprotection of the carboxylate, forming 1H, is evident from the disappearance of the associated benzyl aromatic protons (Bz, 7.3–7.5 ppm) and the CH2–Bz linker (5.4 ppm). 13C NMR also confirmed formation of 1H with the correct number of carbon environments and appearance of 1,2,3-triazole ring signals (144.8 (C) and 123.5 (CH) ppm).
:1 DMF:H2O forming the benzyl protected intermediate (1Bz).32 Removal of the benzyl protecting group was carried out using LiOH following reported procedures32 and resulted in 1H as a off-white solid in 59% yield. All compounds were characterised using 1H-NMR, 13C-NMR, FTIR and Mass Spectrometry. Successful formation of 1H was confirmed by LRMS with peaks at 485.10 m/z and 507.00 m/z corresponding to the [1H + H]+ species (calc. for C25H21N6O5+, 485.16) and [1H + Na]+ species (calc. for C25H20N6O5Na+, 507.14) respectively. 1H-NMR also indicated successful formation of 1H, particularly when comparing the spectra of 1H with intermediate 1Bz and precursors A and B (Fig. S21†). The appearance of a singlet resonance peak at ≈8.0 ppm assigned to the 1,2,3-triazole proton along with the corresponding disappearance of the terminal alkyne proton signal at 3.2 ppm of precursor A and downfield shifts observed in linking methylene groups bonded to the 1,2,3-triazole ring (t) (3.5 to 4.1 ppm for N3–CH2 to t–CH2 and 4.2 to 4.6 ppm for NH–CH2–C CH to NH–CH2–t) post CuAAC reaction is evidence of successful 1,2,3-triazole formation in intermediate 1Bz. Deprotection of the carboxylate, forming 1H, is evident from the disappearance of the associated benzyl aromatic protons (Bz, 7.3–7.5 ppm) and the CH2–Bz linker (5.4 ppm). 13C NMR also confirmed formation of 1H with the correct number of carbon environments and appearance of 1,2,3-triazole ring signals (144.8 (C) and 123.5 (CH) ppm).
Lanthanide complexation and characterisation
Ln(1)3 complexes (Ln = Eu3+ and La3+) were prepared by heating 1H with 0.3 equivalents of Ln(CF3SO3)·xH2O and 1 equivalent of NEt3, in DCM:MeOH:CHCl3 (3:3:2). Vapour diffusion of diethyl ether resulted in off-white solids in good yields (68–83%). Complexes were fully characterised by elemental analysis, FTIR, HRMS and 1H-NMR (see ESI†). Lanthanide coordination within the PDC NO2 pocket is suggested by the notable shift to lower energy for both the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches from 1739 cm−1 (carboxyl) and 1656 cm−1 (amide) to a merged peak at 1632 cm−1, a shift commonly associated with coordination to Ln3+ ions (Fig. S22†).37 While the carbonyl stretch associated with the Nap is seen to remain unchanged at 1693 cm−1, indicating the lack of interaction between the Ln3+ and the Nap as expected. 1H NMR of La(1)3 (Fig. S28†) shows a significant downfield shift in the NH signal (0.71 ppm) and a corresponding upfield shift in the methylene linker (CH2–NH, 0.18 ppm), previously observed in similar PDC systems and indicative of La3+ coordination in the NO2 pocket.38 While proton signals associated with the central pyridyl ring of the PDC motif (which overlapped as one signal in the 1H spectrum) now separate into three distinct proton signals, while all other signals observed no significant shift, further indicative of lanthanide coordination occurring only within the PDC NO2 pocket. HRMS (Fig. S24–S27†) showed ions with the expected masses and isotropic distributions: [Eu(1)3 + 2Na]2+ 824.1980 m/z (calc. for C75H57N18O15EuNa2 824.1630 m/z), and [La(1)3 + 2Na]2+ 817.1818 m/z (calc. for C75H57N18O15LaNa2 817.1549 m/z).
O stretches from 1739 cm−1 (carboxyl) and 1656 cm−1 (amide) to a merged peak at 1632 cm−1, a shift commonly associated with coordination to Ln3+ ions (Fig. S22†).37 While the carbonyl stretch associated with the Nap is seen to remain unchanged at 1693 cm−1, indicating the lack of interaction between the Ln3+ and the Nap as expected. 1H NMR of La(1)3 (Fig. S28†) shows a significant downfield shift in the NH signal (0.71 ppm) and a corresponding upfield shift in the methylene linker (CH2–NH, 0.18 ppm), previously observed in similar PDC systems and indicative of La3+ coordination in the NO2 pocket.38 While proton signals associated with the central pyridyl ring of the PDC motif (which overlapped as one signal in the 1H spectrum) now separate into three distinct proton signals, while all other signals observed no significant shift, further indicative of lanthanide coordination occurring only within the PDC NO2 pocket. HRMS (Fig. S24–S27†) showed ions with the expected masses and isotropic distributions: [Eu(1)3 + 2Na]2+ 824.1980 m/z (calc. for C75H57N18O15EuNa2 824.1630 m/z), and [La(1)3 + 2Na]2+ 817.1818 m/z (calc. for C75H57N18O15LaNa2 817.1549 m/z).
Photophysical properties
UV-visible absorption properties of 1H and Ln(1)3 complexes were measured in MeOH (Fig. S23†). 1H showed a broad absorbance spectrum from 210 to 360 nm, significant absorptions are observed at 220 nm and 370 nm associated with n → π* and π → π* transitions from the Nap,39 and weaker absorptions from 260–280 nm assigned to the n → π* and π → π* transitions from the central pyridyl unit.33,40–42 Upon complexation the absorption spectra of all Ln3+ complexes underwent a slight red shift in the pyridyl associated bands while the Nap bands remained unshifted (Fig. S23†).
Photoluminescence studies of 1H (Fig. S29†) showed the expected Nap monomer emission ranging from 350 to 450 nm, with λmax = 388 nm in both the solution and solid states,20 when excited into the Nap based absorption bands (240 or 340 nm). The fluorescence quantum yield in MeOH was 12.4%, similar to other 1,8-naphthalimide containing systems.20,25,43
Upon coordination, the Nap monomer emission at 388 nm was retained in all complexes (when excited at λex = 240, 274 or 340 nm). However, an additional new broad peak at 490 nm was observed (Fig. 2 and S30–S31†). This is likely from the three Nap moieties being in close proximity within the complex, resulting in an excimer emission band – a known phenomenon in Nap systems.20,44 Excitation spectra showed the excimer bands associated with the Nap motif.
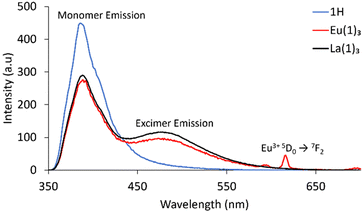 |
| | Fig. 2 Fluorescence spectra of 1H, Eu(1)3 and La(1)3 showing excimer formation upon complexation (0.01 mM, MeOH, λex = 300 nm). | |
Eu(1)3 displayed strong Eu3+ centred emission (λex between 210 and 290 nm) with (J = 0–4) transitions in both steady state and time delayed measurements, including the 581 nm 5D0 → 7F0 transition indicative of an overall C3 symmetry (Fig. S31 and 32†).45 Emission lifetimes of the 5D0 → 7F2 transition were found to be 1.175 ms and were best fit to a single exponential decay, indicative of a single species in solution (Fig. S33†). The overall quantum yield was determined to be 7.5% which is midrange for PDC based Eu3+ complexes.32,38,46–48 Excitation plots indicated the primary antenna for Eu3+ centred emission is the pyridyl group with a major peak at 278 nm (Fig. S31†). Interestingly, there is a much smaller band from 300 to 350 nm associated with the Nap group, which can act as a secondary antenna. This is expected due to the Nap triplet state energy matching the Eu3+ excited state (18500 cm−1 and 17500 cm−1 respectively).10
Self-assembly studies
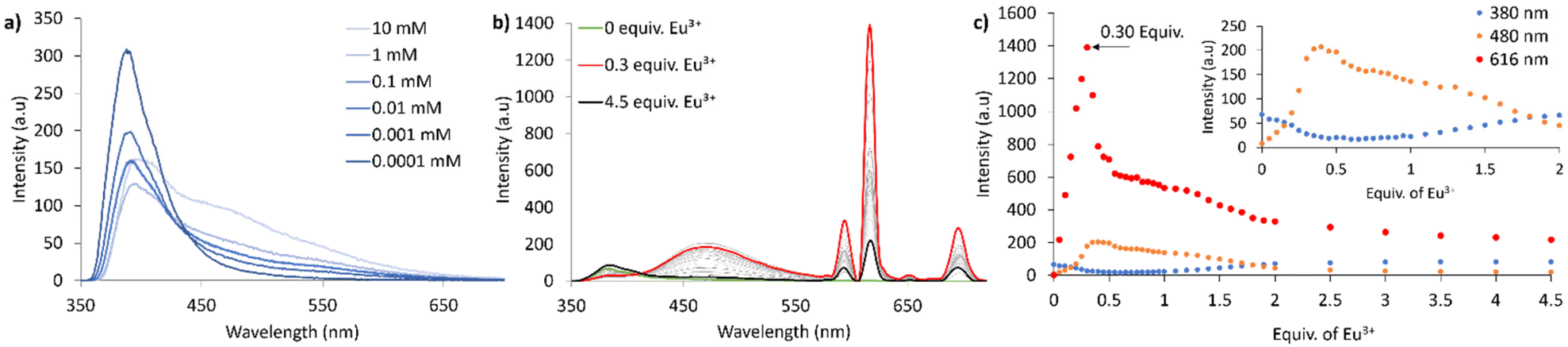
To further confirm that excimer formation was a result of complexation, concentration vs. emission intensity, and self-assembly studies of 1H were carried out. At high concentrations of 1H (10 mM) aggregation induced excimer emission was present. Upon reducing the concentration from 10 mM to 1 × 10−4 mM excimer emission disappeared with monomer emission becoming more intense (Fig. 3a). Fluorescence self-assembly studies between Eu3+ and 1H (0.02 mM) showed that on addition of Eu3+ from 0 to 0.35 equiv. excimer (and Eu3+ centred) emission increased and monomer emission decreased. Excimer emission (and Eu3+ centred emission) peaked at 0.35 equivalents of Eu3+ while monomer emission reached a minimum, signifying the formation of the Eu(1)3 complex and intramolecular interaction between neighbouring Nap, resulting in excimer emission (Fig. 3b and c).44 Further addition of Eu3+ resulted in the formation of Eu(1)2 and Eu(1) species (as seen in other PDC systems)32 which was accompanied by decreased excimer emission and increased monomer emission, suggestive of all three Nap motifs situated on the same side of the Eu(1)3 complex, thus maximising intramolecular interaction between neighbouring Nap (Fig. 3b and c).
 |
| | Fig. 3 (a) Fluorescence emission spectra (λex = 340 nm) of 1H at different concentrations showing aggregation induced excimer formation at higher concentrations in MeOH. (b) Fluorescence titration of 1H with Eu(CF3SO3)3 from 0 to 4.5 equivalents (λex = 279 nm, 0.02 mM, MeOH). (c) Changes in fluorescence emission of 1H (0.02 mM, MeOH) when titrated with Eu(CF3SO3)·6H2O: Nap monomer emission at λ = 380 nm, Nap excimer emission at λ = 480 nm and Eu3+ emission at λ = 616 nm. | |
Colour-tunability
With two well separated luminophores with different excitation wavelengths, the overall emission profile of Eu(1)3 in solution (0.01 mM, MeOH) is dependent on the excitation wavelength (Nap emission at λex = 200–240 nm and 290–350 nm, and Eu3+ emission at λex = 245–290 nm, Fig. 4a, b and S39†). As a result, Eu(1)3 is a rare example of a single component compound displaying colour-tunable emission. By simply altering the excitation wavelength, a large range of colour profiles were achieved. At excitation wavelengths below 240 nm blue emission was observed with Nap-based emission dominating the spectrum. Excitation at 245 nm resulted in increased Eu3+ emission and a colour-profile in the white emission region (CIE x,y = 0.35, 0.26). On increasing the excitation wavelength to 275 nm, where Eu3+ emission was maximised, bright red emission (CIE x,y = 0.54, 0.31) was observed. Further increasing the excitation to >290 nm, where Eu3+ emission reduced and Nap increased, gave the expected blue emission profile.
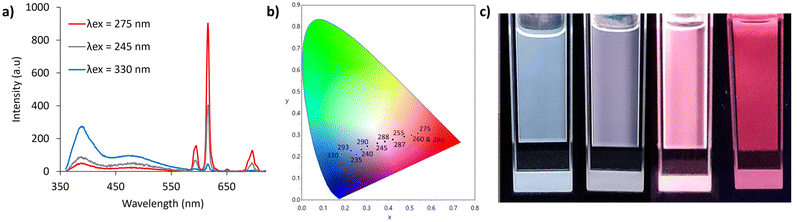 |
| | Fig. 4 (a) Eu(1)3 fluorescent emission (0.01 mM, MeOH), showing Eu3+ centred emission vs.Nap centred emission at critical wavelengths. (b) CIE chromaticity diagram showing the colour-tunable emission range of Eu(1)3 in solution (0.01 mM, MeOH). (c) Different concentrations of Eu(1)3 in MeOH ranging from 10, 5, 1, 0.01 mM left to right, under shortwave UV light (λex = 254 nm). | |
The colour-tunable emission can be further modified by changing the concentration to bring about aggregation-induced intermolecular excimer emission between complexes. Increasing the concentration from 0.01 mM to 1 mM, 5 mM and 10 mM significantly increased excimer emission (Fig. S40–42†). As a result, the colour-tunable window was reduced with the 1 mM solution only able to obtain a light red emission (Fig. S40†), and the 5 mM (Fig. S41†) and 10 mM (Fig. S42†) solutions unable to achieve red emission. The reduction of the colour-tunable window coincides with improved white emission where the 1 mM solution achieved close to pure-white emission when excited at 250 nm (CIE x,y = 0.33, 0.28). The 5 mM solution appeared white under shortwave UV irradiation (Fig. 4c) and achieved emission with CIE coordinates of x,y = 0.31, 0.27 when excited at 270 nm. A further increase to 10 mM caused overall emission to appear blue-white under shortwave UV (Fig. 4c) and excitation at 270 nm gave white emission (CIE x,y = 0.28, 0.26) similar to Ward and co-workers system.10
Solid state emission properties
The colour-tunable emission was also observed in the solid state. As expected, with only a Nap luminophore, solids of 1H and La(1)3 gave bright blue emission under both shortwave (λex = 254 nm) and longwave (λex = 365 nm) irradiation. While Eu(1)3 displayed colour-tunable emission (Fig. 5), switching from bright blue to white under long- and short-wave UV respectively. Solid state fluorescence showed increased excimer emission, and a colour-tunable window similar to that observed in the Eu(1)3 5 mM solution (Fig. S41†). Excitation outside of the 260–288 nm range resulted in overall blue emission while between these excitation wavelengths, overall emission was white with CIE coordinates ranging within x = 0.28–0.32 and y = 0.29. Nearest to pure white was achieved when λex = 275 nm with CIE coordinates x,y = 0.32, 0.29.
 |
| | Fig. 5 Eu(1)3 solid powder, shown (left to right) under ambient light, 254 nm UV irradiation and 365 nm UV irradiation. | |
Thin film fabrication
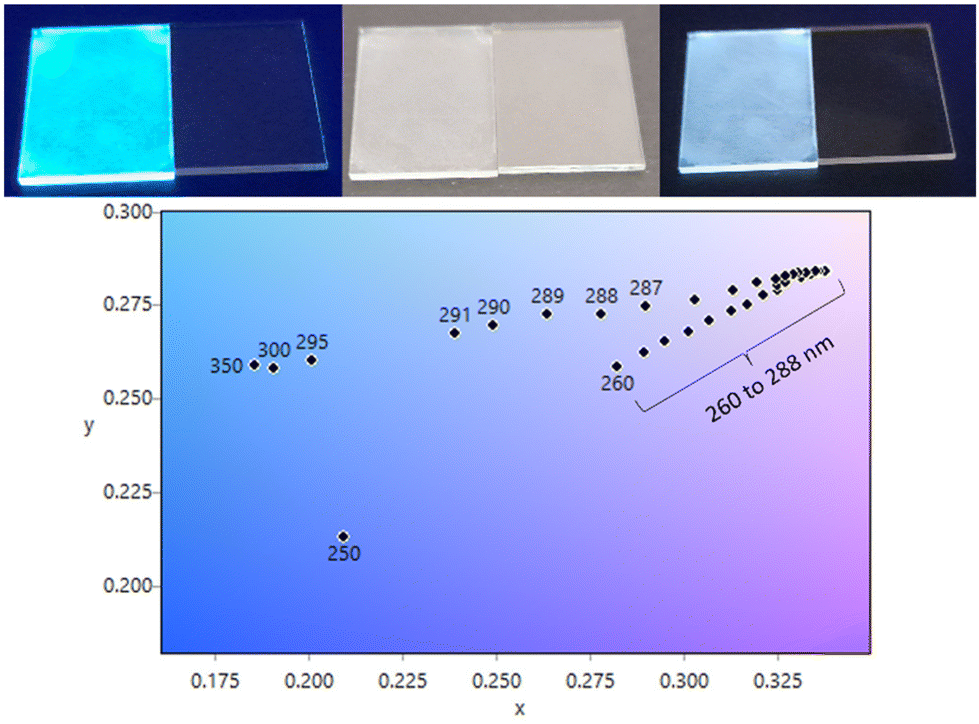
With Eu(1)3 displaying colour-tunable and white emission observed in both solid- and solution-states we next investigated the ability of Eu(1)3 to form thin films through spin coating deposition. A 10 mg ml−1 solution of Eu(1)3 in DCM:MeOH:DMF (4:4:2) was spin coated onto 15 × 20 mm glass and 20 × 20 mm quartz substrates. 50 μL was dynamically deposited onto the substrates at 500 rpm and then spun up to 4000 rpm, resulting in a uniform film to the eye (Fig. 6). AFM measurements showed a stippled coating with an average particle height of 133.5 nm, while also containing some larger aggregates (see Fig. S45 and 46†). Importantly, the film retained the properties exhibited in the solution and bulk solid states. The film appeared bright white under short-wave and light blue under long-wave UV irradiation (Fig. 6). Photophysical measurements were carried out on the film showing that it retained the same absorption profile of Eu(1)3 as measured in solution (Fig. S44†). Fluorescence emission showed a similar colour-tunable emission profile to the solid state (and high concentration solution) with strong excimer emission (Fig. S47†). Excitation wavelengths outside of the 260–288 nm range resulted in overall blue emission as it favours Nap excimer emission. Excitation between 260–288 nm (where Eu3+ centred emission increases) resulted in overall emission within the white region with CIE coordinates ranging within x = 0.28–0.34 and y = 0.26–0.28 (Fig. 6). Closest to pure white emission is achieved over a range of excitation wavelengths (λex = 270–272, 279–283 nm) with CIE coordinates equalling x,y = 0.33, 0.28, close to the emission of the original solid. While λex of 273–278 nm emission is still close to pure white with CIE coordinates x,y = 0.34, 0.28.
 |
| | Fig. 6 (Top) Spin coated films of Eu(1)3 on a 15 × 20 mm glass substrate, under 365 nm, ambient light and 254 nm UV irradiation, respectively. (Bottom) CIE coordinates showing the colour-tunable emission range for the Eu(1)3 spin coated film (see ESI† for CIE coordinates). | |
Conclusions
The discrete lanthanide complex Eu(1)3 resulted in the formation of a multi-emissive assembly capable of colour-tunable emission dependent on the excitation wavelength ranging from blue to red and close to white emission in solution and solid states. With Eu(1)3 being a discrete system, it is easily solubilised in a range of solvents. This allowed Eu(1)3 to form thin films by spin coating. The thin films retained both colour-tunability and white emission, which to the best of our knowledge is the first example of a discrete Ln3+-1,8-naphthalimide complex processed into thin films that exhibit such properties. Additionally, the luminophores were observed to act almost independently from each other at most excitation wavelengths. Such multi-emission is ideal for sensor development, as it allows for self-calibrating ratiometric systems.1,49,50 We are currently investigating the potential for self-calibrating ratiometric oxygen sensors both in solution and as films, in which the organic emission is quenched in the presence of oxygen, while the Ln3+ signal self-calibrates and regulates signal change.
Experimental
General experimental details
All reagents, solvents and starting materials were purchased from Sigma-Aldrich. Precursors A32 and B35,36 were synthesised using literature methods and showed the expected characterisation. NMR spectra were recorded using a Bruker Ultrashield 300, with chemical shifts recorded in parts per million (ppm) downfield from the standard. UV-visible absorption was recorded on a Shimadzu UV-1800 using CH2Cl2 and MeOH solvents in a 3 mL quartz cuvette with a 1 cm path length. Steady state–fluorescence measurements were recorded on a Shimadzu RF-6000 Spectro fluorophotometer and time resolved phosphorescence measurements were carried out using an Agilent Technologies Cary Eclipse fluorescence spectrophotometer, with CH2Cl3 and MeOH solvents in a capped 3.5 mL quartz cuvette with a 1 cm path length. Self-assembly titrations: The formation of 1:1, 1:2 and 1:3 (M:L) species was monitored by measuring changes in the UV-visible absorption and fluorescence spectra (λex = 279 nm) of 1H (2 × 10−5 M in 3 mL volume in MeOH diluted from 0.001 M stock solution in DCM:MeOH (1:1)) when titrated with Eu(CF3SO3)3·6H2O solution (1 mM, MeOH) from 0→4.5 equivalents, with 3 μL additions for 0 to 1 equivalents, 6 μL additions for 1 to 2 equivalents, and 30 μL additions for 2 to 4.5 equivalents. Commission Internationale de l'Eclairage (CIE) plots were generated in OSRAM LED ColorCalculator software. Melting points were obtained on a Electrothermal IA9000 Series Melting Point Apparatus. Fourier-transform infrared spectroscopy (FTIR) of solids were recorded on a Bruker Alpha platinum-ATR. Mass spectrometry of ligands was carried out in HPLC grade solvents, on a Shimadzu LCMS-2020 for ligands and a Bruker Daltonics MicrOTOF™ Spectrometer for complexes. Spin coated films were fabricated on an Ossila spin coater using 1 mm thick quartz substrates (10 mm × 15 mm). Atomic force microscopy (AFM) measurements were performed in tapping mode, on an Asylum Cypher ES (Oxford Instruments, US). AFM tip parameters with silicon probes (nominal spring constant 5 N m−1, resonance frequency 150 kHz, from Budget Sensors, Bulgaria), with a set point at 80% amplitude and measurements run in attractive mode. Safety Note: whilst no issues were encountered here, low molecular weight organic azides are potential explosives and care must be taken during their handling.
Quantum yield measurements
Quantum yield measurements were determined by the dilute comparison method51 using relative standards Cs3[Eu(dpa)3]·8H2O,52,53 complex in a 0.1 M tris-HCl buffer solution (pH ≈ 7.45) and quinine sulfate in 0.5 M H2SO4,54 with known quantum yields of Φref = 24 ± 2.5%, and Φref = 54.6% respectively. Cs3[Eu(dpa)3]·9H2O was used for Eu(1)3 and quinine sulfate was used for the 1,8-naphthalimide emission. Barrier slit widths remained the same between measurements for different compounds with 1.5, 3 nm excitation and emission widths. Excitation wavelength was the same for all measurements with the standard 279 nm excitation wavelength being used for Eu3+ emissions and 366 nm for quinine sulfate and Nap emissions. Complexes were dissolved in a 1:1 MeOH:CH2Cl2 and then diluted into MeOH.| |  | (1) |
Estimated overall quantum yields ΦLLn = Φx were calculated according to eqn (1).51,52 Here grad refers to the slope of plotted emission area vs. absorbance (emission area was taken from specific emission peaks, Eu(1)3 (5D0 → 7F2), and 1,8-naphthalimide excimer and monomer broad emissions vs. quinine sulfate), n refers to refractive index of the solution (a refractive index of n = 1.3295 was found for MeOH:CH2Cl2 solution), and subscripts are ref for reference and x for sample.51,52
Synthesis of N2-bis((1-(N-(3-propyl)-1,8-naphthalimide)-1H-1,2,3-triazol-4-yl)methyl)-O6-((benzyloxy)carbonyl)pyridine-2 carboxamide-6-carboxylate (1Bz)
A (0.400 g, 1.4 mM) was combined with sodium ascorbate (0.269 g, 1.4 mM), CuSO4·5H2O (0.170 g, 0.7 mM) and B (0.381 g, 1.4 mM) in 30 mL of DMF:H2O (4:1). The solution was heated to 80 °C overnight, changing from a light yellow to a dark green. Water was slowly added to the solution until a cloudy precipitate formed, which was filtered and then washed with 0.1 M NaOH/0.5 M EDTA solution yielding 1Bz as an off-white solid (0.558 g, 71%). Melting point = 143.3 °C. LRMS m/z = 575.15 [1Bz + H]+ (calc. for C32H27N6O5+, 575.20). 1H NMR (300 MHz, DMSO-d6, ppm), δ = 9.04 (t, J = 6.0 Hz, 1H, NH), 8.49 (t, J = 7.0, 4H, Nap–H), 8.27 (m, 3H, Py–H), 8.05 (s, 1H, t–H), 7.89 (m, 2H, Nap–H), 7.41 (m, 5H, Bz–H), 5.45 (s, 1H, CH2–Bz), 4.59 (d, J = 5.5 Hz, 2H, NH–CH2–t), 4.48 (t, J = 7.0 Hz, 2H, t–CH2), 4.15 (t, J = 7.0 Hz, 2H, CH2–Nap), 2.23 (m, 2.23, CH2–CH2–CH2); 13C NMR (75 MHz, DMSO-d6, ppm): δ = 164.3 (CO–Py), 164.1 (CO–Nap), 163.6 (CO–Py), 150.6 (CN–Py), 146.9 (CN–Py), 144.8 (C–t), 140.0 (CH–Py), 136.2 (C–Bz), 134.7 (CH–Nap), 131.7 (C–Nap), 131.1 (CH–Nap), 129.0 (CH–Bz), 128.7 (CH–Bz), 128.5 (CH–Bz), 127.9 (C–Nap), 127.9 (CH–Py), 127.6 (CH–Nap), 125.9 (CH–Py), 123.5 (CH–t), 122.6 (C–Nap), 67.3 (CH2–Bn), 48.0 (CH2), 37.8 (CH2), 35.2 (CH2–NH), 29.0 (CH2). FTIR (cm−1) 1779, 1734, 1695, 1651, 1585, 1531, 1438, 1348, 1305, 1305, 1232, 1171, 1124, 1049, 1002, 907, 843, 773, 745, 675, 643, 541, 514.
Synthesis of N2-((1-(N-(3-propyl)-1,8-naphthalimide)-1H-1,2,3-triazol-4-yl)methyl)pyridine-2-carboxamide-6-carboxylate (1H)
1Bz (0.858 g, 1.5 mM) was combined with LiOH (0.143 g, 6.0 mM) in 20 mL of THF:MeOH:H2O (3:1:1). This was stirred overnight under a nitrogen atmosphere, where the solid slowly dissolved into a light-yellow solution. The solvent was then removed resulting in an oily residue. 1 M HCl was then added causing an off-white precipitate to form, which was filtered and washed with water and acetone to remove the 1,8-naphthalimide by-product, yielding 1H as an off-white solid (0.428 g, 59%). Melting point = 148.2 °C. LRMS m/z = 485.10 [1H + H]+ (calc. for C25H21N6O5+, 485.16) and m/z = 507.00 [1H + Na]+ (calc. for C25H20N6O5Na+, 507.14). 1H NMR (300 MHz, DMSO-d6, ppm), δ = 9.71 (t, J = 6.0 Hz, 1H, NH), 8.46 (t, J = 6.0, 4H, Nap–H), 8.23 (m, 3H, Py–H), 8.05 (s, 1H, t–H), 7.87 (m, 2H, Nap–H), 4.58 (d, J = 6.0 Hz, 2H, NH–CH2–t), 4.45 (t, J = 7.0 Hz, 2H, t–CH2), 4.11 (t, J = 7.0 Hz, 2H, CH2–Nap), 2.20 (m, 2H, CH2–CH2–CH2); 13C NMR (75 MHz, DMSO-d6, ppm): 165.4 (CO–Py), 164.1 (CO–Nap), 163.3 (CO–Py), 149.5 (CN–Py), 146.4 (CN–Py), 144.8 (C–t), 140.4 (CH–Py), 134.8 (CH–Nap), 131.8 (C–Nap), 131.2 (CH–Nap), 127.9 (C–Nap), 127.7 (CH–Nap), 127.1 (CH–Py), 125.9 (CH–Py), 123.6 (CH–t), 122.8 (C–Nap), 48.0 (CH2), 37.8 (CH2), 34.9 (CH2–NH), 29.0 (CH2). FTIR (cm−1) 3234, 3138, 2925, 2853, 2394, 1893, 1739 (COOH–Py), 1695 (CO–Nap), 1657 (CONH–Py), 1625, 1587, 1530, 1454, 1440, 1387, 1345, 1306, 1235, 1170, 1147, 1121, 1074, 1054, 1016, 1001, 963, 907, 880, 847, 801, 780, 765, 747, 682, 643, 628, 563, 546, 516, 489, 423.
Synthesis of Eu(1)3
1H (0.0640 g, 0.132 mM) was combined with Eu(CF3SO3)3·6H2O (0.0308 g, 0.044 mM) and 1 equivalent of triethylamine in 5 mL of DCM:MeOH:CHCl3 (3:3:2). The complex precipitated as a white powder, which was filtered giving Eu(1)3 as a white solid (0.058 g, 83%). Melting point = 205–226 °C. HRMS m/z = 824.1980 [Eu(1)3 + 2Na]2+ (calc. for (C75H57N18O15EuNa2)2+, 824.1630), m/z = 1625.3545 [Eu(1)3 + Na]+ (calc. for (C75H57N18O15EuNa)+, 1625.3367). FTIR (cm−1) 3233, 3073, 2965, 1698, 1633, 1587, 1565, 1462, 1430, 1347, 1277, 1235, 1155, 1082, 1048, 1029, 1018, 890, 848, 801, 779, 761, 724, 687, 660, 637, 573, 540, 517, 410. Elemental analysis for C75H57N18O15Eu·3.5H2O·2.5CH2Cl2 (1878.7389 g mol−1) calculated: C 49.55, H 3.70, N 13.42%. Found C 49.74, H 4.00, N 13.45%.
Synthesis of La(1)3
1H (0.0622 g, 0.128 mM) was combined with La(CF3SO3)3·9H2O (0.0317 g, 0.044 mM) and 1 equivalent of triethylamine in 5 mL of DCM:MeOH:CHCl3 (3:3:2). The complex precipitated as a white powder, which was filtered giving La(1)3 as a white solid (0.046 g, 68%). HRMS m/z = 817.1818 [La(1)3 + 2Na]2+ (calc. for (C75H57N18O15LaNa2)2+, 817.1549). FTIR (cm−1) 3270, 3080, 2967, 2337, 2318, 1697, 1624, 1587, 1566, 1436, 1347, 1275, 1235, 1169, 1084, 1049, 1030, 890, 847, 801, 778, 759, 723, 687, 637, 573, 540, 516, 407. Elemental analysis for C75H57N18O15La·2·CHCl3 (1860.0489 g mol−1) calculated: C 49.72, H 3.20, N 13.55%. Found C 49.76, H 3.46, N 13.37%. 1H NMR (300 MHz, DMSO-d6, ppm), δ = 10.43 (t, J = 6.0 Hz, 1H, NH), 8.45 (t, J = 6.0, 4H, Nap–H), 8.37 (m, 1H, Py–H), 8.22 (m, 1H, Py–H), 8.1 (s, 1H, t–H), 7.91 (m, 1H, Py–H), 7.83 (m, 2H, Nap–H), 4.48 (d, J = 6.0 Hz, 2H, CH2–Nap), 4.40 (t, J = 7.0 Hz, 2H, NH–CH2–t), 4.11 (t, J = 7.0 Hz, 2H, t–CH2), 2.20 (m, 2H, CH2–CH2–CH2).
Author contributions
Alex O'Neil: conceptualisation, investigation, methodology, analysis, writing – original draft. Anaïs Chalard: AFM investigation, writing – review & editing. Jenny Malmström: supervision, AFM investigation, writing – review & editing. Jonathan Kitchen: conceptualisation, writing – review & editing, supervision, project administration.
Conflicts of interest
There are no conflicts to declare.
References
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885–4901 RSC.
- Y. Zhou and B. Yan, J. Mater. Chem., 2015, 3, 9353–9358 RSC.
- T. Sun, Y. Gao, Y. Du, L. Zhou and X. Chen, Front. Chem., 2021, 8, 624592 CrossRef PubMed.
- J. Wang, Y. Suffren, C. Daiguebonne, S. Freslon, K. Bernot, G. Calvez, L. Le Pollès, C. Roiland and O. Guillou, Inorg. Chem., 2019, 58, 2659–2668 CrossRef PubMed.
- M. A. Hernández-Rodríguez, C. D. S. Brites, G. Antorrena, R. Piñol, R. Cases, L. Pérez-García, M. Rodrigues, J. A. Plaza, N. Torras, I. Díez, A. Millán and L. D. Carlos, Adv. Opt. Mater., 2020, 8, 2000312 CrossRef.
- D. Kim, K. Jeong, J. E. Kwon, H. Park, S. Lee, S. Kim and S. Y. Park, Nat. Commun., 2019, 10, 3089 CrossRef PubMed.
- S. SeethaLekshmi, A. R. Ramya, M. L. P. Reddy and S. Varughese, J. Photochem. Photobiol., C, 2017, 33, 109–131 CrossRef CAS.
- J. Zhang, H. Li, P. Chen, W. Sun, T. Gao and P. Yan, J. Mater. Chem. C, 2015, 3, 1799–1806 RSC.
- O. Kotova, S. Comby, C. Lincheneau and T. Gunnlaugsson, Chem. Sci., 2017, 8, 3419–3426 RSC.
- A. H. Shelton, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Chem. Commun., 2012, 48, 2749–2751 RSC.
- R. Boddula, J. Tagare, K. Singh and S. Vaidyanathan, Mater. Chem. Front., 2021, 5, 3159–3175 RSC.
- V. Anand, R. Mishra and Y. Barot, Dyes Pigm., 2021, 191, 109390 CrossRef CAS.
- N.-C. Chiu, K. T. Smith and K. C. Stylianou, Coord. Chem. Rev., 2022, 459, 214441 CrossRef CAS.
- N. Kim, J. Jeong and H. Chae, Appl. Sci. Converg. Technol., 2016, 25, 1–6 CrossRef.
- P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., Int. Ed., 2005, 44, 1806–1810 CrossRef CAS PubMed.
- A. R. Ramya, S. Varughese and M. L. P. Reddy, Dalton Trans., 2014, 43, 10940–10946 RSC.
- J.-H. Jou, S. Sahoo, D. K. Dubey, R. A. K. Yadav, S. S. Swayamprabha and S. D. Chavhan, J. Mater. Chem., 2018, 6, 11492–11518 CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS PubMed.
- D. W. Cho and D. W. Cho, New J. Chem., 2014, 38, 2233–2236 RSC.
- A. B. Carter, N. Zhang, I. A. Kühne, T. D. Keene, A. K. Powell and J. A. Kitchen, ChemistrySelect, 2019, 4, 1850–1856 CrossRef CAS.
- D. L. Reger, A. P. Leitner and M. D. Smith, Cryst. Growth Des., 2016, 16, 527–536 CrossRef CAS.
- D. L. Reger, A. Leitner and M. D. Smith, Cryst. Growth Des., 2015, 15, 5637–5644 CrossRef CAS.
- K. R. Johnson and A. de Bettencourt-Dias, Inorg. Chem., 2019, 58, 13471–13480 CrossRef CAS PubMed.
- Z. Wang, N. Liu, H. Li, P. Chen and P. Yan, Eur. J. Inorg. Chem., 2017, 2017, 2211–2219 CrossRef CAS.
- V. F. Plyusnin, A. S. Kupryakov, V. P. Grivin, A. H. Shelton, I. V. Sazanovich, A. J. H. M. Meijer, J. A. Weinstein and M. D. Ward, Photochem. Photobiol. Sci., 2013, 12, 1666–1679 CrossRef CAS PubMed.
- C. S. Bonnet, M. Devocelle and T. Gunnlaugsson, Org. Biomol. Chem., 2012, 10, 126–133 RSC.
- M. A. Alcala, C. M. Shade, H. Uh, S. Y. Kwan, M. Bischof, Z. P. Thompson, K. A. Gogick, A. R. Meier, T. G. Strein, D. L. Bartlett, R. A. Modzelewski, Y. J. Lee, S. Petoud and C. K. Brown, Biomaterials, 2011, 32, 9343–9352 CrossRef CAS PubMed.
- J. Tang, H. Yang, J. Liu, Y. Wang, X. Yin, R. Wang, L. Huang and Z. Huang, Opt. Mater., 2010, 32, 1417–1422 CrossRef CAS.
- J. E. Elbert, S. Paulsen, L. Robinson, S. Elzey and K. Klein, J. Photochem. Photobiol., A, 2005, 169, 9–19 CrossRef CAS.
- S. Dang, J.-H. Zhang and Z.-M. Sun, J. Mater. Chem., 2012, 22, 8868–8873 RSC.
- A. T. O'Neil, J. A. Harrison and J. A. Kitchen, Chem. Commun., 2021, 57, 8067–8070 RSC.
- A. T. O'Neil, N. Zhang, J. A. Harrison, S. M. Goldup and J. A. Kitchen, Supramol. Chem., 2021, 33, 160–173 CrossRef.
- A. T. O'Neil and J. A. Kitchen, Chemistry, 2022, 4, 1457–1465 CrossRef.
- U. H. Sk and S. Bhattacharya, Environ. Toxicol. Pharmacol., 2006, 22, 298–308 CrossRef CAS PubMed.
- Q. M. Chen, Z. Li, G. X. Tian, Y. Chen and X. H. Wu, J. Chem. Res., 2020, 45, 258–264 CrossRef.
- R. Jagannathan and S. Soundararajan, J. Coord. Chem., 1979, 9, 31–35 CrossRef CAS.
- O. Kotova, S. Blasco, B. Twamley, J. O'Brien, R. D. Peacock, J. A. Kitchen, M. Martínez-Calvo and T. Gunnlaugsson, Chem. Sci., 2015, 6, 457–471 RSC.
- J. Gawronski, K. Gawronska, P. Skowronek and A. Holmén, J. Org. Chem., 1999, 64, 234–241 CrossRef CAS PubMed.
- T. Le Borgne, J.-M. Bénech, S. Floquet, G. Bernardinelli, C. Aliprandini, P. Bettens and C. Piguet, Dalton Trans., 2003, 3856–3868 RSC.
- F. Renaud, C. Piguet, G. Bernardinelli, J.-C. G. Bünzli and G. Hopfgartner, Chem. – Eur. J., 1997, 3, 1646–1659 CrossRef CAS.
- A.-S. Chauvin, J.-C. G. Bünzli, F. Bochud, R. Scopelliti and P. Froidevaux, Chem. – Eur. J., 2006, 12, 6852–6864 CrossRef CAS PubMed.
- V. F. Plyusnin, A. S. Kupryakov, V. P. Grivin, A. H. Shelton, I. V. Sazanovich, A. J. Meijer, J. A. Weinstein and M. D. Ward, Photochem. Photobiol. Sci., 2013, 12, 1666–1679 CrossRef CAS PubMed.
- M. Licchelli, A. O. Biroli, A. Poggi, D. Sacchi, C. Sangermani and M. Zema, Dalton Trans., 2003, 4537–4545 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- J.-M. Senegas, G. Bernardinelli, D. Imbert, J.-C. G. Bünzli, P.-Y. Morgantini, J. Weber and C. Piguet, Inorg. Chem., 2003, 42, 4680–4695 CrossRef CAS PubMed.
- B.-L. An, M.-L. Gong, J.-M. Zhang and S.-L. Zheng, Polyhedron, 2003, 22, 2719–2724 CrossRef CAS.
- B.-L. An, M.-L. Gong, K.-W. Cheah, J.-M. Zhang and K.-F. Li, Chem. Phys. Lett., 2004, 385, 345–350 CrossRef CAS.
- A. P. Demchenko, Lab Chip, 2005, 5, 1210–1223 RSC.
- M. Amelia, A. Lavie-Cambot, N. D. McClenaghan and A. Credi, Chem. Commun., 2011, 47, 325–327 RSC.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- A. S. Chauvin, F. Gumy, D. Imbert and J. C. G. Bünzli, Spectrosc. Lett., 2004, 37, 517–532 CrossRef CAS.
- A. S. Chauvin, F. Gumy, D. Imbert and J. C. G. Bünzli, Spectrosc. Lett., 2007, 40, 193–193 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Anaïs
Chalard
bc,
Jenny
Malmström
a,
Anaïs
Chalard
bc,
Jenny
Malmström