
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IrI(η4-diene) precatalyst activation by strong bases: formation of an anionic IrIII tetrahydride†
Paven
Kisten
 ab,
Eric
Manoury
b,
Agustí
Lledós
c,
Adrian C.
Whitwood
a,
Jason M.
Lynam
a,
John M.
Slattery
a,
Simon B.
Duckett
*a and
Rinaldo
Poli
*bd
ab,
Eric
Manoury
b,
Agustí
Lledós
c,
Adrian C.
Whitwood
a,
Jason M.
Lynam
a,
John M.
Slattery
a,
Simon B.
Duckett
*a and
Rinaldo
Poli
*bd
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD UK. E-mail: simon.duckett@york.ac.uk
bCNRS, LCC (Laboratoire de Chimie de Coordination), Université de Toulouse, UPS, INPT, 205 route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France CNRS. E-mail: rinaldo.poli@lcc-toulouse.fr
cDepartament de Química, Universitat Autònoma de Barcelona, 08193 Barcelona, Catalonia, Spain
dInstitut Universitaire de France, 1, rue Descartes, 75231 Paris Cedex 05, France
First published on 26th January 2023
Abstract
The reaction between [IrCl(COD)]2 and dppe in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio was investigated in detail under three different conditions. [IrCl(COD)(dppe)], 1, is formed at room temperature in the absence of base. In the presence of a strong base at room temperature, hydride complexes that retain the carbocyclic ligand in the coordination sphere are generated. In isopropanol, 1 is converted into [IrH(1,2,5,6-η2:η2-COD)(dppe)] (2) on addition of KOtBu, with k12 = (1.11 ± 0.02) × 10−4 s−1, followed by reversible isomerisation to [IrH(1-κ-4,5,6-η3-C8H12)(dppe)] (3) with k23 = (3.4 ± 0.2) × 10−4 s−1 and k32 = (1.1 ± 0.3) × 10−5 s−1 to yield an equilibrium 5:95 mixture of 2 and 3. However, when no hydride source is present in the strong base (KOtBu in benzene or toluene), the COD ligand in 1 is deprotonated, followed by β-H elimination of an IrI–C8H11 intermediate, which leads to complex [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] (4) selectively. This is followed by its reversible isomerisation to 5, which features a different relative orientation of the same ligands (k45 = (3.92 ± 0.11) × 10−4 s−1; k5-4 = (1.39 ± 0.12) × 10−4 s−1 in C6D6), to yield an equilibrated 32:68 mixture of 4 and 5. DFT calculations assisted in the full rationalization of the selectivity and mechanism of the reactions, yielding thermodynamic (equilibrium) and kinetic (isomerization barriers) parameters in excellent agreement with the experimental values. Finally, in the presence of KOtBu and isopropanol at 80 °C, 1 is transformed selectively to K[IrH4(dppe)] (6), a salt of an anionic tetrahydride complex of IrIII. This product is also selectively generated from 2, 3, 4 and 5 and H2 at room temperature, but only when a strong base is present. These results provide an insight into the catalytic action of [IrCl(COD)(LL)] complexes in the hydrogenation of polar substrates in the presence of a base.
:2 ratio was investigated in detail under three different conditions. [IrCl(COD)(dppe)], 1, is formed at room temperature in the absence of base. In the presence of a strong base at room temperature, hydride complexes that retain the carbocyclic ligand in the coordination sphere are generated. In isopropanol, 1 is converted into [IrH(1,2,5,6-η2:η2-COD)(dppe)] (2) on addition of KOtBu, with k12 = (1.11 ± 0.02) × 10−4 s−1, followed by reversible isomerisation to [IrH(1-κ-4,5,6-η3-C8H12)(dppe)] (3) with k23 = (3.4 ± 0.2) × 10−4 s−1 and k32 = (1.1 ± 0.3) × 10−5 s−1 to yield an equilibrium 5:95 mixture of 2 and 3. However, when no hydride source is present in the strong base (KOtBu in benzene or toluene), the COD ligand in 1 is deprotonated, followed by β-H elimination of an IrI–C8H11 intermediate, which leads to complex [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] (4) selectively. This is followed by its reversible isomerisation to 5, which features a different relative orientation of the same ligands (k45 = (3.92 ± 0.11) × 10−4 s−1; k5-4 = (1.39 ± 0.12) × 10−4 s−1 in C6D6), to yield an equilibrated 32:68 mixture of 4 and 5. DFT calculations assisted in the full rationalization of the selectivity and mechanism of the reactions, yielding thermodynamic (equilibrium) and kinetic (isomerization barriers) parameters in excellent agreement with the experimental values. Finally, in the presence of KOtBu and isopropanol at 80 °C, 1 is transformed selectively to K[IrH4(dppe)] (6), a salt of an anionic tetrahydride complex of IrIII. This product is also selectively generated from 2, 3, 4 and 5 and H2 at room temperature, but only when a strong base is present. These results provide an insight into the catalytic action of [IrCl(COD)(LL)] complexes in the hydrogenation of polar substrates in the presence of a base.
Introduction
Catalytic hydrogenation1–6 and transfer hydrogenation7–14 are powerful tools for the reduction of a variety of polar unsaturated compounds, especially when using enantiopure chiral catalysts to convert prochiral substrates enantioselectively. Hydrogenation is of particular interest because of atom economy, especially if combined with H2 production from renewable resources, whereas transfer hydrogenation makes use of safer reagents such as isopropanol or formate as hydrogen sources. Among the many catalysts used to carry out this transformation, those based on iridium have received considerable attention.15–23 One of the most common ways to generate an active catalyst is ligand addition to commercially available [IrCl(COD)]2 (COD = 1,5-cyclooctadiene) in the presence of a strong base. The transformations are commonly carried out in isopropanol, whether just as a solvent in hydrogenation or as a solvent and reagent in transfer hydrogenation. The role of the base has generally been ascribed to the generation of the active hydride species via chloride/isopropoxide exchange, followed by β-H elimination with expulsion of acetone.The precise nature of the active catalyst and the mechanism of the catalytic cycle have remained relatively obscure in these systems, with several possible pathways being viable in principle.24–26 These include inner-sphere coordination/insertion monohydride or dihydride pathways, as well as outer-sphere pathways. A direct transfer of the isopropoxide C-bonded H atom to the ketone substrate (Meerwein–Ponndorf–Verley mechanism) is also possible in principle for transfer hydrogenations. Several experimental and computational contributions have addressed the mechanism of this process.27–40 One crucial question that remains is whether the diene ligand in the precatalyst remains coordinated to the metal in the catalytically active species. Although it seems logical to presume that COD is removed under hydrogenation conditions (i.e. under H2), whether this also occurs under transfer hydrogenation conditions (particularly in warm isopropanol) has been controversial. For instance, the [IrCl(diene)]2/aminosulfide/HCOOH/NEt3 system was reported to show diene-dependent activity in the order COD > 1,5-Me2COD > (COE)2 (COE = cyclooctene) at 60 °C.31 These observations led the authors to propose that the diene or alkene remains metal bound in the active species, but the intimate nature of the active species remained obscure. In a combined experimental and computational investigation of the acetophenone transfer hydrogenation catalysed by [Ir(OMe)(COD)]2/L at 60 °C in isopropanol, where L is a P- or N-donor ligand, the active species was also considered to have the diene in the metal coordination sphere.37 On the other hand, several other studies on similar systems, conducted under similar conditions, have shown evidence for the release of COD (either in a hydrogenated form or not).27,28,38,40 In this contribution, we shall address two issues: (i) the role of the base in Ir complex speciation; (ii) the fate of the COD ligand under transfer hydrogenation conditions.
We have previously used complexes [IrCl(COD)(PSR)], (P,SR = chiral ferrocenyl phosphine thioethers, CpFe[1,2-C5H3(PPh2)(CH2SR)], R = Et, Bz, Ph, tBu), obtained by addition of P,SR to [IrCl(COD)]2,41,42 as precatalysts for ketone hydrogenation in isopropanol in the presence of NaOMe, resulting in excellent activities and enantioselectivities.43 An experimental investigation revealed that COD is quantitatively removed from the catalytic system as a mixture of cyclooctene and cyclooctane,44 which is not surprising under hydrogenation conditions (H2). Thus, [IrH(P,SR)(iPrOH)] is presumably formed, at least initially. In addition, using the alternative [Ir(OMe)(COD)]2 precatalyst still required the addition of a strong base to achieve the same high activities as the [IrCl(COD)]2/(P,SR)/NaOMe system. Thus, the role of the strong base cannot be limited to the generation of an active neutral hydride complex. A parallel DFT study, carried out with inclusion of [MeO(MeOH)5]− as a model of the strong base, revealed that the most stable species, i.e. likely to be the catalyst resting state, is not a neutral complex, but rather the [IrH4(P,SR)]− ion.44 This complex does not contain the mobile proton needed for a classical outer-sphere (Noyori-type)45 mechanism, nor a vacant coordination site for ketone coordination/insertion. Instead, the catalytic action of this complex was rationalized via a new type of ionic mechanism, involving hydride transfer to the counterion-activated ketone to generate alkoxide, followed by ionic H2 activation and alkoxide protonation. The calculated Gibbs energy span of 18.2 kcal mol−1 is in good agreement with the experimental activity.44
Anionic tetrahydride IrIII derivatives have previously been obtained by Morris et al. from the reaction of KH with [IrH5L2] (L = PPh3, PiPr3) in THF (eqn (1)).46,47 However, the above-mentioned DFT study suggested that the [IrH4(P,SR)]− complexes should be quantitatively generated by H2 addition to [IrHL2(iPrOH)] through a sequence of deprotonation, H2 oxidative addition and iPrOH reductive elimination steps (eqn (2)). Less intuitively, the same complexes were suggested by the DFT study to form favourably in the absence of free H2, with the H equivalents needed being provided by isopropanol under transfer hydrogenation conditions (eqn (3)). Open questions, to be addressed in this contribution, are whether COD is removed by transfer hydrogenation and whether an anionic tetrahydridoiridium(III) complex is formed under catalytically relevant conditions, as predicted.
| [IrH5L2] + KH → K+[IrH4L2]− + H2 | (1) |
| [IrHL2(iPrOH)] + iPrO− + 2H2 → [IrH4L2]− + 2iPrOH | (2) |
| [IrHL2(iPrOH)] + iPrO− + 2iPrOH → [IrH4L2]− + 4MeCOMe | (3) |
The extreme sensitivity of the [IrH4L2]− complex with L2 = P,SR has not allowed us to generate and isolate, or spectroscopically detect, this putative anionic tetrahydride complex from [IrCl(COD)]2/(P,SR)/NaOMe via the processes shown in eqn (2) or (3). We could demonstrate that the COD ligand in [IrCl(COD)]2 is indeed hydrogenated to cyclooctene and cyclooctane by isopropanol under transfer hydrogenation conditions (reflux in isopropanol) in the presence of KOtBu and PPh3.48 However, the metallic complex in the final solution was not Morris’ [IrH4(PPh3)2]− but rather a mixture of isomeric fac- and mer-[IrH3(PPh3)3].
In the present contribution, we present a detailed study of metal-complex activation and speciation using the strongly chelating ligand dppe as L2, in order to probe species that would otherwise be too reactive when L2 = P,SR. Successful generation of a tetrahydrido IrIII species, according to the strategy of eqn (3), was indeed observed. We also present a new and thorough NMR investigation of the previously reported [IrH(COD)(dppe)] (two isomers, 2 and 3) which forms by the low-temperature action of the strong base on [IrCl(COD)(dppe)] (1), as well as the generation and characterization of two new hydride compounds (two isomers of [IrH(C8H10)(dppe)], 4 and 5), which result from the deprotonation of COD by the strong base in a non-protic solvent.
Experimental
General
All reactions and purifications were carried out under nitrogen using high-vacuum or Schlenk-line techniques. The [IrCl(COD)]2 and [IrCl(COD)(dppe)] precursors used in the experiments were synthesised according to published procedures.49NMR instrumentation
NMR spectra were recorded using the Bruker Ascend 500 MHz instrument available in the Centre for Hyperpolarization in Magnetic Resonance at York. NMR measurements were carried out using NMR tubes fitted with J. Young's valves. Deuterated solvents were dried using sodium metal. All samples were degassed on a high-vacuum line and the solutions were prepared in a glovebox by the addition of deuterated solvent (0.6 mL) to the solid complexes (∼10 mg). Quantitative data for kinetic plots were obtained from the NMR spectra using inverse gated decoupling methods.Synthesis of [IrH(1,2,5,6-η2:η2-COD)(dppe)] (2) and [IrH(1-κ-4,5,6-η3-C8H12)(dppe)] (3)
KOtBu (77 mg, 0.69 mmol) was added to an isopropanol (10 mL) solution of 1 (100 mg, 0.14 mmol) and stirred at room temperature for 1 h, generating a cream-coloured solution. The solution was concentrated to dryness, the solid residue was dissolved in toluene (8 mL), and the resulting mixture was passed through an alumina lined pipette. The resulting solution was then concentrated in vacuo. Yield = 98 mg (0.14 mmol, 99%). This reaction can also be performed using an equivalent amount of NaOMe. ESI-HRMS (m/z relative intensity %): [M − H]+ calcd: 699.1930; found: 699.1933 (Fig. S1†). The 1H NMR analysis of the product reveals that it contains a mixture of isomers 2 and 3 (see data in Table S3†).Synthesis of [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] (4 and 5)
Compound 1 (102 mg, 0.14 mmol) and KOtBu (71 mg, 0.63 mmol) were added to toluene (15 mL) and the solution was stirred at room temperature for 1 h, yielding a deep-orange solution. The solvent was then removed in vacuo. The orange solid was dissolved in toluene (5 mL) and the resulting mixture was passed through an alumina-lined pipette. The resulting solution was concentrated to dryness. Yield = 40 mg (0.06 mmol, 43%). 1H NMR analysis of the product reveals that it contains a mixture of the isomers 4 and 5 (see data in Table S4†).In situ synthesis of K[IrH4(dppe)]
Compound 1 (10 mg, 0.01 mmol), and KOtBu (7 mg, 0.06 mmol), were added to a solution of isopropanol (0.01 mL) and toluene-d8 (0.6 mL). The solution was heated to 353 K and NMR spectra were recorded (see Results and discussion). The formation of K[IrH4(dppe)] is complete within 30 minutes of heating the mixture. Several attempts were made to isolate the complex as a solid to no avail.Computational details
The computational work undertaken for this study was carried out using the Gaussian09 suite of programs.50 Geometry optimizations were carried out without any symmetry constraints using the B3LYP functional51–53 combined with Grimme's D3 correction to account for dispersion effects.54 Optimizations were performed in toluene (ε = 2.3741) using the SMD continuum model55 with basis set 1 (BS1). BS1 includes the 6-31G(d,p) basis set for the main-group atoms,56,57 and the scalar relativistic Stuttgart-Dresden SDD effective core potential (ECP) and its associated double-ζ basis set,58 complemented with a set of f polarization functions,59 for Ir. Frequency calculations were performed for all the optimized geometries in order to characterise the stationary points as either minima or transition states. Connections between the transition states and the corresponding minima were checked by displacing in both directions, following the transition vector, the geometry of the transition states, and subsequent geometry optimization until a minimum was reached. Energies in toluene were refined by means of single-point calculations at the optimised BS1 geometries using an extended basis set (BS2). BS2 consists of the def2-TZVP basis set for the main-group atoms, and the quadruple-ζ def2-QZVP basis set for Ir, together with the def2 ECP (60 core electrons: [Kr] + 4d + 4f).60 Gibbs energies in toluene were calculated by adding to the BS2 energies in toluene the thermal and entropic corrections obtained with BS1. An additional correction of 1.9 kcal mol−1 was applied to all Gibbs energies to change the standard state from the gas phase (1 atm) to the condensed phase (1 M) at 298.15K (ΔG1 atm→1 M).61 In this way, all the energy values given in the article are Gibbs energies in toluene solution calculated using the formula:| G = E(BS2) + G(BS1) − E(BS1) + ΔG1 atm→1 M |
Results and discussion
The interaction between [IrCl(COD)]2 and dppe, at various P/Ir ratios, in isopropanol solution was investigated sequentially under three different conditions. First, the ligand was added to the complex at room temperature in the absence of a base. Then, a strong base was added at room temperature. Finally, the resulting solution was heated to reflux.[IrCl(COD)]2-dppe interaction. Formation of [IrCl(COD)(dppe)] (1)
The addition of dppe to [IrCl(COD)]2 at room temperature, leading to the characterization of a 1:1 adduct, [IrCl(COD)(dppe)], 1, has already been described in the literature.49 Single crystals of 1, suitable for X-ray diffraction studies, were obtained and the previously unreported crystal structure is reported here (Fig. 1 and Table S1†). The coordination geometry is square pyramidal, as expected, and is analogous to that of other similar compounds, e.g. [IrCl(COD)L2] with L2 = Ph2P(CH2)nPPh2 (n = 3, 4).49
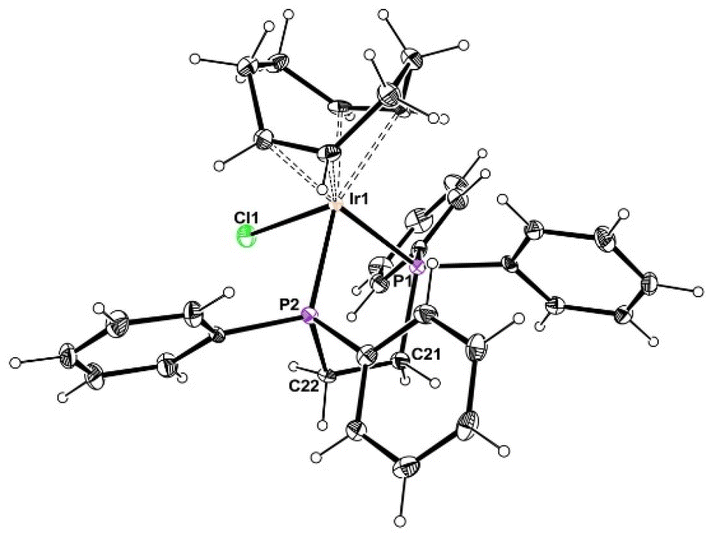 | ||
| Fig. 1 A view of the molecular structure of compound 1. Relevant metric data: Ir1–Cl1; 2.5317(8); Ir1–P1, 2.3117(9); Ir1–P2, 2.3072(9); Ir1–C1, 2.211(4); Ir1–C2, 2.177(3); Ir1–C5, 2.147(4); Ir1–C6, 2.142(4) Å; Cl1–Ir1–P1, 90.64(3), Cl1–Ir2–P2, 83.93(3); P1–Ir1–P2, 81.84(3)°. | ||
We have now studied the interaction at variable dppe/Ir ratios (Fig. 2). The addition of <1 equiv. of dppe per Ir atom yields, in addition to the expected [IrCl(COD)(dppe)] (31P{1H} resonance at δ 33.4 in C6D6), a second product characterized by a resonance at δ 19.5. This second resonance disappears upon addition of further dppe to readjust the dppe/Ir stoichiometry to 1:1. On the basis of the known behaviour for several other bidentate ligands, for instance α-diimines,62 bis- and tris-pyrazoles,63,64 phosphine-thioethers,42 and also for one diphosphine,65 this is assigned to the salt [Ir(dppe)(COD)]+[IrCl2(COD)]−. Addition of more dppe beyond the 1:1 ratio maintained the sharp 31P{1H} resonance of [IrCl(COD)(dppe)], but also generated an additional resonance at δ 50.0 (in C6D6), which we attribute to [IrCl(dppe)2] on the basis of the follow-up reaction in the presence of base (vide infra). Compounds of analogous stoichiometry have been described for the reaction between [IrCl(COD)]2 and excess of other bidentate ligands such as (C6F5)2PCH2CH2P(C6F5)266 and a bis(phosphole).67
 | ||
| Fig. 2 31P{1H} spectra of solutions obtained by addition of dppe to [IrCl(COD)]2 in indicated ratio. Solvent = C6D6. | ||
Compound 1 and base at r.t. in isopropanol. Formation of the hydride complexes 2 and 3
The addition of excess base (5 equiv.) at room temperature to a C6D6 solution spiked with a small amount of isopropanol and with dppe/Ir slightly greater than 1 produced a mixture of thee hydride-dppe compounds (see Fig. S2† for the hydride region of the 1H spectrum and Fig. S3† for the 31P{1H} spectrum, respectively), all of them already reported in the literature. Two of them correspond to a [IrH(COD)(dppe)] stoichiometry and can be assigned the structures [IrH(η2:η2-1,5-C8H12)(dppe)], 2,68 which is formally IrI and [IrH(1-κ-4,5,6-η3-C8H12)(dppe)], 3, which is formally IrIII.69 The stoichiometry of the reaction leading to the formation of these products is shown in eqn (4). The isopropoxide anion is produced by deprotonation of isopropanol by tert-butoxide, the stronger base. | (4) |
The third reaction product, observed in minor amounts, corresponds to compound [IrH(dppe)2].70 It is characterized by a quintet hydride resonance at δ −12.64 (JHP = 8.7 Hz), coupled to a 31P resonance at δ 36.2. This product is presumably derived directly from the [IrCl(dppe)2] that is generated when the dppe/Ir ratio exceeds 1:1 stoichiometry (vide supra). Indeed, it was not formed when the reaction was carried out using a dppe/Ir ratio of slightly less than 1:1. To avoid the formation of this by-product, a toluene solution of 1 was passed through an alumina-lined pipette to remove the bis-dppe species. Crystallization attempts without prior removal of [IrCl(dppe)2] led to single crystals of [IrH(dppe)2] (Table S2†), the structure of which was previously published.71
The generation of compounds 2 and 3 under conditions similar to ours, as well as that of other related Ir complexes with diphosphinomethane (dppm), diphosphinopropane (dppp), diphosphinobutane (dppb), o-C6H4(PPh2)2 and PPh3 in place of dppe, has previously been described.48,68,69,72 Some confusion concerning the nature and spectral assignments for these two isomeric products needs clarification (see Table S3†). In the first report,68 Oro et al. obtained [IrH(COD)(dppe)] from the reaction of [Ir(μ-OMe)(COD)]2 with dppe in MeOH and described it as an IrI complex containing a regularly η2:η2-bonded 1,5-COD ligand, 2, only on the basis of the 1H NMR properties in CDCl3. The compound was mentioned as being unstable in this solvent. We have found a similar instability in CD2Cl2. The same group later reported that the same reaction carried out with dppm yields instead an IrIII product with an isomerized COD ligand, [IrH(1-κ-4,5,6-η3-C8H12)(dppm)], as confirmed by a single crystal X-ray diffraction analysis.72 Subsequently, Farnetti et al. revisited the same reaction with dppe and found that it produces the same type of rearranged product as Oro's dppm compound, i.e. compound 3.69 The NMR properties of this product are different than those reported for 2 by Oro et al. and the observation of 2 was not mentioned. In addition, Farnetti et al. have shown that the rearrangement is disfavoured for diphosphine ligand with a longer backbone chain, Ph2P(CH2)nPPh2 (n = 3, 4): the dppp ligand (n = 3) gave both regular IrI and isomerized IrIII products in an 8:1 ratio and the dppb (n = 4) ligand gave only the regular IrI product, which partially isomerized upon warming.
We found that the addition of excess KOtBu (5 equiv.) to 1 in iPrOH produced both IrI and IrIII products, presumably via the same type of intermediate as the [Ir(μ-OMe)(COD)]2/dppe reaction, namely [Ir(OiPr)(COD)(dppe)], in a 2/3 ratio of 5:95 at equilibrium at 298 K. The NMR spectral parameters in C6D6, where the two products appeared perfectly stable, correspond to those reported for the two isomers respectively by Oro et al. and by Farnetti et al., although a few of their assignments have been revised (see discussion in the ESI following Table S3†) on the basis of detailed multiple resonance and HMQC (H–C, H–P, C–P) experiments (see ESI, Fig. S4 through S10†), and DFT calculations (data also reported in Table S3†).
Compound 2 could, however, be selectively generated by an alternative strategy, namely by the low-temperature addition of L-selectride to 1, as confirmed by the 1H NMR spectrum collected immediately after mixing in THF-d8 at 263 K (single triplet hydride resonance at δ −14.20 with JPH = 21.6 Hz). After 10 minutes, a second hydride peak for the isomer 3 (dd at δ −11.55 with JPH = 14.7, 20.6 Hz) was observed. Increasing the temperature to 298 K resulted in further isomerization to 3 until a stable equilibrium was reached, where the resonances of 2 remained visible. The isomerization process could be monitored by 1H NMR during in situ generation from 1 and NaOMe in THF-d8 at constant temperature (298 K), with resonance integration, see Fig. 3. Kinetic analysis as a sequential process with irreversible first-order transformation of 1 to 2 followed by first-order reversible transformation of 2 to 3, as detailed in the ESI,† yielded k12 = (1.11 ± 0.02) × 10−4 s−1; k23 = (3.4 ± 0.2) × 10−4 s−1 and k32 = (1.1 ± 0.3) × 10−5 s−1 (K23 = k23/k32 = 32 ± 10). The decay of compound 1 could also be treated independently as a clean first order process (Fig. S11†), yielding k12 = (1.13 ± 0.03) × 10−4 s−1, in agreement with the value obtained from the global fit.
 | ||
| Fig. 3 Time evolution of the 31P{1H} data for 1 decay and 2–3 isomerization. The continuous lines are the result of the non-linear least-squares fit as described in the ESI.† | ||
A DFT investigation was carried out, without any structural simplification (see Computational details), to establish the mechanism of the isomerization process. The results are summarized in Fig. 4. The investigation started with the optimization of the two isomers 2 and 3, finding greater stability for 3 by 3.5 kcal mol−1 relative to 2. This is in good agreement with the experimental equilibrium, which returns a Gibbs energy difference of 2.06 ± 0.16 kcal mol−1 in favour of 3 by application of the van ‘t Hoff equation. The optimized structure of 2 is trigonal bipyramidal with the hydride and one of the COD double bonds occupying the axial positions, whereas that of 3 is best described as derived from an octahedron, with a fac arrangement of the hydride and the two P donors and with the 1-κ-C donor trans to one of the two P donors, rendering the two P nuclei inequivalent, which agrees with the NMR data. This ligand arrangement is identical to that observed by X-ray diffraction for the analogous dppm derivative.72 An alternative structural isomer for 3 with the 1-κ-C donor trans to the hydride, which would be symmetrical with magnetically equivalent P nuclei, is located 6.8 kcal mol−1 higher in Gibbs energy. It is probably disfavoured by the placement of two strong σ-donors (the hydride and the 1-κ-C atom) trans to each other.
 | ||
| Fig. 4 Gibbs energy profile (in kcal mol−1) for the isomerization of 2 to 3. | ||
A reasonable working hypothesis for this isomerization consists of a two-step process, proceeding via the 16-electron 1-κ-4,5-η2-cyclooctenyl intermediate, [Ir(1-κ-4,5-η2-C8H13)(dppe)] (I), of Scheme 1. DFT calculations could optimize the geometry of this intermediate, which is square-planar as expected, and of the two transition states relating I to 2 (TS2-I) and to 3 (TSI-3), see Fig. 4. The Ir–H distance lengthens and the C–H distance to the appropriate C atom shortens going from 2 and from 3 to the transition state leading to I. The highest free-energy transition state is TSI-3, located at 19.8 kcal mol−1 from 2 (23.3 kcal mol−1 from 3). This value agrees well with the experimentally determined barrier (ΔG‡2-3 = 22.19 ± 0.04 kcal mol−1 by application of the Eyring equation). Thus, in kinetic terms, the 2/I step is a pre-equilibrium, preceding the rate-determining conversion of I to 3. The Gibbs energy of I is computed as 1.3 kcal mol−1 higher than that of 2, which is consistent with the fact that it is not observed by either 1H or 31P{1H} NMR during the isomerization monitoring.
 | ||
| Scheme 1 Reaction coordinate for the isomerization of 2 to 3 (dppe omitted for clarity). | ||
Reaction between compound 1 and KOtBu at room temperature in toluene. Formation of the hydride complexes 4 and 5
When KOtBu was added to 1 in C6D6 or THF-d8 in the absence of isopropanol, a rapid reaction takes place. The 1H NMR spectrum in THF-d8 at 263 K recorded immediately after addition of the base showed the generation of two new isomeric hydride complexes (4 and 5) in an 86:14 ratio, subsequently evolving to an equilibrium position where 5 slightly predominates (32:68), see Fig. S12.† The NMR properties of these two compounds are summarized in Table S4.† Resonances for complexes 2 and 3 described above were not observed in this experiment, see Fig. S13–S15.†
The hydride resonance of 4 is a doublet of doublets at δ −11.43 (JPH = 14.1, 21.8 Hz), correlating with two resonances at δ 30.6 (d, JPP = 2.4 Hz), and δ 46.7 (d, JPP = 2.4 Hz) in the 31P{1H} NMR spectrum. Two broad peaks at δ 5.94 and δ 5.47 appear for an unbound olefin moiety, confirmed by connections to two 13C signals at δ 146.42 and δ 129.90, respectively (Fig. S15†).
Three additional resonances in the alkene region of the 1H NMR spectrum suggest a cyclooctatriene ligand coordinating through η3 and κ1 interactions, [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] (4), as confirmed by NOE interactions between the CH2 fragments and the alkene CH groups (see Fig. S18†). Hence, the reaction consists of the elimination of HCl from 1via the initial deprotonation of one COD C–H bond, chloride dissociation and transfer of a second COD H atom to the metal centre as a hydride ligand (Scheme 2). The σ CH donor is located trans to one of the P donor atoms and the κ3 moiety is trans to the second phosphine and the hydride, with the uncoordinated double bond placed in the equatorial plane.
 | ||
| Scheme 2 Direct deprotonation of the COD on 1 occurs in the absence of isopropanol. | ||
Compound 5 is characterized by a hydride resonance at δ −11.57 (t, JPH = 19.5 Hz), which is coupled to two 31P resonances at δ 32.4 (d, JPP = 2.7 Hz) and 39.4 (d, JPP = 2.7 Hz). It is an isomer of 4 with the same coordination geometry, [IrH(1-κ-4,5,6-η3-C8H10)(dppe)], except that the saturated ethylene bridge and uncoordinated alkene have exchanged position. This isomer exhibits resonances for an unbound alkene at δ 5.61 and δ 5.52 with connections to 13C signals at δ 127.5 and δ 148.1 (Fig. S15†). The 1-κ-4,5,6-η3 binding mode for a C8H10 ligand was also previously demonstrated for the structurally characterized complex [IrH(1-κ-4,5,6-η3-C8H10)(dppb)],73 which is homologous with complex 4.
The process depicted in Scheme 2 was also analysed kinetically in C6D6 at 298 K. Unlike the production and isomerization of 2/3 shown in Fig. 3, the consumption of 1 was a rapid first step, allowing clean timescale separation of the two steps and analysis of the second step as a standard reversible first-order reaction (Fig. 5), yielding k45 = (2.75 ± 0.06) × 10−4 s−1 and k54 = (1.16 ± 0.08) × 10−4 s−1; K45 = k45/k54 = 2.37 ± 0.17. A separate kinetic monitoring in toluene-d8 (Fig. S19†) gave very similar rate and equilibrium parameters, with k45 = (3.92 ± 0.11) × 10−4 s−1, k5-4 = (1.39 ± 0.12) × 10−4 s−1 and K45 = k45/k54 = 2.83 ± 0.26.
 | ||
| Fig. 5 First-order kinetics of the 4–5 isomerization in C6D6 at 298 K. | ||
The identity and relative energy of the two [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] isomers 4 and 5 was supported by DFT calculations, which could also rationalize the selective formation of 4 by deprotonation, and the mechanism of its conversion to 5. As illustrated in Scheme 3, the deprotonation of 1 is expected to produce, after chloride elimination, a neutral 16-electron [Ir(C8H11)(dppe)] intermediate. Four possibilities may be envisaged, two corresponding to a [Ir(1-κ-5,6-η2-C8H11)(dppe)] structure (II and II′) and two corresponding to a [Ir(η3-C8H11)(dppe)] structure (II′′ and II′′′). Each pair of structures corresponds to the dissociation of a different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C donor function in the COD ligand of 1 and the two conformers in each pair differ in the orientation of the dppe ligand (‘flip’ or ‘flop’ conformation of the IrP2C2 5-membered ring) relative to the C8H11 ligand. The lowest energy was found for conformer II, with II′ being located only 0.9 kcal mol−1 above. The η3 isomer III′ is located at 3.7 kcal mol−1 and III′′′ was not explored. These intermediates may then proceed to a hydride-C8H10 product by β-H elimination.
C donor function in the COD ligand of 1 and the two conformers in each pair differ in the orientation of the dppe ligand (‘flip’ or ‘flop’ conformation of the IrP2C2 5-membered ring) relative to the C8H11 ligand. The lowest energy was found for conformer II, with II′ being located only 0.9 kcal mol−1 above. The η3 isomer III′ is located at 3.7 kcal mol−1 and III′′′ was not explored. These intermediates may then proceed to a hydride-C8H10 product by β-H elimination.
 | ||
| Scheme 3 Reaction scheme for the generation of 4. The phenyl groups on the dppe are omitted for clarity. (V: 5.8; VI: 4.9; VII; 9.7). | ||
Starting from II or II′, the hydride may originate from carbons 4, 5 and 8 to yield in principle three different regioisomers, each one in a flip or flop conformation (Scheme 3). Two of them (unobserved), generated by H transfer from C4 and C5, have two separate methylene groups either between unsaturated C2 and C4 moieties (1,3,6-COT; III and III′) or between two unsaturated C3 moieties (1-κ-5,6,7-η3-C8H10; IV and IV′). The configurations produced by H transfer from C8 (4 and 4′) match the structure observed by NMR. Exploration of all three pathways for II (Fig. 6) and II′ (Fig. S20†) showed that the β-H8 eliminations are associated with the lowest-G transition states (22.5 kcal mol−1 for TSII-4, 20.8 kcal mol−1 for TSII-4′) and yield the most stable products. The other pathways involve higher-energy transition states and yield higher energy hydride complexes, justifying their non-observation. Among the two conformers 4 and 4′, the former is more stable (−1.8 kcal mol−1 relative to II) while the latter (at 1.5 kcal mol−1) is accessed from the higher-energy II′ through a lower barrier. We presume that the barrier for the flip-flop conformational change is even lower, thus the two conformers are at rapid equilibrium on the NMR timescale and only one set of signals is observed.
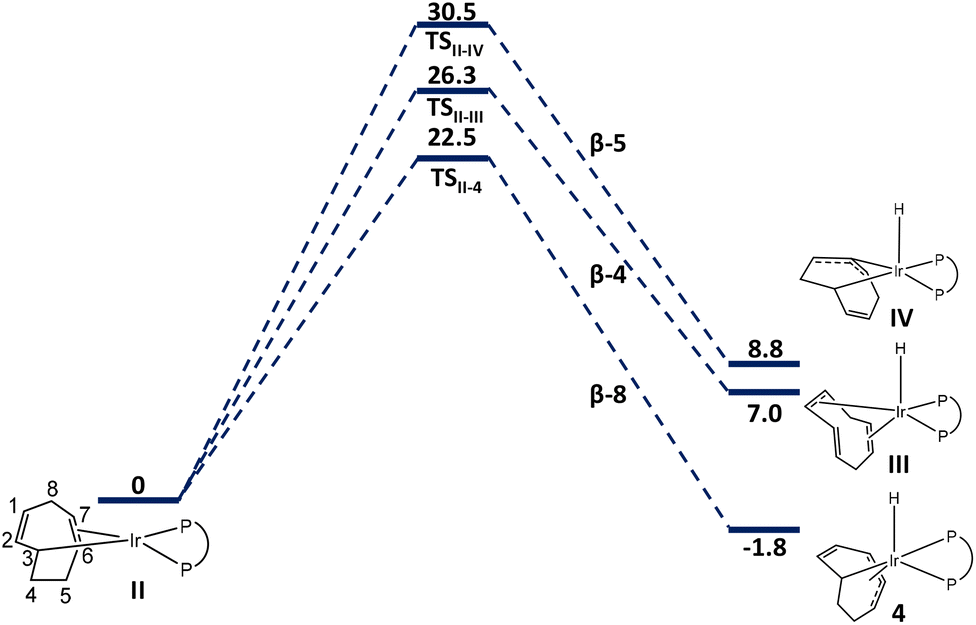 | ||
| Fig. 6 Gibbs-energy profile (in kcal mol−1) for all possible β-H elimination reactions from intermediate II. | ||
The β-H elimination process from II′′ and II′′′ may in principle occur from carbons 4, 7 and 8. The β-elimination of H4 may lead again, upon rearrangement of the C4–C5–C6 electron density, to 4 and 4′, or directly to the new isomers V and V′, in which the COT ligand binds as a conjugated η4 diene. The elimination of H7 leads to VI and VI′, in which the ligand is bonded to the Ir atom as in IV and IV′ but is oriented differently with respect to the H and dppe ligands. Finally, the elimination of H8 leads to another two configurations (VII and VII′) with a 1,3,6-COT ligand bonded as a conjugated η4 diene., like III and III′ but in a different orientation. The geometries of V, VI and VII were optimized and found again at higher energies (5.8, 4.9 and 9.7 kcal mol−1, respectively) with respect to II. Therefore, these processes were not further explored.
Note how the lowest-energy product obtained from II (4) features the uncoordinated alkene moiety syn relative to the hydride ligand. No direct pathway is available to transform intermediate II to isomer 5, for which, on the other hand, the uncoordinated alkene moiety is located anti relative to the hydride ligand. Note also that the higher G value of intermediate II relative to 4 agrees with its non-observation.
The isomerization of 4 to 5 then proceeds via the rearrangement schematically illustrated in Fig. 7. This entails a rotation of the C8H10 ligand relative to the Ir–H axis viaTS4-5′ with simultaneous displacement toward the Ir atom of the two CH groups of the uncoordinated alkene, which become part of the coordinated η3 moiety, and the inverse displacement of the two CH groups of the coordinated η3 moiety that end up forming the uncoordinated alkene moiety. This process, which is rate-determining, has a calculated barrier of 24.0 kcal mol−1 from 4, quite close to that obtained from the measured rate constant k4-5 by application of the Eyring relationship (22.3 ± 0.1 kcal mol−1). The product of this rearrangement, however, is a higher-energy flip-flop conformer (5′) of the most stable geometry for 5. The conformational flip-flop rearrangement can probably occur through a small barrier. We could also find, however, an alternative pathway, via the electronic rearrangement of the C8H10 ligand, as illustrated in Fig. 7. The transition state for this process is located at a relatively low G (10.3 kcal mol−1). Therefore, 5′ converts to the more stable structure 5 very rapidly and the two conformers are expected to be in rapid equilibrium on the NMR timescale at room temperature. The calculated energy difference between 5 and 4 (−0.5 kcal mol−1) agrees with the experimental value of −0.51 ± 0.04 kcal mol−1 in benzene (or −0.62 ± 0.06 kcal mol−1 in toluene).
 | ||
| Fig. 7 Gibbs-energy profile (in kcal mol−1) for the isomerisation of 4 to 5 by electronic rearrangement of the (1-κ-4,5,6-η3-C6H10) ligand. | ||
Of particular interest is the fact that the ligand prefers to adopt a 1-κ-4,5,6-η3-C8H10 geometry with an uncoordinated C2–C3 double bond in both 4 and 5, hence yielding two formally IrIII isomers, rather than an 18-electron 1,3,5-COT structure resulting in a formally IrI 18-electron complex (the optimized higher-energy V). There are literature precedents for the 1,2,5,6-η2,η2 binding mode of COT in iridium chemistry, for instance [IrCl(COT)]2 and CpIr(COT),74 although none has been structurally characterized to the best of our knowledge. Structures featuring this type of COT coordination are, however, available for CoI,75 Mo0,76 and Ru0.77–79
Concerning the 1-κ-4,5,6-η3 binding mode, as already mentioned above, this is documented for the homologous complex [IrH(1-κ-4,5,6-η-C8H10)(dppb)],73 which was obtained by addition of dppb to [Ir(NH2)(COD)]2. However, the presence of more than one isomer was not mentioned in that contribution. That reaction is closely related to ours, because a bridging amido ligand serves as an internal base to deprotonate the COD ligand with elimination of ammonia.73 Thus, the action of a strong base on the COD ligand in compound 1, in the absence of more acidic reagents such as isopropanol that can themselves be deprotonated, coordinate to the metal and then deliver a hydride ligand by β-H elimination (eqn (4)), induces a COD deprotonation with the stoichiometry depicted in eqn (5) after chloride loss and β-H elimination. In this respect, it is also relevant to note the work of Kubiak et al.,80 where the treatment of [Ir(COD)(triphos)]+Cl− (triphos = bis(diphenylphosphinoethyl)phenylphosphine) with a variety of bases gave the [Ir(1-κ-5,6-η-C8H11)(triphos)] product. Here, the tridentate nature of triphos stops the β-H elimination process after deprotonation.
 | (5) |
Compound 1 and KOtBu in warm isopropanol. Formation of [IrH4(dppe)]−
Upon heating the solution of the equilibrium mixture of 2 and 3, either after isolation or in situ generation from 1, in the presence of isopropanol and KOtBu in C7D8 at 353 K, the 31P{1H} resonances of both compounds decrease in intensity while that of the [IrH4(dppe)]− complex (6) increases. After 30 minutes the reaction is complete, with a single peak for 6 being observed (Fig. S21†). This complex is characterized by two distinct hydride resonances in a 1:1 ratio at δ −12.20 and δ −13.60 in the 1H NMR spectrum (Fig. 8) and a singlet at δ 44.2 in the 31P{1H} NMR spectrum, coupled to both hydride resonances (Fig. S22†), while the 13C NMR spectrum (Fig. S23†) only shows the dppe ligand resonances. The equatorial hydrides (Ha) are equivalent, but differ in the way they are coupled to a specific P atom (cis or trans). Therefore, an AA′M2XX′ spin system is observed. The coupling pattern, established by selective decoupling experiments, is summarized in Fig. 9 and Table S5.† For Ha we observe four independent coupling patterns (2JHH′, 2JHHb, 2JHPcis, 2JHPtrans). The HaHb coupling constant can be directly observed from the 1H{31P} NMR spectrum. However, the full set of coupling constants had to be elucidated using NMR simulation tools (gNMR v5.0). The large splitting observed for Ha in Fig. 8 is a result of HPcis ± HPtrans. The Hb resonance, expected as a triplet of triplets, is observed as a septet, but selective decoupling from 31P yields only leaves the residual 2JHH coupling, observed as a binomial triplet.
 | ||
| Fig. 8 Representative 1H spectra in the hydride region for a solution of complex 6 in C7D8 at 298 K. (a) Regular 1H spectrum. (b) 1H{31P} spectrum. (c) Ha resonance with selective decoupling from Hb. | ||
 | ||
| Fig. 9 Schematic representation of the H–H and H–P coupling pattern in [IrH4(dppe)]−. Coupling constants of Ha and Hb are shown in red and blue, respectively. | ||
Complex 6 could also be generated by warming the equilibrated solution of 4 and 5 in the presence of KOtBu and isopropanol. In both cases, the liberation of free cyclooctene, via partial hydrogenation of COD or COT, was revealed by 1H NMR. It should be noted that the addition of H2 to solutions of 2/3 or 4/5 in C6D6 (or THF-d8), without isopropanol, also produces 6 within 18 hours at room temperature, but only in the presence of KOtBu because H2 addition to a neutral hydride species cannot lead to an anionic product unless a proton is removed by a base. To further investigate the role of the base in forming 6, H2 (3 bar) was added to a solution of 1 in THF-d8. Without base, both the 1H (hydride region) and 31P{1H} NMR spectra revealed a very complex pattern indicating the formation of several products, among which 2 and 3 could be identified, but not 6. Equally complex mixtures resulted from the treatment of a THF-d8 solution of pre-formed 2/3 (Fig. S24†) and 4/5 (Fig. S25†) with H2 (3 bar) in the absence of base. The complete removal of the COD ligand with partial hydrogenation was indicated by the 1H spectrum. The subsequent addition of KtOBu to these complex mixtures led to the selective production of 6. When, on the other hand, KtOBu (5 equiv.) was added first, both the 2/3 and 4/5 mixtures were selectively transformed to 6 (Fig. S26 and S27,† respectively). These collective results establish the thermodynamic feasibility of the formation of the anionic tetrahydride complex [IrH4(dppe)]− from 1 under both hydrogenation and transfer hydrogenation (from isopropanol) conditions in the presence of a strong base, according to the stepwise process and stoichiometry shown in Scheme 4.
 | ||
| Scheme 4 Idealized stoichiometry (further cyclooctene hydrogenation may also occur) for the [IrH4(dppe)]− generation from [IrCl(dppe)(COD)] under both hydrogenation and transfer hydrogenation conditions in isopropanol. | ||
Conclusions
We have experimentally demonstrated a route towards the formation of an iridium tetrahydrido complex 6 ([IrH4(dppe)]−) from 1 [IrCl(COD)(dppe)], with elimination of cycloctene under both hydrogenation and transfer hydrogenation (from isopropanol) conditions. This complex is formed in the presence of a strong base, with the cation of the base providing a suitable counterion to the tetrahydride 6. The use of a diphosphine ligand allowed the reaction mechanism to be explored in full due to the strong chelating effect. This has so far not been explored with the ferrocenyl P,S-ligand systems43 because of the higher reactivity and thus higher sensitivity. This is consistent with the computational study on ketone hydrogenation with ‘non-N–H’ ligands,44 suggesting that an anionic tetrahydride species is the active catalyst.Under ambient conditions in the absence of an alcohol in the solvent, the type of alkoxide has a marked effect on 1. When a β-hydrogen is present, the COD ligand maintains its chemical composition and two isomers of [IrH(C8H12)(dppe)], 2 and 3, are produced. When a base with no β-hydrogen is used, on the other hand, deprotonation of the COD fragment occurs with subsequent β-H elimination from the C8H11 fragment to yield two isomers of [IrH(C8H10)(dppe)], 4 and 5. We have fully characterized each product using NMR spectroscopy, while DFT calculations, in combination with the experimental kinetics of the generation and isomerization of the various products, explain the detailed mechanism of formation of each neutral hydride species and their interconversions.
The results of this work provide an insight into the catalytic action of [IrCl(COD)(LL)] complexes in the hydrogenation of polar substrates in the presence of a base.
Author contributions
PK; investigation, writing – original draft. EM; supervision; writing – review and editing. AL; formal analysis; writing – review and editing. ACW; investigation (X-ray diffraction). JML; supervision; writing – review and editing. JS; supervision; writing – review and editing. SBD; formal analysis; supervision; writing – review and editing. RP; conceptualization; funding acquisition; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 860322, and was granted access to the resources of the CICT (Centre Interuniversitaire de Calcul de Toulouse, project CALMIP). We are also grateful to the CNRS (Centre National de la Recherche Scientifique) for additional funding and to S. M. Wahidur Rahaman for a few initial investigations. JML is funded through a Royal Society Industrial Research fellowship.References
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed.
- J. H. Xie, S. F. Zhu and Q. L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- H.-U. Blaser, B. Pugin and F. Spindler, in Organometallics as Catalysts in the Fine Chemical Industry, ed. M. Beller and H. U. Blaser, 2012, vol. 42, pp. 65–102 Search PubMed.
- C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS.
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- J. L. Wen, F. Y. Wang and X. M. Zhang, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051–1069 CrossRef CAS.
- M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045–2061 CrossRef CAS.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- C. Wang, X. F. Wu and J. L. Xiao, Chem. – Asian J., 2008, 3, 1750–1770 CrossRef CAS PubMed.
- B. Stefane and F. Pozgan, Catal. Rev.: Sci. Eng., 2014, 56, 82–174 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- B. Stefane and F. Pozgan, Top. Curr. Chem., 2016, 374, 18 CrossRef PubMed.
- S. J. Roseblade and A. Pfaltz, C. R. Chim., 2007, 10, 178–187 CrossRef CAS.
- X. F. Wu, X. H. Li, A. Zanotti-Gerosa, A. Pettman, J. K. Liu, A. J. Mills and J. L. Xiao, Chem. – Eur. J., 2008, 14, 2209–2222 CrossRef CAS PubMed.
- J. K. Liu, X. F. Wu, J. A. Iggo and J. L. Xiao, Coord. Chem. Rev., 2008, 252, 782–809 CrossRef CAS.
- C. Bianchini, L. Gonsalvi and M. Peruzzini, in Iridium Complexes in Organic Synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH, Weinheim, 2009, pp. 55–106 Search PubMed.
- R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS.
- D. H. Woodmansee and A. Pfalz, in Iridium Catalysis, ed. P. G. Andersson, Springer, Berlin, 2011, vol. 34, pp. 31–76 Search PubMed.
- A. Cadu and P. G. Andersson, J. Organomet. Chem., 2012, 714, 3–11 CrossRef CAS.
- K. H. Hopmann and A. Bayer, Coord. Chem. Rev., 2014, 268, 59–82 CrossRef CAS.
- C. X. Cui, H. H. Chen, S. J. Li, T. Zhang, L. B. Qu and Y. Lan, Coord. Chem. Rev., 2020, 412, 213251 CrossRef CAS.
- A. Fabrello, A. Bachelier, M. Urrutigoity and P. Kalck, Coord. Chem. Rev., 2010, 254, 273–287 CrossRef CAS.
- A. Comas-Vives, G. Ujaque and A. Lledós, Adv. Inorg. Chem., 2010, 62, 231–260 CrossRef CAS.
- R. Poli, Adv. Organomet. Chem. Search PubMed , in press.
- P. Kvintovics, J. Bakos and B. Heil, J. Mol. Catal., 1985, 32, 111–114 CrossRef CAS.
- R. Spogliarich, J. Kaspar, M. Graziani and F. Morandini, J. Organomet. Chem., 1986, 306, 407–412 CrossRef CAS.
- G. Zassinovich and G. Mestroni, J. Mol. Catal., 1987, 42, 81–90 CrossRef CAS.
- M. Bernard, V. Guiral, F. Delbecq, F. Fache, P. Sautet and M. Lemaire, J. Am. Chem. Soc., 1998, 120, 1441–1446 CrossRef CAS.
- D. G. I. Petra, P. C. J. Kamer, A. L. Spek, H. E. Schoemaker and P. W. N. M. Van Leeuwen, J. Org. Chem., 2000, 65, 3010–3017 CrossRef CAS PubMed.
- V. Guiral, F. Delbecq and P. Sautet, Organometallics, 2000, 19, 1589–1598 CrossRef CAS.
- V. Guiral, F. Delbecq and P. Sautet, Organometallics, 2001, 20, 2207–2214 CrossRef CAS.
- F. Delbecq, V. Guiral and P. Sautet, Eur. J. Org. Chem., 2003, 2092–2097 CrossRef CAS.
- J. W. Handgraaf, J. N. H. Reek and E. J. Meijer, Organometallics, 2003, 22, 3150–3157 CrossRef CAS.
- M. V. Jimenez, J. Fernandez-Tornos, J. J. Perez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. J. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef CAS.
- S. A. Popoola, E. A. Jaseer, A. A. Al-Saadi, V. Polo, M. A. Casado and L. A. Oro, Inorg. Chim. Acta, 2015, 436, 146–151 CrossRef CAS.
- M. V. Jimenez, J. Fernandez-Tornos, J. J. Perez-Torrente, F. J. Modrego, P. Garcia-Orduna and L. A. Oro, Organometallics, 2015, 34, 926–940 CrossRef CAS.
- N. Garcia, E. A. Jaseer, J. Munarriz, P. J. Sanz Miguel, V. Polo, M. Iglesias and L. A. Oro, Eur. J. Inorg. Chem., 2015, 4388–4395, DOI:10.1002/ejic.201500853.
- A. Iturmendi, N. Garcia, E. A. Jaseer, J. Munarriz, P. J. S. Miguel, V. Polo, M. Iglesias and L. A. Oro, Dalton Trans., 2016, 45, 12835–12845 RSC.
- R. Malacea, J.-C. Daran, S. B. Duckett, J. P. Dunne, C. Godard, E. Manoury, R. Poli and A. C. Whitwood, Dalton Trans., 2006, 3350–3359 RSC.
- R. Malacea, E. Manoury, L. Routaboul, J.-C. Daran, R. Poli, J. P. Dunne, A. C. Withwood, C. Godard and S. B. Duckett, Eur. J. Inorg. Chem., 2006, 1803–1816 CrossRef CAS.
- E. Le Roux, R. Malacea, E. Manoury, R. Poli, L. Gonsalvi and M. Peruzzini, Adv. Synth. Catal., 2007, 349, 309–313 CrossRef CAS.
- J. M. Hayes, E. Deydier, G. Ujaque, A. Lledós, R. Malacea-Kabbara, E. Manoury, S. Vincendeau and R. Poli, ACS Catal., 2015, 5, 4368–4376 CrossRef CAS.
- C. A. Sandoval, T. Ohkuma, K. Muniz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS PubMed.
- K. Abdur-Rashid, D. G. Gusev, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 1998, 120, 11826–11827 CrossRef CAS.
- S. E. Landau, K. E. Groh, A. J. Lough and R. H. Morris, Inorg. Chem., 2002, 41, 2995–3007 CrossRef CAS PubMed.
- S. M. W. Rahaman, J.-C. Daran, E. Manoury and R. Poli, J. Organomet. Chem., 2017, 829, 14–21 CrossRef CAS.
- T. Makino, Y. Yamamoto and K. Itoh, Organometallics, 2004, 23, 1730–1737 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Hoellwarth, V. Jonas, K. F. Koehler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
- M. Bikrani, L. Fidalgo and M. A. Garralda, Polyhedron, 1996, 15, 83–89 CrossRef CAS.
- L. D. Field, B. A. Messerle, M. Rehr, L. P. Soler and T. W. Hambley, Organometallics, 2003, 22, 2387–2395 CrossRef CAS.
- M. A. Esteruelas, L. A. Oro, M. C. Apreda, C. Foces-Foces, F. H. Cano, R. M. Claramunt, C. Lopez, J. Elguero and M. Begtrup, J. Organomet. Chem., 1988, 344, 93–108 CrossRef CAS.
- C. Laborde, M.-M. Wei, A. van der Lee, E. Deydier, J.-C. Daran, J.-N. Volle, R. Poli, J.-L. Pirat, E. Manoury and D. Virieux, Dalton Trans., 2015, 44, 12539–12545 RSC.
- R. C. Schnabel and D. M. Roddick, Inorg. Chem., 1993, 32, 1513–1518 CrossRef CAS.
- O. Tissot, M. Gouygou, F. Dallemer, J. Daran and G. Balavoine, Eur. J. Inorg. Chem., 2001, 2385–2389 CrossRef CAS.
- M. J. Fernandez, M. A. Esteruelas, M. Covarrubias and L. A. Oro, J. Organomet. Chem., 1986, 316, 343–349 CrossRef CAS.
- M. Fabbian, N. Marsich and E. Farnetti, Inorg. Chim. Acta, 2004, 357, 2881–2888 CrossRef CAS.
- P. Meakin, J. P. Jesson and E. L. Muetterties, J. Am. Chem. Soc., 1972, 94, 5271–5285 CrossRef CAS.
- B. K. Teo, A. P. Ginsberg and J. C. Calabrese, J. Am. Chem. Soc., 1976, 98, 3027–3028 CrossRef CAS.
- M. A. Esteruelas, M. Oliván, L. A. Oro, M. Schulz, E. Sola and H. Werner, Organometallics, 1992, 11, 3659–3664 CrossRef CAS.
- I. Mena, P. Garcia-Orduna, V. Polo, F. J. Lahoz, M. A. Casado and L. A. Oro, Dalton Trans., 2017, 46, 11459–11468 RSC.
- J. Evans, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1972, 2668 RSC.
- R. P. Aggarwal, N. G. Connelly, B. J. Dunne, M. Gilbert and A. G. Orpen, J. Chem. Soc., Dalton Trans., 1991, 1–9, 10.1039/dt9910000001.
- F. J. Liotta, G. Vanduyne and B. K. Carpenter, Organometallics, 1987, 6, 1010–1023 CrossRef CAS.
- T. Suzuki, M. Shiotsuki, K. Wada, T. Kondo and T. A. Mitsudo, J. Chem. Soc., Dalton Trans., 1999, 4231–4237, 10.1039/a907333g.
- M. Shiotsuki, H. Miyai, Y. Ura, T. Suzuki, T. Kondo and T. Mitsudo, Organometallics, 2002, 21, 4960–4964 CrossRef CAS.
- T. Suzuki, M. Shiotsuki, K. Wada, T. Kondo and T. Mitsudo, Organometallics, 1999, 18, 3671–3678 CrossRef CAS.
- A. M. Gull, P. E. Fanwick and C. P. Kubiak, Organometallics, 1993, 12, 2121–2125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2226722 and 2226723. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04036k |
| This journal is © The Royal Society of Chemistry 2023 |