
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational reprogramming of the sesquiterpene synthase BcBOT2 yields new terpenes with presilphiperfolane skeleton†
Vanessa
Nikolaiczyk
a,
Jenny
Irwan
a,
Trang
Nguyen
 a,
Jörg
Fohrer
b,
Philipp
Elbers
c,
Paul
Schrank
c,
Mehdi D.
Davari
*c and
Andreas
Kirschning
*a
a,
Jörg
Fohrer
b,
Philipp
Elbers
c,
Paul
Schrank
c,
Mehdi D.
Davari
*c and
Andreas
Kirschning
*a
aInstitute of Organic Chemistry and Center of Biomolecular Drug Research (BMWZ), Leibniz Universität Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: andreas.kirschning@oci.uni-hannover.de
bDepartment of Chemistry, Technical University Darmstadt, Alarich-Weiss-Straße 4, 64287 Darmstadt, Germany
cDepartment of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, 06120 Halle, Germany. E-mail: mehdi.davari@ipb-halle.de
First published on 24th November 2022
Abstract
Computer-aided rational design of the substrate binding pocket of sesquiterpene synthases BcBOT2 from Botrytis cinerea yielded FPP cyclization products with presilphiperfolane backbone other than the naturally formed sesquiterpene presilphiperfolan-8β-ol. Particularly, amino acids W118 and F138 were found to strongly control the stability and conformation of key cationic intermediates. The W118Q variant forms only presilphiperfolan-9β-ol, whereas the exchange of amino acids at position 138, such as F138V, has a fundamental effect on the course of the cationic cascade. Here, the 1,3-hydride shift en route to presilphiperfolan-8β-ol is suppressed and substituted by a so far unknown 1,2-hydride shift that leads to presilphiperfol-1-ene and presilphiperfolan-1α-ol along with β-caryophyllene and the so far unknown caryophyllene-8β-ol.
Introduction
Sesquiterpene cyclases (STCs) are powerful enzymes capable of converting linear unsaturated farnesyl diphosphate (FPP, 1), into (oligo)cyclic sesquiterpenes via a series of sequentially forming carbocationic intermediates. The last carbocation of this series of intermediates is thereby captured either by proton removal or by reaction with external nucleophiles such as water.1–3The fungal STC presilphiperfolan-8β-ol synthase (BcBOT2) from Botrytis cinerea performs a complex multistep transformation that yields the tricyclic sesquiterpene (−)-presilphiperfolan-8β-ol (PSP, 2) from FPP 1 (Scheme 1A), which is further oxidized to botrydial (3) a phytotoxic metabolite of B. cinerea.4,5
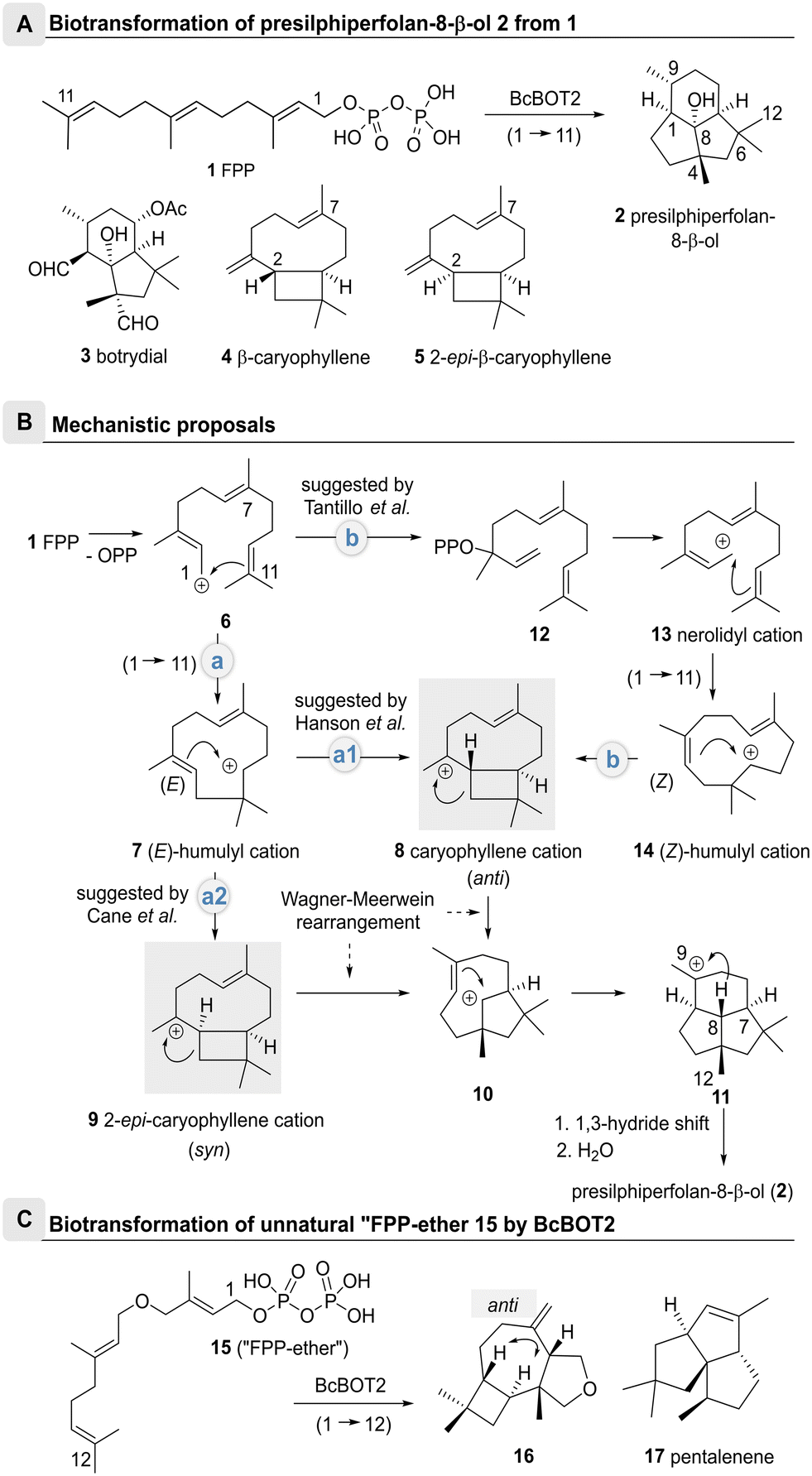 | ||
| Scheme 1 Biotransformations of FPP 1 and “FPP-ether” 15 with the entire cationic cascade (route a2). The caryophyllenyl cation 8 was proposed as an important intermediate with anti-stereochemistry between the cyclobutane and cycloheptane rings in the proposed mechanism of BcBOT2. A: Conversion of farnesyl pyrophosphate (1) into (−)-presilphiperfolan-8β-ol (PSP, 2) by the STC BcBOT2; structures of botrydial (3) and byproduct β-caryophyllene (4); structure of 2-epi-caryophyllene (5) (numbering of carbon atoms is individualized for single compounds); B: proposed mechanistic pathways of BcBOT2-catalyzed formation of (−)-presilphiperfolan-8β-ol (PSP, 2);5,12–14 C: transformation of “FPP-ether” 15 into terpenoid 16 by BcBOT2; anti-stereochemistry is established between the cyclobutane and cycloheptane rings; structure of pentalenene (17). | ||
Mechanistically, there is still some ambiguity about the formation of PSP 2 by BcBOT2. One line of discussion is based on the search for byproducts.4 As such, Bohlmann and colleagues also found β-caryophyllene (4) and were able to propose a possible biocatalytic reaction cascade for the formation of PSP 2 (Scheme 1B).4–11 This was substantiated by Cane and colleagues who used 2H,13C-labelled mevalonic acid in biotransformations with BcBOT2 (route a1). They demonstrated that a 1,3-hydride shift from C8 to C9 must have taken place.12
On the basis of theoretical calculations, Tantillo et al. proposed an alternative route (route b) via the nerolidyl cation 13, that yields the (Z)-configured humulyl cation 14 after cyclization.13 From there, the caryophyllenyl cation 8 may form bearing an anti-annulated bicyclic system that supports Bohlmann's observation of β-caryophyllene (4) formation.12,14 Indeed from there, cation–alkene cyclization, followed by a 1,3-hydride shift and the capture of the intermediate cation by water straightforwardly furnishes PSP 2 (Scheme 1B).14
Additional support for the anti-orientation of the two H atoms in the caryophyllenyl cation 8 was collected from biotransformations with the unnatural “FPP-ether” 15, that yielded the tetrahydrofurano terpenoid 16 in the presence of BcBOT2, whereby the anti-orientation of the H atoms was clearly proven by selected NOE experiments (Scheme 1C).13,15
On the other hand, Cane et al. employed isotope-labelled FPP derivatives and found that the proton at C7 remained in syn-orientation with the deuterated methyl group at position 12 during the entire cationic cascade (route a2). The 2-epi-caryophyllenyl cation 9 was proposed as an important intermediate with syn-stereochemistry between the cyclobutane and cycloheptane rings (Scheme 1B).14 This assumption is contradictory to the original report by Bohlmann et al., since β-caryophyllene (4) and PSP (2) were isolated from the same batch,6–9 but in no case was 2-epi-caryophyllene (5) found in PSP 2-containing extracts.16,17
In the present study, we investigated and rationally engineered the BcBOT2 substrate binding pocket to influence and expand the product spectrum of this STC. To this end, amino acid residues were first selected based on their evolutionary conservation and position in the substrate binding pocket. Subsequently, the selected residues were subjected to mutagenesis experiments to generate improved variants. Finally, the advantageous variants were analyzed by computational studies to gain a better molecular understanding and to propose the role of amino acid residues in the function and control of the putative underlying catalytic mechanism.
Importantly, we verified these computational studies experimentally by focusing on the formation and identification of new and preferably unknown cyclization products.
The formation of the new sesquiterpene derivatives widens the structural diversity of sesquiterpenes and expands their structural space. In principal, this is associated with new biological properties e.g. in the context of scents, which are significant for the fragrance industry.15 Furthermore, we have shown that amino acid exchanges help to provide additional insight into the complex mechanisms of terpene synthases.
Results and discussions
Overview of the selected BcBOT2 variants
First, substitutions in terpene synthases targeting aromatic or hydrophobic residues are of particular importance, as they contour the active site pocket. Moreover, they guide intermediates along the reaction pathway through stabilizing π-interactions with carbocations and influence substrate folding.18–24 Secondly, numerous mutagenesis studies with STCs have shown that metal binding motifs are essential for enzyme activity and can induce a change in the product spectrum.20 Thirdly, studies with the terpene synthases Cop4 and Cop6 from Coprenius cinerea revealed a direct relationship between pocket size and product promiscuity.25,26 Using the hedycaryol synthase, it was demonstrated that D82 of the DDxxD motif is involved in water activation.27 On the other hand, a possible role of the amino acids tryptophan and histidine as bases in PenA from Streptomyces exfoliates UC5319 (PDB ID: 1PS1) could be excluded by targeted mutagenesis, whereas in the STC TEAS from Nicotiana tabacum, tryptophan (W273) was found to act as a base.28,29 Moreover, in the case of PenA, F76 was shown to be essential for the formation of the secondary (E,E)-humulyl cation 7.29To understand the mechanism in more detail and to open up new reaction pathways, we have chosen to rationally describe the BcBOT2 substrate binding pocket. Since no crystal structure of BcBOT2 has been reported so far, a reliable structural model was first constructed using AlphaFold2. The model was built in monomeric state, as this organization is proposedly sufficient for catalytic functionality in BcBOT2 and other STCs.1,30,31 Structural analysis of the model led to the identification of the active site pocket using the Fpocket tool of HotSpot Wizard with standard settings and a probe radius of 3 Å (see Fig. S16 in the ESI†).32 The active site pocket comprises 23 amino acids (see Table S14 in the ESI†) with a relevance score of 100%. It is located within the α-helical fold typical for class I terpene synthase, with the highly conserved DDxxD and NSE motifs. Also, the effector triad was identified by structural alignment of the BcBOT2 model with the selina-4(15),7(11)-diene synthase (PDB ID: 4OKM19) crystal structure (see Fig. S17 in the ESI†).19,33 The three motifs formed the entrance to the active site pocket (Fig. 1A).19,33–35 Its volume is with 567 Å3 quite low compared to other sesquiterpene synthase but it exhibits a high druggability score (0.91), which indicates high affinity binding of small “drug – like” molecules through the active site.25,36 The active site pocket is more polar and hydrophilic in the entrance region, while the part further deep inside is mainly composed of nonpolar and hydrophobic amino acids, similar to the active site pocket of other STCs such as TEAS (Fig. 1B).19,24 Docking of FPP to the wild-type (WT) BcBOT2 model showed orientation of the diphosphate toward the trinuclear Mg2+ and the hydrocarbon chain adopts a spiral conformation toward the nonpolar residues (Fig. 1A). The docking of the intermediates suggests a clear role for F138 in stabilizing the intermediates through significant cation–pi or hydrogen-pi interactions (see Fig. S18 in the ESI†). Additionally, these types of interactions from aromatic residues located deep inside the active site, were shown to guide the different intermediates through the reaction mechanism, further supporting the suggested importance of F138.24
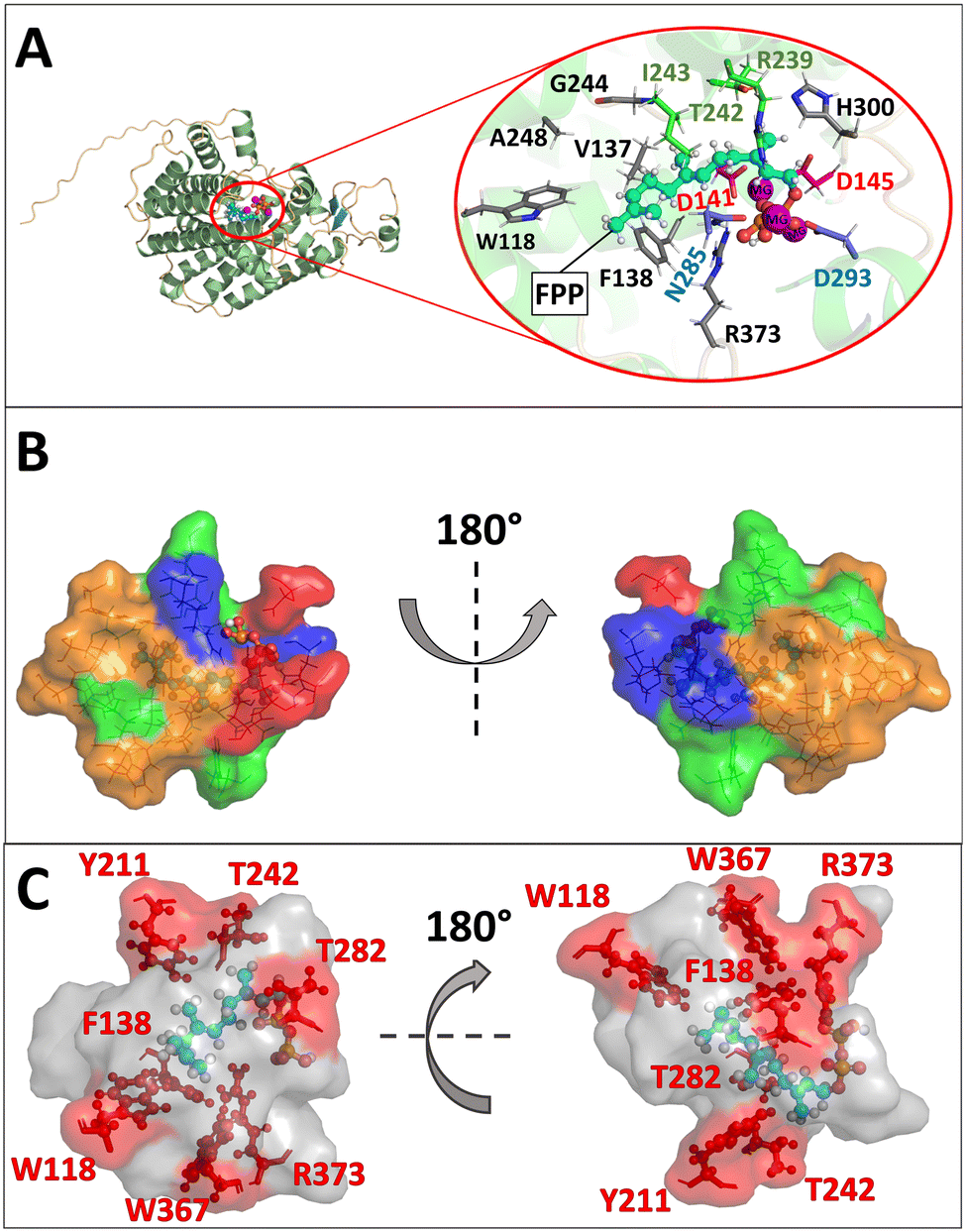 | ||
| Fig. 1 Structural model and active site of BcBOT2; A: BcBOT2 shown in cartoon with alpha helices in green, loops in orange, and beta-sheets in cyan. The active site for the natural substrate FPP is encircled in red. Zoom-in view of the active site shows FPP in ball–stick representation (colored in cyan) and amino acids interacting with the substrate in a rod representation. The residues of effector triad are colored green. Residues of the DDXXD motif are colored red, and those of the NSE/DTE motif in blue. Additional interacting residues are represented in grey. Mg2+ ions are shown as spheres in magenta; B: the active site pocket of BcBOT2 in a surface-line plot. FPP is shown in a cyan sphere-rod representation. The amino acids that form the active site pocket are colored based on their physicochemical properties. Acidic residues are colored in red, while basic residues are colored in blue. Polar and neutral residues are colored in green and nonpolar and hydrophobic residues in orange; C: positions selected for mutagenesis in the active site pocket. The active site pocket is shown as the grey surface, FPP in the cyan ball–stick representation. The amino acid positions selected for mutagenesis are highlighted in the red ball–stick representations and labeled accordingly. | ||
In order to engineer more promiscuous variants of BcBOT2, the reactivity of the cyclization steps had to be altered by introducing new or removing old stabilizing interactions.29,35 At the same time, the unilateral activation of the diphosphate group should not be obscured.1–3,37 To achieve this, polar and aromatic residues of the first shell with moderate conservation scores not involved in the initial activation step were selected for mutagenesis experiments (see Fig. S13 and S14 in the ESI†).
The first preselection of residues was made on the assumption that they exert a significant influence on the intermediates and these include primarily aromatic residues that either contour the active site (W118, Y211, W367) or that are also known to interact with the cations (F138).19,25 Furthermore, polar and basic residues with possible catalytic significance (T242, R373) were included in the selection (Fig. 1C).
Based on our BcBOT2 model, a series of variants were generated by site-directed mutagenesis at positions W118, W367, F138, Y211, T242, T282, and R373. We particularly identified W118 as an important candidate for exchange, as it is located deep in the active site pocket. The exchange W118Q was expected to enlarge the local volume. At the same time, the exchange should increase the enzyme's thermostability, as demonstrated in a previous directed evolution study.38 After collecting the results of this first example, we expanded the number of variants and included W118F, W118H, W118M, W118N, and W118R (Fig. 2B). Once the first results of F138V were at hand, we expanded the number of variants at position 138 to include F138A, F138M, F138N, F138Q, and F138Y into the repertoire. Accordingly, we replaced the aromatic ring with smaller residues to increase the active site pocket and broaden the product promiscuity.25,26 Furthermore, the replacement with polar residues could trigger new interactions between the active site pocket and the intermediates, leading to new reaction pathways and eventually products. In addition, two other variants were generated at position T242, the linker in the effector triad. The rationale for this choice leads to the group of Dickschat, who showed by sequence comparison of 200 bacterial terpene synthases that, in addition to threonine, asparagine and serine can also function as linker amino acids.19 We, therefore, generated variants T242S and T242N (Fig. 2B). With similar reasoning, T282S was chosen, which should also lead to a local enlargement of the catalytic pocket. Afterwards, the replacement of the basic amino acid arginine at position 373 with histidine (R373H) and glycine (R373G) was pursued (Fig. 2B). While an alternative base was introduced with histidine, the choice of glycine resulted in the loss of basicity while locally increasing the size of the active site pocket. Finally, we also selected W367, which is located near R373 and W118 in the deeper part of the active center (Fig. 1B). Based on the observation that W367A resulted in a change in the product spectrum, several additional exchanges were also made at this position. These were W367F, W367H, W367K, W367N, W367T, and W367V (Fig. 2B). Similar to the exchanges described above, an increase in pocket size and a change in polarity should be induced.
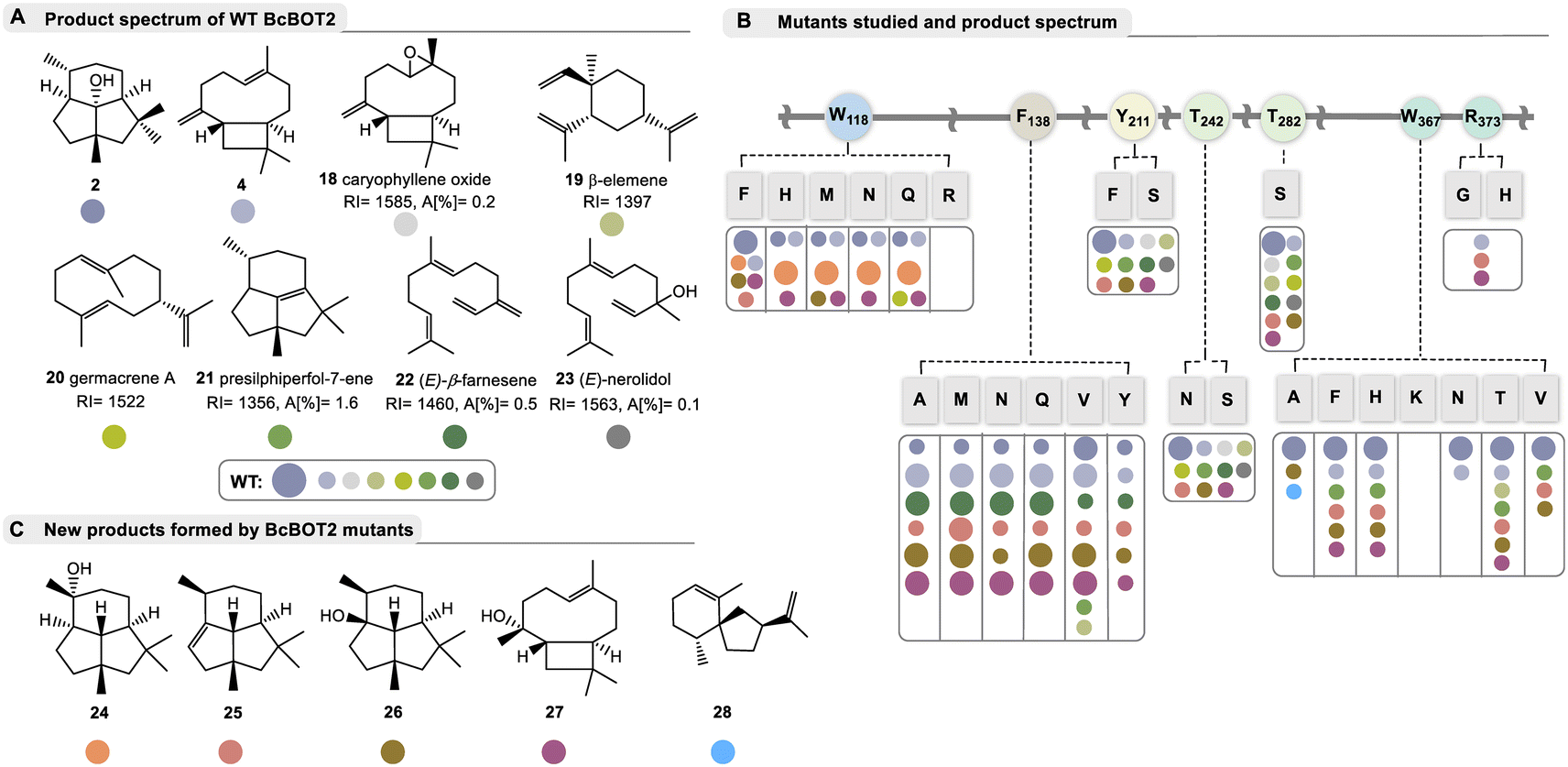 | ||
| Fig. 2 Products formed by WT BcBOT2 and variants chosen in this study; A: by-products 18–23 (with RI values on a DB5-column and area percentages given in %) formed from FPP 1 by BcBOT2 (15 is probably the thermally induced Claisen rearrangement product of germacrene A (20) and may have been formed during GC analysis; absolute configuration of the stereogenic center in 23 not determined); structures were elucidated by alignment with MS databases. The complete GC chromatogram, including details on unknown byproducts are found in the ESI;† B: list of amino acid exchanges in BcBOT2 and overview of products formed from FPP 1. These are listed according to the color code, and the size of dots refers to major and minor abundance (small dots refer to area percentages commonly below 2%, specific data are listed in Table S10 in ESI;† abundance of 19 and 20 varied with respect to GC-temperature); C: list of new sesquiterpenes 24–28 not produced by wild-type BcBOT2 in sufficient amounts to be detected by mass spectrometry. Details on the structure elucidation and the underlying mechanisms are found in the text below and Scheme 2. | ||
Biotransformations
After examining the product profile of the WT BcBOT2 enzyme in more detail (in triplicate) than had been done previously, complemented by two negative controls (FPP only and alternatively enzyme only), we found that BcBOT2 forms a variety of other products in minute amounts not reported so far. The negative controls allowed the determination of impurities and clearer identification of new biotransformation products. In fact FPP 1 gave (−)-presilphiperfolan-8β-ol (2, RIDB5 = 1623, A [%] = 91.2) as the major product and 15 other by-products determined by GC-MS analysis with area percentages between 0.1% and 1.6% (Fig. 2A).Of these, seven compounds were identified by database matching using two columns with different polarities coupled with MS detection. We found β-caryophyllene (4, RIDB5 = 1448, A [%] = 0.7) and its (aut)oxidation product (18).39 In addition, β-elemene (19), the Cope rearrangement product of thermolabile germacrene A (20), was detected, which was not found when employing a different temperature program during chromatographic separation.40 In addition, we found presilphiperfol-7-ene (21), (E)-β-farnesene (22), and (E)-nerolidol (23), whereas the intensity of the other product signals was too low to obtain representative MS spectra of these compounds (see ESI,† Table S10 for details). Negative control experiments confirmed that farnesene and nerolidol were not formed by spontaneous hydrolysis of FPP 1. With this now expanded product portfolio for WT BcBOT2 in hand, we next performed analytical enzyme assays with the variants proposed by our BcBOT2 model (Fig. 2B).
Comparison with WT BcBOT2 showed that the variants Y211S, Y211F, T242N, T242S, and T282S yielded nearly identical product spectra (2, 4, 18–23). A semiquantitative comparison of the biotransformation products of the wildtype with those of the enzyme variant using an internal standard revealed only minor differences for the amount of each product. Thus, we determined yields in the range of 43.9 ± 0.5% and 50.1 ± 3.3% for the production of (−)-presilphiperfolan-8-ol (2), comparable to the yield of WT BcBOT2 with 52.6 ± 4.2%. The results are also similar for the biotransformed by-products (see Table S10 in the ESI†). Interestingly, substitutions in the effector triad (T242) of fungal BcBOT2 did not exert a strong effect, quite consistent with studies on bacterial STCs.19
In addition, we examined the thermostability of these enzyme variants in more detail in the hope of achieving higher reaction rates without loss of activity or denaturation as noted by Arnold et al. in related studies with STCs.38 Indeed, the yield of 2 increased for all five variants with elevating temperatures with maxima for Y211S, Y211F, and T242N at approximately 40 °C (see ESI,† Fig. S5–S7), so these variants can be considered as starting points for further mutagenesis studies in the future.
Replacement of arginine by histidine (R373H) or removal of the basic function by introducing glycine (R373G) suppresses the formation of (−)-presilphiperfolan-8β-ol (2) and also most of the other by-products. The formation of β-caryophyllene (4) demonstrates that FPP 1 is still accepted to a small degree. Therefore, R373 appears to play an important role only after the formation of the caryophyllenyl cation (8, see Fig. 1B). The nearby residue W367 also appears to be relevant for the formation of terpenoid 2. Introduction of smaller residues resulted in significantly reduced activity (see Table S10 in the ESI†). In addition, replacement with the positively charged lysine (W367K) caused complete inactivation of the enzyme (see Fig. 2B). A less drastic change, such as the W367F variant, resulted in a yield of 24 ± 5.58%. Apart from the enzymatic activity, remarkable observations were made in the product spectrum in W367A. In addition to the major product 2 and presilphiperfolan-1α-ol (26), another unknown by-product was found with a low yield of 0.9 ± 0.09%. Accordingly, the retention index and mass spectrum of this compound were matched in databases. Similarities were found with the sesquiterpene vetispiradiene (28), which occurs as an FPP cyclization product of the two STCs Hvs1 from Hyoscyamus muticus and Tps32 from Solanum lycopersum.15,41 Moreover, these conjectures were demonstrably confirmed by co-injection experiments. For this purpose, extracts from the biotransformation of FPP 1 were combined with BcBOT2 W367A or FPP 1 with Tps32 and analyzed by GC-MS.
As suggested by our model studies, tryptophan 118 may also play an important role. With the exception of one transmutation (W118R) that resulted in complete loss of catalytic activity, all other variants (W118F, W118H, W118M, W118N, and W118Q) remained active. Of particular note, two new presilphiperfolanols 24 and 26 were formed. This effect was minor for W118F, which was still able to generate 2 as the major product. In contrast, substitution of smaller residues at this position resulted in higher specificity for the formation of presilphiperfolan-9β-ol (24), which is formed by premature addition of water molecules. This effect was most evident with variants W118Q and W118N.
Semiquantitative product formation analysis for W118Q and W118N yielded 18.7 ± 0.1% and 20.29 ± 1.01% for 24, respectively. Presilphiperfolan-1α-ol (26) occurred only as a minor constituent in the W118F and W118M variants, with yields of 8.17 ± 0.52% and 3.78 ± 0.25%, respectively. Presilphiperfolan-9β-ol (24) is a known constituent of the plant genera Artemisia lacinata and Artemisia chamaemelifolia. Its structure has already been elucidated by NMR spectroscopy, molecular modeling, and total synthesis.42,43 Olfactory analysis characterized a strong woody and resinous note for the racemate.44 However, in our case, 24 did not show a sensory profile, so it can be assumed that only one of the two enantiomers was probably formed in our case. Variant W118Q also produced low levels of germacrene A (20) as well as the caryophyllene derivative 27.
The point mutations at position F138 also significantly altered product spectrum. Interestingly, the exchange of the amino acid phenylalanine (for alanine, methionine, asparagine, glutamine, valine, and tyrosine) transforms BcBOT2 from a relatively product-specific to a multi-product synthase, which in addition to PSP (2) yields up to seven further products that could only be detected in very small amounts in the wild type. Four of these products were formed in sufficient amounts so we could isolate them for the first time and determine their structure. Two of these new products carry the presilphiperfolane backbone, namely presilphiperfol-1-ene (25) and presilphiperfolan-1α-ol (26). In addition, a new sesquiterpene caryophyllene-8β-ol (27) was isolated along with the known β-caryophyllene (4). Determination of the thermostability of the BcBOT2-F138V variant revealed that the maximum yield of all five major products is obtained at temperatures above 30 °C (see Fig. S7 in the ESI†).
The structures of the major products 24–27 were elucidated after the biotransformations of BcBOT2-F138V and BcBOT2-W118Q were repeated on a semipreparative scale as a fed-batch process (Scheme 2A). For the W118Q variant, the crude product was purified by preparative GC and reached a purity of 94.1%. Interestingly, a much higher crude product yield (73%) was obtained with this variant than with the wild-type enzyme (46%). 1D- and 2D-NMR spectroscopic experiments allowed us to identify the known presilphiperfolan-9β-ol 24 and confirmed the published relative stereochemistry (Scheme 2B).42,44,45 The crude product obtained from a semipreparative scaled transformation with the F138V variant was 54.4%, and thus a higher yield was also found for this variant compared with WT BcBOT2. To allow complete structural elucidation, products 25–27 as well as presilphiperfolan-9β-ol (2) and β-caryophyllene (4) were isolated by preparative GC (pGC), with purities ranging from 78.8% to 96.6%.
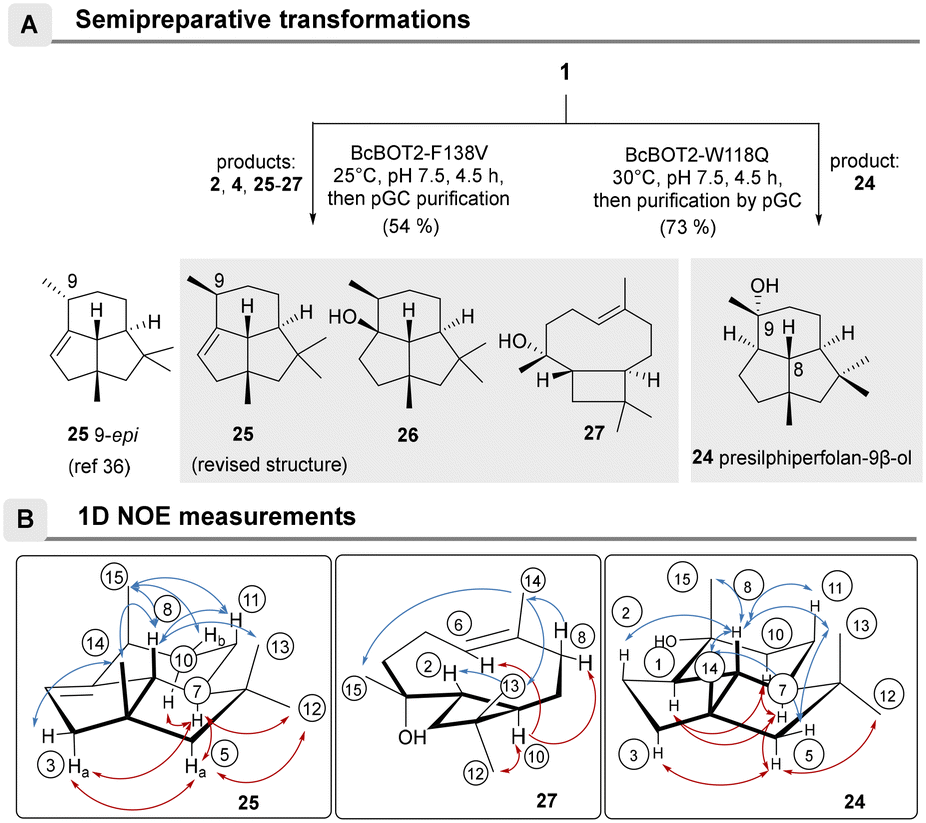 | ||
| Scheme 2 Semipreparative biotransformations with BcBOT2 variants W118Q and F138V and selected 1D-NOE correlations for terpenes 24, 25, and 27 A: enzymatic biotransformation of FPP 1 using the single point-mutated sesquiterpene synthases BcBOT2-W118Q and F138V; B: 1D-NOE correlation observed in presilphiperfolan-9β-ol (24), presilphiperfol-1-ene (25), and caryophyllene-8-ol (27) (positions are circled, carbon atom numbering was analogous to the FPP used). | ||
NMR spectroscopic analyses and comparisons with published literature data confirmed the formation of PSP (2) and β-caryophyllene (4).12,46,47 The formation of the diastereomeric 2-epi-β-caryophyllene (5) could be excluded by NMR analyses and retention index (RI) comparisons. It should be noted that until this time, presilphiperfolan-1-ene (25) was not known to be a biotransformation or natural product, but only as a semisynthetic product.48
Determination of the relative stereochemistry of 25 was also done by 1D-NOE experiments, but the collected data differed from the reported ones (Scheme 2B).48 Thus, magnetization transitions between the axially positioned proton H-8 with the protons of methyl groups 13, 14, and 15 could be identified, clearly showing that the two methyl groups at positions 14 and 15 are directed in the same direction in space. On the other hand, the proton at position 7, points in the opposite direction, as confirmed by the correlations with protons H-10a, H-5a, and H-3a and the methyl group at position 12. Further evidence for our proposal that the isolated presilphiperfol-1-ene (25) is indeed a 9-epimer of the reported structure was obtained from the fifth biotransformation product presilphiperfolan-1-ol (26), a known sesquiterpene originally collected from Concocephalum conicum and Anemia tomentosa var. anthriscifolia, respectively.48,49
The constitution of caryophyllene-8β-ol (27) could only be elucidated by NMR spectroscopy at low temperature (253.3 K in toluene-d8), because interconvertible conformers led to signal too broad in the 1H and 13C NMR spectra at room temperature.50 The two conformers are probably related to the orientation of the methyl group 14, which can point either upward (α) or downward (β). Rotation within the mobile C-4/C-9 segment allows the two conformational isomers to interconvert. Its structure was unambiguously determined from the COSY and the J2,3(C,H) coupling constants in the HMBC spectrum. However, only one of the two conformers could be unambiguously determined at lower temperatures; the other was present only in very small amounts. A corresponding behavior has already been discussed in detail for iso-fermacrene A.16
27 was further analyzed by 1D-NOE experiments, which led to deciphering the relative stereochemistry of the ring alkene and the anti-annulation between the cyclobutane and cyclononane rings. These structural features are also found in β-caryophyllene (4).12 The configuration at C-3 was determined from the magnetization transitions of H-14 with H-13 and H-15 and the absence of this effect for H-15 with H-10 and H-6, respectively. Since the proton H-10 should be β-aligned according to the postulated catalytic mechanism to (−)-presilphiperfolan-8β-ol (2) and thus oriented opposite to the methyl group 15, the addition of water should occur from below the ring system, the α-face.
With respect to the methyl group 14, an α-orientation was found for the present conformer at 253.3 K in toluene-d8 based on the observed magnetization transitions. Of note, caryophyllen-8β-ol (27), although known, has not yet been isolated from natural sources. Instead, it was chemically prepared from caryophyllene oxide,51 where it served as an intermediate in the total synthesis of presilphiperfolan-1-ol (26).50,51
Mechanistic considerations
Our mechanistic considerations are based on the model discussed above, the docking experiments with various intermediates in models of wildtype and selected variants, and especially on the products formed, isolated, and characterized, and previously proposed mechanisms.4–11 We are aware that such considerations always have a certain degree of speculative character. Nevertheless, it is worth generating an overall mechanistic picture of the results discussed so far. In particular, it focuses on the breakdown into divergent mechanistic pathways when amino acids W118 and F138 are specifically exchanged (Scheme 3). This proposal also includes stereochemical considerations.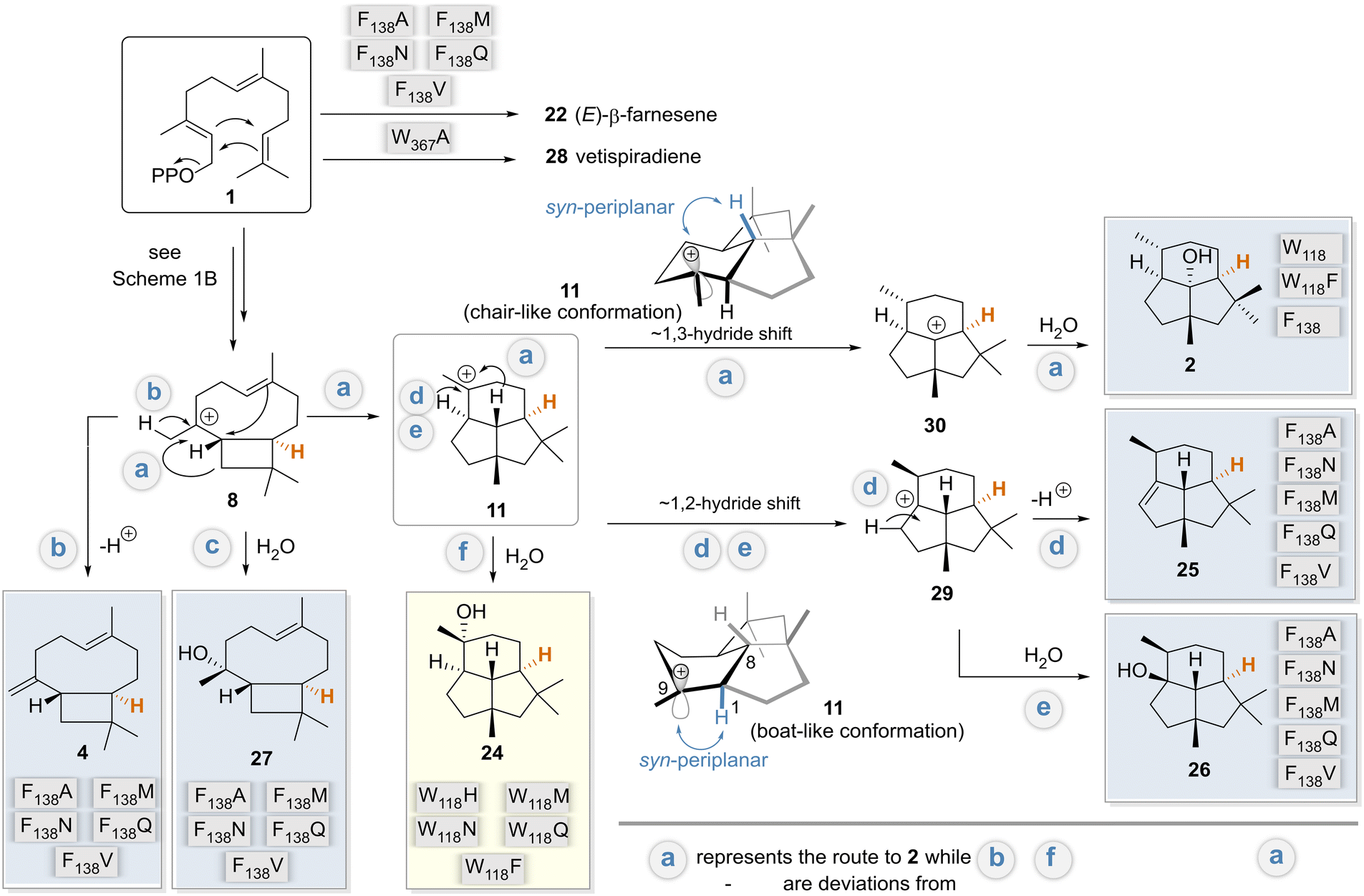 | ||
| Scheme 3 Unified mechanism in relation to variants (routes a–f): postulated mechanisms for the formation of biotransformation products 2, 4 and 24–28 formed by the BcBOT2-W118 and -F138 variants, respectively. Variants are assigned to the main products they form. These were related to the postulated mechanism for the formation of PSP 2 by BcBOT2 discussed in Scheme 1B. The major products formed by variant F138V are colored in light blue, whereas the product 24 isolated from biotransformation with variant W118Q is highlighted in light yellow. The reformulated pathway deviations from the biosynthesis of PSP 2 (pathway a) are marked with b–f. Protons marked in orange are the stereocenter that can be used as a reference point for determining the absolute stereochemistry of the other biotransformation products. | ||
The docking results show that F138 is a key amino acid for interaction with cationic intermediates in WT BcBOT2 and thus can be considered a major reason for its high fidelity (see Fig. S18 in the ESI†). Docking of intermediates in the different variants also revealed a novel way of stabilizing carbocationic intermediates by the D141 residue (see Fig. S19 in the ESI†). The expansion of the active site pocket and the position-selective elimination of the cation–π interaction by BcBOT2-F138V and other variants opened the doors to multiple reaction pathways and consequently to pronounced product promiscuity. Based on the postulated mechanisms, intermediates 8 and 11 serve as starting points for unfolding these new routes that correspond to the natural biosynthetic pathway of BcBOT2.4,5,9 While premature termination of the reaction cascade leads to (E)-farnesene (22) and further downstream to 4 and 27, which are usually induced by exchange for polar amino acids, an entirely new option is opened that is initiated by a stereocontrolled 1,2-hydride shift (hydride is transferred from below; Scheme 3 pathways d and e). For the latter to take place, the variant, unlike the wild type, should have an additional stabilization option for the resulting intermediate 29, from which products 25 and 26 are subsequently formed.
The biosynthesis of presilphiperfolan-9β-ol (24) has been discussed extensively.42,45,49 Remarkably, in our case, a single point mutation paves the way to uncover the biosynthetic route f. The biosynthetic pathways to 24 and PSP (2) separate at the step of the carbocationic intermediate 11, which apparently is assigned a pivotal role in this reaction cascade.
The results of intermediate docking with selected variants suggest that D141 of the DDxxD motif and F138 play an important role in driving the response. In the absence of F138 and with the change in the size of the active site pocket, a new stabilizing interaction with D141 is evident for key intermediates 8 and 11 (see Fig. S19 in the ESI†). This is particularly illustrated in Fig. 3A. Only in the WT BcBOT2 does the carbocation point away from the D141 side chain, resulting in only small amounts of early termination products. In comparison, the variants with a larger yield of 24 (W118F, W118H, F138V) showed a carbocation orientation toward the D141 side chain. Moreover, the expression of this orientation appears to correlate with the amount of termination product formed (Fig. 1A). In the F138V and W118H variants, the carbocation and D141 strongly interact, which correlates with a higher production of 24. In contrast, this interaction is less pronounced in W118F, and less termination product is formed compared to the other two variants. Probably the size of the pocket in the active site of W118F has changed only slightly, so that partial interaction with F138 is still possible (see Fig. S19 in the ESI†).27 In other terpene synthases, aspartic acid is known to play an essential role in the activation of water, which would explain the occurrence of a larger number of early termination products in the BcBOT2 variants considered.
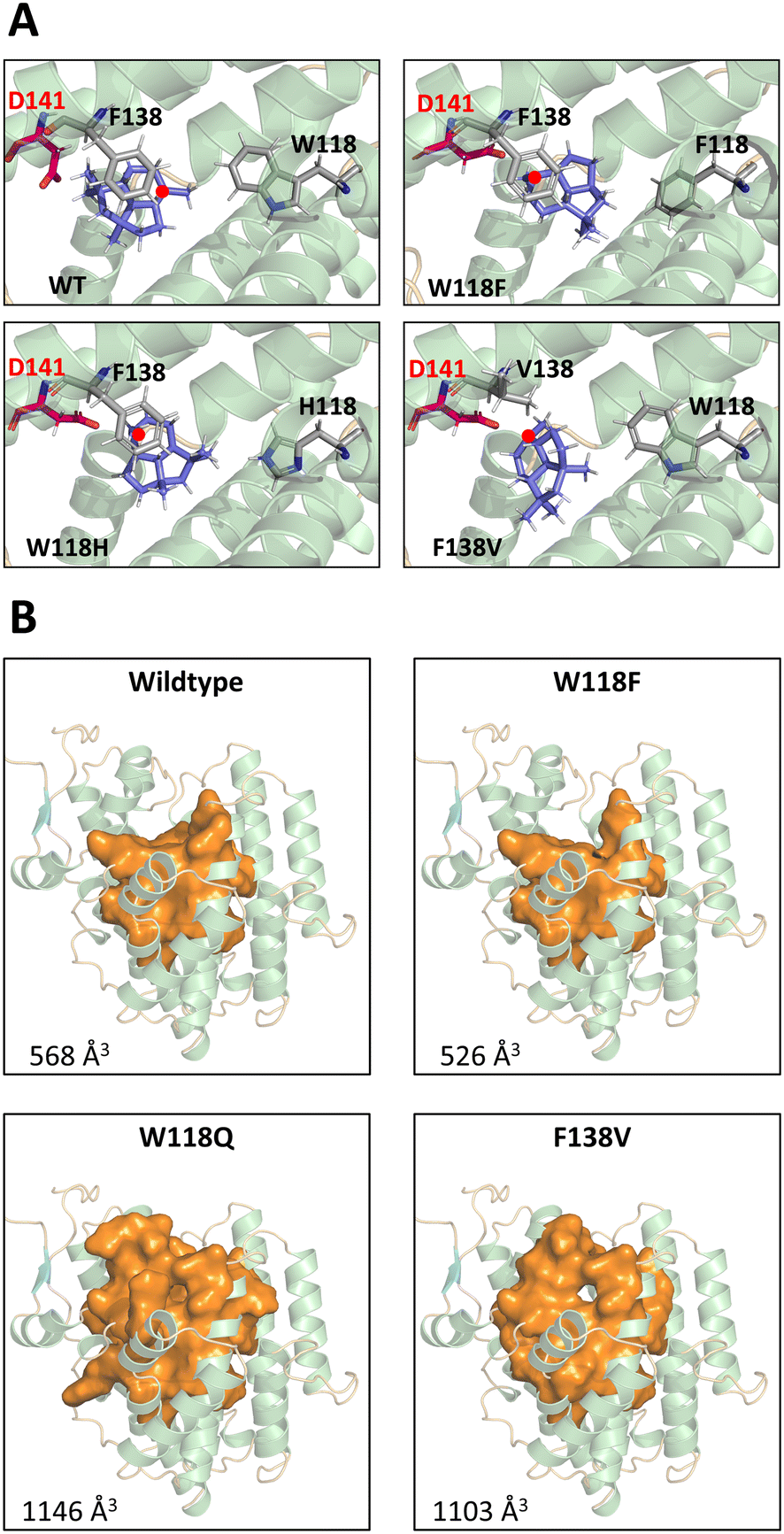 | ||
| Fig. 3 Active site pocket analysis and molecular docking of intermediates for BcBOT2 wildtype and variants. A: The docking pose of intermediate 11 for wildtype, and BcBOT2 variants. The residues of interacting positions (118, 138, 141) and the intermediate are shown in stick representation. Residues 118 and 138 are colored in grey, D141 as part of the DDxxD-motif is colored in red and the intermediate is colored in blue. For a better visualization of the intermediate, the carbocation atom is marked by a red dot. B: Structural and quantitative comparison of the identified active pockets for BcBOT2 wildtype and the single point variants F138V, W118F, and W118Q. The illustration shows an overhead view of each pocket in orange surface representation inside the BcBOT2 structure shown in cartoon representation. Alpha helices in the model are colored in green, beta sheets in blue, and loops in orange. The calculated volumes of each pocket are given. | ||
An increase in the size of the active site pocket, as seen in the W118Q and the F138V variants (Fig. 3B), facilitates additional diffusion of water molecules and thus increases the likelihood of nucleophilic attack and the formation of alcohols. This trend can also be observed in the substitution of W118. As the side chain size decreases, the free space in the active pocket increases (Fig. 3B), giving the substrate and intermediates more space and less guidance toward F138 (Fig. 3A). Additionally, this change in the amino acid side chain impacts the interaction network of the active site pocket residues. The altered interactions lead to different conformations of the first and second shell residues of the active site pocket, thus explaining a partial reshaping of the active site pocket. We propose that these conformational changes partially “open up” the active site pocket for the originally second shell residues, thus leading to a further increase in active site volume. This is nicely illustrated by comparing the W118F and W118Q docking positions (see Fig. S19 in the ESI†). The W118F variant continues to produce PSP as the main product. Docking in the active site of WT BcBOT2 enzyme (567 Å3) shows the interaction of intermediate 11 with F138 (see Fig. S18 in the ESI†). This changes for W118Q as the active site pocket volume increases to 1146 Å3 and the interaction with F138 can no longer be found. Instead, only D141 interacts with the carbocation (see Fig. S19 in the ESI†). The point mutation W118Q almost completely suppresses the 1,3-hydride shift and thus the formation of the intermediate 30. Instead, the tertiary cation 11 is trapped by adding water and the reaction cascade breaks off at an earlier point in the cation cascade.
Since the formation of PSP (2) is almost completely suppressed in the W118Q variant, the conformation of intermediate 11 plays a key role with respect to the possibility of proceeding mechanistically from there via either a 1,2- or 1,3-hydride shift. The flexibility of this tricyclic intermediate 11 is severely limited, except for the cyclohexane ring, which can adopt either a boat- or chair-like conformation (Scheme 3). This has consequences for the alignment of the empty p orbital at C9 with respect to the two pseudo-axially aligned hydrogen atoms at positions 1 and 8, which in principle may be responsible for the reported hydride shifts.
Close examination of the syn-planar alignment between the two C–H sigma bonds and the empty p orbital, an essential stereoelectronic requirement for hydride shifts, reveals that the boat-like conformation should favor a 1,2-hydride shift, while the chair-like conformation should favor the 1,3-hydride shift. When calculating the steric energies of the boat- and chair-like conformation of 11in silico, we observe only small energy differences, with the steric energies being 71.9 and 65.8 kcal mol−1, respectively (see Fig. S20A in the ESI†). Furthermore, we could identify that 11 adapts a more twist-boat-like conformation in its energy minimum (41.4 kcal mol−1) (see Fig. S20A in the ESI†). These findings support the argument that a subtle change in the interaction environment in the variants could lead to a conformational change of 11 in favor of an alternate hydride shift. Moreover, the observed conformations of 11 in the wildtype and in variant F138A fit to this proposition (see Fig. S20B in the ESI†). The more chair-like conformation of 11 observed for the wildtype would indeed favor the 1–3 hydride shift and thus the production of 2 as the main product. Moreover, the more twist boat-like conformation of 11 in variant F138A should also favor the 1–2 hydride shift and the production of 25 and 26 as the main product. However, these conformational changes remain small and subtle, so it remains a question if these trends could be directly correlated with the product spectrum. In summary, these mechanistic considerations also explain the opposite configuration of carbon atom C9 in PSP 2 and the pairs 25 and 26.
Statements on the absolute stereochemistry of sesquiterpenes 24–27 can also be made on the basis of mechanistic considerations. The absolute stereochemistry of PSP 2 has been unequivocally demonstrated by isotopic labeling studies, crystallographic analysis, and total synthesis.5,52,53 Since all isolated compounds as well as PSP 2 proceed mechanistically via the intermediate 8, and the (R)-configured stereocenter at C-10 (highlighted in orange) in 8 is conserved across all pathways (a–f), this information allows us to propose the absolute stereochemistry for 24–27 as shown in Scheme 3.
Conclusions
Based on a structural model of STC BcBOT2, a total of 26 variants were selected, successfully heterologously expressed and purified. Detailed analysis of the product spectrum of wild-type BcBOT2 with FPP 1 allowed us to identify previously unpublished byproducts using databases, retention indices, and MS spectra. These initial studies were essential as an analytical basis for subsequent studies with variants. Analysis of the product spectrum formed by BcBOT2 variants with FPP 1 revealed that the replacement of amino acid F138 with the amino acids tested in this study converted STC BcBOT2 to a multiproduct sesquiterpene synthase. In addition, replacement of W118 with Q118 and N118 resulted in the formation of a new biotransformation major product 24, which is not present in the product spectrum of wild-type BcBOT2. Interestingly, temperature profile analysis also showed increased thermal stability for all variations examined. Semipreparative biotransformations with variants W118Q and F138V yielded sufficiently large amounts of crude products to follow a pGC-based isolation protocol and collect the individual biotransformation products. Based on our results, the replacement of W118 can be attributed to a premature termination of the cascade at the intermediate 11 stage, indicated by the formation of the new major product presilphiperfolan-9β-ol (24). The docking experiments of intermediates 8 and 11 points to the common importance of the occurring D141 interactions and the increased volume of the active site pocket for the early water accumulation.As a side aspect of this work, we were able to revise the reported relative stereochemistry of presilphiperfol-1-ene (25). This work opens new possibilities for the use of terpene cyclases in the preparation of new natural products in an efficient sense.54 Finally, the present work paves the way to combine the rational reprogramming with the use of unnatural FPP derivatives thereby dramatically expanding the synthetic potential of terpene cyclases.15–17
Experimental procedures
Biotransformations
Isolation and structure elucidation
Product 4 was isolated by pGC on a DB1 column. The purified product (∼1 mg) was dissolved in C6D6; HR-GC-MS: 4: exact mass: 204.1878 [M]+, found: 204.1878 [M]+; RIDB5HT: 1456, RIFFAP: 1599 1H NMR (600 MHz, C6D6) δ [ppm]: 5.37 (dd, J = 9.5, 5.8 Hz, 1H, H-6), 5.04–5.03 (m, 1H, H-15b), 4.87 (d, J = 0.9 Hz, 1H, H-15a), 2.47–2.38 (m, 1H, H-1b), 2.36–2.31 (m, 1H, H-5b), 2.29 (q, J = 9.1 Hz, 1H, H-2), 2.18–2.13 (m, 1H, H-4b), 2.06–2.02 (m, 1H, H-8b), 2.02–1.96 (m, 1H, H-5a), 1.98–1.93 (m, 1H, H-4a), 1.94–1.88 (m, 1H, H-8a), 1.73 (d, J = 9.4 Hz, 1H, H-1a), 1.71–1.67 (m, 1H, H-10), 1.57 (s, 3H, H-14), 1.43–1.37 (m, 1H, H-9b), 1.36–1.30 (m, 1H, H-9a), 1.01 (s, 3H, H-12), 0.95 (s, 3H, H-13); 13C NMR (151 MHz, C6D6) δ [ppm]: 154.6 (C-3), 135.3 (C-7), 124.9 (C-6), 112.2 (C-15), 53.8 (C-10), 48.9 (C-2), 40.7 (C-8), 40.3 (C-1), 35.1 (C-4), 33.1 (C-11), 30.2 (C-12), 29.7 (C-9), 28.8 (C-5), 22.7 (C-13), 16.4 (C-14).
Product 25 was isolated by pGC on a DB1 column. The purified product (∼1 mg) was dissolved in C6D6. HR-GC-MS: 25: exact mass: 204.1878 [M]+, found: 204.1876 [M]+; RIDB5HT: 1351, RIFFAP: 1415; 1H NMR (500 MHz, C6D6) δ [ppm]: 4.95 (t, J = 2.3 Hz, 1H, H-2), 2.77–2.70 (m, 1H, H-9), 2.43 (dd, J = 16.5, 1.8 Hz, 1H, H-3b), 2.35 (dt, J = 16.5, 2.5 Hz, 1H, H-3a), 2.26 (dd, J = 11.9, 2.6 Hz, 1H, H-8), 1.65–1.59 (m, 1H, H-10b), 1.62 (d, J = 12.9 Hz, 1H, H-5b), 1.55 (d, J = 12.9 Hz, 1H, H-5a), 1.57–1.48 (m, 1H, H-10a), 1.45 (ddt, J = 12.3, 4.7, 2.5 Hz, 1H, H-11b), 1.31 (qd, J = 12.3, 4.3 Hz, 1H, H-11a), 1.17 (s, 3H, H-14), 1.10 (dd, J = 7.1, 0.3 Hz, 3H, H-15), 0.97 (td, J = 12.0, 2.5 Hz, 1H, H-7), 0.96 (s, 3H, H-12), 0.89 (s, 3H, H-13); 13C NMR (126 MHz, C6D6) δ [ppm]: 151.6 (C-1), 113.8 (C-2), 63.5 (C-7), 63.4 (C-8), 59.8 (C-5), 52.0 (C-3), 43.5 (C-4), 42.6 (C-6), 36.9 (C-10), 33.0 (C-9), 30.4 (C-14), 28.1 (C-12), 22.4 (C-11), 21.6 (C-13), 19.0 (C-15) (see ESI,† Table S11).
Product 26 (ref. 44 and 45) was isolated by pGC on DB1 column. The purified product (∼1 mg) was dissolved in C6D6. HR-GC-MS: 26: exact mass: 222.1984 [M]+; 204.1878 [M–H2O]+, found: 204.1875 [M–H2O]+; RIDB5HT: 1564, RIFFAP: 1943; 1H NMR (600 MHz, C6D6) δ [ppm]: 1.88–1.81 (m, 1H, H-3b), 1.86–1.80 (m, 1H, H-2b), 1.72–1.65 (m, 1H, H-2a), 1.62–1.58 (m, 1H, H-3a), 1.62–1.56 (m, 1H, H-9), 1.56 (d, J = 12.0 Hz, 2H, H-8), 1.51 (s, 1H, H-5), 1.47–1.40 (m, 1H, H-10b), 1.33 (s, 3H, H-14), 1.36–1.27 (m, 1H, H-10a), 1.34–1.27 (m, 1H, H-11b), 1.18–1.09 (m, 1H, H-11a), 1.15–1.07 (m, 1H, H-7), 0.95 (s, 3H, H-12), 0.90 (d, J = 7.0 Hz, 3H, H-15), 0.81 (s, 3H, H-13); 13C NMR (151 MHz, C6D6) δ [ppm]: 84.5 (C-1), 63.9 (C-8), 59.4 (C-5), 51.7 (C-7), 46.2 (C-4), 42.7 (C-3), 40.5 (C-6), 39.7 (C-2), 36.9 (C-9), 31.4 (C-14), 29.9 (C-10), 28.8 (C-12), 22.3 (C-13), 20.3 (C-11), 15.7 (C-15).
Product 27 was isolated by pGC on DB1 column. The purified product (∼1 mg) was dissolved in toluene-d8; HR-GC-MS: 27: exact mass: 222.1984 [M]+; 204.1878 [M–H2O]+, found: 204.1879 [M–H2O]+; RIDB5HT: 1625, RIFFAP: 2034; 1H NMR (600 MHz, toluene-d8, 253.3 K) δ [ppm]: 5.37 (t, J = 7.9 Hz, 1H, H-6), 2.14–2.07 (m, 1H, H-5b), 2.02–1.97 (m, 1H, H-5a), 1.89 (dt, J = 7.3, 3.6 Hz, 1H, H-8b), 1.84 (qd, J = 12.3, 3.9 Hz, 1H, H-8a), 1.64 (t, J = 10.0 Hz, 1H, H-1b), 1.61–1.56 (m, 1H, H-10), 1.49–1.46 (m, 1H, H-1a), 1.47–1.46 (m, 3H, H-14), 1.45–1.43 (m, 1H, H-2), 1.43–1.41 (m, 1H, H-4b), 1.41–1.38 (m, 1H, H-4a), 1.34–1.30 (m, 1H, H-9b), 1.18 (dddd, J = 14.5, 12.8, 10.7, 4.0 Hz, 1H, H-9a), 0.96 (s, 3H, H-12), 0.87 (s, 3H, H-13), 0.82 (s, 3H, H-15); 13C NMR (151 MHz, toluene-d8, 253.3 K) δ [ppm]: 135.8 (C-7), 123.5 (C-6), 73.2 (C-3), 52.6 (C-2), 46.8 (C-10), 41.7 (C-4), 40.7 (C-8), 37.5 (C-1), 31.8 (C-11), 31.1 (C-15), 30.6 (C-9), 30.2 (C-12), 23.8 (C-13), 23.4 (C-5), 16.4 (C-14) (see ESI,† Table S12).
Biotransformations of natural substrate FPP and BcBOT2-variants F138V and W118Q on a semi-preparative scale
| Crude product (BcBOT2-F138V) = 22.5 mg (101.1 μM, 73%); |
| Crude product (BcBOT2-W118Q) = 45.6 mg (207.0 μM, 54%) |
Computational methods
X-ray crystal structure of the sesquiterpene synthase BcBOT2 is not available. The structure of BcBOT2 was generated using Alphafold2.0.55 Further, models were subjected to energy minimization using AMBER14 force field.56 The Enzyme's active site and tunnels were computed and localized by Caver Web 1.0 (ref. 57) with default settings. YASARA (version 20.12.24.L.64)58 was applied for model visualization and analysis. Molecular docking of substrates was performed using Molecular Operating Environment (MOE).59 MOE tools was used to generate 2D substrate–enzyme interaction diagrams. Figures were created with PyMOL 2.3.3 (ref. 60) (Schrödinger, LLC.). Further details on computational methods are described in the ESI.†Author contributions
V. Nikolaiczyk, J. Irwan and T. Nguyen performed the protein expressions and biotransformations as well as isolation and characterization of products. J. Fohrer provided NMR support. P. Elbers and P. Schrank performed the computational modeling and analysis. A. Kirschning and M. D. Davari supervised the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Gerald Dräger (Leibniz Universität Hannover, Hannover, Germany) for excellent support in the structural analysis of MS spectrometric data. MDD is supported through funds from IPB Halle.Notes and references
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS.
- J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87–110 RSC.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef.
- D. Tantillo, Chem. Soc. Rev., 2020, 39, 2847–2854 RSC.
- F. Bohlmann, C. Zdero, J. Jakupovic, H. Robinson and R. M. King, Phytochemistry, 1981, 20, 2239–2244 CrossRef.
- F. Bohlmann and C. Zdero, Phytochemistry, 1981, 20, 2529–2534 CrossRef.
- F. Bohlmann and J. Jakupovic, Phytochemistry, 1980, 19, 259–265 CrossRef.
- P. Weyerstahl, H. Marschall, I. Seelmann and J. Jakupovic, Eur. J. Org. Chem., 1998, 1998, 1205–1212 CrossRef.
- J. Hanson, Pure Appl. Chem., 1981, 53, 1155–1162 CrossRef.
- A. P. W. Bradshaw, J. R. Hanson and R. Nyfeler, J. Chem. Soc., Perkin Trans. 1, 1981, 1469–1472 RSC.
- A. P. W. Bradshaw, J. R. Hanson, R. Nyfeler and I. H. Sadler, J. Chem. Soc., Perkin Trans. 1, 1982, 2187–2192 RSC.
- C.-M. Wang, R. Hopson, X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 8360–8361 CrossRef PubMed.
- S. C. Wang and D. J. Tantillo, Org. Lett., 2008, 10, 4827–4830 CrossRef.
- S. F. Hinkley, N. B. Perry and R. T. Weavers, Phytochemistry, 1994, 35, 1489–1494 CrossRef.
- C. Oberhauser, V. Harms, K. Seidel, B. Schröder, K. Ekramzadeh, S. Beutel, S. Winkler, L. Lauterbach, J. S. Dickschat and A. Kirschning, Angew. Chem., Int. Ed., 2018, 57, 11802–11806 CrossRef.
- V. Harms, B. Schröder, C. Oberhauser, C. D. Tran, S. Winkler, G. Dräger and A. Kirschning, Org. Lett., 2020, 22, 4360–4365 CrossRef.
- V. Harms, A. Kirschning and J. S. Dickschat, Nat. Prod. Rep., 2020, 37, 1080–1097 RSC.
- M. D. Toscano, K. J. Woycechowsky and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 3212–3236 CrossRef.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Angew. Chem., Int. Ed., 2014, 53, 7652–7656 CrossRef PubMed.
- L. S. Vedula, J. Jiang, T. Zakharian, D. E. Cane and D. W. Christianson, Arch. Biochem. Biophys., 2008, 469, 184–194 CrossRef.
- D. J. Miller and R. K. Allemann, Nat. Prod. Rep., 2012, 29, 60–71 RSC.
- A. Deligeorgopoulou, S. E. Taylor, S. Forcat and R. K. Allemann, Chem. Commun., 2003, 2162–2163 RSC.
- A. Deligeorgopoulou and R. K. Allemann, Biochemistry, 2003, 42, 7741–7747 CrossRef CAS.
- B. Weigel, J. Ludwig, R. A. Weber, S. Ludwig, C. Lennicke, P. Schrank, M. D. Davari, M. Nagia and L. A. Wessjohann, ChemBioChem, 2022, 23, e202200211 CrossRef CAS.
- F. Lopez-Gallego, S. A. Agger, D. Abate-Pella, M. D. Distefano and C. Schmidt-Dannert, ChemBioChem, 2010, 11, 1093–1106 CrossRef CAS.
- F. López-Gallego, G. Wawrzyn and C. Schmidt-Dannert, Appl. Environ. Microbiol., 2010, 76, 7723–7733 CrossRef.
- P. Baer, P. Rabe, C. A. Citron, C. C. de Oliveira Mann, N. Kaufmann, M. Groll and J. S. Dickschat, ChemBioChem, 2014, 15, 213–216 CrossRef CAS.
- C. M. Starks, K. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815–1820 CrossRef CAS.
- M. Seemann, G. Zhai, J.-W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681–7689 CrossRef CAS.
- C. Pinedo, C.-M. Wang, J.-M. Pradier, B. Dalmais, M. Choquer, P. Le Pêcheur, G. Morgant, I. G. Collado, D. E. Cane and M. Viaud, ACS Chem. Biol., 2008, 3, 791–801 CrossRef CAS.
- C. Contreras-Martel, C. Dahout-Gonzalez, A. D. S. Martins, M. Kotnik and A. Dessen, J. Mol. Biol., 2009, 387, 899–909 CrossRef CAS PubMed.
- V. Le Guilloux, P. Schmidtke and P. Tuffery, BMC Bioinf., 2009, 10, 1–11 CrossRef.
- Z.-Y. Huang, R.-Y. Ye, H.-L. Yu, A.-T. Li and J.-H. Xu, Bioresour. Bioprocess., 2021, 8, 1–27 CrossRef.
- E. Y. Shishova, L. Di Costanzo, D. E. Cane and D. W. Christianson, Biochemistry, 2007, 46, 1941–1951 CrossRef CAS PubMed.
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13543–13548 CrossRef.
- P. Schmidtke and X. Barril, J. Med. Chem., 2010, 53, 5858–5867 CrossRef CAS PubMed.
- Y. Yoshikuni, T. E. Ferrin and J. D. Keasling, Nature, 2006, 440, 1078–1082 CrossRef CAS.
- R. Lauchli, K. S. Rabe, K. Z. Kalbarczyk, A. Tata, T. Heel, R. Z. Kitto and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 5571–5574 CrossRef CAS PubMed.
- M. Sköld, A.-T. Karlberg, M. Matura and A. Börje, Food Chem. Toxicol., 2006, 44, 538–545 CrossRef.
- J. de Kraker and M. Franssen, Plant Physiol., 1998, 117, 1381–1392 CrossRef CAS PubMed.
- K. Back and J. Chappell, J. Biol. Chem., 1995, 270, 7375–7381 CrossRef CAS.
- J. A. Marco, J. F. Sanz-Cervera, M. Morante, V. García-Lliso, J. Vallès-Xirau and J. Jakupovic, Phytochemistry, 1996, 41, 837–844 CrossRef CAS.
- P. Weyerstahl, H. Marschall, M. Schröder, H. C. Wahlburg and V. K. Kaul, Flavour Fragrance J., 1997, 12, 315–325 CrossRef CAS.
- P. Weyerstahl, H. Marschall, M. Schulze and I. Schwope, Liebigs Ann., 1996, 1996, 799–807 CrossRef.
- A. Y. Hong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 5248–5260 CrossRef CAS.
- P. Rabe, T. Schmitz and J. S. Dickschat, Beilstein J. Org. Chem., 2016, 12, 1839–1850 CrossRef CAS PubMed.
- R. Chiba, A. Minami, K. Gomi and H. Oikawa, Org. Lett., 2013, 15, 594–597 CrossRef CAS.
- S. Melching and W. A. König, Phytochemistry, 1999, 51, 517–523 CrossRef CAS.
- S. C. Pinto, G. G. Leitão, H. R. Bizzo, N. Martinez, E. Dellacassa, F. M. dos Santos Jr, F. L. P. Costa, M. B. de Amorim and S. G. Leitão, Tetrahedron Lett., 2009, 50, 4785–4787 CrossRef CAS.
- L.-D. Syntrivanis, I. Némethová, D. Schmid, S. Levi, A. Prescimone, F. Bissegger, D. T. Major and K. Tiefenbacher, J. Am. Chem. Soc., 2020, 142, 5894–5900 CrossRef CAS PubMed.
- S. Shankar and R. M. Coates, J. Org. Chem., 1998, 63, 9177–9182 CrossRef CAS.
- R. M. Coates, Z. Ho, M. Klobus and S. R. Wilson, J. Am. Chem. Soc., 1996, 118, 9249–9254 CrossRef CAS.
- P. Hu and S. A. Snyder, J. Am. Chem. Soc., 2017, 139, 5007–5010 CrossRef CAS PubMed.
- V. Harms, V. Ravkina and A. Kirschning, Org. Lett., 2021, 23, 3162–3166 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Nature, 2021, 596, 583–589 CrossRef CAS.
- D. A. Case, V. Babin, J. Berryman, R. Betz, Q. Cai, D. Cerutti, T. Cheatham Iii, T. Darden, R. Duke and H. E. Gohlke, Amber14, University of California, San Francisco, 2014 Search PubMed.
- J. Stourac, O. Vavra, P. Kokkonen, J. Filipovic, G. Pinto, J. Brezovsky, J. Damborsky and D. Bednar, Nucleic Acids Res., 2019, 47, W414–W422 CrossRef CAS.
- E. Krieger, G. Koraimann and G. Vriend, Proteins, 2002, 47, 393–402 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2022.02, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022 Search PubMed.
- The PyMOL Molecular Graphics System, Version 2.0, Schrodinger, LLC Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of mutants, biotransformations and analytical and spectroscopic data including copies of NMR spectra of synthetic intermediates can be found with this article online. See DOI: https://doi.org/10.1039/d2cy01617f |
| This journal is © The Royal Society of Chemistry 2023 |