
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Link-INVENT: generative linker design with reinforcement learning†
Jeff
Guo‡
a,
Franziska
Knuth‡
 ab,
Christian
Margreitter§
a,
Jon Paul
Janet
c,
Kostas
Papadopoulos§
a,
Ola
Engkvist
ad and
Atanas
Patronov§
*a
ab,
Christian
Margreitter§
a,
Jon Paul
Janet
c,
Kostas
Papadopoulos§
a,
Ola
Engkvist
ad and
Atanas
Patronov§
*a
aMolecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden. E-mail: patronov@gmail.com
bDepartment of Physics, Norwegian University of Science and Technology, Trondheim, Norway
cMedicinal Chemistry, Research and Early Development, Cardiovascular, Renal and Metabolism (CVRM), BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
dDepartment of Computer Science and Engineering, Chalmers University of Technology, Gothenburg 41756, Sweden
First published on 4th February 2023
Abstract
In this work, we present Link-INVENT as an extension to the existing de novo molecular design platform REINVENT. We provide illustrative examples on how Link-INVENT can be applied to fragment linking, scaffold hopping, and PROTAC design case studies where the desirable molecules should satisfy a combination of different criteria. With the help of reinforcement learning, the agent used by Link-INVENT learns to generate favourable linkers connecting molecular subunits that satisfy diverse objectives, facilitating practical application of the model for real-world drug discovery projects. We also introduce a range of linker-specific objectives in the Scoring Function of REINVENT. The code is freely available at https://github.com/MolecularAI/Reinvent.
1. Introduction
Deep learning (DL) offers potential to accelerate drug design by efficiently traversing chemical space, defined as the set of all possible biologically relevant molecules, and estimated to be on the order of 1023 to 1060.1–3 The task is challenging as candidate drug molecules must satisfy a multi-parameter optimization (MPO) objective where parallel optimization of the individual objectives can be difficult. Thus, a brute-force molecular search approach is often infeasible. DL-augmented molecular design combined with computational oracles to approximate physico-chemical properties has enabled candidate drug molecules to be designed in an accelerated manner.4 DL-based approaches to the drug design task include deep generative models with reinforcement learning (RL),5–10 learning a molecular latent space,11 and genetic algorithms,12–14 which generate molecular ideas as Simplified Molecular-Input Line-Entry System (SMILES) strings15 or molecular graphs.9,10Recently, the application of DL-based methods to join two molecular subunits via a chemical linker has gained considerable interest.16–23 Generating suitable linkers is important for fragment-based drug discovery (FBDD)24,25 and scaffold hopping,26 and fundamental for the design of proteolysis targeting chimeras (PROTACs).27–29 The former two techniques are avenues to discover and optimize novel small molecule drugs, while the latter is a relatively new therapeutic modality able to achieve targeted protein degradation. Therefore, linker design represents a relevant problem in drug discovery.
FBDD is an alternative to traditional high-throughput screening (HTS) and virtual screening (VS) which screens ‘Lipinski compliant’ small molecules. In contrast, FBDD screens ‘fragments’, typically with a molecular weight (MW) under 260 Da. Although ‘fragment’ hits typically exhibit weaker binding affinities than small molecules, they often form polar interactions with the receptor and possess favourable lipophilicity, limiting entropically driven binding.24,25,30 Thus, ‘fragments’ can be an advantageous starting point for drug design and techniques to optimize their potency and physico-chemical properties include fragment growing and fragment linking.24,25,31,32 The latter is of particular interest as proper linking of two ‘fragments’ such that the linked molecule does not perturb the constituents' interactions can lead to significant potency gain. This is attributed to favourable entropic effects and known as ‘super-additivity’. In practice, fragment linking is challenging and ‘super-additivity’ is rarely achieved, owing to incompatible linkers disrupting the fragments' binding poses.31,32 Thus, improvements in linker design are critical to unlock the full potential of FBDD.
Scaffold hopping refers to modifying the core structure of a molecule to improve physico-chemical properties while retaining potency.26 The task can be formulated as a linker design problem if the scaffold itself is defined as the linker between two molecular subunits. Scaffold hopping is challenging as retaining potency requires 3D structural awareness of the interactions formed between the molecule and its receptor. Similar to fragment linking, improvements in linker design can enhance the ability to generate novel scaffold ideas.
PROTACs are heterobifunctional molecules in which a linker joins a ligand binding to a protein of interest (POI), conferring specificity, and an E3 ubiquitin ligase. The formation of the ternary complex leads to subsequent ubiquitination, achieving POI degradation and thus, targeted knockdown.27–29 While the unique mechanism of action provides promising therapeutic applicability beyond traditional small molecules, PROTAC design is challenging. PROTACs are comparably large molecules, typically existing beyond ‘Lipinski's rules' and thereby posing a design challenge since experience is limited.33–35 Moreover, linker design is challenging due to the relatively high conformational flexibility present in longer linkers and has mostly deferred to empirical structure–activity relationship (SAR) studies, often necessitating numerous iterations of design-make-test-analyze (DMTA) cycles.36 Therefore, there is a need for improved linker design to improve the overall PROTAC design.
Previously developed computational tools for linker design involve searching a database, making the generalizability of proposed linkers inherently limited.37–40 While success has been demonstrated when using these methods combined with filtering steps, one would ideally want to generalize the task such that plausible linker ideas can be proposed given any molecular subunits.37–40 Recently, DL-based linker design models have been proposed that circumvent database searches.16–23 DeLinker is a graph-based model proposed by Imrie et al. which explicitly incorporates 3D information via the distance and angle between the molecular subunits to augment the feature vector.16 Imrie et al. further improve DeLinker and introduce DEVELOP which couples DeLinker with a convolutional neural network (CNN) operating on the 3D structure of the starting fragments.20 SyntaLinker is a conditional transformer model proposed by Yang et al. which treats linker generation as a natural language processing (NLP) task using SMILES.15,17 SyntaLinker was further extended by Hu et al. to perform kinase scaffold hopping after focusing the model via transfer learning.18 Similarly, Feng et al. introduce the SyntaLinker-Hybrid workflow which performs transfer learning on a base SyntaLinker model using known active compounds to focus the generative model.23 Moreover, Langevin et al. proposed the Scaffold Constrained Molecular Generation (SAMOA) algorithm based on recurrent neural networks (RNNs) where one of the capabilities of the model is linker generation.19 Recently, equivariant models including 3DLinker21 and DiffLink22 operating on the coordinates of fragments have been applied for linker generation. Equivariance enforces that symmetry operations applied to the input transforms the output in the same way, and thus model performance is independent of the initial coordinates. However, while these models are capable of generating linker ideas, a major drawback is the limited support to optimize explicitly for desired physico-chemical properties. The current models only allow users to control for the desired linker length16–19 and a select number of physico-chemical properties, e.g., number of hydrogen-bond donors (HBDs).17 To encourage wide adoption of DL-based linker design, increased flexibility to define tailored MPO objectives and better generalizability is needed.
In this work, we present Link-INVENT as an extension to the existing de novo design platform REINVENT, which has previously identified experimentally validated nM potent inhibitors.6,41 The suggested algorithm shares some similarities with the SAMOA algorithm as proposed by Langevin et al.19 in that the code builds upon REINVENT's existing codebase and uses policy-based RL for MPO.6 However, our algorithm has three crucial differences compared to earlier work. Firstly, the prior trained by Langevin et al. is based on ChEMBL compounds and follows the protocol as reported for REINVENT, which was purposed to sample small molecules as SMILES.6,19,42 Consequently, in their linker generation solution, linkers are sampled when the “*” token (the model's internal representation of characters in a SMILES string), denoting the attachment point, is reached, and based on the conditional probabilities of the SMILES sequence so far. The limitation is that linkers should be generated in the context of both molecular subunits. In the extreme case, the SAMOA algorithm may struggle to generate plausible linkers if the SMILES sequence was “CC*C…” where the length of the SMILES on the right side of the “*” token is greater than that on the left side, as the conditional probabilities for linker generation would only be based on the sequence so far, i.e., “CC”. In contrast, Link-INVENT is trained based on the conditional probabilities of observing a linker given both molecular subunits, similar to the SyntaLinker model reported by Yang et al.17 Secondly, the data preparation to train the Link-INVENT prior was based on reaction-splicing of the ChEMBL compounds similar to the Lib-INVENT library design model we reported previously.42,43 Our training set contains linkers that join molecular subunits ranging from a few atoms in size to larger moieties with rings. As a result, a single Link-INVENT prior is suited for diverse linker generation tasks. Finally, Link-INVENT was built on the latest version of REINVENT (3.2) and supports an extensive selection of physico-chemical properties that can be optimized through RL. Moreover, we have implemented additional linker specific properties that can be optimized (in the form of additional Scoring Function components), ranging from physico-chemical properties to flexibility and rigidity, allowing one to explicitly optimize linker properties. We demonstrate the use of Link-INVENT in fragment linking, scaffold hopping, and PROTAC design case studies. Through RL, the Link-INVENT agent learns to generate favourable linkers connecting molecular subunits that satisfy diverse MPO objectives, facilitating practical application of the model for real-world drug discovery projects. The code is freely available at https://github.com/MolecularAI/Reinvent.
2. Methods
2.1 Model overview
Link-INVENT takes as input a pair of warheads, i.e., two molecular subunits with exit vectors defined, generates a linker, and returns the linked molecule in the SMILES format (Fig. 1).15 The model is adapted from Lib-INVENT, our previously reported generative model for library design by Fialková et al. which in turn is based on work by Arús-Pous et al.43,44 Specifically, Link-INVENT features an encoder-decoder architecture consisting of identical RNNs with embedding size 256 and three hidden layers of 512 long short-term memory cells (LSTM).45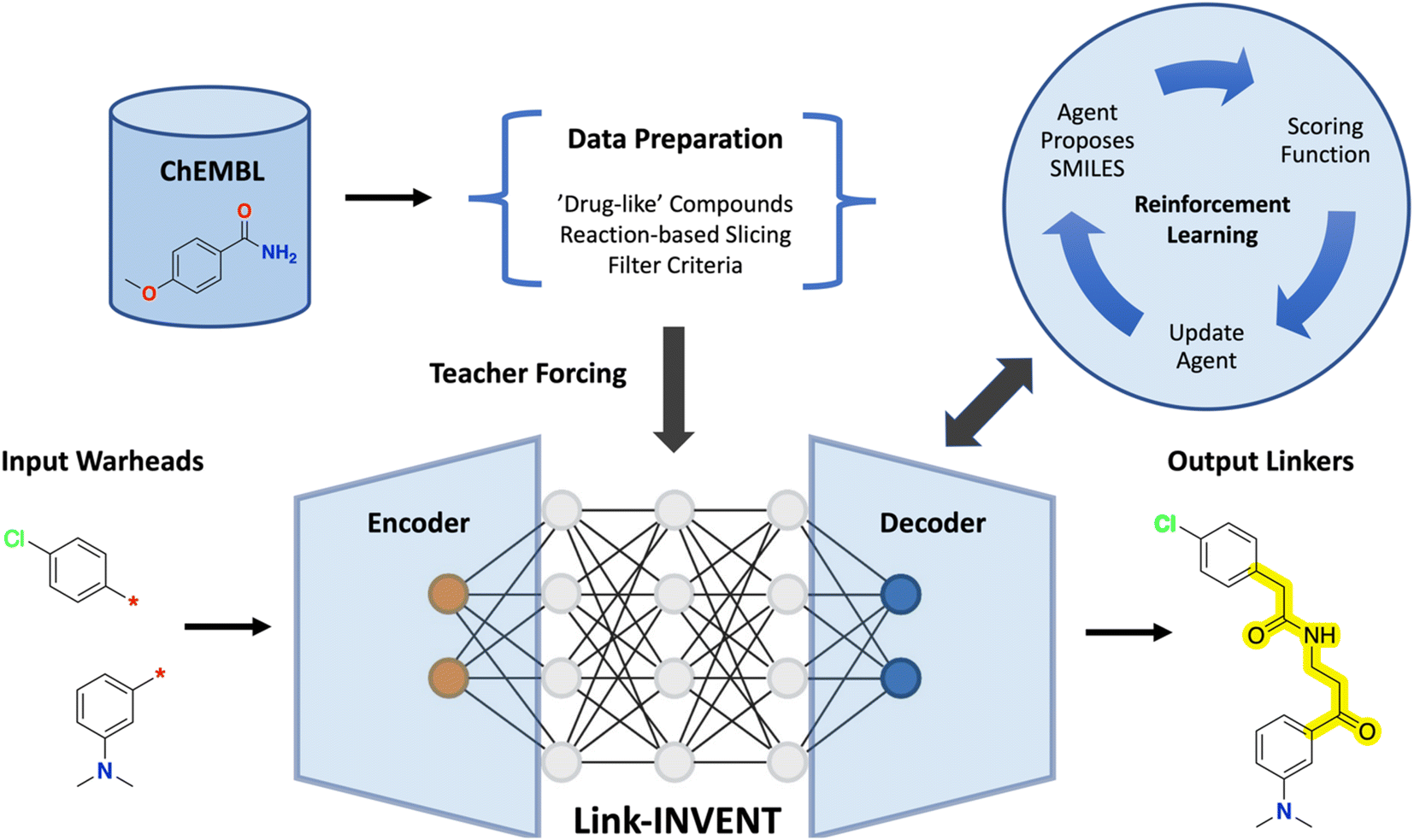 | ||
| Fig. 1 Link-INVENT training and inference overview. ChEMBL data were processed to generate the training data for Link-INVENT which features an encoder-decoder architecture of recurrent neural networks (RNNs). Link-INVENT takes as input a pair of warheads and generates output linkers (highlighted), yielding connected molecules. One crucial capability of Link-INVENT is the flexible scoring. | ||
2.2 Data preparation
The training data were generated from ChEMBL via the following steps:421. Initial filtering: filter the raw ChEMBL data (version 27) to keep ‘drug-like’ compounds only (see the ESI† for details). Lenient filtering criteria were applied such that the training data are effective for PROTAC applications where the warheads can be larger in size compared to traditional ‘fragments’.34,35
2. Reaction-based slicing: slice the filtered ChEMBL compounds following the protocol from our Lib-INVENT work using the reaction SMIRKS.43 The result is a dataset of tuples with the structure: (linker, warheads pair, full molecule).
3. Sliced data filtering: filter the tuples to remove unrealistic data points, e.g., linkers with a molecular weight greater than 500 Da.
4. Generate training and validation sets: a validation set containing 287 Bemis–Murcko scaffolds was held out.46
5. SMILES randomization: data augmentation for the training and validation sets was performed via SMILES randomization. At each training epoch, the model is provided with datasets composed of the same sliced tuples (linker, warheads pair, full molecule) but with a different SMILES representation. The purpose was to improve the chemical space generalizability of the generative model as shown by Arús-Pous et al.47
For full details of the data preparation, see the ESI.†
2.3 Model training
First, a vocabulary was generated that maps characters present in the training set (and validation set) SMILES to tokens (see the ESI† for token details). Querying Link-INVENT requires tokenization of the input warheads by the encoder and the output linker tokens from the decoder are then transformed into their SMILES equivalent.15 The initial generative model, denoted the prior, was trained by maximizing the likelihood of generating a linker conditioned on the input pair of warheads. Teacher forcing was used such that the ground-truth labels were fed back to the model at each token sampling step to improve training stability.48 The trained prior is a generative model that has learned the SMILES syntax and is thus capable of generating syntactically valid linkers given a pair of input warheads.2.4 Model inference and multi-parameter optimization
Following REINVENT's protocol, the agent is initialized to have the same parameters as the prior and serves two purposes:6,49 ensuring that the agent is also capable of generating syntactically valid linker SMILES and anchoring the sampled linkers to relevant chemical space as defined by the training data derived from ChEMBL.15,42 Subsequently, the agent is tasked to generate linkers that satisfy MPO objectives, given by the Scoring Function. The Scoring Function specifies all components to be optimized and is formulated as a weighted geometric mean in this work: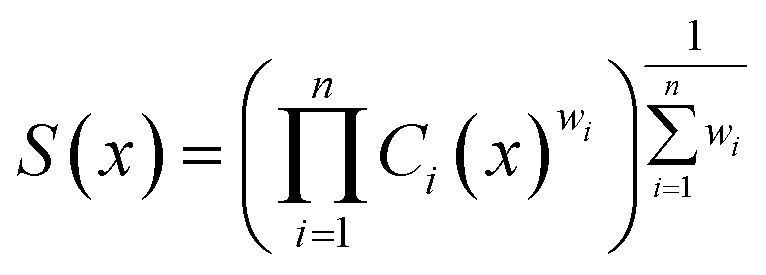 | (1) |
1. Agent sampling: generate a batch size (128 in this work) number of linkers conditioned on an input pair of warheads. Thus, 128 linked molecules were generated at each epoch.
2. Assess linked molecules' desirability: combine the warheads and linkers to form the linked molecules and compute their desirability based on the satisfaction of the Scoring Function.
3. Update the agent policy: compute the loss and update the agent's policy to steer sampling towards favourable linkers. The specific loss function used in Link-INVENT was previously introduced by Fialková et al. in our Lib-INVENT work and defined as the difference between the augmented and posterior likelihoods (DAP).43 Correspondingly, the same loss function was used in this work and is constructed by first defining the augmented log likelihood:
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) πaugmented = logπprior + σS(x) πaugmented = logπprior + σS(x) | (2) |
| J(θ) = (logπaugmented − logπagent)2 | (3) |
Steps 1–3 are repeated until the permitted number of epochs has elapsed. All favourable linkers (and the corresponding full molecules) that achieve a total score (computed by aggregating the scores achieved on each composite objective defined in the Scoring Function) exceeding a user-defined threshold (typically 0.4) are outputted. In this work the threshold was set to 0 to store all molecules generated. The purpose of this was to compare the profiles of molecules generated towards the beginning of the experiment and how RL gradually guides the generation of favourable molecules.
2.5 Balancing chemical space exploration and exploitation
Link-INVENT offers full control over chemical space exploration and exploitation by leveraging Diversity Filters (DFs) as implemented in REINVENT.6,50 Buckets can be defined with limited size that keeps track of unique scaffolds. Agent sampling of molecules containing identical scaffolds populates the same bucket. If a bucket is full, further sampling of the scaffold will cause the agent to receive a score of 0, regardless of the corresponding molecule's desirability, as assessed by the Scoring Function. This mechanism encourages agent exploration of diverse minima. For more details regarding DFs, see the work by Blaschke et al.6,50 The specific DF used in this work features buckets of size 25 and penalizes repeated sampling of Bemis–Murcko scaffolds.46 We note that as Link-INVENT generates linkers conditioned on a pair of input warheads, the warheads themselves are held constant. Thus, the DF effectively penalizes repeated sampling of the Bemis–Murcko scaffolds of the linker themselves.462.6 Scoring Function: controlling linker properties
In addition to the previously supported Scoring Function properties in REINVENT that operate on the full molecule, Link-INVENT offers control over the linker itself (Fig. 2). One can control the linker length and branching (Fig. 2a) via the following properties: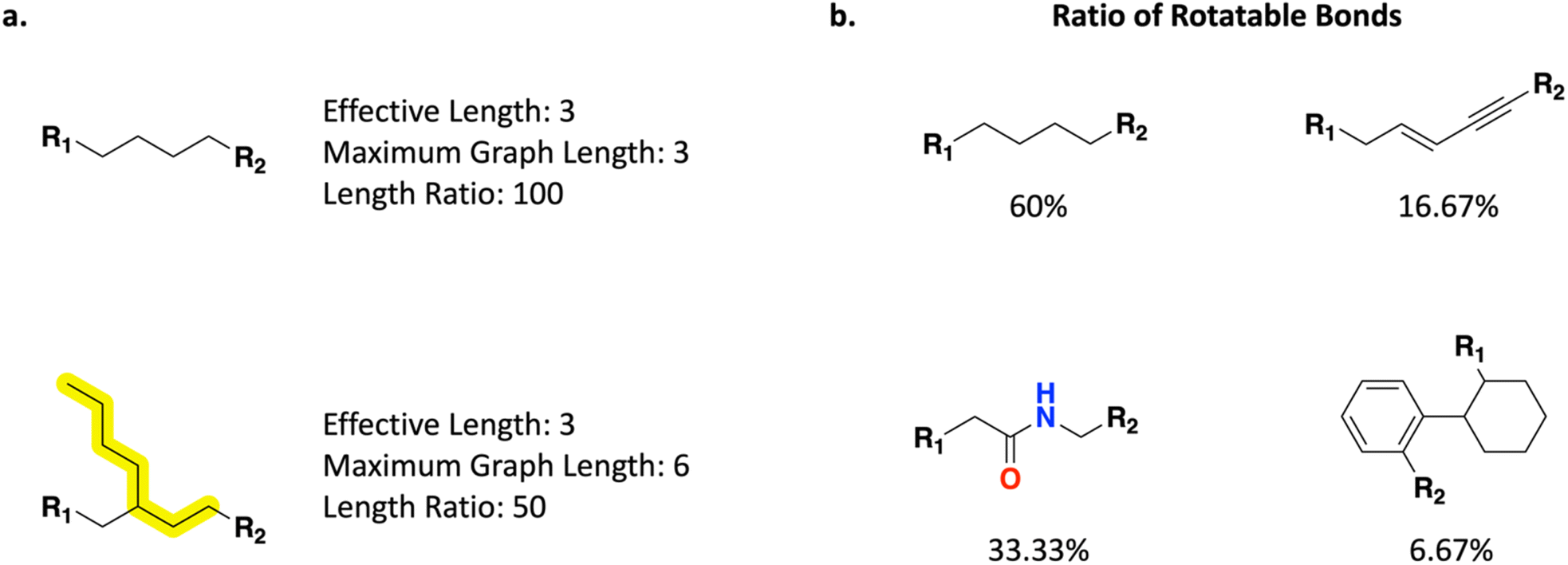 | ||
| Fig. 2 Link-INVENT Scoring Function selected linker specific properties. Attachment points are denoted by R-groups. (a) “Linker effective length”, “linker maximum graph length”, and “linker length ratio” provide direct control over the distance between attachment atoms in a linker and the degree of branching permitted. The maximum graph length bond traversal is highlighted in the bottom example. (b) “Linker ratio of rotatable bonds” provides control over the flexibility of proposed linkers. | ||
1. Linker effective length: the number of bonds between the attachment atoms.
2. Linker maximum graph length: the number of bonds encompassed in the longest molecular graph traversal path.
3. Linker length ratio: the ratio of the “linker effective length” over the “linker maximum graph length”.
Moreover, one can control linker flexibility through the “linker ratio of rotatable bonds” component which is defined as the number of rotatable bonds (as calculated by using RDKit51) over the total number of bonds (Fig. 2b). We note that this treatment of flexibility is not the only valid definition and inherent limitations exist such as being completely agnostic to intra-molecular hydrogen bonds. Furthermore, RDKit's calculation of rotatable bonds does not consider bonds to terminal atoms rotatable as it depends on the hybridization of the atom they are attached to. Consequently, bonds to attachment points are always considered non-rotatable. This is exemplified in Fig. 2b where the butane linker receives a ratio of 60/100 (60%). Consequently, a linker can never achieve a ratio of rotatable bonds of 100% and to achieve a higher ratio, and linkers must become increasingly longer which can lead to unrealistic ideas being proposed. In practice, this is not a limitation in guiding Link-INVENT towards flexible/rigid linkers as one can introduce appropriate score transformations that provide meaningful agent feedback (discussed in the Results section). For a full list of properties available in the Link-INVENT Scoring Function, see the ESI.†
3. Results
We demonstrate the application of Link-INVENT by devising the following experiments:1. Illustrative example: a simple experiment to illustrate how Link-INVENT gradually learns to satisfy MPO objectives.
2. Experiment 1a: fragment linking: link two fragment hits and satisfy a hydrogen-bond molecular docking constraint.
3. Experiment 1b: comparison fragment linking: link two fragment hits and satisfy a core constrained molecular docking protocol. Results are compared to the existing DL-based linker design tools DeLinker and SyntaLinker.16,17
4. Experiment 2: scaffold hopping: generate new scaffold ideas to improve physico-chemical properties while retaining potency by satisfying a hydrogen-bond molecular docking constraint.
5. Experiment 3: PROTACs: demonstrate the flexibility of Link-INVENT to generate linkers with diverse properties. The focus in this section is to showcase the linker specific properties implemented for the Link-INVENT Scoring Function.
The same prior was used for all the experiments and demonstrates the versatility of the single trained generative model in addressing diverse tasks.
Illustrative example. As an initial illustrative example, we devise an experiment to link two benzene rings with the objective of limiting the number of HBDs and the linker possessing exactly one ring (Fig. 3). Correspondingly, the Scoring Function contains two components:
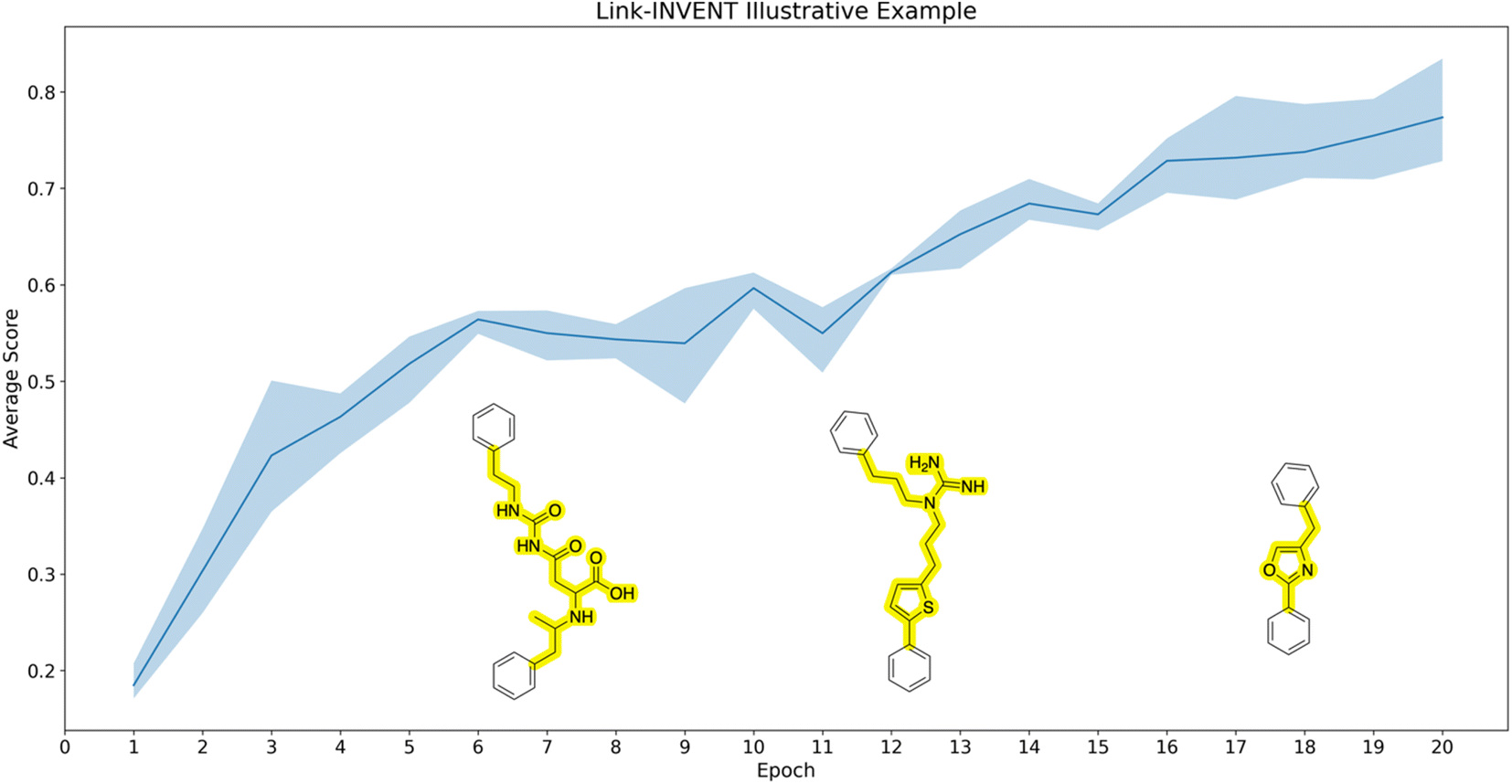 | ||
| Fig. 3 Illustrative example. The experiment was run in triplicate. The curve shows the average score achieved by the batch of molecules sampled at a given epoch and the upper and lower bounds of the shaded region represent the maximum and minimum scores, respectively. The objective is to link two benzene rings while limiting the number of hydrogen bond donors (HBDs) and the linker containing exactly one ring. Example structures (linkers highlighted) are superimposed on the plot at various stages of training to illustrate how Link-INVENT gradually learns to generate molecules that satisfy the desired MPO objective. | ||
1. Linker number of hydrogen bond donors: maximum reward is given if the linker contains no HBDs. See ESI Fig. S1† for the score transformation.
2. Linker number of rings: reward is only given if the linker contains exactly one ring.
Fig. 3 shows the Link-INVENT training progress over 20 epochs. The average score over triplicate runs shown in the curve is gradually increasing. Example molecules generated over the course of training are superimposed on the plot. The first molecule on the left possesses multiple HBDs and the linker does not contain a ring. Consequently, this molecule receives low reward. As training progresses, the example molecules start to satisfy our MPO objective. Towards the end of the 20 epochs, the example molecule not only possesses no HBDs, but the linker also has exactly one ring. The purpose of this experiment was to illustrate how the Link-INVENT agent learns via RL to generate molecules that increasingly satisfy the target objective.
3.1 Experiment 1a: fragment linking
Fusco and Brear, et al. applied a fragment linking strategy (Fig. 4) to design casein kinase 2 inhibitors for the alpha catalytic site (CK2α).52,53 CK2 is overexpressed in cancers and causes apoptosis evasion, leading to poor cancer prognosis and is therefore a therapeutic target of interest. Fig. 4a shows the fragment linking strategy envisioned by Fusco and Brear, et al. The gray fragment binds by forming hydrogen-bond interactions with Lys68 and Asp175 and the green fragment binds primarily through hydrophobic interactions (Fig. 4a). The difference in the binding modes of the constituent fragments were judged to be suitable for linking. Following this strategy, a potent and selective CK2α inhibitor (CAM4066) was discovered that retains the important Lys68 hydrogen-bond interaction (Fig. 4b). CAM4066 was demonstrated to exhibit in vivo efficacy and is thus an example of a successful fragment linking campaign.52,53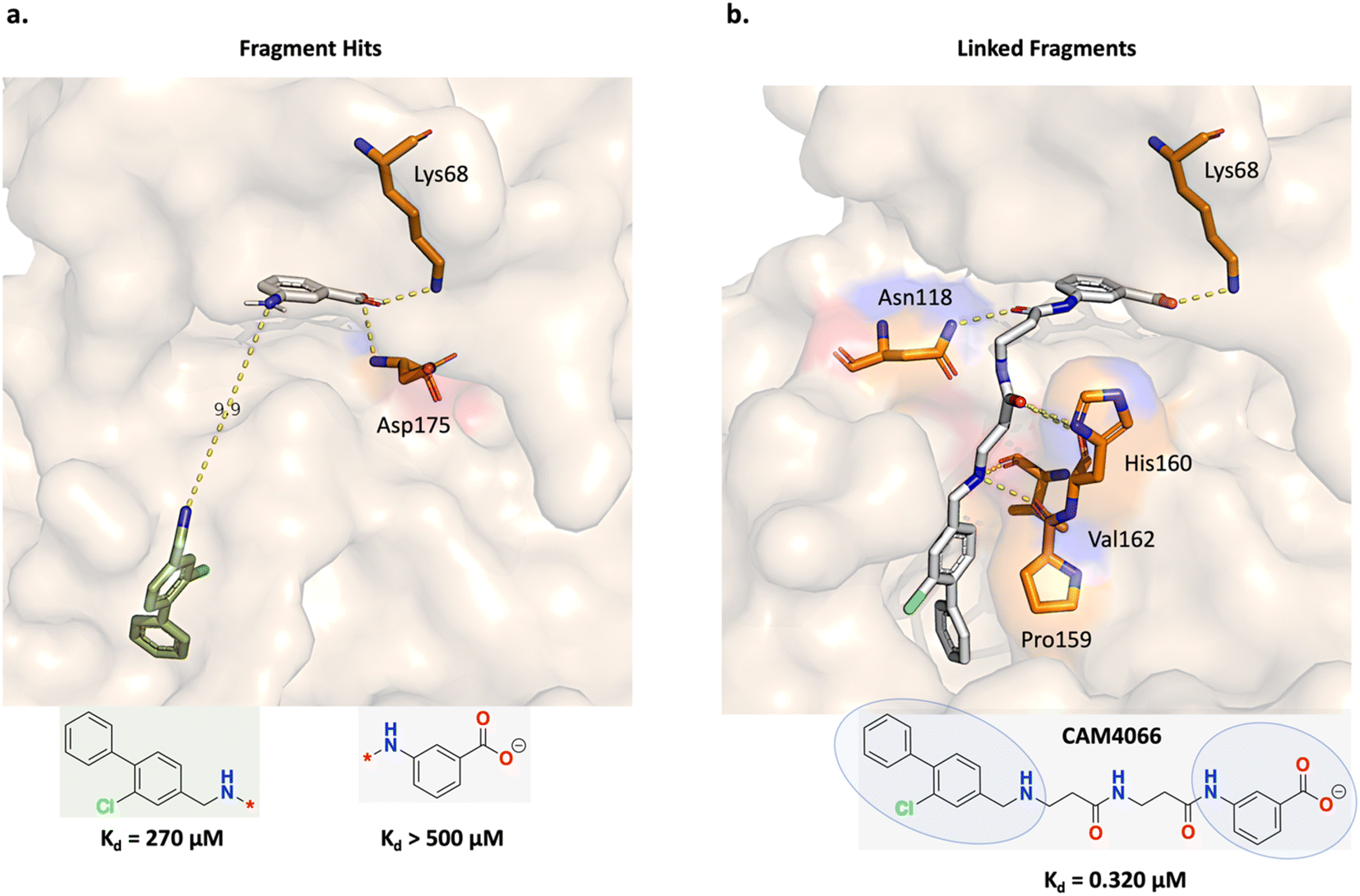 | ||
| Fig. 4 Experiment 1a: fragment linking strategy for casein kinase 2 inhibitors for the alpha catalytic site (CK2α). (a) Initial fragment hits. The fragment structures are colour-coded: gray fragment PDB ID: 5CSV and green fragment PDB ID: 5CSH. The gray fragment binds by forming hydrogen-bond interactions with Lys68 and Asp175 while the green fragment binds via hydrophobic interactions. The fragment linking strategy was to leverage the nitrogen atoms on both fragments to design a linear linker, separated by 9.9 Å. (b) Fragment linking led to the discovery of the linked molecule, CAM4066 (PDB ID: 5CU4). The constituent fragments are circled in the structure. The linear linker features amide bonds that modulate the linker flexibility and rigidity which the authors attribute to its binding potency.52,53 | ||
In this section, we adopt the fragment linking strategy devised by Fusco and Brear, et al. (Fig. 4a) and task Link-INVENT with generating plausible linked molecules that retain the Lys68 hydrogen-bond interaction.52,53 Moreover, while Fusco and Brear, et al. exclusively evaluated linear linker ideas, we allow Link-INVENT to explore linkers with rings and branching (to a certain extent). Correspondingly, we devise a Scoring Function composed of the following components (see ESI S2 and S3† for Scoring Function transformations):
1. DockStream: this component is a molecular docking package that is fully compatible with Link-INVENT. DockStream supports docking using a variety of backends. In this work, we use Glide and LigPrep which we previously identified to yield the best average performance over a variety of receptor targets.54–59 A docking constraint was enforced to retain the Lys68 hydrogen-bond interaction.52,53
2. Linker length ratio ≥70: this component prevents linkers with branching that is significantly longer than the effective length (number of bonds between the linker attachment atoms).
3. Linker molecular weight ≤200 Da: this component also prevents linkers with extensive branching but more importantly, prevents the Link-INVENT agent from exploiting the weaknesses of molecular docking, e.g., generating linkers that possess a large number of HBDs which may achieve a favourable docking score but at the expense of limited permeability.60
The fragment linking experiment was run in triplicate and the results are shown in Fig. 5 (see ESI Fig. S4† for all training plots). Over the course of 100 epochs, the average Glide docking score of the batch of molecules generated by Link-INVENT gradually becomes more favourable (Fig. 5a). The docking score distributions of the triplicate runs are essentially identical and demonstrate a reproducible experimental outcome (Fig. 5b). The relatively few molecules that possess a docking score of 0 do not satisfy the docking constraint and were generated towards the beginning of the Link-INVENT run at a timestep where the agent has received minimal feedback. Furthermore, some molecules proposed by Link-INVENT exhibit a more favourable docking score than the reference ligand (−15.20 kcal mol−1, black dotted line in Fig. 5b). The majority of the remaining molecules dock similar to the reference ligand (approximately −14 kcal mol−1) and demonstrates that Link-INVENT at the very least proposes chemical ideas that can satisfy the docking constraint. Subsequently, the interplay between the agent and the DF is exemplified in Fig. 5c. The DF encourages balance between agent exploration and exploitation by penalizing repeated sampling of identical Bemis–Murcko scaffolds.46 The triplicate runs yield a large number of unique scaffolds with minimal overlap, demonstrating diversity in the results and showing that replicate experiments explore different areas in chemical space (Fig. 5c). Next, the plausibility of generated molecules was investigated by comparing their binding poses with the reference ligand. Fig. 5d shows the binding pose of an example top scoring molecule (based on the satisfaction of the composite Scoring Function) superimposed with the reference ligand (see ESI Fig. S5† for more examples). Firstly, the proposed linker is similar to the ground-truth linker, differing only by a single atom shift of an amide bond and the presence of an additional nitrogen. It is important to note that information about the reference ligand was not available to the Link-INVENT agent during the generative process and is not present in the training set (see ESI† for more details). Fusco and Brear, et al. posited that the flexibility and rigidity of the reference ligand linker are crucial to its potency.52,53 The similarity in the linker proposed by Link-INVENT suggests that the docking constraint implicitly guides the agent towards 3D structural awareness, in agreement with our previous results.54 This is further supported by the predicted polar interactions of the generated molecule (Fig. 5d turquoise dotted lines) being mostly identical to those of the reference ligand (Fig. 5d yellow dotted lines) with the only exception being His160. Consequently, the structural similarity between the linkers naturally results in significant overlap of the binding poses and is exemplified in the docking score in which the generated molecule is predicted to dock more favourably than the reference ligand. Taken together, the results in this section demonstrate that Link-INVENT is able to generate plausible chemical ideas spanning diverse minima and is easily tuned for bespoke applications via the Scoring Function.
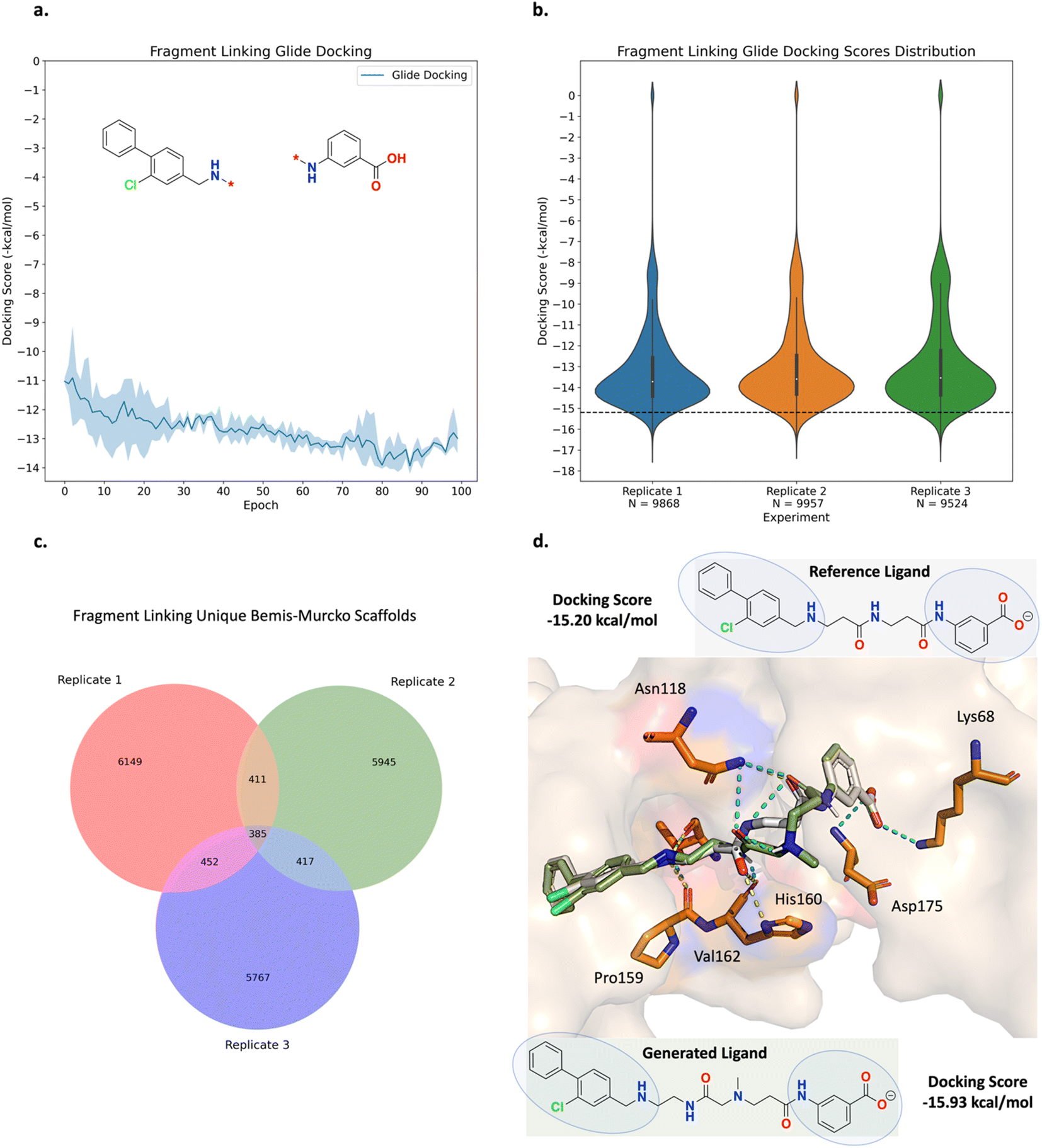 | ||
| Fig. 5 Experiment 1a: fragment linking Link-INVENT results. The experiment was run in triplicate. The curve shows the average score achieved by the batch of molecules sampled at a given epoch and the upper and lower bounds of the shaded region represent the maximum and minimum scores, respectively. (a) Glide LigPrep docking score optimization. The average docking score achieved by the batch of compounds generated by Link-INVENT gradually becomes more favourable (lower score in the case of Glide). (b) Violin plots showing the distribution of docking scores for the triplicate runs. ‘N’ is the number of molecules generated over 100 epochs. The distributions are nearly identical, demonstrating a reproducible experimental outcome. The black dotted line shows the docking score possessed by the reference ligand (CAM4066, −15.20 kcal mol−1). (c) Venn diagram plots showing the overlap between unique Bemis–Murcko scaffolds in the triplicate runs. (d) The binding pose of a selected generated molecule (green) superimposed with the reference ligand (gray) and the constituent fragments are circled. PDB ID: 5CU4. The structure of the generated molecule is similar to the reference ligand. The yellow and turquoise dotted lines show the interactions formed by the reference ligand and generated ligand, respectively. The generated molecule retains the Lys68 interaction as enforced by the docking constraint and forms the same polar interactions as the reference ligand, largely attributed to the extensive overlap between the binding poses and supporting plausibility. | ||
3.2 Experiment 1b: comparison fragment linking
Trapero et al. applied a fragment linking strategy to design inosine 5′-monophosphate dehydrogenase (IMPDH) inhibitors for tuberculosis (TB).61 A hit identified from fragment screening binds at different locations and is well positioned to facilitate self-linking (Fig. 6a). Following this strategy, linear linkers were designed which led to the discovery of a linked molecule possessing significantly enhanced in vitro potency (Fig. 6b reference ligand shown in gray).61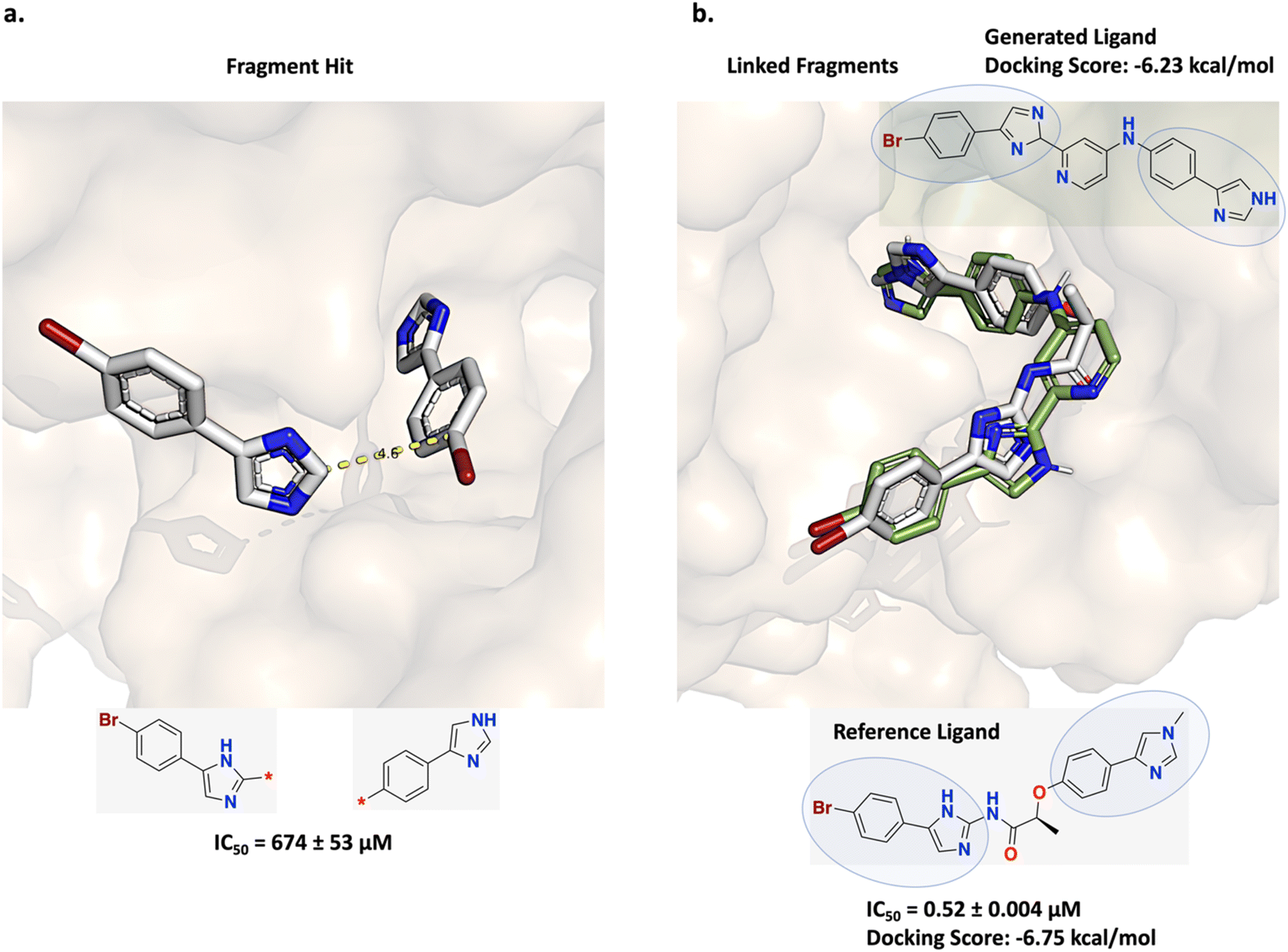 | ||
| Fig. 6 Experiment 1b: comparison fragment linking inosine 5′-monophosphate dehydrogenase (IMPDH) inhibitors for tuberculosis (TB). (a) Initial fragment hits (PDB ID: 5OU2). Trapero et al. linked copies of the fragment hit separated by 4.6 Å via a linear linker.61 (b) Fragment linking led to the discovery of the linked molecule shown in gray (PDB ID: 5OU3) and possessing significantly enhanced in vitro potency. The constituent fragments are circled in the structure. The methyl substituent on the imidazole ring of the right fragment in (b) is not present in the initial fragment hit structure and was added post linker design.61 A Link-INVENT generated molecule is superimposed with the reference ligand (green), showing excellent pose overlap of the constituent fragments and with a comparable docking score. | ||
This specific case study was also investigated in DeLinker and SyntaLinker DL-based linker design studies.16,17 To assess the prospective compatibility with the protein, DeLinker and SyntaLinker dock their generated molecules with AutoDock Vina62 and MOE docking,63 respectively, post hoc, as proxies for binding affinity. However, an important criterion in any fragment linking campaign is good agreement between the constituent fragment poses of the generated molecule to the reference fragment poses. DeLinker does not show any binding poses of their generated molecules while SyntaLinker shows only three example poses where none of the fragment poses overlap with the reference fragment poses even though they recover the reference ligand.16,17 This suggests that neither docking protocol was able to capture the constituent fragments' binding poses. To address this problem, we task Link-INVENT with generating linker ideas by adopting the strategy envisioned by Trapero et al.61 In particular, we perform core constrained docking with Glide to enforce that the binding pose of at least one fragment is within 0.3 Å to the reference fragment pose (see the ESI† for full details).55–59 This is in contrast to the docking protocol applied in the previous experiment 1a: fragment linking as π-interactions contributes extensively to the binding affinity of the fragment hit in the IMPDH binding pocket (Fig. 6a). For a fair comparison, we apply our core constrained docking protocol to the example generated molecules provided in DeLinker and SyntaLinker studies.16,17 We note that the training data used in DeLinker, SyntaLinker, and Link-INVENT are different which can contribute to differences in performance. We devise a Scoring Function composed of the following components (see ESI S6 and S7† for Scoring Function transformations):
1. DockStream: core constrained docking in Glide was applied through DockStream to prevent significant binding pose deviation of the constituent fragments in the linked molecule compared to the reference fragment pose (see the ESI† for full details).54–59
2. 3 ≤ linker effective length ≤ 5: this component enforces linkers to possess an effective length between 3 and 5 bonds. The specific interval was chosen so that proposed linkers generally span 4.6 Å, capturing the a priori knowledge from fragment screening (Fig. 6a).
3. Linker length ratio ≥70: this component prevents linkers with branching that is significantly longer than the effective length (number of bonds between the linker attachment atoms). In contrast to Trapero et al. where only linear linkers were evaluated, we allow Link-INVENT to explore moderately branched linkers.61
4. Linker molecular weight ≤150 Da: similar to the Scoring Function in the previous fragment linking experiment, this component prevents the Link-INVENT agent from exploiting the weaknesses of molecular docking,60 the only difference is the upper limit of the linker molecular weight being 150 Da instead of 200 Da. The rationale is that the constituent fragments here possess a greater MW compared to the previous fragment linking case study and thus, a lower upper limit is enforced to keep the linked molecules within a reasonable MW range.
The fragment linking experiment was run in triplicate for 70 epochs, generating a total of 8960 SMILES, which is similar to the 9000 molecular graphs generated in the DeLinker work, and facilitating a fair comparison.16 The training plots are shown in Fig. S8.† An example binding pose of a generated molecule is shown in Fig. 6b (green). The 4-aminopyridine linker facilitates extensive overlap of the binding poses of the constituent fragments with the reference poses. Moreover, the docking score is comparable to that of the reference ligand, demonstrating that Link-INVENT is able to generate plausible ideas within a relatively narrow solution space (linkers were enforced to possess an effective linker length between 3 and 5 bonds). DeLinker and SyntaLinker example molecules also show good pose agreement when docked with our protocol (Fig. S10†). SyntaLinker also recovers the reference ligand. However, we note that in the experimental design for SyntaLinker, the authors introduced bias by providing their model with information from the reference ligand. Specifically, in one of the fragments, Trapero et al. included a methyl substituent on the imidazole ring due to synthetic accessibility and the linker with the greatest in vitro potency featured an ether linkage (Fig. 6b reference ligand shown in gray).61 Correspondingly, the methyl substituent and the ether linkage information was provided to the SyntaLinker model during the generative process.17
Next, we assess the docking scores of the generated ideas by Link-INVENT, DeLinker, and SyntaLinker (Table S2†). Across triplicate runs, Link-INVENT generates molecules with a more favourable docking score than the reference ligand (see Fig. S9a† for an example binding pose). By contrast, none and only one (the recovered reference ligand) of the molecules provided in the DeLinker and SyntaLinker studies dock better than (or equal to) the reference ligand. We acknowledge that it is possible that some DeLinker and SyntaLinker proposed molecules do indeed possess more favourable docking scores than the reference ligand and the analysis performed is based on what the authors have provided (20 and 3 example molecules for DeLinker and SyntaLinker, respectively). We note that the reference linker is present in the training data. However, Link-INVENT generates a large number of ideas with comparable docking scores to the reference ligand and also possesses high diversity as shown by the number of unique Bemis–Murcko scaffolds in the generated molecules (Fig. S8f†). Specifically, on average, of the 5000 generated molecules by Link-INVENT, there are around 3000 unique Bemis–Murcko scaffolds. We further note that since the fragments are held constant, this means that the unique scaffolds pertain to the linker itself. Therefore, Link-INVENT generates diverse linker ideas that satisfy the core constrained docking protocol.
Accordingly, we show that the Link-INVENT Scoring Function steers the agent to generate molecules that satisfy the desired MPO objective. By including docking explicitly as a learning objective, Link-INVENT is able to generate molecules with favourable docking scores and outperforms DeLinker and SyntaLinker which dock their generated ideas post hoc.
3.3 Experiment 2: scaffold hopping
The c-Jun N-terminal kinase (JNK) pathway is implicated in neuronal injury and neurodegeneration and is a therapeutic target of interest. Patel et al. aimed to develop a small molecule inhibitor to modulate this pathway via targeting dual leucine zipper kinase (DLK) which is an upstream JNK regulator. Initial efforts led to the development of a potent and selective inhibitor but with high in vivo clearance (Fig. 7a).64,65 Subsequently, in a more recent study, Patel et al. applied a scaffold hopping strategy to improve the physico-chemical properties of their initial inhibitor to achieve central nervous system (CNS) penetration (Fig. 7b). Scaffold hopping from the pyridine core to a pyrazole core led to the discovery of a DLK inhibitor with in vivo efficacy (Fig. 7b).65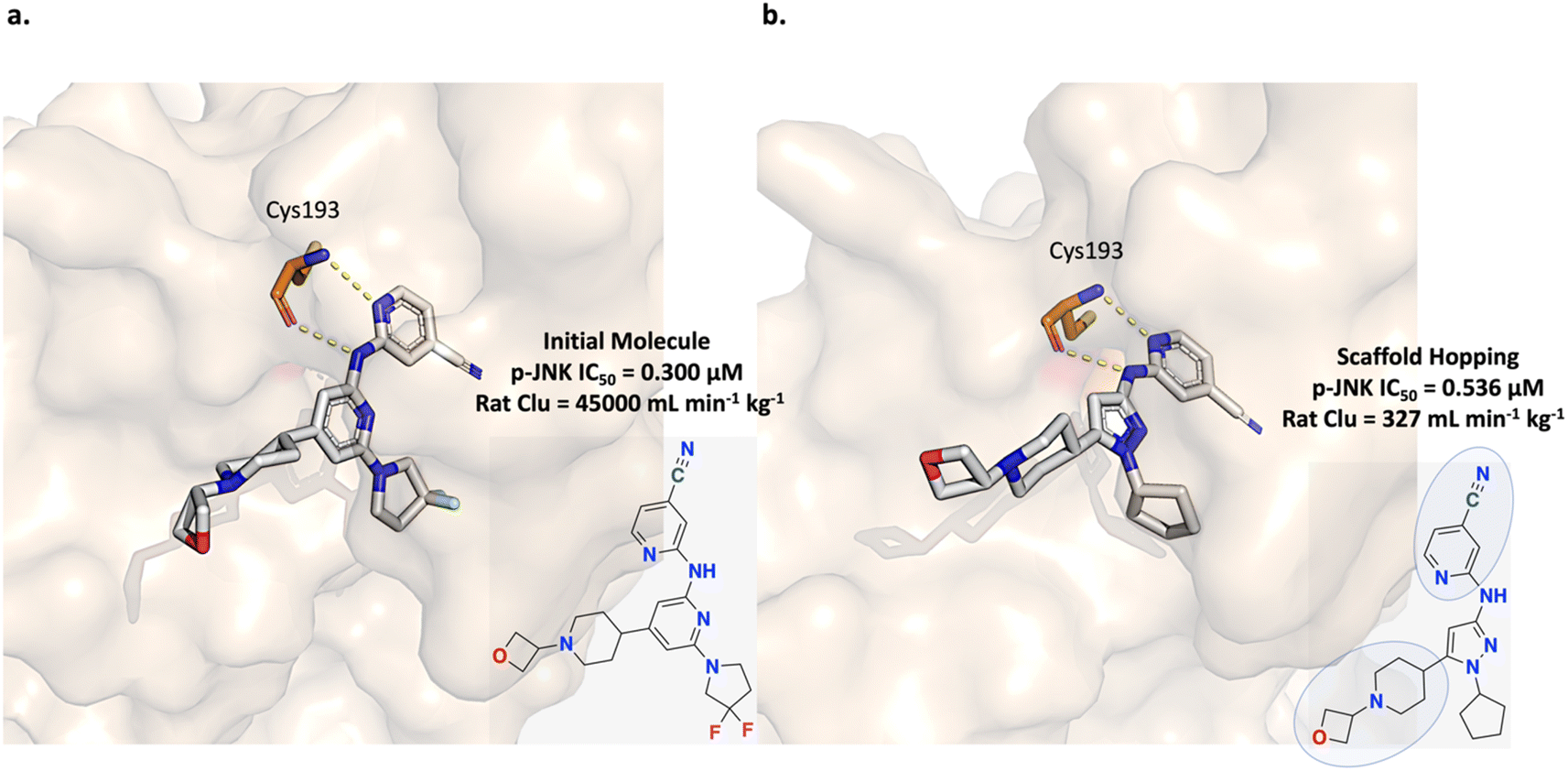 | ||
| Fig. 7 Experiment 2: scaffold hopping strategy for dual leucine zipper kinase (DLK) inhibitor optimization. (a) Initial inhibitor possessing poor physico-chemical properties causing in vivo high clearance (PDB ID: 5CEO). The two hydrogen bonds in the hinge region with Cys193 are crucial for potency. The goal was to replace the pyridine core while retaining the Cys193 interactions. (b) Scaffold hopping led to the discovery of a DLK inhibitor with a pyrazole core and with demonstrated in vivo efficacy (PDB ID: 5CEQ).64,65 The retained molecular sub-units are circled in the structure. | ||
In this section, we adopt the scaffold hopping strategy devised by Patel et al. and task Link-INVENT with generating novel core ideas with a focus on improving CNS properties. A docking constraint to enforce the Cys193 hydrogen-bond interactions is applied to retain predicted potency and the following specific physico-chemical properties, adopted from Patel et al., were enforced:65 the number of HBDs must be less than 2, the topological polar surface area (tPSA) must be less than 90 Å2, and the CNS MPO score must be greater than or equal to 4. The CNS MPO is an algorithm developed from analysis of CNS drugs and candidates as a predictor for CNS efficacy and encompasses six physico-chemical properties (ClogP, ClogD, MW, tPSA, number of HBDs, and pKa).66 In the devised experiment, we do not account for all six CNS MPO properties and only enforce logP, MW, tPSA, and number of HBDs. Correspondingly, we define the Scoring Function with the following components (see ESI S11 and S12† for Scoring Function transformations) and note that the reference linker is not present in the training set:
1. DockStream: this component is identical to the usage described in the fragment linking section. The only exception was that the docking constraint was enforced to retain the Cys193 hydrogen-bond interactions in the hinge region.65
2. Number of hydrogen bond donors <2: this component is included in the CNS MPO algorithm and enforces the overall linked molecule to possess less than two HBDs. This quantity was specifically desired by Patel et al.65
3. Molecular weight ≤450 Da: this component is included in the CNS MPO algorithm and is enforced to be in an interval in agreement with CNS penetration but with some leniency to allow more Link-INVENT exploration of chemical space.66
4. 3 ≤ SlogP ≤ 4: this component is included in the CNS MPO algorithm and is enforced to be in an interval in agreement with CNS penetration.66
5. tPSA ≤ 90 Å2: this component is included in the CNS MPO algorithm and is enforced to be in an interval in agreement with CNS penetration.66 The interval was also specifically desired by Patel et al.65
6. 1 ≤ linker number of aromatic rings ≤2: this component was specifically desired by Patel et al. as the binding site geometry is most compatible with a planar ring present in the core scaffold.65
The scaffold hopping experiment was run in triplicate and the results are shown in Fig. 8 (see ESI Fig. S13† for all training plots). Over the course of 100 epochs, the average Glide docking score of the batch of molecules generated by Link-INVENT gradually becomes more favourable (Fig. 8a) and the similarity in the docking score distributions demonstrates a reproducible experimental outcome (Fig. 8b). In contrast to the fragment linking experiment, relatively few molecules possess a more favourable docking score than the reference ligand (shown by the black dotted line). Instead, the majority of molecules score slightly worse (approximately −9.5 kcal mol−1). This is not completely unexpected as the MPO objective is significantly more challenging than the previous fragment linking case study. Consequently, the solution space is much narrower. It is important to note, however, that the objective of the scaffold hopping experiment is not strictly to propose novel cores that dock better than the initial inhibitor (Fig. 6a). Patel et al. noted that their initial inhibitor, while potent, exhibits high in vivo clearance.64,65 Therefore, an inhibitor with sufficient binding affinity and good CNS penetration could achieve in vivo efficacy. The narrower solution space in the scaffold hopping experiment is further supported by Fig. 8c where the absolute counts of unique Bemis–Murcko scaffolds is less than the fragment linking experiment.46 This is not a limitation of Link-INVENT but rather the nature of the MPO objective. Nonetheless, the absolute count for the generated scaffolds is still high and demonstrates Link-INVENT samples with diverse minima. Similar to the fragment linking results, minimal overlap between replicate runs shows that replicate experiments explore different areas in chemical space (Fig. 8c). The plausibility of the proposed scaffolds was investigated by comparing their binding poses with the reference ligand. Fig. 8d shows the binding pose of an example top scoring molecule (based on the satisfaction of the composite Scoring Function) superimposed with the reference ligand (see ESI Fig. S14† for more examples). Firstly, the proposed scaffold features planar aromatic rings, as enforced by the Scoring Function, and as desired by Patel et al.65 Secondly, the Cys193 hydrogen-bond interactions are retained, as enforced by the docking constraint. The proposed ligand is predicted to form an additional hydrogen bond with Gln195, owing to the hydrocarbon chain that extends the spatial occupancy of the overall molecule (Fig. 8d). This suggests that the application of a docking constraint can guide the Link-INVENT agent towards 3D structural awareness, learning to exploit the binding site geometry and electronics. Finally, the binding poses of the generated ligand and the reference ligand overlap significantly, supporting plausibility. Taken together, the results in this section demonstrate the flexibility of the Link-INVENT Scoring Function to optimize relatively complex MPO objectives and that the agent learns to propose plausible chemical ideas.
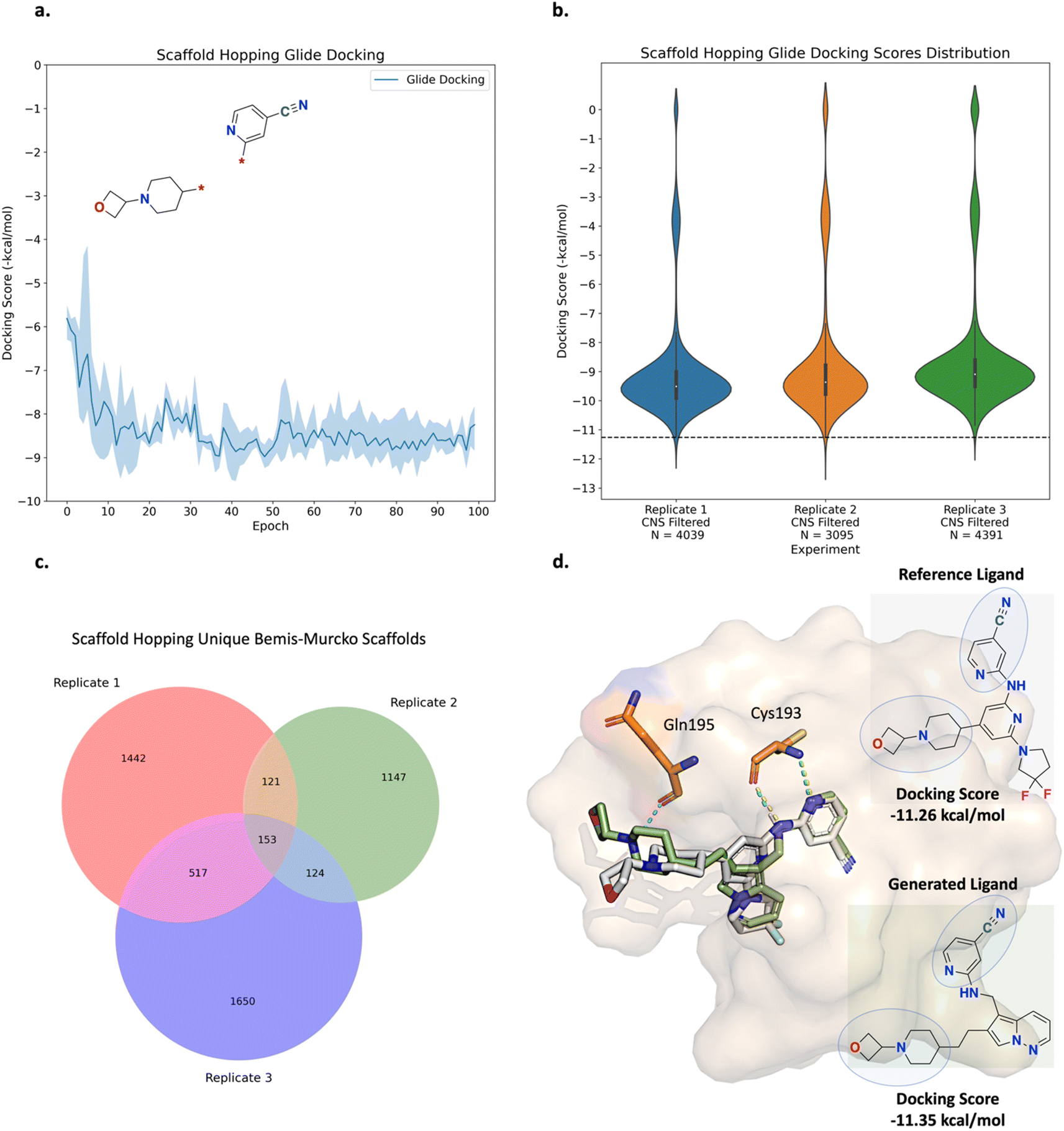 | ||
| Fig. 8 Experiment 2: scaffold hopping Link-INVENT results. The experiment was run in triplicate. The curve shows the average score achieved by the batch of molecules sampled at a given epoch and the upper and lower bounds of the shaded region represent the maximum and minimum scores, respectively. The analysis is identical to the fragment linking experiments. (a) Glide LigPrep docking score optimization. The average docking score achieved by the batch of compounds generated by Link-INVENT. (b) Violin plots showing the distribution of docking scores for the triplicate runs. ‘N’ is the number of molecules generated over 100 epochs that satisfy all the CNS criteria. The black dotted line shows the docking score possessed by the reference ligand (−11.26 kcal mol−1). (c) Venn diagram plots showing the overlap between unique Bemis–Murcko scaffolds in the triplicate runs. (d) The binding pose of a selected generated molecule (green) superimposed with the reference ligand (gray) and the retained molecular sub-units are circled. PDB ID: 5CEO. The yellow and turquoise dotted lines show the interactions formed by the reference ligand and generated ligand, respectively. The generated molecule retains the Cys193 interaction as enforced by the docking constraint and is predicted to form an additional hydrogen-bond interaction with Gln195. The extensive overlap between the binding poses of the generated ligand and the reference ligand supports plausibility. | ||
3.4 Experiment 3: PROTACs
B-cell lymphoma 2 (Bcl-2) and myeloid leukemia 1 (Mcl-1) are anti-apoptotic proteins which can inhibit intrinsic apoptosis, i.e., induced by mitochondrial stress, and are therapeutic targets of interest. Wang et al. designed linkers between a naphthalimide-based µM inhibitor and pomalidomide which is a cereblon (CRBN) binding ligand (Fig. 9).67 The resulting ternary complex would undergo ubiquitination and lead to targeted dual degradation of Bcl-2 and Mcl-1. Due to challenges in obtaining crystal structures as a basis to form hypotheses on optimal linker lengths, PROTAC linker design has mostly been empirical.36 Wang et al. adopted an iterative SAR approach to investigate the effect of the linker length on Bcl-2 and Mcl-1 dual degradation, whereby a linker too short would cause steric clash and prevent the formation of a stable ternary complex and a linker too long could result in too much conformational entropy to overcome.36,67 Following this approach, Wang et al. successfully transformed a low-affinity and non-selective ligand for Bcl-2 and Mcl-1 into PROTACs stabilized via protein–protein interactions (PPIs) and show potent and selective dual degradation in vitro.67Fig. 9a and b show the MD simulated ternary complex of one of the discovered PROTACs performed by Wang et al. and the general linking strategy envisioned, respectively.67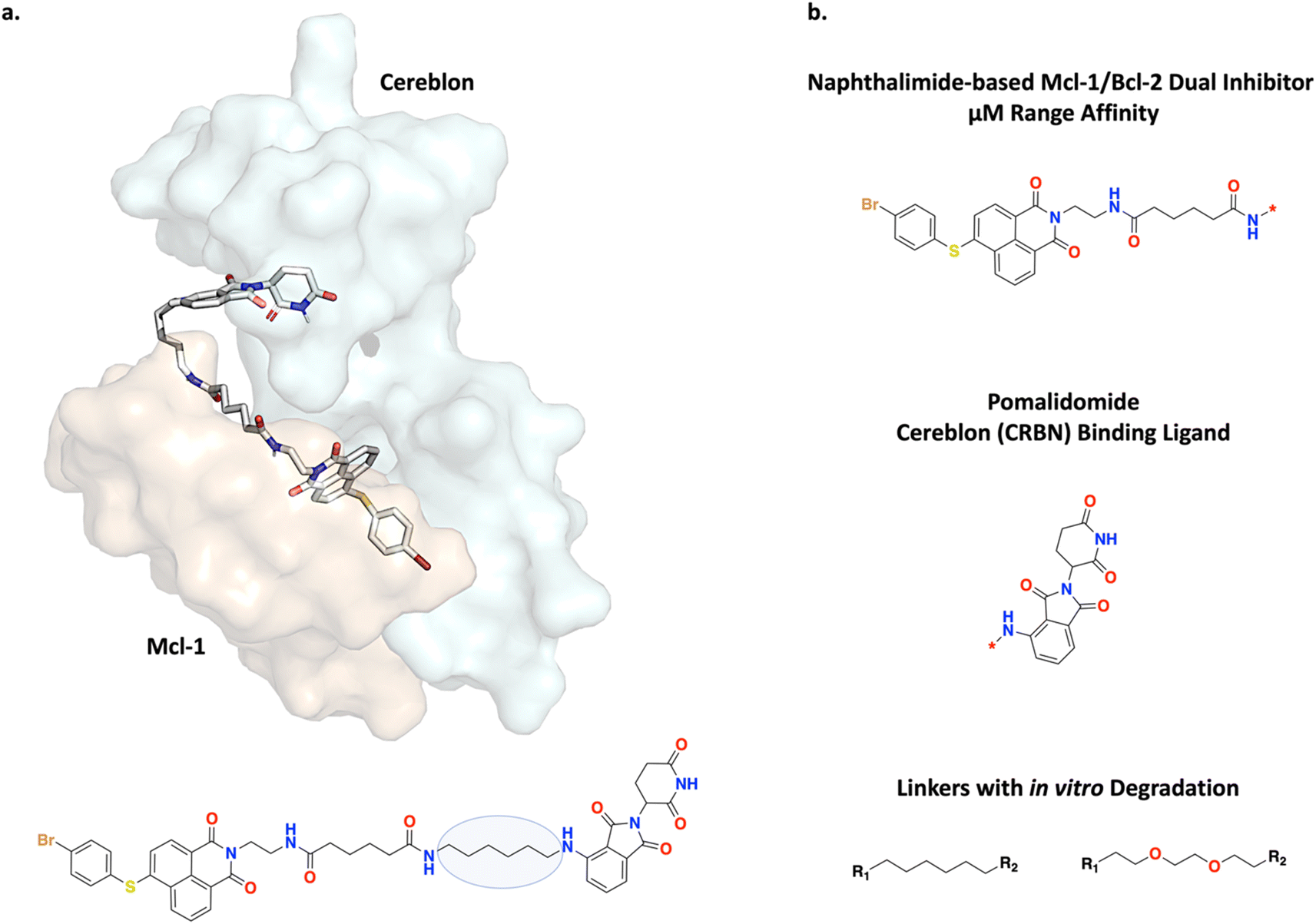 | ||
| Fig. 9 Experiment 3: PROTAC strategy for Bcl-2 and Mcl-1 dual degradation. (a) Molecular dynamics (MD) simulated ternary complex of one of the developed PROTACs binding to Mcl-1 (orange) and cereblon (CRBN, blue). The ternary complex is stabilized by protein–protein interactions (PPIs). The linker is an alkyl chain and is circled in the structure. Mcl-1 PDB ID: 2PQK and CRBN PDB ID: 4TZ4. (b) The naphthalimide-based µM inhibitor which was linked with pomalidomide to form the ternary complex. The R-groups denote the linker attachment points. The two linkers that achieved potent and selective in vitro degradation are shown. | ||
In this section, we use the PROTAC design strategy by Wang et al. to demonstrate Link-INVENT's linker specific components for the Scoring Function. In select experiments, a fixed set of physico-chemical properties was enforced and based on observed values from compiled PROTAC databases.34,35 Correspondingly, we define the Scoring Function with the following components (see ESI Fig. S10† for the Scoring Function transformations):
1. tPSA ≤ 250 Å2.
2. 3.5 ≤ logP ≤ 6.0.
3. Number of hydrogen bond acceptors ≤ 16.
4. Number of hydrogen bond donors ≤ 6.
5. Number of rotatable bonds < 25.
We demonstrate control over the properties of generated linkers while keeping physico-chemical properties of the PROTAC within the specified intervals described above. Subsequently, we devise three Sub-Experiments:
1. Sub-Experiment 1: fix physico-chemical properties and control the linker length. We show that Link-INVENT can generate linkers within a specified narrow length interval. In addition to including the physico-chemical properties listed above, the Scoring Function contains the following components:
(1) Linker effective length = [4, 6], [7, 9], [10, 12], or [13, 15]: this component enforces linkers to possess an effective length within the specified intervals. See ESI S15† for the Scoring Function transformation.
(2) Linker length ratio = 100; this component prevents linker branching.
The combination of components 1 and 2 enforces Link-INVENT to generate linkers without branching.
2. Sub-Experiment 2: fix physico-chemical properties and the linker length within the interval [7, 9], and control linker linearity, i.e., linkers with and without rings. We show that Link-INVENT can generate linkers within a specified narrow length interval and control for the presence of rings. In addition to including the physico-chemical properties listed above, the Scoring Function contains the following component (see ESI Fig. S22† for the Scoring Function transformations):
(1) Linker effective length = [7, 9]: this component enforces linkers to possess an effective length within the specified interval of [7, 9].
(2) Linker length ratio = 100; this component prevents linker branching.
(3) Linker number of rings = 0; this component enforces linkers to possess no rings, i.e., the linker is linear. In the experiment where we want to generate linkers with rings, we simply omit this component in the Scoring Function.
Similar to Sub-Experiment 1, components 1 and 2 enforce Link-INVENT to generate linkers without branching.
3. Sub-Experiment 3: in this Sub-Experiment, no length or physico-chemical properties are enforced. Instead, we task Link-INVENT with generating linkers with variable flexibility, which is defined by the “linker ratio of rotatable bonds” component, i.e., ratio between the number of rotatable bonds over the total number of bonds. Correspondingly, the Scoring Function contains only one component:
(1) Linker ratio of rotatable bonds = [0, 30], [40, 60], and [70, 100]: the defined intervals correspond to “Low”, “Moderate”, and “High” flexibility (see ESI S26† for the Scoring Function transformation).
PROTAC Sub-Experiment 1: controlling the linker length. Link-INVENT was tasked with generating linker ideas of variable length while keeping physico-chemical properties within a specified range (Fig. 10a, see ESI Fig. S17–S21† for all training plots). The baseline experiment does not enforce a specific effective linker length interval and the distribution of lengths spans a large range (Fig. 10a). In contrast, one can enforce the Link-INVENT agent to explore effective linker lengths within a certain interval, as shown by the enrichments observed in Fig. 10a, e.g., the ‘enforce 4–6’ experiment enforced effective linker lengths in the interval [4–6] and the corresponding bar is enriched relative to other lengths. The purpose of this Sub-Experiment is to show the ease with which one can control effective linker length exploration, mimicking a real-world PROTAC linker design campaign.36,67
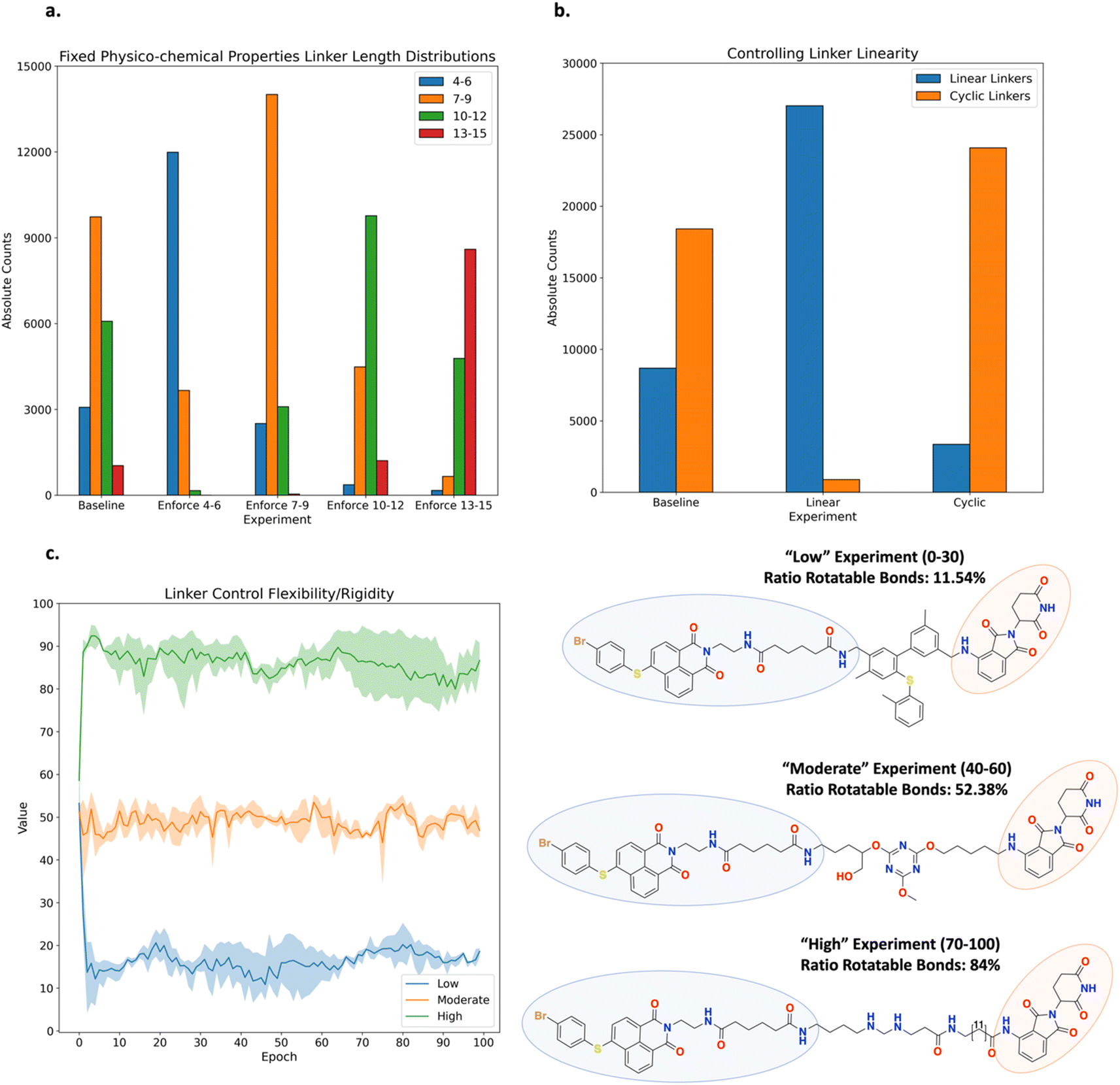 | ||
| Fig. 10 Experiment 3: PROTAC Link-INVENT results. The experiments were run in triplicate. The curve in (c) shows the average score achieved by the batch of molecules sampled at a given epoch and the upper and lower bounds of the shaded region represent the maximum and minimum scores, respectively. (a) Experiment that fixes physico-chemical properties and tasks Link-INVENT with generating linkers with an effective length within the specified intervals: [4, 6], [7, 9], [10, 12], and [13, 15]. The baseline experiment does not enforce the linker length. Consequently, the effective linker lengths resemble a broad distribution. The generated linkers from the triplicate runs which also satisfy all the physico-chemical property criteria are aggregated in the plot. (b) Experiment that fixes physico-chemical properties and the effective linker length within the interval [7–9]. Link-INVENT is tasked with generating linear and cyclic linkers. The baseline experiment does not enforce linker linearity and approximately a 1:2 ratio of linear:cyclic is observed. The generated linkers from the triplicate runs which also satisfy all the physico-chemical property criteria are aggregated in the plot. (c) Experiment that tasks Link-INVENT with generating linkers containing a ratio of rotatable bonds within the specified intervals: “Low” [0, 30], “Moderate” [40, 60], and “High” [70, 100]. The agent implicitly learns that linkers containing rings and sp2 hybridized atoms achieve a low ratio of rotatable bonds. Conversely, linear linkers with sp3 hybridized atoms achieve a high ratio of rotatable bonds. | ||
PROTAC Sub-Experiment 2: controlling linker linearity. Link-INVENT was tasked with generating linker ideas with an effective length in the interval [7, 9], while keeping physico-chemical properties within a specified range and controlling linearity (Fig. 10b, see ESI Fig. S23–S25† for all training plots). The baseline experiment does not enforce linearity and the resulting ratio of linear linkers to cyclic linkers, i.e., linkers containing at least one ring, is approximately 1:2. In contrast, one can enforce the Link-INVENT agent to explore linear linkers or cyclic linkers, shown by the enrichments observed in Fig. 10b. The purpose of this Sub-Experiment is to further showcase the user flexibility in specifying desired linker properties.
PROTAC Sub-Experiment 3: controlling linker flexibility. This Sub-Experiment showcases Link-INVENT's “linker ratio of rotatable bonds” component which can be specified in the Scoring Function. We note that while the component itself is meant to be a descriptor of linker flexibility, inherent limitations exist, e.g., not accounting for intra-molecular hydrogen-bonding interactions which would rigidify the linker. Link-INVENT was tasked with generating linker ideas with variable ratios of rotatable bonds where we define ‘Low’, ‘Moderate’, and ‘High’ as the intervals [0, 30], [40, 60], and [70, 100], respectively (Fig. 9c, see ESI Fig. S27† for all training plots). Examples of linkers possessing variable degrees of flexibility are shown in Fig. 10c. The agent implicitly learns that linkers containing rings and sp2 hybridized atoms are more rigid. A clear transition from “Low” flexibility to “High” flexibility is marked by increasing linearity and sp3 hybridized atoms. Without enforcing any length constraints, the proposed linkers become increasingly longer to achieve a high “linker ratio of rotatable bonds” value. This is exemplified in the example linker in the “High” experiment (Fig. 10c). Naturally, the linker shown is likely unrealistic and this Sub-Experiment was an extreme example to showcase the flexibility of Link-INVENT's Scoring Function. In practice, one could constrain the linker length within a specified interval as was done in Sub-Experiments 1 and 2 and explore variable flexibility. In this regard, the “linker ratio of rotatable bonds” provides some control over the conformational entropy of proposed linker ideas.
4. Conclusions
In this work, we introduced Link-INVENT as an extension to the de novo design platform, REINVENT.6 Link-INVENT is a recurrent neural network (RNN)-based generative model trained to propose linker ideas given two input molecular subunits. In contrast to previous methods for linker design involving database searching which are inherently limited to a pre-defined collection of chemical ideas,37–40 Link-INVENT builds linkers at the token level, proposing linkers as SMILES and can generalize in chemical space.15,47 Moreover, Link-INVENT's Scoring Function provides users with the ability to optimize bespoke multi-parameter optimization (MPO) objectives via reinforcement learning (RL), offering control that is not present in existing deep learning (DL)-based approaches to linker generation.16–19 A vast number of molecular properties can be optimized, ranging from previously implemented components available in REINVENT that operate on the entire molecule to newly implemented linker specific components which provide control over linker properties.We demonstrate the application of Link-INVENT in three case studies encompassing fragment linking,24,25 scaffold hopping,26 and PROTAC design.27–29 The Scoring Functions for the experiments were devised based on the corresponding fragment linking,52,53,61 scaffold hopping,64,65 and PROTAC design67 studies. We illustrate the practical adoption of Link-INVENT to real-world drug discovery projects by showcasing how to translate experimental insights into an informative Scoring Function for Link-INVENT. Subsequently, the agent learned to satisfy the desired MPO objective via RL. Specifically, in Experiment 1a: fragment linking,52,53 we showed that Link-INVENT can propose plausible linker ideas that satisfy a molecular docking constraint with an additional constraint over the permitted linker spatial occupancy by controlling for branching. More than 5000 unique Bemis–Murcko scaffolds were generated by the Link-INVENT agent, demonstrating that diverse linker ideas were explored.46 Similarly, in Experiment 1b: comparison fragment linking, we showed that the Link-INVENT agent can learn to generate molecules that satisfy a core constrained docking protocol. Furthermore, by including docking explicitly as a component in the Scoring Function, Link-INVENT is able to generate molecules that possess generally more favourable docking scores than DeLinker and SyntaLinker which are previously reported DL-based methods for linker design.16,17 In the scaffold hopping experiment,64,65 we showed that Link-INVENT can simultaneously optimize a relatively complex MPO objective encompassing a molecular docking constraint and favourable central nervous system (CNS) compatible physico-chemical properties. In this experiment, Link-INVENT navigated a narrow solution space and proposed plausible scaffold ideas which satisfy all desired properties and are diverse as shown by the number of unique Bemis–Murcko scaffolds.46 In the PROTAC experiment,67 we further showed Link-INVENT's extensive user control on the linker properties. We demonstrated the ability to enforce the Link-INVENT agent to explore effective linker lengths within a specified interval while keeping physico-chemical properties within a specified range. Moreover, linker linearity can be controlled, enforcing the agent to explore only linear linkers or linkers containing rings. Finally, we prove that linker flexibility can be controlled via the “linker ratio of rotatable bonds” component which provides users with the ability to modulate the conformational entropy of proposed linker ideas. These series of PROTAC Sub-Experiments mimic real-world PROTAC linker design, which typically investigate linkers of variable length and flexibility.36,67
Link-INVENT is a ready-to-use generative model for linker design with the capability to optimize bespoke MPO objectives via the flexible Scoring Function. The case studies in this work show how Link-INVENT can be applied to real-world drug discovery projects and that the agent proposes plausible and diverse linker ideas. The code is freely available at https://github.com/MolecularAI/Reinvent.
Data availability
The data used to train the Link-INVENT prior model are publicly available in the following GitHub repository: https://github.com/MolecularAI/ReinventCommunity. Provided files include the reaction SMIRKS used to slice the raw ChEMBL data and the corresponding training and validation data sets. The unique Bemis–Murcko scaffolds held out in the validation set are provided in an additional file. Moreover, the pre-trained prior presented in this work and an accompanying Link-INVENT Jupyter tutorial notebook are provided.The Link-INVENT code is publicly available in the following GitHub repository: https://github.com/MolecularAI/Reinvent. Finally, molecular docking was performed using proprietary software licensed by Schrödinger (version 2019-4): LigPrep and Glide.55–59 Reproducing experiments 1 and 2 require a Schrödinger license.
Conflicts of interest
The authors declare no competing interests.Notes and references
- P. Schneider, W. P. Walters, A. T. Plowright, N. Sieroka, J. Listgarten, R. A. Goodnow, J. Fisher, J. M. Jansen, J. S. Duca, T. S. Rush, M. Zentgraf, J. E. Hill, E. Krutoholow, M. Kohler, J. Blaney, K. Funatsu, C. Luebkemann and G. Schneider, Nat. Rev. Drug Discovery, 2020, 19, 353–364 CrossRef CAS PubMed.
- J. Jiménez-Luna, F. Grisoni, N. Weskamp and G. Schneider, Expert Opin. Drug Discovery, 2021, 1–11 Search PubMed.
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675–679 CrossRef CAS PubMed.
- Beam Search for Automzated Design and Scoring of Novel ROR Ligands with Machine Intelligence** – Moret – 2021, Angew. Chem., Int. Ed., Wiley Online Library, https://onlinelibrary.wiley.com/doi/full/10.1002/anie.202104405, accessed 19 March 2022 Search PubMed.
- M. Popova, O. Isayev and A. Tropsha, Sci. Adv., 2018, 4, eaap7885 CrossRef CAS PubMed.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, J. Chem. Inf. Model., 2020, 60, 5918–5922 CrossRef CAS PubMed.
- M. Thomas, R. T. Smith, N. M. O'Boyle, C. de Graaf and A. Bender, J. Cheminf., 2021, 13, 39 CAS.
- Y. Li, L. Zhang and Z. Liu, J. Cheminf., 2018, 10, 33 Search PubMed.
- R. Mercado, T. Rastemo, E. Lindelöf, G. Klambauer, O. Engkvist, H. Chen and E. Jannik Bjerrum, Mach. Learn.: Sci. Technol., 2021, 2, 025023 Search PubMed.
- S. R. Atance, J. V. Diez, O. Engkvist, S. Olsson and R. Mercado, J. Chem. Inf. Model., 2022, 62(20), 4863–4872 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- Q. Bai, S. Tan, T. Xu, H. Liu, J. Huang and X. Yao, Briefings Bioinf., 2021, 22(3), bbaa161 CrossRef PubMed.
- J. Choi and J. Lee, Int. J. Mol. Sci., 2021, 22(21), 11635 CrossRef CAS PubMed.
- A. Nigam, R. Pollice and A. Aspuru-Guzik, Digital Discovery, 2022, 1(4), 390–404 RSC.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- F. Imrie, A. R. Bradley, M. van der Schaar and C. M. Deane, J. Chem. Inf. Model., 2020, 60, 1983–1995 CrossRef CAS PubMed.
- Y. Yang, S. Zheng, S. Su, C. Zhao, J. Xu and H. Chen, Chem. Sci., 2020, 11, 8312–8322 RSC.
- L. Hu, Y. Yang, S. Zheng, J. Xu, T. Ran and H. Chen, J. Chem. Inf. Model., 2021, 61, 4900–4912 CrossRef CAS PubMed.
- M. Langevin, H. Minoux, M. Levesque and M. Bianciotto, J. Chem. Inf. Model., 2020, 60, 5637–5646 CrossRef CAS PubMed.
- F. Imrie, T. E. Hadfield, A. R. Bradley and C. M. Deane, Chem. Sci., 2021, 12, 14577–14589 RSC.
- Y. Huang, X. Peng, J. Ma and M. Zhang, 3DLinker: An E(3) Equivariant Variational Autoencoder for Molecular Linker Design, arXiv, 2022, preprint, arXiv:2205.07309, DOI:10.48550/arXiv.2205.07309.
- I. Igashov, H. Stärk, C. Vignac, V. G. Satorras, P. Frossard, M. Welling, M. Bronstein and B. Correia, arXiv, 2022, preprint, arXiv:2210.05274, DOI:10.48550/arXiv.2210.05274.
- Y. Feng, Y. Yang, W. Deng, H. Chen and T. Ran, Artif. Intell. Life Sci., 2022, 2, 100035 Search PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- H.-J. Böhm, A. Flohr and M. Stahl, Drug Discovery Today: Technol., 2004, 1, 217–224 CrossRef PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- X. Sun, H. Gao, Y. Yang, M. He, Y. Wu, Y. Song, Y. Tong and Y. Rao, Signal Transduction Targeted Ther., 2019, 4, 1–33 CrossRef PubMed.
- J. D. Chodera and D. L. Mobley, Annu. Rev. Biophys., 2013, 42, 121–142 CrossRef CAS PubMed.
- A. Bancet, C. Raingeval, T. Lomberget, M. Le Borgne, J.-F. Guichou and I. Krimm, J. Med. Chem., 2020, 63, 11420–11435 CrossRef CAS PubMed.
- H. S. Yu, K. Modugula, O. Ichihara, K. Kramschuster, S. Keng, R. Abel and L. Wang, J. Chem. Theory Comput., 2021, 17, 450–462 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- H. J. Maple, N. Clayden, A. Baron, C. Stacey and R. Felix, Med. Chem. Commun., 2019, 10, 1755–1764 RSC.
- G. Weng, C. Shen, D. Cao, J. Gao, X. Dong, Q. He, B. Yang, D. Li, J. Wu and T. Hou, Nucleic Acids Res., 2021, 49, D1381–D1387 CrossRef CAS PubMed.
- T. A. Bemis, J. J. La Clair and M. D. Burkart, J. Med. Chem., 2021, 64, 8042–8052 CrossRef CAS PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, J. Chem. Inf. Model., 2006, 46, 665–676 CrossRef CAS PubMed.
- P. Maass, T. Schulz-Gasch, M. Stahl and M. Rarey, J. Chem. Inf. Model., 2007, 47, 390–399 CrossRef CAS PubMed.
- D. C. Thompson, R. A. Denny, R. Nilakantan, C. Humblet, D. Joseph-McCarthy and E. Feyfant, J. Comput.-Aided Mol. Des., 2008, 22, 761–772 CrossRef CAS PubMed.
- M. J. Vainio, T. Kogej, F. Raubacher and J. Sadowski, J. Chem. Inf. Model., 2013, 53, 1825–1835 CrossRef CAS PubMed.
- A. Yoshimori, Y. Asawa, E. Kawasaki, T. Tasaka, S. Matsuda, T. Sekikawa, S. Tanabe, M. Neya, H. Natsugari and C. Kanai, ChemMedChem, 2021, 16, 955–958 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- V. Fialková, J. Zhao, K. Papadopoulos, O. Engkvist, E. Bjerrum, T. Kogej and A. Patronov, J. Chem. Inf. Model., 2022, 62(9), 2046–2063 CrossRef PubMed.
- J. Arús-Pous, A. Patronov, E. J. Bjerrum, C. Tyrchan, J.-L. Reymond, H. Chen and O. Engkvist, J. Cheminf., 2020, 12, 38 Search PubMed.
- S. Hochreiter and J. Schmidhuber, Neural Comput., 1997, 9, 1735–1780 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- J. Arús-Pous, S. V. Johansson, O. Prykhodko, E. J. Bjerrum, C. Tyrchan, J.-L. Reymond, H. Chen and O. Engkvist, J. Cheminf., 2019, 11, 71 Search PubMed.
- R. J. Williams and D. Zipser, Neural Comput., 1989, 1, 270–280 CrossRef.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminf., 2017, 9, 48 Search PubMed.
- T. Blaschke, O. Engkvist, J. Bajorath and H. Chen, J. Cheminf., 2020, 12, 68 CAS.
- RDKit, Open-source cheminformatics, 2019, https://www.rdkit.org Search PubMed.
- C. De Fusco, P. Brear, J. Iegre, K. H. Georgiou, H. F. Sore, M. Hyvönen and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 3471–3482 CrossRef CAS PubMed.
- P. Brear, C. De Fusco, K. Hadje Georgiou, N. J. Francis-Newton, C. J. Stubbs, H. F. Sore, A. R. Venkitaraman, C. Abell, D. R. Spring and M. Hyvönen, Chem. Sci., 2016, 7, 6839–6845 RSC.
- J. Guo, J. P. Janet, M. R. Bauer, E. Nittinger, K. A. Giblin, K. Papadopoulos, A. Voronov, A. Patronov, O. Engkvist and C. Margreitter, J. Cheminf., 2021, 13, 89 Search PubMed.
- Schrödinger Release 2019-4: Glide, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- Schrödinger Release 2019-4: LigPrep, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- A. Alex, D. S. Millan, M. Perez, F. Wakenhut and G. A. Whitlock, Med. Chem. Commun., 2011, 2, 669–674 RSC.
- A. Trapero, A. Pacitto, V. Singh, M. Sabbah, A. G. Coyne, V. Mizrahi, T. L. Blundell, D. B. Ascher and C. Abell, J. Med. Chem., 2018, 61, 2806–2822 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- Molecular Operating Environment (MOE), 2020.09 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022 Search PubMed.
- S. Patel, F. Cohen, B. J. Dean, K. De La Torre, G. Deshmukh, A. A. Estrada, A. S. Ghosh, P. Gibbons, A. Gustafson, M. P. Huestis, C. E. Le Pichon, H. Lin, W. Liu, X. Liu, Y. Liu, C. Q. Ly, J. P. Lyssikatos, C. Ma, K. Scearce-Levie, Y. G. Shin, H. Solanoy, K. L. Stark, J. Wang, B. Wang, X. Zhao, J. W. Lewcock and M. Siu, J. Med. Chem., 2015, 58, 401–418 CrossRef CAS PubMed.
- S. Patel, S. F. Harris, P. Gibbons, G. Deshmukh, A. Gustafson, T. Kellar, H. Lin, X. Liu, Y. Liu, Y. Liu, C. Ma, K. Scearce-Levie, A. S. Ghosh, Y. G. Shin, H. Solanoy, J. Wang, B. Wang, J. Yin, M. Siu and J. W. Lewcock, J. Med. Chem., 2015, 58, 8182–8199 CrossRef CAS PubMed.
- Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties, ACS Chem. Neurosci., https://pubs.acs.org/doi/pdf/10.1021/cn100008c, accessed 31 January 2022 Search PubMed.
- Z. Wang, N. He, Z. Guo, C. Niu, T. Song, Y. Guo, K. Cao, A. Wang, J. Zhu, X. Zhang and Z. Zhang, J. Med. Chem., 2019, 62, 8152–8163 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dd00115b |
| ‡ These authors contributed equally. |
| § Present address: Odyssey Therapeutics, Cambridge, MA, USA. |
| This journal is © The Royal Society of Chemistry 2023 |