
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural insights into latency of the metallopeptidase ulilysin (lysargiNase) and its unexpected inhibition by a sulfonyl–fluoride inhibitor of serine peptidases
Arturo
Rodríguez-Banqueri
,
Marina
Moliner-Culubret
,
Soraia R.
Mendes
,
Tibisay
Guevara
,
Ulrich
Eckhard
 * and
F. Xavier
Gomis-Rüth
*
* and
F. Xavier
Gomis-Rüth
*
Proteolysis Laboratory; Department of Structural and Molecular Biology; Molecular Biology Institute of Barcelona (IBMB), Higher Scientific Research Council (CSIC); Barcelona Science Park; c/Baldiri Reixac 4-8, Tower R, 08028 Barcelona, Catalonia, Spain. E-mail: ueccri@ibmb.csic.es; xgrcri@ibmb.csic.es
First published on 24th February 2023
Abstract
Peptidases are regulated by latency and inhibitors, as well as compatibilization and cofactors. Ulilysin from Methanosarcina acetivorans, also called lysargiNase, is an archaeal metallopeptidase (MP) that is biosynthesized as a zymogen with a 60-residue N-terminal prosegment (PS). In the presence of calcium, it self-activates to yield the mature enzyme, which specifically cleaves before basic residues and thus complements trypsin in proteomics workflows. Here, we obtained a low-resolution crystal structure of proulilysin, in which 28 protomers arranged as 14 dimers form a continuous double helix of 544 Å pitch that parallels cell axis b of the crystal. The PS includes two α-helices and obstructs the active-site cleft of the catalytic domain (CD) by traversing it in the opposite orientation of a substrate, and a cysteine blocks the catalytic zinc according to a “cysteine-switch mechanism”. Moreover, the PS interacts through its first helix with an “S-loop” of the CD, which acts as an “activation segment” that lacks one of two essential calcium cations. Upon PS removal during maturation, the S-loop adopts its competent conformation and binds the second calcium ion. Next, we found that in addition to general MP inhibitors, ulilysin was competitively and reversibly inhibited by 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF; Ki = 4 μM). This is a compound that normally forms an irreversible covalent complex with serine peptidases but does not inhibit MPs. A high-resolution crystal structure of the complex revealed that the inhibitor penetrates the specificity pocket of ulilysin. A primary amine of the inhibitor salt-bridges an aspartate at the pocket bottom, thus mimicking the basic side chain of substrates. In contrast, the sulfonyl fluoride warhead is not involved and the catalytic zinc ion is freely accessible. Thus, the usage of inhibitor cocktails of peptidases, which typically contain AEBSF at ∼25-fold higher concentrations than the determined Ki, should be avoided when working with ulilysin. Finally, the structure of the complex, which occurred as a crystallographic dimer recurring in previous mature ulilysin structures, unveiled an N-terminal product fragment that delineated the non-primed side of the cleft. These results complement prior structures of ulilysin with primed-side product fragments and inhibitors.
1. Introduction
The pappalysins are a family of zinc-dependent metallopeptidases (MPs) whose founding member is a ∼180 kDa multidomain protein first found in the bloodstream of pregnant women termed pregnancy-associated plasma protein-A or pappalysin-1.1,2 They form family M43 in the MEROPS peptidase database (https://www.ebi.ac.uk/merops;3) and are included within the metzincin clan of mononuclear MPs, which englobes at least 12 structurally-distinct families from all kingdoms of life.4–8 Metzincin catalytic domains (CDs) span ∼130–350 residues and share structural features including a five-stranded β-sheet, a “backing helix” and an “active-site helix” within an N-terminal subdomain (NSD); and a “Met-turn” centred on a conserved methionine residue plus a “C-terminal helix” within a C-terminal subdomain (CSD).7–10 The two subdomains frame the top and bottom of the active-site cleft, respectively, when viewed in the typical standard orientation of MPs,11 with the lowermost β-strand of the five-stranded β-sheet featuring the upper rim of the active-site cleft. Moreover, a conserved zinc-binding consensus sequence (H–E–x–x–H–x–x–G/N–x–x–H/D; amino-acid one-letter-code, x for any residue), which is contained in the second half of the active-site helix and beyond, forms the interface between the NSD and the CSD. It includes three histidine/aspartate zinc ligands and the general base/acid glutamate for catalysis.8,9The structure of pappalysin-1 has been recently elucidated.10,12 It reveals that its CD resembles that of the subfamily of the “lower pappalysins”, formerly the “unicellular pappalysins”,13,14 which encompasses related sequences from archaea, bacteria, and, as more recently ascertained, also unicellular and multicellular eukaryotes like fungi and algae. They span a short N-terminal prosegment (PS) engaged in latency, as described for many MPs,9 followed by the CD and, optionally, downstream segments and domains. Several subfamily members have been biochemically, functionally or transcriptionally characterized (Table 1). In contrast, experimental structural information of these pappalysins is restricted to mature ulilysin from the archaeon Methanosarcina acetivorans, as well as mature and latent mirolysin from the periodontopathogenic bacterium Tannerella forsythia.13–18
| Name | Organism | Ref. |
|---|---|---|
| Cytophagalysin | Collagenolytic bacterium Cytophaga sp. L.43–1 | 69–71 |
| Fpp2, Fpcol and related MPs | Bacterial fish pathogen Flavobacterium psychrophilum and the related psychrophilic Antarctica bacterium Flavobacterium frigidimaris | 72–76 |
| PoMTP | Edible hiratake mushroom Pleurotus ostreatus | 77 |
| MEP1 | Fungal pathogen Coccidioides posadasii, which causes the respiratory San-Joaquín-Valley fever | 78 |
| Ulilysin (lysargiNase) | Archaeon Methanosarcina acetivorans | 13, 15–17 and 19 |
| Mirolysin | periodontopathogenic bacterium Tannerella forsythia | 14, 18, 43 and 79 |
| Mrmep1, Mrep2 and orthologues | Filamentous entomopathogenic fungi Metarhizium robertsii (formerly M. anisopliae), which is utilized in pest control, and Metarhizium rileyi | 80 and 81 |
| RcMEP1 | Fungal plant pathogen Rhizonia cerealis | 82 |
| Mep2 | Fungal plant pathogen Verticillium longisporum/dahlia | 83 |
| Tryp-N | Thermophilic filamentous fungus Chaetomium thermophilum | 84 |
| Leptolysin | Spirochaete bacterium Leptospira interrogans, which causes leptospirosis and Well's disease | 46 |
| M43-type MP | Human dermatophytic fungal pathogen Trichophyton violaceum | 85 |
| M43-type MP | Mutualistic root-symbiotic fungus Piriformospora indica | 86 |
| M43-type MP | Hemibiotrophic fungal plant pathogens Colletotrichum spp. | 87 and 88 |
| M43-type MP | Nematodeegg-parasitic fungus Pochonia chlamydosporia | 89 |
Ulilysin is biosynthesized as a zymogen with a 60-residue PS for latency that is self-activated in trans at bond S60–R61 (ulilysin residue numbers in superscript according to UniProt database entry [UP] Q8TL28) in the presence of calcium as the indispensable cofactor.13 In addition, a 20-residue negatively-charged flexible C-terminal peptide is further removed upon cleavage at bond A322–R323, eventually releasing the mature 262-residue CD (R61–A322). Functionally, ulilysin efficiently cleaved insulin, insulin-like growth-factor-binding proteins 2 to 6, several types of denatured collagen and gelatin, as well as other model and extracellular-matrix proteins.13,15 Regarding inhibitors, ulilysin was only inhibited by the general metal chelators 1,10-phenanthroline and EDTA, as well as excess zinc.13 Some inhibition (∼75%) was observed with the broad-spectrum inhibitor of matrix metallopeptidases, batimastat, at ∼50-fold molar excess.13,16 A comprehensive analysis of ulilysin's substrate specificity revealed strict N-terminal selectivity for lysine and arginine residues, including methylated and dimethylated species, and further proved the suitability of the MP, which is also referred to as “lysargiNase”, as a tool for proteomics.19 It is currently marketed by Merck/Millipore and, by generating peptides with N-terminal positively-charged side chains, it is complementary to trypsin, which generates peptides with C-terminal positively-charged residues. Thus, by using both enzymes, more peptides can be covered during proteomics studies. Moreover, ulilysin operates at temperatures of up to 55 °C while proteomics-grade trypsin usually works at 37 °C, which further enlarges the palette of biotechnological applications of the archaeal MP.
Structurally, mature ulilysin includes all the aforementioned hallmarks of the metzincins, with H228–H238 featuring the zinc-binding consensus sequence.13,16,17 Moreover, like astacins and serralysins, ulilysin possesses a fourth zinc ligand in the form of a tyrosine (Y292) two positions downstream of the Met-turn methionine (M290), which participates in substrate and reaction–intermediate binding following a “tyrosine switch”.20 Among the specific characteristics of lower pappalysins, ulilysin further contains a Lin12-Notch-repeat-like loop within the upper β-strand of the NSD β-sheet, a β-ribbon inserted after the central β-strand of this sheet, and a segment encompassing two short α-helices, which is found between the backing helix and the top β-strand of the sheet and lines the back of the CSD. A C-terminal product fragment bound to the primed-side of the cleft revealed that the basic side chains in the P′1 position of substrates (active-site cleft sub-site nomenclature according to ref. 11 and 21) are bound by D295 at the bottom of the deep S′1 pocket. This pocket is the main driver of specificity in most MPs11 and in ulilysin it is further lined by the L188, F220, T225 and M298 side chains and the main chain of segment Y292–D295.15
Sulfonyl fluorides (SFs) were identified in the 1930s as toxic to animals through their capacity to inhibit serine esterases including acetylcholinesterase.22,23 They were found to also inhibit the serine peptidases α-chymotrypsin, trypsin, neutrophil elastase, cathepsin G and Staphyloccus aureus V8 protease, as well as complement, fibrinolytic and coagulation serine peptidases.24–27 These enzymes operate through covalent acylenzyme reaction intermediates resulting from the nucleophilic attack of the Oγ atoms of the catalytic residue onto the scissile carbonyl carbon of a substrate and the concomitant release of the C-terminal product half.28,29 In the second part of the reaction, this intermediate is resolved by the attack of a solvent molecule, which frees the N-terminal half of the product and restores the catalytic residue. SFs mimic the first part of the reaction inasmuch as the catalytic serine performs a nucleophilic attack onto the fluorosulfonyl group of the inhibitor, which produces a sulfonic ester and causes a fluoride anion to leave.27 In contrast to substrates, however, this reaction product is highly stable and cannot be hydrolysed, which causes irreversible inhibition of the enzyme.23,27 This property has been taken advantage of to develop probes for target identification and validation, as well as the mapping of protein–protein interactions, substrates and binding sites.30 Finally, in addition to catalytic serine residues, reactive threonine, lysine, tyrosine, cysteine and histidine residues, as well as N-terminal α-amino groups of proteins, may likewise be sulfonylated by SFs.30
Among the most common SFs in protein chemistry are phenylmethylsulfonyl fluoride (PMSF) and 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), which is commercialized under the brand Pefabloc® SC (Fig. 1A).27,30–32AEBSF is more soluble and stable in water than PMSF, which results in higher inhibitory efficiency and duration, as well as storability in solution.26,30,32,33 Moreover, contrary to PMSF, AEBSF is not toxic and does not inhibit cysteine peptidases. Currently, AEBSF and/or PMSF are included in commercial peptidase inhibitor cocktails that are routinely employed during protein purification protocols to prevent degradation.30,34
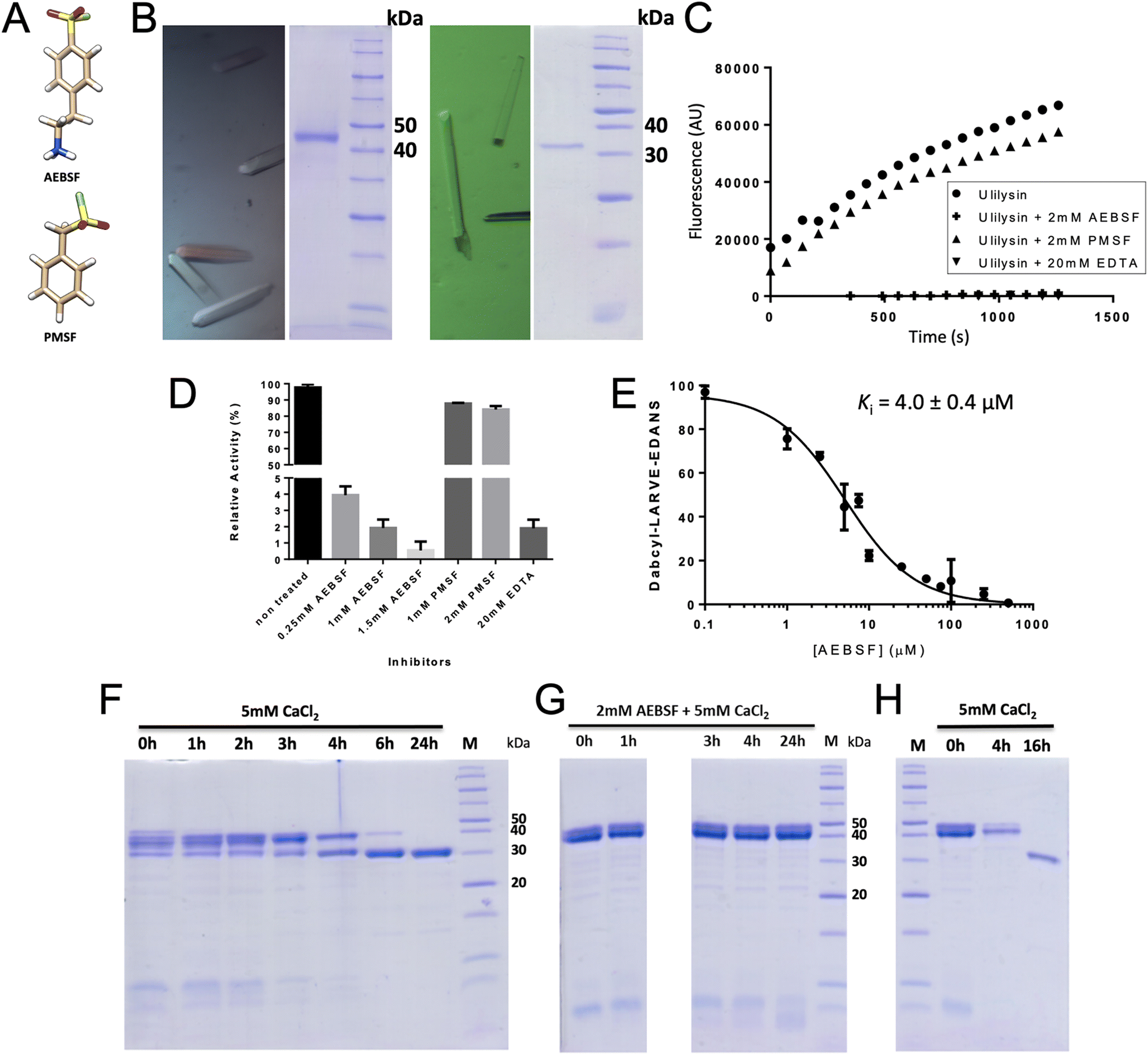 | ||
| Fig. 1 Biochemical studies. (A) Chemical structures of 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) and phenylmethylsulfonyl fluoride (PMSF). (B) Centred orthorhombic crystals of proulilysin E229A (left panel) contained the intact zymogen (middle-left panel) while primitive orthorhombic crystals of ulilysin C269A (middle-right panel) contained the mature enzyme only (right panel). (C) Inhibition of ulilysin cleavage of the fluorogenic peptide Dabcyl-L–A–R–V–E-Edans by AEBSF or EDTA but not by PMSF as shown by the fluorescence response. (D) Relative residual activity plot of ulilysin showing that AEBSF inhibition is concentration dependent. (E) Graphical determination of the apparent inhibition constant Ki. (F) Time-dependent autolytic activation of proulilysin to mature ulilysin in the presence of 5 mM calcium chloride. Conversion is complete after 6–24 h under the conditions assayed. (G) Activation of proulilysin (at 1 mg mL−1) is abrogated by AEBSF at 1 mM. (H) Removal of AEBSF from (G) in a PD-10 desalting column results in complete activation before 16 h in the presence of calcium chloride. Thus, AEBSF inhibition is reversible. | ||
Here, we aimed to understand the regulation of ulilysin activity through zymogenicity, and, in the process, we serendipitously discovered that AEBSF – but not PMSF – leads to reversible inhibition, which we further explored through biochemical and structural approaches. Finally, we dissected the in crystallo oligomerization behaviour of the mature enzyme and the zymogen.
2. Results and discussion
2.1. Molecular structure of proulilysin
We prepared proulilysin with the E229A mutation in which the general base/acid for catalysis was replaced with alanine. This is a common strategy to prevent autolysis and obtain intact MP zymogens for structural studies because this mutation generally does not affect the fold.9 The construct spanned segment M1–R342, which thus included the aforementioned 20-residue flexible C-terminal peptide, and was flanked by a disordered 20-residue tail on the N-terminus owing to the cloning strategy. This notwithstanding, we managed to obtain well-shaped crystals (Fig. 1B, left), which diffracted to 4.5 Å resolution (see Table 2). Exhaustive efforts to obtain higher-diffracting crystals with a shorter construct lacking these tails (S14–L321) were unsuccessful. Next, the structure solution proved challenging owing to highly anisotropic diffraction, low resolution of the diffraction data, presence of translational non-crystallographic symmetry, very high solvent content (71%) and large cell constants (max. 544 Å). Eventually, 14 protomers totalling 4284 residues (33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 586 atoms) were found in the asymmetric unit (a.u.) of the crystals (see section 2.5) by recursive molecular replacement employing a predicted model for the searches (see section 3.5).
586 atoms) were found in the asymmetric unit (a.u.) of the crystals (see section 2.5) by recursive molecular replacement employing a predicted model for the searches (see section 3.5).
| Dataset | Proulilysin E229A | Ulilysin C269A/AEBSF |
|---|---|---|
| a Abbreviations: AES, 4-(2-aminoethyl)benzenesulfonyl fluoride; a.u., crystallographic asymmetric unit; GOL, glycerol; GSS, tripeptide of sequence glycine-serine-serine; RSRZ, real-space R-value Z-score. b Values in parentheses refer to the outermost resolution shell. c For definitions, see ref. 90. d Average intensity is 〈I/σ(I)〉of unique reflections after merging according to Xscale.49 e According to the wwPDB validation Service (https://wwpdb-validation.wwpdb.org/validservice). | ||
| Beam line (synchrotron) | XALOC (ALBA) | XALOC (ALBA) |
| Space group/protomers per a.u.a | C2221/14 | P21212/2 |
| Cell constants (a, b, and c in Å) | 192, 544, 186 | 50.13, 124.66, 86.90 |
| Wavelength (Å) | 0.97926 | 0.97866 |
| Measurements/unique reflections | 254017/57824 |
874866/66338 |
| Resolution range (Å) (outermost shell)b | 94.7–4.50 (4.77–4.50) | 86.9–1.65 (1.75–1.65) |
| Completeness (%)/Rmergec |
99.5 (98.9)/0.277 (0.934) | 99.9 (89.2)/0.120 (1.655) |
|
R
measc/CC(½)c |
0.314 (1.054)/0.994 (0.874) | 0.125 (1.723)/0.999 (0.892) |
| Average intensityd | 5.3 (2.0) | 11.8 (1.8) |
| B-Factor (Wilson) (Å2)/Aver. multiplicity | 121.7/4.4 (4.5) | 53.7/13.2 (13.1) |
| Resolution range used for refinement (Å) | 94.7–4.50 | 62.3–1.65 |
| Reflections used (test set) | 56408 (842) |
65256 (752) |
| R cryst (Rfree)c | 0.289 (0.302) | 0.207 (0.243) |
| Non-H protein atoms/ionic ligands /waters/non-ionic ligands per a.u. | 33586/14 Zn2+, 14 Ca2+/-/- |
4120/2 Zn2+, 5 Ca2+/688/2 GOL, 2 AES, 2 GSS |
| Rmsd from target values | ||
| bonds (Å)/angles (°) | 0.008/0.875 | 0.006/0.901 |
| Average B-factor (Å2) | 171.2 | 29.1 |
| Analysis of protein contacts and geometrye | ||
| Ramachandran favoured/outliers/all analysed | 4026 (89%)/0/4256 | 509 (97%)/0/524 |
| Bond-length/bond-angle/chirality/plan. outliers | 1/0/0/0 | 0/0/0/0 |
| Side-chain outliers | 412 (11.2%) | 7 (1.5%) |
| All-atom clashes/clashscoree | 1446/22.1 | 15/1.7 |
| RSRZ outliers e/Fo:Fc correlation |
176 (4.1%)/0.86 | 6 (1.1%)/0.98 |
| PDB access code | 8CDB | 8CD8 |
The final refined structure of proulilysin (see Table 2 for statistics) includes residues P18–R323 plus a zinc and a calcium cation for each protomer (A–N). No further features were modelled due to the low resolution of the data. Segment P18–S60 constitutes the PS, which runs across – and thus blocks – the active-site cleft in the opposite orientation of a substrate (Fig. 2A). This is a general strategy found in MP zymogens to prevent untimely autolytic cleavage in cis as the PS:CD interaction does not conform to a Michalis complex.9 The PS includes two α-helices (α1p and α2p) and a cysteine (C23), which blocks the catalytic zinc following a “cysteine-switch mechanism”. Within metzincins, such mechanisms have been previously reported for matrix metallopeptidases,35,36 a-disintegrin-and-metallopeptidase (ADAM) enzymes37 and bacterial – but no holozoan – astacins.38,39 The mature part of proulilysin (R61–R323) is largely indistinguishable from the mature CD (Fig. 2B), which has been previously reported.13,15,16 The only exception is an “S-loop” (I239–D264) within the CSD, which adopts a different trace in the zymogen for the first “S” turn (G241–P249). It is hereafter termed “activation segment” in analogy to similar elements for latency in peptidases from the astacin family within metzincins39–41 and trypsin-type serine peptidases in general.42 The S-loop is a key regulatory element for activity of mature ulilysin as it binds two nearby calcium cations, which are ∼9 Å apart (Ca997 and Ca998 in Fig. 2C) and are essential for protein stability as a “calcium switch”. Indeed, treatment with EDTA, a broad-spectrum calcium chelator, abolishes activity, but it can be reversibly restored by removing the chelate and adding calcium.13 In the zymogen, the activation segment lacks Ca997 (Fig. 2C) and is partially flexible. Through segment D242–R245, it participates in interactions with M26–H30 from α1p (Fig. 2A). Moreover, it is engaged in packing contacts in the crystal, which would at least partially explain the low resolution of the diffraction data.
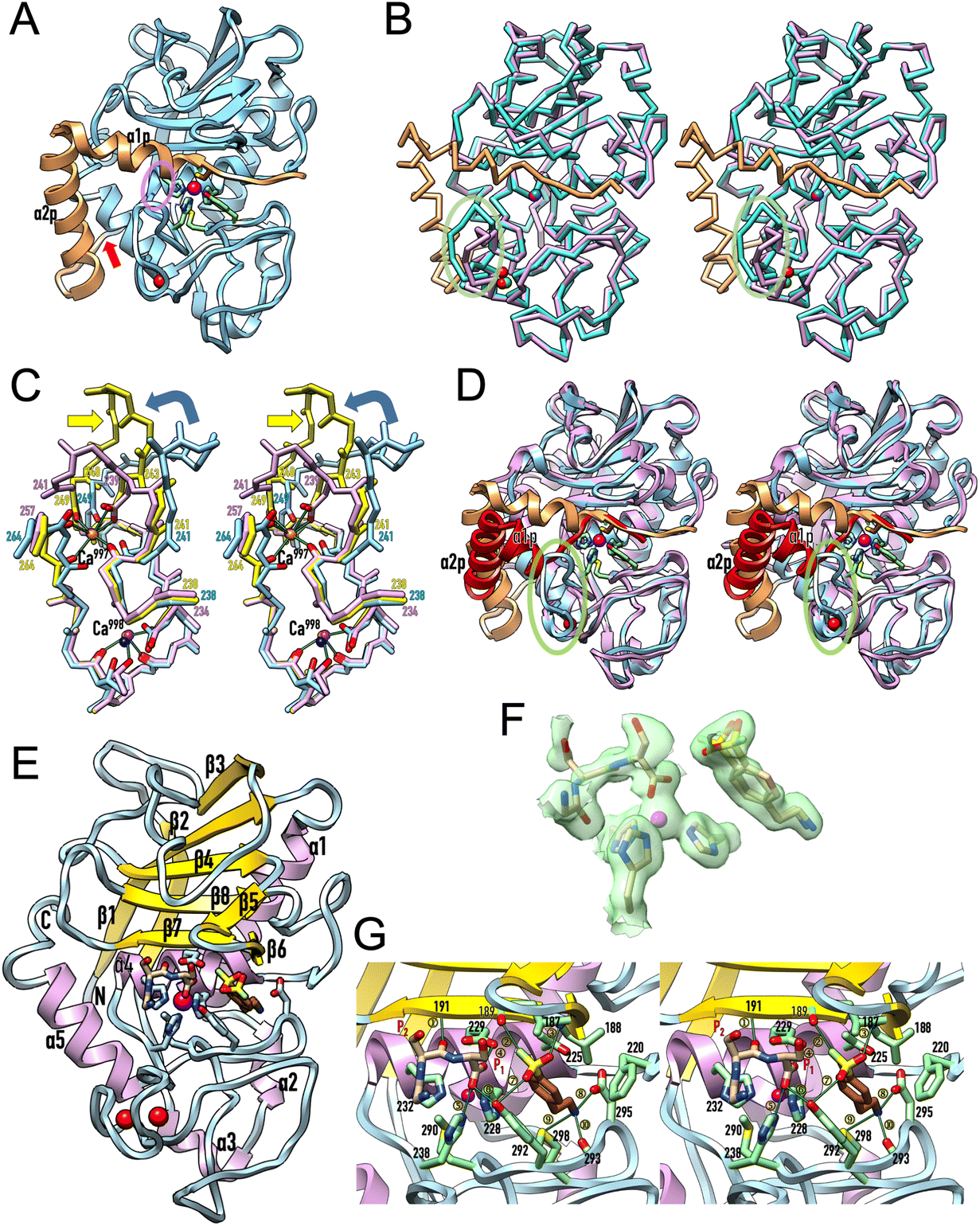 | ||
| Fig. 2 Protomer structures. (A) Ribbon-type plot of proulilysin, with the prosegment featuring helices α1p and α2p in orange and the mature moiety in light blue. The “cysteine-switch” cysteine C23 of the prosegment and the zinc-binding residues, as well as M290 and Y292, are shown for their side chains. The calcium cation (red sphere) and the catalytic zinc ion (magenta sphere) are further displayed. A red arrow pinpoints the final maturation site (S60–R61) and a semi-transparent ellipse in purple highlights the interaction between α1p and the “activation segment” at the tip of the “S-loop”. (B) Superposition in cross-eye stereo of proulilysin (colours as in (A); calcium ion in green, zinc in blue) and the mature ulilysin CD (in plum; calcium ions in red, zinc in magenta) as Cα-atom traces. The only noteworthy differences occur in the activation segment (encircled by a semi-transparent green ellipse) and the associated absence of the second calcium cation in the zymogen. (C) Superposition in stereo of the main chain of the S-loops of proulilysin (carbons and residue numbers in light blue, the calcium ion as a dark blue sphere) and mature ulilysin (carbons and residue numbers in yellow, calcium ions as orange spheres), both spanning segment H238–D264, as well as the S-loop of promirolysin (carbons and residue numbers in plum, calcium ions as purple spheres), which spans segment H234–G257. Side chains participating in calcium binding are further displayed, all calcium-binding oxygens from the protein moieties are coloured red and green lines denote the calcium–oxygen bonds of ulilysin for reference. The metal-coordinating solvent molecules have been omitted for clarity. Both ulilysin and promirolysin share both metal-binding sites (Ca997 and Ca998) and possess equivalent chain traces for their S-loops, except for E243–D248 (ulilysin) and D239–G241 (promirolysin) owing to a three-residue insertion in the archaeal enzyme (yellow arrow). In contrast, proulilysin lacks Ca997 and adopts a chain trace differing from the mature enzyme for the activation segment (G241–P249). Upon binding of the second calcium and maturation, the enzyme acquires its competent conformation (blue curved arrow). In turn, mirolysin already adopts the competent conformation of the mature moiety in the zymogen, and activation entails mere removal of the blocking prosegment (see also Fig. 4 in ref. 14). (D) Superposition in stereo of proulilysin (colouring as in panel (A)) and promirolysin (prosegment in red, mature moiety in plum) in the same view as in (A). The main differences are found in the arrangement of the two prosegment helices (labelled α1p and α2p for the Tannerella zymogen only) and, concomitantly, the S-loop (semi-transparent green ellipse). (E) Ribbon-type plot of mature ulilysin (protomer A) in complex with an upstream product fragment in S1, S2 and one position upstream (stick model with carbons in tan), as well as AEBPS (present in double occupancy; carbons in sienna) fitting into the S′1 pocket. The view follows the standard orientation of MPs.11 Selected protein side chains are depicted with carbons in light blue, the zinc and calcium cations are shown as magenta and red spheres, respectively. The regular secondary structure elements of the peptidase are presented as arrows for β-strands (labelled β1–β8) and ribbons for α-helices (α1–α5). The N- and the C-terminus are labelled. (F) Experimental (2mFobs–DFcalc)-type Fourier omit map contoured at 0.7σ above threshold superposed on the catalytic zinc, the three zinc-binding histidine residues, the AEBSF molecule (present in double conformation) and the upstream product fragment. (G) Close-up of (E) in stereo centred on the active site. Selected protein residues are shown for their side chains or relevant main-chain atoms with carbons in light green, and labelled with their residue number. Product residues in P1 and P2 are likewise labelled. Only the conformation of AEBSF that performs most interactions with the protein is shown for clarity. Thin dark green lines labelled ① to ⑩ denote electrostatic interactions (see Table 3 for the numerical values). | ||
2.2. Mechanism of maturation and comparison with mirolysin
Maturation cleavage before R61 releases the PS so that the interaction between α1p and the activation segment is disrupted. A subsequent rotation of the latter under maximal displacement of ∼9 Å (at R245) reorients E243 from its surface location to a position compatible with Ca997 binding and adoption of an overall competent conformation of the calcium switch (Fig. 2C). In the mature enzyme, the cation is further tightly bound by the main-chain oxygens of W240, P249, Q262 and A263, which are in a comparable position in the incompetent zymogen (Fig. 2C). Accordingly, calcium binding and the associated enzyme activation is mainly triggered by the repositioning of E243, which thus functions as a “glutamate switch”.To date, structural information on lower pappalysin zymogens is restricted to promirolysin14,18 whose CD shares ∼50% sequence identity with ulilysin.43 Both proulilysin E229A and promirolysin E225A (residue numbers in subscript according to UP A0A0F7IPS1/G8ULV1) were crystallized in the absence of calcium (see section 3.4 and ref. 14) but both structures contain the calcium site around Ca998 (Fig. 2C). In contrast to the archaeal zymogen, however, the Tannerella zymogen further bears the calcium site around Ca997, which in turn is found in mature ulilysin and mirolysin.14 This results from the S-loop adopting an equivalent conformation in mature and latent mirolysin. When compared with mature ulilysin (Fig. 2C), the chain trace is also comparable bar a three-residue insertion within the first turn of the S-loop, which causes the tip of this turn to flip outward in the archaeal enzyme without affecting the calcium-binding sites. Most noteworthy, this state of the arts obeys to deviating PS structures in promirolysin and proulilysin (Fig. 2D). Indeed, while both zymogens share the general topology of two tandem helices (α1p and α2p) blocking the left, non-primed side of the cleft and a cysteine binding the catalytic zinc ion, the particular arrangement of the helices largely deviates (Fig. 2D). This causes helix α1p of the bacterial enzyme to be incompatible with the activation segment of the archaeal zymogen. In mirolysin, the topologically equivalent segment, which is three residues shorter (see above), adopts the orientation of the mature CD, i.e., it is preformed in the zymogen.
This remarkable difference in so closely related enzymes correlates with slight but significant differences in function: in contrast to ulilysin (see section 2.1), in mirolysin calcium acts first as an activator of the zymogen to yield the mature form by autolysis and then as a protector from further degradation, thus providing structural – though not thermal – stabilization to the CSD.43 Moreover, calcium is an activity enhancer but it is not essential.
2.3. AEBSF is a ulilysin inhibitor
During purification of the distinct proulilysin variants, an EDTA-free inhibitor cocktail was added after cell lysis to prevent degradation by most peptidases except MPs.30,34 Thus, this crude extract should have permitted proulilysin autoactivation upon addition of calcium, as observed for the zymogen once purified,13 but we found this was not the case. We hypothesized that any of the cocktail components might have an unexpected inhibitory effect on ulilysin, which led us to analyse SFs. These covalently target the catalytic Oγ atoms of serine peptidases and are normally not inhibitors of MPs, which, like aspartic and glutamic peptidases, operate through a polarized solvent as the attacking nucleophile.9,44,45 This notwithstanding, we found that mature ulilysin was inhibited by AEBSF in a competitive dose-dependent manner, with an apparent inhibition constant (Ki) of 4.0 μM (Fig. 1C–E). Similarly, the enzyme was inhibited by the general metal chelator EDTA (Fig. 1C and D), as previously reported.13 In contrast, it was not inhibited by other reversible and irreversible serine- and cysteine-peptidase inhibitors such as aprotinin, E-64, iodacetamide or benzamidine (Fig. 1C, D and ref. 13). Moreover, PMSF, which is also an SF and shares with AEBSF the warhead and an aromatic ring (Fig. 1A), did not inhibit ulilysin (Fig. 1C and D), thus confirming earlier findings.13 Overall, this contrasts with mirolysin, which was not inhibited by either PMSF or AEBSF (M. Ksiazek and J. Potempa, personal communication, and ref. 43). Finally, another lower pappalysin, leptolysin, was also insensitive to PMSF but AEBSF was not tested.46We next studied the effect of AEBSF on the autolytic activation of proulilysin in vitro. In the presence of 5 mM calcium chloride, the MP zymogen underwent activation, which was complete after <6 hours (Fig. 1F). In contrast, activation was ablated if a further 2 mM AEBSF was present in the reaction mixture (Fig. 1G). This inhibition was reverted upon removal of the inhibitor (Fig. 1H). Thus, both mature and latent ulilysin are efficiently inhibited by AEBSF in a dose-dependent, competitive and reversible fashion, i.e. not according to the conventional covalent mechanism of SFs.
2.4. Structure of the ulilysin/AEBSF complex
To study the modus of inhibition of AEBSF, we obtained its high-resolution structure in complex with mature ulilysin by soaking of unbound protein crystals (Fig. 1B, right), which revealed that the inhibitor molecule is present in two very similar conformations (Fig. 2E and F). It is tightly bound in the deep S′1 specificity pocket of the enzyme, so that the terminal primary amine establishes a salt bridge with D295 at the bottom of the pocket and further interacts with M298Sδ and V293O (see Fig. 2G and Table 3). The aromatic/aliphatic part of AEBSF is sandwiched by the hydrophobic side chains of L188, F220, T225, H228 and Y298, which frame the S′1 pocket. The SF warhead is not involved in covalent binding but establishes electrostatic interactions with L188N, G189O and Y292Oη (Fig. 2G and Table 3). Most noteworthy, the catalytic zinc is not targeted, so AEBSF operates through competitive inhibition of substrate binding but not catalytic hindrance. Overall, this inhibitory modus also explains the lack of inhibition by PMSF, as this SF does not possess an amine on the end of the aromatic ring opposite to the warhead to block the S′1 pocket (Fig. 1A).| A + a | A + b | B + a | B + b | |
|---|---|---|---|---|
| Indicated are the values in Å of the ten distances (①–⑩) displayed in Fig. 2G for the two ulilysin protomers (A and B) and the two alternate conformations of AEBSF (a and b), respectively. | ||||
| ① | 2.83 Å | — | 2.84 Å | — |
| ② | 2.76 Å | — | 2.83 Å | — |
| ③ | 2.66 Å | 2.79 Å | 2.91 Å | 2.93 Å |
| ④ | 2.48 Å | — | 2.47 Å | — |
| ⑤ | 2.58 Å | — | 2.59 Å | — |
| ⑥ | 2.87 Å | — | 2.85 Å | — |
| ⑦ | 3.15 Å | 3.34 Å | 3.06 Å | 3.29 Å |
| ⑧ | 2.70 Å | 2.59 Å | 2.58 Å | 2.65 Å |
| ⑨ | 3.42 Å | 3.22 Å | 3.32 Å | 3.22 Å |
| ⑩ | 2.62 Å | 3.20 Å | 2.82 Å | 3.08 Å |
The structure of the complex further revealed a tripeptide of tentative sequence G–S–S in the non-primed side of the cleft, thus mimicking a left-behind upstream product fragment after catalysis (Fig. 2E, G and Table 3). The two C-terminal serine residues occupy positions S2 and S1 of the cleft, respectively, in extended conformation owing to an inter-main-chain interaction of the P2-serine with A191N from the upper-rim β-strand, as expected for a product. The C-terminal carboxylate binds the zinc in a (distorted) bidentate fashion, again as expected for a product. However, the upstream glycine of the tripeptide points out of the cleft owing to a 90°-rotation of the polypeptide chain at the P2 serine. This is reminiscent of the chain trace of the prosegments of proulilysin and promirolysin (Fig. 2D), although in reverse orientation (see sections 2.1 and 2.2). Overall, this upstream product complex complements prior structures of ulilysin with downstream product fragments and primed-side inhibitors.13,15
2.5. Considerations about ulilysin oligomerization
Despite migrating as a monomer in size-exclusion chromatography,13,15,16 all previously reported and present crystal structures of mature ulilysin (Table 4) revealed identical types of dimers (Fig. 3A). Indeed, the protein has been previously crystallized indistinctly in four crystal forms from space groups P21212 and C2221, with disparate cell constants, and crystals contained either two or four protomers per a.u. The crystals with four protomers (Protein Data Bank entry [PDB] 3LUM;17) included one dimer formed by chains A and C, while chains B and D formed respective dimers with crystallographic symmetry mates, thus totalling three identical dimers formed by variable combinations of crystallographic and non-crystallographic chains. Dimers are generated by a vertical twofold relating the two protomers joined by the respective front surfaces of Fig. 2E after a relative rotation in the plane of ∼35°. Thus, mature ulilysin has a strong propensity to associate symmetrically in an equivalent fashion despite differences in crystal packing.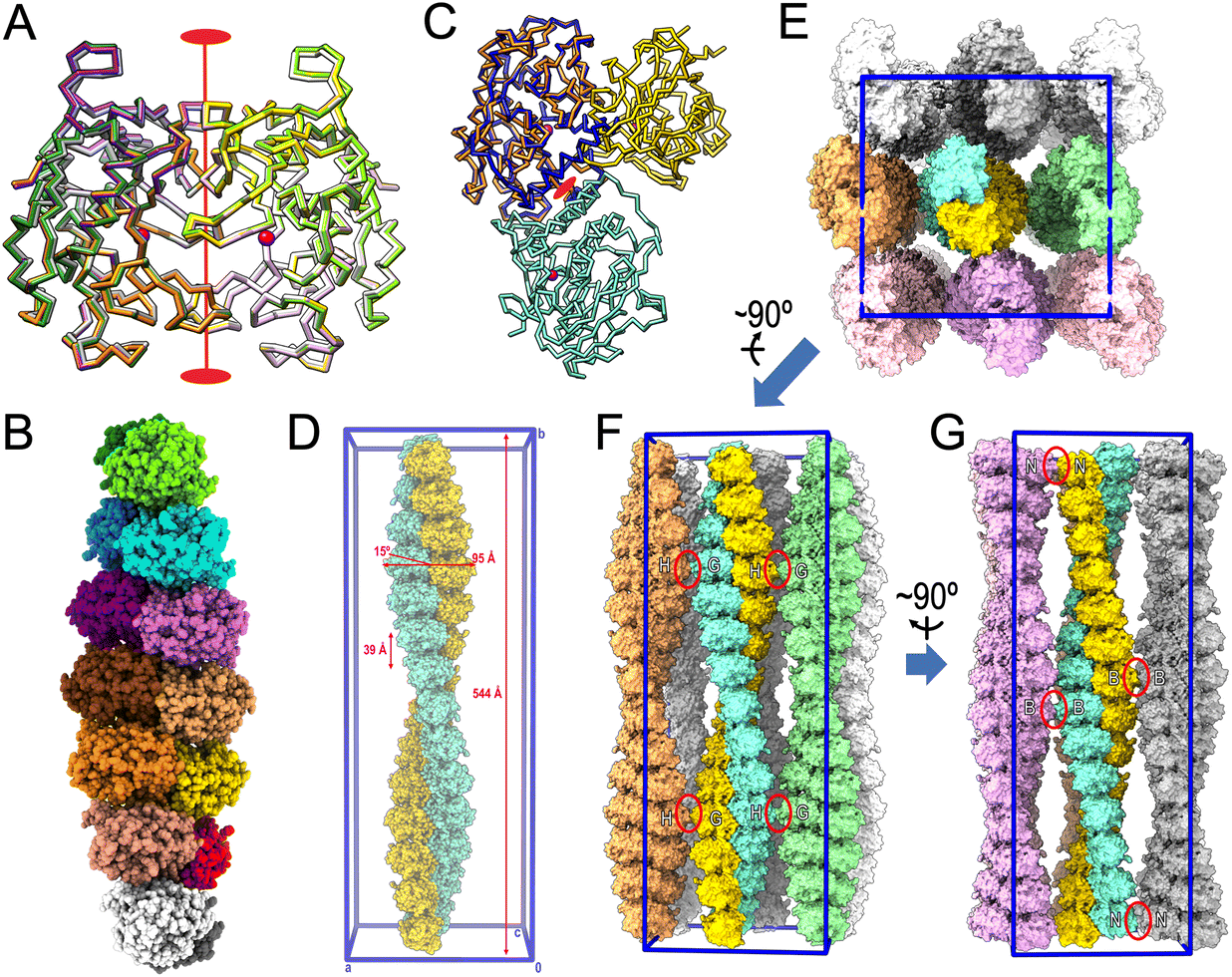 | ||
| Fig. 3 Oligomerization within the crystals. (A) Superposition of the Cα-traces of the mature ulilysin dimers found in PDB 2J83 (dark green/chartreuse), PDB 2LUM (plum/purple), PDB 2LUN (white/grey) and the complex with upstream product and AEBSF (protomers in gold/orange; PDB 8CD8). See also Table 4. The view is perpendicular to the twofold axis (in red) relating both protomers. The overlapping zinc cations are shown as magenta spheres to mark the active site. (B) Full-atom model of the seven identical dimers (protomers in, bottom to top, white/gray, salmon/red, orange/gold, brick/coral, orchid/magenta, dodger blue/cyan and dark green/chartreuse) present in the asymmetric unit of the proulilysin crystals. (C) Same as (A) showing only the ulilysin dimer of the product + AEBSF complex (gold/orange) after a horizontal 45°-rotation downward. The orange protomer was superposed onto the mature moiety of one of the protomers of the proulilysin dimer (dodger blue), which interacts with a second protomer (in aquamarine) over a different interface that is created by its own twofold (red ellipse). This twofold has its axis rotated 45° towards the reader when compared with the twofold of mature dimers. (D) Two dimeric heptamers of (B) from vertically vicinal asymmetric units associate to a continuous left-handed double helix of ∼544 Å pitch and ∼95 Å width, whose unbent axis is parallel to and spans the entire length of cell axis b of the crystal. The unit cell is further shown in blue for reference (0 stands for the origin; a, b and c are the unit-cell axes). The two congruent intertwined helices (shown as gold and aquamarine full-atom models) are translated relative to each other along the helix axis by half the helix pitch (∼272 Å). The two protomers give rise to each step of the double helix are not perpendicular to the helix axis but inclined by ∼15°. The double helix extends ∼39 Å and rotates ∼26° per step. (E) View resulting from a horizontal 90°-rotation towards the reader of (D), i.e., along cell axis b, of the full content of the unit cell (framed in blue), which includes eight helices further to the one shown in (D). (F) View roughly perpendicular to (E) from which the three bottom double helices (in pink tones) were removed to provide insight into crystal packing contacts. The only lateral interactions are observed between G and H protomers from vicinal double helices (red ellipses). (G) Roughly perpendicular view to (F) depicting the two left columns of double helices of (E). The only interactions are observed between B protomers and N protomers, respectively, from vicinal double helices (red ellipses). | ||
| PDB code [ref.] | Symmetry | Cell constants (a, b, c in Å) | Content of a.u. |
|---|---|---|---|
| a The mature dimers are all equivalent and differ from zymogen dimers, which are also all equal. | |||
| Mature ulilysin (all C269A) | |||
| 2CKI 13 | P21212 | 50, 126, 87 | One dimera |
| 8CD8 | P21212 | 50, 126, 87 | One dimer |
| 2J83 16 | C2221 | 119, 61, 169 | One dimer |
| 3LUM 17 | C2221 | 97, 174, 125 | Two dimers |
| 3LUN 17 | C2221 | 59, 116, 165 | One dimer |
| Prolulilysin (E229A) | |||
| 8CDB | C2221 | 192, 544, 186 | Seven dimersa |
With respect to the proulilysin crystals, fourteen protomers were found in the a.u. forming seven dimers (Fig. 3B). These dimers, which follow a twofold at the interface between respective PS helices α2p, are distinct from the mature dimers owing to the presence of the PS, which would interfere in the latter dimerization (Fig. 3C). Here, protomers do not associate over the front surface of Fig. 2A but rather through the lower left surface, and the twofold has its axis rotated 45° when compared with the axis relating mature dimers. Two dimeric heptamers from vertically vicinal a.u.s associate to a continuous left-handed double helix of ∼544 Å pitch and ∼95 Å width, whose straight axis is parallel to and spans the entire length of the crystal cell axis b (Fig. 3D). The two congruent intertwined helices are translated relative to each other along the helix axis by half the helix pitch (∼272 Å). Moreover, the two protomers from each of the two intertwined helices that interact and give rise to each step of the double helix are not perpendicular to the helix axis but inclined by ∼15°. In addition, the double helix extends ∼39 Å and rotates ∼26° per step. Overall, the crystal is formed through the parallel association of such continuous double helices (Fig. 3E–G). Superhelices establish only very few contacts with neighbours, namely among G and H protomers from laterally contacting double helices (Fig. 3F) and among B and N protomers from frontally/posteriorly interacting double helices (Fig. 3G). These findings explain the low overall diffraction resolution (few crystal contacts) and the strongly anisotropic diffraction of the crystals (protein fibers within the crystals).
3. Materials and methods
3.1. Protein production and purification
Wild-type proulilysin and the E229A and C269A mutants, all spanning residues M1–R342 (see UniProt database [UP] entry Q8TL28 for residue numbering), were expressed in BL21 (DE3) Star Escherichia coli cells transformed with a pET28a-derived vector encoding the respective protein fused with an N-terminal hexahistidine (His6)-tag, similarly to reported previously for the C269A variant.13,15,16,47 The E229A mutation was introduced by site-directed mutagenesis using the Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) employing 5′-CCATGCAATCGGACACTGGTTA-3′ as the forward primer and 5′-CCGATTGCATGGGTTGCCGT-3′ as the reverse primer with wild-type proulilysin as the template. A shorter construct of proulilysin E229A spanning residues S14–L321 was obtained by PCR using the aforementioned polymerase with 5′-ATATCATATGTCATCTGAAGTGCCAA-3′ as the forward primer and 5′-ATATCTCGAGTTACAGGAACGATGATCTTGGTCC-3′ as the reverse primer, and the vector encoding proulilysin E229A as the template.Single bacterial colonies transformed by heat shock with the respective vectors were grown overnight in 5 mL starter pre-cultures of Luria–Bertani medium (LB) supplemented with 50 μg mL−1 kanamycin at 20 °C under gentle agitation at 200 rpm in an INFORS orbital incubator. Subsequently, 500 mL of medium were inoculated with a single pre-culture, and cells were grown at 37 °C under agitation until the culture reached an OD600 of ∼0.8. Protein expression was then induced by adding 0.1 mM of isopropyl-β-D-1-thiogalactopyranoside, and the culture was further incubated overnight at 20 °C under agitation. Cells were harvested by centrifugation (3500g; 20 min; 4 °C) and washed twice with washing buffer (50 mM Tris·HCl, 250 mM sodium chloride, pH 7.5), resuspended in the same buffer plus 10–20 mM imidazole and “cOmplete” EDTA-free protease inhibitor cocktail tablets (Roche Life Sciences), and lysed with a cell disruptor (Constant Systems) operated at 1.35 kbar. The cell debris was removed by centrifugation (50000g; 30 min; 4 °C), and the supernatant containing proulilysin was subsequently filtered, loaded onto a 5 mL nickel-nitrilotriacetic acid HisTrap HP column (GE Healthcare) and washed with washing buffer plus 20–40 mM imidazole. The protein was subsequently eluted by passing 2–3 times 6 mL elution buffer (washing buffer plus 250–300 mM imidazole). After this affinity chromatography step, protein containing fractions were concentrated using centricons of 10 kDa molecular-mass cutoff and applied onto a Superdex 75 Increase 10/300 GL column attached to an ÄKTA Purifier chromatography apparatus (both from Cytiva) for size-exclusion chromatography at 20 °C with purification buffer (20 mM Tris·HCl, 150 mM sodium chloride, pH 7.5). The purified protein was collected in 0.5 mL fractions, and its purity was assessed by SDS-PAGE on custom-made 14% glycine gels followed by staining with Coomassie Brilliant Blue (Sigma-Aldrich).
3.2. Autolytic activation studies of proulilysin
Purified proulilysin E229A was incubated with activation buffer (purification buffer plus 5 mM calcium chloride), with or without 2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), for 0 h, 1 h, 2 h, 3 h, 4 h, 6 h and overnight, and the reactions were stopped by boiling the samples in SDS reducing buffer for SDS-PAGE analysis. In a second round of experiments, protein was first incubated with activation buffer containing AEBSF for 24 h, then buffer-exchanged to purification buffer in a PD-10 desalting column, and incubated again with activation buffer without AEBSF. Aliquots were taken after 0 h, 4 h and overnight, and reactions were stopped by boiling the samples with SDS reducing buffer for SDS-PAGE monitoring.3.3. Activity, inhibition and kinetic analyses
Enzymatic activity of ulilysin C269A was measured with the fluorescence-based EnzCheck Assay Kit (Thermo Fisher Scientific) at 4 μg mL−1 (140 nM) mature enzyme concentration, as well as with the fluorescein-conjugate DQ Gelatin from Pig Skin at 5 μM (λexc = 480 nm; λem = 520 nm; Thermo Fisher Scientific). Reactions were performed in the presence of 10 μM zinc chloride and 5 mM calcium chloride and monitored for the fluorescence response at 37 °C in an Infinite M200 microplate fluorimeter (Tecan). Inhibition of proteolytic activity against DQ Gelatin was assessed by incubation of the enzyme with EDTA (at 20 mM), AEBSF (at 0.25 mM, 1 mM, 1.5 mM and 2 mM) or phenylmethylsulfonyl fluoride (PMSF; at 1 mM and 2 mM) 5 min at 37 °C before adding the substrate.The apparent constant (Ki) of ulilysin (at 140 nM) inhibition by AEBSF (at 0.1 mM, 1 mM, 2.5 mM, 10 mM, 50 mM, 250 mM and 500 mM) was determined with the fluorogenic peptide Dabcyl–L–A–R–V–E–Edans (λexc = 340 nm; λem = 460 nm) as the substrate (at 18.4 μM) after preincubating the enzyme with the inhibitor for 1 h at room temperature.
All activity and inhibition experiments were carried out in triplicate, and data were analysed with the GraphPad software.48
3.4. Crystallization and diffraction data collection
Crystals of mature ulilysin C269A formed at 20 °C in crystallization sitting drops containing 1 μL zymogen solution at 5.5 mg mL−1 and 1 μL reservoir solution (100 mM 2-(N-morpholino)ethanesulfonic acid pH 6.5, 200 mM calcium chloride, 18% polyethylene glycol 8000), as reported previously.13,16 These crystals, which resulted from autolytic maturation at bonds S60–R61 and A322–R323 in the crystallization drops, were incubated for 16 h in soaking solution (reservoir solution plus 10 mM AEBSF) to form a complex with AEBSF. Crystals were harvested with 0.1–0.2 mm nylon loops (Hampton Research), cryoprotected by rapid passage through a drop containing soaking solution plus 20% (v/v) glycerol, and flash-vitrified in liquid nitrogen prior to storage and transport for diffraction data collection.For proulilysin E229A, we searched for initial crystallization conditions through the sitting-drop vapour diffusion method at the joint IBMB/IRB Automated Crystallography Platform (https://www.ibmb.csic.es/en/platforms/automated-crystallographic-platform). Reservoir solutions were prepared with a Tecan Freedom EVO robot and dispensed into the reservoir wells of 96 × 2-well MRC plates (Innovadyne Technologies). Crystallization nanodrops containing 100 nL each of protein and reservoir solution were dispensed by a Phoenix/RE robot (Art Robbins) into the respective protein wells. Subsequently, crystallization plates were incubated at 4 or 20 °C in thermostatic crystal farms (Bruker) and remotely inspected for crystal growth. Whenever possible, successful conditions were scaled up to the microliter range in 24-well Cryschem crystallization dishes (Hampton Research). The best crystals of proulilysin E229A were obtained at 20 °C in drops consisting of 1 μL each of protein solution at 5.5 mg mL−1 and reservoir solution (100 mM sodium acetate pH 4.5, 1 M diammonium hydrogen phosphate, 10 mM AEBSF). Crystals were harvested as above and cryoprotected by rapid passage through a drop containing reservoir solution plus 20% glycerol [v/v] prior to flash-vitrification.
X-ray diffraction data were recorded on a Pilatus 6 M pixel detector (Dectris) at 100 K at the XALOC beamline of the ALBA synchrotron (Cerdanyola, Catalonia) and processed using Xds49 and Xscale. Structure-factor amplitudes were transformed with Xdsconv to MTZ-format for the Phenix50 and CCP451 program suites. The asymmetric unit (a.u.) of the ulilysin/AEBSF crystals contained one non-crystallographic dimer and that of the proulilysin E229A crystals encompassed 14 molecules arranged as seven non-crystallographic dimers. Analysis of the diffraction data with Pointless52 within CCP4 and Xtriage53 within Phenix discarded twinning but revealed significant off-origin peaks in both cases, at fractional cell coordinates 0.50, 0.50, 0.50 (ulilysin C269A/AEBSF; 95% of the origin peak, p = 3 × 10−9) and 0.62, 0.00, 0.50 (proulilysin E229A; 33% of the origin peak, p = 9 × 10−4). These accounted for the presence of (pseudo)translational non-crystallographic symmetry (NCS), which results in distortions of the mean intensity distribution.54,55Table 2 provides essential statistics on data collection and processing.
3.5. Structural solution and refinement
The structure of the ulilysin C269A/AEBSF complex, which was obtained in the same space group (P21212) of and similar cell constants to Protein Data Bank (PDB) entry 2CKI,13 was solved by Fourier synthesis employing the dimer of protomers A and B found in the a.u. of this structure. The coordinates were subjected to crystallographic refinement using the Phenix56 and Buster/Tnt57 programs, which included translation/libration/screw-motion refinement. Subsequently, several rounds of refinement alternated with manual model building with Coot58 until the final model was obtained. The latter comprised ulilysin residues R61–A322, one zinc and two calcium cations, one AEBSF moiety (in alternate occupancy) and a tripeptide of tentative sequence G–S–S for each ulilysin protomer. An extra, functionally irrelevant calcium cation was found at the dimerization interface, and 688 solvent molecules plus two glycerol molecules were placed on the accessible surfaces of the protein molecules. The final structure displays slightly above-average Rcryst and Rfree values considering the high resolution of the data and the quality of the final Fourier map, as previously reported for other cases with translational NCS.59–62Table 2 provides essential statistics of the final model.The structure of proulilysin E229A was solved by molecular replacement using the Phaser crystallographic software63 and a computational model for residues V17–L321 obtained with AlphaFold.64 The prediction suggested that, out of the full-length sequence, this segment would correspond to a well-defined structure based on the per-residue values of the predicted local-distance difference test.65 Superposition of this model onto the experimental structure of PDB 2J83,16 which spans the mature moiety (E63–L321), enabled us to implement the catalytic zinc ion and one of the structural calcium cations into the searching model. Molecular replacement calculations with this model found 12 solutions, which formed a helical staircase with seven steps whose axis paralleled the cell axis c. Each step featured an identical dimer bar two, which only evinced one protomer. Superposition of one such dimer onto the two isolated protomers allowed us to complete the 14 protomers actually present in the a.u. Given the low resolution of the data (4.50 Å), only protomer A was manually adjusted to the electron density with Coot and then copied onto the other 13 protomers. Crystallographic refinement was painstakingly carried out with Phenix applying Cartesian-based NCS restraints, secondary-structure restraints, restrains to the AlphaFold model to steer refinement, translation/libration/screw-motion refinement considering each protomer a separate group, and one thermal displacement parameter per residue. The final model consisted of residues P18–R323 plus a zinc and a calcium cation for each of the 14 protomers (chains A–N). As for the ulilysin C269A/AEBSF structure, the final Rcryst and Rfree values were also artificially higher than expected due to the aforementioned strong translational NCS. Moreover, albeit reflections for Rfree monitoring had been selected in thin shells, the final values of Rcryst and Rfree were nonetheless partially coupled owing to the 14-fold NCS. Table 2 provides essential statistics of the final model.
3.6. Miscellaneous
Structural superpositions and structure-based sequence alignments were calculated using the SSM routine66 in Coot. Figures were prepared using Chimera67 and/or ChimeraX.68 The final refined models of ulilysin C269A/AEBSF and proulilysin E229A were validated using the wwPDB Validation Service at https://validate-rcsb-1.wwpdb.org/validservice (see Table 2) and deposited with the PDB at https://www.pdb.org (access codes 8CD8 and 8CDB).Author contributions
F.X.G.R. and U.E. conceived and supervised the project; A.R.B. and M.M.C. produced and purified proteins, A.R.B. generated mutants, A.R.B. and M.M.C. performed in vitro studies, A.R.B. and S.R.M. crystallized proteins, A.R.B. and U.E. collected diffraction data, analysed data and supervised workers; F.X.G.R. solved and refined crystal structures and wrote the manuscript with contributions from all authors.Conflicts of interest
The authors declare no financial or non-financial conflicts of interest with the contents of this article.Acknowledgements
We are grateful to the joint IBMB/IRB Automated Crystallography Platform and the Protein Purification Service for assistance during purification and crystallization. The authors also would like to thank the ALBA synchrotron in Catalonia for beamtime allocation and the beamline staff for assistance during diffraction data collection. This study was supported in part by grants from public bodies (grant references PID2019-107725RB-I00, RYC2020-029773-I, PID2021-128682OA-I00, PDC2022-133344-I00 and PID2022-137827OB-I00 from the State Agency of Research [MCIN/AEI/10.13039/501100011033; may include “Next Generation EU/PRTR” and/or “cofunded by the EU” and/or “ERDF – A way of making Europe”]; and 2021SGR00423 from the Catalan Government).References
- J. B. Lawrence, C. Oxvig, M. T. Overgaard, L. Sottrup-Jensen, G. J. Gleich, L. G. Hays, J. R. Yates, 3rd and C. A. Conover, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3149–3153 CrossRef CAS PubMed.
- C. Oxvig, J. Cell Commun. Signaling, 2015, 9, 177–187 CrossRef PubMed.
- N. D. Rawlings and A. Bateman, Protein Sci., 2021, 30, 83–92 CrossRef CAS PubMed.
- W. Bode, F. X. Gomis-Rüth and W. Stöcker, FEBS Lett., 1993, 331, 134–140 CrossRef CAS PubMed.
- H. B. Boldt, M. T. Overgaard, L. S. Laursen, K. Weyer, L. Sottrup-Jensen and C. Oxvig, Biochem. J., 2001, 358, 359–367 CrossRef CAS PubMed.
- F. X. Gomis-Rüth, Mol. Biotechnol., 2003, 24, 157–202 CrossRef PubMed.
- F. X. Gomis-Rüth, J. Biol. Chem., 2009, 284, 15353–15357 CrossRef PubMed.
- N. Cerdà-Costa and F. X. Gomis-Rüth, Prot. Sci., 2014, 23, 123–144 CrossRef PubMed.
- J. L. Arolas, T. Goulas, A. Cuppari and F. X. Gomis-Rüth, Chem. Rev., 2018, 118, 5581–5597 CrossRef CAS PubMed.
- S. D. Kobberø, M. Gajhede, O. A. Mirza, S. Kløverpris, T. R. Kjaer, J. H. Mikkelsen, T. Boesen and C. Oxvig, Nat. Commun., 2022, 13, 6084 CrossRef PubMed.
- F. X. Gomis-Rüth, T. O. Botelho and W. Bode, Biochim. Biophys. Acta, 2012, 1824, 157–163 CrossRef PubMed.
- R. A. Judge, J. Sridar, K. Tunyasuvunakool, R. Jain, J. C. K. Wang, C. Ouch, J. Xu, A. Mafi, A. H. Nile, C. Remarcik, C. L. Smith, C. Ghosh, C. Xu, V. Stoll, J. Jumper, A. H. Singh, D. Eaton and Q. Hao, Nat. Commun., 2022, 13, 5500 CrossRef CAS PubMed.
- C. Tallant, R. García-Castellanos, J. Seco, U. Baumann and F. X. Gomis-Rüth, J. Biol. Chem., 2006, 281, 17920–17928 CrossRef CAS PubMed.
- T. Guevara, A. Rodríguez-Banqueri, M. Ksiazek, J. Potempa and F. X. Gomis-Rüth, IUCrJ, 2020, 7, 18–29 CrossRef CAS PubMed.
- C. Tallant, R. García-Castellanos, A. Marrero, F. Canals, Y. Yang, J. L. Reymond, M. Solà, U. Baumann and F. X. Gomis-Rüth, Biol. Chem., 2007, 388, 1243–1253 CrossRef CAS PubMed.
- R. García-Castellanos, C. Tallant, A. Marrero, M. Solà, U. Baumann and F. X. Gomis-Rüth, Arch. Biochem. Biophys., 2007, 457, 57–72 CrossRef PubMed.
- C. Tallant, R. García-Castellanos, U. Baumann and F. X. Gomis-Rüth, J. Biol. Chem., 2010, 285, 13951–13957 CrossRef CAS PubMed.
- K. M. Zak, M. J. Bostock, I. Waligorska, I. B. Thøgersen, J. J. Enghild, G. M. Popowicz, P. Grudnik, J. Potempa and M. Ksiazek, J. Enzyme Inhib. Med. Chem., 2021, 36, 1267–1281 CrossRef CAS PubMed.
- P. F. Huesgen, P. F. Lange, L. D. Rogers, N. Solis, U. Eckhard, O. Kleifeld, T. Goulas, F. X. Gomis-Rüth and C. M. Overall, Nat. Methods, 2015, 12, 55–58 CrossRef CAS PubMed.
- I. Yiallouros, S. Vassiliou, A. Yiotakis, R. Zwilling, W. Stöcker and V. Dive, Biochem. J., 1998, 331, 375–379 CrossRef CAS PubMed.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS PubMed.
- G. Schrader, in British Intelligence Objectives Sub-committee Final Report 714, ed. S. A. Mumford and E. A. Perren, H.M. Stationery Office, London (UK), 1945, p. 8 Search PubMed.
- D. K. Myers and A. Kemp, Jr., Nature, 1954, 173, 33–34 CrossRef CAS PubMed.
- D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS.
- A. M. Gold and D. E. Fahrney, Biochemistry, 1964, 3, 2911–2913 Search PubMed.
- G. S. Salvesen and H. Nagase, in Proteolytic enzymes, ed. R. J. Beynon and J. S. Bond, Oxford University Press, Oxford (UK), 2nd edn, 2001, pp. 105–130 Search PubMed.
- J. C. Powers, J. L. Asgian, O. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- L. Polgár, in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. S. Salvesen, Academic Press, Oxford, 3rd edn, 2013, vol. 3, pp. 2524–2534 Search PubMed.
- L. Polgár, in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. S. Salvesen, Academic Press, Oxford, 3rd edn, 2013, vol. 2, pp. 1773–1784 Search PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- R. J. Beynon and G. S. Salvesen, in Proteolytic enzymes, ed. R. J. Reynon and J. S. Bond, Oxford University Press, Oxford, 2nd edn, 2001, pp. 317–330 Search PubMed.
- P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 3648–3666 CrossRef CAS.
- M. J. North and R. J. Beynon, in Proteolytic enzymes, ed. R. J. Beynon and J. S. Bond, Oxford University Press, Oxford, 2001, pp. 211–232 Search PubMed.
- R. L. Lundblad, in Handbook of biochemistry and molecular biology, ed. R. L. Lundblad and F. M. Macdonald, CRC Press, Taylor & Francis Group, 5th edn, 2018, p. 1017, DOI:10.1201/b21846.
- E. B. Springman, E. L. Angleton, H. Birkedal-Hansen and H. E. Van Wart, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 364–368 CrossRef CAS PubMed.
- G. Rosenblum, S. Meroueh, M. Toth, J. F. Fisher, R. Fridman, S. Mobashery and I. Sagi, J. Am. Chem. Soc., 2007, 129, 13566–13574 CrossRef CAS PubMed.
- F. Loechel, M. T. Overgaard, C. Oxvig, R. Albrechtsen and U. M. Wewer, J. Biol. Chem., 1999, 274, 13427–13433 CrossRef CAS PubMed.
- T. Guevara, A. Rodríguez-Banqueri, W. Stöcker, C. Becker-Pauly and F. X. Gomis-Rüth, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 1347–1357 CrossRef CAS PubMed.
- F. X. Gomis-Rüth and W. Stöcker, Front. Mol. Biosci., 2023, 9, 1080836 CrossRef PubMed.
- T. Guevara, I. Yiallouros, R. Kappelhoff, S. Bissdorf, W. Stöcker and F. X. Gomis-Rüth, J. Biol. Chem., 2010, 285, 13958–13965 CrossRef CAS PubMed.
- F. X. Gomis-Rüth, S. Trillo-Muyo and W. Stöcker, Biol. Chem., 2012, 393, 1027–1041 CrossRef PubMed.
- R. Huber and W. Bode, Acc. Chem. Res., 1978, 11, 114–122 CrossRef CAS.
- L. Koneru, M. Ksiazek, I. Waligorska, A. Straczek, M. Lukasik, M. Madej, I. B. Thøgersen, J. J. Enghild and J. Potempa, Biol. Chem., 2017, 398, 395–409 CAS.
- A. Wlodawer, A. Gutschina and M. N. G. James, in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. S. Salvesen, Academic Press, Oxford, 3rd edn, 2013, vol. 1, pp. 19–26 Search PubMed.
- L. del Amo-Maestro, S. R. Mendes, A. Rodríguez-Banqueri, L. Garzon-Flores, M. Girbal, M. J. Rodríguez-Lagunas, T. Guevara, A. Franch, F. J. Pérez-Cano, U. Eckhard and F. X. Gomis-Rüth, Nat. Commun., 2022, 13, 4446 CrossRef CAS PubMed.
- D. d. S. Courrol, C. C. F. d. S. da Silva, L. G. Prado, R. M. Chura-Chambi, L. Morganti, G. O. de Souza, M. B. Heinemann, L. Isaac, F. P. Conte, F. C. V. Portaro, R. N. Rodrigues-da-Silva and A. S. Barbosa, Front. Cell. Infect. Microbiol., 2022, 12, 966370 CrossRef PubMed.
- U. Baumann and F. X. Gomis-Rüth, in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. Salvesen, Academic Press, Oxford, 2013, vol. 1, pp. 1208–1211 Search PubMed.
- M. L. Swift, J. Chem. Inf. Comput. Sci., 1997, 37, 411–412 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 282–292 CrossRef CAS PubMed.
- P. H. Zwart, R. W. Grosse-Kunstleve and P. D. Adams, in CCP4 Newsletter on Protein Crystallography, ed. F. Remacle, Daresbury Laboratory, Daresburt, Warrington (UK), 2005, vol. 43 (Winter 2005), pp. 27–35 Search PubMed.
- Y. M. Chook, W. N. Lipscomb and H. Ke, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 822–827 CrossRef CAS PubMed.
- R. J. Read, P. D. Adams and A. J. McCoy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 176–183 CrossRef CAS PubMed.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L.-W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 861–877 CrossRef CAS PubMed.
- O. S. Smart, T. O. Womack, C. Flensburg, P. Keller, W. Paciorek, A. Sharff, C. Vonrhein and G. Bricogne, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 368–380 CrossRef CAS PubMed.
- A. Casañal, B. Lohkamp and P. Emsley, Protein Sci., 2020, 29, 1069–1078 CrossRef PubMed.
- F. F. Vajdos, S. Yoo, M. Houseweart, W. I. Sundquist and C. P. Hill, Protein Sci., 1997, 6, 2297–2307 CrossRef CAS PubMed.
- T. R. M. Barends and B. W. Dijkstra, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 2237–2241 CrossRef PubMed.
- D. Jani, S. Lutz, N. J. Marshall, T. Fischer, A. Köhler, A. M. Ellisdon, E. Hurt and M. Stewart, Mol. Cell, 2009, 33, 727–737 CrossRef CAS PubMed.
- Q. Zhao, D. Saro, A. Sachpatzidis, T. R. Singh, D. Schlingman, X. F. Zheng, A. Mack, M. S. Tsai, S. Mochrie, L. Regan, A. R. Meetei, P. Sung and Y. Xiong, Nat. Commun., 2014, 5, 2987 CrossRef PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski, A. Zidek, A. Bridgland, A. Cowie, C. Meyer, A. Laydon, S. Velankar, G. J. Kleywegt, A. Bateman, R. Evans, A. Pritzel, M. Figurnov, O. Ronneberger, R. Bates, S. A. A. Kohl, A. Potapenko, A. J. Ballard, B. Romera-Paredes, S. Nikolov, R. Jain, E. Clancy, D. Reiman, S. Petersen, A. W. Senior, K. Kavukcuoglu, E. Birney, P. Kohli, J. Jumper and D. Hassabis, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2256–2268 CrossRef CAS PubMed.
- C. C. Huang, E. C. Meng, J. H. Morris, E. F. Pettersen and T. E. Ferrin, Nucleic Acids Res., 2014, 42, W478–W484 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- Y. Sasagawa, Y. Kamio, Y. Matsubara, Y. Matsubara, K. Suzuki, H. Kojima and K. Izaki, Biosci. Biotechnol. Biochem., 1993, 57, 1894–1898 CrossRef CAS PubMed.
- Y. Sasagawa, K. Izaki, Y. Matsubara, K. Suzuki, H. Kojima and Y. Kamio, Biosci. Biotechnol. Biochem., 1995, 59, 2068–2073 CrossRef CAS PubMed.
- Y. Kamio and Y. Sasagawa, in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. S. Salvesen, Academic Press, Oxford, 3rd edn, 2013, vol. 1, pp. 1207–1208 Search PubMed.
- P. Secades, B. Álvarez and J. A. Guijarro, FEMS Microbiol. Lett., 2003, 226, 273–279 CrossRef CAS PubMed.
- D. Pérez-Pascual, E. Gómez, B. Álvarez, J. Méndez, P. Reimundo, R. Navais, E. Duchaud and J. A. Guijarro, Microbiology, 2011, 157, 1196–1204 CrossRef PubMed.
- S. Hesami, D. S. Metcalf, J. S. Lumsden and J. I. Macinnes, Appl. Environ. Microbiol., 2011, 77, 1593–1600 CrossRef CAS PubMed.
- H. Nakayama, K. Tanaka, N. Teramura and S. Hattori, Biosci. Biotechnol. Biochem., 2016, 80, 135–144 CrossRef CAS PubMed.
- C. Peralta-Figueroa, J. Martínez-Oyanedel, M. Bunster and G. González-Rocha, Antarctic Sci., 2021, 33, 633–644 CrossRef.
- J.-H. Joh, B.-G. Kim, W.-S. Kong, Y.-B. Yoo, N.-K. Kim, H.-R. Park, B.-G. Cho and C.-S. Lee, FEMS Microbiol. Lett., 2004, 239, 57–62 CrossRef CAS PubMed.
- C.-Y. Hung, K. R. Seshan, J.-J. Yu, R. Schaller, J. Xue, V. Basrur, M. J. Gardner and G. T. Cole, Infect. Immun., 2005, 73, 6689–6703 CrossRef CAS PubMed.
- M. Jusko, J. Potempa, D. Mizgalska, E. Bielecka, M. Ksiazek, K. Riesbeck, P. Garred, S. Eick and A. M. Blom, J. Immunol., 2015, 195, 2231–2240 CrossRef CAS PubMed.
- R. Zhou, X. Zhou, A. Fan, Z. Wang and B. Huang, Front. Microbiol., 2018, 9, 1528 CrossRef PubMed.
- N. S. Iwanicki, I. D. Júnior, L. L. B. de Carvalho, J. Eilenberg and H. H. d. F. Licht, Fungal Genet. Biol., 2023, 164, 103766 CrossRef PubMed.
- L.-J. Pan, L. Lu, Y.-P. Liu, S.-X. Wen and Z.-Y. Zhang, J. Integr. Agric., 2020, 19, 2044–2055 CrossRef CAS.
- M. Leonard, A. Kühn, R. Harting, I. Maurus, A. Nagel, J. Starke, H. Kusch, O. Valerius, K. Feussner, I. Feussner, A. Kaever, M. Landesfeind, B. Morgenstern, D. Becher, M. Hecker, S. A. Braus-Stromeyer, J. W. Kronstad and G. H. Braus, Front. Microbiol., 2020, 11, 1876 CrossRef PubMed.
- J. P. Wilson, J. J. Ipsaro, S. N. del Giudice, N. S. Turna, C. M. Gauss, K. H. Dusenbury, K. Marquart, K. D. Rivera and D. J. Pappin, J. Proteome Res., 2020, 19, 1459–1469 CrossRef CAS PubMed.
- K. Giddey, M. Monod, J. Barblan, A. Potts, P. Waridel, C. Zaugg and M. Quadroni, J. Proteome Res., 2007, 6, 3081–3092 CrossRef CAS PubMed.
- A. Zuccaro, U. Lahrmann, U. Güldener, G. Langen, S. Pfiffi, D. Biedenkopf, P. Wong, B. Samans, C. Grimm, M. Basiewicz, C. Murat, F. Martin and K.-H. Kogel, PLoS Pathog., 2011, 7, e1002290 CrossRef CAS PubMed.
- P. Gan, K. Ikeda, H. Irieda, M. Narusaka, R. J. O'Connell, Y. Narusaka, Y. Takano, Y. Kubo and K. Shirasu, New Phytol., 2013, 197, 1236–1249 CrossRef CAS PubMed.
- R. Baroncelli, D. B. Amby, A. Zapparata, S. Sarrocco, G. Vannacci, G. Le Floch, R. J. Harrison, E. Holub, S. A. Sukno, S. Sreenivasaprasad and M. R. Thon, BMC Genomics, 2016, 17, 555 CrossRef PubMed.
- E. Larriba, M. D. L. A. Jaime, J. Carbonell-Caballero, A. Conesa, J. Dopazo, C. Nislow, J. Martïn-Nieto and L. V. López-Llorca, Fungal Genet. Biol., 2014, 65, 69–80 CrossRef CAS PubMed.
- H. M. Einspahr and M. S. Weiss, in International Tables for Crystallography. Volume F: Crystallography of biological macromolecules, ed. E. Arnold, D. M. Himmel and M. G. Rossmann, John Wiley & Sons, Inc., Hoboken (NJ), 2nd edn, 2012, pp. 64–74, DOI:10.1107/97809553602060000809.
| This journal is © The Royal Society of Chemistry 2023 |