
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation of steric influence on the reactivity of FeV(O)(OH) tautomers in stereospecific C–H hydroxylation†
Mainak
Mitra
 ab,
Alexander
Brinkmeier
a,
Yong
Li
a,
Margarida
Borrell
c,
Arnau
Call
c,
Julio
Lloret Fillol
d,
Michael G.
Richmond
e,
Miquel
Costas
*c and
Ebbe
Nordlander
*a
ab,
Alexander
Brinkmeier
a,
Yong
Li
a,
Margarida
Borrell
c,
Arnau
Call
c,
Julio
Lloret Fillol
d,
Michael G.
Richmond
e,
Miquel
Costas
*c and
Ebbe
Nordlander
*a
aChemical Physics, Department of Chemistry, Lund University, Box 124, SE-221 00, Lund, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se
bDepartment of Chemistry, Burdwan Raj College, Aftab Avenue, W.B. 713104, India
cQBIS-CAT, Department of Chemistry and Institut de Quimica Computacional i Catàlisi, University of Girona, Campus Montilivi, E-17071 Girona, Spain. E-mail: miquel.costas@udg.edu
dInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avinguda Paisos Catalans 16, 43007, Tarragona, Spain
eDepartment of Chemistry, University of North Texas, Denton, Texas 76203, USA
First published on 5th January 2023
Abstract
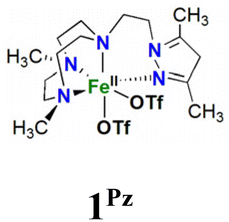
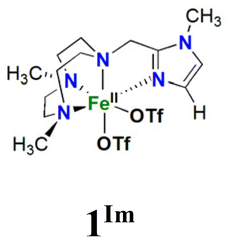
Two new tetradentate N4 ligands (LN4), LN4 = Me2,Me2PyzTACN (1-(2-(3,5-dimethyl-1H-pyrazol-1-yl)ethyl)-4,7-dimethyl-1,4,7-triazacyclononane) and Me2,MeImTACN (1-((1-methyl-1H-imidazol-1-yl)methyl)-4,7-dimethyl-1,4,7-triazacyclononane) have been synthesized and their corresponding Fe(II) complexes [FeII(Me2,Me2PyzTACN)(CF3SO3)2], 1Pz, and [FeII(Me2,MeImTACN)(CF3SO3)2], 1Im, have been prepared and characterized. Complexes 1Pz and 1Im catalyse the hydroxylation of C–H bonds of alkanes with excellent efficiencies, using hydrogen peroxide as oxidant. The high H/D kinetic isotope effect values for C–H hydroxylation, large normalized tertiary/secondary C–H (C3/C2) bond selectivities in adamantane oxidation, and high degrees of stereoretention in the oxidation of cis-1,2-dimethylcyclohexane are indicative of metal-based oxidation processes. The complexes also catalyse the oxidation of cyclooctene to form its corresponding epoxide and syn-diol. For 1Pz the epoxide is the main product, while for the analogous complex 1Im the syn-diol predominates. The active oxidant is proposed to be an [(LN4)FeV(O)(OH)]2+ species (2Pz, LN4 = Me2,Me2PyzTACN and 2Im, LN4 = Me2,MeImTACN) which may exist in two tautomeric forms related by a proton shift between the oxo and hydroxo ligands. Isotope labelling experiments show that the oxygen atom in the hydroxylated products originates from both water and hydrogen peroxide, and labelling experiments involving oxygen atom transfer to sterically bulky substrates provide indirect information on the steric influence exerted by the two ligands in the relative reactivities of the two hypervalent iron tautomers. Based on these labelling studies, the steric influence exerted by each of the ligands towards the relative reactivity of the oxo ligands of the corresponding pair of Fe(V)(O)(OH) tautomers can be derived. Furthermore, this steric influence can be gauged relative to related complexes/ligands.
Introduction
Over the last two decades, a number of transition metal-based complexes have been developed as catalysts for selective oxidations of hydrocarbons.1,2 Iron is often a metal of choice for such catalytic transformations because it is inexpensive, non-toxic and highly abundant.3,4 In nature, oxygenases carry out a wide range of organic substrate oxidations, and many of them contain iron in their active sites.5–8 These iron oxygenases are usually highly selective towards specific C–H bonds and utilize atmospheric oxygen as the ultimate oxidant.6 An example is the Rieske oxygenase family of non-heme iron enzymes, which contains an Fe(II) center bound to two histidine residues and one aspartate in the active site.8 This family of bacterial enzymes catalyzes the cis-dihydroxylation of arenes via activation of dioxygen (Scheme 1) as the first step in the biodegradation of pollutants.6 Although the identity of the active species remains a matter of debate, one of the mechanistic proposals favours a formally high valent Fe(V) center bound to an oxo and a hydroxo groups that are cis to each other (Scheme 1).5,6,9 These species hydroxylate aliphatic C–H bonds and syn-dihydroxylate alkenes and arenes. The mechanism of C–H hydroxylation is proposed to consist of a two-step process that is directly related to the canonical “oxygen rebound” mechanism of the cytochrome P450 family of heme oxygenases; this mechanism involves (i) abstraction of a hydrogen atom from a substrate C–H bond by the oxo group to form a transient substrate-based alkyl radical and an Fe(IV)(OH)2 moiety, and (ii) interaction of the alkyl radical with one of the hydroxyl groups of the Fe(IV)(OH)2 moiety to form the product.10 The reaction with alkenes is envisioned to occur via a 3 + 2 reaction of the Fe(V)(O)(OH) moiety with the olefin site, akin to syn-dihydroxylations performed by OsO4, RuO4 or MnO4−. With the reactivities of non-heme iron oxygenases such as Rieske oxygenases serving as an inspiration, several mononuclear non-heme iron catalysts have been developed to address the challenging oxidations of poorly reactive C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.11–15
C bonds.11–15
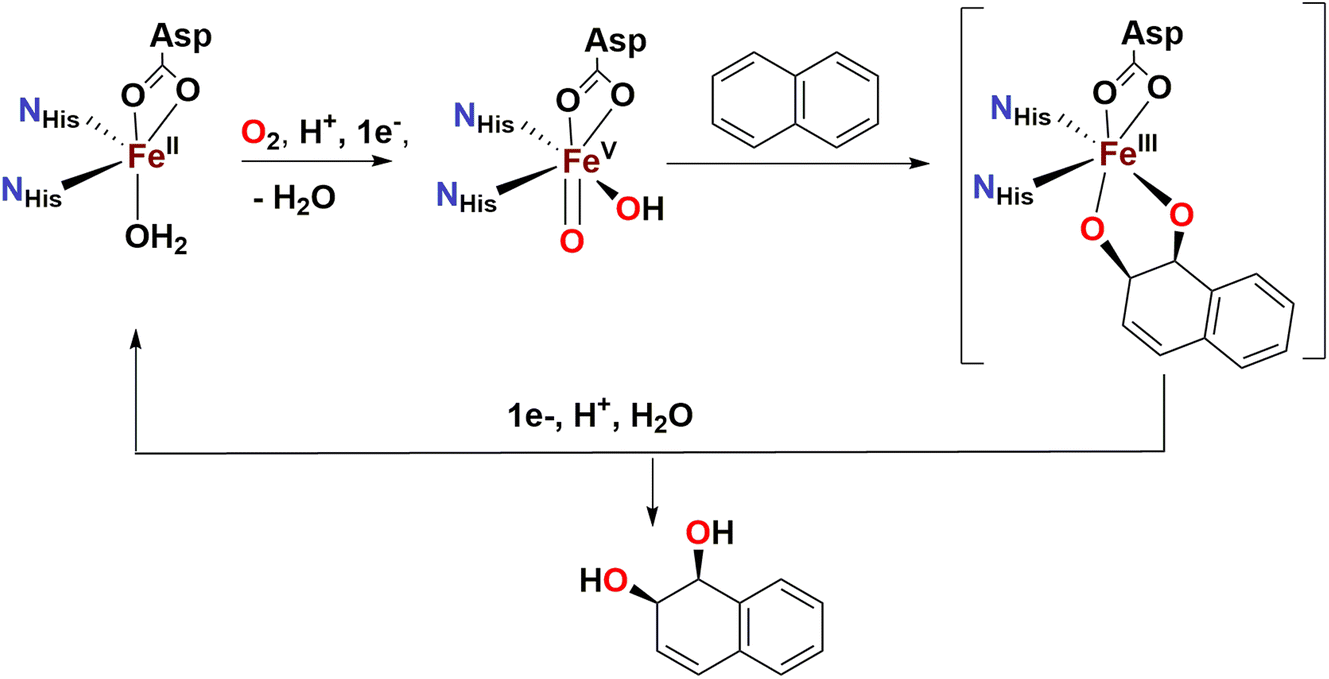 | ||
| Scheme 1 Proposed reaction mechanism for cis-dihydroxylation by naphthalene dioxygenase. | ||
The Fe(PyTACN) family of complexes (PyTACN = 1-(2-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane) has been shown to mediate stereospecific C–H hydroxylation with excellent efficiencies.16–18 Isotope labelling and theoretical studies strongly indicate that the oxidations occur via the involvement of high valent Fe(V) oxo intermediates.16–18 The reactive intermediate in C–H hydroxylation reactions has been identified as a highly electrophilic [FeV(O)(OH)(LN4)]2+ oxidant (LN4 corresponds to tetradentate TACN-based ligands), which is formed via water-assisted O–O cleavage of the hydroperoxo precursor [FeIII(OOH)(OH2)(LN4)]2+.19 The [FeV(O)(OH)(LN4)]2+ oxidant can exist in two tautomeric forms, corresponding to structures OA and OB in Scheme 2, because of the unsymmetric nature of the tetradentate TACN-derivatives that serve as ligands.18 The two tautomers differ in the relative positions of the cis-coordinated oxo and hydroxo groups, and are connected through a prototopic oxo-hydroxo tautomerism. Investigations have been made on the influence on C–H hydroxylation reactions exerted by manipulating the electronic and steric properties via introduction of different groups (e.g. Me, F, NO2, NMe2) in α and γ positions of the pyridine ring of the PyTACN ligand.18,20 These studies indicated that electronic properties of the groups in γ position on the pyridine ring have minor influence on the relative reactivities between the two tautomers OA and OB while, instead, the steric properties of the groups in the α position of the pyridine ring do.18,20 The discrimination between the relative reactivities of OA and OB was found to be more pronounced in cases of substrates containing tertiary C–H bonds.18 Replacement of the pyridyl arm of the PyTACN ligand with a corresponding (N-methyl)benzimidazolyl arm led to the formation of the complex [FeII(Me2,MeBzImTACN)(CF3SO3)2], 1BzIm.21 Reactivity (catalytic oxidation) studies on this complex were entirely in keeping with the proposed formation of an active Fe(V)(O)(OH) oxidant, and it was found that the (N-methyl)benzimidazolyl moiety exerts an effect in the relative reactivities of OA and OB that lies in between that of pyridyl and α-Me-pyridyl moieties. This effect parallels the steric demand of the heterocyclic ligand, as deduced from an inspection of the crystal structures of the corresponding ferrous complexes. Importantly, 1BzIm was found to maintain high efficiencies and selectivities in the hydroxylation of alkanes and olefins.21
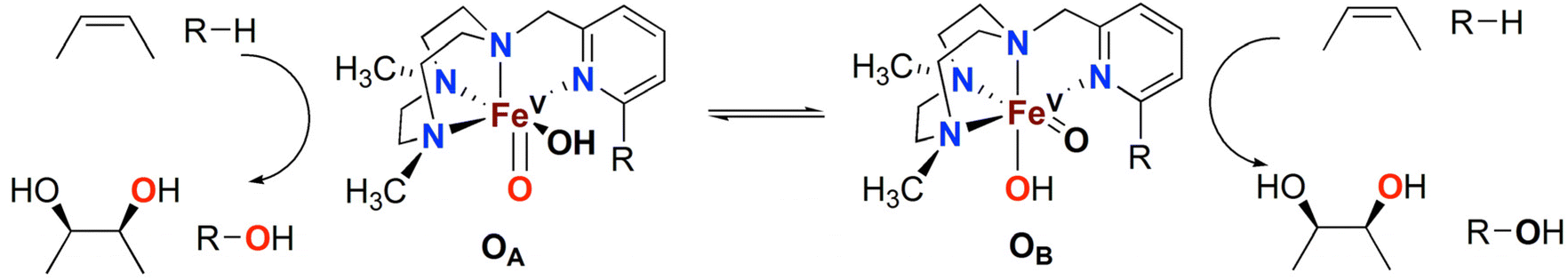 | ||
| Scheme 2 Oxidation of alkanes (R–H) and olefins by the two tautomers, OA and OB, observed in reactions catalysed by the Fe(PyTACN) family of complexes.18,20,21 | ||
In this work, we describe [FeII(LN4)(CF3SO3)2] complexes bearing two new tetradentate N4 ligands, Me2,Me2PyzTACN and Me2,MeImTACN (Fig. 1). These ligands are based on the PyTACN ligand scaffold, where the pyridyl side arm is replaced by (N-methyl)imidazolyl and 3,5-dimethylpyrazolyl arms, respectively. The ligand Me2,Me2PyzTACN contains an ethylene spacer connecting one of the amines of the triazacyclononane (TACN) ring and the N(2) atom of the 3,5-dimethylpyrazole, whereas the corresponding spacer is a methylene in Me2,MeImTACN, in direct correspondence to the ligand frameworks of BzImTACN and PyTACN. The (N-methyl)imidazolyl moiety (pKa of conjugate acid: 7.06) is more basic than (N-methyl)benzimidazolyl (pKa of conjugate acid: 5.41) and pyridine (pKa of conjugate acid: 5.22), while 3,5-dimethylpyrazole is less basic (pKa of conjugate acid: 4.12). Collectively, the different ligands discussed above are thus expected to span a range of steric and electronic demands, while yielding structurally similar Fe(II) complexes that in turn may give rise to analogous Fe(V)(O)(OH) complexes that will exist in tautomeric forms (cf.Scheme 2). Thus, the ligands are expected to exert not only electronic but also steric influence on the relative reactivities of the Fe(V)(O)(OH) OA and OB tautomers operating in the C–H hydroxylation reactions. An investigation of the catalytic hydroxylation and olefin oxidation reactions effected by these two iron complexes is described herein. Isotope labelling studies and computational methods have also been performed to elucidate the reaction mechanism(s) and to assess the steric and electronic influence of the ligands on the reactivities of the iron catalysts.
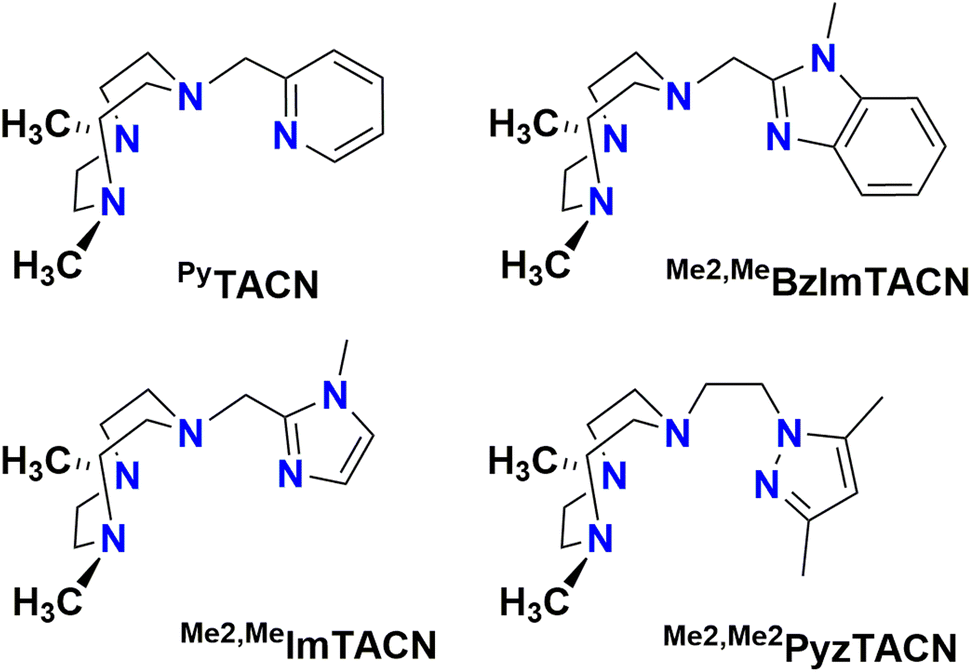 | ||
| Fig. 1 Structures of the tetradentate ligands PyTACN, Me2,MeBzImTACN, Me2,MeImTACN and Me2,Me2PyzTACN discussed in this work. | ||
Results and discussion
Syntheses and characterization
The two new tetradentate N4 ligands Me2,Me2PyzTACN and Me2,MeImTACN were synthesized by reaction of TACN·3HBr with the corresponding chloromethylene/chloroethylene precursor complex of the five membered heterocyclic ring (pyrazole and imidazole) as described in Scheme 3 (cf. Experimental section and ESI† for detailed characterization).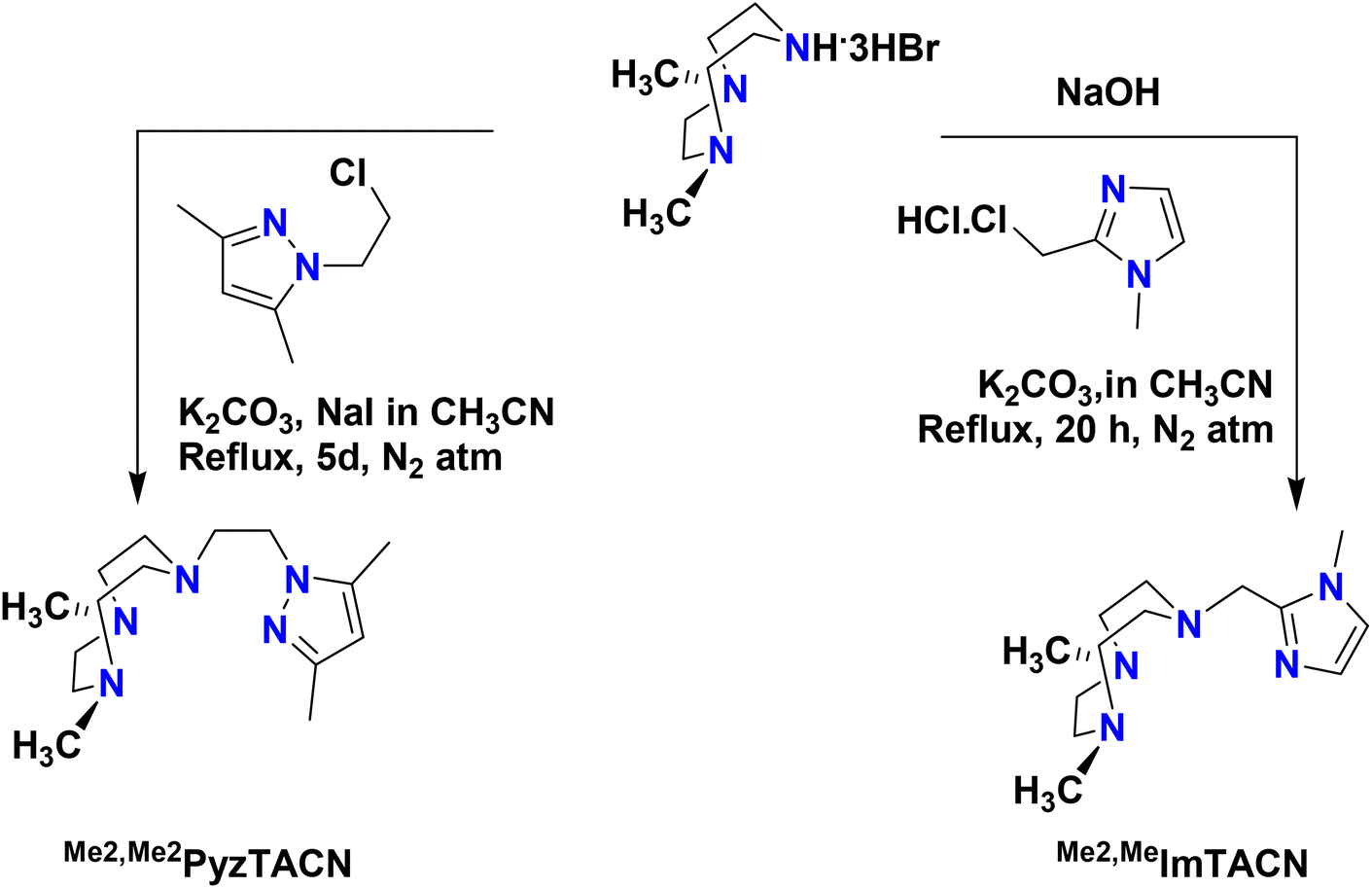 | ||
| Scheme 3 Schematic synthetic routes to the ligands Me2,Me2PyzTACN and Me2,MeImTACN. The free base 1,4-dimethyl-1,4,7-triazacyclononane was used for the synthesis of Me2,Me2PyzTACN. | ||
The corresponding Fe(II) complexes were synthesized inside a dry atmosphere box. Reaction of one equiv. of [FeII(CH3CN)2(CF3SO3)2] with one equiv. of Me2,Me2PyzTACN in dry THF resulted in precipitation of the corresponding complex [FeII(Me2,Me2PyzTACN)(CF3SO3)2] (1Pz), which was collected by filtration, washed with a small amount of THF and dried under vacuum (cf. Experimental section for a detailed description of the synthesis). The corresponding reaction with the Me2,MeImTACN ligand in dry THF led to the formation of a complex that mostly remained in solution. The reaction solution was evaporated under vacuum and a small amount of a CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (4:1) mixture was added to the resultant residue. Diffusion of diethyl ether into the resultant solution resulted in precipitation of the complex [FeII(Me2,MeImTACN)(CF3SO3)2] (1Im, Scheme 4).
:CH3CN (4:1) mixture was added to the resultant residue. Diffusion of diethyl ether into the resultant solution resulted in precipitation of the complex [FeII(Me2,MeImTACN)(CF3SO3)2] (1Im, Scheme 4).
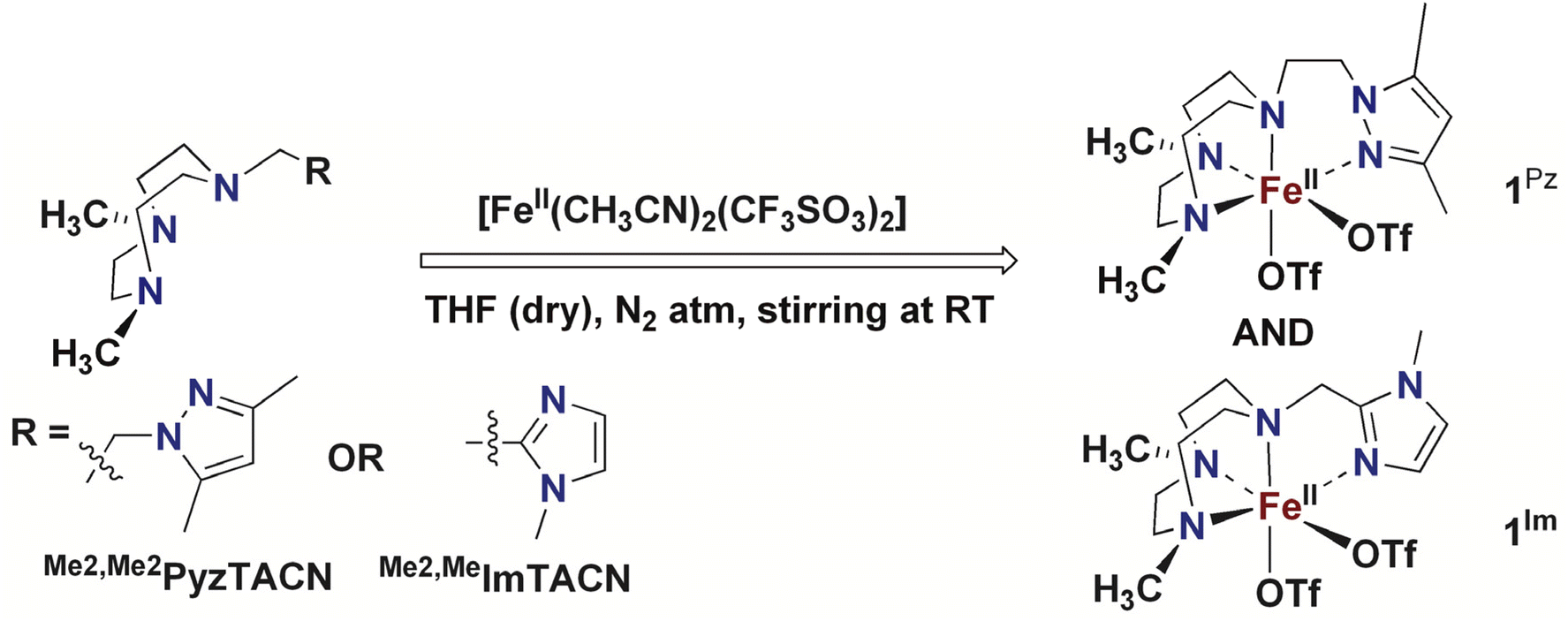 | ||
| Scheme 4 Synthesis of Fe(II) complexes 1Pz and 1im. | ||
The complexes 1Pz and 1Im were characterized by mass spectrometry and 1H NMR spectroscopy. The high resolution mass spectrum (HRMS) of 1Pz in CH3CN showed prominent mass peaks at m/z = 167.5903 and 484.1303, corresponding to the formulations [FeII(Me2,Me2PyzTACN)]2+ (calc. 167.5881) and [FeII(Me2,Me2PyzTACN)(CF3SO3)]+ (calc. 484.1287), respectively (Fig. S1–S3, ESI†). Analogously, the HRMS of complex 1Im in CH3CN showed prominent mass peaks at m/z = 153.5751 and 456.0970, corresponding to the formulations [FeII(Me2,MeImTACN)]2+ (calc. 153.5724) and [FeII(Me2,MeImTACN)(CF3SO3)]+ (calc. 456.0974), respectively (Fig. S4–S6†). The 1H NMR spectra of the two complexes were measured in CD3CN solvent (cf. Fig. S7 and S8†). Both complexes gave broad paramagnetically shifted spectra in spectral windows ranging from −10 to 120 ppm, indicative of the presence of high spin ferrous ions.
In order to confirm its identity, the solid-state structure of 1Pz was established by X-ray crystallography. The details of the structure determination are collated in Table S1 (ESI†). The X-ray structure (Fig. 2) shows that the Fe(II) center is in a slightly distorted octahedral coordination geometry. The ligand Me2,Me2PyzTACN is tetradentate with the three nitrogens of the triazacyclononane ring bound facially to the metal center and the pyrazole ring providing a fourth coordinated nitrogen. The two triflate anions are coordinated in cis positions on the Fe(II) ion. The Fe–N bond distances lie in the range 2.2–2.25 Å, which are typical distances observed for high spin Fe(II) complexes.22–26 The Fe–OOTf bond distances are 2.088(3) and 2.199(3) Å. In order to ensure that coordination of the pyrazole nitrogen would be geometrically possible, the Me2,Me2PyzTACN ligand was designed to contain an ethylene spacer between the TACN ring and the pyrazole moiety, distinguishing this ligand from the related ligands discussed here, all of which contain methylene spacers in the corresponding positions. The Fe–N (pyrazole) bond distance is 2.209(3) Å while the Fe–N(pyridine) bond distance is 2.165(4) Å in [FeII(Me,HPyTACN)(CF3SO3)2] (1Py)16 and 2.246(2) Å in [FeII(Me,MePyTACN)(CF3SO3)2] (1MePy),27 and the Fe–N (benzimidazole) bond distance is 2.134(7) Å in [FeII(Me2,MeBzImTACN)(CF3SO3)2] (1BzIm).21 The OTfO–Fe–OOTf angle is larger in 1Pz (93.6(1)°) than that of 3OTf (91.64(14)°)16 and 1BzIm (89.1(3)°),21 but shorter than that of 1MePy (94.9(7)°),27 implying different steric environments around the Fe(II) center. It should be noted that one of the methyl groups of the pyrazole moiety lies in the same plane as the oxygen of one triflate anion (O4) and thus also in a plane roughly perpendicular to the iron-oxygen axis of the second triflate anion (cf.Fig. 2).
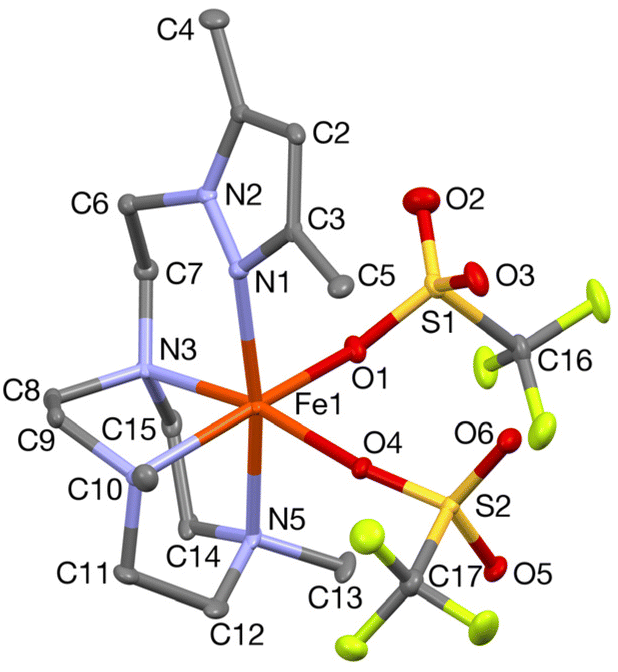 | ||
| Fig. 2 A Mercury plot of the crystal structure of complex [FeII(Me2,Me2PyzTACN)(CF3SO3)2] (1Pz) with 50% probability ellipsoids. The hydrogens have been omitted for clarity. Selected bond distances (Å) and bond angles (°): Fe(1)–N(1) 2.209(3), Fe(1)–O(1) 2.088(3), Fe(1)–N(3) 2.227(4), Fe(1)–O(4) 2.199(3), Fe(1)–N(4) 2.208(4), Fe(1)–N(5) 2.249(3), N(1)–Fe(1)–N(5) 171.0(1), O(1)–Fe(1)–N(4) 166.7(1), N(3)–Fe(1)–O(4) 169.1(1). | ||
Catalytic C–H bond oxidation studies
The catalytic properties of 1Pz and 1Im were tested in the hydroxylation of several alkanes employing H2O2 as the oxidant. In a typical catalytic experiment, 10 equiv. of H2O2 (from a stock solution of 70 mM H2O2 in CH3CN) were delivered by a syringe pump to a CH3CN solution containing the Fe(II) complex (1 mM), alkane substrate (1 M) and H2O (1 M) over a period of 30 min. The catalysis experiments were performed under air at room temperature. Under these conditions, both complexes oxidized cyclohexane into cyclohexanol (1Pz: TON 6.5; 1Im: TON 7.7) as the main product with high alcohol/ketone (A/K) ratios (1Pz: A/K 9.4; 1Im: 12) and high conversions of the oxidant (H2O2) into products (1Pz: 72%; 1Im: 83%). Such high conversions and high A/K ratios are comparable to those obtained with related iron complexes under analogous experimental conditions.16,21,25–31 The catalytic efficiencies of these two complexes with different alkane substrates are summarized in Table 1.| Catalyst | Substrate | TN (A) | TN (K) | A/K | Yield (%) |
|---|---|---|---|---|---|
| 1Pz | Cyclohexane | 6.5 | 0.7 | 9.3 | 72 |
| 1Im | Cyclohexane | 7.7 | 0.64 | 12 | 83 |
| 1Pz | Cyclohexane-d12 | 4 | 0.5 | 8 | 45 |
| 1Im | Cyclohexane-d12 | 5 | 0.5 | 10 | 55 |
| 1Pz | Cyclooctane | 6 | 0.6 | 10 | 66 |
| 1Im | Cyclooctane | 6.2 | 0.6 | 10.3 | 69 |
| 1Pzf | n-Hexane | 3 | 0.8 | 3.8 | 38 |
| 1Im | n-Hexane | 3.6 | 0.64 | 5.6 | 43 |
| 1Pz | 2,3-Dimethylbutane | 2.6 | — | — | 26 |
| 1Im | 2,3-Dimethylbutane | 3.5 | — | — | 35 |
The high A/K ratios observed for these complexes suggest the involvement of a metal-based oxidant, as proposed previously.32 Several other mechanistic substrate probes were used to verify the involvement of a metal-based active oxidant. The kinetic isotope effect (KIE) values obtained in the oxidation of a mixture of cyclohexane and its deuterated isotopologue (1:3 molar ratio) were 4.0 for 1Pz and 4.6 for 1Im. Both of the complexes showed a preference for oxidation of tertiary C–H bonds over secondary C–H bonds in the oxidation of adamantane (normalized 3°/2° ratios were 12 for 1Pz and 26 for complex 1Im). Finally, in the oxidation of cis-1,2-dimethylcyclohexane (cis-DMCH), the complexes provided the corresponding tertiary alcohol with a high degree of stereoretention (RC 83% for 1Pz and 100% for 1Im), excluding any significant involvement of long-lived carbon-centered radicals or cations in the C–H oxidation reactions.
Isotope labelling studies
Isotope labelling experiments were performed using H218O2 and H218O to gain insight into the hydroxylation mechanism by determining the origin of the oxygen atom incorporated into the (alcohol) products. In the oxidation of cyclohexane (1000 equiv.), using 10 equiv. H2O2 in the presence of 1000 equiv. of H218O (Table 2), 1Pz (1 equiv.) was found to introduce only <3% labelled oxygen into the alcohol product. The complementary experiment performed with reverse labelling, i.e. 10 equiv. H218O2 and 1000 equiv. H2O resulted in an 18O-label incorporation of 86% into the alcohol. It may thus be concluded that peroxide is the main source of oxygen in the alcohol product and only 11% of oxygen atoms are incorporated from air/water (according to the oxygen mass balance) for this specific hydroxylation reaction. In the oxidation of other alkane substrates containing secondary C–H bonds, complex 1Pz with 10 equiv. H2O2 and 1000 equiv. of H218O gave rise to similar low percentages of incorporated 18O (from water) into the alcohol (3% for cyclohexane-d12 and cyclooctane).| Substrate |
|
|
|
|
|
|---|---|---|---|---|---|
|
a Reaction conditions: catalyst:H2O2:H218O:substrate = 1:10:1000:1000, CH3CN, RT, air.
b
cis-Cyclooctene was employed as the substrate.
c Reaction performed under N2.
d Catalyst:H2O2:H2O:substrate = 1:10:1000:1000, CH3CN, RT, under 18O2.
e ref. 16–18,20.
f ref. 21.
|
|||||
| Cyclohexane | 3 | 39 | 45 | 11 | 48 |
| 1c | 36c | ||||
| 11d | 3d | ||||
| Cyclohexane-d12 | 3 | NA | 40 | NA | 48 |
| Cyclooctane | 3 | 43 | 44 | NA | 41 |
| cis-DMCH | 0 | 31 | 79 | 2 | 26 |
| Adamantane | 0 | 39 | 74 | 3 | 28 |
| cis-Cycloocteneb epoxide | 2 | 36 | 77 | 5 | 24 |
| 11d | 36c | ||||
| 8d | |||||
| syn-Cyclooctane-1,2-diolb | 97 | 92 | 97 | 78 | 98 |
| 4d | 91c | ||||
| 3d | |||||
On the other hand, 1Im gave rise to considerably greater incorporation of oxygen from water into the products. In the presence of 10 equiv. H2O2 and 1000 equiv. of H218O, complex 1Im (1 equiv.) gave 39 and 43% 18O-alcohols in the oxidation of cyclohexane and cyclooctane, respectively (Table 2). The complementary experiment with 10 equiv. H218O2 and 1000 equiv. H2O resulted in the formation of 55% 18O-cyclohexanol in oxidation of cyclohexane. Thus, for this complex, both peroxide and H2O are the sources of oxygen in the alcohol products. Again, the lack of significant incorporation of oxygen atoms from air rules out the transient formation of long-lived carbon-centered radicals.
In the oxidation of tertiary C–H bonds (cis-DMCH and adamantane), 1Pz did not incorporate any oxygen from water into the products (cf.Table 2). On the other hand, the equivalent experiments with 1Im led to 39% and 31% oxygen incorporations from water into product(s) in the oxidation of adamantane and cis-DMCH, respectively (cf.Table 2). For comparison, results from oxidation with structurally related catalysts are included in Table 2.
cis-Dihydroxylation vs. epoxidation in the oxidation of alkenes
Complexes 1Pz and 1Im catalyzed the oxidation of alkenes in the presence of H2O2 and H2O to give both epoxide and cis-dihydroxylated products. Under catalytic conditions (1:10:1000 for catalyst:oxidant:substrate), 1Pz oxidized cis-cyclooctene to give cis-cyclooctene epoxide (TON 8.2) as a major product with the concomitant formation of a minor amount of syn-cyclooctane-1,2-diol (TON 0.5), i.e. an epoxide:diol (E/D) ratio of 16.5. Addition of H2O (1000 equiv.) slightly increased the formation of syn-diol (TON 1.1), but had almost no influence in the yield of epoxide (TON 8.2). When 100 equiv. of H2O2 was employed, the yield of both epoxide (TON 53) and cis-diol (TON 4.8) was increased. In the oxidation of 1-octene, using 100 equiv. H2O2, 1Pz yielded 1-octene epoxide (TON 16.5) as the major product together with a minor amount of syn-dihydroxylated product (TON 2.5). Complex 1Im, on the other hand, preferred formation of syn-diol over epoxide in the oxidation of cis-cyclooctene; in this reaction, complex 1Im (1 equiv.) together with H2O2 (10 equiv.) produced syn-cyclooctane-1,2-diol with a TON of 5.9 and cis-cyclooctene epoxide with a TON of 3.9 (E/D = 0.6) and an overall conversion of 96% (w.r.t. the oxidant). In the oxidation of the terminal alkene 1-octene, complex 1Im (1 equiv.) together with H2O2 (10 equiv.) produced cis-diol with a TON of 4.4 and epoxide with a TON of 1.5 (E/D = 3) and a conversion of approximately 36%.
In order to investigate the identity of the active oxidant, isotope labelling studies were made for both complexes, employing cis-cyclooctene as substrate. In the presence of 10 equiv. H2O2 and 1000 equiv. H218O, 1Pz (1 equiv.) introduced 97% (100% and 200% account for one and two 18O atoms per diol product, respectively) 18O into the product syn-cyclooctane-1,2-diol during the oxidation of cis-cyclooctene, while complex 1Im gave 92% label incorporation under the same conditions. These results are in full agreement with previous isotope labelling results on the Fe(PyTACN) family of complexes and of the related complex [Fe(Me2,MeBzImTACN)(OTf)2], 1BzIm.21 This isotopic labelling pattern is indeed a distinctive feature of the so called class I type of iron catalysts.18 These catalysts are proposed to operate via a high valent FeV(O)(OH) active species, formed via water assisted O–O cleavage of an FeIII(OOH)(OH2) precursor.19 In accordance with this scheme, syn-dihydroxylation of olefins by FeV(O)(OH) results in diols where one of the two oxygen atoms originates from water and the other from hydrogen peroxide. Consequently, the isotopic labelling data derived from the syn-dihydroxylation reactions (Table 2) indicate that the series of complexes 1Pz, 1Im, 1BzIm, 1Py and 1MePy can all be categorized as class I type of iron catalysts, operating via a FeV(O)(OH) species.
On the other hand, the epoxidation reaction presents a completely different pattern to the syn-dihydroxylation. The isotopic labelling in the epoxidation actually parallels the alkane hydroxylation reaction, a similarity that may be attributed to the fact that in these reactions only the oxygen atom of the oxo ligand is transferred to the substrate, while the hydroxide ligand remains in the complex. Thus, interrogation of the 18O labelling in the epoxidation reaction may be used to establish the origin of the oxygen atoms in the reactive oxo ligand.
Interestingly, when the incorporation of water into cis-cyclooctene epoxide was analyzed, complex 1Pz behaved drastically different from 1Im. The incorporation of 18O using H218O was 36% for the latter, while only ∼2% for the former. For comparison, for the related complexes 1BzIm and 1Py, the amount of 18O-labeled epoxides formed were 24 and 77%, respectively. Therefore, the origin of the oxo ligand differs in a substantial manner in the series of complexes from 1Pz, where the oxo ligand originates from H2O2 in a practically quantitative manner, to 1Py where it predominantly (77%) originates from water. We thus conclude that the incorporation of water in the reactive oxo ligand differs substantially within the series of complexes. It may be noted that the chemoselectivity patterns and the isotopic labelling data collated in Tables 1 and 2 for the five catalysts are not consistent with reactions where FeIV(O) species are the main oxidants.16–18,21,24,33–37
Finally, control experiments were conducted to investigate the possible involvement of O2 from air in the reactions. The O-inventory in the reactions catalyzed by 1Py, 1MePy and 1BzIm has been studied previously by performing oxidation reactions with 18O-labelled peroxide and water, under air. In these reactions it was shown that the oxygen atoms in the oxidized products are derived from the peroxide or water and that atmospheric oxygen atoms account for <10% of incorporated oxygen.16,18,21 In order to address the possible implication of O2 in the reactions of 1Pz and 1Im, the following experiments were performed, the results of which are included in Table 2; – oxidation of cyclohexane with 1Pz was performed under a N2 atmosphere and also under an 18O2 atmosphere. The data show no significant changes between air and N2 atmospheres and a small (11%) incorporation of oxygen from 18O2 in the C–H oxidation. Oxidation of cyclooctene with 1Pz was also performed under 18O2 and the data were compared with reactions performed under air. The epoxide shows a small incorporation of 18O from 18O2 (11%), resembling the C–H oxidation reaction. However, the cis-diol shows no significant incorporation of 18O from 18O2. It can be concluded that O2 participates to a minor extent in the epoxidation and C–H hydroxylation reaction, but not in the cis-dihydroxylation.
Oxidation of cyclohexane with 1Im was performed under a N2 atmosphere and also under an 18O2 atmosphere. The data shows no significant changes between air and N2 atmospheres and marginal (3%) incorporation of oxygen from 18O2 in the C–H oxidation. Oxidation of cyclooctene with 1Pz was then performed under 18O2, under N2 and the data were compared with reactions performed under air. The epoxide shows a small incorporation of 18O from 18O2 (8%). However, as was the case for 1Im, the cis-diol shows again no significant incorporation of 18O from 18O2. We conclude again that in the catalytic oxidations conducted using 1Im as a (pre)catalyst, only a minor extent of O2 participation is detected in the epoxidation reaction but not in the cis-dihydroxylation and C–H hydroxylation reactions. Overall, the data indicate that atmospheric O2 is either not implicated in the reactions, or represents only a minor pathway in the epoxidation reactions for both catalysts, and in the C–H hydroxylation catalyzed by 1Pz.
In summary, the very minor involvement of O2 in the reactions is consistent with the mechanistic interpretation for the C–H and CC oxidation reactions detailed above, where a metal based FeV(O)(OH) oxidant is responsible for substrate oxidation. Radical intermediates must be short lived and react within the solvent cage.
It should be borne in mind that intramolecular C–H bond activation/ligand oxidation in the course of the above-mentioned oxidation reactions cannot be ruled out, but such reactions were not specifically studied. Mass spectrometric measurements did not provide any evidence of such side reactions.
Computational modelling
In order to gain an understanding of the relative energies of the postulated FeV(O)(OH) tautomers, computational modelling of the two tautomers generated from [FeII(Me2,MeBzImTACN)(CF3SO3)2] (1BzIm)21 was undertaken. The ground-state stability ordering of the two Me2,MeBzIm compounds was computationally investigated by density functional theory (DFT), the tautomeric species were labelled in accordance with Scheme 2, i.e. the species that contains an oxo moiety coplanar with the benzimidazole ligand is labelled as OA, while OB represents the isomer whose oxo moiety is perpendicular to the heterocyclic ligand (Fig. 3). The DFT analyses confirm the quartet state (S = 3/2) as the thermodynamically favoured spin state for both isomers, with 4OA found to lie 2.4 kcal mol−1 lower in energy than 4OB. The locus of the unpaired spin density in both tautomers is similar, and the largest contributions of unpaired spin populations (Fig. 3) are found on the iron and oxo oxygen atoms. Interestingly, each tautomer displays a small amount of spin density at the hydroxyl oxygen atom.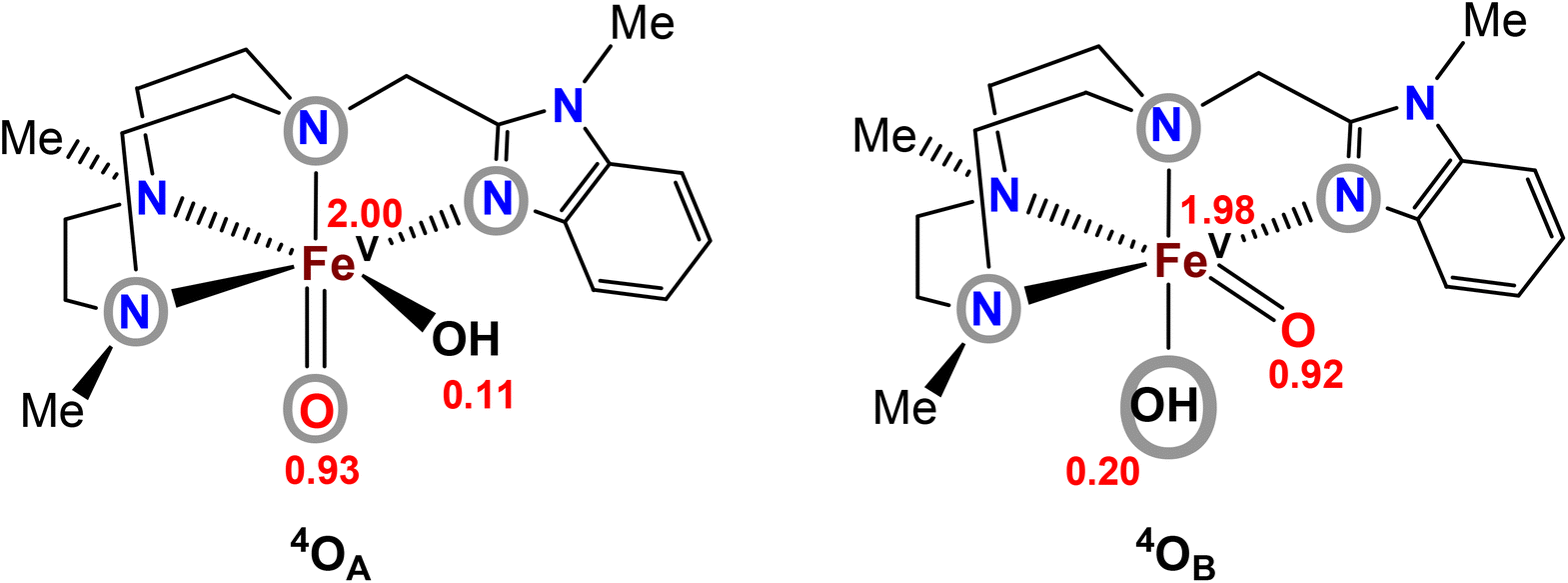 | ||
| Fig. 3 Computed spin-density populations in the two tautomers of complex 2BzIm (cf.Scheme 2). Atoms/groups inside grey circles indicate the ligand entities that lie in the equatorial planes of the tautomers. | ||
The corresponding doublet spin states of 2OA and 2OB (S = 1/2) lie 11.5 and 9.1 kcal mol−1 higher in energy than their respective quartet counterparts. Having established the ground-state stability ordering and spin-state preference for the two isomeric oxo-hydroxyl compounds, the lability of proton transfer between these tautomers was next explored. Proton transfer between the different oxygen centers in 4OA and 4OB occurs through transition structure 4TS4OA4B, as depicted in Fig. 4, and basically involves the synchronous transfer of the hydrogen between the two oxygen centers. The free-energy barrier is high and in excess of 30 kcal mol−1, and these data are in keeping with the reported configurational stability of the related pyridyl-derivatives prepared by Company and Costas.18 The absence of facile proton transfer between the two oxygen centers in 4OA and 4OB suggests that the oxo-based C–H abstractions observed in these studies must originate from structurally different oxidants that do not allow equilibration of the oxo moiety on the time scale of the chemical reactions, a feature supported by the isotopic labelling studies.
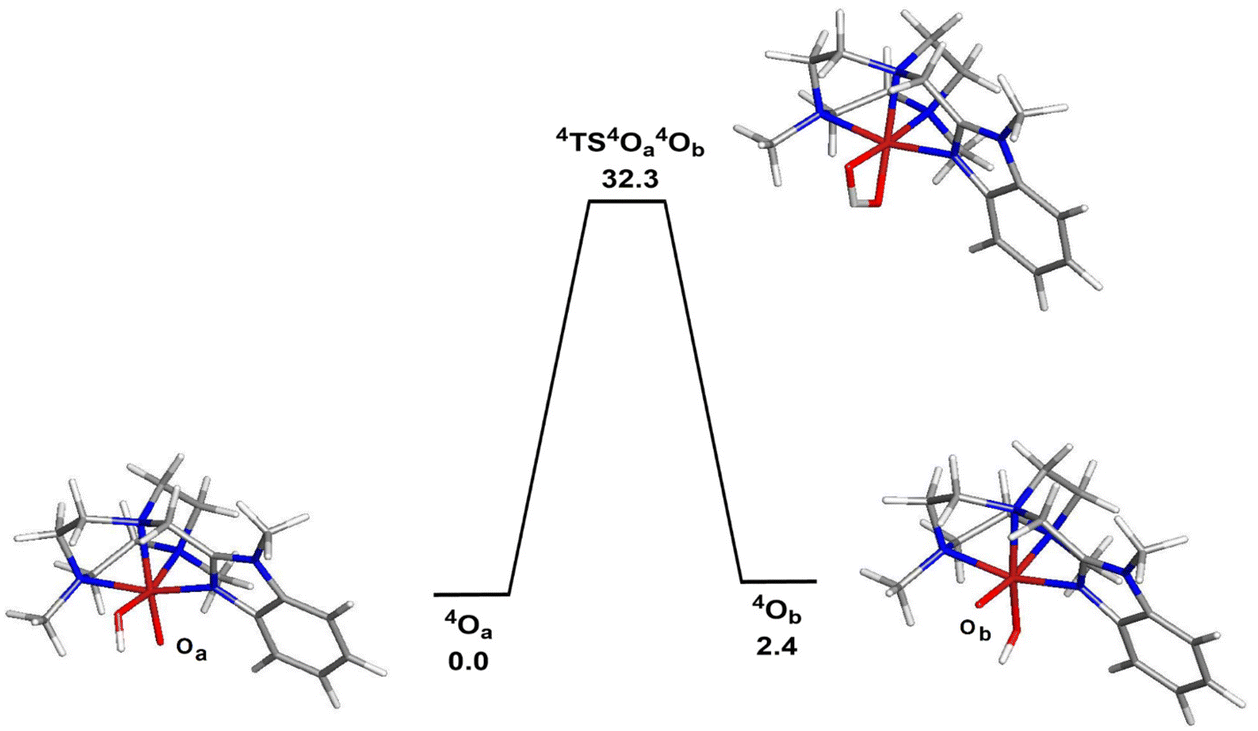 | ||
| Fig. 4 B3LYP-optimized structures and potential energy surface for the isomerization of 4OA (4A) to 4OB (4B). Energy values are in ΔG in kcal mol−1 relative to 4OA. | ||
In order to elucidate the mechanism of the hydrogen-atom transfer (HAT) of alkanes by 4OA and 4OB, DFT calculations were carried out using methane (C) as the substrate.38 This particular substrate was chosen in order to simplify the calculations and reduce the computational time. Fig. 5 shows the geometry-optimized transition-state structures involved in the H-abstraction and the resulting MeOH-substituted compounds, while Fig. 6 shows the potential energy surface for these reactions. Hydrogen abstraction is mediated by the oxo moiety in 4OA and 4OB, leading to the respective transition-state structures 4TS4OAC6D and 4TS4OBC6E, which lie 25.1 and 22.9 kcal mol−1, respectively, above the pertinent tautomer and methane reactants. The transition state for methane activation from the less stable tautomer lies 2.2 kcal mol−1 lower in energy from the corresponding transition state 4TS4OAC6D formed from the thermodynamically favoured tautomer 4A. The expected geminate radical pairs that follow each H-abstraction could not be found by IRC calculations, presumably a manifestation of the flat nature of the reaction surface en route to the alcohol product. Collapse of the geminate radical pair in each reaction via a rebound process, coupled with a spin crossover to the thermodynamically favoured high-spin sextet species (S = 5/2), completes the hydroxylation sequence and affords the MeOH-substituted products 6D and 6E. Both hydroxylation routes are exergonic in nature, and the MeOH-substituted product having the alcohol ligand disposed perpendicular to the heterocyclic ligand is more stable (by a 3.1 kcal mol−1) due to reduced interactions between the coordinated alcohol and ligand scaffold.
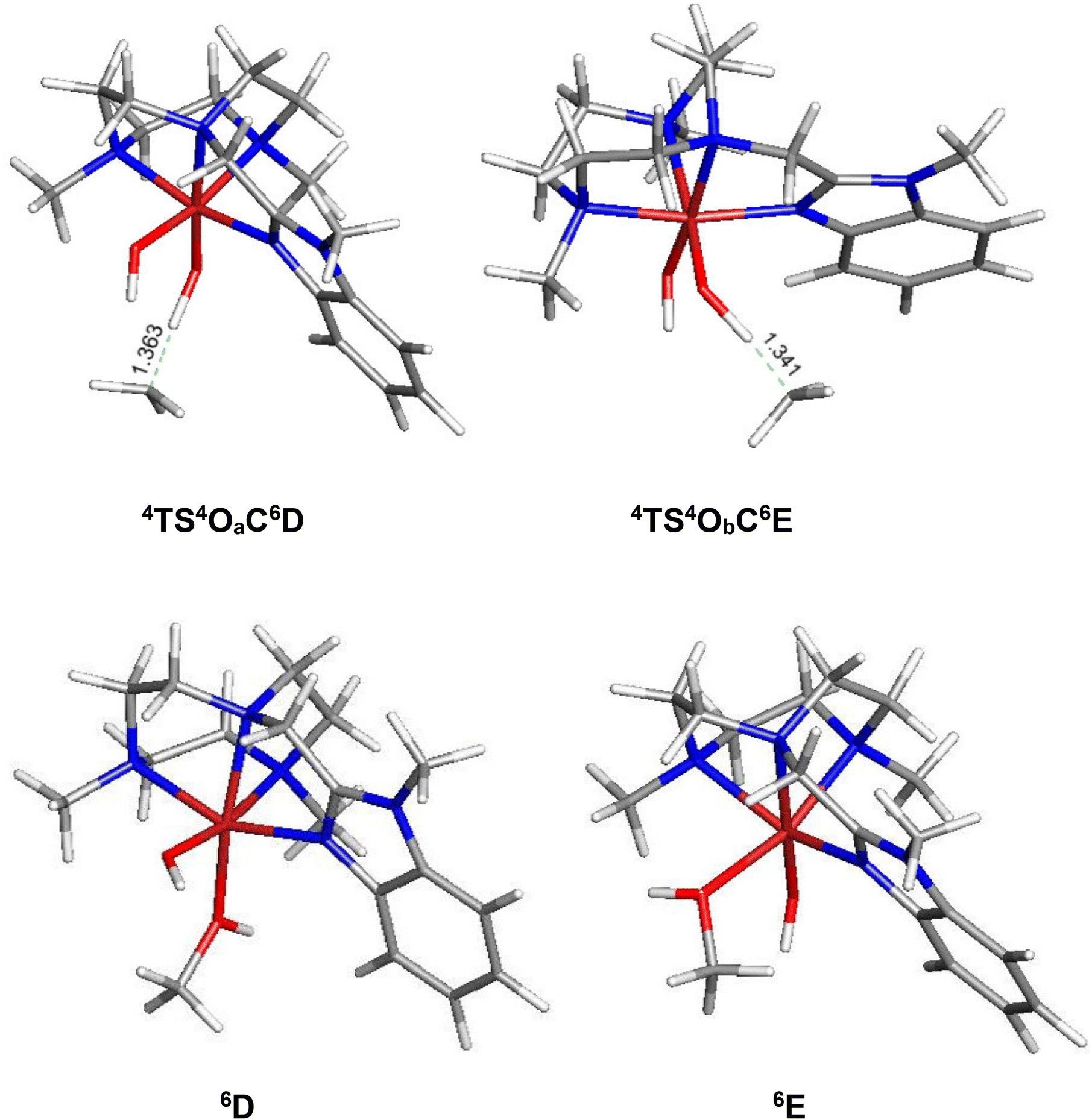 | ||
| Fig. 5 B3LYP-optimized transition structures 4TS4OaC6D and 4TS4ObC6E and the MeOH-substituted products 6D and 6E from methane (C) hydroxylation reaction using 4Oa and 4Ob. | ||
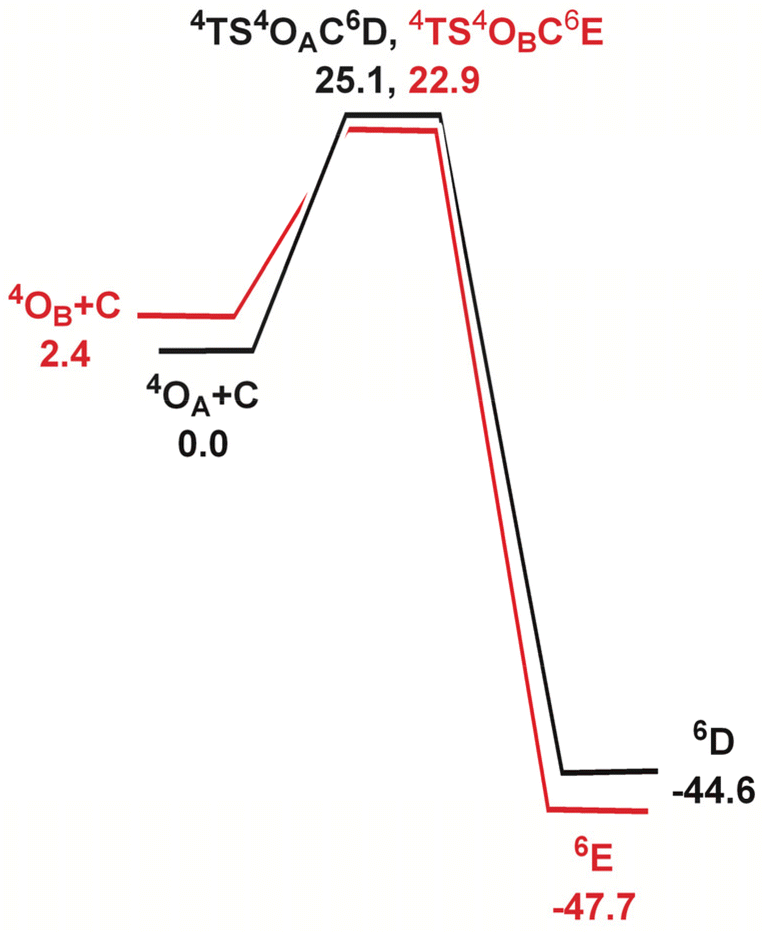 | ||
| Fig. 6 Potential energy surface for the reaction of 4OA and 4OB with methane (C). Energy values are in ΔG in kcal mol−1 relative to the respective Fe(oxo)(hydroxo) tautomer and C. | ||
The same calculations were carried out for 2Im and 2Pz. For both complexes, the OB tautomer is the least favoured (by 1.4 kcal mol−1 for 2Im, and by 3.7 kcal mol−1 for 2Pz). Furthermore, in agreement with the results obtained for 2BzIm that are discussed above, HAT from methane is favoured for this least stable tautomer with a transition state that is stabilized by 2.5 kcal mol−1 for 2Im (21.9 vs. 24.4 kcal mol−1) and by 4.9 kcal mol−1 for 2Pz (29.3 vs. 24.4 kcal mol−1) (cf. ESI†). Overall, a general trend in relative energy between the isomers was observed. In all three cases, the computed ground state energy for OB lies higher than that for OA. This trend can be rationalized by considering that the iron coordination site coplanar with the heterocycle is sterically more congested, as rapidly deduced from an inspection of the two coordination sites in Fig. 3. Presumably, the higher steric demand of the OH ligand when compared with the terminal oxo, arising from the larger Fe–O(H) distance, destabilizes isomer OB, where the hydroxide and the heterocycle are coplanar. The more stable isomer OA thus contains the reactive oxo ligand buried in the sterically more congested site, which may disfavour reaction against an external substrate. Furthermore, OB is relatively more destabilized for 2Pz when compared to 2BzIm and 2Im; the order of this relative destabilization is 2Pz > 2BzIm > 2Im, in keeping with the relative steric hindrance for the three ligands. In addition, the computed HAT profiles for the three complexes (Fig. 6 and ESI†) show that the relative stabilization in the HAT reaction, going from the transition state for OA (4TS4OAC6D, cf. Fig. 6) to the more favoured transition state for OB (4TS4OBC6D) also follows the order 2Pz > 2BzIm > 2Im. The calculations thus indicate that the least stable tautomer is also the most reactive against the C–H bond, but differences in energy are relatively small and may be dependent on computational details. Consequently, the calculations support the idea that both isomers can participate in the oxidation reactions. A perspective analysis of the results from the computations and the experimental data suggest that indeed the observed outcome in the isotopic labelling experiments reflects the relative participation of the two tautomers in the catalytic oxidation reactions, which in turn result from a balance between their relative ground state energies, and their relative reactivities toward C–H and CC bonds (activation energies).
Summary and conclusions
We have previously demonstrated the steric influence exerted by ligands on the relative reactivities of iron(V) oxo-hydroxo tautomers operating in C–H hydroxylation reactions.21 This study further addresses and strengthens the fact that the ligand exerts a profound influence on the relative oxygen atom transfer reactivity, with the steric bulk of the donor moiety of the pendant arm attached to the TACN ligand deciding the relative ease of approach of a substrate towards the reactive oxo-ligand at position A (trans to N-CH2R, tautomer OA) or at position B (trans to a N-Me, tautomer OB) of the proposed active Fe(V)(O)(OH) oxidant. An inspection of the space-filling diagrams generated for the series of complexes 2Py, 2MePy, 2Im, 2BzIm, and 2Pz (cf.Fig. 7) shows that position A is sensitive to the nature of the ligand heterocycle. The crystal structures of the Fe(II) catalysts, which are direct precursors to the active perferryl (FeV(O)) oxidants, provide a structural basis for the steric influence of the ligands.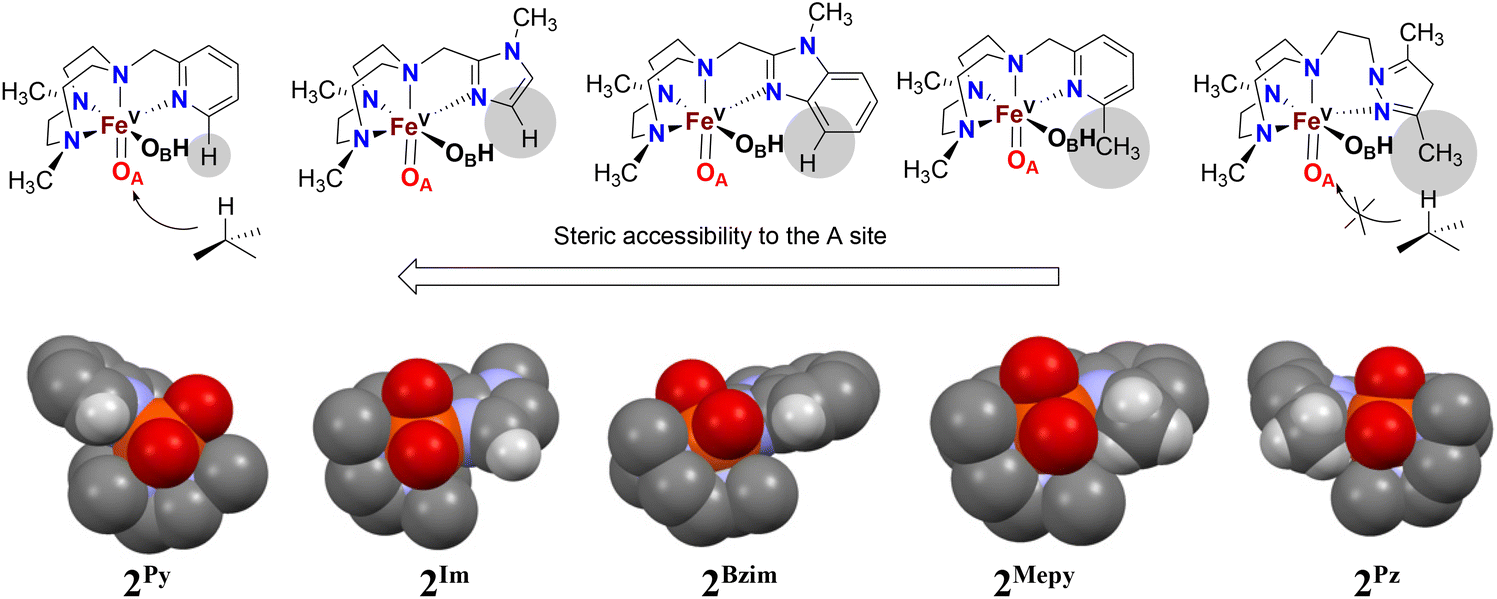 | ||
| Fig. 7 Schematic depictions (top) and space filling models (bottom) of the Fe(V)(O)(OH) species 2Py, 2Im, 2BzIm, 2MePy, and 2Pz obtained by modifying the crystal structures of 1Py, IBzIm, 1MePy and 1Pz by excluding most hydrogens and only displaying the oxygen atoms of the trifluoromethylsulfonate ligands. The space-filling structure of 2Im is derived from the structure of 1BzIm, with the imidazolyl hydrogen being placed in the position benzimidazolyl carbon atom. | ||
As illustrated in Fig. 7, the oxygen atom that lies in the N3-donor plane of the ligand(s) in which the “pendant” N-donor ligand is situated (i.e. corresponding to the oxo ligand in tautomer OA, cf. Fig. 3) is positioned in a sterically more encumbered environment than the oxygen atom in the OB position, which protrudes from the ligand plane. On steric grounds, it may therefore be expected that sterically hindered C–H entities, e.g. tertiary C–H bonds, will react preferentially with the least sterically hindered oxygen atom/oxido ligand, i.e. that of the OB tautomer. As there is evidence that the oxo ligand in OA originates from water,18,19 the relative incorporation of oxygen from H218O into a (sterically bulky) alkane substrate (especially those containing tertiary C–H bonds) may be used as a guide to the steric influence of the ligand on substrate access to the OA oxo ligand – low incorporation of labelled oxygen into the oxidation product indicates steric hindrance by the ligand. The levels of incorporation of 18O from water into the substrates (cyclohexane, cyclooctane, adamantane, cis-DMCH, cis-cyclooctene) are listed in Table 2. For complex 1Pz, the very low percentage of 18O incorporation (<3%) from H218O in the products suggests that the OB tautomer predominates over OA (irrespective of substrates containing secondary or tertiary C–H bonds) in the C–H hydroxylation reaction. On the other hand, the levels of 18O incorporation (within the range 30–45%) from H218O into products for complex 1Im indicate that both OA and OB are equally reactive towards substrates with little preference of OB over OA for substrates containing tertiary C–H bonds.
While the high percentage (above 90%) 18O incorporation in the syn-diol product in oxidation of cis-cyclooctene by complexes 1Pz and 1Im (cf.Table 2) demonstrates the involvement of Fe(V)(O)(OH) oxidant as observed for Fe(PyTACN) complexes, the different ranges of 18O incorporation into the epoxide product also discriminate the relative reactivity between OA and OB. The amounts of labelled epoxide were 2% and 36% for complexes 1Pz and 1Im, respectively, clearly implying that epoxidation reaction catalyzed by complex 1Pz is performed exclusively by OA and in complex 1Im, OA and OB are active in an approximate 2/1 ratio.
On the basis of these experimental results, the order of steric interference of the ligand with the OA oxo ligand (cf.Scheme 2 and Fig. 7) is 1Pz > 1MePy > 1BzIm > 1Im > 1Py. Indeed, it is interesting to notice that the steric bulk inside the first coordination sphere of the different Fe(II) precursor complexes obtained from percent buried volume (%Vbur)39 calculations based on the crystallographic data for 1Pz, 1MePy, 1BzIm and 1Py follows the same order. Fig. 7 shows space filling models for the ligand pockets and the corresponding calculated %Vbur values for the crystal structures of 1Pz, 1MePy, 1BzIm and 1Py (cf. Experimental section and ESI† for details). There are no crystal data for 1Im, but the percent buried volume for this complex is anticipated to lie between the values obtained for 1BzIm and 1Py. The N-methyl imidazole ring has a sterically less pronounced effect on the metal center compared to the N-methyl benzimidazole ring in 1BzIm, but has a higher steric effect compared to the pyridine ring present in 1Py (Fig. 8).
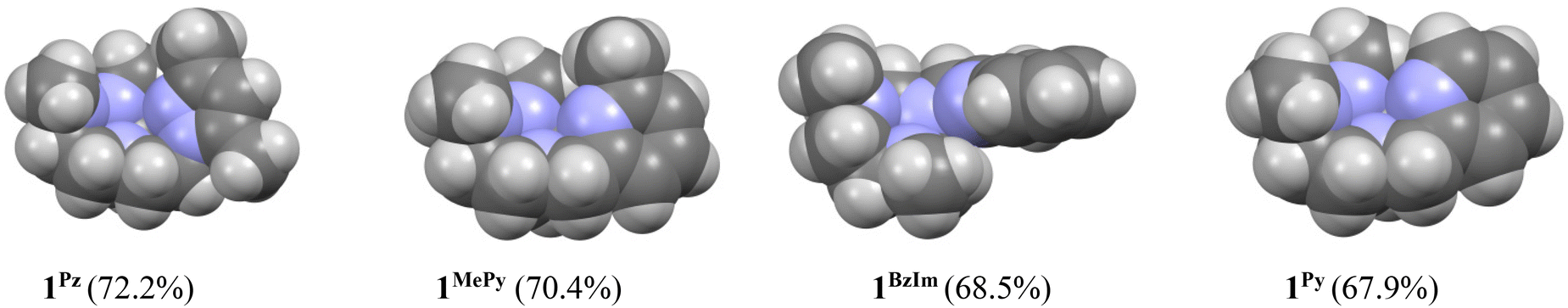 | ||
| Fig. 8 Space filling models and the corresponding percent buried volume (%Vbur) values of the ligand pockets, within a 3.5 Å radius of the centrally located iron ion, in the Fe(II) precursor complexes 1Pz, 1MePy, 1BzIm and 1Py. | ||
The results described above indicate that the steric bulk of the tetradentate TACN-based ligands exerts a significant influence on the relative HAT reactivities of the postulated tautomeric FeV(O)(O) oxidants. It should be noted that different basicities or donor properties possessed by the side arms ((N-methyl)imidazole, (N-methyl)benzimidazole and 3,5-dimethylpyrazole) of the tetradentate ligands might have some influence on the reactivity of Fe(V)(O)(OH) oxidant in general, and the relative reactivities of the two tautomers in particular. However, the electronic effects of the side arms are not as apparent or conclusive as the steric properties of the side arms from the present investigation. Thus, the possible effects of electronic properties of the ligands on the reactivities need to be clarified, and further studies are undertaken towards this direction.
Experimental section
Reagents and materials
Reagents and solvents were of at least 99% purity and used as received without any further purification. H218O2 (90% 18O-enriched, 2% solution in H218O) and H218O (95% 18O-enriched) were purchased from ICON isotopes. All reagents and solvents were purchased from Sigma Aldrich or Fisher Scientific. Dichloromethane and acetonitrile were dried by distillation from CaH2; diethyl ether was dried by distillation from Na/benzophenone. The starting materials 1-(2-chloroethyl)-3,5-dimethyl-1H-pyrazole,40 1-(2-chloromethyl)-2-methylimidazole hydrochloride,41 and 4,7-dimethyl-1,4,7-triazacyclononane42 or its trihydrobromide salt43 were synthesized according to literature procedures.Instrumentation
Infrared spectra were collected on a Nicolet Avatar 360 FTIR spectrometer. UV-Visible spectroscopy was performed in a 1 cm quartz cell using an Agilent Technology 8453 UV-Vis spectrophotometer equipped with a diode-array detector. NMR spectra were recorded a Bruker DPX 400 MHz and Varian Inova 500 MHz spectrometers in CDCl3 or CD3CN solvent using standard conditions, and were referenced to the residual proton signal of the solvent. Elemental analysis was performed on a 4.1 Vario EL 3 elemental analyzer from Elementar. The ESI-MS experiments were performed with a Bruker Esquire 6000 LC/MS chromatograph, using acetonitrile as a mobile phase. The product analyses after catalysis experiments were carried out on an Agilent Technology 7820A gas chromatograph equipped with a 16-sample automatic liquid sampler, flame ionization detector and EzChrom Elite Compact software. GC-MS analyses were performed on an Agilent Technology 7890A GC system equipped with a 5975C inert XL EI/CI MSD with triple-axis detector. The products were identified by comparison of their GC retention times and, in the case of GC/MS, with those of authentic compounds.Syntheses
:1 mixture) and filtered through Celite. Diffusion of ether into the resultant solution resulted in precipitation of the complex as a light yellowish white solid. Yield: 137 mg (45%). HRMS (m/z) 153.5751 [FeII(Me2,MeImTACN)]2+ (calc. 153.5724) (z = 2); 456.0970 [FeII(Me2,MeImTACN)(CF3SO3)]+ (calc. 456.0974) (z = 1); Elemental analysis C15H25F6N5O6S2Fe (MW = 605.353 g mol−1) Calc. (%) C 29.76, H 4.16, N 11.57; Found (%) C 30.55, H 4.28, N 11.08; 1H-NMR (500 MHz, CD3CN) 99.43, 82.93, 69.52, 64.56, 55.36, 50.00, 33.54, 21.24, 6.72, 4.06, 3.65, 3.59, 3.05–3.02, 2.88–2.82, 2.77–2.74, 2.66, 2.59, 2.51–2.48, 2.21, 1.94, 1.81, 1.56, 1.32, 1.24, 1.21, 0.05; UV/Vis λ (nm) 365 (ε = 3100 M−1 cm−1), 512 (ε = 60 M−1 cm−1); FTIR (KBr) ν (cm−1) 2921, 1634, 1458, 1259, 1176, 1032, 756, 642, 580, 519.
Crystal structure determination for complex 1Pz
Colourless crystals of 1Pz were grown from slow diffusion of ethyl ether in a CH2Cl2 solution of the compound, and used for low temperature (100(2) K) X-ray structure determination. The measurement was carried out on a BRUKER SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from an X-ray tube. The measurements were made in the range 1.636 to 28.083° for θ. Hemi-sphere data collection was carried out with ω and φ scans. A total of 16414 reflections were collected of which 6388 [R(int) = 0.0502] were unique. Programs used: data collection, Smart;44 data reduction, Saint+;45 absorption correction, SADABS.46 Structure solution and refinement was done using SHELXTL.47
The structure was solved by direct methods and refined by full-matrix least-squares methods on F2. The non-hydrogen atoms were refined anisotropically. The H-atoms were placed in geometrically optimized positions and forced to ride on the atom to which they are attached. A considerable amount of electron density attributable to a disordered ethyl ether solvent molecule per asymmetric unit was removed with the SQUEEZE option of PLATON.48 Those solvent molecules are, however, included in the reported chemical formula and derived values (e.g. formula weight, F(000), etc.).
Reaction conditions for catalysis experiments
In a typical reaction, 360 μL of H2O2 (25 μmol), taken from a 70 mM H2O2 stock solution in CH3CN together with 45 μL of water (2500 μmol), was delivered by syringe pump over 30 min at room temperature under air to a vigorously stirred CH3CN solution (2.14 ml) containing the Fe-catalyst (2.5 μmol) and the alkane substrate (2500 μmol). The final concentrations were 1 mM for catalyst, 10 mM for the oxidant, 1000 mM for H2O and 1000 mM for substrate (1:10:1000:1000 for cat:ox:H2O:sub). For adamantane, due to the low solubility, only 50 μmol of the substrate was used and so the final concentration was 20 mM. At the conclusion of the syringe pump addition, 500 μL of a biphenyl solution of a known concentration (∼25 mM) was added to the reaction mixture as an internal standard. The reaction mixture was then passed through a small silica column (to remove the iron complex), followed by elusion with 2 ml ethyl acetate. Finally, the solution was subjected to GC analysis. The organic products were identified and their yields were calculated by using authentic compounds as quantitative standards.
For the measurement of kinetic isotope effects (KIE), a substrate mixture of cyclohexane:cyclohexane-d12 in a ratio of 1:3 was used to improve the accuracy of the obtained KIE value.
Isotope labeling studies
:10:1000:1000 for cat:H2O2:H218O:sub). For adamantane, due to the low solubility, only 50 μmol of the substrate was used and so the final concentration was 20 mM.
:10:1000:1000 for cat:H2O218:H2O:sub).
In the oxidation of adamantane and cis-1,2-dimethylcyclohexane, the solution (after syringe pump addition) was passed through a small silica column to remove the Fe-catalyst, followed by elution with 2 ml ethyl acetate. For other substrates, the reaction solution was treated with 1 ml acetic anhydride and 0.1 ml of 1-methylimidazole to esterify the alcohol products for GC-MS analyses (tertiary alcohols are not esterified under these conditions). Samples were concentrated by removing part of the solvent under vacuum and subjected to GC-MS analyses.
Computational details and modeling
All DFT calculations were carried out with the Gaussian 09 package of programs49 using the B3LYP hybrid functional. This functional is comprised of Becke's three-parameter hybrid exchange functional (B3)50 and the correlation functional of Lee, Yang, and Parr (LYP).51 The iron atom was described with the Stuttgart–Dresden effective core potential and SDD basis set,52,53 and the 6-31G(d′) basis set54,55 was employed for all remaining atoms.All reported geometries were fully optimized, and analytical second derivatives were evaluated at each stationary point to determine whether the geometry was an energy minimum (no negative eigenvalues) or a transition structure (one negative eigenvalue). Unscaled vibrational frequencies were used to make zero-point and thermal corrections to the electronic energies. The resulting potential energies and enthalpies are reported in kcal mol−1 relative to the specified standard. Standard state corrections were applied to all species to convert concentrations from 1 atm to 1 M according to the treatise of Cramer.56 The geometry-optimized structures have been drawn with the JIMP2 molecular visualization and manipulation program.57,58
Calculation of buried volume
Calculations of buried volume were carried out using the online version of the SambVca 2.1 program (https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html).59 Atomic radii were chosen as Bondi radii scaled by a factor of 1.17; the sphere radius was 3.5 Å around origo; the mesh spacing for numerical integration was 0.10; all hydrogen atoms of the ligand were included in the calculation. For each molecule, the position of the iron ion was chosen as origo, the iron-oxygen (OB) axis was chosen as the positive z-axis direction, and the iron-N(heterocycle) axis was chosen as the X-axis.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This research has been carried out within the frameworks of the International Research Training Group Metal sites in biomolecules: structures, regulation and mechanisms and COST Action CM1003. MM thanks the European Commission for an Erasmus Mundus fellowship. YL thanks the Chinese Scholarship Council for a predoctoral fellowship. MGR acknowledges financial support from the Robert A. Welch Foundation (grant B-1093-MGR). Computational resources through the High-Performance Computing Services and CASCaM at the University of North Texas are acknowledged. This paper is dedicated to the memory of Professor Gábor Speier, a pioneer in bio-inspired coordination chemistry and biomimetic oxidation catalysis.References
- P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- A. Furstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef.
- L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- C. Perry, E. L. C. de los Santos, L. M. Alkhalaf and G. L. Challis, Nat. Prod. Rep., 2018, 35, 622–632 RSC.
- S. M. Barry and G. L. Challis, ACS Catal., 2013, 3, 2362–2370 CrossRef.
- E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef PubMed.
- S. Chakrabarty, R. N. Austin, D. Deng, J. T. Groves and J. D. Lipscomb, J. Am. Chem. Soc., 2007, 129, 3514–3515 CrossRef PubMed.
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- G. Olivo, O. Cusso, M. Borrell and M. Costas, J. Biol. Inorg. Chem., 2017, 22, 425–452 CrossRef.
- O. Lyakin, K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2019, 384, 126–139 CrossRef.
- S. Kal, S. Xu and L. Que Jr., Angew. Chem., Int. Ed., 2020, 59, 7332–7349 CrossRef PubMed.
- J. Chen, Z. Jiang, S. Fukuzumi, W. Nam and W. Bing, Coord. Chem. Rev., 2020, 421, 213443 CrossRef.
- A. A. Shteinman and M. Mitra, Inorg. Chim. Acta, 2021, 523, 120388 CrossRef.
- A. Company, L. Gomez, M. Guell, X. Ribas, J. M. Luis, L. Que Jr. and M. Costas, J. Am. Chem. Soc., 2007, 129, 15766–15767 CrossRef PubMed.
- A. Company, L. Gomez, X. Fontrodona, X. Ribas and M. Costas, Chem. – Eur. J., 2008, 14, 5727–5731 CrossRef CAS PubMed.
- I. Prat, A. Company, V. Postils, X. Rivas, L. Que Jr., J. M. Luis and M. Costas, Chem. – Eur. J., 2013, 19, 6724–6738 CrossRef PubMed.
- I. Prat, J. S. Mathienson, M. Guell, X. Ribas, J. M. Luis, M. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef PubMed.
- I. Prat, A. Company, T. Corona, T. Parella, X. Ribas and M. Costas, Inorg. Chem., 2013, 52, 9229–9244 CrossRef PubMed.
- M. Mitra, J. Lloret-Fillol, M. Haukka, M. Costas and E. Nordlander, Chem. Commun., 2014, 50, 1408–1410 RSC.
- A. Diebold and K. S. Hagen, Inorg. Chem., 1998, 37, 215–223 CrossRef.
- D. W. Blakesley, S. C. Payne and K. S. Hagen, Inorg. Chem., 2000, 39, 1979–1989 CrossRef PubMed.
- K. Chen and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England and A. P. J. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- A. J. Simaan, S. Dopner, F. Banse, S. Bourcier, G. Bouchoux, A. Boussac, P. Hildebrandt and J. J. Girerd, Eur. J. Inorg. Chem., 2000, 1627–1633 CrossRef CAS.
- P. Spannring, I. Prat, M. Costas, M. Lutz, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 708–716 RSC.
- J. England, G. J. P. Britovsek, N. Rabadia and A. J. P. White, Inorg. Chem., 2007, 46, 3752–3767 CrossRef CAS.
- J. England, C. R. Davies, M. Banaru, A. J. P. White and G. J. P. Britovsek, Adv. Synth. Catal., 2008, 350, 883–897 CrossRef CAS.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef CAS.
- M. Costas, K. Chen and L. Que Jr., Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS.
- P. Comba and G. Rajaraman, Inorg. Chem., 2008, 47, 78–93 CrossRef CAS.
- P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef CAS.
- K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que Jr., J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS PubMed.
- K. B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- K. B. Cho, X. Wu, Y. M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef CAS PubMed.
- V. Postils, A. Company, M. Sola, M. Costas and J. M. Luis, Inorg. Chem., 2015, 54, 8223–8236 CrossRef CAS PubMed.
- A. Gomez-Suarez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- G. Esquius, J. Pons, R. Yanez, J. Ros, R. Mathieu, B. Donnadieu and N. Lugan, Eur. J. Inorg. Chem., 2002, 2999–3006 CrossRef CAS.
- C. Robinson, J. Zhang, D. Garrod, T. Perrior, G. Newton, K. Jenkins, R. Beevers, M. Major and M. Stewart, Pyruvamide Compounds as Inhibitors of Dust Mite Group 1 Peptidase Allergen and their Use, US2012/0322722Al, 2011.
- D. A. Valyaev, S. Clair, L. Patrone, M. Abel, L. Porte, O. Chuzel and J.-L. Parrain, Chem. Sci., 2013, 4, 2815–2821 RSC.
- C. Flassbeck and K. Wieghart, Z. Anorg. Allg. Chem., 1992, 608, 60–68 CrossRef CAS.
- Bruker Advanced X-ray Solutions. SMART: Version 5.631, 1997–2002 Search PubMed.
- Bruker Advanced X-ray Solutions. SAINT+, Version 6.36A, 2001 Search PubMed.
- G. M. Sheldrick, Empirical Absorption Correction Program, Universität Göttingen, 1996 Search PubMed; Bruker Advanced X-ray Solutions. SADABS Version 2.10, 2001 Search PubMed.
- G. M. Sheldrick, Program for Crystal Structure Refinement, Universität Göttingen, 1997 Search PubMed; Bruker Advanced X-ray Solutions. SHELXTL Version 6.14, 2000–2003 Search PubMed; G. M. Sheldrick, SHELXL-2013, 2013 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- S. P. Walch and C. W. Bauschlicher, J. Chem. Phys., 1983, 78, 4597–4605 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- C. J. Cramer, Essentials of Computational Chemistry, Wiley, Chichester, UK, 2nd edn, 2004 Search PubMed.
- M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS.
- J. Manson, C. E. Webster and M. B. Hall, JIMP2, version 0.091, a free program for the visualization and manipulation of molecules, Texas A&M University, College Station, TX, 2006, https://www.chem.tamu.edu/jimp2/index.html Search PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS, 1H-NMR, UV/Vis and FT-IR spectra of complexes 1Pz and 1Im. CCDC 2154313 (1Pz). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00725h |
| This journal is © The Royal Society of Chemistry 2023 |