
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic and optical studies of a new family of multidimensional and multiproperty PO-lanthanide(III) derived systems†
Evangelos
Pilichos
a,
Ànnia
Tubau
a,
Saskia
Speed
a,
Mercè
Font-Bardia
b,
Albert
Escuer
 ac,
Arnald
Grabulosa
*ac and
Júlia
Mayans
*ac
ac,
Arnald
Grabulosa
*ac and
Júlia
Mayans
*ac
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Marti i Franques 1-11, Barcelona-08028, Spain
bDepartament de Mineralogia, Cristal·lografia i Dipòsits Minerals and Unitat de Difracció de R-X, Centre Científic i Tecnològic de la Universitat de Barcelona (CCiTUB), Universitat de Barcelona, Solé i Sabarís 1-3, 08028 Barcelona, Spain
cInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, Barcelona-08028, Spain
First published on 25th January 2023
Abstract
A new family of lanthanide compounds has been synthesized using 1,2-bis(diphenylphosphino)ethane dioxide (dppeO2) as an O-donor ligand through the phosphoryl group to lanthanide(III) cations and structurally, magnetically and optically studied. Depending on the lanthanide, two different topologies appear: the two-dimensional structure [LnIII(dppeO2)1.5(NO3)3(H2O)0.5]n (Ln = Ce (1), Sm (2) and Dy (6)) and the one-dimensional structure [Ln(dppeO2)(NO3)3DMF]n (Ln = Eu (3), Gd (4) and Tb (5)). Some of the Ln-derived complexes have been used as structural probes, while others have been synthesized to use the specific characteristics of each cation to take advantage of their magnetic/luminescence properties. Complex 6 presents slow relaxation of the magnetization while 2, 3 and 5 present emitting properties in the visible range.
Introduction
During the last few years, molecular lanthanide(III)-derived compounds climbed to the top of different new applications like luminescent sensors,1,2 slow-relaxing magnetic systems3,4 spintronics5,6 or anticounterfeit technologies.7,8 One of the most exploited applications of lanthanide molecular compounds is their use as potential Single Molecule Magnets (SMMs) or Single Ion Magnets (SIMs) due to their comparatively high single ion anisotropy arising from their Spin–Orbit Coupling (SOC) and from the crystal field splitting of the lowest J multiplet when they are forming a coordination complex.4,9,10 Among this, lanthanide cations present extraordinary emitting properties originating from the electronic transitions within the partially filled 4f orbitals except for CeIII, which presents 5d–4f electronic transitions. However, due to the very low sensitivity of lanthanide(III) to their surroundings, their hybridization is limited and, consequently, LnIII exhibits very narrow emission lines, which can be considered as a fingerprint of each cation. The main drawback of this approach to the luminescence of lanthanide-derived complexes is that transitions in-between 4f orbitals are partially Laporte-forbidden. Therefore, any strategy to increase the luminescence of lanthanide coordination compounds must consider the sensitization of lanthanide(III) cations.11,12 To this end, a useful procedure is the coordination of lanthanide(III) ions to phosphine oxides, known to be excellent ligands.13 Phosphine oxides are ideal for this purpose because they can be easily prepared by oxidation of the parent phosphines, a huge family of ligands extensively studied due to their application in organometallic homogeneous catalysis. Hence, this kind of ligand allows the fine-tuning of the system's properties.Some extended systems derived from the diphosphine oxide bis(diphenylphosphino)ethane dioxide (dppeO2), Scheme 1, were previously reported by other authors14–19 but their magnetic properties remain practically unexplored.
 | ||
| Scheme 1 Structural formula of the ligand employed in this work, 1,2-bis(diphenylphosphino)ethane dioxide (dppeO2). | ||
Noteworthily, the stereochemistry or dimensionality of the dppO2/LnIII system can show a multifactorial dependence on factors such as the size of the cation, the counterions present in the reaction media, reaction conditions or the solvent employed.20 In the present paper, we report a new family of Ln-derived systems using dppeO2. Single crystal X-ray diffraction demonstrated that two different structures could be obtained with no changes in the reaction conditions: bidimensional networks with the general formula [LnIII(dppeO2)1.5(NO3)3(H2O)0.5]n (Ln = Ce (1), Sm (2) and Dy (6)) and monodimensional compounds with the general formula [Ln(dppeO2)(NO3)3DMF]n (Ln = Eu (3), Gd (4) and Tb (5)), which are apparently independent of the above-mentioned factors affecting the final structures. Complexes 1, 3 and 4 were used as structural probes to test the isostructurality with other family members by X-ray powder diffraction. Noteworthily, the reported systems provide the first examples of 1-D arrangement for the dppeO2/LnIII system. The anisotropic derivative 6 has been magnetically characterized (both statically and dynamically) while complexes 2, 3 and 5, the typical visible-range emitters, have been studied to check their emitting properties.
Experimental
X-ray crystallography
White (1, 3 and 4) prism-like specimens were used for single crystal X-ray crystallographic analysis. The X-ray intensity data were measured on a D8 Venture system equipped with a multilayer monochromator and a Mo microfocus (Table 1).| 1 | 3 | 4 | |
|---|---|---|---|
| Formula | C78H74Ce2N6O25P6 | C29 H31EuN4O12P2 | C29 H31GdN4 O12 P2 |
| FW | 1961.49 | 841.48 | 846.77 |
| System | Trigonal | Monoclinic | Monoclinic |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
P21/c | P21/n |
| a/Å | 15.5252(3) | 9.1723(5) | 17.0768(7) |
| b/Å | 15.5252(3) | 17.776(1) | 9.9525(5) |
| c/Å | 60.112(2) | 20.858(1) | 20.509(1) |
| α/° | 90 | 90 | 90 |
| β/° | 90 | 102.952(2) | 104.076(2) |
| γ/° | 120 | 90 | 90 |
| V/Å3 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 547.9(7) 547.9(7) |
3314.4(3) | 3380.9(3) |
| Z | 6 | 4 | 4 |
| T, K | 100(2) | 100(2) | 100(2) |
| λ(MoKα), Å | 0.71073 | 0.71073 | 0.71073 |
| p cal, g cm−3 | 1.557 | 1.686 | 1.664 |
| μ(MoKα), mm−1 | 1.269 | 2.057 | 2.123 |
| R | 0.0480 | 0.0316 | 0.0271 |
| ωR2 | 0.1138 | 0.0724 | 0.0530 |
The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The structures were solved and refined using the Bruker SHELXTL software.21
Powder X-ray diffraction was performed with a PANalytical X'Pert PRO MPD θ/θ powder diffractometer of 240 millimetre radius, in a configuration of the convergent beam with a focalizing mirror and a transmission geometry with flat samples sandwiched between low absorbing films and Cu Kα radiation (λ = 1.5418 Å). Comparison between the calculated spectrum from the single crystal structures of 1 and 4 and the experimental powder X-ray spectrum for compounds 2, 5 and 6 confirms the isostructurality among the two different groups, Fig. S1.†
Crystal data and refinement details for complexes 1, 3 and 4 are summarized in Table 1. Further crystallographic details can be found in the corresponding CIF files provided in the ESI.†
Physical measurements
Magnetic susceptibility measurements were carried out on pressed polycrystalline samples with an MPMS5 Quantum Design susceptometer working in the range 30–300 K under magnetic fields of 0.3 T and under a field of 0.03 T in the 30–2 K range to avoid saturation effects at low temperature. Diamagnetic corrections were estimated from Pascal Tables. Infrared spectra (4000–400 cm−1) were recorded from KBr pellets on a Bruker IFS-125 FT-IR spectrophotometer.Solid-state fluorescence spectra of compounds 2, 3 and 5 were recorded on a Horiba Jobin Yvon SPEX Nanolog fluorescence spectrophotometer (Fluorolog-3 v3.2, HORIBA Jovin Yvon, Cedex, France) equipped with a three-slit, double-grating excitation and emission monochromator with dispersions of 2.1 nm mm−1 (1200 grooves per mm) at room temperature. The steady-state luminescence was excited with unpolarized light from a 450 W xenon CW lamp and detected at an angle of 22.5° for solid-state measurement with a red-sensitive Hamamatsu R928 photomultiplier tube. The instrument was adjusted to obtain the highest background-to-noise ratio. The sample was mounted between two quartz plates. Spectra were corrected for both the excitation source light intensity variation (lamp and grating) and the emission spectral response (detector and grating). Lifetime decay curves were measured in the same instrument monitored in the phosphoresce mode using a 450 W xenon pulsed lamp (1.5 ns pulse). The measured decays were analysed using the OriginPro 8 software package. The decay curves of compounds 3 and 5 and the fit quality were determined by the χ2 method of Pearson. A band pass of 0.5 nm was employed in order to not surpass 106 CPS to make sure that we are not measuring in the non-linear optical zone.
The 1H and 31P{1H} NMR spectra of dppeO2 were recorded at room temperature in a cryoprobe-equipped, 400 MHz Bruker Avance III spectrometer in dmso-d6.
Syntheses
Results and discussion
Description of the structures
 | ||
| Fig. 1 Top, labelled plot of the CeIII environment for complex 1. Bottom, intermediate Oh/TP polyhedron around the decacoordinated CeIII cation and in-plane arrangement of the dppeO2 ligands. Color key for all figures, CeIII, light blue; GdIII, firebrick; P, orange; O, red; N, navy; and C, black. The positions indicated as green balls correspond to the mean position between the two O-donors of each bidentate nitrate ligand. | ||
 | ||
| Fig. 2 Left, a view of the honeycomb layers of 1 along the c-axis; right, view of the network along the a-axis. | ||
| Ce1–O1 | 2.641(4) | Ce1–O1′ | 2.35(1) |
| Ce1–O2 | 2.592(5) | Ce1–O3′ | 2.52(3) |
| Ce1–O5 | 2.74(1) | Ce1–O4 | 2.428(2) |
| P(1)–O(4) | 1.503(2) | ||
| O1–Ce1–O2 | 48.5(1) | O1–Ce1–O5 | 63.36(8) |
| O1–Ce1–O4 | 68.49(9) | O2–Ce1–O5 | 76.3(1) |
| O2–Ce1–O4 | 75.0(2) | P(1)–O(4)–Ce(1) | 165.2(2) |
| O4–Ce1–O5 | 131.71(6) | O4–Ce1–O4′ | 80.55(9) |
| (3) Ln = Eu | (4) Ln = Gd | |
|---|---|---|
| Ln(1)–O(1) | 2.315(2) | 2.313(1) |
| Ln(1)–O(2) | 2.323(2) | 2.314(1) |
| Ln(1)–O(3) | 2.531(3) | 2.473(2) |
| Ln(1)–O(5) | 2.503(3) | 2.468(2) |
| Ln(1)–O(6) | 2.498(3) | 2.525(2) |
| Ln(1)–O(7) | 2.538(3) | 2.439(2) |
| Ln(1)–O(9) | 2.474(3) | 2.573(2) |
| Ln(1)–O(10) | 2.523(3) | 2.506(2) |
| Ln(1)–O(12) | 2.386(3) | 2.338(2) |
| P(1)–O(1) | 1.508(2) | 1.505(1) |
| P(2)–O(2) | 1.506(3) | 1.503(1) |
| O(1)–Ln(1)–O(2) | 149.27(8) | 149.26(5) |
| O(1)–Ln(1)–O(12) | 84.13(9) | 85.41(5) |
| O(2)–Ln(1)–O(12) | 85.05(9) | 83.97(5) |
| P(1)–O1–Ln(1) | 166.4(2) | 153.46(9) |
| P(2)–O(2)–Ln(1) | 168.9(2) | 166.43(9) |
The structure consists of a 1D arrangement of GdIII cations bridged by bis-monodentate dppeO2 ligands. The enneacoordinated environment around the GdIII cation is formed by three bidentate nitrato ligands, two O-donors from two dppeO2 ligands and one dimethylformamide molecule. The two dppO2 ligands are coordinated in trans positions to the lantanoid cation (O–Gd–O bond angle of 149.3°) whereas the nitrato and DMF ligands are roughly linked in the equatorial plane, Fig. 3-top. The Gd–O bond distances are in the range of 2.313–2.506 Å, with the shorter ones being those involving the Ophosphine bonds. The dppeO2 ligands link the GdIII cations resulting in a zig-zag chain along the a-axis of the cell, Fig. 3. SHAPE23 analysis of the coordination sphere around the GdIII cation shows that, as for 1, the minimum CShM value of 1.79 indicates a strong distortion from any ideal polyhedron whereas with regard to the six ligands, their arrangement is clearly octahedral, Fig. 3, right.
 | ||
| Fig. 3 Top, partially labelled plot and polyhedral environment of complex 4. Bottom, plot of the 1D chain of GdIII cations (left) and network arrangement of the chains along the a-axis of the unit cell. | ||
Structural and synthetic comments
The chemistry of bis(diphenylphosphine)-dioxide ligands with lanthanide cations is strongly dependent on several factors that can modify the denticity of the ligands and/or the dimensionality of the resulting complexes. This kind of ligand can act as a bidentate or bis-monodentate donor Scheme 2-top, allowing for molecular or extended systems, respectively. The length of the flexible (CH2)n spacer between the phosphor atoms is a determinant resulting in always a bidentate coordination for n = 1 and consequently exclusively molecular compounds have been reported whereas both modes have been characterized for n = 2–4. Interestingly, the bis-monodentate mode is preferred for the n = 2 case yielding 2D networks whereas the bidentate mode is preferred for the n = 4 case due to the flexibility of the butane spacer. | ||
Scheme 2 Top, bidentate (left) and bis-monodentate (right) coordination modes of the dppeO2 ligand. Bottom, previously reported fac (a) and mer (b) arrangement of the ligands around the lanthanide cations. Arrangement of the ligands for the 2D (c) and 1D (d) structures reported in this paper. X for (a) and (b): O,O′-NO3− or Cl− allowing for coordination 9 or 6, respectively; X for (c) and (d): O,O′-NO3−; P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O represent one of the donors of the dppeO2 ligand; S: solvent molecule. O represent one of the donors of the dppeO2 ligand; S: solvent molecule. | ||
The simultaneous presence of both coordination modes has been found in dinuclear systems for the dppeO2 ligand and PrIII, DyIII and LuIII cations18 but all the remainder complexes are 2D systems with exclusive presence of the bis-monodentate mode, Table 4. These 2D networks with the [Ln(dppeO2)1.5(X)3]n formula are equally found for X = O,O′-NO3− (resulting LnO9 coordination), or for X = Cl−, (resulting LnO3Cl3) hexacoordination. In the two cases the arrangement of the ligands can adopt a fac- or mer-arrangement around the lanthanide (Scheme 2a and b), which allows for different 2D network architectures (parquet-floor, brick-wall or honeycomb).
As can be extracted from Table 4, any relationship can be extracted from factors like the cation size or the size or denticity of the coordinated anions. In the same way, the syntheses under soft conditions (room temperature) or solvothermal conditions at high temperatures and large reaction times become also irrelevant.18 In light of these data, the solvent employed as reaction medium becomes a determinant as was proposed by Spichal et al.15 This assumption has been corroborated by means of the MeOH/DMF medium employed in the present work that allows for unprecedented coordination numbers or dimensionality of the reported systems, Table 4.
Magnetic properties
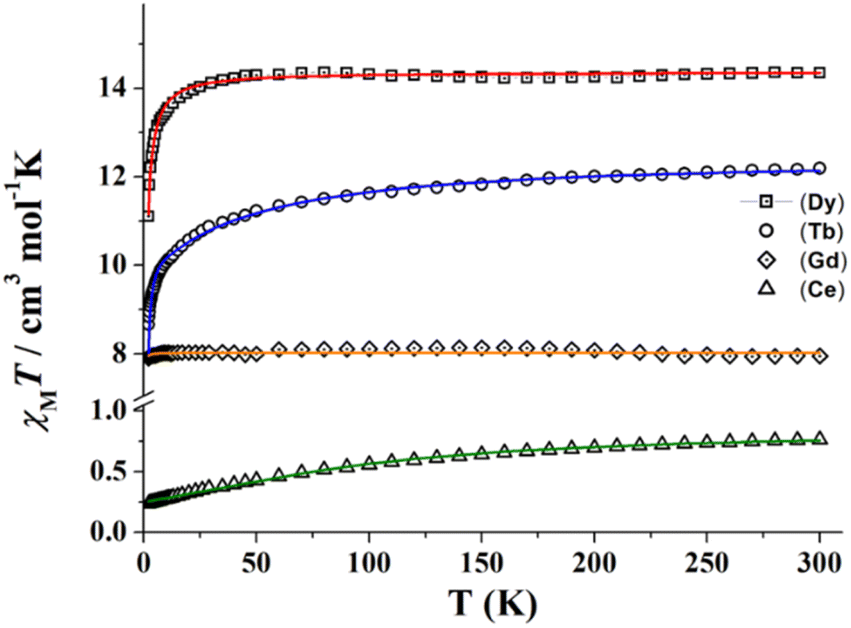 | ||
| Fig. 4 χ M T vs. temperature plots of complexes 1 (triangles), 3 (diamonds), 5 (circles) and 6 (squares). Solid lines show the best fit of the experimental data. | ||
Due to the topology of all the studied structures, the magnetic behaviour of the paramagnetic carriers in the compounds corresponds to single ion contributions. χMT values for complexes 1, 5 and 6 are very close to the corresponding values for the isolated lanthanide cations 2F5/2 (CeIII, 0.80 cm3 mol−1 K), 7F6 (TbIII, 11.82 cm3 mol−1 K), and 6H15/2 (DyIII, 14.17 cm3 mol−1 K), respectively. For these three compounds, χMT slowly decreases when lowering the temperature due to the progressive depopulation of the mJ levels, with a more pronounced decay below 50 K, probably due to antiferromagnetic intramolecular interactions in-between the extended structures. The isotropic compound 4 (GdIII, 8S7/2) shows a room temperature χMT value of 7.95 cm3 mol−1 K close to the expected value of 7.875 cm3 mol−1 K that follows a Curie law down to low temperature.
Experimental data from Fig. 4 was fitted using PHI software24 and using a simple, but effective model reported by Lloret and co-workers for axial symmetry environments in lanthanide derived compounds, which has been previously used by us.25,26 For compounds 1, 5 and 6, the used Hamiltonian (eqn (1)), in which Ŝ are the spin operators, L is the orbit operator, and λ is the spin–orbit coupling, while Δ describes the energy gap between mL components. The value for the orbital reduction parameter (κ) was assumed as 1 (lanthanide ions behave as purely ionic). The important point about this fitting is the different shapes of the χMT curves depending on the sign of Δ: if Δ is greater than 0, the smallest value of mL corresponds to the ground states, so the lowest mL will be the ground state. For Δ smaller than 0, occurs the opposite and the highest mL corresponds to the ground state. The fact that only the shape of the curve is relevant here is because the low symmetry of the compounds tends to mix the ground state. The first term of the Hamiltonian describes the spin–orbit coupling, while the second term is related to the ligand field around the lanthanide cation while the third term describes the Zeeman effect.
| H = (λŜL) + Δ[Lz2 − L(L + 1)/3] + [βH(−κL + 2Ŝ)] | (1) |
A very good fit of the experimental data was obtained (solid lines in Fig. 4) with Δ parameters of −8.3, −12.1 and −6.2 cm−1, for 1, 5 and 6, respectively. Magnetization experiments performed at 2 K show quasi saturated plots with Nμβ values of 1.05 (1), 4.83 (5) and 5.31 (6) that confirm the negative Δ value for these cations and 7.09 for the isotropic gadolinium system (4).
 | ||
| Fig. 5 Temperature (top) and frequency (bottom) dependence of χ′′M of complex 6 at an optimal applied external magnetic field of 0.1 T. | ||
Plots of the maxima of χ′′Mvs. inverse of the temperature, the so-called Arrhenius plot, evidence that their dependence is not linear and only a rough linearity can be found for higher temperatures, indicating the presence of more than one relaxation mechanism (Fig. S4†). Because for a Kramers ion the magnetic relaxation should occur through excited Kramers doublets with a sequential number of transitions induced by an emission of a quantum of energy lattice (a phonon), a preliminary fit of the high temperature region using the equation ln(1/(2πω) = ln(1/τ0) − Ueff/(kBT) yields in the fitting parameters Ueff = 21.77 K and reasonable τ0 = 2.00 10−7 s (Fig. S4†). However, on one hand, these values are poorly precise due to the low number of points in the linear region and on the other hand, the low Ueff values are probably much lower than the gap with the DyIII first excited doublet.
A more precise analysis of the χ′M and χ′′M behaviour was performed using the generalized Debye model,27,28 where these two variables are analysed jointly with τ (the relaxation rate) and α (the parameter that defines the broadness of the spectra involving the distribution in the relaxation times) represented as Argand plots (Fig. 6, left).
 | ||
| Fig. 6 Left, Argand plot of the magnetic susceptibility of 6. Right, temperature dependence of the relaxation time as a function of the temperature plotted as ln(τ) vs. inverse of T. Solid lines show the best fit of the experimental data (Orbach plus Raman fitting) and dotted lines show the slope of the Orbach high temperature regime. | ||
The data extracted from the fitting (CCfit program), is represented in the form of ln(τ) vs. 1/T (Fig. 6, right). Two different relaxations can be observed: the data were simulated following the combination of Orbach and Raman relaxations because the high temperature region clearly follows an Arrhenius-like behaviour while the low temperature region follows the two-phonon relaxation path (eqn (2)).
| τ−1 = τ0−1e−Ea/KT + CTn | (2) |
The alpha parameter has a value of 0.06, indicating a narrow distribution of the relaxation times, while the Orbach parameters Ueff and τ0 of 21.77 K and 2.0 × 10−7 s are in good agreement with those obtained from the χ′′M(T−1) fit. The Raman relaxation path was fitted with C = 0.06 and n = 7.3. This value of the Raman coefficient is close to the ones expected for Kramers ions (typically 9–11). However, lower Raman coefficients have been reported commonly relating them to the participation of optical phonons in the spin–phonon interaction.29,30
To demonstrate the intervention of the lattice in the magnetic relaxation of complex 6, ultra-low frequency Raman measurements have been performed (Fig. 7).31,32 The spectrum evidences the presence of low frequency vibrations appearing at 27, 59 and 84 cm−1, which are compatible with the energy required for a spin–phonon relaxation path.
 | ||
| Fig. 7 Ultralow frequency Raman spectrum of complex 6 showing low energy vibrations. | ||
Photoluminescence characterization
The emissive properties of compounds 2, 3 and 5 have been studied in polycrystalline samples. As a previous step, the first absorption and emission of the free ligand, dppeO2, was measured in a chloroform solution (Fig. S5†). The phosphine oxide ligand shows an absorption band at 265 nm. Additionally, the excitation and emission spectra of the free ligand have been recorded at an excitation wavelength (λexc) of 270 nm (an λexc of 260 nm cannot be used due to instrumental limitations) and at an emission wavelength (λem) of 320 nm, respectively.Excitation of the lanthanide samples at 270 nm induces an emission response for the three measured compounds leading to the characteristic luminescence for each lanthanide cation. Nevertheless, the sensitization effect is not efficient since the emission intensity is rather low. Furthermore, in the excitation spectra recorded at λem values of 597 nm (4G5/2 → 6H7/2) for 2, 619 nm (5D0 → 7F2) for 3 and 545 nm (5D4 → 7F5) for 5 the signals arising from the excitation f–f transitions are more intense and can be well differentiated with respect to the bands corresponding to the π–π* and n–π* transitions from the benzene groups of the ligand moieties. Also, the free ligand absorption and emission wavelengths are found in the 270–300 nm UV range suggesting that the phosphine molecule in each LnIII compound would also absorb light in this range. Then, the whole excitation of the ligand under 300 nm is difficult to observe in all the excitation spectra due to instrumental limitations.
Besides, the energy gaps between the excited singlet and triplet states of the ligand and the respective lanthanide emitting state ions (5D0 for EuIII, 4D4 for TbIII, 4G5/2 for SmIII) are too large. Nevertheless, the distortion due to crystal field perturbation in these structures makes the f–f transitions less forbidden by Laporte's rule and excitation of each sample at their respective f–f excitation transitions, induced more intense emission signals than when exciting the sample in the ligand excitation wavelength, especially for the EuIII and TbIII compounds that the emission color could be seen in the naked eye (Fig. S6†).33,34
 | ||
| Fig. 8 Excitation (top) and emission (bottom) spectra of compound 2. | ||
Nevertheless, when 2 is excited at 403 nm, corresponding to the 6H5/2 → 4F7/2 excitation transition, a more intense spectrum with the expected SmIII emission bands is acquired, Fig. 8. In this manner, there is a rapid non-radiative deactivation from the 4F7/2 energy level to the emitting 4G5/2 state due to the closely spaced energy levels of these spectroscopic terms. Then the 4G5/2 → 6H5/2–11/2 radiative transitions take place. The band at 564 nm is assigned to 4G5/2 → 6H5/2 which is a pure magnetic dipole transition (ΔJ = 0). The more intense signal at 597 nm corresponds to 4G5/2 → 6H7/2 (ΔJ = ± 1) and along with the selection rule, the transition is partially magnetic and partially forced electric dipole allowed. Next the electric dipole transition (ΔJ = ± 2) 4G5/2 → 6H9/2, more susceptible to the crystal field (hypersensitive transition), is discerned at 643 nm followed by 4G5/2 → 6H11/2 as a low intensity band. It is known that the intensity ratio between the magnetic dipole and electric dipole transitions can be an indicator of the symmetry site of the lanthanide cation inside the structure. For compound 2, the integrated area of the 4G5/2 → 6H9/2 band is larger than that of the 4G5/2 → 6H5/2 band, indicating certain asymmetry in the SmIII coordination environment37–39 (the symmetry point group of the coordination polyhedron of 2 does not present an inversion symmetry).
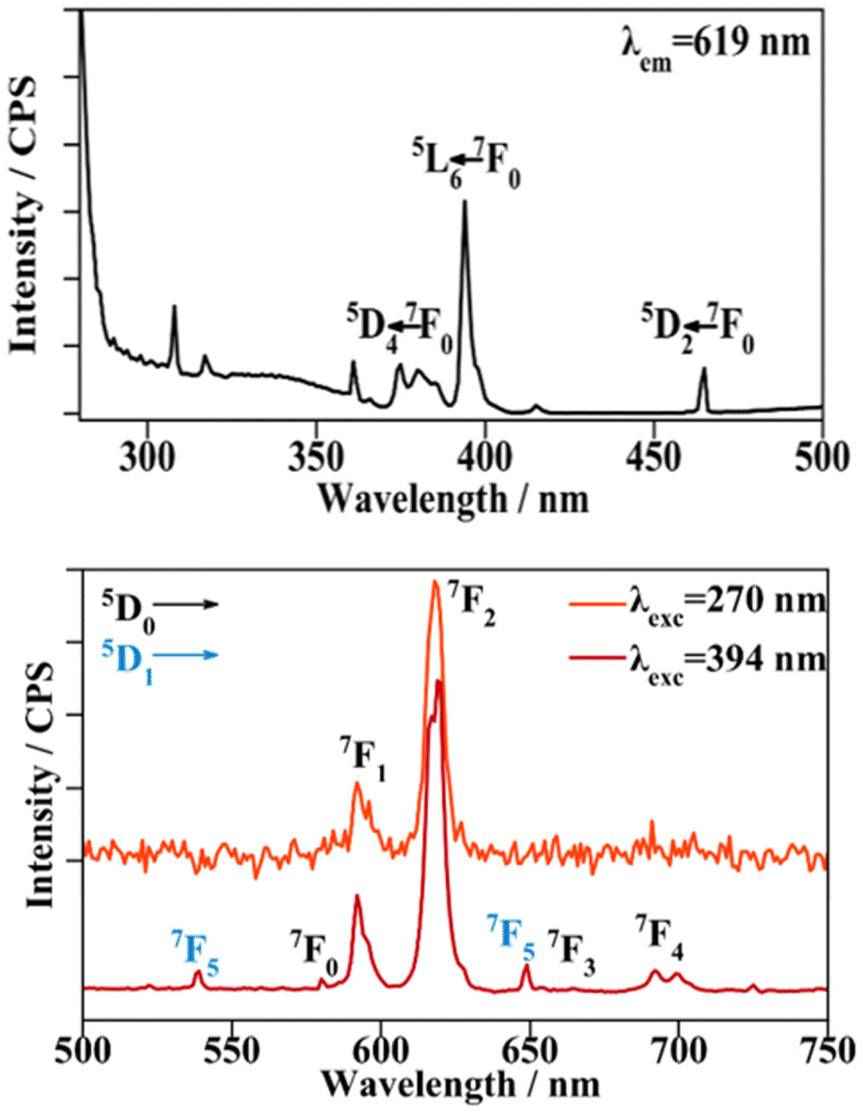 | ||
| Fig. 9 Excitation (top) and emission (bottom) spectra of compound 3. | ||
When the sample is excited at the ligand excitation wavelength (270 nm) the emission spectrum (shown in Fig. 9, top) is recorded. The signal to noise ratio is high and only two bands are observed. Still, when the λexc employed is 394 nm, corresponding to the highest metal centred f–f excitation transition 5L6 ← 7F0, an emission spectrum with a higher resolution could be recorded (Fig. 9, bottom). The bands corresponding to the emission from the 5D0 emitting energy level at 580, 592, 619, 654 and 696 nm are in good agreement with the 5D0 → 7F(0–4) transitions. The magnetic dipole 5D0 → 7F1 transition, the intensity of which is independent of its environment, is split due to the crystal field in the spectrum recorded at λexc = 270 nm. This splitting is manifested as a shoulder for the λexc = 394 nm spectrum. Next, the most intense band corresponding to the electric dipole 5D0 → 7F2 transition presents a small split due to the crystal field effect and the 5D0 → 7F2/5D0 → 7F1 ratio is 4.7, suggesting that the EuIII cation does not belong to a point group with inversion symmetry inside the structure. When exciting the sample at 394 nm, the 5D4 excited level deactivates through radiationless transitions to the 5D0 emitting level, leading to the above-mentioned transitions. In addition, radiative transitions take place from the 5D1 energy level, higher in energy than the 5D0 level. The bands corresponding to these transitions are located at 539 nm corresponding to 5D1 → 7F1 and at 649 nm, they overlapped with the 5D0 → 7F3 transition, assigned to 5D1 → 7F5.40
 | (3) |
Luminescence lifetime decay was measured for the europium compound. The experiment was monitored at an emission wavelength of 614 nm and the sample was excited at 395 nm. The decay curve presented in the semi-log plot is shown in Fig. 10 and it could be fitted with a mono-exponential equation (eqn (3)) which is consistent with one deactivation mechanism. The luminescence lifetime (τobs) of 3 is 1.65 ms. Furthermore, the pure magnetic dipole character of europium's 5D0 → 7F1 transition allows the calculation of the radiative rate constant krad from the corrected emission spectrum, using a simplified equation presented by Werts et al. (eqn (4)).41 The τrad (krad = 1/τrad) parameter accounts for the lifetime of luminescence deactivation from the emitting level if all the deactivation transitions were of radiative nature and for 4 it was found to be 2.9 ms. The calculated τrad value is higher than the observed lifetime obtained from the measurement elucidating that faster non-radiative deactivation processes take place reducing the τobs. Additionally, the ratio between the radiative rate and the luminescence deactivation rate of the compound defines the intrinsic quantum yield (ϕLnLn) which is the emission efficiency once the EuIII emitting level is populated. For 3, (ϕLnLn) is 55%.
 | (4) |
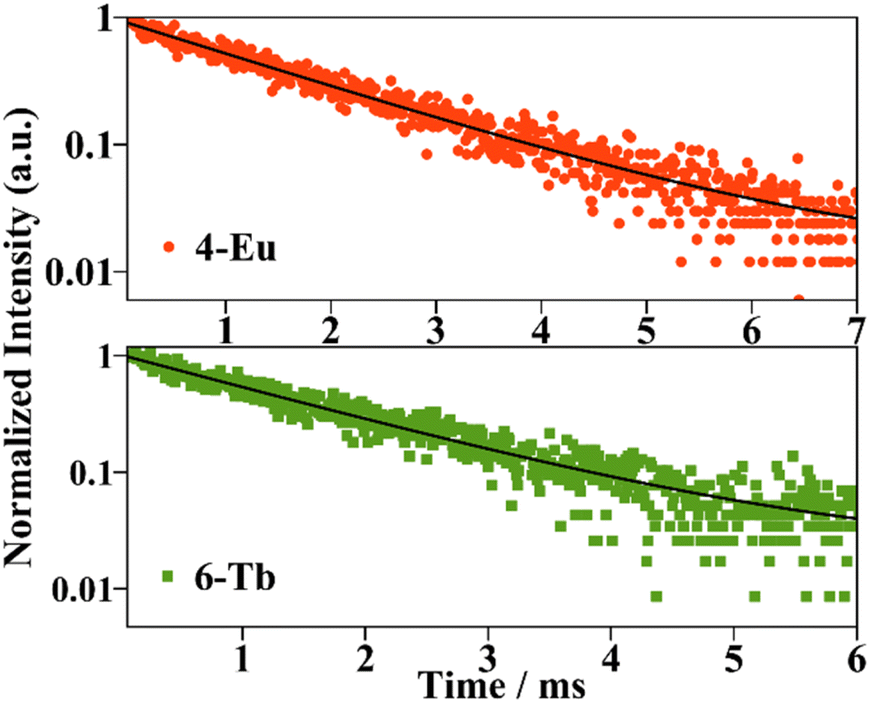 | ||
| Fig. 10 Luminescence lifetimes curves of compounds 3 and 5. Solid lines represent mono-exponential fittings using eqn (3). | ||
 | ||
| Fig. 11 Excitation (top) and emission (bottom) spectra of compound 5. | ||
The luminescence lifetime decay of 5, monitored at an emission wavelength of 545 nm and at an excitation wavelength of 350 nm that agrees with the TbIII centred 7F6 → 5D2 transition, yielded 1.51 ms. For the terbium compound, the decay curve also followed a mono-exponentially function suggesting a single deactivation mechanism as the EuIII compound.
Conclusions
A new family of phosphine oxide lanthanide-derived compounds has been synthesized and characterized by structural, magnetic and luminescence techniques. Two different structures can be achieved while using the same synthesis, and the final topology of the products seems independent of the previously proposed factors like lanthanide size or co-ligands but determined by the solvents. Moreover, the first monodimensional chain-like structure has been achieved for dppeO2 lanthanide coordination compounds. All the family members have been magnetically characterized, but only Dy presents a field-induced SIM response following an Orbach-like plus Raman relaxation path. Among these, the visible emitters SmIII, EuIII and TbIII systems have been studied in their specific emission range, and all of them present good emission properties.Author contributions
E. P.: synthesis, magnetic measurements, and data analysis; A. T.: luminescence measurements, data analysis, and original draft writing; M. F.-B.: single crystal X-ray crystallography; S. S.: preliminary luminescence spectra; A. E.: structural analysis, conceptualization, writing and editing. A. G.: funding acquisition and project administration; and J. M.: supervision of the experimental work and measurements, final manuscript writing, and conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JM and AE are thankful for the funding from MICINN (Project PGC2018-094031-B-100). AG is thankful for the support from MICINN (Project PID2020-115658GB-I00) and IN2UB (Project ART_2021_3).References
- C. M. G. Dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS.
- S. Shinoda and H. Tsukube, Analyst, 2011, 136, 431 RSC.
- A. Dey, P. Kalita and V. Chandrasekhar, ACS Omega, 2018, 3, 9462 CrossRef CAS PubMed.
- R. A. Layfield and M. Murugesu, Lanthanides and Actinides in Molecular Magnetism, John Wiley & Sons, 2015 Search PubMed.
- R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728 CrossRef CAS PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- A. M. Kaczmaret, Y.-Y. Liu, C. Wang, B. Laforce, L. Vincze, P. Van Der Vort, K. Van Hecke and R. Van Deun, Adv. Funct. Mater., 2017, 27, 1700258 CrossRef.
- W. Ren, G. Lin, C. Clarke, J. Zhou and D. Jin, Adv. Mater., 2020, 32, 1901430 CrossRef CAS PubMed.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092 RSC.
- J. Luzón and R. Sessoli, Dalton Trans., 2012, 41, 13556 RSC.
- H. Zhang, Z.-H. Chen, X. Liu and F. Zhang, Nano Res., 2020, 13, 1795 CrossRef CAS.
- A. D'Aléo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Coord. Chem. Rev., 2012, 256, 1604 CrossRef.
- A. W. G. Platt, Coord. Chem. Rev., 2017, 340, 62 CrossRef CAS.
- R. D. Bannister, W. Levason, G. Reid and W. Zhang, Polyhedron, 2017, 133, 264 CrossRef CAS.
- Z. Spichal, M. Necas and J. Pinkas, Inorg. Chem., 2005, 44, 2074 CrossRef CAS PubMed.
- Z. Spichal, M. Nekas, J. Pankas and J. Novosad, Inorg. Chem., 2004, 43, 2776 CrossRef CAS PubMed.
- P. Pekarkova, Z. Spichal, P. Taborsky and M. Nekas, Luminescence, 2011, 26, 650 CrossRef CAS PubMed.
- W. Q. Lin, D. R. Wang, W. J. Long, L. S. Lin, Z. X. Tao, J. L. Liu, Z. Q. Liu and J. D. Leng, Dalton Trans., 2022, 51, 5159 Search PubMed.
- S. A. Cotton and P. R. Raithby, Coord. Chem. Rev., 2017, 340, 220 CrossRef CAS.
- A. M. J. Lees and A. W. G. Platt, Inorg. Chem., 2003, 42, 4673 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2014/7: Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 2014 Search PubMed.
- F. Marchetti, C. Pettinari, A. Pizzabiocca, A. A. Drozdov, S. I. Troyanov, C. O. Zhuravlev, S. N. Semenov, Y. A. Belousov and I. G. Timokhin, Inorg. Chim. Acta, 2010, 363, 4038 CrossRef CAS.
- M. Lunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE v.2.0, Barcelona, 2010. The program can be obtained by request to the authors Search PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- M. V. Marinho, D. O. Reis, W. X. C. Oliveira, L. F. Marques, H. O. Stumpf, M. Déniz, J. Pasán, C. Ruíz-Pérez, J. Cano, F. Lloret and M. Julve, Inorg. Chem., 2017, 56, 2108 CrossRef CAS PubMed.
- A. Worrell, D. Sun, J. Mayans, C. Lampropoulos, A. Escuer and T. C. Stamatatos, Inorg. Chem., 2018, 57, 13944 CrossRef CAS PubMed.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS.
- P. Debye, Phys. Z., 1938, 39, 616 CAS.
- L. Gu and R. Wu, Phys. Rev. B, 2021, 103, 01441 CrossRef.
- K. N. Shivatava, Phys. Status Solidi B, 1983, 117, 437 CrossRef.
- M. Atzori, S. Benci, E. Morra, L. Tesi, M. Chiesa, R. Torre, L. Sorace and R. Sessoli, Inorg. Chem., 2018, 57, 731 CrossRef CAS PubMed.
- M. Briganti, F. Santanni, L. Tesi, F. Totti, R. Sessoli and A. Lunghi, J. Am. Chem. Soc., 2021, 143, 13633 CrossRef CAS PubMed.
- Y.-S. Meng, Y.-S. Qiao, Y.-Q. Zhang, S.-D. Jiang, Z.-S. Meng, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. – Eur. J., 2016, 22, 4704 CrossRef CAS PubMed.
- M. Latva, H. Takalo, V. M. Mukkala, C. Matachescu, U. J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149 CrossRef CAS.
- F. A. Mautner, F. Bierbaumer, R. Vicente, S. Speed, A. Tubau, R. C. Fischer and S. S. Massoud, Magnetochemistry, 2022, 8, 72 CrossRef CAS.
- C. K. Jayasankar and E. Rukmini, Opt. Mater., 1997, 8, 193 CrossRef CAS.
- R. Yu, N. Xue, T. Wang, Z. Zhao, J. Wang, Z. Hei, M. Li, H.-M. Noh and J.-H. Jeong, Ceram. Int., 2015, 41, 6030 CrossRef CAS.
- J. Bolton, J. Lumin., 2021, 231, 117717 CrossRef CAS.
- I. J. Al-Busaidi, R. Ilmi, D. Zhang, J. D. L. Dutra, W. F. Oliveira, N. K. Al Rasbi, L. Zhou, W.-Y. Wong, P. R. Raithby and M. S. Kahn, Dyes Pigm., 2022, 197, 109879 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- Y.-C. Li, Y.-H. Chang, Y.-J. Lin and C.-H. Laing, J. Phys. Chem. C, 2007, 111, 210682 Search PubMed.
- A. J. Kanimozdhi and V. Alexander, Dalton Trans., 2017, 46, 8562 RSC.
- X. Zhou, X. Zhao, Y. Wang, B. Wu, J. Shen, L. Li and Q. Li, Inorg. Chem., 2014, 53, 12275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural and magnetic data. CCDC 2215834–2215836. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03621e |
| This journal is © The Royal Society of Chemistry 2023 |