
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvBERT for solvation free energy and solubility prediction: a demonstration of an NLP model for predicting the properties of molecular complexes†
Jiahui
Yu
a,
Chengwei
Zhang
b,
Yingying
Cheng
b,
Yun-Fang
Yang
 b,
Yuan-Bin
She
b,
Fengfan
Liu
a,
Weike
Su
a and
An
Su
*b
b,
Yuan-Bin
She
b,
Fengfan
Liu
a,
Weike
Su
a and
An
Su
*b
aNational Engineering Research Center for Process Development of Active Pharmaceutical Ingredients, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, PR China
bCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310014, PR China. E-mail: ansu@zjut.edu.cn
First published on 30th January 2023
Abstract
Deep learning models based on NLP, mainly the Transformer family, have been successfully applied to solve many chemistry-related problems, but their applications are mostly limited to chemical reactions. Meanwhile, solvation is an important concept in physical and organic chemistry, describing the interaction of solutes and solvents. In this study, we introduced the SolvBERT model, which reads the solute and solvent through the SMILES representation of their combination. SolvBERT was pre-trained in an unsupervised learning fashion using a large database of computational solvation free energies. The pre-trained model could be used to predict the experimental solvation free energy or solubility, depending on the fine-tuning database. To the best of our knowledge, this multi-task prediction capability has not been observed in previously developed graph-based models for predicting the properties of molecular complexes. Furthermore, the performance of our SolvBERT in predicting solvation free energy was comparable to the state-of-the-art graph-based model DMPNN, mainly due to the clustering feature of the pre-training phase of the model, as demonstrated using the TMAP visualization algorithm. Last but not least, our SolvBERT outperformed the recently-developed GNN–Transformer hybrid model, GROVER, in predicting a set of experimentally evaluated solubility data with out-of-sample solute–solvent combinations.
Introduction
The use of deep learning models to study the chemical sciences is rapidly increasing, especially in subfields including synthetic planning1,2 and automatic chemical designing.3,4 From a model architecture perspective, the two most commonly used deep learning architectures for chemistry-related problems are graph-based models and text-based natural language processing (NLP) models. Graph-based models, including graph convolutional networks (GCN),5,6 message passing neural networks (MPNN),7 and the recently developed directed-MPNN (D-MPNN),8 have mostly been applied to the prediction of molecular properties.9–13 On the other hand, NLP models, mainly the Transformer14 family, have been mainly applied to the study of chemical reactions, such as the prediction of forward reaction outcomes15–17 and retrosynthetic pathways18–20 as well as inferring reaction mechanisms21–23 and experimental procedures.24,25 Previously, the application of NLP models in chemistry was mostly limited to chemical reactions, one reason being that early NLP models were sequence-to-sequence – the models could only read in and output text-based representations. However, due to the rapid development of the Transformer family, a model called Bidirectional Encoder Representations from Transformers (BERT)26 was built for classification and regression tasks. Two recent studies by Schwaller et al. used BERT to predict the classifications27 and yields28 of chemical reactions, demonstrating the potential of NLP models for predicting numerical properties.In addition to reactions, solvation is another type of molecular interaction that describes the interaction of a solvent with a dissolved molecule, in which the solute and solvent reorganize into a solvation complex.29,30 Solvation free energy and solubility are two physical properties commonly used to describe solvation.31,32 Solvation free energy is the change in free energy associated with the transfer of a molecule between the ideal gas and a solvent at a given temperature and pressure.33 Solubility is a parameter used to assess how much of a substance can remain in solution without precipitating and is defined as the maximum amount of a solute that can dissolve in a solvent under given physical conditions (pressure, temperature, pH, etc.).34 The prediction of these solvation properties can facilitate solvent screening in drug design, synthesis route design, process intensification, and crystallization.31,35
Using machine learning and deep learning to predict the relevant properties of solvation can save the expensive time and cost of performing experiments or computations, and also help to find important features that contribute to the solvation properties.36 Recent studies by Vermeire et al. and Zhang et al. both pre-trained graph-based neural networks on computational solvation free energy datasets and then used smaller experimental solvation free energy datasets for transfer learning. A potential drawback of using supervised learning in the pre-training phase is that it may lead to negative transfer (i.e., result in undesirable degradation of model performance) when transferring knowledge from pre-training on one attribute (e.g., solvation free energy) to the prediction of another attribute (e.g., solubility).37–39 In contrast, BERT, a new generation of NLP models, does not have this drawback because its pre-training phase is completely unsupervised (does not depend on data labels). Furthermore, to our knowledge, few NLP-based models have been used to study solvation.40,41 Given the success of the above NLP models in predicting chemical reactions, it is likely that NLP models can also study different types of molecular interactions, such as solvation.
Therefore, in this study, we introduced a BERT-based regression model, SolvBERT, to predict two properties of solvation, namely solvation free energy and solubility. Instead of inputting the solute and solvent separately, SolvBERT reads the SMILES of solute–solvent combinations and converts the SMILES combination into vectorized representations. We trained SolvBERT using three different databases computed or curated in previous studies.42,43 We also compared this model with state-of-the-art graph-based models, one graph-input NLP model, and a traditional machine learning model to further discuss the impact of model architecture in predicting the properties of molecular complexes. Also, TMAP, an advanced tree-based algorithm for visualizing high-dimensional data, was used to show the benefits of SolvBERT in clustering solvent–solute combinations. Finally, we measured the solubility of 21 solute–solvent combinations as out-of-sample data to experimentally evaluate the performance of SolvBERT.
Methods
Datasets
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 029 solutes. A detailed description can be retrieved from ref. 44.
145 different solute and solvent combinations from Vermeire et al.44 The dataset was curated from multiple sources, including the Minnesota solvation database,45 the FreeSolv database,46 the CompSol database,47 and a dataset published by Abraham et al.48 Unfortunately, to the best of our knowledge, only 8780 of these 10145 data instances are publicly available. Therefore, we downloaded data from these 8780 instances and renamed this dataset as CombiSolv-Exp-8780 to distinguish it from the original CombiSolv-Exp dataset. We compared the distribution of solvation free energy for our CombiSolv-Exp-8780 and the original CombiSolv-Exp in Vermeire et al.’s study44 and found no significant differences in their distributions (Fig. S1 in the ESI†).
S.
029 solutes. A detailed description can be retrieved from ref. 44.
145 different solute and solvent combinations from Vermeire et al.44 The dataset was curated from multiple sources, including the Minnesota solvation database,45 the FreeSolv database,46 the CompSol database,47 and a dataset published by Abraham et al.48 Unfortunately, to the best of our knowledge, only 8780 of these 10145 data instances are publicly available. Therefore, we downloaded data from these 8780 instances and renamed this dataset as CombiSolv-Exp-8780 to distinguish it from the original CombiSolv-Exp dataset. We compared the distribution of solvation free energy for our CombiSolv-Exp-8780 and the original CombiSolv-Exp in Vermeire et al.’s study44 and found no significant differences in their distributions (Fig. S1 in the ESI†).
S.
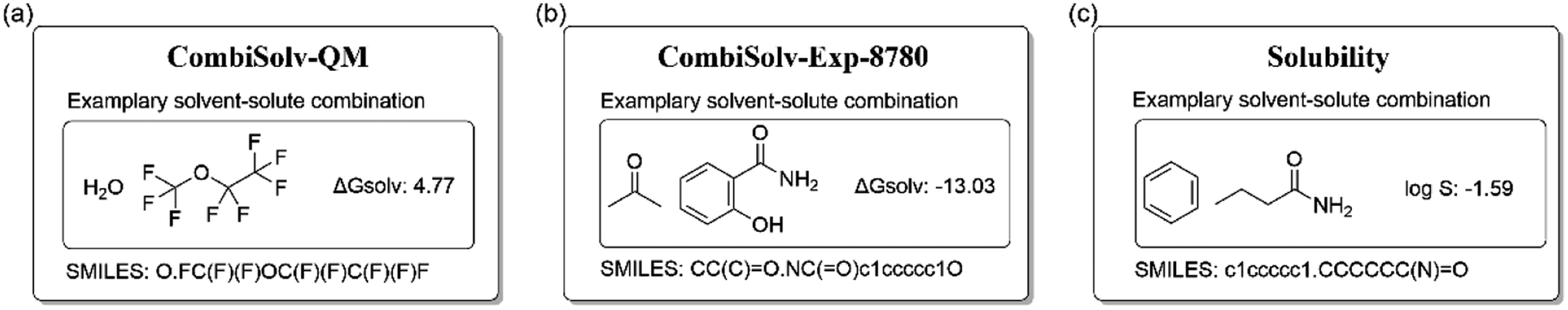 | ||
| Fig. 1 SMILES representation of solvent–solute combinations. | ||
Model names and architectures
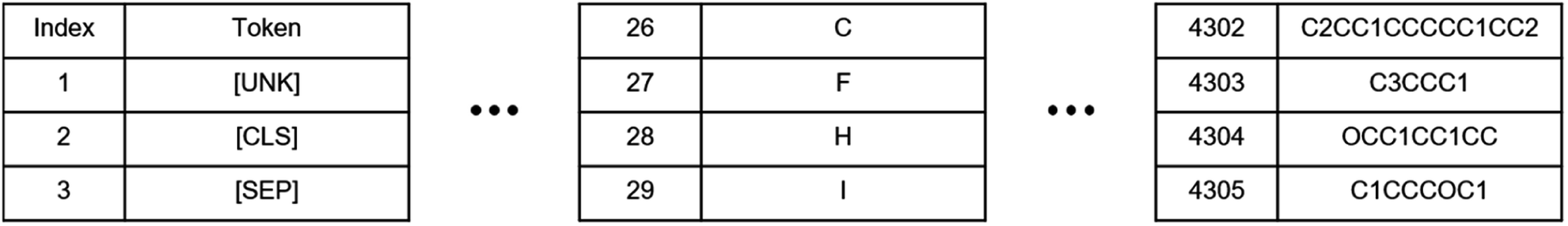 | ||
| Fig. 2 Vocabulary created by using tokenizer based on the combine-QM database. | ||
Similar to other BERT models for chemistry-related problems,27,49 SolvBERT performs a masked language model (MLM) learning task in the pre-training phase. MLM is a fill-in-the-blank task in which the model predicts the masked words based on the contextual words surrounding the mask tokens.50 SolvBERT was pre-trained by performing the MLM task of the SMILES combinations from CombiSolv-QM, where individual atoms of the input SMILES were masked with a probability of 0.15. In addition, a special class token [CLS] was prepended to the tokenized SMILES but was never masked during this pre-training phase. In contrast to the original rxnfp that uses the [CLS] as a classification header for reaction classification, SolvBERT uses the [CLS]-labeled embedding as the input for the regression head for predicting solvation-related properties. Furthermore, based on the default parameters of the original rxnfp framework, the hyperparameters of SolvBERT were further optimized, including batch size, learning rate, hidden dropout rate, and the number of training epochs.
| Model architecture | Molecular representation | Model name | Dataset for pre-training | Dataset for fine-tuning |
|---|---|---|---|---|
| SolvBERT | SMILES | SolvBERT-QM-Exp | CombiSolv-QM | CombiSolv-Exp-8780 |
| SolvBERT-Exp | CombiSolv-Exp-8780 | |||
| SolvBERT-QM | CombiSolv-QM | |||
| SolvBERT-QM-logS | CombiSolv-QM | Solubility | ||
| SolvBERT-logS | Solubility | |||
| GCN | Graphs | GCN-QM-Exp | CombiSolv-QM | CombiSolv-Exp-8780 |
| GCN-Exp | CombiSolv-Exp-8780 | |||
| D-MPNN | Graphs | D-MPNN-QM-Exp | CombiSolv-QM | CombiSolv-Exp-8780 |
| D-MPNN-Exp | CombiSolv-Exp-8780 | |||
| GROVER | Graphs | GROVER-Exp | CombiSolv-Exp-8780 | |
| GROVER-logS | Solubility | |||
| RF | MHFP | RF-Exp | CombiSolv-Exp-8780 |
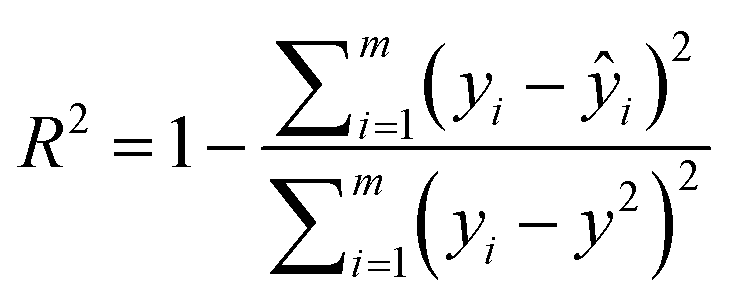
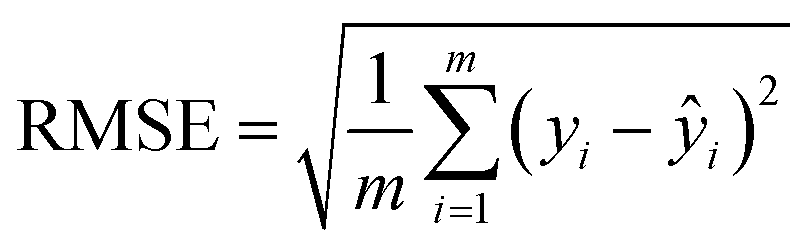
Results
Training SolvBERT
To fully evaluate the performance of the SolvBERT model without the potential interference of data scarcity, we first pre-trained and fine-tuned the model using the CombiSolv-QM dataset, which contained the computed solvation free energy of ∼1 million solute–solvent pairs (Fig. 3, left part). In the pre-training phase, the model was trained with SMILES of solute–solvent combinations without any property data. Fig. S2† in the ESI† provides learning curves showing the losses of the training and validation sets in relation to the training steps. The losses in both the training and evaluation set decreased sharply in 1000 steps and then gradually decreased to a stable minimum by 8000 steps. No overfitting is observed since the validation loss does not show a significant increase with decreasing training loss.59 Afterward, the same solute–solvent complexes were provided along with the corresponding free energy data for the fine-tuning stage. The final RMSE/MAE for test sets are shown in Fig. 4d (where the dataset size is 106).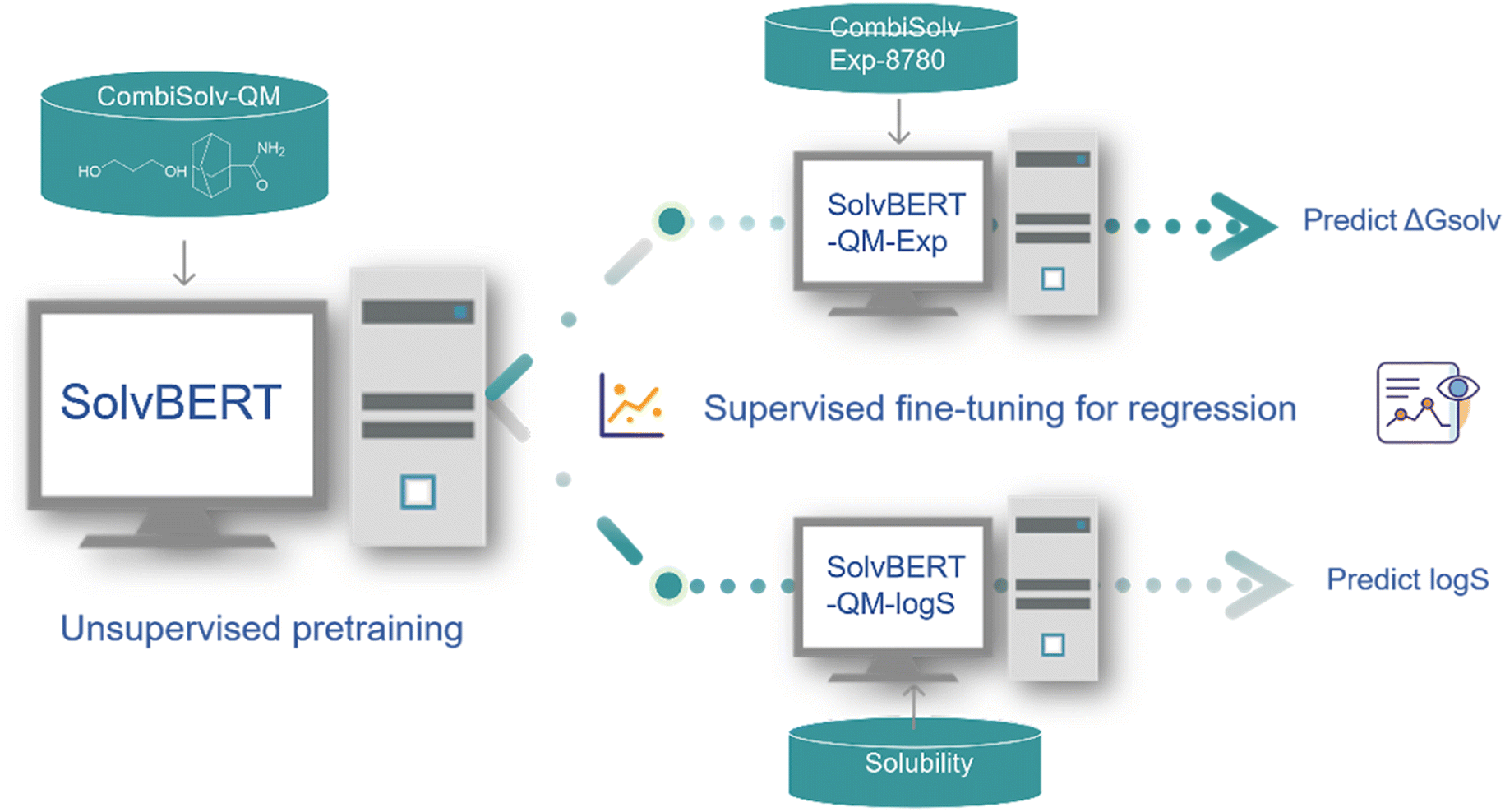 | ||
| Fig. 3 Workflow of SolvBERT, including pre-training, finetuning, and property prediction. | ||
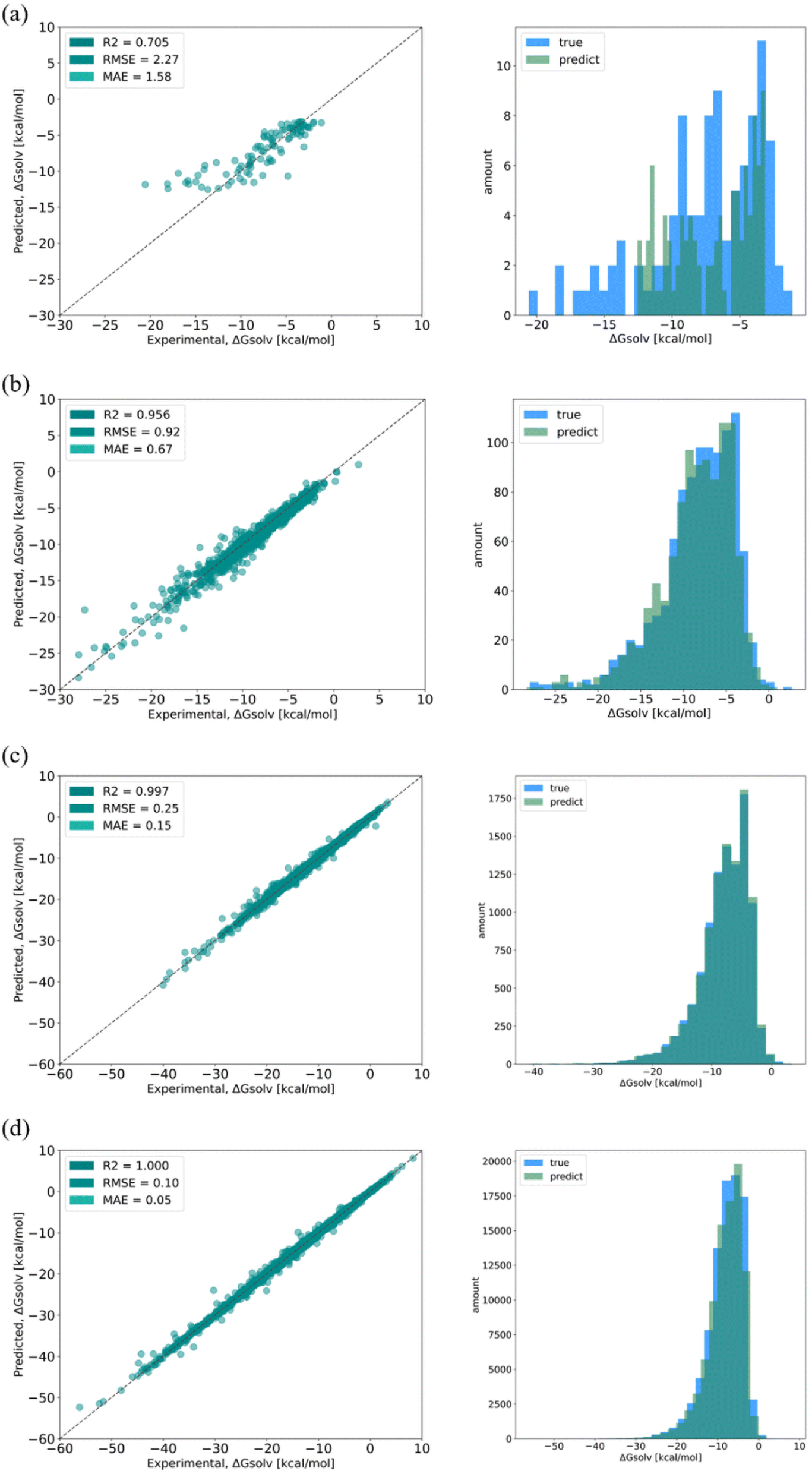 | ||
| Fig. 4 Regressions (left column) and distributions (right column) of true and predicted values of solvation free energy from the SolvBERT model trained on the CombiSolv-QM dataset of different sizes: (a) 103, (b) 104, (c) 105, and (d) 106. | ||
In addition, the effect of the size of the training dataset was evaluated by using different proportions of the CombiSolv-QM dataset (Fig. 4). Considering that the mean value of the solvation free energy in the dataset is −8.10 kcal mol−1, the size of the training dataset needs to be more than 105 to reach a relative error of less than 10% when using the same dataset in the pre-training and fine-tuning phases. The effect of training set size was also assessed using the distribution of predicted values and true values (Fig. 4, right column). As the training sizes increased, the overlap of the distributions became more pronounced, with significant overlap observed when the size does not fall below 105 (Fig. 4c and d).
SolvBERT-QM-Exp is a SolvBERT model pre-trained on the CombiSolv-QM dataset and fine-tuned on the CombiSolv-Exp-8780 dataset (Fig. 3, upper right). After optimization, hyperparameters including an epoch of 20, a batch size of 16, a learning rate of 0.00008 and a hidden dropout rate of 0.4 were selected (Table S4 in the ESI†). To demonstrate the benefits of pre-training with CombiSolv-QM, we pre-trained and fine-tuned another SolvBERT model using only the CombiSolv-Exp-8780 dataset, resulting in the SolvBERT-Exp model for comparison. It was found that SolvBERT-QM-Exp outperformed SolvBERT-Exp – SolvBERT-QM-Exp had a higher R2 and lower RMSE and MAE in both training and validation results (Table 2). For the test set, the distributions of predicted and true values of SolvBERT-QM-Exp overlapped slightly more than those of SolvBERT-Exp, while the regression plot diverges less (Fig. 5).
| Model | Training | Validation | ||||
|---|---|---|---|---|---|---|
| R 2 | RMSE (kcal mol−1) | MAE (kcal mol−1) | R 2 | RMSE (kcal mol−1) | MAE (kcal mol−1) | |
| SolvBERT-QM-Exp | 0.990 | 0.45 | 0.30 | 0.981 | 0.60 | 0.37 |
| SolvBERT-Exp | 0.984 | 0.58 | 0.38 | 0.964 | 0.83 | 0.51 |
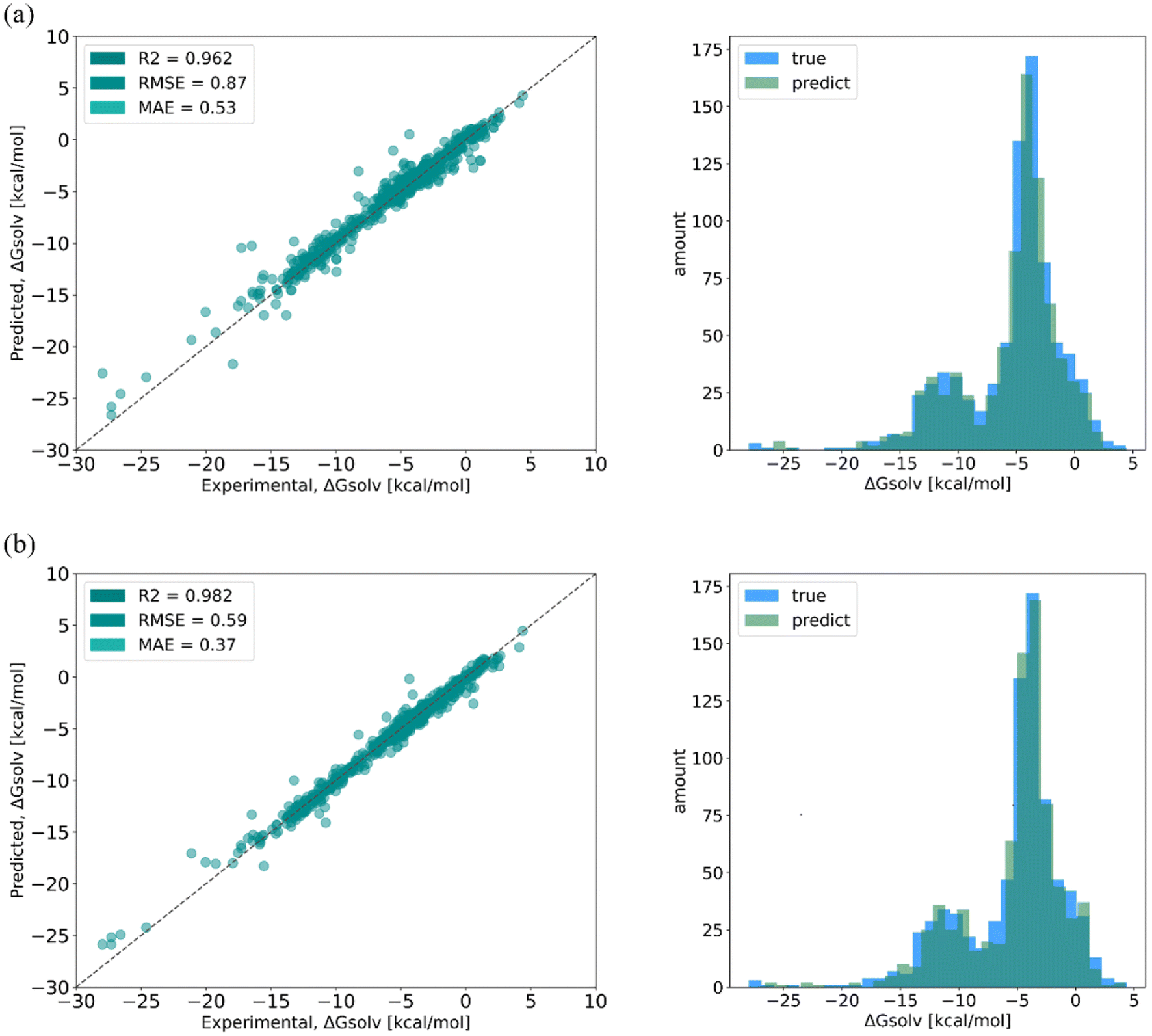 | ||
| Fig. 5 Regressions (left column) and distributions (right column) of true and predicted values of solvation free energy from (a) SolvBERT-QM-Exp and (b) SolvBERT-Exp. | ||
Previous studies have found that the benefits of pre-training on large datasets are more pronounced when the size of the fine-tuning dataset is small. To see if this phenomenon existed in our case, we extracted different proportions from the CombiSolv-Exp-8780 dataset for fine-tuning the SolvBERT-QM-Exp model and training and fine-tuning the SolvBERT-Exp model. The results show that the prediction error of SolvBERT-Exp is significantly higher than that of SolvBERT-QM-Exp when the size of CombiSolv-Exp-8780 is less than 20% of its original size, which indicates that pre-training the model using CombiSolv-QM has a more obvious advantage when the size of the fine-tuning dataset is small (Fig. 6).
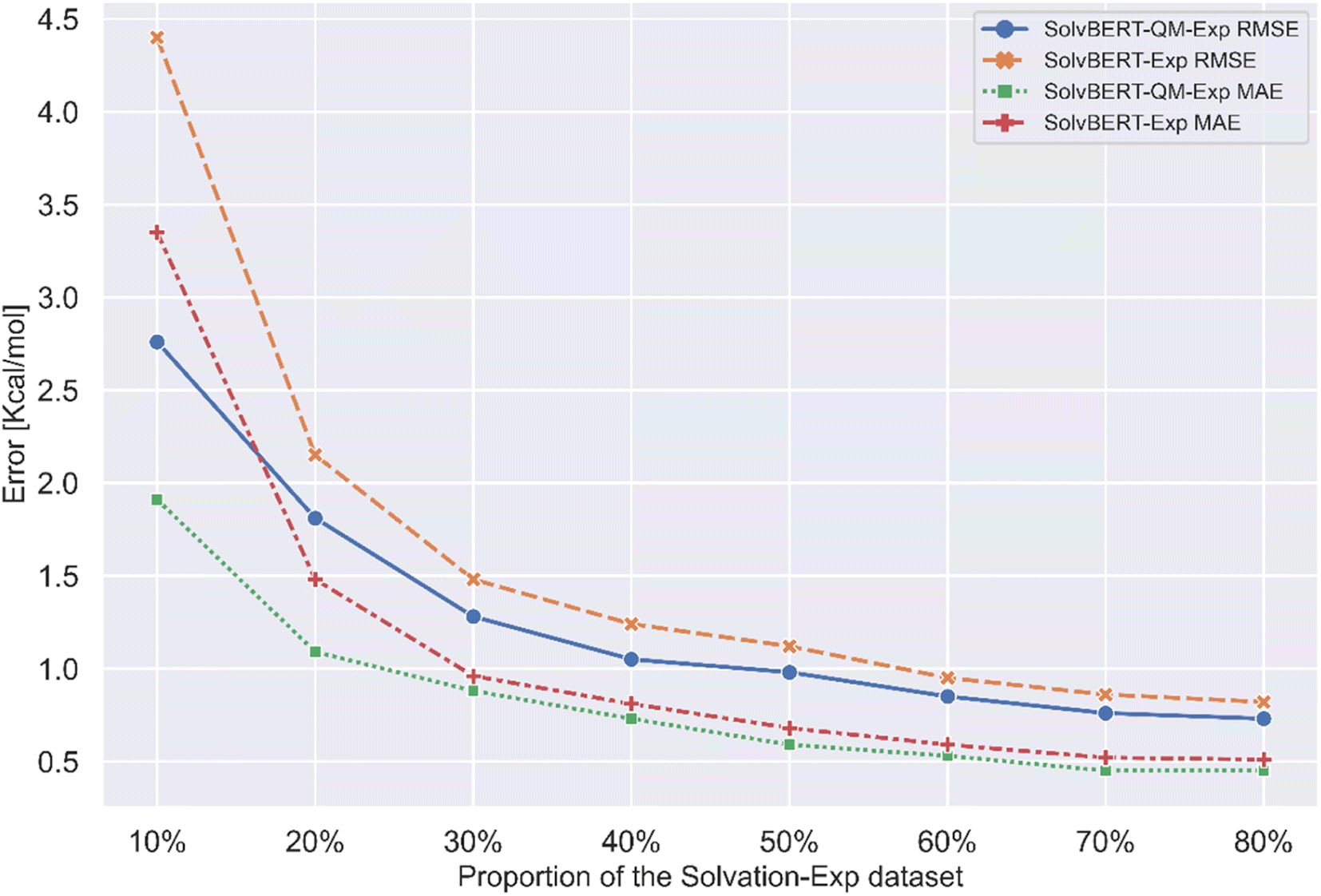 | ||
| Fig. 6 Performance of the SolvBERT-QM-Exp and SolvBERT-Exp models fine-tuned with different proportions of the CombiSolv-Exp-8780 dataset. | ||
Since the pre-training stage of SolvBERT was unsupervised, which means the pre-training does not require any property data, it is possible to pre-train the model using the data from property A (e.g., the CombiSolv-QM dataset, size = 1 million) and fine-tune using the data from property B (e.g., the solubility dataset, size = 2511), as long as both properties are for the same target (e.g., solvation), which is important when the data size of property B is significantly smaller than that of property A. Here, we proposed two additional combinations of models and datasets – SolvBERT-logS using the solubility dataset for pre-training and fine-tuning, and SolvBERT-QM-logS using the CombiSolv-QM dataset for pre-training and the solubility dataset for fine-tuning (Fig. 2, lower right). Since the solubility dataset was significantly smaller than the CombiSolv-Exp-8780 dataset, we increased the epochs to 60, while keeping the other hyperparameters the same as in the SolvBERT-QM-Exp model. The results show that the solubility prediction performance of the model is significantly improved with lower RMSE and MAE and higher R2 by pre-training with the CombiSolv-QM dataset (Table 3).
| Model | Training | Validation | Test | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R 2 | RMSE | MAE | R 2 | RMSE | MAE | R 2 | RMSE | MAE | |
| SolvBERT-logS | 0.870 | 0.69 | 0.54 | 0.65 | 1.08 | 0.82 | 0.73 | 1.10 | 0.86 |
| SolvBERT-QM-logS | 0.921 | 0.54 | 0.38 | 0.926 | 0.52 | 0.38 | 0.925 | 0.47 | 0.36 |
| GROVER-logS | 0.903 | 0.60 | 0.43 | 0.908 | 0.59 | 0.44 | 0.935 | 0.49 | 0.38 |
| Model | Training | Validation | Test | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R 2 | RMSE | MAE | R 2 | RMSE | MAE | R 2 | RMSE | MAE | |
| a The transfer learning of the DMPNN model is by a fine-tuning approach rather than by a feature-based approach. Both the method and the results of transfer learning differ from those of the study by Vermeire et al.44 | |||||||||
| GCN-Exp | 0.896 | 1.36 | 1.01 | 0.893 | 1.50 | 1.04 | 0.905 | 1.36 | 0.97 |
| GCN-QM-Exp | 0.906 | 1.4 | 0.88 | 0.926 | 1.24 | 0.91 | 0.929 | 1.18 | 0.88 |
| DMPNN-Expa | 0.901 | 1.44 | 0.96 | 0.894 | 1.47 | 1.00 | 0.885 | 1.55 | 1.07 |
| DMPNN-QM-Expa | 0.989 | 0.48 | 0.29 | 0.988 | 0.51 | 0.32 | 0.974 | 0.73 | 0.43 |
| GROVER-Exp | 0.982 | 0.61 | 0.37 | 0.984 | 0.61 | 0.37 | 0.979 | 0.64 | 0.38 |
| RF-Exp | 0.636 | 2.26 | 1.56 | 0.742 | 1.79 | 1.36 | 0.694 | 1.95 | 1.40 |
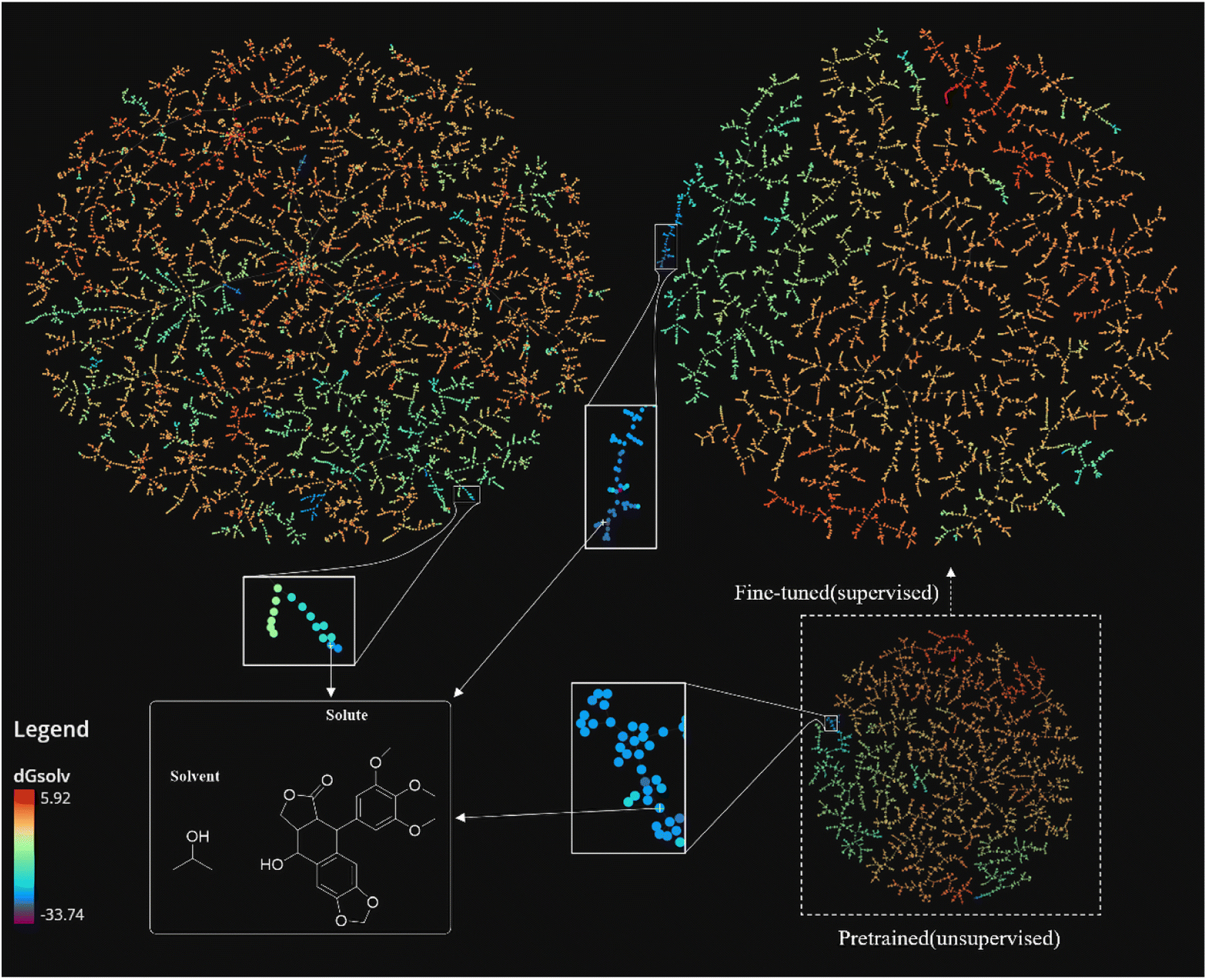 | ||
| Fig. 7 TMAP visualization of the CombiSolv-Exp-8780 dataset based on three different types of fingerprints: the MHFP fingerprint on the upper left without SolvBERT processing, the fingerprint generated by the fine-tuning model based on SolvBERT (SolvBERT-fp-finetuned) on the upper right, and the dashed box on the lower right shows the fingerprints generated by the unsupervised learning model during the pre-training process (SolvBERT-fp-pre-trained). The color bars are from high (red) to low (blue) depending on the value of solvation free energy. | ||
While the solute–solvent combinations described by MHFP do not show a clear relationship between clustering and solvation free energy (Fig. 7, left) those described by SolvBERT-fp-pre-trained show a significantly improved clustering of solute–solvent combinations. Clustering is broadly divided into four regions, red (0–5 kcal mol−1), red-orange (−10 to 0 kcal mol−1), green-blue (−20 to −10 kcal mol−1), and blue-purple (−20 to −30 kcal mol−1). Obviously, the vectorized representation of the pre-training phase output using SolvBERT, has a better association between clustering on TMAP and the free energy, although we did not train with the solvation free energy data in the pre-training phase.
The SolvBERT-fp-finetuned shows an even better clustering closely related to the value of solvation free energy. We can see the exemplary solvent–solute combination in the solid line box at the bottom left of Fig. 7, in which the solute is 9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuro[3′,4′:6,7]naphtho[2,3-d][1,3]dioxol-6(5aH)-one and the solvent is propan-2-ol, with the solvation free energy being −25.12 kcal mol−1. This combination, represented by blue color, is trapped in a green cluster in the MHFP chemical space (Fig. 7, upper left). In contrast, the combination is located in a clearly demonstrated blue cluster in both SolvBERT-fp-pre-trained and SolvBERT-fp-finetuned.
| In-sample solvent | Out-of-sample solute | |||
|---|---|---|---|---|
| O-Methyl-N-nitroisourea | 3-Methyl-2-nitrobenzoic acid | 2-Amino-5-chloro-3-methylbenzoic acid | ||
| Acetone | Measured | 2.5148 | 4.8715 | 5.3016 |
| SolvBERT-QM-logS | 1.1233 | 2.6173 | 2.1327 | |
| GROVER-logS | 0.0066 | 3.6388 | 0.6401 | |
| Ethanol | Measured | 1.4266 | 4.8422 | 3.6682 |
| SolvBERT-QM-logS | 1.9220 | 4.0771 | 3.2472 | |
| GROVER-logS | 0.0026 | 0.6187 | 0.5715 | |
| Out-of-sample solvent | In-sample solute | |||
|---|---|---|---|---|
| 1,4-Naphthoquinone | Anthracene | 4-Chlorophthalic anhydride | ||
| Propanol | Measured | 2.3501 | 0.1924 | 3.7477 |
| SolvBERT-QM-logS | 1.9579 | 0.0785 | 3.2751 | |
| GROVER-logS | 0.1417 | 0.2818 | 0.0427 | |
| Dichloromethane | Measured | 4.0199 | 1.3657 | 5.0050 |
| SolvBERT-QM-logS | 4.6786 | 3.2678 | 5.1182 | |
| GROVER-logS | 0.3627 | 0.0672 | 0.4066 | |
| Out-of-sample solvent | Out-of-sample solute | |||
|---|---|---|---|---|
| O-Methyl-N-nitroisourea | 3-Methyl-2-nitrobenzoic acid | 2-Amino-5-chloro-3-methylbenzoic acid | ||
| Methanol | Measured | 2.6491 | 2.5303 | 3.2882 |
| SolvBERT-QM-logS | 2.8350 | 3.8242 | 4.2751 | |
| GROVER-logS | 0.0014 | 0.4522 | 0.5031 | |
| Propanol | Measured | 2.0917 | 0.6267 | 1.4661 |
| SolvBERT-QM-logS | 4.4350 | 0.7406 | 1.4601 | |
| GROVER-logS | 0.0012 | 0.3509 | 0.4434 | |
| Ethyl acetate | Measured | 5.7051 | 3.5050 | 2.4918 |
| SolvBERT-QM-logS | 1.8873 | 2.7925 | 2.6461 | |
| GROVER-logS | 0.0026 | 1.0075 | 0.2499 | |
The results show that out of the total 21 out-of-sample solute–solvent combinations, SolvBERT gives 11 predictions with relative errors less than 25% and 8 predictions with relative errors between 25% and 75%. In contrast, GROVER gives 0 predictions with relative errors less than 25%, but 16 predictions with relative errors higher than 75%. Thus, our SolvBERT model significantly outperforms GROVER in predicting out-of-sample solubility data.
Discussion
One feature that distinguishes SolvBERT from graph-based models5,10,44 and a previously developed NLP model for solvation free energy or solubility prediction is that SolvBERT reads SMILES representation of solute–solvent pairs instead of reading solutes and solvents separately. Previously developed model architectures, such as chemprop and Delfos, use separate series of feature extraction layers for solutes and solvents, and then concatenate the extracted features into fully connected layers. In contrast, SolvBERT, as a BERT-derived model, treats the solute and solvent as a combination and maps a large number of such combinations into a chemical space, as is visualized by TMAP in Fig. 5. Such an approach not only enables a rapid search of nearest neighbors (i.e., similar solute–solvent pairs), but also enables a more flexible representation of molecular complexes. Similar flexible representations have been seen in the classification27 and yields prediction28 of chemical reactions in the study of Schwalleret al. In their BERT-based model, no split of reactants, reagents, catalysts, and solvent is required, and these components of the reaction are mapped as a whole into a chemical space.
Another unique feature of SolvBERT is its unsupervised pre-training phase, which does not require any property data of the molecular complex. This feature has two benefits. First, larger and cheaper databases, such as CombiSolv-QM, can be used to pre-train the model without considering the potential impact of different data fidelity (i.e., computational vs. experimental) or difference property types (i.e., ΔG for the CombiSolv-QM dataset and logS for the solubility dataset), since no target data are required in unsupervised learning. In addition, the model being pre-trained will be familiar with the molecular structure of the system and can be fine-tuned to predict multiple properties of the system (i.e., solvation free energy and solubility for the solvation system), which requires a much smaller dataset than training a model from the beginning. This unique feature is particularly useful in cases where training datasets of different properties are significantly different in size or have little overlap in their chemical space, making it difficult for traditional multi-task deep learning to merge all these datasets into a single database.
We conclude this study by noting the recently published GNN–Transformer hybrid model GROVER,39 which emerged as a new state-of-the-art model for molecular property prediction. Similar to our SolvBERT, GROVER includes a self-supervised pre-training phase and a down-stream supervised fine-tuning phase.39 Although GROVER was comparable to SolvBERT in predictions from the CombiSolv-Exp-8780 and solubility datasets, it performed significantly worse in the out-of-sample predictions from our experimentally-measured dataset. One possible reason for this divergence between the two models in prediction of out-of-sample data is the nature of pre-training datasets. The GROVER model was pre-trained on 11 million (M) unlabeled molecules39 selected from ZINC15 and ChemBL, while our SolvBERT model was pre-trained on the CombiSolv-QM dataset that contains ∼1 M combinations of solvents and solutes. In addition, the impressive clustering ability of solute–solvent combinations during the pre-training phase, which has been demonstrated in this study using TMAP visualization, may also help SolvBERT to better understand the solvation system. After all, we have to admit that since the model architecture of GROVER is significantly larger39 than that of SolvBERT (48.8 M vs. 7.6 M), it is difficult to retrain GROVER on the CombiSolv-QM dataset with our current computational resources to give an absolutely fair comparison. Since SolvBERT takes only 12 hours to fully pre-train and fine-tune the model using only 1 Nvidia RTX 3060 GPU, while the GROVER-base requires 2.5 days using 250 Nvidia V100 GPUs,39 SolvBERT can be considered as a more efficient model for predicting solvation-related properties.
Conclusion
An NLP-based deep learning model, SolvBERT, was developed to predict solvation free energy and solubility. Unlike graph-based models that read solutes and solvents separately, SolvBERT reads solute–solvent pairs through a combined SMILES representation. The model was pre-trained in an unsupervised learning manner, using a computational solvation free energy database containing 1 million combinations of solutes and solvents. The model was then fine-tuned with a regression layer on either the experimental solvation free energy database or the solubility database, depending on the type of regression task. The results showed that pre-training SolvBERT with a large computational solvation free energy database was beneficial for predicting the experimental solvation free energy, especially when the experimental fine-tuning dataset was small in size. Moreover, the performance of SolvBERT was comparable to that of the state-of-the-art graph-based model, DMPNN, and a recently-developed graph-Transformer hybrid model, GROVER. In addition, TMAP visualization of solute–solvent combination clustering showed the benefits of the unsupervised learning phase of SolvBERT in facilitating the clustering of solvent combinations with similar solvation properties. Finally, an out-of-sample solubility dataset was experimentally measured, and SolvBERT was found to have better prediction performance than GROVER on this dataset.We also summarized two unique features of SolvBERT compared to graph-based models and non-BERT NLP models. SolvBERT is more flexible in representing molecular complexes, allowing not only the search for similar complexes by mapping in chemical space, but also the extension of molecular complexes beyond two components. In addition, the pre-training phase of SolvBERT does not require any attribute data, suggesting that it is possible to use the BERT-based model to predict multiple properties regardless of the differences in the size, fidelity, or attributes of training datasets. We recommend that researchers apply SolvBERT to at least three situations in future studies: (1) predicting different properties of chemical reactions, including the selectivity, conversion rates, and environmental impacts; (2) predicting different biological activities of molecules; (3) predicting the properties of molecular complexes containing more than two components.
Code availability
The code supporting the finding of this study has been deposited at figshare43 and GitHub (https://github.com/su-group/SolvBERT). All codes required for SolvBERT and TMAP, as well as repeating data pre-processing, is included in the “solv-bert” folder. The folder also contains a detailed SolvBERT instruction manual, and the code for TMAP is placed in the “tmapfiles” folder, where the “tmapfiles/TMAP” folder has high-resolution images.The open source version we use is as follows: torch 1.11.0 + cu113, python 3.7.13, rxnfp 0.0.7, tokenizers 0.7.0, wandb 0.12.15, tmap1.0.6, fearun 0.3.20, mhfp1.9.2, sklearn 0.23.1, matplotlib 3.2.2, Pandas 1.3.4.
Data availability
The authors declare that the main data supporting the finding of this study are available within the article. All the supporting data have been deposited at figshare43 and GitHub (https://github.com/su-group/SolvBERT). The supporting data are in the “solv-bert/data” folder, while the data used for training are placed in the “solv-bert/data/training_files” folder.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge Zhejiang Province Science and Technology Plan Project (No. 2019- ZJ-JS-03), National Natural Science Foundation of China (No. 22108252), and Zhejiang Province Science and Technology Plan Project (No. 2022C01179) for financial support.References
- C. W. Coley, W. H. Green and K. F. Jensen, Machine learning in computer-aided synthesis planning, Acc. Chem. Res., 2018, 51(5), 1281–1289 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler and F. Glorius, Machine learning the ropes: principles, applications and directions in synthetic chemistry, Chem. Soc. Rev., 2020, 49(17), 6154–6168 RSC.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic chemical design using a data-driven continuous representation of molecules, ACS Cen. Sci., 2018, 4(2), 268–276 CrossRef PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Inverse molecular design using machine learning: generative models for matter engineering, Science, 2018, 361(6400), 360–365 CrossRef CAS PubMed.
- D. K. Duvenaud; D. Maclaurin; J. Iparraguirre; R. Bombarell; T. Hirzel; A. Aspuru-Guzik and R. P. Adams, Convolutional networks on graphs for learning molecular fingerprints, in Advances in Neural Information Processing Systems, 2015, vol. 28, pp. 2224–2232 Search PubMed.
- T. Lei; W. Jin; R. Barzilay and T. Jaakkola, Deriving neural architectures from sequence and graph kernels, in Proceedings of the 34th International Conference on Machine Learning, ed. P. Doina and T. Yee Whye, PMLR: Proceedings of Machine Learning Research, 2017, vol. 70, pp. 2024–2033 Search PubMed.
- J. Gilmer; S. S. Schoenholz; P. F. Riley; O. Vinyals and G. E. Dahl, Neural message passing for quantum chemistry, in Proceedings of the 34th International Conference on Machine Learning, ed. P. Doina and T. Yee Whye, PMLR: Proceedings of Machine Learning Research, 2017, vol. 70, pp. 1263–1272 Search PubMed.
- H. Dai; B. Dai and L. Song, Discriminative embeddings of latent variable models for structured data, in Proceedings of The 33rd International Conference on Machine Learning, ed. B. Maria Florina and Q. W. Kilian, PMLR: Proceedings of Machine Learning Research, 2016, vol. 48, pp. 2702–2711 Search PubMed.
- C. W. Coley, R. Barzilay, W. H. Green, T. S. Jaakkola and K. F. Jensen, Convolutional embedding of attributed molecular graphs for physical property prediction, J. Chem. Inf. Model., 2017, 57(8), 1757–1772 CrossRef CAS PubMed.
- W.-L. Chiang; X. Liu; S. Si; Y. Li; S. Bengio and C.-J. Hsieh, Cluster-GCN, in Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2019, pp. 257–266 Search PubMed.
- S. Ryu, Y. Kwon and W. Y. Kim, A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification, Chem. Sci., 2019, 10(36), 8438–8446 RSC.
- L. Hirschfeld, K. Swanson, K. Yang, R. Barzilay and C. W. Coley, Uncertainty quantification using neural networks for molecular property prediction, J. Chem. Inf. Model., 2020, 60(8), 3770–3780 CrossRef CAS PubMed.
- A. P. Soleimany, A. Amini, S. Goldman, D. Rus, S. N. Bhatia and C. W. Coley, Evidential deep learning for guided molecular property prediction and discovery, ACS Cent. Sci., 2021, 7(8), 1356–1367 CrossRef CAS PubMed.
- A. Vaswani; N. Shazeer; N. Parmar; J. Uszkoreit; L. Jones; A. N. Gomez; Ł. Kaiser and I. Polosukhin, Attention is all you need, in Advances in Neural Information Processing Systems, 2017, vol. 30, pp. 5998–6008 Search PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS PubMed.
- G. Pesciullesi, P. Schwaller, T. Laino and J.-L. Reymond, Transfer learning enables the molecular transformer to predict regio- and stereoselective reactions on carbohydrates, Nat. Commun., 2020, 11(1), 4874 CrossRef CAS PubMed.
- Y. Zhang, L. Wang, X. Wang, C. Zhang, J. Ge, J. Tang, A. Su and H. Duan, Data augmentation and transfer learning strategies for reaction prediction in low chemical data regimes, Org. Chem. Front., 2021, 8(7), 1415–1423 RSC.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11(12), 3316–3325 RSC.
- I. V. Tetko, P. Karpov, R. Van Deursen and G. Godin, State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis, Nat. Commun., 2020, 11(1), 5575 CrossRef CAS PubMed.
- S. Zheng, J. Rao, Z. Zhang, J. Xu and Y. Yang, Predicting retrosynthetic reactions using self-corrected transformer neural networks, J. Chem. Inf. Model., 2020, 60(1), 47–55 CrossRef CAS PubMed.
- A. Su, X. Wang, L. Wang, C. Zhang, Y. Wu, X. Wu, Q. Zhao and H. Duan, Reproducing the invention of a named reaction: zero-shot prediction of unseen chemical reactions, Phys. Chem. Chem. Phys., 2022, 24(17), 10280–10291 RSC.
- J. Xu, Y. Zhang, J. Han, A. Su, H. Qiao, C. Zhang, J. Tang, X. Shen, B. Sun, W. Yu, S. Zhai, X. Wang, Y. Wu, W. Su and H. Duan, Providing direction for mechanistic inferences in radical cascade cyclization using a transformer model, Org. Chem. Front., 2022, 9(9), 2498–2508 RSC.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of organic chemistry grammar from unsupervised learning of chemical reactions, Sci. Adv., 2021, 7(15), eabe4166 CrossRef PubMed.
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Automated extraction of chemical synthesis actions from experimental procedures, Nat. Commun., 2020, 11(1), 3601 CrossRef PubMed.
- A. C. Vaucher, P. Schwaller, J. Geluykens, V. H. Nair, A. Iuliano and T. Laino, Inferring experimental procedures from text-based representations of chemical reactions, Nat. Commun., 2021, 12(1), 2573 CrossRef CAS PubMed.
- J. Devlin, M.-W. Chang, K. Lee and K. J. A. Toutanova, Bert: Pre-training of deep bidirectional transformers for language understanding, 2018, arXiv preprint arXiv:1810.04805.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J.-L. Reymond, Mapping the space of chemical reactions using attention-based neural networks, Nat. Mach. Intell., 2021, 3(2), 144–152 CrossRef.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Prediction of chemical reaction yields using deep learning, Mach. Learn. Sci. Technol., 2021, 2(1), 015016 CrossRef.
- K. Kwak, S. Park and M. D. Fayer, Dynamics around solutes and solute–solvent complexes in mixed solvents, Proc. Natl. Acad. Sci., India, 2007, 104(36), 14221–14226 CAS.
- K. Kwak, D. E. Rosenfeld, J. K. Chung and M. D. Fayer, Solute−solvent complex switching dynamics of chloroform between acetone and dimethylsulfoxide−two-dimensional ir chemical exchange spectroscopy, J. Phys. Chem. B, 2008, 112(44), 13906–13915 CrossRef CAS PubMed.
- S. Boobier, D. R. J. Hose, A. J. Blacker and B. N. Nguyen, Machine learning with physicochemical relationships: solubility prediction in organic solvents and water, Nat. Commun., 2020, 11(1), 5753 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49(W1), W5–W14 CrossRef CAS PubMed.
- G. D. R. Matos, D. Y. Kyu, H. H. Loeffler, J. D. Chodera, M. R. Shirts and D. L. Mobley, Approaches for calculating solvation free energies and enthalpies demonstrated with an update of the FreeSolv database, J. Chem. Eng. Data, 2017, 62(5), 1559–1569 CrossRef PubMed.
- B. Guo; S. Song; J. Chacko and A. Ghalambor, CHAPTER 15 – flow assurance, in Offshore Pipelines, ed. B. Guo, S. Song, J. Chacko, and A. Ghalambor, Gulf Professional Publishing, Burlington, 2005, pp. 169–214 Search PubMed.
- F. Eckert and A. Klamt, Fast solvent screening via quantum chemistry: COSMO-RS approach, AIChE J., 2002, 48(2), 369–385 CrossRef CAS.
- I. T. Ho, M. Matysik, L. M. Herrera, J. Yang, R. J. Guderlei, M. Laussegger, B. Schrantz, R. Hammer, R. A. Miranda-Quintana and J. Smiatek, Combination of explainable machine learning and conceptual density functional theory: applications for the study of key solvation mechanisms, Phys. Chem. Chem. Phys., 2022, 24(46), 28314–28324 RSC.
- W. Zhang, L. Deng, L. Zhang and D. Wu, A survey on negative transfer, IEEE/CAA J. Automat. Sin., 2022, 305–329 Search PubMed.
- Z. Wang; Z. Dai; B. Póczos and J. Carbonell, Characterizing and avoiding negative transfer, in IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 15-20 June 2019, 2019, pp. 11285–11294 Search PubMed.
- Y. Rong; Y. Bian; T. Xu; W. Xie; Y. Wei; W. Huang and J. Huang, Self-supervised graph transformer on large-scale molecular data, in Advances in Neural Information Processing Systems, 2020, vol. 33, pp. 12559–12571 Search PubMed.
- H. Lim and Y. Jung, Delfos: deep learning model for prediction of solvation free energies in generic organic solvents, Chem. Sci., 2019, 10(36), 8306–8315 RSC.
- S. Jaeger, S. Fulle and S. Turk, Mol2vec: unsupervised machine learning approach with chemical intuition, J. Chem. Inf. Model., 2018, 58(1), 27–35 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Machine learning for molecular and materials science, Nature, 2018, 559(7715), 547–555 CrossRef CAS PubMed.
- S. Boobier, D. R. J. Hose, A. J. Blacker and B. N. Nguyen, Machine learning with physicochemical relationships: solubility prediction in organic solvents and water, Nat. Commun., 2020, 11(1), 5753 CrossRef CAS PubMed.
- F. H. Vermeire and W. H. Green, Transfer learning for solvation free energies: from quantum chemistry to experiments, Chem. Eng. J., 2021, 418, 129307 CrossRef CAS.
- A. V. Marenich; C. P. Kelly; J. D. Thompson; G. D. Hawkins; C. C. Chambers; D. J. Giesen; P. Winget; C. J. Cramer and D. G. Truhlar, Minnesota Solvation Database (MNSOL) Version 2012, 2020 Search PubMed.
- D. L. Mobley and J. P. Guthrie, FreeSolv: a database of experimental and calculated hydration free energies, with input files, J. Comput.-Aided Mol. Des., 2014, 28(7), 711–720 CrossRef CAS PubMed.
- E. Moine, R. Privat, B. Sirjean and J.-N. Jaubert, Estimation of solvation quantities from experimental thermodynamic data: development of the comprehensive compsol databank for pure and mixed solutes, J. Phys. Chem. Ref. Data, 2017, 46(3), 033102 CrossRef.
- L. M. Grubbs, M. Saifullah, N. E. De La Rosa, S. Ye, S. S. Achi, W. E. Acree and M. H. Abraham, Mathematical correlations for describing solute transfer into functionalized alkane solvents containing hydroxyl, ether, ester or ketone solvents, Fluid Phase Equilib., 2010, 298(1), 48–53 CrossRef CAS.
- X. C. Zhang, C. K. Wu, Z. J. Yang, Z. X. Wu, J. C. Yi, C. Y. Hsieh, T. J. Hou and D. S. Cao, MG-BERT: leveraging unsupervised atomic representation learning for molecular property prediction, Briefings Bioinf., 2021, 22(6), bbab152 CrossRef PubMed.
- J. Devlin, M.-W. Chang, K. Lee and K. J. A. Toutanova, BERT: pre-training of deep bidirectional transformers for language understanding, 2019, arXiv:1810.04805.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing learned molecular representations for property prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, A probabilistic molecular fingerprint for big data settings, J. Cheminf., 2018, 10(1), 66 CAS.
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand discovery for everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337 CrossRef CAS PubMed.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Krüger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, The ChEMBL bioactivity database: an update, Nucleic Acids Res., 2014, 42(D1), D1083–D1090 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, Visualization of very large high-dimensional data sets as minimum spanning trees, J. Cheminf., 2020, 12(1), 12 Search PubMed.
- A. Andoni; I. P. Razenshteyn and N. S. Nosatzki, in LSH Forest: Practical Algorithms Made Theoretical, ACM-SIAM Symposium on Discrete Algorithms, 2017 Search PubMed.
- J. B. Kruskal, in On the Shortest Spanning Subtree of a Graph and the Traveling Salesman Problem, 1956 Search PubMed.
- D. Probst and J.-L. Reymond, FUn: a framework for interactive visualizations of large, high-dimensional datasets on the web, Bioinformatics, 2018, 34(8), 1433–1435 CrossRef CAS PubMed.
- X. Ying, An overview of overfitting and its solutions, J. Phys.: Conf. Ser., 2019, 1168 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dd00107a |
| This journal is © The Royal Society of Chemistry 2023 |