
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the roughness of structure–property relationships using pretrained molecular representations†
David E.
Graff
 ab,
Edward O.
Pyzer-Knapp
c,
Kirk E.
Jordan
d,
Eugene I.
Shakhnovich
a and
Connor W.
Coley
*be
ab,
Edward O.
Pyzer-Knapp
c,
Kirk E.
Jordan
d,
Eugene I.
Shakhnovich
a and
Connor W.
Coley
*be
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA
bDepartment of Chemical Engineering, MIT, Cambridge, MA 02139, USA. E-mail: ccoley@mit.edu
cIBM Research Europe, Warrington WA4 4AD, UK
dIBM Thomas J. Watson Research Center, Cambridge, MA 02142, USA
eDepartment of Electrical Engineering and Computer Science, MIT, Cambridge, MA 02139, USA
First published on 3rd August 2023
Abstract
Quantitative structure–property relationships (QSPRs) aid in understanding molecular properties as a function of molecular structure. When the correlation between structure and property weakens, a dataset is described as “rough,” but this characteristic is partly a function of the chosen representation. Among possible molecular representations are those from recently-developed “foundation models” for chemistry which learn molecular representation from unlabeled samples via self-supervision. However, the performance of these pretrained representations on property prediction benchmarks is mixed when compared to baseline approaches. We sought to understand these trends in terms of the roughness of the underlying QSPR surfaces. We introduce a reformulation of the roughness index (ROGI), ROGI-XD, to enable comparison of ROGI values across representations and evaluate various pretrained representations and those constructed by simple fingerprints and descriptors. We show that pretrained representations do not produce smoother QSPR surfaces, in agreement with previous empirical results of model accuracy. Our findings suggest that imposing stronger assumptions of smoothness with respect to molecular structure during model pretraining could aid in the downstream generation of smoother QSPR surfaces.
Introduction
The development of quantitative structure–property relationships (QSPRs) is central to molecular discovery, as they help rationalize trends in molecular properties and suggest to chemists how they can or should modify certain structural motifs to achieve a target property. A key challenge in building QSPRs arises when similar molecules possess divergent property labels. These scenarios, so-called “activity cliffs,”1–5 can pose challenges in downstream modeling tasks depending on the choice of molecular representation. As the assumption that similar molecules have similar properties breaks down, a dataset will contain both more and sharper activity cliffs, resulting in a “rougher” structure–activity landscape. In turn, these rougher QSPR landscapes make generalization harder due to the increasingly complex relationship between molecular structure and properties.Dataset roughness is typically assessed qualitatively, although there are metrics that attempt to quantify this, such as the structure–activity relationship index (SARI)6 and the modelability index (MODI).7 However, these metrics are primarily intended for application to bioactivity datasets (SARI) or to classification datasets (MODI), and extending these metrics to arbitrary regression tasks remains a challenge. To address this, we have recently proposed the ROuGhness Index (ROGI),8 a scalar metric that captures global surface roughness by measuring the loss in the dispersion of molecular properties as a dataset is progressively coarse-grained. Briefly, we are given an input representation for each molecule x ∈ ![[Doublestruck R]](https://www.rsc.org/images/entities/char_e175.gif) d and a distance metric d : d × d → , then (1) the dataset is clustered using complete linkage clustering at a given distance threshold t, (2) the dataset is coarse-grained by replacing the property label yi of each point with the mean of its respective cluster ȳj, (3) the standard deviation of the coarse-grained dataset σt is calculated, (4) steps (1)–(3) are repeated for t ∈ [0, …, max
d and a distance metric d : d × d → , then (1) the dataset is clustered using complete linkage clustering at a given distance threshold t, (2) the dataset is coarse-grained by replacing the property label yi of each point with the mean of its respective cluster ȳj, (3) the standard deviation of the coarse-grained dataset σt is calculated, (4) steps (1)–(3) are repeated for t ∈ [0, …, max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dx], (5) the area under the curve of 2(σ0 − σt) vs. t is measured to yield the ROGI. Datasets with larger ROGI values result in larger cross-validated model errors, consistent with intuition. Across a variety of datasets from GuacaMol,9 TDC,10 and ChEMBL11 and machine learning (ML) model architectures, the ROGI correlates strongly with cross-validated model root-mean-square error (RMSE) and generally outperforms alternative metrics.8
dx], (5) the area under the curve of 2(σ0 − σt) vs. t is measured to yield the ROGI. Datasets with larger ROGI values result in larger cross-validated model errors, consistent with intuition. Across a variety of datasets from GuacaMol,9 TDC,10 and ChEMBL11 and machine learning (ML) model architectures, the ROGI correlates strongly with cross-validated model root-mean-square error (RMSE) and generally outperforms alternative metrics.8
Given these strong correlations, we sought to broadly examine recent claims about the superiority of molecular representations learned by “foundation models” for chemistry12–17 through the lens of QSPR surface roughness (Fig. 1). Foundation models are a class of ML models that are trained on large, unlabeled datasets via self-supervised learning (sometimes supervised learning) and are in principle capable of adapting rapidly to downstream tasks with very few labeled data points.18 Pretrained foundation models are now standard practice in several domains, such as natural language processing,19–21 computer vision,22,23 and protein modeling.24,25 Given the abundance of unlabeled chemical data and the limited amount of data encountered in many property prediction tasks, foundation models may benefit chemistry by learning meaningful molecular representations suitable for property prediction tasks in the low data regime.
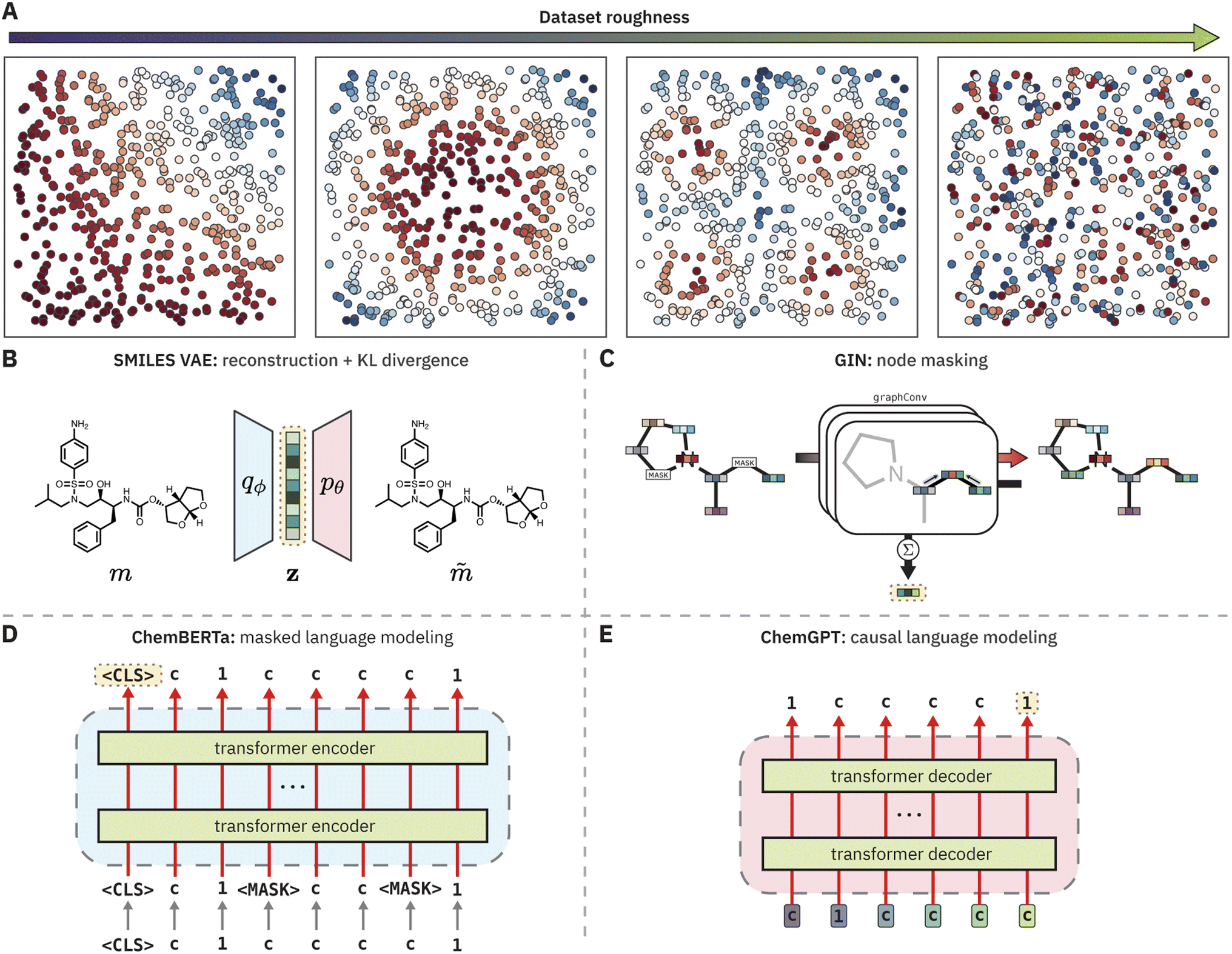 | ||
| Fig. 1 (A) Examples of increasingly rough datasets. (B, C, D, E) Schematics of pretrained chemical models evaluated in this study. Yellow, dotted boxes indicate the source of the pretrained representation. (B) A SMILES variational autoencoder (VAE) uses the mean latent representation after encoding. (C) A graph isomorphism network (GIN) uses the sum of the node hidden representations after graph convolutions. (D) ChemBERTa uses the embedding of the 〈CLS〉 token after the final transformer encoder block. (E) ChemGPT uses the embedding of the last token in the input sequence after the final transformer decoder block. | ||
Despite this interest, empirical evaluation of proposed chemical foundation models has shown mixed results. Recent work from Deng et al.26 assessed the performance both SMILES- and graph-based pretrained chemical models (PCMs), MolBERT14 and GROVER,15 respectively, on a variety of benchmark tasks from MoleculeNet27 and opioid bioactivity datasets from ChEMBL.11 For each task, they compared the performance of these proposed chemical foundation models to a random forest model trained on radius 2, 2048-bit Morgan fingerprints. The authors found that this baseline was competitive for many benchmark tasks and even superior in several of the opioid tasks. This finding is consistent with results reported in the PCM literature where learned representations offer inconsistent improvement over baseline approaches.
In this work, we complement this analysis by characterizing the roughness of the QSPR surfaces generated by PCMs on both toy and experimental modeling tasks. To do so, we reformulate ROGI as ROGI-XD to enable cross-representation comparison. While the original ROGI correlates strongly with cross-validated RMSE across datasets when holding the representation constant, it does not necessarily provide a meaningful basis for comparison among representations due to the relationship between distances and the dimensionality of a given representation. We show that for a variety of PCMs (VAE,28 GIN,29,30 ChemBERTa,12 and ChemGPT31) and a variety of molecular tasks, learned molecular representations do not provide smoother structure–property relationships than simple descriptor and fingerprint representations. The failure of PCMs to learn a continuous embedding of molecular structures that smoothly correlates with various properties of interest both (a) explains their poor empirical performance in property prediction tasks without fine-tuning and (b) motivates the use of ROGI-XD to evaluate smoothness when new pretraining strategies are proposed.
Results and discussion
Reformulation of the ROGI as ROGI-XD enables cross-dimensional comparisons
In its original formulation, ROGI values are not comparable across representations of different dimensionality. Distances between randomly-sampled points generally increase with representation size, so even when normalizing distances to the same range (e.g., [0, 1]), higher-dimensional representations do not coarse-grain until larger values of normalized distanced threshold (Fig. 2A). Ultimately, this will result in artificially low ROGI values for QSPR datasets with high-dimensional representations. To illustrate this, consider N points sampled uniformly from the unit hypercube of dimension d with random property labels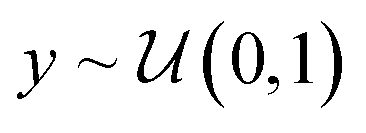 . As d increases, the normalized distance distribution of these points will become more tightly peaked and centered closer to 1 (Fig. 2B), which results in the delayed coarse-graining phenomenon mentioned earlier. This delayed coarse-graining causes the curve of loss of dispersion 2(σ0 − σt) vs. normalized distance threshold t to be depressed at lower values of t, producing lower ROGI values for higher dimensional representations (Fig. 2C). It could be argued that a higher-dimensional representation may result in a “smoother” representation due to the larger distances between points, but for large differences in d, the ROGI essentially becomes a proxy for the inverse of representation size rather than differences in the underlying SPR surface. The datasets sampled from the unit hypercube abstractly represent the “same” dataset in hyperspace as N → ∞, so they should possess roughly equal roughness values when controlling for d.
. As d increases, the normalized distance distribution of these points will become more tightly peaked and centered closer to 1 (Fig. 2B), which results in the delayed coarse-graining phenomenon mentioned earlier. This delayed coarse-graining causes the curve of loss of dispersion 2(σ0 − σt) vs. normalized distance threshold t to be depressed at lower values of t, producing lower ROGI values for higher dimensional representations (Fig. 2C). It could be argued that a higher-dimensional representation may result in a “smoother” representation due to the larger distances between points, but for large differences in d, the ROGI essentially becomes a proxy for the inverse of representation size rather than differences in the underlying SPR surface. The datasets sampled from the unit hypercube abstractly represent the “same” dataset in hyperspace as N → ∞, so they should possess roughly equal roughness values when controlling for d.
 | ||
| Fig. 2 Reformulation of ROGI as ROGI-XD enables cross-representation and cross-dimension comparisons. (A) The dendrograms produced by complete linkage clustering of eight points sampled uniformly from the domain [0, 1]d. As the size of the domain increases, the clustering steps become more compressed and happen closer to 1 (normalized distance). The minor ticks and vertical gridlines in each subplot correspond to a step in the dendrogram. (B) The distribution of normalized distance for 1000 points drawn from the domain [0, 1]d. As the dimensionality increases, the distance distribution sharpens and centers closer to 1. (C) Higher dimensional representations produce lower ROGI values. (D) Redefining the coarse-graining domain to 1 − logNclusters/logN results in similar ROGI values regardless of representation size. | ||
To minimize the impact of dimensionality on the ROGI, we change its integration variable to capture the degree of coarse-graining independent of representation size. Procedurally, “coarse-graining” entails taking a step up in the dendrogram produced during the clustering routine. Whereas originally we scan along the distance required to take such a step, we now opt to use 1 − logNclusters/logN, where Nclusters is the number of clusters at the given step in the dendrogram and N is the dataset size. This new formulation, which we refer to as ROGI-XD, produces similar values for each toy dataset regardless of its dimensionality (Fig. 2D). We note that while there are other formulations that reflect a similar concept, they must possess a constant integration domain. For example, using 1 − Nclusters/N as the x-axis produces a similar trend as above (Fig. S1†), but it is defined on the domain [0, 1 − 1/N], thus making the score dependent on N and confounding comparisons across datasets with large differences in size.
The ROGI-XD correlates strongly across representations
We next sought to evaluate how well the ROGI-XD correlates with model error across chemical representations. First, we measured ROGI-XD and cross-validated RMSE for all combinations of task, model, and representation then calculated the Pearson correlation coefficient r between ROGI-XD and cross-validated RMSE across representations for the same task and model. We analyze a variety of regression tasks using experimental ADMET datasets from the TDC10 and datasets generated using GuacaMol9 oracle functions, and we use the same ML models as in our previous study.8 We look at two fixed representations: molecular descriptors and Morgan fingerprints; four pretrained representations: SMILES variational autoencoder (VAE),28 graph isomorphism network (GIN)29 pretrained with node attribute masking,30 ChemBERTa,12 and ChemGPT;31 and 128-dimensional random embeddings. For more details, see the Materials and methods section below.The ROGI-XD produces strong correlations with model error across molecular representation for the majority of tasks and ML models tested (Fig. 3). The median correlation across all combinations of model and task ranges between 0.72 and 0.88, with the best correlations observed for both the random forest (RF) and k-nearest neighbors (KNN) models. This is in contrast to the original ROGI, which generally produces weak correlations (median r ∈ [−0.32, 0.28]) when subjected to the same analysis (Fig. S2†). As shown in the toy example above, the original ROGI is affected by representation size, so the range of dimensionalities in the representations tested (14 to 2048, Table S1†) negatively impacts correlation strength.
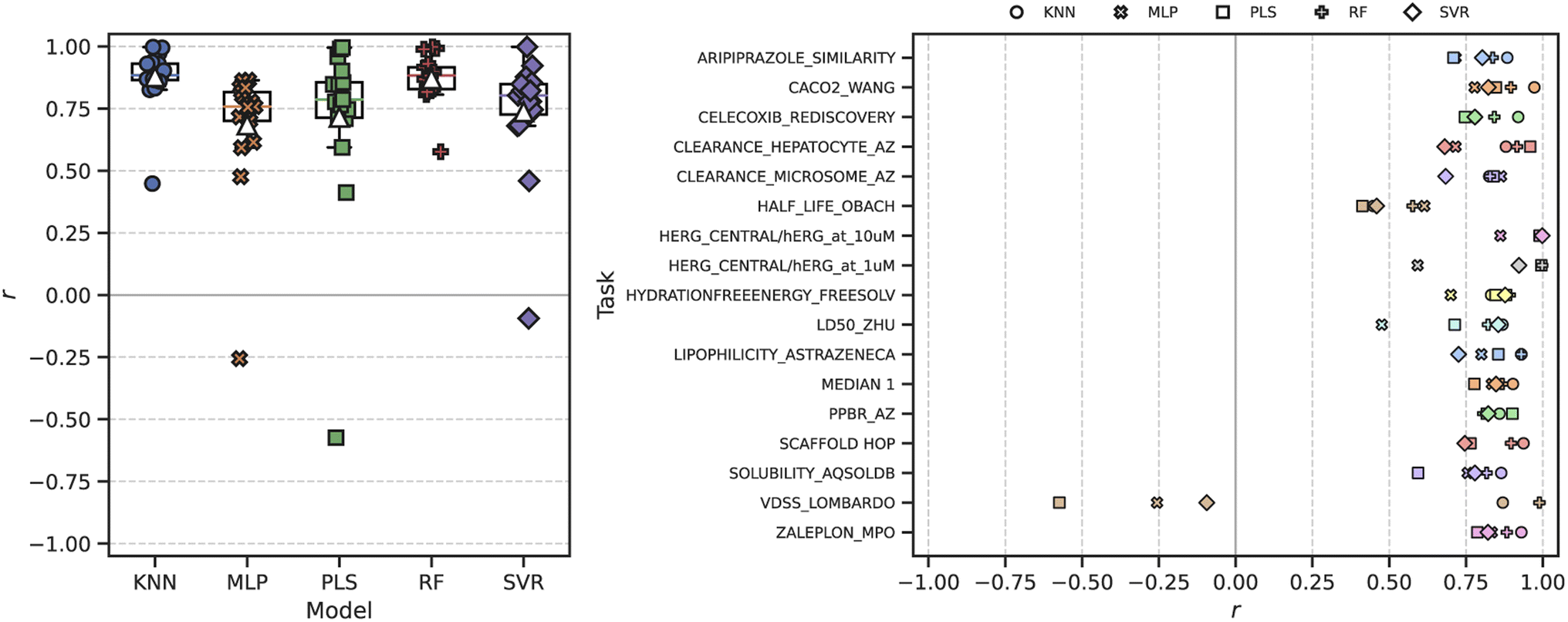 | ||
| Fig. 3 Distribution of Pearson correlation coefficients r between ROGI-XD and cross-validated RMSE across all representations evaluated for a given ML model and task. Left: Box plot of correlations grouped by ML model architecture with individual data points plotted above. The median is depicted via the solid, colored line, and the mean by the white triangle (△). Right: Correlations grouped by task. KNN: k-nearest neighbors; MLP: multilayer perceptron; PLS: partial least squares; RF: random forest; SVR: support vector regression. | ||
Of note are the generally strong correlations between ROGI-XD and the RMSE from a KNN model. This is perhaps not surprising due to the thematic similarities between these two algorithms. However, the RMSE of a KNN model on the full dataset provides worse correlations with the cross-validated RMSE of other models than does ROGI-XD for all values of k tested (Fig. S3†). For a more detailed discussion on the fundamental differences between the two, we refer a reader to the Differences between ROGI-XD and k-nearest neighbors section in the ESI† text.
When we measure the correlation between ROGI-XD and RMSE across tasks for a given model using molecular descriptors (as in our original study), we see similarly strong correlations (Fig. S5†). These correlations remain strong when we measure correlation over both representations and tasks, whereas they decrease significantly with the original ROGI (Table 1). In turn, this allows for the direct comparison of ROGI values measured for two datasets with differing representations.
| Metric | Model | ||||
|---|---|---|---|---|---|
| KNN | MLP | PLS | RF | SVR | |
| ROGI | 0.800 | 0.675 | 0.835 | 0.809 | 0.771 |
| ROGI-XD | 0.990 | 0.913 | 0.983 | 0.985 | 0.958 |
It is also possible to measure the correlation between ROGI or ROGI-XD and the minimum model error for a given task and representation. In other words, rather than treating each ML model separately as above, we now (1) measure the model error and roughness metric for all combinations of task, representation, and ML model; (2) take the minimum model error for each combination of task and representation; and (3) measure the correlation between the roughness metric and this “best-case” model error for each task. The ROGI-XD again produces strong correlations across all datasets (median r = 0.82) compared to the original ROGI (median r = 0.16) (Fig. 4). This discrepancy in correlation strength is expected because this analysis still relies on comparisons across representations.
 | ||
| Fig. 4 Distribution of Pearson correlation coefficients r between roughness metric and minimum cross-validated RMSE for a given task across all representations. Each point corresponds to an individual task. The median is depicted via the solid, colored line, and the mean by the white triangle (△). | ||
Performing a similar analysis using the RMSE of a KNN model, as above, produces competitive correlations with ROGI-XD (Fig. S4†). As one model decreases in RMSE, it is likely that so too will the RMSEs of other models. Despite this, the distribution of these correlations of KNN RMSE is much broader than that of ROGI-XD with more frequent worst-case performance.
The ROGI-XD's strong correlation with best model error across representations thus allows a user to quickly get an idea of best-case model performance for a variety of representations without resorting to empirical testing. This can further be extended to comparing best-case modelability among datasets given a set of possible representations by calculating the ROGI-XD for each representation and then selecting the lowest one for the task. For example, by selecting the representation with the lowest ROGI-XD and then optimizing over model architecture in each of our 17 tasks, the average relative increase in best-case model error would be only 6.8%. In 8 out of 17 tasks, selecting the lowest ROGI-XD identifies the optimal representation with respect to best-case model error.
Pretrained molecular representations do not provide smoother structure–activity landscapes than fingerprints and descriptors
The ROGI-XD formulation's strong correlation with model error across representations thus allows us to compare the smoothness of learned representations to that of fixed representations. We use the same pretrained representations as before to broadly survey different strategies of learning molecular representations: a recurrent neural network-based encoder-decoder framework (VAE), graph-based pretraining (GIN), encoder-only large language model (LLM; ChemBERTa), and decoder-only LLM (ChemGPT). For each task, we calculate the relative difference between ROGI-XD values for each pair of pretrained and fixed representations (i.e., ROGI-XDp/ROGI-XDf − 1, where ROGI-XDp and ROGI-XDf are the ROGI-XD values of a given pretrained and fixed representation, respectively, for the same task). While there are other pretraining techniques and model architectures available, we do not intend for this analysis to be exhaustive. Rather, our goal is to provide a supplementary technique by which to understand trends in model performance.We find that across all tasks tested above, PCMs do not generate quantitatively smoother QSPR surfaces when compared to those generated via molecular descriptors or fingerprints (Fig. 5). In more than 50% of the tasks evaluated, both descriptors and fingerprints generated smoother QSPR surfaces. The median relative ROGI-XD values for each pretrained representation compared to descriptors and fingerprints range between 9.1–21.3% and 2.3–10.1%, respectively. Indeed, these ROGI-XD values are consistent with the cross-validation results of descriptors and fingerprints being generally lower in RMSE than the pretrained representations (Fig. S6 and S7†). An extreme case is the Scaffold Hop task, where the GIN, ChemGPT, and ChemBERTa representations produce ROGI-XDs of 0.150, 0.174, and 0.172, respectively, compared to the 0.085 of descriptors. However, we emphasize that PCMs do not generate bad representations, but rather that these learned representations are not smoother than simple, fixed representations.
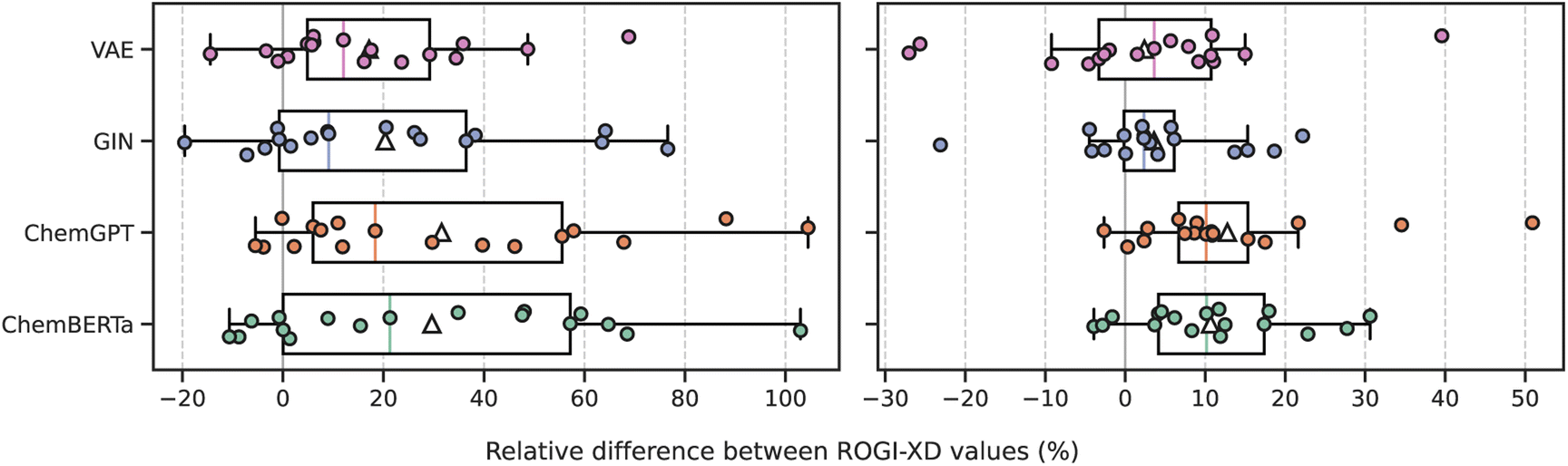 | ||
| Fig. 5 Distribution of relative difference between ROGI-XD values of the given pretrained representation (y-axis) and fixed representation (top) for each task. Positive values indicate that the pretrained representation produces a rougher QSPR surface, and negative values indicate the opposite. Individual data are plotted above and box plot is plotted above. The median is depicted via the solid, colored line, and the mean by the white triangle (△). FP: Morgan fingerprint; VAE: variational autoencoder; GIN: graph isomorphism network. | ||
One potential benefit of learned representations is their ability to be finetuned using task-specific data. Given the nature of the learning task, we would naturally expect this to smooth the corresponding QSPR surface. We tested this approach with our VAE model through a contrastive loss between the latent representations and target properties on the Lipophilicity_AstraZeneca task from the TDC.10 Finetuning the VAE on 80% of the dataset (Ntot = 4200) improves the ROGI-XD from 0.254 to 0.107 (±0.02), considerably smoother than that of descriptors at 0.227. Attempting the same strategy on the CACO2_WANG task (Ntot = 910) yields a ROGI-XD of 0.143 (±0.05), no smoother than descriptors (0.132). The impact of finetuning on smoothness varies and is sensitive to both the task and the number of labeled examples.
Studies that introduce new pretraining techniques or model architectures rarely, if ever, analyze the smoothness of the underlying QSPR surfaces. Rather, they benchmark their method on a variety of property prediction tasks and frequently report mixed results; on some tasks, the new technique outperforms the current state-of-the-art, but on others, it fails to compete with simple baselines. In our evaluations, we find that baseline representations outperform learned representations in 10 of the 17 tasks tested. The relative roughness observed for the QSPR surfaces generated by these learned representations is consistent with their generally mixed performance in property prediction tasks. Thus, we believe that this lack of smoothness at least partially explains their inability to consistently outperform established molecular representations on supervised learning benchmarks.
While it is intuitive that worse model performance could be due to a rougher QSPR surface, such analysis has not previously been conducted. We attribute this to the former lack of metrics that can (i) quantify QSPR surface roughness and (ii) directly compare these quantities across representations. Our analysis using the ROGI-XD allows us to quantitatively show that this is typically not the case.
Conclusion
We have described ROGI-XD, a reformulation of the ROGI that enables comparison of structure–activity roughness values across representations via changing the integration variable. The ROGI-XD correlates strongly with cross-validated model error both across representations for a given task (median r = 0.72–0.88) and over both representations and tasks (median r = 0.91–0.99). We then use ROGI-XD to evaluate several recently-reported pretrained chemical representations from SMILES VAE, GIN, ChemGPT, and ChemBERTa. Across all of the tasks evaluated, the ROGI-XD values of these representations were no smoother than those of fingerprints and descriptors. These results are consistent with the empirical results of various benchmark studies which show that pretrained models are not universally superior to fingerprints or descriptors.Taken together, these observations suggest that more work remains in developing chemical foundation models. Though it is unreasonable to expect that any single pretrained representation will produce a smoother QSPR surface in every task, a reasonable desideratum is that such a representation is of comparable smoothness to simple baseline representations for a majority of useful properties. The ROGI-XD is thematically similar to a contrastive loss, as both will scale proportionally with the frequency and severity of activity cliffs in a given dataset. Imposing stronger assumptions of smoothness with respect to molecular structure during model pretraining by weak supervision on simple, calculable properties could aid in producing smoother QSPR surfaces.
A limitation of our analysis is that we have treated the pretrained representations as static for downstream modeling; an alternative is to fine-tune them by training the model on additional, labeled data, in turn helping to smooth the corresponding QSPR surface. In a sense, the evaluations here have demonstrated the need for fine-tuning in the absence of a universally smooth representation. This introduces many additional design choices, so we leave this evaluation for future work.
Materials and methods
All code and data used in this work is available at: https://github.com/coleygroup/rogi-xd.Representations
Tasks
As in our previous work,8 we used two groups of tasks, (1) ADME and toxicity datasets from the TDC10 and (2) toy datasets generated by sampling 10000 molecules from the ZINC250k dataset and then calculating the following GuacaMol9 oracle function values for these molecules: Scaffold Hop, Median 1, Aripiprazole_Similarity, Zaleplon_MPO, Celecoxib_Rediscovery. We exclude many of the original GuacaMol tasks, as their oracle functions use descriptor values in the scoring function that overlap with our descriptor representation. For the hERG_at_1uM and hERG_at_10uM tasks from the TDC, datasets were downsampled to 10000 molecules; in these instances, reported ROGI values are the mean of five random subsamples.
Cross-validation
As in previous work,8 we performed 5-fold cross-validation using the following five regression models from Scikit-learn:39k-nearest neighbors (KNN), multilayer perceptron (MLP), partial least squares (PLS), random forest (RF), and support vector regression (SVR). All models utilized default settings except for the RF model, which used an n_estimators value of 50. We scale the property labels to the range [0, 1] before cross-validation and report the mean root-mean-squared error (RMSE) of all five folds.Finetuning
We finetune the VAE model by adding a contrastive loss term to the loss function:where
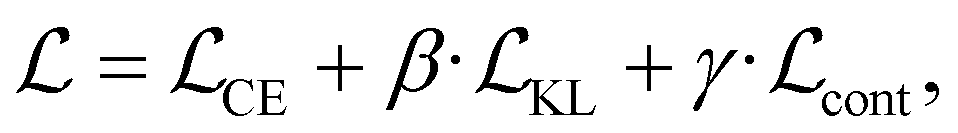
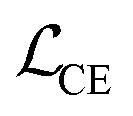 ,
, 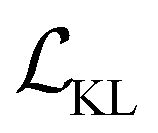 , and
, and  are the cross-entropy, KL divergence, and contrastive terms, respectively, and β and γ are loss weights. We set these weights to 0.1 and 50, respectively. For two points i and j, the contrastive term is defined as the squared difference between their distance in the latent space and their distance in the target space:
are the cross-entropy, KL divergence, and contrastive terms, respectively, and β and γ are loss weights. We set these weights to 0.1 and 50, respectively. For two points i and j, the contrastive term is defined as the squared difference between their distance in the latent space and their distance in the target space:where z and y are the latent representation and target value, respectively, of a given point and d :
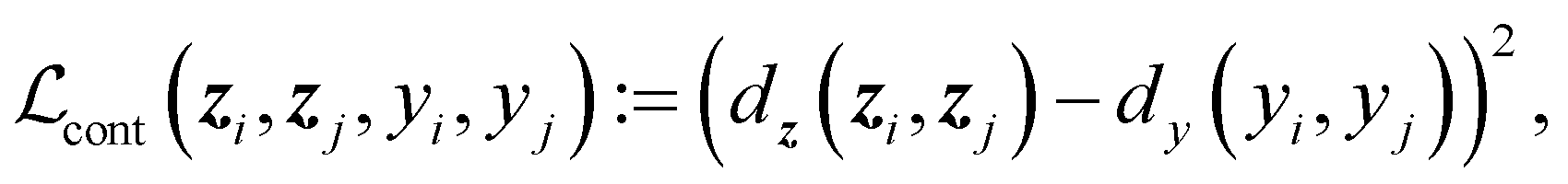 D × D → ≥0 is a (pseudo)metric. For latent space distances dz, we use the cosine distance, and for target space distances dy, we use the absolute value. We minimize the mean of all pairwise differences across an entire batch.
D × D → ≥0 is a (pseudo)metric. For latent space distances dz, we use the cosine distance, and for target space distances dy, we use the absolute value. We minimize the mean of all pairwise differences across an entire batch.
Software
This work was performed using Python 3.9, fastcluster 1.2.6, NumPy 1.2.4, PyTorch Lightning 1.9.0, RDKit 2022.9.4, Pandas 1.5.3, PyTDC 0.3.9, PyTorch 1.13.0, Scikit Learn 1.2.1, SciPy 1.8.1, SELFIES 2.1.1, TorchDrug 0.2.0, and Transformers 4.26.0.Hardware
This work was performed on a workstation with two AMD Ryzen Threadripper PRO 3995WX CPUs, four Nvidia A5000 GPUs, and 512 GB of RAM running Ubuntu 20.04 LTS.Data availability
All code, data, and scripts used in this work is available at: https://github.com/coleygroup/rogi-xd. All results can be regenerated automatically using make all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the MIT-IBM Watson AI Lab. The authors thank Jenna Fromer and Itai Levin for commenting on the manuscript.References
- QSAR: Rational Approaches to the Design of Bioactive Compounds: Proceedings of the VIII European Symposium on Quantitative Structure-Activity Relationships, Sorrento, Italy, 9-13 September 1990, ed. Silipo, C. and Vittoria, A., Elsevier Science, Amsterdam; New York, 1991 Search PubMed.
- G. M. Maggiora, J. Chem. Inf. Model., 2006, 46, 1535 CrossRef CAS PubMed.
- D. Stumpfe and J. Bajorath, J. Med. Chem., 2012, 55, 2932–2942 CrossRef CAS PubMed.
- D. Stumpfe, Y. Hu, D. Dimova and J. Bajorath, J. Med. Chem., 2014, 57, 18–28 CrossRef CAS PubMed.
- D. Stumpfe, H. Hu and J. Bajorath, ACS Omega, 2019, 4, 14360–14368 CrossRef CAS PubMed.
- L. Peltason and J. Bajorath, J. Med. Chem., 2007, 50, 5571–5578 CrossRef CAS PubMed.
- A. Golbraikh, E. Muratov, D. Fourches and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 1–4 CrossRef CAS PubMed.
- M. Aldeghi, D. E. Graff, N. Frey, J. A. Morrone, E. O. Pyzer-Knapp, K. E. Jordan and C. W. Coley, J. Chem. Inf. Model., 2022, 62, 4660–4671 CrossRef CAS PubMed.
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Nat. Chem. Biol., 2022, 18, 1033–1036 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- W. Ahmad, E. Simon, S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2022, preprint, arXiv:2209.01712 [cs.LG], DOI:10.48550/arXiv.2209.01712.
- O. Méndez-Lucio, C. Nicolaou and B. Earnshaw, arXiv, 2022, preprint, arXiv:2211.02657[cs, q-bio], DOI:10.48550/arXiv.2211.02657.
- B. Fabian, T. Edlich, H. Gaspar, M. Segler, J. Meyers, M. Fiscato and M. Ahmed, arXiv, 2020, preprint, arXiv:2011.13230 [cs], DOI:10.48550/arXiv.2011.13230.
- Y. Rong, Y. Bian, T. Xu, W. Xie, Y. Wei, W. Huang and J. Huang, arXiv, 2020, preprint, arXiv:2007.02835 [cs, q-bio], DOI:10.48550/arXiv.2007.02835.
- S. Wang, Y. Guo, Y. Wang, H. Sun and J. Huang, in Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, ACM: Niagara Falls, NY, USA, 2019, pp. 429–436 Search PubMed.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Nat. Mach. Intell., 2022, 4, 1256–1264 CrossRef.
- R. Bommasani, et al., arXiv, 2022, preprint, arXiv:2108.07258 [cs], DOI:10.48550/arXiv.2108.07258.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, arXiv, 2019, preprint, arXiv:1810.04805 [cs], DOI:10.48550/arXiv.1810.04805.
- C. Raffel, N. Shazeer, A. Roberts, K. Lee, S. Narang, M. Matena, Y. Zhou, W. Li and P. J. Liu, arXiv, 2020, preprint, arXiv:1910.10683 [cs, stat], DOI:10.48550/arXiv.1910.10683.
- T. B. Brown, et al., arXiv, 2020, preprint, arXiv:2005.14165 [cs], DOI:10.48550/arXiv.2005.14165.
- L. Yuan, et al., arXiv, 2021, preprint, arXiv:2111.11432 [cs], DOI:10.48550/arXiv.2111.11432.
- A. Radford, J. W. Kim, C. Hallacy, A. Ramesh, G. Goh, S. Agarwal, G. Sastry, A. Askell, P. Mishkin, J. Clark, G. Krueger and I. Sutskever, arXiv, 2021, preprint, arXiv:2103.00020 [cs], DOI:10.48550/arXiv.2103.00020.
- N. Brandes, D. Ofer, Y. Peleg, N. Rappoport and M. Linial, Bioinformatics, 2022, 38, 2102–2110 CrossRef CAS PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli, A. dos Santos Costa, M. Fazel-Zarandi, T. Sercu, S. Candido and A. Rives, Science, 2023, 379, 1123–1130 CrossRef CAS PubMed.
- J. Deng, Z. Yang, H. Wang, I. Ojima, D. Samaras and F. Wang, arXiv, 2022, preprint, arXiv:2209.13492 [cs, q-bio], DOI:10.48550/arXiv.2209.13492.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- K. Xu, W. Hu, J. Leskovec and S. Jegelka, How Powerful are Graph Neural Networks?, 2019 Search PubMed.
- W. Hu, B. Liu, J. Gomes, M. Zitnik, P. Liang, V. Pande and J. Leskovec, Strategies for Pre-training Graph Neural Networks, 2020 Search PubMed.
- N. Frey, R. Soklaski, S. Axelrod, S. Samsi, R. Gomez-Bombarelli, C. Coley and V. Gadepally, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-3s512.
- RDKit: Open-source cheminformatics, https://rdkit.org/, accessed 01/05/2023 Search PubMed.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J.-L. Reymond, Nat. Mach. Intell., 2021, 3, 144–152 CrossRef.
- Z. Zhu, C. Shi, Z. Zhang, S. Liu, M. Xu, X. Yuan, Y. Zhang, J. Chen, H. Cai, J. Lu, C. Ma, R. Liu, L.-P. Xhonneux, M. Qu and J. Tang, arXiv, 2022, preprint, arXiv:2202.08320 [cs.LG], DOI:10.48550/arXiv.2202.08320.
- DeepChem/ChemBERTa-77M-MLM, Hugging Face, https://huggingface.co/DeepChem/ChemBERTa-77M-MLM, accessed 03/27/2023 Search PubMed.
- ncfrey/ChemGPT-1.2B, Hugging Face, https://huggingface.co/ncfrey/ChemGPT-1.2B, accessed 03/27/2023 Search PubMed.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn.: Sci. Technol., 2020, 1, 045024 Search PubMed.
- S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2020, preprint, arXiv:2010.09885 [cs.LG], DOI:10.48550/arXiv.2010.09885.
- F. Pedregosa, et al. , J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00088e |
| This journal is © The Royal Society of Chemistry 2023 |