
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst ternary tungsten tellurate(IV) WTe2O7 with unique crystal structure type†
Raimund
Ziegler
 ,
Felix R. S.
Purtscher
,
Heidi A.
Schwartz
,
Thomas S.
Hofer
and
Gunter
Heymann
*
,
Felix R. S.
Purtscher
,
Heidi A.
Schwartz
,
Thomas S.
Hofer
and
Gunter
Heymann
*
University of Innsbruck, Department for General, Inorganic and Theoretical Chemistry, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: Gunter.Heymann@uibk.ac.at; Fax: +43 (512) 507-57099
First published on 12th December 2022
Abstract
At multianvil high-pressure/high-temperature conditions of 10 GPa and 1273 K, the first ternary tungsten tellurate WTe2O7 is formed, starting from a stoichiometric mixture of WO3 and TeO2. The compound crystallizes triclinic in a hitherto unknown crystal structure type with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; (no. 2), and was refined from single-crystal X-ray diffractometer data: a = 538.3(1), b = 687.5(1), c = 802.3(1) pm, α = 72.4(1)°, β = 85.7(1)°, γ = 68.1(1)°, wR2 = 0.0323, GooF = 1.048, 3157 F2 values, and 106 variables. The main motifs of the crystal structure are pairs of edge-linked [WO6]6− octahedra and fourfold oxygen-coordinated Te4+ atoms. The oxidation state of W6+ and Te4+ was further verified by measuring the characteristic binding energy values for the W 4f and the Te 3d core levels via X-ray photoelectron spectroscopy (XPS). In addition, DFT calculations of the structure, the associated electron localisation functions (ELF) and vibrational spectra have been carried out. The theoretical data clearly demonstrates the impact of the residual electron density located at the Te4+ ions, which can be directly interpreted as the presence of lone electron pairs within the solid structure.
; (no. 2), and was refined from single-crystal X-ray diffractometer data: a = 538.3(1), b = 687.5(1), c = 802.3(1) pm, α = 72.4(1)°, β = 85.7(1)°, γ = 68.1(1)°, wR2 = 0.0323, GooF = 1.048, 3157 F2 values, and 106 variables. The main motifs of the crystal structure are pairs of edge-linked [WO6]6− octahedra and fourfold oxygen-coordinated Te4+ atoms. The oxidation state of W6+ and Te4+ was further verified by measuring the characteristic binding energy values for the W 4f and the Te 3d core levels via X-ray photoelectron spectroscopy (XPS). In addition, DFT calculations of the structure, the associated electron localisation functions (ELF) and vibrational spectra have been carried out. The theoretical data clearly demonstrates the impact of the residual electron density located at the Te4+ ions, which can be directly interpreted as the presence of lone electron pairs within the solid structure.
I. Introduction
For various reasons, tellurate compounds have attracted increasing interest in recent years. From a structural point of view, tellurium with its +IV and +VI oxidation states in oxotellurates reveals a diverse variety of coordination environments and Te–O bond lengths.1,2 A comprehensive overview about the structural abundance of oxotellurates(IV) and -(VI) has recently been provided by Christy et al.3 Moreover, the Te4+ atom and the electron lone pair (Ψ) have a decisive contribution to the structural diversity of tellurates. Low symmetrical and one-sided open coordination polyhedra (space requirement of the Ψ) are the consequence and also the origin of the highly interesting physicochemical properties of oxotellurates. The low symmetric and polar structure-directing building blocks often lead to non-centrosymmetric crystal structures with non-linear optical, pyro-, piezo-, or ferroelectrical properties,4–7 which is one of the main reasons, why tellurates are currently attracting a lot of interest.Our approach to expand the broad field of oxotellurates by further representatives is to add and vary another synthetic parameter – namely pressure. By multianvil high-pressure/high-temperature (HP/HT) experiments, maximum conditions of 1700 K and 15 GPa are accessible for us. The high-pressure influences coordination environments, oxidation states and bond lengths as well as physicochemical properties.8–13 In addition to high-pressure phase transformations and the synthesis of polymorphs, new compositions can also be stabilized that would not be accessible at ambient pressure conditions.14–16 Using this technique, we recently reported on the synthesis of HP-Mg3TeO6, an SHG active and UV-transparent material,17 HP-Sc2TeO6,18 and HP-Co3TeO6,19 which shows magnetoelectric behavior. All the aforementioned tellurates are oxotellurates(VI) without exception, because a stabilization of oxotellurates(IV) at HP/HT conditions is challenging. Within this work, we accomplished the synthesis of an oxotellurate(IV), namely WTe2O7, the first ternary tungsten tellurate. To the best of our knowledge, we are not aware of any structurally investigated ternary tungsten tellurate. This is all the more astonishing, since several representatives of the almost identically sized molybdenum are known. From Mo5TeO16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and MoTe2O7,21,22 a monoclinic as well as an orthorhombic modification were synthesized. Via a hydrothermal route, MoTe2O7 (Pna21) and MoTeO5,21 a third composition in the ternary system was accessible. Remarkably, a rare tetravalent cation conduction of Te4+ ions is observed for monoclinic MoTe2O7.22 In this work, we report about the multianvil HP/HT synthesis, structural elucidation and theoretical calculations of the first oxotellurate(IV) WTe2O7.
20 and MoTe2O7,21,22 a monoclinic as well as an orthorhombic modification were synthesized. Via a hydrothermal route, MoTe2O7 (Pna21) and MoTeO5,21 a third composition in the ternary system was accessible. Remarkably, a rare tetravalent cation conduction of Te4+ ions is observed for monoclinic MoTe2O7.22 In this work, we report about the multianvil HP/HT synthesis, structural elucidation and theoretical calculations of the first oxotellurate(IV) WTe2O7.
II. Results and discussion
At ambient pressure conditions, no reaction of WO3 occurred in combination with Te(OH)6 or TeO2. Telluric acid dehydrated to TeO3 and decomposed to TeO2 and O2. Using corundum crucibles, side reactions and the insertion of aluminum was observed. By adding the synthesis parameter pressure, the title compound WTe2O7 was synthesized from WO3 and TeO2 at 1273 K and 10 GPa in closed platinum capsules. EDX analyses confirmed the elemental composition of WTe2O7, in particular concerning the tungsten to tellurium ratio. No additional elemental impurities were visible. The respective data can be found in the Experimental section.II.1 Crystal structure of WTe2O7
The compound WTe2O7 crystallizes triclinic in the space group P (no. 2) with the corresponding lattice parameters a = 539(1), b = 689(1), c = 803(1) pm, α = 72.4(1)°, β = 85.6(1)°, γ = 68.1(1)° and a volume of V = 0.263(1) nm3 (selected crystallographic data are listed in Table 1).
| Empirical formula | WTe2O7 |
| Molar mass, g mol−1 | 551.05 |
| Crystal system | Triclinic |
| Space group |
P (no. 2) |
| Powder data | |
| Powder diffractometer | Stoe Stadi P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| a, pm | 538.650(6) |
| b, pm | 688.515(8) |
| c, pm | 802.649(9) |
| α, ° | 72.41(1) |
| β, ° | 85.62(1) |
| γ, ° | 68.06(1) |
| V, nm3 | 0.263(1) |
| T, K | 293(2) |
| Single-crystal data | |
| Single-crystal diffractometer | Bruker D8 quest, photon 100 |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| a, pm | 538.31(2) |
| b, pm | 687.49(3) |
| c, pm | 802.30(4) |
| α, ° | 72.44(1) |
| β, ° | 85.70(1) |
| γ, ° | 68.11(1) |
| V, nm3 | 0.2624(1) |
| Formula units per cell, Z | 2 |
| Calculated density, g cm−3 | 6.97 |
| Temperature, K | 223(2) |
| Absorption coefficient, mm−1 | 32.88 |
| F(000), e | 468 |
| 2θ range, ° | 2.67–39.42 |
| Range in hkl | ±9, ±12, ±14 |
| Total no. of reflections | 22468 |
| Independent reflections/Rint | 3157/0.0324 |
| Reflections with I > 2σ(I) | 2932 |
| Data/ref. parameters | 3157/106 |
| Goodness-of fit on Fo2 | 1.048 |
| Absorption correction | Multi-scan |
| Final R1/wR2 (I ≥ 2σ(I)) | 0.0155/0.0316 |
| Final R1/wR2 (all data) | 0.0187/0.0323 |
| Largest diff. peak/hole, e Å−3 | 1.93/−1.88 |
A unit cell of WTe2O7 comprises two formula units and 20 atoms. The main motifs of the crystal structure are pairs of edge-linked [WO6]6− octahedra and fourfold oxygen-coordinated tellurium atoms. The tungsten site revealed a minor positional disorder of about 1.9%. Consequently, the W–W distance within the [W2O10]8− pairs varies from majority WAA = 316 pm to WAB = 330 pm to possible WBB = 347 pm, and is thus significantly longer than the W–W distance of 273 pm in metallic tungsten.23 There are two crystallographically distinct tellurium positions Te1 and Te2, of which the Te1 position has also been refined as a split position with Te1A and Te1B occupying 96.8% and 3.2%, respectively. Despite the low splitting, refinement of the split positions significantly improved the residual electron density and the quality of the final results. Possible refinement of disordered oxygen positions, resulting from the displacement of the W and Te atoms was omitted, since this enhanced the quality factors of the structure optimization only marginally. All Te4+ sites show fourfold oxygen coordination in a first approximation with Te–O distances from 190 pm to 230 pm. The next and fifth coordinated oxygen atoms, involved in secondary bonding interactions (SBIs), are found only at the following distances: Te1A–O1 = 248 pm, Te1B–O6 = 264 pm and Te2–O7 = 247 pm. An exact determination of the threshold value for a coordination is difficult and was made on the basis of MAPLE (Madelung part of lattice energy) calculations, which showed only a negligible coordinative contribution to the respective tellurium atom for a fifth oxygen atom. More details on the MAPLE calculations can be found below. Christy et al. also proposed a cut off at 245 pm in their review about the structural architecture of tellurium oxy-compounds.3 Coordination geometries and distances are shown in Fig. 1. Table SI1† contains the atomic coordinates, site occupation, and isotropic displacement parameters. Anisotropic displacement parameters, bond length and angles are given in Tables SI2–SI4 of the ESI.†
 | ||
| Fig. 1 (top) Common and single coordination spheres of Te1A and Te1B atoms. (bottom) Te2 is also coordinated bisphenoidal and tungsten shows a distorted octahedral oxygen environment. All Te atoms exhibit a unilateral coordination with corresponding free space for the positioning of the electron lone pair. Te1B and W1B atoms with occupancies of 3.2% and 1.9%, respectively, are plotted in transparent. Bond lengths are given in pm. | ||
Fig. 1 (top center) displays the common coordination geometry of the disordered atoms Te1A and Te1B. According to the lower site-occupation, the Te1B atom is drawn in transparent mode. Both Te4+ atoms together show a unilateral nutshell-shaped coordination, whereas each individual atom has a distorted trigonal bipyramidal coordination with one of the equatorial positions left unoccupied, creating free space for the electron lone pair of Te4+ (Fig. 1, top left and top right). The Te2 atoms exhibit a comparable bisphenoidal coordination geometry. Within these units, three shorter and one longer Te4+–O contacts are observed. Since Te4+ has a wide range of bond lengths and coordination geometries to oxygen, reference is made here to a comprehensive review.3
The tungsten atoms are surrounded octahedrally by six oxygen atoms. Due to second-order Jahn–Teller (SOJT) distortions, for which d0 W6+ cations are predisposed, the tungsten cations are off-center coordinated. SOJT effects occur from mixing of vacant cation d orbitals with the filled p orbitals of the ligands.24,25 The geometrical changes of the coordination polyhedra are not predictable but depend on the extended structure of the crystal. For WTe2O7, this is evident in three ‘short’ (173–189 pm) and three ‘long’ (195–229 pm) W1A–O bonds and in the case of the octahedral W1B out-of-center coordination it is apparent in two ‘short’ (168 and 183 pm) and four ‘longer’ (201–213 pm) spacings. The distances agree well with the bond lengths reported in literature for triclinic WO3 (176 pm to 218 pm)26 or W17O47 (143 pm to 239 pm).27
A database search in the ICSD28 based on the Wyckoff sequence i10 yielded 152 hits of triclinic compounds. To the best of our knowledge, none of these compounds showed pairs of edge-linked octahedrally coordinated atoms in combination with bisphenoidal atomic coordinations. Consequently, the crystal structure of the title compound WTe2O7 can be considered as a unique structure.
After this description of the local configuration, the topology of the crystal structure is described in more detail. In Fig. 2, the WTe2O7 structure with view along the crystallographic a- and c-axis is depicted. Here, isolated [W2O10]8− units can be identified as well as single chains built up from bisphenoidal coordinated and edge linked Te2 atoms running along the a-axis. These are so-called zweier edge-sharing chains of CN4 Te forming electrostatically neutral chains [Te2O4]0. Fig. 3 shows more clearly the chains described according to Christy et al.3 in the terminology of Liebau29 as (… = ◊ = ◊ = …), where ◊ and = each represent a [TeO4]4− unit with edge-sharing of such units.
 | ||
| Fig. 2 Crystal structure of WTe2O7 with view along the a- (top) and c-axis (bottom). Along a, the pairs of edge-linked [W2O10]8− units can be identified, which are connected by dimers of bisphenoidal coordinated Te1A polyhedra ([Te2O6]4− units, (Liebau:29 (◊ = ◊)). In addition, the polyhedra of the Te1B dimers are also depicted, but with higher transparency. With view along the c-axis, the zweier edge-sharing chains of CN4 Te2 atoms ([Te2O4]0 units, (… = ◊ = ◊ = …)) are shown. | ||
 | ||
| Fig. 3 Crystal structure of WTe2O7 with view along the b-axis. [W2O10]8− dimers as well as zweier edge-sharing chains of Te2 atoms running along a are depicted. By increased transparency, the disorder of the Te1 atoms is highlighted and leads to a z-shaped linkage of the chains. On the right side, a structural detail of this linkage is shown. | ||
A similar structural feature is known from the compound Ag(TeO2)(NO3) ≡ Ag2(Te2O4)(NO3)230 with chains parallel to the c-axis. Due to the higher symmetry of the compound Ag(TeO2)(NO3) (S.G. Pbcn), the Te–O distances within the chains are more uniformly distributed and are determined as 192 pm and 207 pm, respectively. The Te1 atoms in WTe2O7 form edge-sharing dimers of bisphenoidal [TeO4]4−. The resulting [Te2O6]4− units (Liebau:29 (◊ = ◊)) are isolated and share corners with the [W2O10]8− octahedral dimers (see Fig. 2 (top), 3) and in the case of [(Te1B)2O6]4− dimers, additional linkages to the O1 atoms of the chains are formed (see Fig. 3). Comparable [Te2O6]4− units can also be found in the mixed-metal oxide halide material Pb3Te2O6X2 (X = Cl, Br).31 Te–O distances in this compound are reported from 194 pm to 205 pm. In the following a closer look on the disorder in the WTe2O7 crystal structure is given. It is noteworthy that the occupancies of W1B (1.9%) and Te1B (3.2%) are comparable, indicating a possible mutual dependence and the formation of a local alternative structural arrangement. Fig. 4 (top) illustrates the changing situation in the coordination environments with different occupation of the A- and B-sites. The top right part shows the most common situation and the top left part of Fig. 4 the exclusive occupation of the B-sites. In between, the superposition of both coordination possibilities is given, because not always both A or B atoms of the [Te2O6]4− units have to be present, but also an A and B occupation is possible.
 | ||
| Fig. 4 (top) Illustration of the different coordination environments when switching from only occupied B-sites (top left) to only occupied A-sites (top right). The center of the top figure shows the overlay of the two coordination options. The secondary bonding interactions (Te4+⋯O2−) formed in WTe2O7 are given as dashed bonds. All distances are given in pm. (bottom) Layers within the crystal structure of WTe2O7 connected via [Te2O4]0 chains running along a. The disorder takes place within the layers or in the type of stacking disorder. | ||
By switching from A to B sites, the largest structural changes occur at the following locations. The shortest W1A–O4 distance (173 pm) increases to 201 pm for W1B–O4, since O4 is now additionally bonded to Te1B at a distance of 217 pm. The shortest W1B–O distance is now formed to the O2 site (168 pm), which subsequently coordinates only with Te1B at a distance of 277 pm. An additional stabilizing effect by crosslinking of the structure is achieved by the formation of a new connection between Te1B and O1 atoms at a distance of 203 pm. This leads to a z-shaped linkage of the chains running along a (see Fig. 3). Overall, the oxygen surrounding of the tungsten atoms becomes more uniform for the W1B coordination. The long W1A–O5 distance of 229 pm decreases to 203 pm (W1B–O5). Apparently, however, the off-center coordination of tungsten atoms is structurally preferred due to the SOJT effects. If the oxygen surrounding of the Te1 atoms is compared, a similar situation is found. The bisphenoidal coordination is retained and the Te1–O distances become more uniform, too. The average bond length increases from 201 pm (Te1A–O) to 207 pm (Te1B–O). However, due to the unilateral coordination and the open crystal structure, both Te1A- and Te1B-atoms have enough space for the orientation of their lone-pair electrons. The question of the cause for the disorder in the WTe2O7 crystal structure is difficult to answer and will be a combination of stabilizing SOJT effects as well as a similar stabilization of the lone-pair electrons in their different orientations. It is obvious that the structural changes all take place within one layer of the crystal structure. As the bottom part of Fig. 4 shows, the layers are connected only by the [Te2O4]0 chains. These layers can stack differently, which can lead to stacking faults of the heavy atoms’ tellurium and tungsten at its different A- and B-sites with different probabilities. The disorder within one layer or the formation of alternative local structural arrangements (clustering) appear to be purely statistical. Also, no higher order was observed in the sequence of layers, which should have become visible by superstructure reflections or diffuse scattering. In addition, so-called secondary bonding interactions (SBIs) are formed, which also have structure-stabilizing effects. These Te4+⋯O2− interactions are common in rare-earth metal(III) oxotellurates(IV) such as RE2Te3O9 (RE = La, Ce, Pr, Nd) in a space region of d (Te⋯O) = 250–297 pm.32 Oxotellurate(IV) groups, and in particular the Ψ-bisphenoidal [TeO4]4− groups, are known for their pronounced Te4+⋯O2− interactions to other [TeO4]4− groups as well as [W2O10]8− octahedral dimers in WTe2O7. The theoretical concept of SBIs was established by Alcock et al.,33 and the bonding interactions originate from the hyperconjugation of a σ*-Te–O molecular orbital with a non-bonded p-orbital of an oxygen atom.34 Another common name for this specific interaction is called “chalcogen bonding”.35Fig. 4 (top) displays these additional Te⋯O contacts in WTe2O7. On the right part of the top graphic, the Te1A and Te2 SBIs are shown. The bisphenoidal [(Te1A)O4]4− group establishes a weak interaction with the O1 atom of the [Te(2)O4]4− group in a distance of 248 pm. Vice versa, Te2 also has a secondary bonding interaction via the O7 atom of the [Te2O6]4− dimer with a distance of 247 pm. Two additional weaker contacts are formed to O4 (288 pm) and O3 (265 pm) of the [W2O10]8− octahedral dimers. In the left part of Fig. 4 (top), the [(Te1B)O4]4− groups are displayed. Even in this case of site occupation, the formation of SBIs can occur.
Concerning the charge distribution in WTe2O7, the reliability of the crystal structure model was checked by bond-length/bond-strength (BLBS),36–38 CHARDI (Charge Distribution),39 and MAPLE40–42 calculations. The obtained values are in good agreement with the formal charges W6+, Te4+, and O2− derived by X-ray crystal structural analysis. The MAPLE value of WTe2O7 was calculated and compared to the sum of the MAPLE values of the binary high-pressure polymorphs WO343 and 2× TeO244 (for details, see Table SI5 of the ESI†). The values are in good agreement with a deviation of 0.55%. For the MAPLE and CHARDI calculations, the disorder of the tungsten and tellurium atoms was neglected, because the method does not support split sites. Consequently, the calculated valences (∑Q) of these atoms are less accurate. The BLBS model was optimized to consider split sites by multiplying the valence contribution of each bond between atoms i and j (vij) by the site occupation factor (SOF) of the split site according to 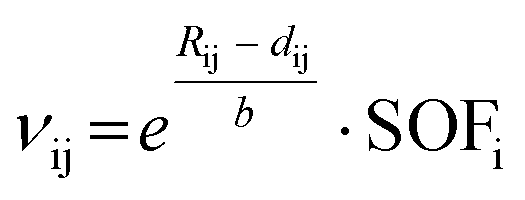 . Rij is the bond valence parameter, b is a “universal” constant and dij is the distance between the atoms i and j.36 These results are shown in the Table 2.
. Rij is the bond valence parameter, b is a “universal” constant and dij is the distance between the atoms i and j.36 These results are shown in the Table 2.
), calculated with the bond-length/bond-strength concept (∑V) and the CHARDI concept (∑Q)
| Atom | W1A | W1B | Te1A | Te1B | Te2 | O1 | O2 | O3 | O4 | O5 | O6 | O7 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ∑Q | +6.20 | +3.86 | +3.93 | −2.29 | −2.20 | −1.65 | −1.92 | −1.77 | −2.25 | −1.91 | ||
| ∑V | +6.00 | +5.90 | +3.93 | +3.28 | +3.67 | −2.13 | −2.02 | −1.74 | −1.68 | −1.93 | −2.03 | −1.93 |
The low BLBS values for the atoms O3 and O4, which show only one coordination to tungsten, are noticeable. Two O3 atoms form the edge linkage between the tungsten octahedra dimers. O4 is a terminal oxygen atom, which just coordinates to W1A and W1B, respectively. If the split position Te1B is occupied, it is also coordinated to O4. The influence of the tellurium lone pair electrons and secondary bonding interactions on the O3 and O4 atoms are not considered in the BLBS calculation and could be an explanation for the low valences.
The crystal structure model of WTe2O7 was verified on a powdered sample by Rietveld analysis depicted in Fig. 5. Starting atomic parameters were derived from the single crystal solution. The fitting was performed in good quality and exhibited an almost X-ray pure specimen. Only a marginal unknown side phase, marked with an asterisk, is visible in the diffraction pattern and the difference plot. Refined cell parameters of the powder data are given in Table 1.
 | ||
| Fig. 5 X-ray powder diffraction pattern (Mo-Kα1 radiation) of WTe2O7 and Rietveld refinement (measured (black) and calculated (red) patterns). The calculated reflection positions are indicated by the vertical green bars below the difference pattern (Rexp = 1.94%, Rwp = 3.98%, Rp = 3.03%, GooF = 2.05). A small unknown side phase is indicated by an asterisk. | ||
II.2. XPS studies
X-ray photoelectron spectroscopy (XPS) was applied for the determination of the binding energy values of W 4f and Te 3d core levels. To make a reliable statement about the position of the binding energies, the averaged values for the O 1s level in WO3 and TeO2 were used to correct for charging effects during the measurement procedure. Detailed information on this can be found in the Experimental Part. In Fig. 6, the characteristic energy core levels for Te 3d and W 4f are depicted. Table 3 lists the results of the peak fits for the respective species. | ||
| Fig. 6 XP spectra of the Te 3d (top) and W 4f (bottom) core levels with experimental data (dots) and fit (purple and green, respectively). | ||
| Te 3d3/2 | Te 3d5/2 | W 4f5/2 | W 4f7/2 | |
|---|---|---|---|---|
| Position/eV | 586.41 | 576.02 | 37.62 | 35.44 |
| FWHM | 1.81 | 1.81 | 1.49 | 1.49 |
| % Lorentz:Gaussian | 65 | 65 | 48 | 48 |
For tellurium, the positions of the 3d core levels are 586.41 eV and 576.02 eV, respectively. The 4f energy core levels for tungsten are located at 37.62 eV and 35.44 eV. In previous studies, the position of the Te 3d5/2 and the W 4f7/2 core levels were determined for tellurium and tungsten oxides. Here, Te 3d5/2 of Te4+ is positioned in the range of 575.6 eV to 576.9 eV.45–51 For W 4f7/2 with an oxidation state of W6+, these values range from 35.2 eV to 36 eV.47,52–57 By comparing these values to the experimental data obtained under consideration of the overall structural motif of WTe2O7, oxidation states of Te4+ and W6+ are reasonable. The oxidation states determined agree with those obtained from crystal structure analysis.
II.3 Theoretical calculations and analysis
The optimized lattice parameters a, b and c as well as the lattice angles α, β and γ show good agreement with the experimental measurements and deviate only by ≤2.5% (PBESOL) and 1% (HSESOL), respectively. The associated values are listed and compared to the experimental ones in Table 4. As expected, the range-separated hybrid functional HSESOL leads to slightly better results, which is also reflected in the calculated powder XRD patterns depicted in Fig. SI1.†| a | b | c | α | β | γ | V/nm3 | |
|---|---|---|---|---|---|---|---|
| HSESOL | 535.38 | 694.82 | 795.39 | 72.58° | 85.90° | 67.81° | 0.2611 |
| PBESOL | 538.61 | 705.83 | 790.14 | 72.18° | 86.10° | 67.38° | 0.2635 |
| Single crystal data | 538.31(2) | 687.49(3) | 802.30(4) | 72.44(1)° | 85.70(1)° | 68.11(1)° | 0.2624(1) |
| Powder data | 538.650(6) | 688.515(8) | 802.649(9) | 72.41(1)° | 85.62(1)° | 68.06(1)° | 0.2629(1) |
Band structure and density of states calculations lead to viable results as well, although the band gap values determined by the functionals seems unusual as the employment of the hybrid functional HSESOL leads to a larger band gap (3.49 eV) as observed in case of PBESOL (2.28 eV), which in general tends to overestimate band gap energies. It is likely that the unusual character of the tungsten tellurate(IV) featuring residual electron density (Ψ) at the tellurium atoms is a particular challenge in the calculations. Fig. 7 shows the HSESOL band structure and density of states. The corresponding PBESOL band structure is given in the ESI (see Fig. SI2†). It is evident that the valence band is almost exclusively formed from Te- and O-orbitals, whereas the bottom edge of the conduction bands is dominated by W-, Te-, and O-orbitals, which determine the band gap. The Fermi-level is placed in the center of the band gap, determined as 3.49 eV. From diffuse UV/Vis reflectance data, the optical bandgap was determined experimentally via the Kubelka–Munk function (F(R)). In Fig. SI3,† the UV/Vis spectrum, the plot (F(R)hν)1/2/(hν) for the indirect band gap (3.0 eV) and the plot (F(R)hν)2/(hν) for the direct band gap (3.3 eV) are depicted. A wide transparency range up to an absorption edge of 260 nm is visible from the spectrum.
 | ||
| Fig. 7 Band structure and density of states (right part of the figure) of WTe2O7 determined at the HSESOL/double-zeta level. | ||
To visualize the electron lone pair contributions to the overall electron density, electron localization functions (ELFs) have been calculated, which are depicted in Fig. 8. Three independent values (0.55, 0.65, 0.75) in arbitrary units were chosen for the isosurfaces, resembling the ELF around the atoms in the unit cell. While the oxygen and tungsten atoms feature electron densities resembling a spherical shape centered at the respective core, the residual electron density near the tellurium atom is clearly represented via the hemisphere-like shaped functions pointed at by the arrows in Fig. 8. Due to the associated crystal symmetry, two pairs of tellurium atoms can be distinguished, showing a slightly different residual density (Ψ). Nevertheless, it can be seen that these density contributions do not form bonds within the crystal, but can be interpreted as electron lone pairs being localized within the unit cell. For better comparison, a corresponding structural section is shown in the lower part of Fig. 8. The lone pair electrons point exactly into the non-coordinated region of the tellurium(IV) atoms. Projections of the ELF density as well as an animation of the electron localization function for WTe2O7 obtained at HSESOL level is provided at the ESI.†
 | ||
| Fig. 8 (top) Calculated electron localization functions (ELF). (bottom) Corresponding structural section of WTe2O7. Residual electron density can be attributed to the lone pair electrons of Te4+ atoms. | ||
In addition, the results of the infrared and Raman vibration calculations at the optimized geometries are shown in Fig. 9 and SI6.† While the overall shapes of both calculated infrared spectra are similar, the values of the GGA functional PBESOL seem to feature a shift compared to results generated by the more demanding hybrid functional HSESOL (see Fig. SI6,† top). A comparison between experimental and calculated IR spectrum on HSESOL level is depicted in Fig. 9. Again, the overall shape of the spectrum is comparable, but the calculated absorptions are shifted around 90 cm−1 to lower wavenumbers. The observed shifts in the calculated vibrational wavenumbers are not uncommon and also occurred in spectral data of previously investigated systems.58–60 Both Te atoms in WTe2O7 are bisphenoidal coordinated with two longer axial (ax = axial) and two shorter equatorial (eq = equatorial) bonded oxygen atoms forming [TeO4]4− units. A similar situation exists for α-TeO2, where the stronger Te–Oeq vibrations have been assigned above 600 cm−1, while the weaker Te–Oax modes appear between 400 and 500 cm−1.61 The free TeO2 molecule with two Te![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds (TeO distance: 184 pm, ∡OTeO: 110°), which is close to the Te–O2,eq geometry in the [TeO4]4− bisphenoidal units exhibit three normal vibrations at 849, 830 and 292 cm−1.62 Therefore, the modes of the two different [TeO4]4− units in WTe2O7 can be assigned according to Te–Oeq: 750–880 cm−1 and Te–Oax: 520–750 cm−1 (see Fig. 9) using the calculated IR spectrum (see Table SI6†). If one takes a closer look, such an assignment is not very meaningful in the case of the special structural situation in WTe2O7. The bisphenoidal [TeO4]4− units do not occur in isolation, but are linked via edges to form either chains or pairs in the crystal structure (see Fig. 4). This results in a highly asymmetric bridge Te–axOeq–Te and equatorial and axial vibrations are mutually dependent. The infrared spectra of known molybdenum tellurates(IV) are similar.21 MoTe4O11·KCl shows [Te4O9]2− chains consisting of alternating [TeO3]2− trigonal pyramids and [TeO4]4− bisphenoidal units also causing strong bands in the range of 600–800 cm−1, which were attributed to the antisymmetric stretching vibrations of the Te–O bonds. The bands occurring above 900 cm−1 were assigned as antisymmetric stretches of the Mo–O bonds. An analogous assignment of the absorption bands for W–O can also be made here. An exact attribution of the absorption bands to the corresponding symmetric and antisymmetric stretching or bending modes is difficult in the solid state. Therefore, a list of all modes with their equivalent vibrational components derived from the calculated IR spectrum is given in Table SI6.† In addition, the calculated Raman spectrum obtained at PBESOL level is depicted in Fig. SI6† (bottom).
O double bonds (TeO distance: 184 pm, ∡OTeO: 110°), which is close to the Te–O2,eq geometry in the [TeO4]4− bisphenoidal units exhibit three normal vibrations at 849, 830 and 292 cm−1.62 Therefore, the modes of the two different [TeO4]4− units in WTe2O7 can be assigned according to Te–Oeq: 750–880 cm−1 and Te–Oax: 520–750 cm−1 (see Fig. 9) using the calculated IR spectrum (see Table SI6†). If one takes a closer look, such an assignment is not very meaningful in the case of the special structural situation in WTe2O7. The bisphenoidal [TeO4]4− units do not occur in isolation, but are linked via edges to form either chains or pairs in the crystal structure (see Fig. 4). This results in a highly asymmetric bridge Te–axOeq–Te and equatorial and axial vibrations are mutually dependent. The infrared spectra of known molybdenum tellurates(IV) are similar.21 MoTe4O11·KCl shows [Te4O9]2− chains consisting of alternating [TeO3]2− trigonal pyramids and [TeO4]4− bisphenoidal units also causing strong bands in the range of 600–800 cm−1, which were attributed to the antisymmetric stretching vibrations of the Te–O bonds. The bands occurring above 900 cm−1 were assigned as antisymmetric stretches of the Mo–O bonds. An analogous assignment of the absorption bands for W–O can also be made here. An exact attribution of the absorption bands to the corresponding symmetric and antisymmetric stretching or bending modes is difficult in the solid state. Therefore, a list of all modes with their equivalent vibrational components derived from the calculated IR spectrum is given in Table SI6.† In addition, the calculated Raman spectrum obtained at PBESOL level is depicted in Fig. SI6† (bottom).
 | ||
| Fig. 9 Comparison of calculated and experimental IR spectrum of WTe2O7. The theoretical data are based on the HSESOL functional. A shift of the calculated data towards lower energies is visible. | ||
III. Conclusions
WTe2O7 is the first compound in the ternary system W–Te–O and was synthesized via a multianvil high-pressure/high-temperature route. The crystal structure contains edge linked [W2O10]8− octahedra, dimers of bisphenoidal coordinated [Te2O6]4− units (◊ = ◊), and zweier edge-sharing chains of [Te2O4]0 units (… = ◊ = ◊ = …). The lone pair character of the two Te4+ sites was analyzed by ELF and high values appeared in nonbonding regions. A confirmation of the charge distribution according to W6+, Te4+, and O2− was done by BLBS and CHARDI as well as experimentally by XPS analysis. Theoretical DFT calculations support the experimental data and band structure calculations indicate that WTe2O7 is a wide-band gap semiconducting material.IV. Experimental section
IV. 1. Synthesis
Bulk material with embedded small single-crystals of WTe2O7 was synthesized via a high-pressure/high-temperature synthetic approach. The starting materials WO3 (Alfa Aesar, Haverhill, USA, 99+%) and TeO2 (TCI Chemicals, Tokio, Japan, 98+%) were weight in with a stochiometric ratio of 1:2 and homogenized in an agate mortar in air. The mixture was filled into a platinum capsule (99.95%, Ögussa, Vienna, Austria), inserted in a boron-nitride crucible (Henze Boron Nitride Products AG, Lauben, Germany) and placed in an 18/11 assembly (further details of the multianvil experiment are described elsewhere19,63–65). The sample was compressed (1000 t multianvil press, Walker-type module (Max Voggenreiter GmbH, Mainleus, Germany)) to 10 GPa within 275 min, and held at this pressure until the heating phase was completed. For sample heating, the temperature was increased to 1273 K within 10 min, remained for another 15 min at this temperature, and cooled down to 673 K within 30 min. Subsequently, the sample was quenched to room temperature and decompressed within 825 min. Afterwards, it was mechanically separated from the surrounding assembly material. It appeared colourless, polycrystalline and was stable in air.
IV. 2. X-ray structure determination
X-ray powder diffraction analysis of WTe2O7 was carried out at ambient temperature using a Stoe Stadi P powder diffractometer in transmission geometry with the WinXPOW 3.07 software package.66 Ge(111) monochromated Mo-Kα1 (λ = 70.93 pm) radiation was applied to the sample, mounted between two thin acetate foils with vacuum grease, and diffraction intensities were collected from a Mythen 2 DCS4 detector. The measurement was performed in the 2θ range of 2.0–77.5° with a step size of 0.015° and an exposure time of 30 s per step. Fig. 5 shows the Rietveld refinement plot performed with Diffracplus-Topas 4.2 (Bruker AXS, Karlsruhe, Germany). As starting model, the single-crystal structure reported here was used and the peak shapes were modeled using modified Thompson-Cox-Hastings pseudo-Voigt profiles.67,68 Instrumental contributions on reflection profiles were corrected from the refinement of a LaB6 standard.69 The background was fitted with Chebychev polynomials up to the 12th order.Colorless single-crystals were isolated under a polarization microscope using perfluoropolyalkylether (viscosity 1800) and mounted on the tip of MicroMounts™ (MiTeGen, LLC, Ithaca, NY, USA) with a diameter of 30 μm. The intensity data were collected at 223(2) K on a Bruker D8 Quest single-crystal diffractometer (BRUKER, Billerica, USA) with Mo-Kα radiation (λ = 71.073 pm), Incoatec microfocus X-ray tube (Incoatec, Geesthacht, Germany), Photon 100 detector system and Apex3 program package.70 The multi-scan absorption correction of the intensity data was performed with Sadabs 2016/2. Because of missing additional symmetry except inversion symmetry, P (no. 2) was considered and found to be correct for the structure solution and refinement process (SHELXTL-XT-2018/2). Full-matrix least-squares refinements against F2 were carried out with Shelxl-2017/1.71–73 All sites were refined with anisotropic displacement parameters and the refinement process finally led to values of 0.0187 and 0.0323 for R1 and wR2 (all data), respectively. A verification of the space group was done with the ADDSYM74 routine of the PLATON program package.75 In the case of refinement in space group P1, PLATON indicated additional inversion symmetry. Moreover, no weak superstructure reflections or possible diffuse scattering was observed on the diffraction patterns.
IV. 3. Energy-dispersive X-ray spectroscopy (EDX)
EDX investigations on several crystals of WTe2O7 were performed in a semiquantitative approach using a scanning electron microscope (JSM-6010LV, Jeol, Freising, Germany) with an energy-dispersive Quantax X-ray detector-system (Bruker AXS GmbH, Billerica, MA, USA) for element identification. The crystals were placed on a carbon platelet on an aluminium sample carrier and sputtered with carbon. Three crystals and several measurement points on each were analyzed for 60 s at an excitation energy of 15 kV in high vacuum regarding to their W to Te ratio. All values were arithmetically averaged. Due to the preparation method and the low sensitivity of light atoms like oxygen next to tellurium and tungsten, only the ratio of the heavy atom was considered, and the absolute values are neglected. The measured ratio of W/Te = 14 ± 2 at% W/31 ± 2 at% Te agrees well with the theoretical ratio of W/Te = 1:2 in WTe2O7.
IV.4. X-ray photoelectron spectroscopy (XPS)
For XPS measurements, bulky WTe2O7 was placed on an adhesive copper foil. Measurements were performed in a multi-chamber UHV system at a pressure of 5 × 10−10 mbar using a Phoibos 100 hemispherical analyzer (Specs). A Mg-Kα anode was used as an excitation source (h × ν = 1252.6 eV, probing depth ∼10 nm).Due to charging effects during the measurement caused by the low conductivity of the powder sample, the binding energy scale as measured by XPS was shifted to higher values by a few eV. To account for this, the binding energies were corrected in a way that the O 1s energy level is positioned at 530.7 eV. This value was chosen by a careful evaluation of the O 1s binding energy values within WO3 and TeO2, which are in the range of 530.30 eV and 530.8 eV for the first, and 530.1 eV to 530.7 eV for the latter (data taken from NIST X-ray Photoelectron Spectroscopy Database,76 and from the following publications.45–57
IV.5. Infrared spectroscopy
Infrared spectra were recorded from powdered samples of WTe2O7 in the spectral range of 400–4000 cm−1 using a Bruker Alpha Platinum FTIR-ATR spectrometer (Bruker, Billerica, USA) equipped with a 2 × 2 mm diamond ATR-crystal. A DTGS detector collected the intensity during 24 scans and atmospheric influences were corrected via a reference measurement using the Opus 7.2 software.77IV.6. Quantum chemical calculations, ELF
All DFT calculations of the tungsten tellurate WTe2O7 have been carried out using Crystal1778 employing the dedicated solid-state functionals PBESOL79 and HSESOL80 in conjunction with the W_cora_199681 the Te_m-pVDZ-PP_Heyd_200582 basis and the O_8-411d11f_mahmoud_201383 basis set for tungsten, tellurium and oxygen, respectively. The convergence criteria for energy and forces were set to 10−8 Hartree and 4.5 × 10−4 Hartree bohr−1. In order to ensure the viability of the treatment, Brillouin-zone sampling employing a shrinking factor of 8 was employed. To compare the structures determined by experimental method and theoretical calculation, powder-diffraction patterns of the DFT optimized structures were generated using RIETAN-FP84 included in the VESTA program.85 To assess the ionic character of the compound, partial charges have been calculated employing a Hirshfeld population analysis86 as implemented in Crystal17.
In addition, band structure and density of states (DOS) calculations have been carried out at the optimized geometries using Crystal17. As the tungsten tellurate features residual electron density located near the Te4+ atom (“lone pair”), electron localization functions (ELFs) have been calculated via the Multiwfn program87 and were visualized using VMD.88
Infrared and Raman spectra at the optimized geometries have been determined via the harmonic approximation.89 As Crystal17 does not support calculations employing coupled-perturbed Hartree–Fock/Kohn–Sham equations in conjunction with range-separated hybrid functionals such as HSESOL, the Raman intensities were only calculated using the PBSESOL functional. A weighted kernel density estimation using a Gaussian kernel was applied to the line spectra for better comparison with the experimental results.90
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Martina Tribus for collecting the EDX data and Markus Rödl for measuring the UV/Vis spectrum. Further, we acknowledge H. Huppertz for continuous support and usage of all the facilities of the Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck.References
- J. Zemann, Monatsh. Chem., 1971, 102, 1209–1216 CrossRef CAS.
- A. V. Marukhnov, D. V. Pushkin and V. N. Serezhkin, Russ. J. Inorg. Chem., 2007, 52, 203–208 CrossRef.
- A. G. Christy, S. J. Mills and A. R. Kampf, Mineral. Mag., 2016, 80, 415–545 CrossRef.
- K. M. Ok, E. O. Chi and P. S. Halasyamani, Chem. Soc. Rev., 2006, 35, 710–717 RSC.
- E. O. Chi, K. M. Ok, Y. Porter and P. S. Halasyamani, Chem. Mater., 2006, 18, 2070–2074 CrossRef CAS.
- B. Vidyavathy and K. Vidyasagar, Inorg. Chem., 1999, 38, 1394–1400 CrossRef.
- P. S. Halasyamani and W. Zhang, Inorg. Chem., 2017, 56, 12077–12085 CrossRef CAS PubMed.
- C. T. Prewitt and R. T. Downs, Rev. Mineral. Geochem., 1998, 37, 283–317 CAS.
- H. Huppertz, Chem. Commun., 2011, 47, 131–140 RSC.
- P. F. McMillan, Chem. Commun., 2003, 919–923, 10.1039/B300963G.
- P. F. McMillan, Chem. Soc. Rev., 2006, 35, 855–857 RSC.
- G. Demazeau, H. Huppertz, J. A. Alonso, R. Pöttgen, E. Moran and J. P. Attfield, Z. Naturforsch., B: Chem. Sci., 2006, 61, 1457–1470 CrossRef CAS.
- U. Schwarz, R. Giedigkeit, R. Niewa, M. Schmidt, W. Schnelle, R. Cardoso, M. Hanfland, Z. Hu, K. Klementiev and Y. Grin, Z. Anorg. Allg. Chem., 2001, 627, 2249–2256 CrossRef CAS.
- A. Haberer, G. Heymann and H. Huppertz, J. Solid State Chem., 2007, 180, 1595–1600 CrossRef CAS.
- H. Huppertz and B. von der Eltz, J. Am. Chem. Soc., 2002, 124, 9376–9377 CrossRef CAS PubMed.
- H. Huppertz, S. Altmannshofer and G. Heymann, J. Solid State Chem., 2003, 170, 320–329 CrossRef CAS.
- E. Selb, L. Declara, L. Bayarjargal, M. Podewitz, M. Tribus and G. Heymann, Eur. J. Inorg. Chem., 2019, 2019, 4668–4676 CrossRef CAS.
- R. Ziegler, M. Tribus, C. Hejny and G. Heymann, Crystals, 2021, 11, 1554 CrossRef CAS.
- E. Selb, T. Buttlar, O. Janka, M. Tribus, S. G. Ebbinghaus and G. Heymann, J. Mater. Chem. C, 2021, 9, 5486–5496 RSC.
- S. Vallar and M. Goreaud, J. Solid State Chem., 1997, 129, 303–307 CrossRef CAS.
- J. Ling, H. Zhang, K. Yuan, D. Burgess, J. Hu and M. Hu, J. Solid State Chem., 2020, 287, 121317 CrossRef CAS.
- N. Nunotani, S. Tamura and N. Imanaka, Eur. J. Inorg. Chem., 2013, 2013, 4300–4304 CrossRef CAS.
- H. Hartmann, F. Ebert and O. Bretschneider, Z. Anorg. Allg. Chem., 1931, 198, 116–140 CrossRef CAS.
- H.-S. Ra, K. M. Ok and P. S. Halasyamani, J. Am. Chem. Soc., 2003, 125, 7764–7765 CrossRef CAS PubMed.
- M. Kunz and I. D. Brown, J. Solid State Chem., 1995, 115, 395–406 CrossRef CAS.
- P. M. Woodward, A. W. Sleight and T. Vogt, J. Phys. Chem. Solids, 1995, 56, 1305–1315 CrossRef CAS.
- M. M. Dobson and R. J. D. Tilley, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 474–480 CrossRef.
- CSD Crystallographic Database, FIZ Karlsruhe GmbH, ICSD Web version 4.7.1, data release 2021.2 Search PubMed.
- F. Liebau, Structural Chemistry of Silicates, Springer Verlag, Berlin, Heidelberg, 1st edn, 1985 Search PubMed.
- C. Olsson, L.-G. Johansson and S. Kazikowski, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 427–429 CrossRef.
- Y. Porter and P. S. Halasyamani, Inorg. Chem., 2003, 42, 205–209 CrossRef CAS PubMed.
- S.-C. Chou, P. Höss, P. L. Russ, S. Strobel and T. Schleid, Z. Anorg. Allg. Chem., 2021, 647, 134–150 CrossRef.
- N. W. Alcock, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1972, vol. 15, pp. 1–58 Search PubMed.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- S. J. Mills and A. G. Christy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 145–149 CrossRef CAS PubMed.
- R. Hoppe, S. Voigt, H. Glaum, J. Kissel, H. P. Müller and K. Bernet, J. Less-Common Met., 1989, 156, 105–122 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1966, 5, 95–106 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1970, 9, 25–34 CrossRef CAS.
- R. Hübenthal, MAPLE, Universität Gießen Search PubMed.
- Y. Xu, S. Carlson and R. Norrestam, J. Solid State Chem., 1997, 132, 123–130 CrossRef CAS.
- T. G. Worlton and R. A. Beyerlein, Phys. Rev. B: Condens. Matter, 1975, 12, 1899–1907 CrossRef CAS.
- B. V. R. Chowdari and P. Pramoda Kumari, J. Non-Cryst. Solids, 1996, 197, 31–40 CrossRef CAS.
- T. S. Sun, S. P. Buchner and N. E. Byer, J. Vac. Sci. Technol., 1980, 17, 1067–1073 CrossRef CAS.
- Y. A. Teterin, V. I. Nefedov, M. F. Churbanov, A. Y. Teterin, K. I. Maslakov and E. V. Zorin, Inorg. Mater., 2007, 43, 888–896 CrossRef CAS.
- H. Kong, J.-B. Yeo and H.-Y. Lee, J. Korean Phys. Soc., 2015, 66, 1744–1749 CrossRef CAS.
- A. B. Christie, I. Sutherland and J. M. Walls, Surf. Sci., 1983, 135, 225–242 CrossRef CAS.
- F. Garbassi, J. C. J. Bart and G. Petrini, J. Electron Spectrosc. Relat. Phenom., 1981, 22, 95–107 CrossRef CAS.
- M. F. Al-Kuhaili, S. M. A. Durrani, E. E. Khawaja and J. Shirokoff, J. Phys. D: Appl. Phys., 2002, 35, 910–915 CrossRef CAS.
- O. Y. Khyzhun, Y. M. Solonin and V. D. Dobrovolsky, J. Alloys Compd., 2001, 320, 1–6 CrossRef CAS.
- R. J. Colton, A. M. Guzman and J. W. Rabalais, J. Appl. Phys., 1978, 49, 409–416 CrossRef CAS.
- A. Marin, C. P. Lungu and C. Porosnicu, J. Vac. Sci. Technol., A, 2017, 35, 021403 CrossRef.
- P. J. C. Chappell, M. H. Kibel and B. G. Baker, J. Catal., 1988, 110, 139–149 CrossRef CAS.
- V. I. Nefedov, Y. V. Salyn, G. Leonhardt and R. Scheibe, J. Electron Spectrosc. Relat. Phenom., 1977, 10, 121–124 CrossRef CAS.
- D. D. Sarma and C. N. R. Rao, J. Electron Spectrosc. Relat. Phenom., 1980, 20, 25–45 CrossRef CAS.
- E. Hinteregger, G. Heymann, T. S. Hofer and H. Huppertz, Z. Naturforsch., B: Chem. Sci., 2012, 67b, 605–613 CrossRef.
- E. Hinteregger, T. S. Hofer, G. Heymann, L. Perfler, F. Kraus and H. Huppertz, Chem. – Eur. J., 2013, 19, 15985–15992 CrossRef CAS PubMed.
- E. Hinteregger, K. Kocsis, T. S. Hofer, G. Heymann, L. Perfler and H. Huppertz, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 951–959 CrossRef CAS.
- A. P. Mirgorodsky, T. Merle-Méjean, J. C. Champarnaud, P. Thomas and B. Frit, J. Phys. Chem. Solids, 2000, 61, 501–509 CrossRef CAS.
- M. Spoliti, V. Grosso and S. N. Cesaro, J. Mol. Struct., 1974, 21, 7–15 CrossRef CAS.
- H. Huppertz, Z. Kristallogr., 2004, 219, 330–338 CAS.
- D. Walker, M. A. Carpenter and C. M. Hitch, Am. Mineral., 1990, 75, 1020–1028 Search PubMed.
- D. Walker, Am. Mineral., 1991, 76, 1092–1100 Search PubMed.
- WinXPOW Powder Diffraction Software, STOE & CIE GmbH Search PubMed.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- R. A. Young and P. Desai, Arch. Nauki Mater., 1989, 10, 71–90 Search PubMed.
- M. C. Morris, H. F. McMurdie, E. H. Evans, B. Paretzkin, H. S. Parker and N. P. Pyrros, NBS Monogr. (U. S.), 1984, 25, 62 Search PubMed.
- APEX3 (v. 2017.3-0), CELL_NOW (v. 2008/4), SAINT (v. 8.38A), TWINABS (v. 2012/1), and SADABS (v. 2016/2), Bruker AXS GmbH, Karlsruhe (Germany) Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, ShelXL – Crystal Structure Refinement – Multi-CPU Version, University of Göttingen Search PubMed.
- Y. Le Page, J. Appl. Crystallogr., 1988, 21, 983–984 CrossRef.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2000, NIST X-ray Photoelectron Spectroscopy Database, (retrieved: 18.08.2022) Search PubMed.
- OPUS (v 7.2), Bruker AXS GmbH Search PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- L. Schimka, J. Harl and G. Kresse, J. Chem. Phys., 2011, 134, 024116 CrossRef PubMed.
- F. Corà, A. Patel, N. M. Harrison, R. Dovesi and C. R. A. Catlow, J. Am. Chem. Soc., 1996, 118, 12174–12182 CrossRef.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- A. Mahmoud, A. Erba, K. E. El-Kelany, M. Rérat and R. Orlando, Phys. Rev. B: Condens. Matter, 2014, 89, 045103 CrossRef.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- Y. Ozaki, K. B. Beć, Y. Morisawa, S. Yamamoto, I. Tanabe, C. W. Huck and T. S. Hofer, Chem. Soc. Rev., 2021, 50, 10917–10954 RSC.
- M. A. Wolters and W. J. Braun, Statistics, 2018, 7, e202 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CSD 2212710. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03419k |
| This journal is © The Royal Society of Chemistry 2023 |