
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence4′-Fluoro-nucleosides and nucleotides: from nucleocidin to an emerging class of therapeutics
Phillip T.
Lowe
 * and
David
O’Hagan
*
* and
David
O’Hagan
*
School of Chemistry and Biomedical Sciences Research Centre, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: pl49@st-andrews.ac.uk; do1@st-andrews.ac.uk
First published on 6th December 2022
Abstract
The history and development of 4′-fluoro-nucleosides is discussed in this review. This is a class of nucleosides which have their origin in the discovery of the rare fluorine containing natural product nucleocidin. Nucleocidin contains a fluorine atom located at the 4′-position of its ribose ring. From its early isolation as an unexpected natural product, to its total synthesis and bioactivity assessment, nucleocidin has played a role in inspiring the exploration of 4′-fluoro-nucleosides as a privileged motif for nucleoside-based therapeutics.
 Phillip T. Lowe | Dr Phillip Lowe received an MChem degree at the University of Manchester and continued his postgraduate research at the Manchester Institute of Biotechnology, where he developed RNA-based gene expression tools capable of dynamic control of gene expression, for which he was awarded a PhD in Biological Chemistry. He is currently a Postdoctoral Research Fellow in the O'Hagan group at the University of St Andrews where his research has focused on the biotechnological development of the fluorinase enzyme, elucidating the biosynthesis of fluorometabolites and the production of novel bio-based fluorochemicals. |
 David O’Hagan | Professor David O'Hagan studied for his first degree in chemistry at the University of Glasgow and then PhD at the University of Southampton. He carried out postdoctoral research at the Ohio State University before being appointed to a Lectureship at the University of Durham. At Durham he combined his interests in natural products biosynthesis with organo-fluorine chemistry and began a programme exploring the biosynthesis of the rare naturally occurring fluorine containing compounds. In 2000 he moved to his current location at the University of St Andrews in Scotland where he is associated with both the School of Chemistry and the Centre for Biomolecular Sciences (BMS). At St Andrews his laboratory isolated the fluorinase, a native C–F bond forming enzyme and they have an active interest in exploring nucelocidin biosynthesis. Professor O’Hagan is the current President of the Organic Chemistry Community of the Royal Society of Chemistry. |
1. Introduction
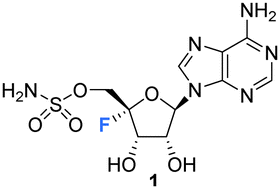
Organofluorine compounds constitute around 25% of all pharmaceuticals that have been licensed since the development of modern medicinal chemistry and that number is increasing for products in development.1 The ratio is even higher, at around 30%, for organofluorines found in agrochemical products including pesticides and herbicides.2,3 The incorporation of fluorine has been weaponised to fine tune pharmacokinetic profiles and other performance indicators to the point where the bioactives industries rely on the fluorine as a tool for optimisation.1,4–7 Thus, it is something of a paradox that nature has essentially ignored fluorine as an element in the biosynthesis of natural products, that myriad of compounds which are often characterised by their structural complexity, and which have largely evolved as bioactives to improve the fitness of the producing organism. Fluorinated natural products are exceedingly rare, to the point that new isolations attract significant interest. Fluoroacetate is the most common metabolite of this class having been identified widely in tropical and sub-tropical plants and in some bacteria. It has the obvious attribute of being exceedingly toxic and has presumably been harnessed by these organisms to fend off predators. Almost all the other fluorometabolites that have been identified so far are biosynthetically related to fluoroacetate and can be deduced to have arisen as products, branching from the same biosynthetic pathway. The only exception to this is nucleocidin 1 (Fig. 1), which was originally isolated from the actinomycete bacterium, Streptomyces calvus.8Fluorination has been extensively utilised in medicinal chemistry to imbue desired characteristics on nucleoside and nucleotide analogues towards a wide range of applications. For instance, the incorporation of fluorine motifs within nucleobases, and structural analogues thereof, has been investigated as a tool to mechanistically probe nucleic acids and proteins.9–13 Incorporation of fluorine within a non-canonical sugar motif of a nucleoside and a nucleotide has been used to generate analogues with tailored conformation and properties when incorporated into oligonucleotides and thus optimise potential biological applications.14–16 Additionally, modification of the nucleic acid backbone using fluorinated PNA monomers has been successfully explored,17,18 as has the development of selectively fluorinated nucleoside amidate prodrugs as antivirals.19,20
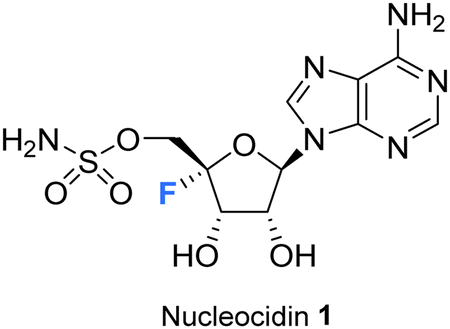 | ||
| Fig. 1 Structure of nucleocidin. | ||
In pharmaceuticals research, nucleosides modified by selective fluorination on the ribose ring have proven to be a rich source of bioactives and a diversity of such compounds has been developed for a variety of applications, most prominently as antiviral agents.21–24 Significant compounds of this class are illustrated in Fig. 2. Sofosbuvir 9 (anti Hep C virus) is a spectacular case in point and has been one of the most successful drugs of all time in commercial terms,25 but there are many other therapeutics of this class such as Lodenosine 3 (FddA) now discontinued,26 and Gemcitabine 4,27 and Clofarabine 528 which are currently used as clinical chemotherapeutics. It is noteworthy however that in all of these cases the fluorine atom is located at either the 2′ and 3′ positions of the ribose ring and not the 4′-position found in nucleocidin 1.29
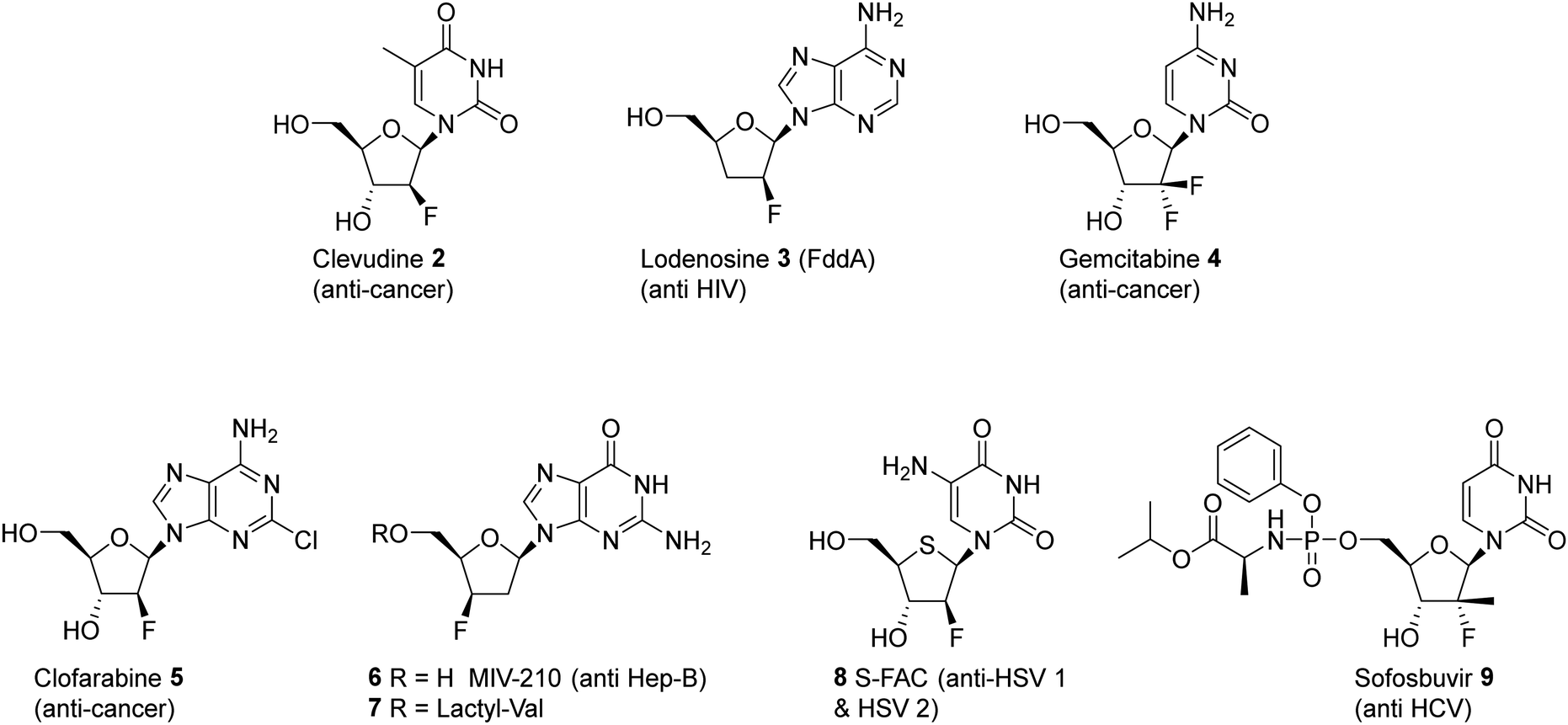 | ||
| Fig. 2 Examples of clinically significant fluorinated nucleosides and their target applications. | ||
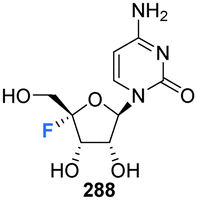
The utility of nucleosides incorporating a variety motifs at C4′ of the ribose, such as azides and alkyl groups, have been quite widely studied.30,31 It emerges that substituents incorporated at the C4′ position become located along the backbone edge of a nucleic acid duplex when assembled into higher ordered structures, and modifications at this position avoid notable steric clashes.31,32 Consequently, C4′-modified nucleosides have found a range of biomedical applications from oligomeric nucleotide therapeutics to antiviral agents.30,33,34 It is notable however that nucleosides with fluorine at the 4′ position of the ribose moiety have featured far less over the years, certainly relative to the abundance of analogues with fluorine at C2′ and C3′. There are perhaps some obvious reasons for this. The chemistry required to introduce a C4′ tertiary fluorine is particularly challenging and with the added complexity of controlling stereochemistry. Also, when fluorine is placed at C4′ of the ribose, it is potentially labile as it has an anomeric relationship with the ether oxygen and becomes susceptible to fluoride ion elimination. This can be contrasted with the relative stability of fluorine at C2′ and C3′. However, with these caveats, such compounds can be made stable and there have been recent disclosures in both the primary and patent literature outlining the preparation of synthetic 4′-fluoro-nucleosides in order to asses them as antivirals, and in that context there has been a particular focus on anti-hepatitus-C therapies.35,36
Nucleocidin 1 is the only fluorine containing nucleoside so far isolated from a natural source and it is an intriguing contradiction that the fluorine is located at C4′, the chemically most challenging site relative to the more common C2′ and C3′ fluoro-nucleosides which have emerged from med-chem programmes.37 This review aims to survey the literature to date on this class of nucleosides, highlighting recent developments in the identification of novel 4′-fluoro-nucleoside metabolites associated with nucleocidin production, as well as outlining progress in the synthesis and development of 4′-fluoro-nucleosides in chemical biology and as candidate antiviral agents among other biological applications.
2. Natural 4′-fluoro-nucleosides
2.1 Nucleocidin isolation and structural elucidation
Nucleocidin 1 was first isolated from the bacterium Streptomyces calvus in 1956, an actinomycete cultured from an Indian soil sample.8 It took over a decade to establish that nucleocidin contained a fluorine atom and another decade before the correct 4′-fluoro-5′-O-sufamyl adenosine structure was confirmed by total synthesis.Initial attempts at structure elucidation by Waller et al. (1957) utilised a variety of chemical and spectroscopic techniques (IR, UV and chemical hydrolysis) and provided a promising yet incomplete structural for nucleocidin (Fig. 3).38 Structure 11 was proposed with adenine bound to a carbohydrate moiety (specifically a 9-adenyl glycoside) and with a sulfamyl moiety attached. However, due to instrument limitations and inaccurate elemental analysis (C11H16N6SO8), the presence of a fluorine was overlooked. Hydrolysis, chromatography and pKa studies confirmed the hexavalent nature of the sulfur, and a sulfamyl group was proposed, however a complete structure was still not confirmed at this point.38 At the time of publication few synthetic examples had been reported of esters of N-unsubstituted sulfamic acids,38 and to date the existence of natural products containing a sulfamate group is rare.39,40 Morton et al., proposed a correct structure for nucelocidin in 1969 using 1H-NMR, 19F-NMR and mass spectrometry.37 The advent of more powerful NMR spectrometers identified coupling constants between the C3′H and C5′H methylene protons and fluorine and this, with accurate mass (C10H18N6SO6F) determination and fragmentation, allowed deduction of the established structure as 9-(4-fluoro-5-O-sulfamoylpento-furanosyl)adenine (Fig. 3). This placed nucleocidin 1 among the exceedingly rare fluorine containing natural products and set it apart from fluoromethyl related structures such as fluoroacetate, as the only fluoro-nucleoside derivative.37 The absolute D-configuration of the ribose sugar was subsequently confirmed with the total synthesis of nucleocidin 1.41,42
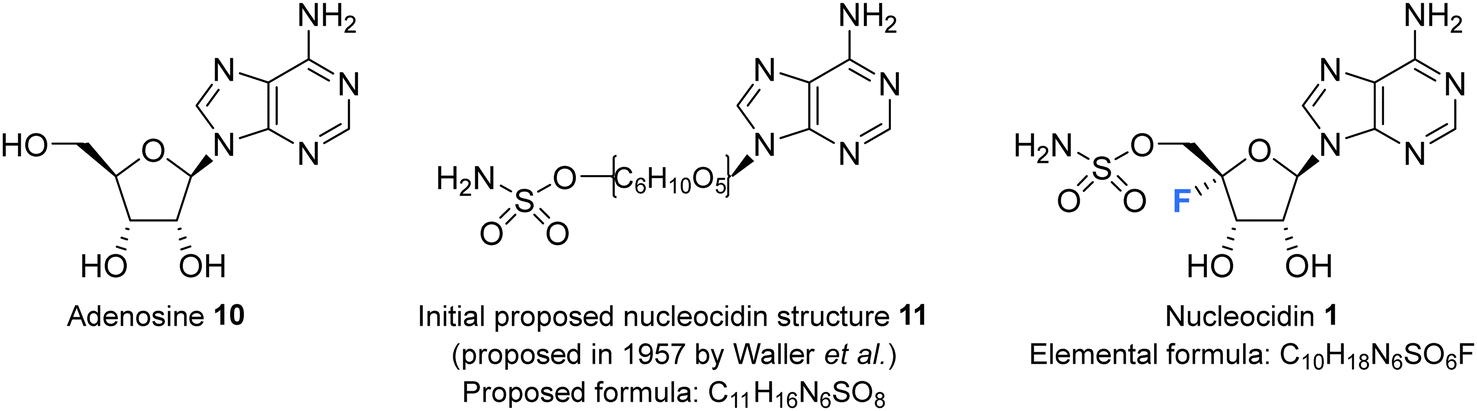 | ||
| Fig. 3 Adenosine 10, the initially proposed structure 11 for nucleocidin38 and the corrected structure of nucleocidin 1 containing a C4′-fluorine.37 | ||
2.2 Biological activity of nucleocidin
Nucleocidin 1 was soon recognised to have significant anti-trypanosomal activity.38 Hewitt et al. revealed a remarkable activity against T. equiperdum in 1956,43 reporting that it was considerably more potent than the structurally related antibiotic puromycin.44 Subsequent assessments found it active against a number of trypanosoma species; in 1957 Tobie et al. found it to be highly effective against T. congolense, T. equinum and T. gambiense in mice and rats, at a dose of 0.15 mg per kg, although it was also demonstrated to display a significant toxicity.45 In a study by Stephen et al., in 1960 it was demonstrated to be active against T. vivax in West African Fulani zebu cattle at low concentrations (0.025 mg per kg), however relapses occurred within one month after treatment.46 Nucleocidin was also found to be active in inhibiting the growth of several insect parasites such as Leptomonas and C. fasciculata, with a comparable potency to established trypanocides and leishmanicides.47In terms of understanding nucleocidin's mode of action, Florini et al. in 1966 and later, Sherman et al., in 1976, demonstrated a potent inhibition of protein synthesis. This occurred in vivo at doses much lower than that of other available antibiotics at the time,48,49 although the demonstrated potency was considerably diminished in cell free systems.48 At the molecular level, nucleocidin appears to inhibit translation and specifically the transfer of amino acids from tRNA's into the growing polypeptide chain at a stage subsequent to the formation of the aminoacyl-RNA. This is a consequence of an unusually slow but reversible formation of a co-complex between nucleocidin and the ribosome.48
The effect of nucleocidin on the fine structure of a monomorphic strain of Trypanosoma rhodesiense has also been examined by Williamson et al.50 This study revealed that nucleocidin induced electron-lucent cytoplasmic clefts in the cytoplasm of T. rhodesiense which possessed a close relationship with the rough endoplasmic reticulum and also resulted in excessive lysosomal vacuolation. Also noted was nucleocidin's ability to provoke nucleolar fragmentation and segregation, an outcome which may be related to interference with RNA synthesis.50
Given its potency and palatability to animals, nucleocidin appeared at the outset to offer a promising treatment for trypanosomal infections, however as a broader trypanocidal and leishmanicidal activity was demonstrated, so too was its toxicity. Numerous investigations revealed high toxicity in mice (LD50 – injection: 0.2 mg kg−1, oral: 2 mg kg−1), rabbits (lethal subcutaneous doses of 5.0 mg kg−1), rats (lethal injection: 0.8 mg kg−1) and most strikingly in young bovines (lethal – 0.05 mg kg−1) and thus any therapeutic promise was impeded by its high toxicity. This was exacerbated too by its lack of availability as it was not yet synthetically accessible, and titres were low from fermentation.43
2.3 Nucleocidin co-produced metabolites
The original strains of S. calvus deposited in public culture collections at the time of isolation subsequently lost their capacity to produce nucleocidin in culture. This hindered biosynthetic investigations as there were no available producing cultures. It was not until relatively recently that Zechel and Bechthold recognised from genome sequencing that the publicly available strain S. calvus ATCC 13382 associated with nucleocidin production was carrying a mutation in a bldA gene.51 This gene codes for the production of Leu-tRNAUUA which is required to translate leucine into selected proteins when instructed by the genome with the relatively rare TTA codon in some genes. Several genes implicated in nucleocidin biosynthesis utilise this codon and when the bldA mutation was corrected nucleocidin production became re-established in culture. This clearly indicated the importance of that particular codon usage for the incorporation of leucine into at least one of the biosynthetic enzymes for nucleocidin assembly.51,52 The original industrial S. calvus T-3018 strain held by Wyeth and then Pfizer had retained an ability to produce nucleocidin and with these producing strains available there has been a renewed interest in exploring nucleocidin biosynthesis.53,54 In particular, this has resulted in a deeper interrogation into the structures of minor fluorine containing metabolites co-produced with nucleocidin in S. calvus.Fig. 4 indicates the current metabolite profile of nucleocidin and its related structures from S. calvus. Most notable are the recently identified 3′-glucosylated metabolites F-Met I 12 and F-Met II 13. These compounds become apparent by 19F-NMR in extracts of S. calvus taken from the fermentation after a few days (days 5–6) but before nucleocidin production (day 8).55 As nucleocidin 1 begins to accumulate both F-Met I 12 and F-Met II 13 disappear in supernatant extracts, giving some sense that they are biosynthetic precursors to the finally formed nucleocidin 1.55
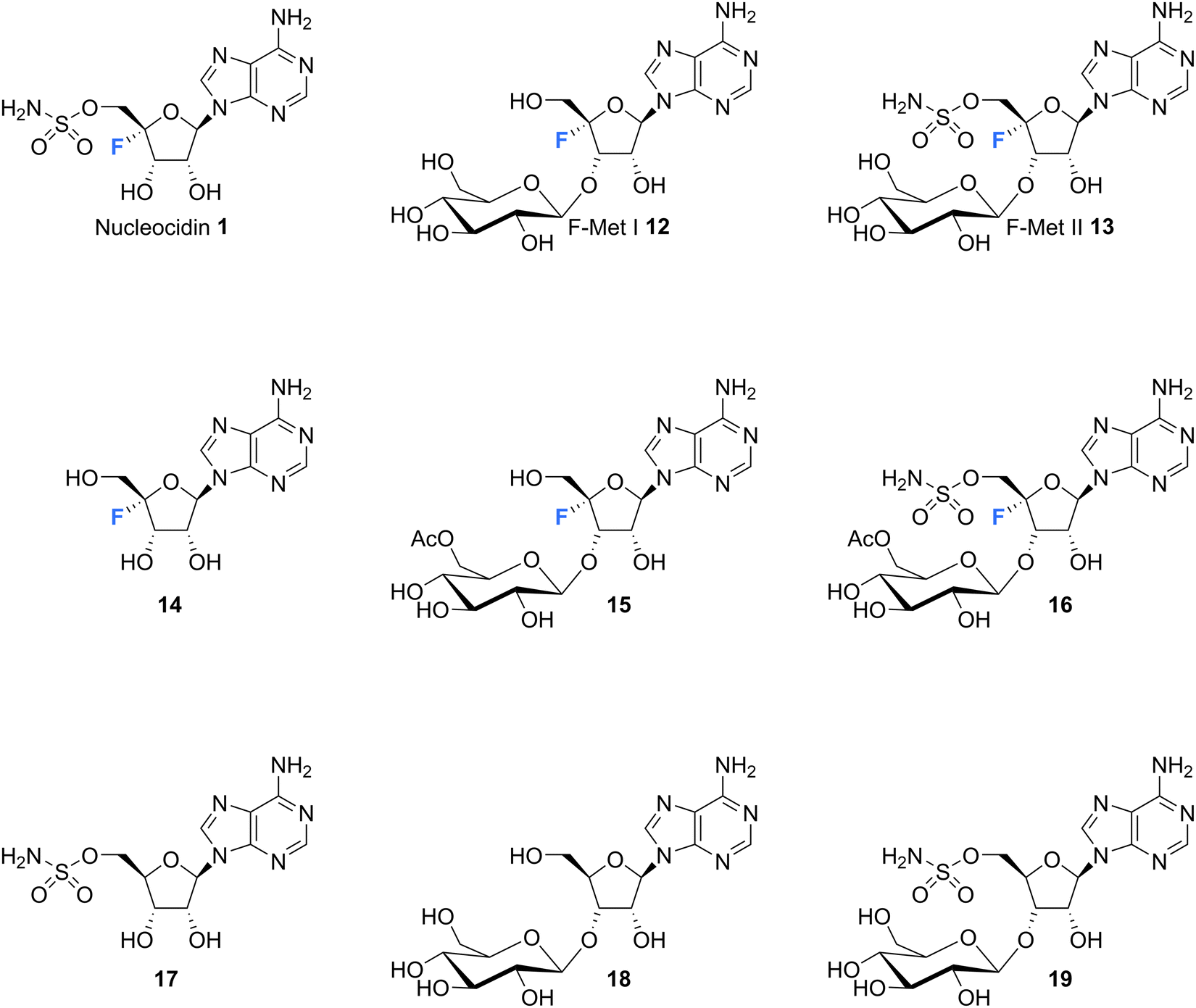 | ||
| Fig. 4 Structures of co-produced metabolites with nucleocidin from S. calvus and also S. virens B-24331 and S. aureorectus B-24301. | ||
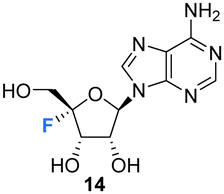
Isolation and structure elucidation has established that F-Met I 12 and F-Met II 13 are both β-glucosylated on the 3′O-alcohol of the ribose, which is a rare regiospecific glucosylation of nucleosides in metabolism. Genome mining identified a candidate glucosyl transferase gene (nucGT)55 which, when over-expressed, was shown to efficiently β-glucosylate the 3′-hydroxyl of adenosine 10 and 5′-O-sulfamyl adenosine 17 in in vitro assays with uridine diphosphate glucose (UDP-glucose) as the glucose donor. Such an unusual regiochemistry indicates that this enzyme is responsible for the biosynthesis of F-Met 1 12 and F-Met II 13. Glucosylation is a general strategy used by microorganisms to export toxic metabolites extracellularly and this may be a means of removing toxic 4′-fluoro-nucleosides from the cell. The recent discovery of nucleocidin producers by genome comparisons revealed Streptomyces virens B-24331 and Streptomyces aureorectus B-24301 as close relatives to S. calvus, and these organisms produce greater titres of nucleocidin and its related derivatives. Two O-acetylated metabolites 15 and 16 (see Fig. 4) were identified as minor metabolites from S. virens, and a glycosyl-O-acetyltransferase in the genome appears to be responsible for their production from F-Met I 12 and F-Met II 13 respectively.39 These acetylated metabolites are not obvious in S. calvus and they, along with their F-Met I 12 and F-Met II 13 precursors, appear to be metabolites of the first formed fluorometabolite, the nature of which is not clear at present. Another recent nucleocidin related metabolite is 4′-fluoroadenosine 14. This was identified in S. virens as a minor metabolite which appears very late in the batch fermentation cycle, after nucleocidin 1 production.56 This timing leaves open the prospect that 4′-fluoro-adenosine 14 is a metabolite rather than a biosynthetic precursor to nucleocidin, although this remains to be determined.
Intriguingly, a selection of nucleocidin analogues without a fluorine have been identified in wild type cultures of S. calvus.57 Defluoro-nucleocidin 17 as well as the corresponding β-glucosylated analogues 18 and 19 were recently isolated and their structures established relative to synthetic and enzymatically prepared reference compounds. These defluorohydra-analogues are present at levels between 10–50% relative to their fluorinated counterparts. This metabolite profile suggests that the biosynthetic assembly of the sulfamyl moiety of nucleocidin appears to be entirely independent of the fluorination event, developing adenosines that are electively fluorinated or not at C4′. In vitro enzyme assays have demonstrated too that both classes of adenosines (fluorinated and not) are also substrates of the β-glucosylation enzyme Nuc-GT.55,57 There is evidence too from gene knockouts that sulfamylation and fluorination are independent processes.39,54
3. Conformation of the 4′-fluororibose ring
Selective fluorination of ribonucleosides, and particularly deoxofluorination at 2′C or 3′C of the ribose ring, is well known to influence ribose ring pucker in a predictable but opposite manner. In solution the ribose ring equilibrates between energy minima that approximate North and South ring conformations, in a dynamic pseudo-rotation process.58,59 The North ring conformation has C-3′ carbon ‘up’ and endo relative to the C2′ carbon, which is ‘down’ and exo. The South conformation has the opposite arrangement, with C3′ carbon down and C2′ carbon up as illustrated in the inset in Fig. 5.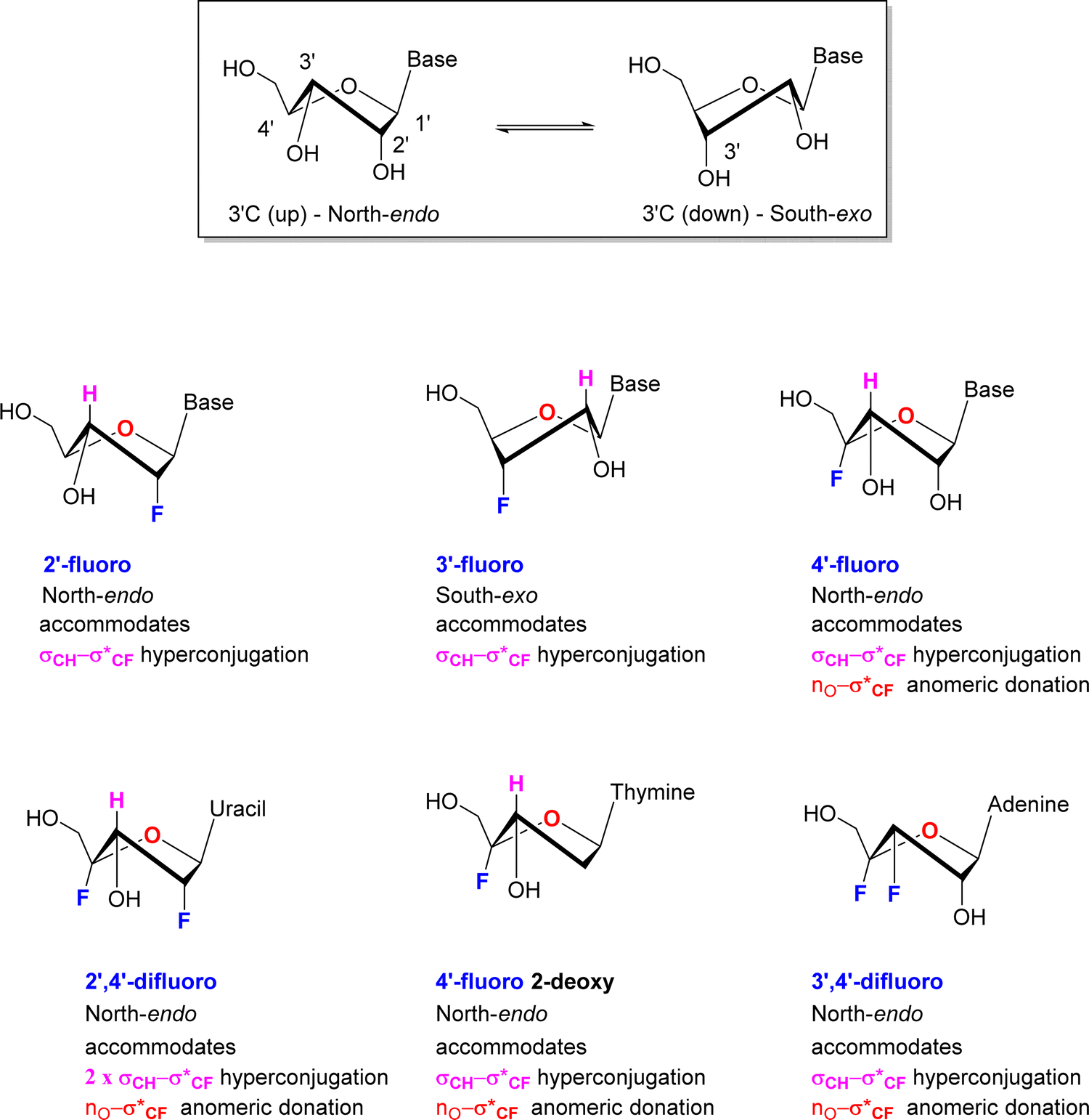 | ||
| Fig. 5 Predominant sugar conformations of deoxyfluororibose nucleosides. | ||
Vicinal H1′–H2′ coupling constants from NMR experiments in solution can be used as a proxy to distinguish North-type from South-type biased conformations. A value close to 3JHH ∼ 0 Hz for the H1′–H2′ vicinal coupling, represents a dihedral angle of 90° and is indicative of a pure North type conformation, whereas a vicinal H1′–H2′ value of 3JHH ∼ 8 Hz represents a dihedral angle approaching 180° and is indicative of a pure South type conformation.58,59 Of course, experimental values represent an average of all population contributions in the pseudorotation interconversion.
For 2′-fluororibose several studies have concluded a predominant North-type conformation in solution.60 This conformation accommodates an anti-periplanar arrangement between the 2′-C–F bond and the vicinal 3′-C–H bond, and thus a stabilising hyperconjugative interaction, typical of the classical gauche effect. Conversely for 3′-fluororibose systems a South-type conformation predominates.61,62 This accommodates a stabilising hyperconjugative interaction between the 2′-C–H bond which can arrange antiperiplanar to the 3′-C–F bond.
For those examples studied so far, when fluorine is placed at C4′ of the ribose in a nucleoside, then the North-type conformation dominates. The vicinal H1′–H2′ coupling constants found in nucleocidin (1.9 Hz),42,63 F-Met-I 12 (2.0 Hz),55 FMet-II 13 (0.9 Hz)55 and 4′-fluoro-adenosine (2.9Hz)56 lie in the 3JFH = 0.9–2.9 Hz range, suggesting only between ∼10–30% of South-type conformer contributions. This is supported too by the vicinal 4′F-3′H coupling constants which lie consistently between 3JFH = 16–18 Hz for 4′-fluoro-adenosine 14, nucleocidin 1 and metabolites F-Met-I 12 and F-Met II 13, and the value equates to an antiperiplanar relationship between the C–F bond and the vicinal 3′-C–H bond. This preference is further reinforced when a fluorine is placed at C-2′ in the case of 2′,4′-difluoro-dideoxy ribose nucleotides.64 In this case the vicinal H1′–H2′ coupling constant equals 3JHH = 0 Hz, consistent with 100% North-type conformation, and the vicinal 4′F–3′H coupling constant increases to 3JFH = 21 Hz further reinforcing an antiperiplanar arrangement between fluorine and the 3′ hydrogen and the North-type conformer. Within this series a conformational analysis65 of 4′-fluoro-2′-deoxythymidine by NMR also concluded a North-type conformation both as the free nucleotide with a vicinal H1′–H2′ coupling of 3JHH = 2.7 Hz and assembled into an oligiodeoxynucleoside. As the free nucleotide only 3% of the South-type conformation contributed, whereas within the oligiodeoxynucleoside this increased to 37%, suggesting that the tertiary and quarternary structures were beginning to override the preferred stereoelectronic preferences of the isolated nucleotide, but again the North-type conformer predominated.
Finally, a study66 on 3′,4′-difluoro-3-deoxyadenosine concluded a North-type preference for this nucleoside. Notably this overturns the South-type preference of 3′-fluoro-deoxynucleotides, and thus introduction of a fluorine at the 4′ position appears to dictate the bias in favour of North-type. The clear predisposition to a North-type conformation for 4′-fluoro-nucleotides has an obvious origin in accommodating an anomeric type interaction between a ring oxygen lone pair and the pseudo axial C–F bond and given the observation that placing a fluorine at 3′-deoxyfluoro ribose switches the conformational bias, suggests that this anomeric interaction is more dominant, and overrides any σCH–σ*CF hyperconjugative interactions between antiperiplanar hydrogens and the C3′-fluorine.
4. Inhibition of RNA polymerases induced by 4′-fluoro-nucleotides
The predominant antiviral mode of action for most of the 4′-fluoro-nucleosides discussed in this review ocurrs via inhibition of viral RNA polymerases, and this necessitates that the 4′-fluoro-nucleosides (or their corresponding prodrug) are substrates for cellular kinases so that the 5′-triphosphates derivatives are formed in vivo. This manner of inhibition has been demonstrated in vitro for example by Wang et al.,35 whereby the incorporation of a 4′-fluoro-nucleoside triphosphate into a growing RNA chain by a viral RNA polymerase results in transcriptional stalling and the termination of chain elongation (Fig. 6). This mode of termination is presumably a consequence of the conformational North-type puckering of the 4′-fluororibose, and the resulting interferance this has upon the incorporation of subsequent nucleoside triphophates (NTPs).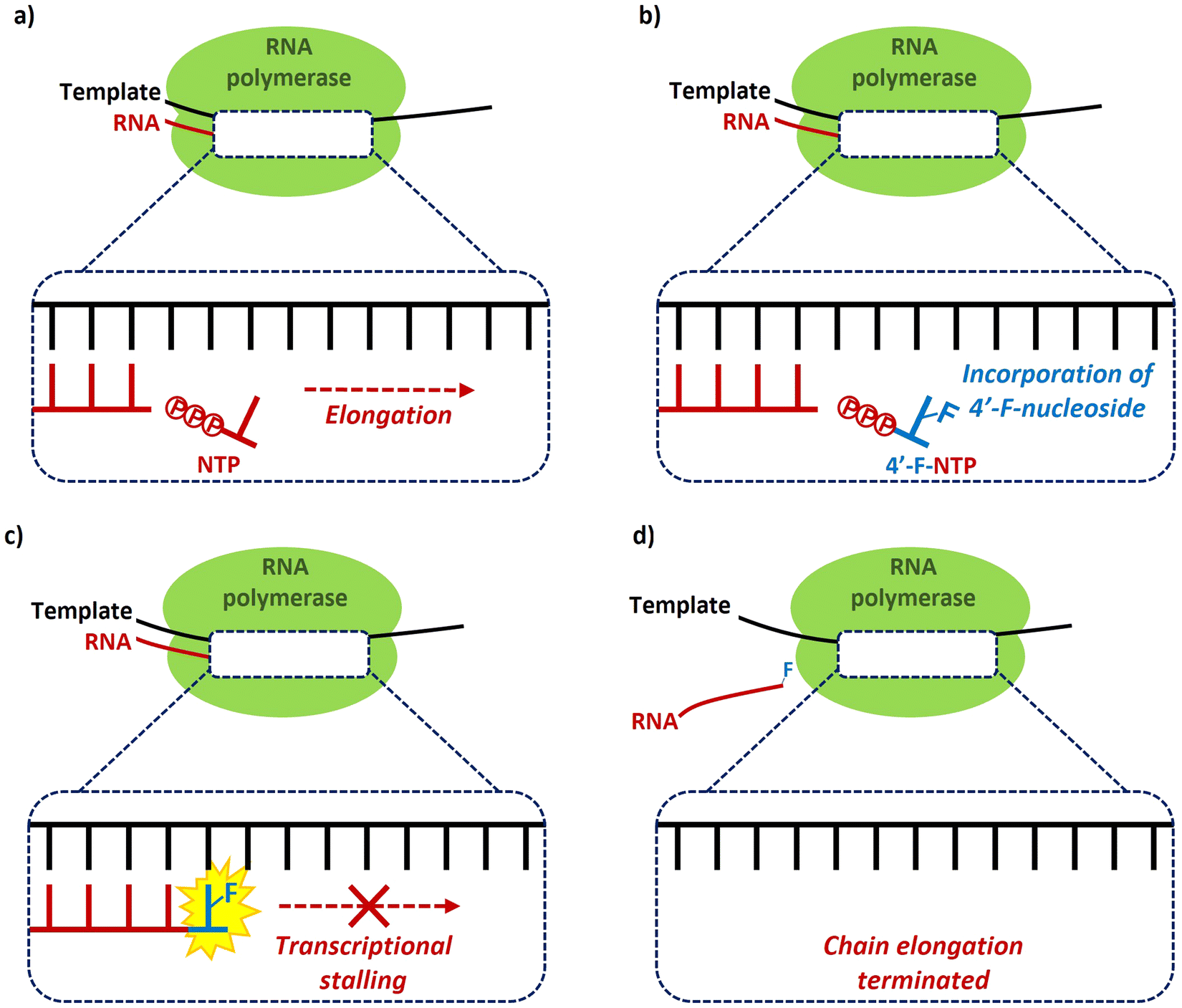 | ||
| Fig. 6 Chain termination mechanism of inhibition of RNA polymerases by 4′-fluoro-nucleoside triphosphates, as exemplified by Wang et al.35 | ||
5. Preparation of 4′-fluoro-nucleosides and nucleotides
5.1 Total syntheses of nucleocidin 1
The first synthesis of nucleocidin 1, was reported as a ‘preliminary account’ by Jenkins et al. in 197167 and is summarised in Scheme 1. A more complete account of this route was reported in 1976.42 The report is of contemporary significance as it remains the most popular method for the synthesis of 4′-C-fluoro-nucleosides. The strategy centred on the introduction of the 4′-fluoro moiety to a protected 4′,5′-dehydro-adenosine derivative, using an appropriate fluorinating pseudohalogen, in this instance iodine fluoride (Scheme 1).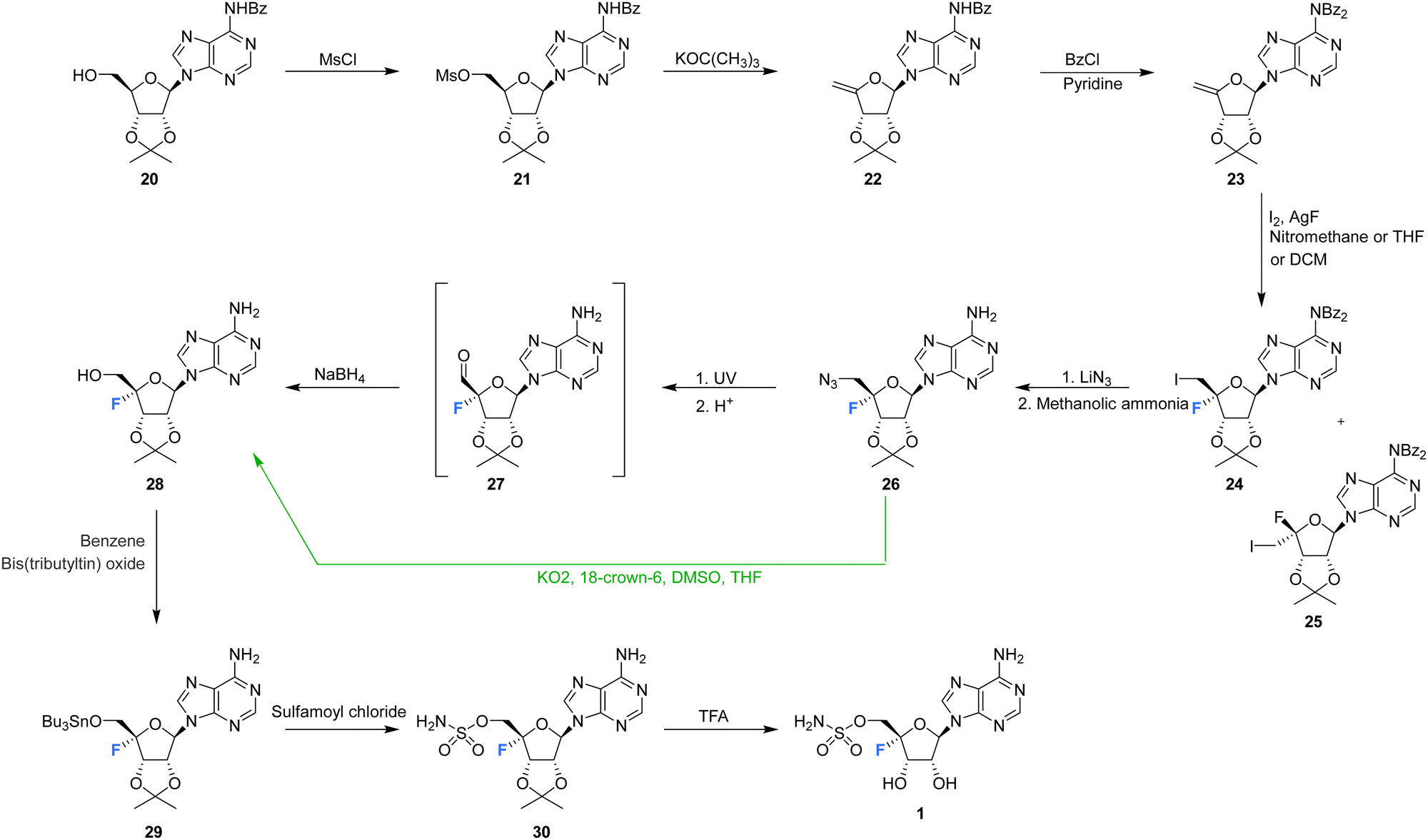 | ||
| Scheme 1 First total synthesis of nucleocidin 1.42,67 (distinct strategic deviations developed by Maguire et al.63 are shown in green). | ||
The synthesis began with mesylation of N6-benzoyl-2′,3′-O-isopropylidene-adenosine 20 and then a base induced elimination to generate the exocyclic enol ether 22. Further benzoyl protection of the adenine ring was followed by iodofluorination of 23 using iodine and silver fluoride. This generated an epimeric mixture of 5′-deoxy-4′-fluoro-5′-iodonucleosides 24 and 25, in a ratio which varied markedly depending on the reaction conditions.42,63 Isomer 24 was separated by chromatography and was then progressed by nucleophilic substitution with lithium azide. Azide was one of the few nucleophiles capable of displacing this primary iodide and the presence of the β-fluorine appears to suppress such substitution reactions. Debenzoylation of the adenine was followed by conversion of the 5′-azide of 26 to alcohol 28 under UV irradiation, and via an intermediate aldehyde which was directly reduced using borohydride. Finally, installation of the sulfamyl group and removal of the ribose acetal afforded nucleocidin 1. In 1993 Maguire et al. improved this general route (Scheme 1, green arrow), most notably by addressing the epimer ratio of 24, which improved after slow addition of iodine and also using potassium superoxide in the conversion of azide 26 to alcohol 28, circumventing the capricious photolytic conversion.63
5.2 4′-Fluoro-adenosines
The first synthesis of 4′-fluoro-adenosine 14 was reported in 1995 by Guillerm et al.,68 (Scheme 2) in a study to investigate whether it would function as an inhibitor of the antiviral target S-adenosyl-L-homocysteine (AdoHcy) hydrolase. After a five step protocol to N6-benzoyl-9-(5-deoxy-2,3-O-dibenzoyl-β-D-erythro-pent-4-enofuranosyl)adenine 32, the fluorine was introduced using IF following a similar approach to the original total synthesis of nucleocidin. This generated the desired iodo-fluoro epimer 33, although only in moderate yield. Conversion of the primary iodine to an alcohol was achieved with mCPBA and was followed by a global deprotection using sodium carbonate in methanol to generate 14. 4′-Fluoro-adenosine 14 was found to be stable in both methanol (CD3OD, no decomposition) and buffered aqueous solutions (5% decomposition over a period of 4 d),68,70 although it was observed to decompose rapidly in water (D2O, 80% in 1 day). 4′-Fluoro-adenosine 14 was shown to impart a time dependent loss of AdoHcy hydrolase activity (Kinact = 0.24 min−1 and Ki = 166 μM), although with a significantly lower affinity for the enzyme (100 fold) than adenosine. This was attributed to the C3′ endo geometry of the ribosyl moiety imposed by the 4′-C fluorine substituent.68 Guillerm et al. proposed a “cofactor depletion mechanism’ of inactivation of AdoHcy hydrolase by 4′-fluoro-adenosine 14.68 The 4′-fluoro-adenosine-5′-O-triphosphate 35 was acquired using established methods71 by Mayes et al.69 and was assayed for inhibitory activity against purified HCV polymerase where it was demonstrated to possess inhibitory activity with an IC50 > 10 μM.69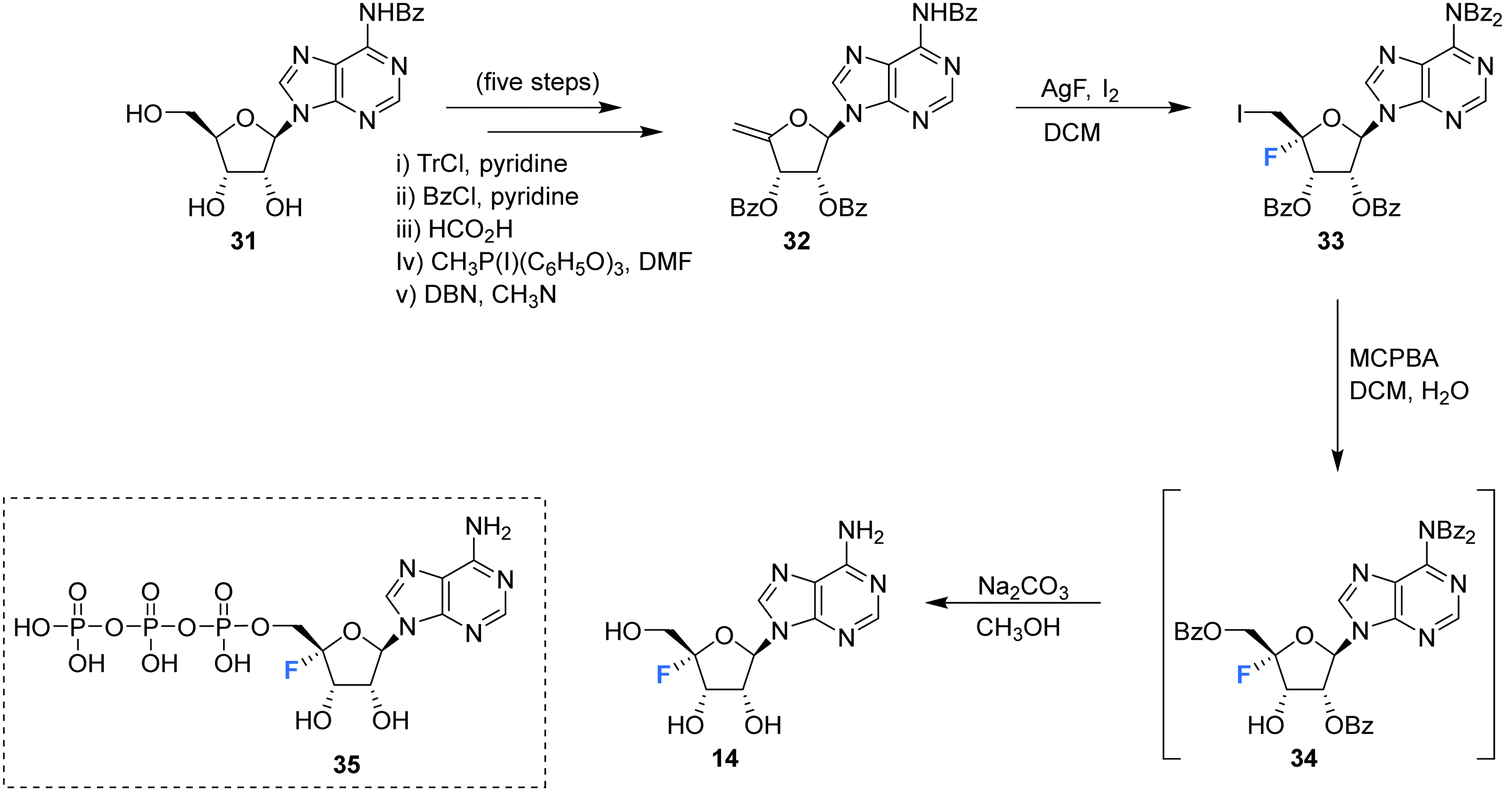 | ||
| Scheme 2 Synthesis of 4′-fluoro-adenosine 14 as described by Guillerm et al.68 and structure of 5′-O-triphosphate 35 synthesised by Mayes et al.69 | ||
The synthesis and biological evaluation of 4′-fluoro-adenosine-glycerodiphosphate 43 has been reported in the patent literature by Xu et al.72 Protected adenosine derivative 36 was acquired as a mixture of isomers in five steps from 31 as illustrated in Scheme 3. Isomers 36 were then processed to the monophosphate 39 after treatment with methanolic ammonia and H2. The resultant monophosphate isomers 39 were coupled to morpholine using DCC to generate the morpholinophosphonate 40. Subsequent coupling of 40 with protected glycerophosphate 41, followed by TFA deprotection generated 43 as a single isomer after HPLC purification. Both 4′-fluoro-adenosine-glycerodiphosphate 43 and precursor 42 were assessed in tissue culture media of HEK293 cells and were shown to act as agonists for alpha protein kinase 1 (ALPKl) in vitro. Alpha protein kinase 1 plays an important role in the immune response and modulation of ALPKl activity is a desired strategy for treating some cancers.
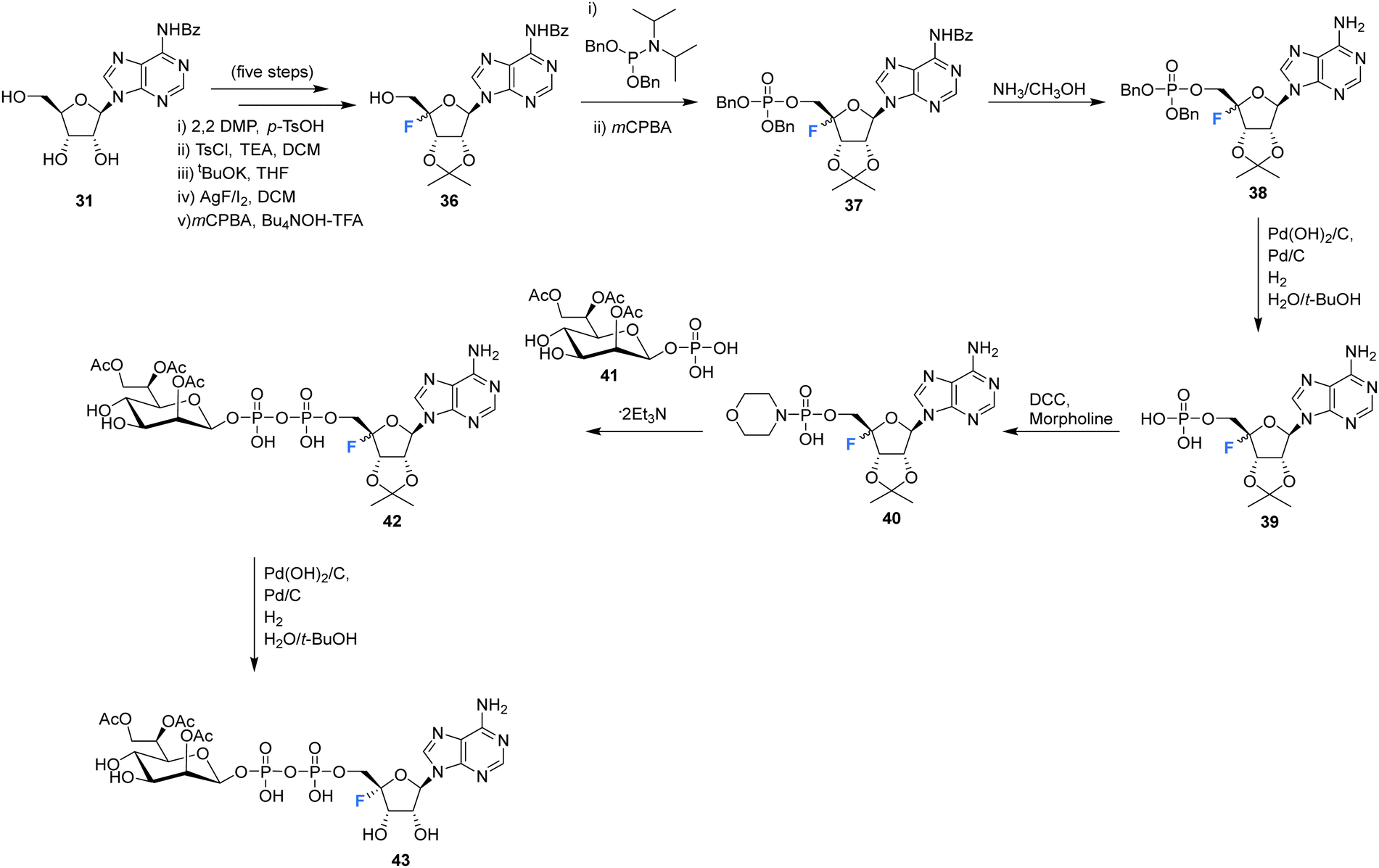 | ||
| Scheme 3 Synthesis of 4′-fluoro-adenosine-glycerodiphosphate 43 described by Xu et al.72 | ||
5.3 4′-Fluoro-uridines and cytidines
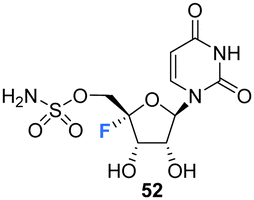
Owen et al. described the synthesis of 4′-fluoro-uridines 52, 55 and 59 in 1976, see Scheme 4.73 Experience with the introduction of substituents at C4′ of the ribose ring in related systems suggested a 2′,3′-cyclic carbonate to enable the correct stereochemical outcome from the addition of iodinefluoride to 44. With this in mind, (5-deoxy-2,3-O-isopropylidene-β-D-erythro-pent-4-enofuranosyl)uracil 44 was subjected to a gradual treatment with iodine in the presence of silver fluoride. This generated 5′-deoxy-4′-fluoro-5′-iodo-2′3′-O-isopropylideneuridine 47 as a single isomer. The remarkable stereoselectivity suggested anchimeric assistance from the uracil carbonyl occluding the top face of the ribose. Displacement of the relatively resistant 5′-iodo substituent with azide required “forcing conditions” (lithium azide in DMF) to yield 48, which was then treated with nitrosyl tetrafluoroborate to give O2,5′-cyclonucleoside 49. The lability of O2,5′-anhydro-4′-fluoro-2′,3′-O-isopropylideneuridine 49 to undergo acidic hydrolysis was then exploited to afford alcohol 50. Introduction of the sulfamyl group to generate 51 was achieved by treatment with sulfamoyl chloride, and this was followed with isopropylidene deprotection using formic acid. Deprotection was accompanied by significant glycosidic cleavage, although preparative TLC allowed isolation of 4′-fluoro-5′-deoxy-O-sulfamoyluridine 52 in a reasonable yield. 52 was found to exhibit considerably reduced antibacterial and cytotoxic properties when compared nucleocidin 1. The electron withdrawing nature of the 5′-O-sulfamyl group seems to impart a favourable stability to nucleocidin, suppressing 4′-fluoride elimination. Similarly, the phosphate ester 55 was investigated as an alternative stabilising electron withdrawing substituent. Accordingly 50 was reacted with bis(2,2,2-tri-chloroethyl)phosphoric acid to generate phosphate 53 and then treatment with formic acid gave diol 54. Finally, 55 was prepared after treatment of 54 with zinc in acetic acid and with catalytic silver acetate. The stability of these 4′-fluoro-uridine derivatives is strongly dependent on the nature of substituents attached to the 2′-,3′- and 5′-ribose hydroxyl groups with the protecting groups in 51, 53, and 57, rendering the compounds more resistant to fluoride elimination, however removal of these substituents generated far less stable compounds. Phosphate ester 53 was found to be susceptible to alkaline hydrolysis (much more so that isopropylideneuridine 50), although it was more stable in acidic conditions. The 5′-azido derivative 48 was sufficiently stable to allow a synthesis of 5′-azido-5′-deoxy-4′-fluoro-uridine 56, and this was then used to access 2′,3′-O-methoxymethylene 57. A treatment with nitrosyl tetrafluoroborate unexpectedly afforded O2,2′-cyclo,4′-fluoronucleoside 59 in a process proceeding through an oxocarbenium ion.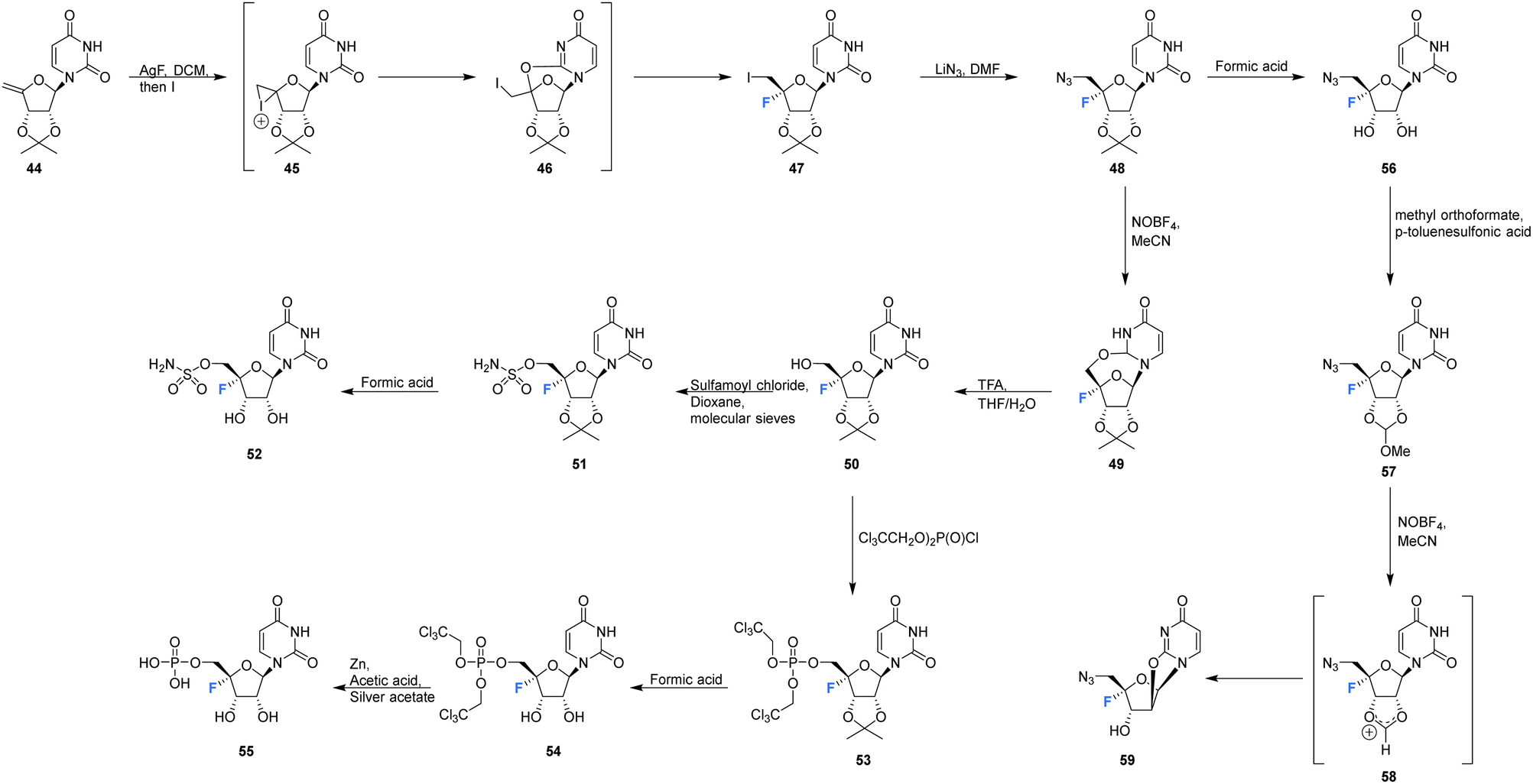 | ||
| Scheme 4 Synthesis of 4′-fluoro-uridines 52, 55 and 59 described by Owen et al.73 | ||
Ivanov et al., reported the synthesis of 4′-fluoro-uridine,5′-O-triphosphate 68, and investigated its efficacy against the hepatitis C virus, see Scheme 5.71 The target 68 was prepared from 4′-fluoro-2′,3′-O-isopropylideneuridine 50, itself prepared from 44 in a 4-step protocol involving fluorination (AgF/I2), azidation (NaN3), treatment with nitrosyl tetrafluoroborate and then hydrolysis to introduce the 5′-OH by a modification of the general method of Owen et al.73 Product 50 was then coupled with the tris-triazolide of phosphoric acid to generate 67, and then deprotection with formic acid gave 5′-monophosphate 55. Activation of 55 with carbonyldiimidazole and then reaction with the tert-butylammonium salt of pyrophosphoric acid generated 68, although the process was reported to be rather inefficient. The stability of 4′-fluoro-uridine,5′-O-monophosphate 55 and triphosphate 68 were assayed in PBS buffer at 37 °C, and this revealed the formation of uracil with a half-life of approximately 18 h for both compounds, although both could be stored at −20 °C without degradation.
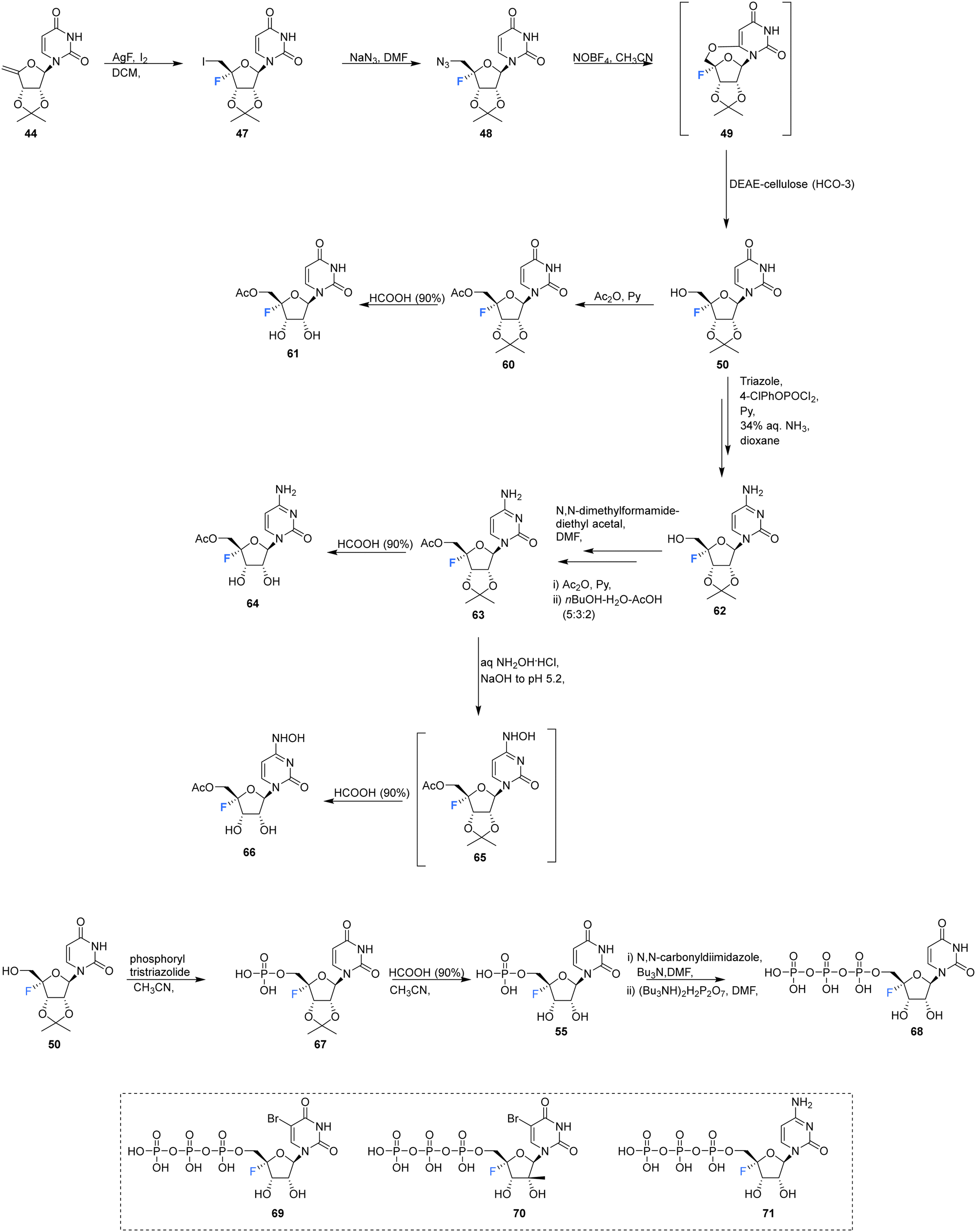 | ||
| Scheme 5 Synthesis of 4′-fluoro-uridine analogue 61, 4′-fluoro-cytidine analogue 64, 4′-fluoro-N4-hydroxycytidine 66, 5′-O-triphosphate 68 described by Ivanov et al.71 and structures of 5′-O-triphosphate 69–71 synthesised by Mayes et al.69 | ||
4′-Fluoro-uridine,5′-O-triphosphate 68 was assayed as a potential inhibitor of two key hepatitis-C viral enzymes: Nucleoside triphosphate (NTP)-dependent RNA polymerase (NS5B) and NTP dependent NTPase/helicase (NS3). In the event 68 proved to be an effective inhibitor of the polymerase with an IC50 of 2 μM.71 It was accepted as a substrate of the helicase but did not display any inhibition. Interestingly 68 acted as a substitute for ATP in its capacity as an allosteric activator of the helicase, but it was a significantly weaker binder than ATP.71 Triphosphates 69–71 were also assayed against purified HCV polymerase by Mayes et al.6971 displayed an IC50 between 250 nM–1 μM, whereas 69 and 70 both displayed IC50's between 1–10 μM.69
The acetyl derivatives 61, 64 and 66 were synthesised from 4′-fluoro-2′,3′-O-isopropylideneuridine 50. Each was found to be more stable that their non-acylated derivative. Acetylation of 50 with acetic anhydride in pyridine, followed by deprotection with formic acid, afforded acetylated 4′-fluoro-uridine 61. Protected 50 was converted to its triazole, followed by aminolysis and then the same acteylation/deprotection treatment to generate acetylated 4′-fluoro-cytidine 64. 4′-Fluoro-5′-O-acetyl-2′,3′-O-isopropylidenecytidine 63 was subjected to aqueous hydroxlyamine solution to generate 65, which was deprotected with formic acid, giving 4′-fluoro-N4-hydroxycytidine 66. The ease at which 5′-O-acetate is hydrolyzed by cell esterases allowed 61, 64 and 66 to be assayed in cell cultures as prodrugs for their non-acylated counterparts. Antiviral activity can then be introduced through in vivo transformation to their triphosphate and then the anticipated inhibition of HCV RNA-dependent RNA polymerase. However in this study, no cytotoxic effects were observed in human hepatocyte Huh7 cell cultures (up to a concentration of 500 μM).71
The synthesis of 4′-fluoro-uridine prodrugs 73 and 74 have been reported in the patent literature by Mayes et al.69 5′-Deoxy-4′-fluoro-5′-iodo-2′3′-O-isopropylideneuridine 50 was generated using conventional methodology and was then converted with the appropriate chlorophosphoramidate followed by treatment with formic acid, as illustrated in Scheme 6. This afforded phosphoramidate diastereomers 73 and 74, which could be separated by semi-prep HPLC, Scheme 6. Prodrugs 73 and 74 were assayed for HCV replicon activity and cytotoxicity. Diastereoisomers 73 and 74 both displayed CC50 values within the range of 1–10 μM, with 73 possessing higher EC50 value (>10 μM) than 74 (with the range of 1–10 μM). The pharmacokinetics of these prodrugs were studied, with the active nucleoside triphosphate for each compound measure by LC-MS/MS. Both 73 and 74 were found to readily accumulate in mouse liver samples.69
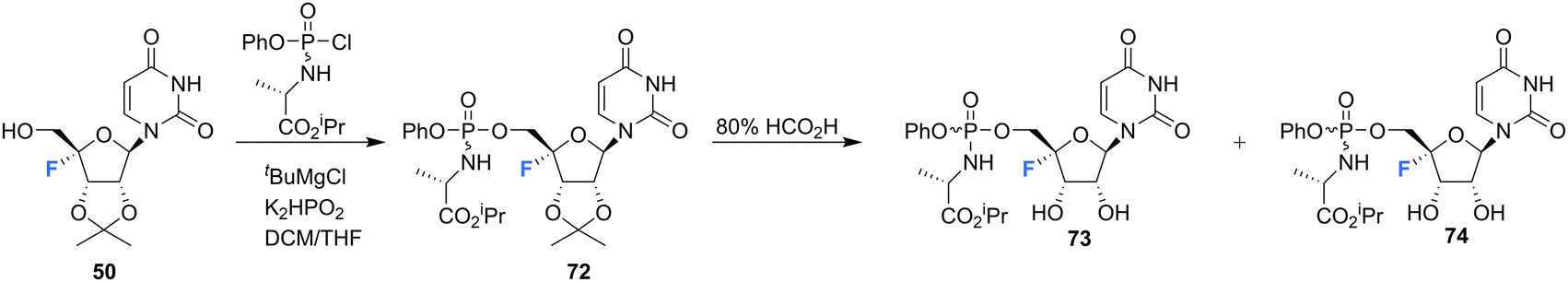 | ||
| Scheme 6 Synthesis of 4′-fluoro-uridine prodrugs 73 and 74 described by Mayes et al.69 | ||
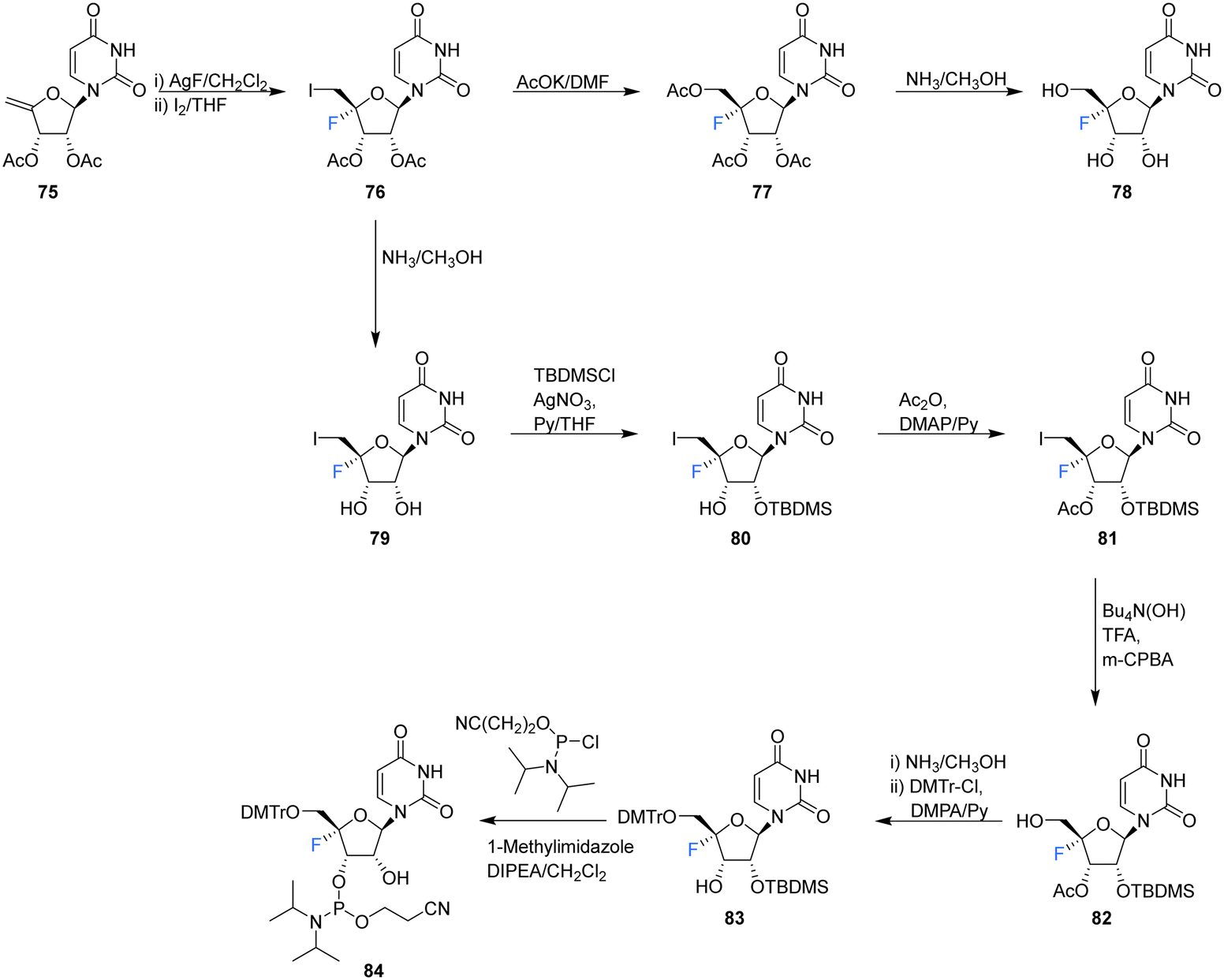 | ||
| Scheme 7 Synthesis of 4′-fluoro-uridine 78 and phosphoramidite derivative 84 as described by Li et al.74 | ||
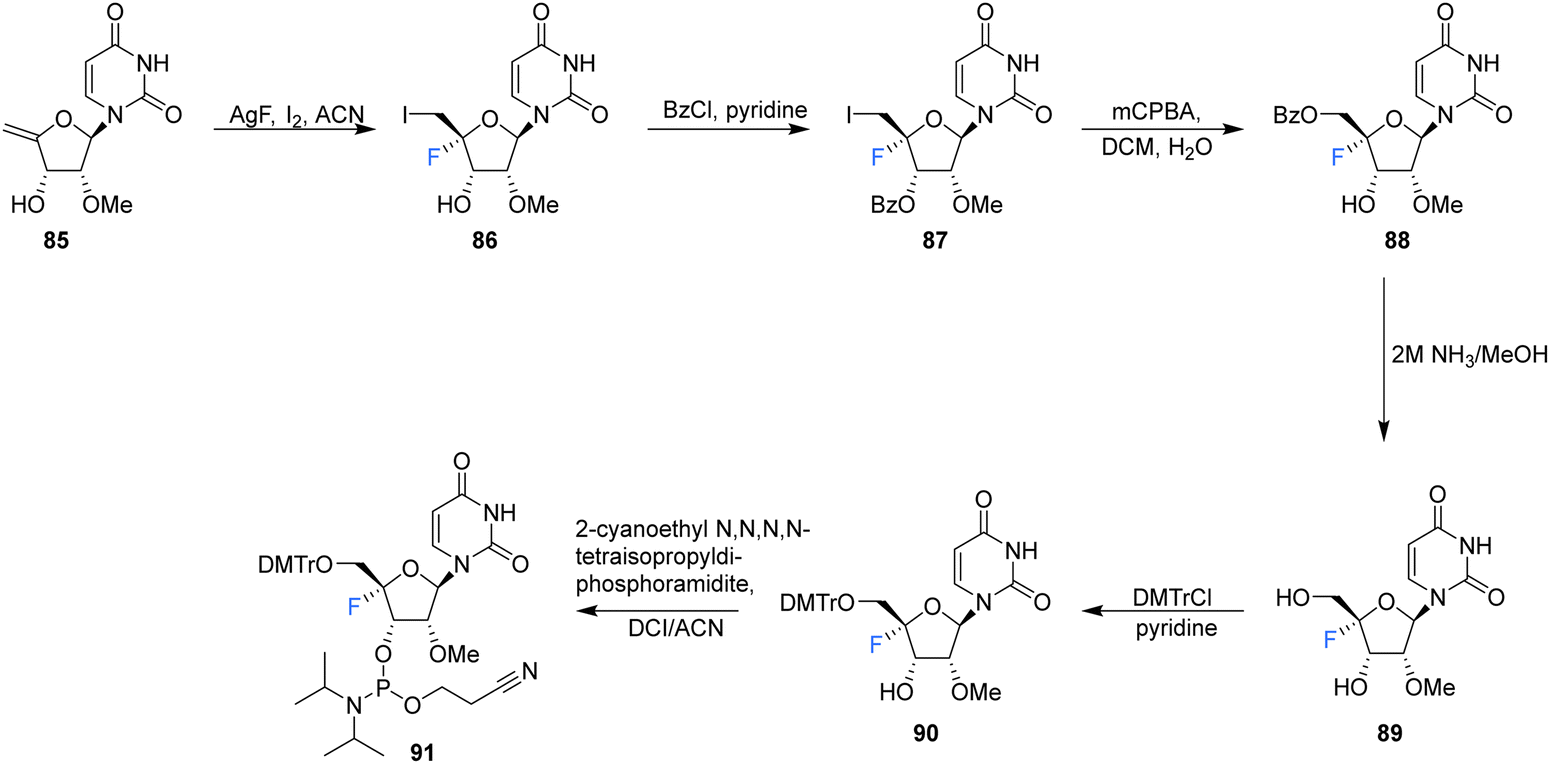 | ||
| Scheme 8 Synthesis of 2′-OMe,4′-fluoro-uridine 89 and phosphoramidite derivative 91 as described by Malek-Adamian et al.76 | ||
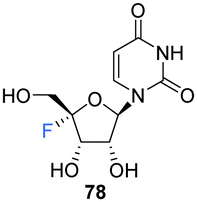
Recently, Li et al., sought to investigate the use of 4′-fluoro-ribonucleosides as 19F NMR probes in studies aiming to elucidate the structure and function of 4′-F-modified RNA.74 With this in mind, the synthesis of phosphoramidite derivative 84 (along with 4′-fluoro-uridine 78) was pursued (Scheme 7). The approach used a similar strategy to that of Owen et al.73 with iodofluorination of 4′,5′-dehydrouridine 75. The desired 4′-fluoro-uridine 78 was obtained through displacement of the 5′-iodine by acetate, followed by deprotection of 77 with methanolic ammonia, although the general instability of 78 did not allow this route to progress further. Inspired by the inferred stability after modification at the 3′-OH of the nucleocidin co-metabolite F-Met II 13, a step wise protection strategy was envisaged. Selective silylation of the 2′-OH using TBDMSCl, followed by acetyl at the 3′-OH afforded 81. Treatment then of 81 with mCPBA under phase transfer conditions offered an efficient transformation to 82. A deprotection/protection strategy and finally phosphitylation at the 3′-OH afforded 84. Phosphoramidite 84 was used to synthesise 4′FU-modified RNA with comparable yields to that of their unmodified counterparts, indicating the stability of 4′FU during RNA synthesis protocols. NMR and biophysical analysis revealed the 4′FU in an RNA strand adopts the typical North-type sugar conformation, with no observable distortion of the RNA structure. 4′FU is capable of being recognised and processed in the same manner as unmodified uridine (by RNase H1 and RNase H2 for instance). The magnitude of the 19F NMR chemical shift was shown to be sensitive to secondary structure (though not sequence context), and therefore can usefully be used to discriminate between changes in the RNA secondary structure. 4′-F-Modified RNA is advantageous over 2′-F-modified RNA for monitoring RNA structural dynamics and enzyme-mediated processing as the 2′-OH group is retained as a key feature of biologically relevant RNA.74
Recently, 4′-fluoro-uridine 78 has been reported by Sourimant et al.,75 to be a candidate broad spectrum antiviral for RNA viruses, showing activity against several strains of respiratory syncytial virus (RSV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) alpha, gamma and delta variants.75 In this study, 4′-Fluoro-uridine triphosphate 68 was assayed in vitro against RSV and SARS-CoV-2 RNA-dependent RNA polymerase (RdRP) and its mode of action was revealed to be via transcriptional stalling after incorporation into the growing oligomer. 4′-Fluoro-uridine 78 was also shown to possess high metabolic stability in human airway epithelial (HAE) cells and to be orally efficacious in animal models (5 mg kg−1, in RSV-infected mice and 20 mg kg−1 in ferrets infected with different SARS-CoV-2 variants).75
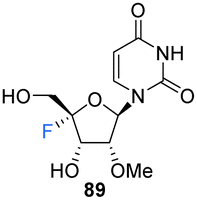
2′-OMe,4′-Fluoro-uridine 89 was prepared Malek-Adamian et al.76 in order to evaluate the effect of fluorine incorporation at the 4′ position of uridine and to investigate the consequences on the subsequent thermal stability of DNA and RNA complexes. Fluorination of 2′-OMe,4′,5′-dehydrouridine 85, prepared from Appel iodination of 2′-OMe-rU followed by elimination with DBU, was accomplished by slow addition of I2 to a suspension of AgF and 85. The resultant 86 was subjected to benzoylation to afford 87, which after treatment with mCPBA generated 88, after migration of the benzoyl group to the 5′-OH position. Treatment of 88 with methanolic ammonia gave the unprotected 2′-OMe,4′-fluorouridine 89, which was converted to its phosphoramidite derivative 91 (Scheme 8). The resultant 91 was incorporated into DNA and RNA oligonucleotides. As expected, computational studies on 2′-OMe,4′-fluoro-uridine suggested that it adopted a North sugar pucker, influenced by both electronic and steric effects. The 2′-OMe,4′-fluoro-uridine analogue modulated the binding affinity of parent 2′-modified homo and heteroduplexes and was found to destabilise DNA:DNA duplexes, although it had a slightly stabilising effect on DNA:RNA hybrids and RNA:RNA duplexes. These observations for DNA:RNA duplexes in particular allow a potential use in CRISPR/Cas9 technologies. The authors proposed application for siRNA and guide RNAs, allowing minimal disruption of structure and thermal stability upon incorporation, while allowing the introduction of this foreign nucleotide to serum nucleases and immunostimulatory receptors. The effect of the 2′-OMe,4′-fluoro-uridine analogue 89 on the gene silencing activity of siRNA duplexes has been explored.77 siRNA modified with 89 was transfected into HeLa cells and was shown to silence both firefly luciferase and renal cell carcinoma (DRR) gene targets, especially when located at the 3′-overhang of the guide strand.77 The authors hypothesised that enhanced nuclease stability along with a favourable interaction with the PAZ domain of human argonaute-2 protein improved the activity of the modified siRNA.
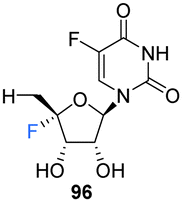
5′-Deoxyfluorouridine has been investigated as a prodrug to 5-fluorouracil, a cytotoxic chemotherapy medication used to treat a variety of cancers.79–81 The lack of a 5′-hydroxyl on 5′-deoxyfluorouridine prevents its intracellular conversion to nucleotides and its cytotoxicity is dependent on enzyme mediated (uridine phosphorylase) cleavage of its glycosidic bond. In an effort to improve on the design of this prodrug, Ajmera et al., sought to obtain a derivative with a greater Vmax for glycosidic bond cleavage by uridine phosphorylase, such that it would enhanced the rate of accumulation of 5-fluorouracil in tumour tissues.78 Ultimately a strategy utilising benzyl protection provided a suitable route to the targeted 5′-deoxy-4′,5-difluorouridine 96, see Scheme 9. Briefly, 96 was prepared through benzylation and then trityl deprotection and mesylation at C5′ of 92. Treatment of mesylate 93 with potassium tert-butoxide afforded 4′-exo-olefin 94, which was then hydrofluorinated with pyridinium poly(hydrogen fluoride) to generate 95 in very good yield, free from the α-L-lyxo isomer. Hydrogenation of 95 with Pearlman's catalyst in anhydrous dioxane furnished 5′-deoxy-4′,5-difluorouridine 96. In the event fluorouracil base release from uridine 96 was approximately 500 times more rapid than from the non-fluorinated analogue under acidic conditions, although it showed reasonable stability at neutral pH. The Vmax for hydrolysis by uridine phosphorylase was 5-fold greater than that for 5′-deoxyfluorouridine, and a Km 10-fold lower, thus a Vmax/Km ratio 50-fold greater. Preliminary data revealed 5′-deoxy-4′,5-difluorouridine 96 was able to inhibit the growth of L1210 cells (mouse lymphocytic leukemia cell line), in line with the favourable uridine phosphorylase catalysed hydrolysis and release of the cytoxic parent drug, 5-fluorouracil.78
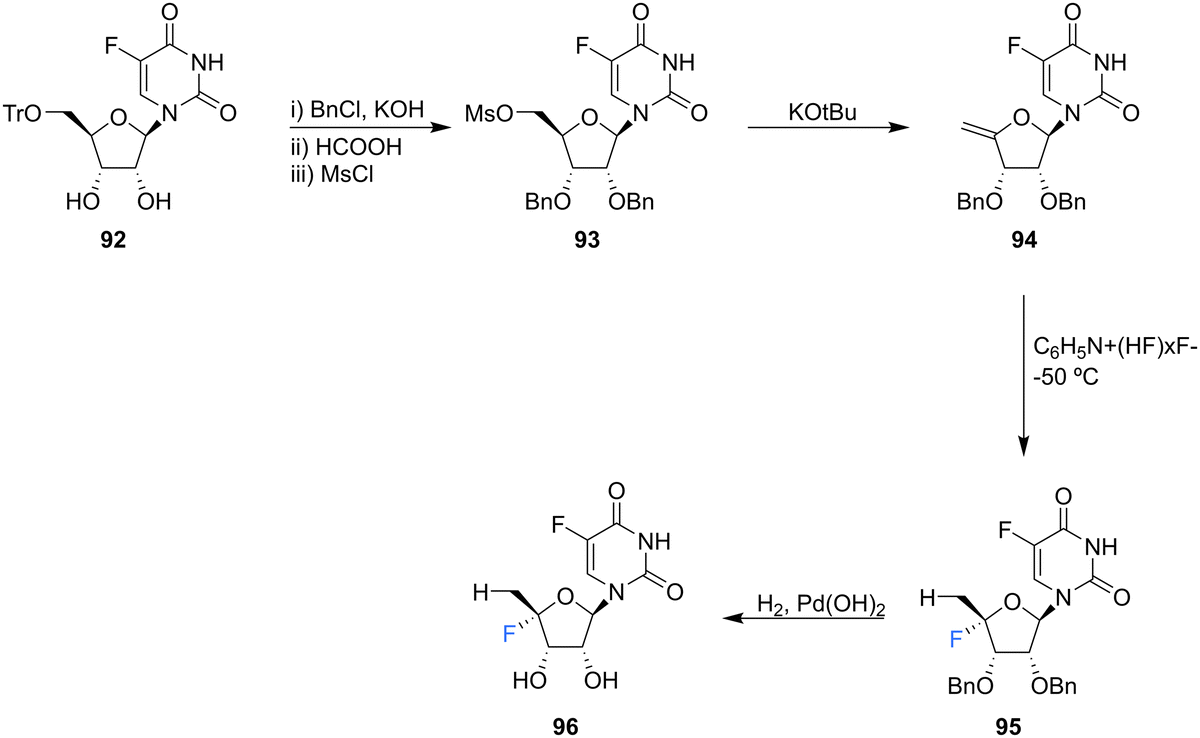 | ||
| Scheme 9 Synthesis of 5′-deoxy-4′,5-difluorouridine 96 as described by Ajmera et al.78 | ||
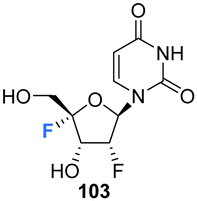
The syntheses of 2′,4′-difluorinated nucleoside analogues, 2′-deoxy-2′,4′-difluoro-uridine (2′,4′-diF-rU) 103 and 2′-deoxy-2′,4′-difluoro-cytidine (2′,4′-diF-rC) 107 were accomplished by Martínez-Montero et al., in 2014 using a common synthesis strategy.64 2′-Deoxy-2′-fluoro-uridine 97 was iodinated under Appel conditions and the resulting iodo-adenosine 98 was then susceptible to elimination in the presence of sodium methoxide, followed by iodofluroination after gradual addition of iodine/AgF. The route is illustrated in Scheme 10. The 5′-iodine was then displaced with mCPBA to afford 102 after benzoylation at the 3′ position, a modification which appears to assist displacement through migration to the 5′ position. Deprotection of 102 gave 2′,4′-diF-rU 103. The cytidine analogue 2′,4′-diF-rC 107 was accessed after conversion of uracil 102 to a cytosine by benzoyl protection followed by installation of an amine at the 4 position of the base via a triazole intermediate and deprotection with ammonia. A combination of NMR and computational studies confirmed that installation of a 4′-fluorine onto 2′-fluoro-uridine provides a tool, driven by stereoelectronic effects, to promote a strong conformational “lock” toward a pure North-type conformation.64 Previous attempts to enforce such a “lock” had required a bicyclic framework linking the 2′- and 4′-positions at the ribofuranose sugar. 2′,4′-DiF-rU 103 revealed a minimally destabilising character when studied in DNA:RNA duplexes, offering potential for gene editing technologies76 The triphosphate of 2′,4′-diF-rU, 108, was acquired commercially by custom synthesis and evaluated as a substrate for HCV NS5B RNA polymerase. 2′,4′-DiF-rUTP 108 was found to inhibit RNA synthesis primarily at the at the level of initiation in dinucleotide-primed reactions, with an IC50 of 54.7 μM.
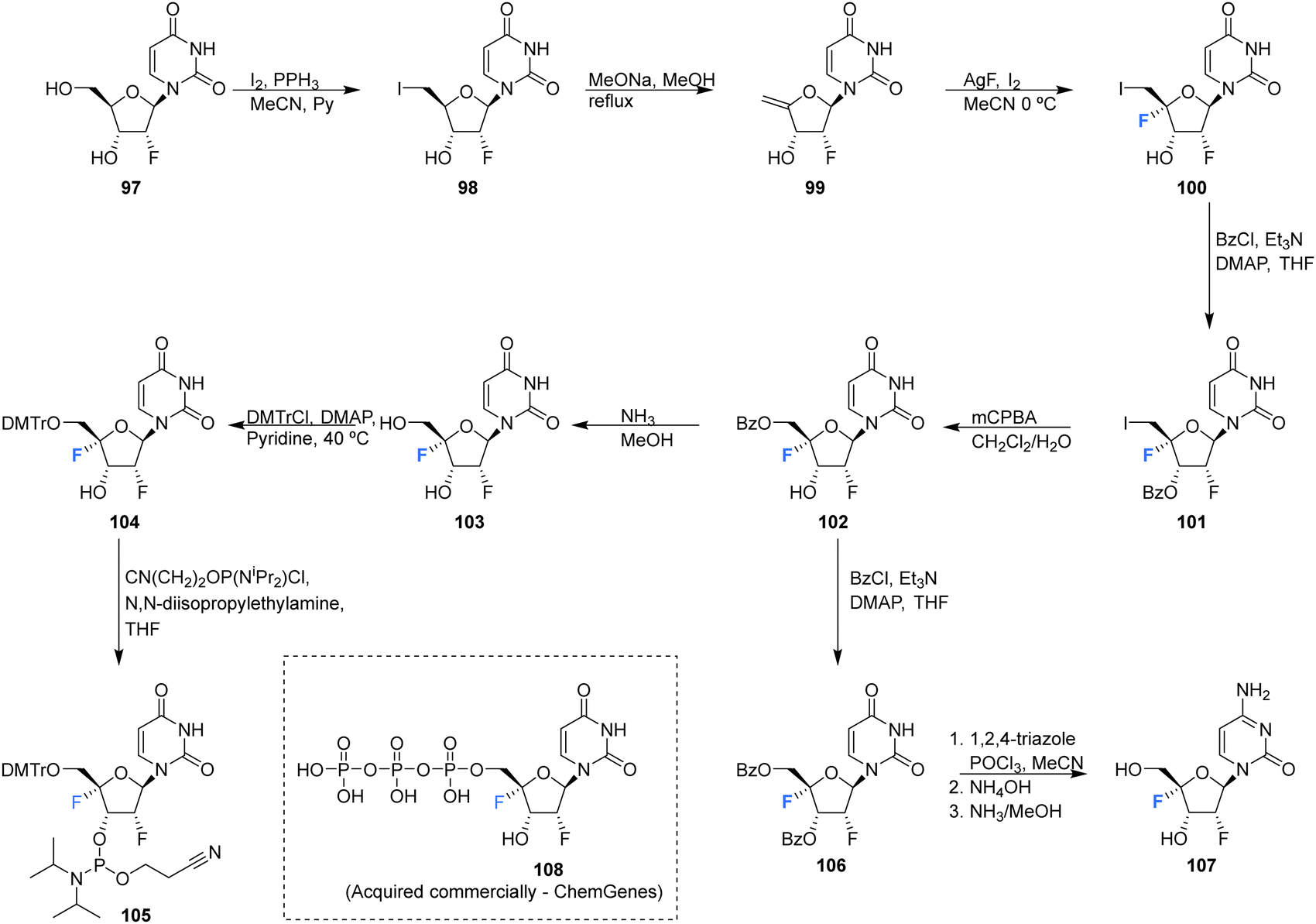 | ||
| Scheme 10 Synthesis of 2′-deoxy-2′,4′-difluoro-uridine (2′,4′-diF-rU) 103, 2′,4′-deoxy-2′,4′-difluoro-cytidine (2′,4′-diF-rC) 107 and phosphoramidite 105 as described by Martínez-Montero et al.64,82 | ||
The phosphoramidite derivative of 103 was prepared by Martínez-Montero et al., in 2015 following standard procedures as summarised in Scheme 10.82 Protection of the 5′-hydroxyl of 103 using DMTr chloride generated 104, which was then subjected to phosphitylation using ClP(OCEt)N(iPr)2 to furnish phosphoramidite 105. Solid phase synthesis was then used to synthesise DNA and RNA oligonucleotides containing 2′,4′-DiF-rU 103 units, with reported coupling yields of ∼80% for phosphoramidite 105. UV thermal duplex denaturing studies, along with molecular dynamics simulations, and NMR observations, revealed the North conformation adopted by 2′,4′-DiF-rU 103 was maintained in DNA and RNA oligonucleotide duplex structures. The 2′,4′-diF-RNA modification generated greater distortions in DNA:RNA duplexes when compared to 2′-F-RNA, and was not well tolerated in DNA:DNA duplexes. This modification was met with a relatively unique neutral response when incorporated into RNA:RNA duplexes, as it is capable of reinforcing the North sugar conformation without the increase thermal stability associated with the incorporation of locked bicyclic nucleoside analogues.82 Such properties are particularly advantageous for siRNA-mediated gene silencing applications.83
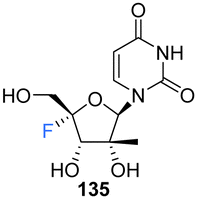
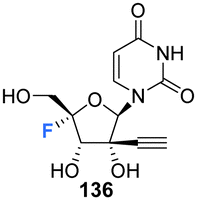
2′-C-Modified-4′-fluoro-uridine analogues 135–137, along with their phosphoramidate 141–146 and triphosphate deriviatives 138–140, were prepared by Wang et al., to investigate their use as inhibitors of dengue virus (DENV) RNA-dependent RNA polymerase (RdRp).84 Firstly, the 2′-C-modified uridines were synthesised from 109, involving Dess-Martin oxidation and subsequent treatment with the appropriate Grignard reagent to acquire 111–113, as shown in Scheme 11a. Conversion of the 2′-OH to its benzoate, followed by coupling with uracil, under Vorbrüggen glycosylation conditions and deprotection, furnished 2′-C-modified-uridines 117–119. 2′-C-Modified-4′-fluoro-uridine analogues 135–137 were then acquired from 117–119, using the general strategy of fluorinating 2′-C-modified-4′,5′-olefins 123–125, followed by oxidative hydrolysis and deprotection to afford 135–137, as shown in Scheme 11a. Triphosphates 138–140 were synthesised using a single step strategy and the phosphoramidate prodrug analogues 141–146 were acquired using the phosphoryl agents as shown in Scheme 11b and c.
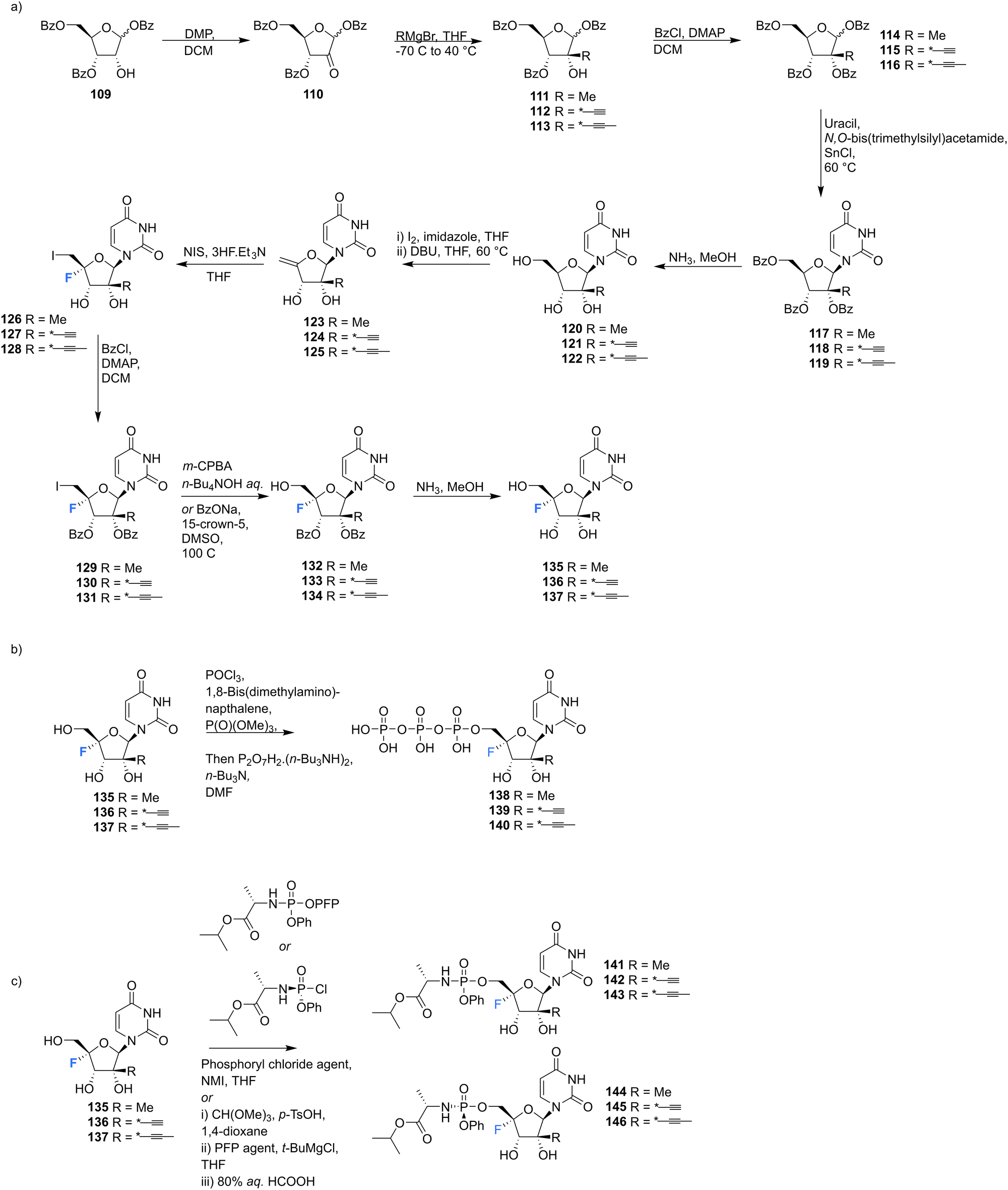 | ||
| Scheme 11 Synthesis of 2′-C-modified-4′-fluoro-uridine analogues 135–137, triphosphates 138–140 and phosphoramidates 141–146 as described by Wang et al.84 | ||
135–137 and their analogues were assayed for their ability to both inhibit RNA replication by DENV RNA-dependent RNA polymerase (RdRp) and to be incorporated into a host mitochondrial DNA-dependent RNA polymerase (POLRMT), which potentually leads to mitochondrial dysfunction in vivo. 2′-C-Modified-4′-fluoro-uridine analogues 135–137 were found to be inactive against dengue virus in cell-based assays. 4′-Fluorination had an overall less deletous effect on RdRp IC50 than 2′-fluorination, whilst reducing POLRMT single nucleotide incorporation rate (SNIR). Triphosphate 139 exhibited an SNIR by POLRMT at background levels, whilst effecting a 2-fold improvement in DENV RdRp inhibition when compared to its non fluorinated derivative. Triphosphate 139 was also shown to be a poor substrate for mitochondrial DNA polymerase and the phosphoramidate prodrug 145 was revealed to be a promising candidate as it displayed potent anti-DENV cellular activity (EC50 = 0.57 μM) whilst showing no cytotoxicity in any cell lines tested and it did not effect mitochondrial protein synthesis in prostate metastatic carcinoma cells (CC50 > 200 μM). Thus this study revealed that 4′-fluorination of 2′-C-modified uridine analogues can lead to phosphoramidate prodrugs with significantly reduced SNIR by POLRMT and a maintained ability to inhibit DENV RdRp inhibition.84
2′-C-Substituted-4′-fluoro-uridine analogues 157, 158, 135, 168, 170 and 179–182 were synthesised and assayed for HCV NS5B inhibition, host polymerase inhibition, and HCV replicon activity.35 To access 154, the synthesis began with PMB protection of the uracil base of 147 and benzyl protection of the ribose hydroxyls to afford 149. Oxidative cleavage and reduction of 149 afforded 150, which was subjected to fluorinatation, via mesylation, with TBAF. Deprotection of 151 generated 152, which was used to generate olefin 154 and this was progressed in a similar manner to analogues 155 and 156, to generate the 4′-fluorinated anologues 157, 158 and 135, as summarised in Scheme 12.
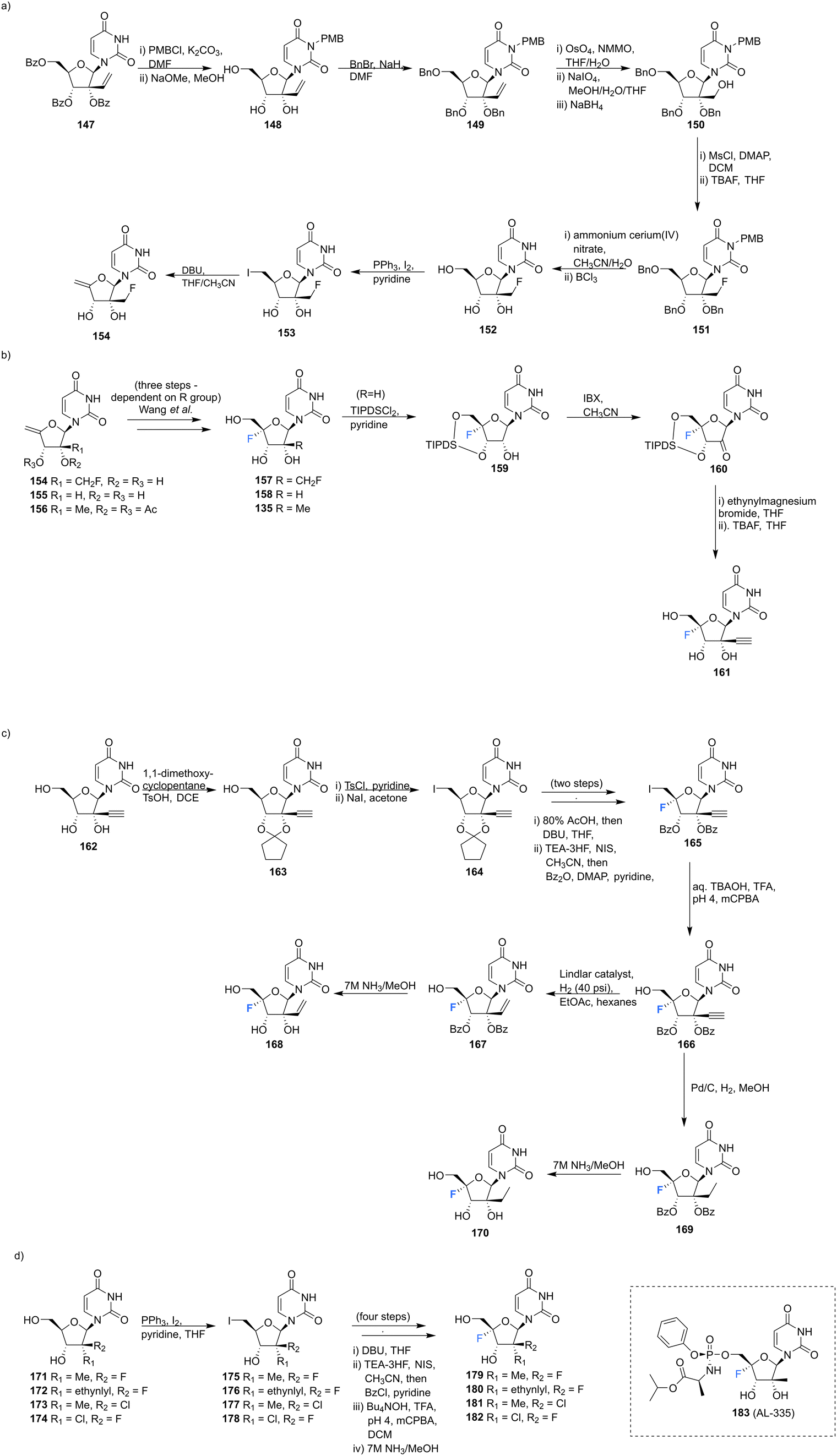 | ||
| Scheme 12 Synthesis of 2′-C-modified-4′-fluoro-uridine analogues 157, 158, 135, 168, 170, 179–182 and 183 (AL-335) as described by Wang et al.35 | ||
3′-Ethynyl-5′-fluoro-uridine 161 was prepared from 158 by generating ketone 160 and subjecting this to a Grignard reaction, followed by subsequent desilylation. 2′-C-Substituted-4′-fluoro-uridine analogues 168 and 170, were acquired from 162. Cyclopentylidene protection of the 2′ and 3′ hydroxyls, followed by iodination at the 5′ position afforded 164. The established strategy of olefin generation then iodofluorination followed by hydroxydeiodination generated 165, which was progressed to either 168 or 170 by controlled hydrogenation followed by hydrolysis. Uridine analogues 179–182 were then synthesised from 171–174 folowing established routes as summarised in Scheme 12.35 Several uridines generated in this study were explored as their 5′-triphosphate derivatives in viral and human polymerase assays. Many of these were shown to be potent inhibtors of HCV NS5B polymerase (most notably the triphosphate of 135 and 181 with IC50 values of 0.14 and 0.11 μM respectively), and showed little or no inhibition of human DNA and RNA polymerases. The 5′-triphosphate of 135 was used to investigate the mechanism of inhibition and was shown to be effective at terminating chain elongation by HCV NS5B polymerase. These results indicate that fluorination at the 4′-position does not alter the nucleosides inhibitory properties of NS5B dramatically and can lead to more potent NTPs targeting this polymerase. A range of 5′-phosphoramidate and 5′-bisphosphoramidate derivatives of uridines 157, 158, 135, 168, 170 and 179–182 were used in cell-based assays. From this study the 5′-phosphoramidate analogue of 135, 183 (AL-335), was established as a lead compound, demonstrating high potency in HCV subgenomic replicon assays (EC50 = 0.07 μM) and a promising cytotoxicity profile (CC50 > 96 μM in six cell lines). In light of its excellent in vitro and in vivo properties, 183 was advanced to clinical development where it showed promising results in Phase 1 and 2 trials.35
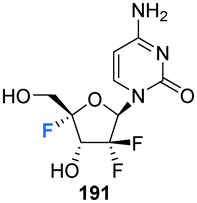
The 4′-fluoro derivative of gemcitabine 184, an antitumor drug with broad spectrum activity against RNA viruses, along with its phosphoramidate prodrug was recently synthesised by Zheng et al., and assessed for its antiviral activity against the varicella zoster virus (VZV), the human cytomegalovirus (HCMV) and the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).85 The rational being that modifcation at the 4′-position will increase its specificity as an antiviral agent by reducing its activity against host ribonucleotide reductases and polymerases.
4′-Fluoro-gemcitabine 191 was acquired from gemcitabin 184 in a route summarised in Scheme 13. It began with a three step sequence involving functional group protections, nucleobase deamination, and O-debenzoylation to obtain 187. 187 was then modified using established methods to install the 4′-fluoro motif using AgF and restore the 5′-O functionality with m-CPBA to generate protected 4′-fluoro-uridine 189. Deprotection of 189 generated the 4′-fluoro-uridine analogue 192 whilst protection along with nucleobase conversion generated 4′-fluoro-gemcitabine 191. The prodrug 195 of 4′-fluoro-gemcitabine was synthesised by selective protection at the 3′-position followed by treatment with phenyl aminoacyl phosphorochloridate and Boc deprotection, as illustrated in Scheme 13. 4′-Fluoro-gemcitabine 191 was observed to exhibited potent activity against VZV, HCMV, and SARS-CoV-2. Notably, when assayed against VZV the EC50 was 0.042 μM, although it displayed significant cytotoxicity (CC50 = 0.11 μM). The prodrug 195 showed reduced anti-VZV activity, though with an improved selectivity index (SI = 36). A similar, although slightly reduced, effect was observed against HCMV. When assayed against SARS-CoV-2 it displayed comparable antiviral activity (EC50 = 0.73 μM) to its cytotoxic concentration in measurements of cell growth.85
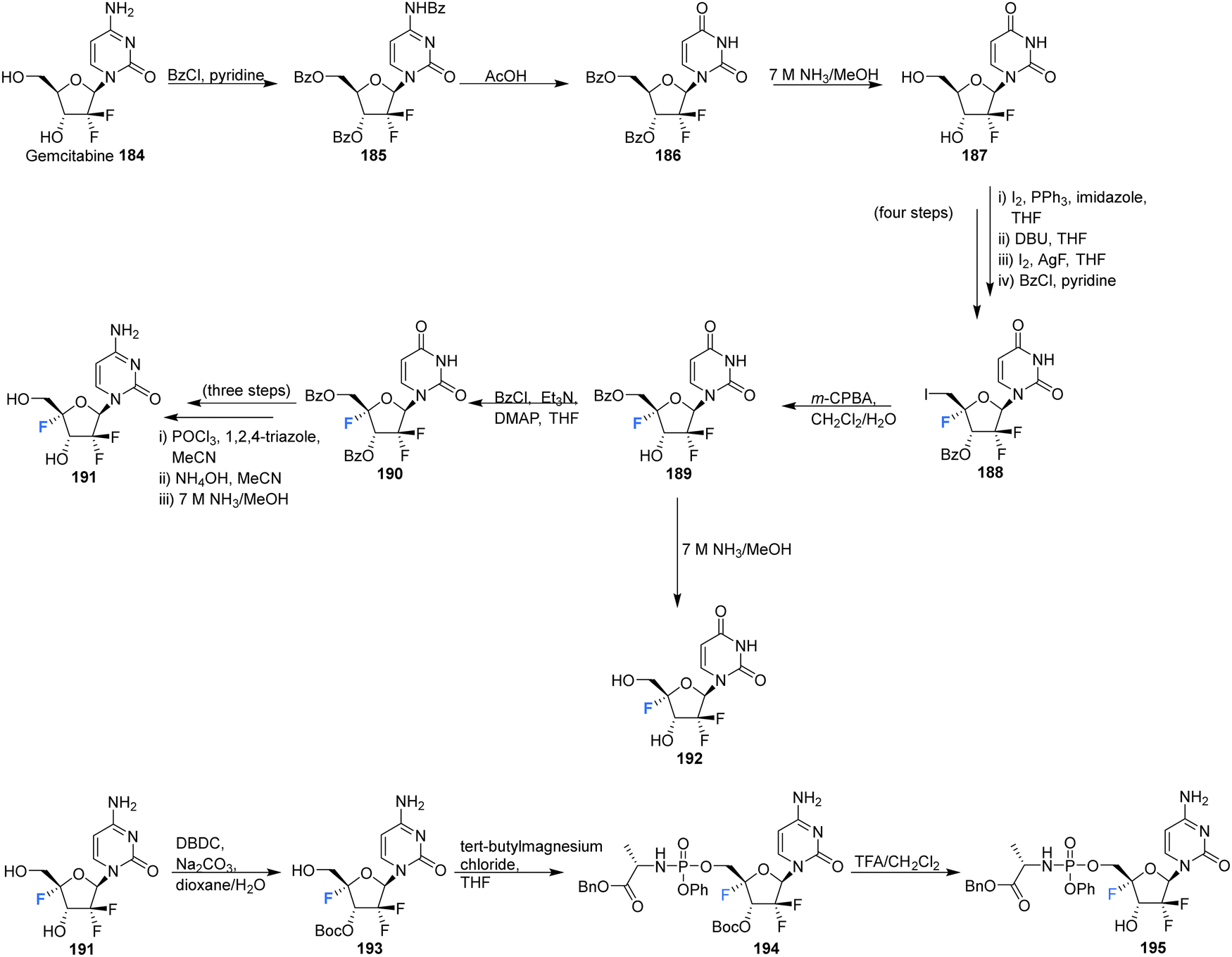 | ||
| Scheme 13 Synthesis of 4′-fluoro-gemcitabine 191, 2′-difluoro-4′-fluoro-uridine 192 and phosphoramidate 195 as described by Zheng et al.85 | ||
5.4 4′-Fluoro-deoxythymidines
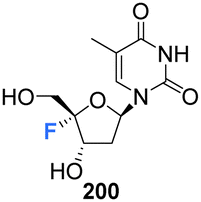
Zhou et al. prepared 4′-fluoro-deoxythymidine 200 and the phosphoramidite 201, as illustrated in Scheme 14,65 using a similar strategy to that for the synthesis of 4′-fluoro-uridine 78 and its phosphoramidite derivative 84. 4′-Fluoro-deoxythymidine 200 was found to adopt a North-type ribose ring pucker, which persisted when introduced to oligodeoxyribnucleotides (ODNs). This conformational tendency was maintained in ODN/cDNA and ODN/cRNA duplexes. The half-life for decomposition for 4′-fluoro-deoxythymidine 200 in pure water was recorded as 4 h, shorter than 4′-fluoro-uridine 81 (t1/2 = 9 h). 4′-Fluoro-deoxythymidine 200 modified ODNs were found to be stable in neutral buffers, although the glycosidic bond of 200 was prone to scission under alkaline conditions. Through techniques such as circular dichroism spectroscopy, thermal denaturing and RNase H1 footprinting studies, 4′-fluoro-deoxythymidine 200 has been shown to resemble 2′-fluoro-deoxyribonucleotides but it imparts less structural perturbation to ODN/cDNA and ODN/cRNA duplexes, thus 4′-fluoro-deoxythymidine 200 represents a good 19F-NMR probe for investigating RNA secondary structure and function.65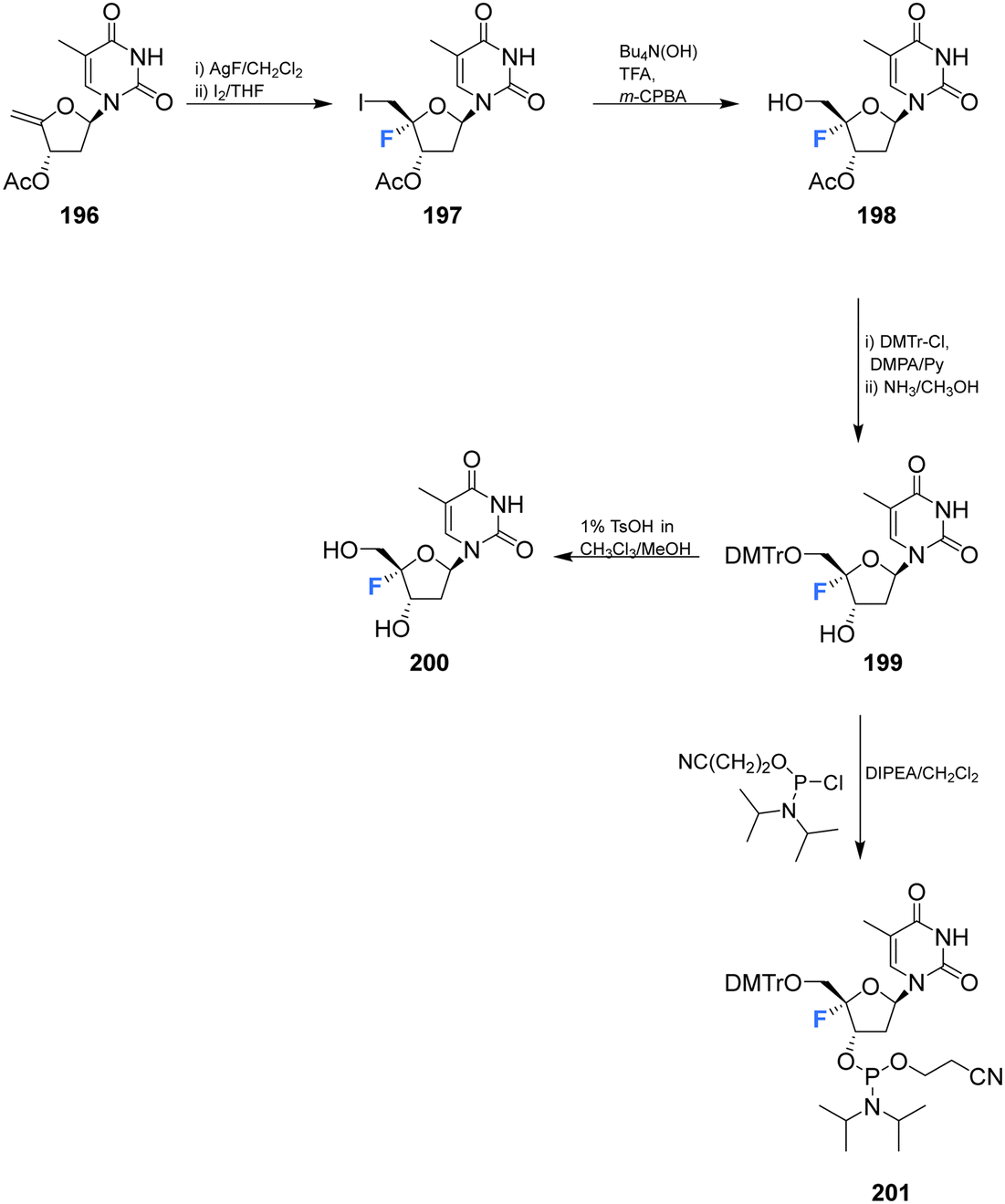 | ||
| Scheme 14 Synthesis of 4′-fluoro-deoxythymidine 200 and phosphoramidite derivative 201 as described by Zhou et al.65 | ||
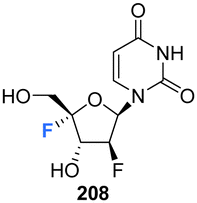
5.5 2′-Deoxy-2′,4′-difluoro-arabinouridines
2′-Deoxy-2′,4′-difluoro-arabinouridine (2,′4′-diF-araU) 208 was synthesised in a similar manner to that of 103 and 200, but commencing with 2′-deoxy-2′-fluoro-arabinouridine 202, an epimer of 97, as shown in Scheme 15.86 Incorporation into oligonucleotide duplexes required activation to phosphoramidite 210. Briefly, 208 was treated with DMTr chloride to generate 209 and then subsequent phosphitylation with ClP(OCEt)N(iPr)2 afforded 210 enabling it's use in solid-phase oligonucleotide synthesis. 2,′4′-DiF-araU 210 was found to be well tolerated in DNA:RNA hybrid oligonucleotides, providing some improvement in thermal stability. The effect of the North-type pucker adopted by 2,′4′-diF-araU 210 was observed in RNase H recruitment studies, where cleavage sites differed from the unmodified duplexes and degradation rates for the RNA strand were lower. Such results are thought to be promising for gene silencing applications.86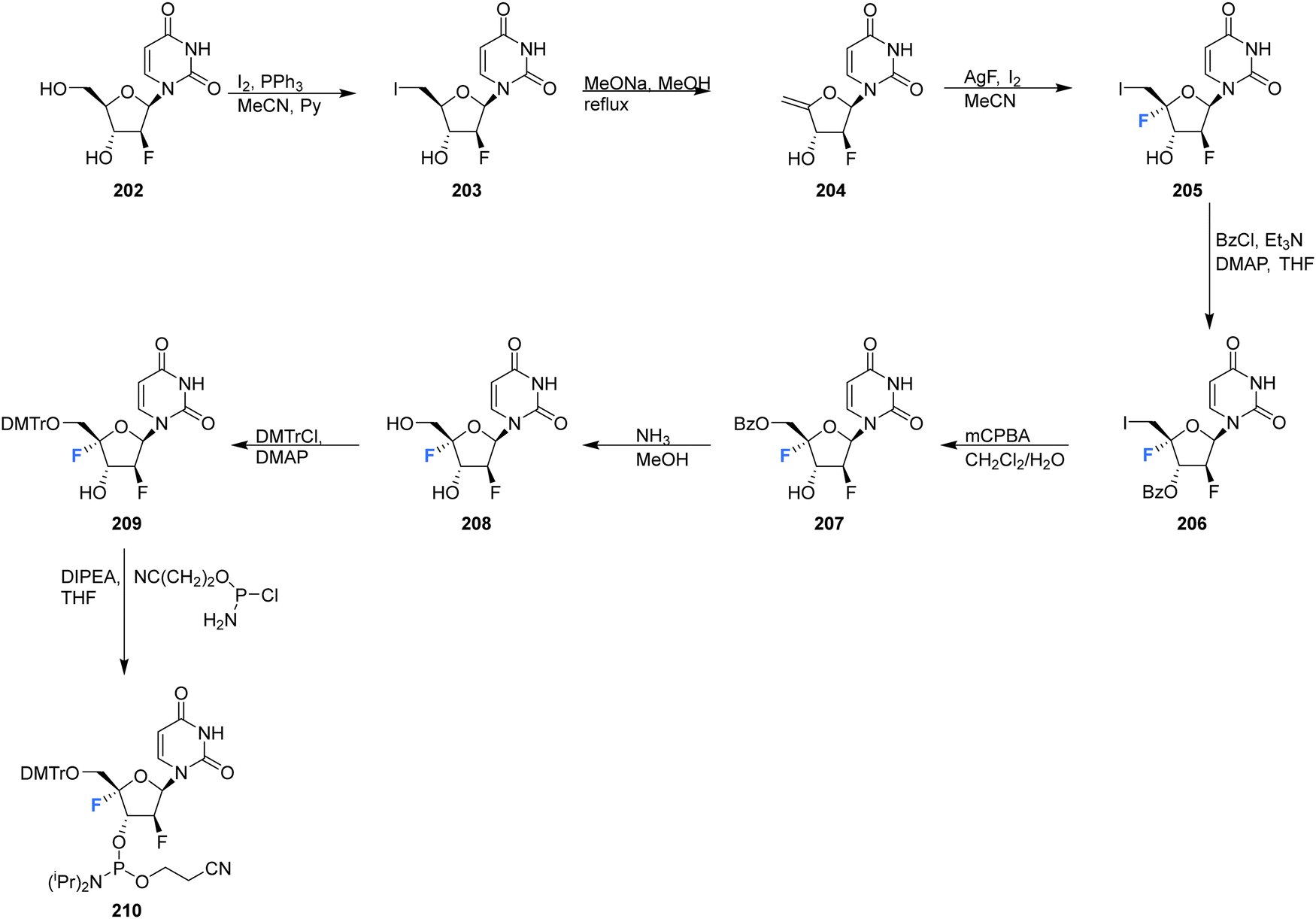 | ||
| Scheme 15 Synthesis of 2′-deoxy-2′,4′-difluoro-arabinouridine (2,′4′-diF-araU) 208 and phosphoramidite derivative 210 as described by Martinez-Montero et al.86 | ||
5.6 3′,4′-Difluoro-3′-deoxyribonucleosides.
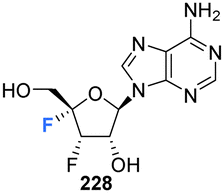
Shimada et al., prepared several 3′,4′-difluoro-3′-deoxyribonucleosides using a strategy involving di-fluorination of 3′,4′-dehydro cytosine and adenine nucleosides with XeF2/BF3·OEt2.66 Briefly, for 215, 216 and 219, protected nucleosides 212 and 217 were first acquired through de-acylation and silylation of 211, and then treated with XeF2 and BF3·OEt2 in Et2O at 0 °C. In order to separate the stereoisomers produced, the silyl-protecting groups were exchanged for acetates, and this allowed for isolation of α/β-anti/syn adducts as illustrated in Scheme 16. The α/β preferrence in these di-fluorination reactions was rationalised by the shielding of the α face by the sterically demanding protecting groups. Finally, deprotection of 213, 214 and 218 using NaOMe in MeOH furnished 3′,4′-difluoro-3′-deoxyribonucleosides 215, 216 and 219. Access to the difluorinated adenine nucleosides 223, 224, 228 and 229 was achieved in a similar manner by di-fluorination of 220 and 225, which, after N6-pivolyation, de-acylation and silylation allowed the isolation of stereoisomers 221, 222, 226 and 227. Deprotection then provided the di-fluorinated adenosines 223, 224, 228 and 229. The stereochemical outcome of the adenine derivatives was largely consistent with that observed for the 3′,4′-difluoro-3′-deoxycytidines, although the authors noted that the α-syn-adduct selectivity was unexpected after fluorination of 225, suggesting a role for the sterically demanding adenine base. 3′,4′-Difluoro-3′-deoxyadenosines 223, 224, 228 and 229 were assayed for activity against murine leukemia cells and human cervix carcinoma, although none showed any inhibitory activities. Additionally, no antiviral activity was observed for 223, 224, 228 and 229 against human cytomegalovirus, herpes simplex virus, vesicular stomatitis virus and varicella-zoster-virus, though no cytotoxicity against the host cells (at 100 μM) was observed either. Promisingly, 3′,4′-difluoro-3′-deoxyadenosine 228 exhibited inhibitory activity in anti-HCV assays (EC50 = 4.7 μM). Interestingly the parent candidate, cordycepin, a non-fluorinated analogue which has shown antitumour activity, exhibited no such significant anti-HCV activity, whilst 3′-fluoro-cordycepin exhibits anti-HCV activity (EC50 = 1.2 μM) but is highly toxic to host cells. Thus the incorporation of fluorine to the 4′-position of 3′-fluoro-cordycepin can be rationalised to have decreased its cytoxicity, whilst retaining its inhibitory activity against HCV.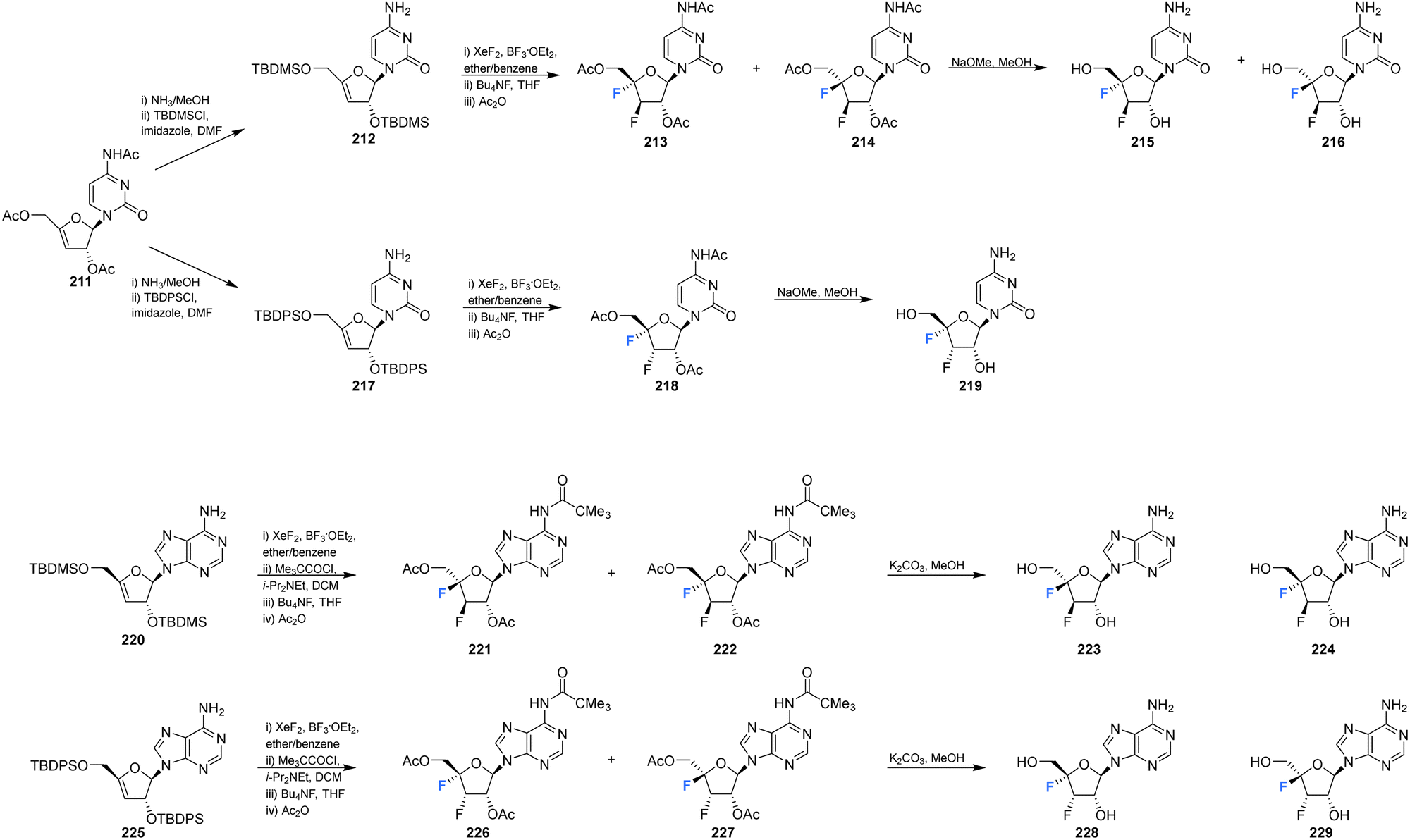 | ||
| Scheme 16 Synthesis of 3′,4′-difluoro-3′-deoxycytidines 215, 216 and 219 and 3′,4′-difluoro-3′-deoxyadenosines 223, 224, 228 and 229 as described by Shimada et al.66 | ||
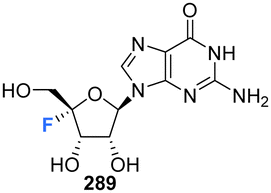
5.7 4′-Fluoro-guanosines
4′-Fluoro-guanosine prodrugs 235, 236 and 238, reported in the patent literature by Mayes et al.,69 were synthesised using a strategy outlined in Scheme 17. Protected guanosine 231 was generated from 230 in six steps using established methods. Reaction of 231 with mCPBA followed by n-butyl amine generated 233, which was then coupled with the appropriate chlorophosphoramidate to afforded 235, 236 and 238, after deprotection. These compounds were then purified by semi prep HPLC. Prodrugs 235, 236 and 238 were assayed for HCV replicon activity and cytotoxicity Diastereoisomers 235 and 236 both demonstrated CC50 values with the range of 1–10 μM, though differed substantially with regards to EC50 with 235 having a value >10 μM and 236 between 250 nM–1 μM. Single isomer 238 demonstrated a CC50 value within the range of 1–10 μM and a EC50 > 10 μM. The pharmacokinetics of these prodrugs were also studied for each compound measured by LC-MS/MS in mouse liver samples. 235 and 238 were found to accumulate in these studies, significantly less so when compared to 73 and 74 (uracil analogues reported in the same study, Scheme 6), and 236 was not detected at all. The triphosphate 239 was also synthesised and was assayed for inhibitory activity against purified HCV polymerase, demonstrating an IC50 ≤ 250 nM.69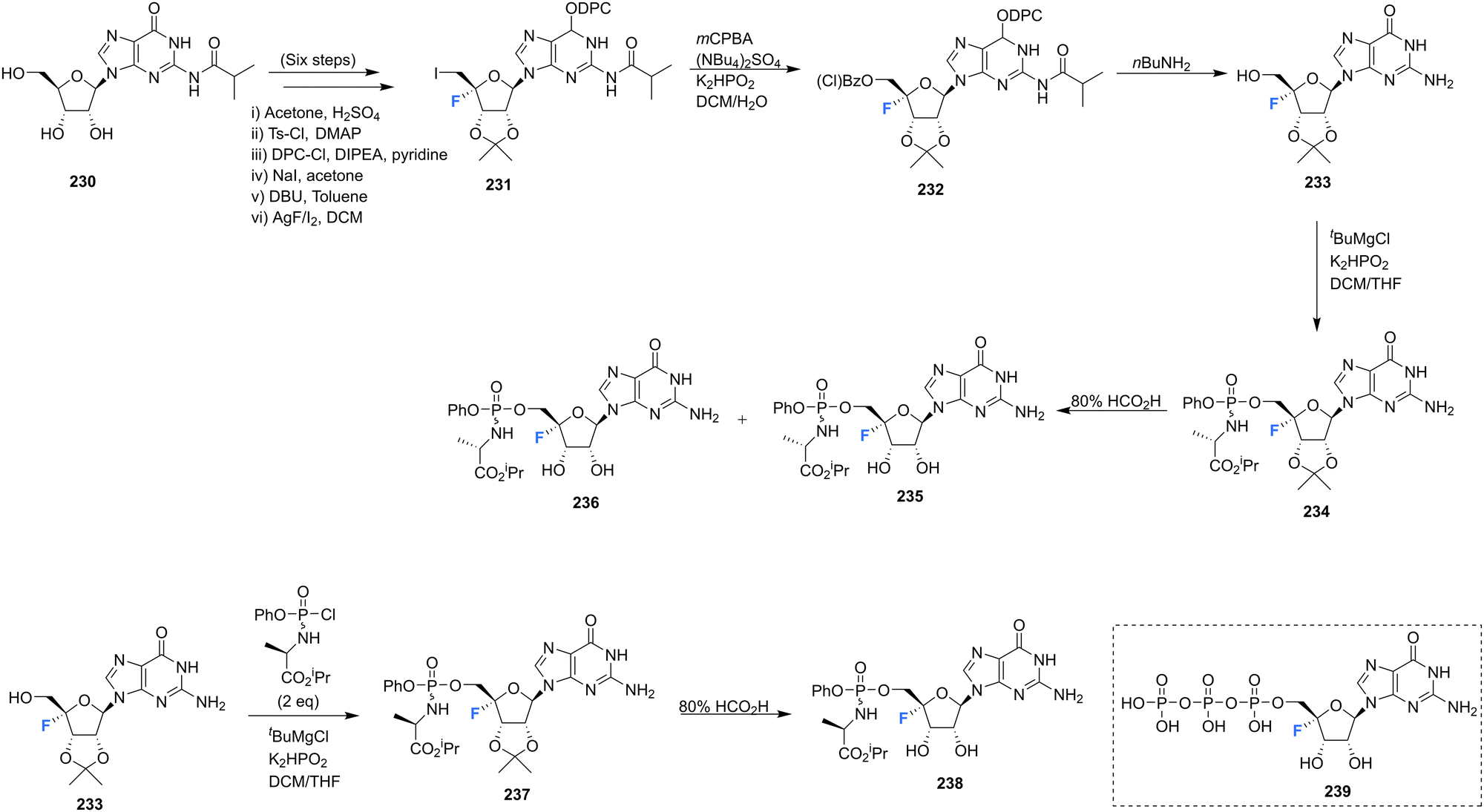 | ||
| Scheme 17 Synthesis of 4′-fluoro-guanosine prodrugs 235, 236, 238 and 5′-O-triphosphate 239 synthesised by Mayes et al.69 | ||
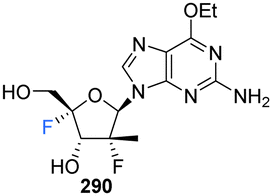
A series of 2′-C-substituted-4′-fluoro-guanosine analogues were also prepared by Wang et al., and assayed for HCV NS5B inhibition, host polymerase inhibition, and HCV replicon activity (Scheme 18).87260–263 were prepared using already established procedures from N2-monomethoxytrytilated guanosines 240–243. The 5′-O-triphosphate derivatives of each 2′-C-substituted-4′-fluoro-guanosine were also synthesised and assayed against HCV NS5B and human polymerases. Each 2′-C-substituted-4′-fluoro-guanosine 260–263 was a potent inhibitor of the HCV NS5B polymerase, with the triphosphate of 261 revealing an increased potency (IC50 = 0.14 μM) when compared to its non-fluorinated analogue (IC50 = 0.42 μM) and the triphosphate of 263 (IC50 = 0.16 μM) a comparable potency relative to its non-fluorinated analogue (IC50 = 0.099 μM). Chain termination assays indicated that the triphosphate of 263 induced immediate chain termination. Triphosphates of 260–262 did not demonstrate any inhibition of human DNA polymerases α, β, and γ and RNA polymerase II. The triphosphate of 263 showed low level inhibition of human DNA pol-α and no inhibition of human DNA polymerases β, γ and RNA polymerase II.87
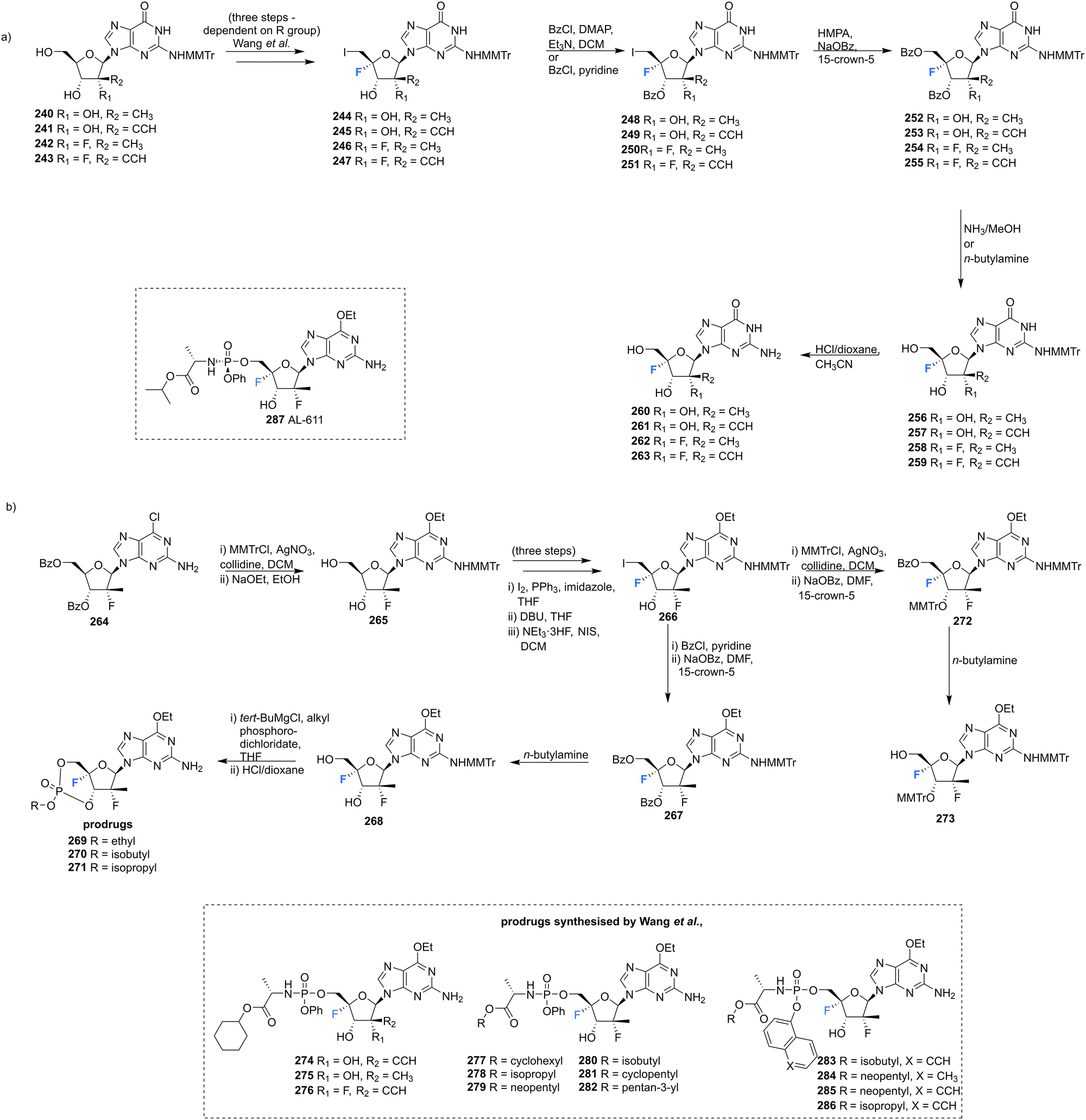 | ||
| Scheme 18 Synthesis of 2′-C-modified-4′-fluoro-guanosine analogues 260–263, cyclic phosphates 269–271 and phosphoramidates 274–286 as described by Wang et al.87 | ||
In order to effectively test in cell-based assays, and to circumvent poor in vivo, monophosphorylation, nucleosides 269–271 and 274–286 were synthesised as 6-OEt monophosphate prodrugs. Monomethoxytritylation of the amino group of 264, followed by fluorine installation at the 4′-position provided 266. 266 was then progressed by nucleophilic substition of the iodine, followed by protecting group protections/deprotections to furnish 268 and 273. 268 was used to acquire cyclic phosphates 269–271 using the appropriate phosphodichloridate, and 273 was used to prepare the phosphoroamidate prodrugs 277–286. 5′-Phosphoroamidate prodrugs of 2′-C-substituted-4′-fluoro-guanosine analogues 274–276 were acquired via their 3′-O,N2-bismonomethoxytrityl-O6-ethyl analogues using standard methods. Most prodrugs in this study demonstrated potent HCV subgenomic replicon activity and the 2′,4′-difluoro-2′-C-methyl scaffold was prioritised for prodrug development, given the promising in vitro data of the precursor 262. Ultimately from this scaffold the Sp-diastereoisomer 287 (AL-611, EC50 = 0.005 μM, CC50 > 100 μM for both) was selected as a lead candidate for preclinical toxicology studies (Table 1).87
| Compound (core scaffold) | Biological activity/application | Ref. |
|---|---|---|
|
Antibiotic via inhibition of protein synthesis. | 38 and 43–50 |
| Highly toxic to mammals. | ||
| Significant anti-trypanosomal and leishmanicidal activity. | ||
|
Inhibitor of S-adenosyl-L-homocysteine hydrolase activity (Kinact = 0.24 min−1 and Ki = 166 μM). | 68, 69 and 72 |
| Prodrug 43 – agonist for protein kinase 1 (ALPKl). | ||
| Triphosphate 35 – inhibitor of HCV NTP-dependent RNA polymerase (NS5B, IC50 > 10 μM) | ||
|
Exhibited considerably reduced antibacterial and cytotoxic properties when compared nucleocidin 1. | 73 |
|
Broad spectrum antiviral for RNA viruses (RSV and SARS-CoV-2) | 69, 71, 74 and 75 |
| Prodrugs 73 and 74 – HCV replicon activity, 73 (EC50 > 10 μM) and 74 (EC50 between 1–10 μM) and cytotoxicity (CC50 values within the range of 1–10 μM). | ||
| Triphosphate 68 – inhibitor of HCV NTP-dependent RNA polymerase (NS5B, IC50 = 2 μM). | ||
| Phosphoramidite 84 – used as a 19F NMR probe to impart conformational lock and to investigate RNA structure. | ||
|
Phosphoramidite 91 – used as a 19F NMR probe to investigate DNA and RNA complexes. | 76 |
| Gene silencing activity of siRNA duplexes modified with 89. | ||
|
Inhibits growth of L1210 cells (mouse lymphocytic leukemia cell line), upon realise of cytotoxic parent drug, 5-fluorouracil. | 78 |
|
Triphosphate 108 – inhibitor of HCV NTP-dependent RNA polymerase (NS5B, IC50 = 54.7 μM). | 64 and 82 |
| Phosphoramidite 105 – used as a 19F NMR probe to impart conformational lock and to investigate DNA and RNA complexes. | ||
|
Prodrug 185 (AL-335) – demonstrated high potency and selectivity in HCV subgenomic replicon assays (EC50 = 0.07 μM, CC50 >96 μM). | 35 |
|
Prodrug 145 – displayed anti-DENV cellular activity (EC50 = 0.57 μM) and no cytotoxicity. | 84 |
|
Triphosphate 71 – inhibitor of HCV NTP-dependent RNA polymerase (NS5B, IC50 between 250 nM–1 μM). | 69 |
|
Inhibitory activity against VZV, HCMV, and SARS-CoV-2. Though significant cytotoxicity (CC50 = 0.11 μM). | 85 |
| Prodrug 195 – inhibitory activity against VZV, HCMV, and SARS-CoV-2 with improved SI (36). | ||
|
Phosphoramidite 201 –19F-NMR probe for investigating RNA secondary structure and function. | 65 |
|
Phosphoramidite 210 – 19F NMR probe to impart conformational lock and stability to investigate DNA and RNA complexes for applications in gene silencing. | 86 |
|
Inhibitory activity in anti-HCV assays (EC50 = 4.7 μM), with no cytotoxicity against the host cells. | 66 |
|
Triphosphate 239 – inhibitor of HCV polymerase (IC50 ≤ 250 nM) | 69 |
| Prodrugs 235, 236 and 238 – inhibitor of HCV replicon, 235 (EC50 >10 μM, CC50 between 1–10 μM) and 236 (EC50 between 250 nM–1 μM, CC50 between 1–10 μM). 235 (EC50 > 10 μM, CC50 between 1–10 μM). | ||
|
Prodrug 287 (AL-611) – potent HCV subgenomic replicon activity (EC50 = 0.005 μM, CC50 > 100 μM). | 87 |
5.8 Non-canonical 4′-fluoro-ribonucleosides
This review has focused principally on canonical nucleosides modified at the 4′-position of the ribose with a fluorine atom, however it is important to note that there are examples in both the published and patent literature of 4′-fluoro-nucleosides containing non-canonical bases. For instance Wang et al., prepared the 4′-fluoro-pyrimidine C-nucleoside analogue 300, an of antiviral favipiravir, in 2016.88 The coupling of ribolactone 291 with pyridine 292 generated hemiacetal 293, the subsequent reduction and deprotection of which gave the desired β-epimer 294. The nitrile was then partially hydrolysed to amide 295, followed by desilylation to afford C-nucleoside 296. The introduction of the 4′-fluorine was accomplished after dehydration to enol ether 297 and then iodofluorination following a standard approach. Hydroxide displacement of 298 followed by aminolysis furnished 4′-fluoro-pyrimidine C-nucleoside 300, see Scheme 19.88 A neuraminidase activity-based assay using Darby canine kidney (MDCK) epithelial cells infected with influenza strain A/WSN/33 (H1N1) revealed 300 to possess only weak anti-influenza activity (EC50 = 98 μM), CC50 = 185 μM. In vivo nucleoside 5′-triphosphate (NTP) levels, a critical factor for in vivo efficacy, were assessed in A549 cellls treated with 300, and revealed very low to undectectable amount of NTPs.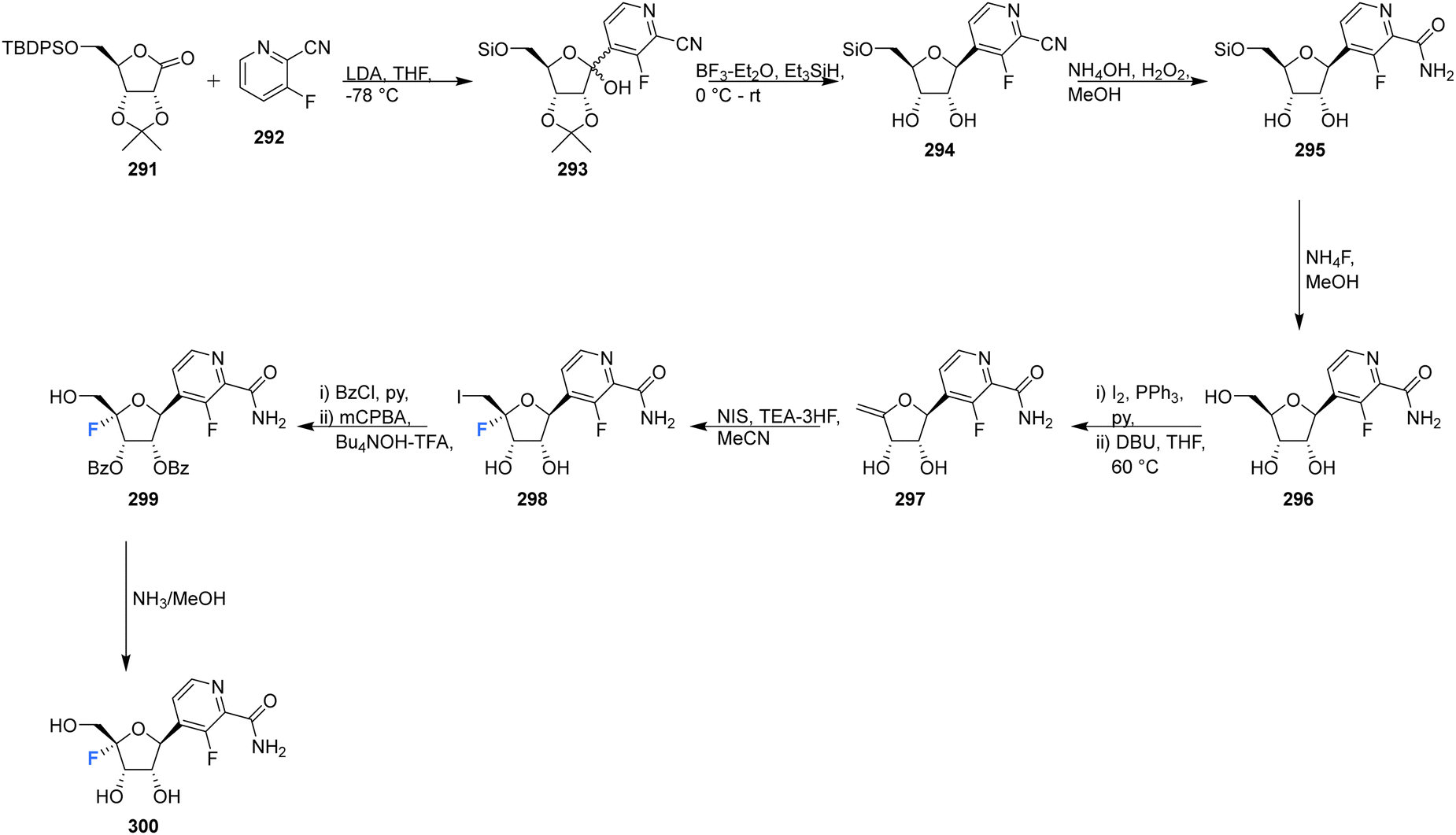 | ||
| Scheme 19 Synthesis of 4′-fluoro-pyrimidine C-nucleoside 300 as described by Wang et al.88 | ||
4′-Fluoro-pyrrolo triazine adenosine analogue 305, along with its triphosphate 312 and various produgs 306–311, as shown in Scheme 20, have been reported in the patent literature.89 Installation of 4′-fluorine in the 1′-cyano adenosine analogue 301 was accomplished using the established protocol, from which the prodrugs 306–311 and triphosphate 312 were acquired using the appropriate phosphoryl agents. Prodrugs 306–311 displayed inhibitory activity against numerous viruses (EBOV, HRV 1B, OC43CoV, DENV, RSV), in many cases displaying IC50's ≤ 1 μM, whilst presenting with CC50's > 100 μM. Triphosphate 312 was assayed against a polymerases derived from HRV16, HCV, DENV and RSV and displayed inhibition in all cases, with IC50's of 0.13, 0.4, 1.1, 0.03 μM respectfully.89 These examples serve to highlight the broadening attention afforded to the 4′-fluorination of nucleosides and demonstrate the potential that the 4′-fluoro-ribose motif has in the exploration of biologically active nucleosides.
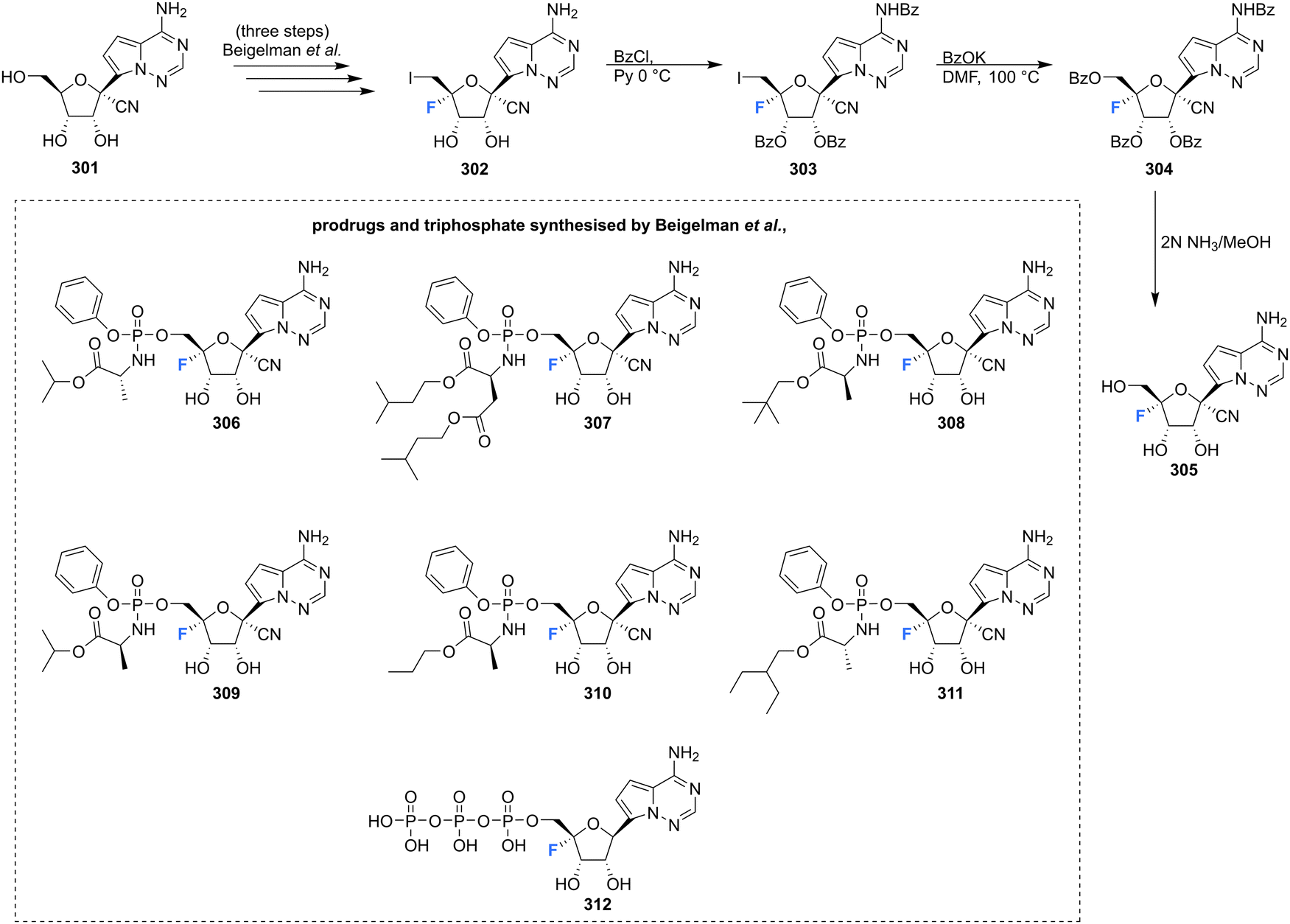 | ||
| Scheme 20 Synthesis of 4′-fluoro-pyrimidine C-nucleoside 305 and prodrugs 306–311 and triphosphate 312 as described by Beigelman et al.89 | ||
6. Additional strategies for the preparation of 4′-fluorinated nucleosides
Many of the synthetic approaches outlined so far have relied on the fluorination of a protected 4′,5′-dehydro-nucleoside using an appropriate fluorinating pseudohalogen. Whilst this has proven to be a relatively successful and widely used approach, other approaches have been developed over the years to access the 4′-fluororibose moiety.Ferrier et al., reported a strategy involving the photobromination of protected β-D-riboses, followed by fluorination of the subsequent 4′-bromoribose product, see Scheme 21.90 Photobromination at the 4′ position of adenosine 313 gave the unstable 4′-bromo-derivative 314, which was isolated from a complex mixture but in low yield. The lyxo-fluoride 315 was subsequently generated from 314 upon treatment with silver fluoride although the conversions were also modest. In a similar manner, treatment of 4-bromo-D-ribofuranose 317 with silver fluoride, afforded the L-lyxo-fluoride 319. The correctly configured epimer 318, was accessed from 317 after treatment with silver tetrafluoroborate, however only in a modest yield.
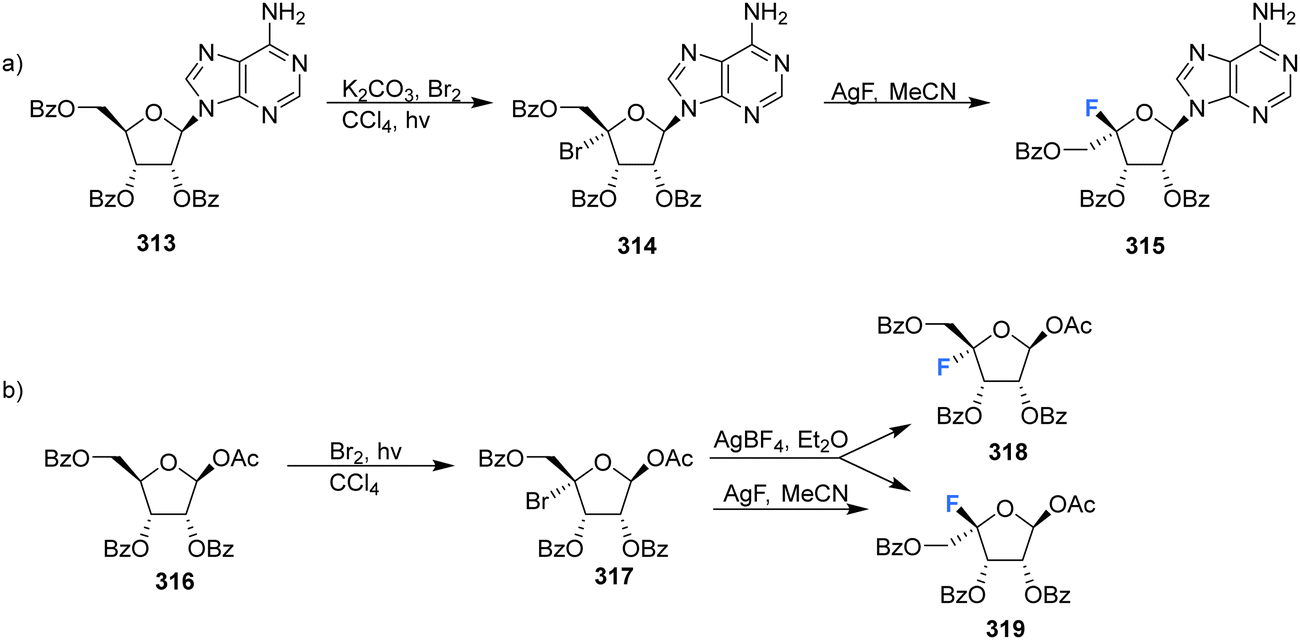 | ||
| Scheme 21 Synthesis of 4′-fluoro-nucleosides by a bromination-fluorination protocol developed by Ferrier et al.90 | ||
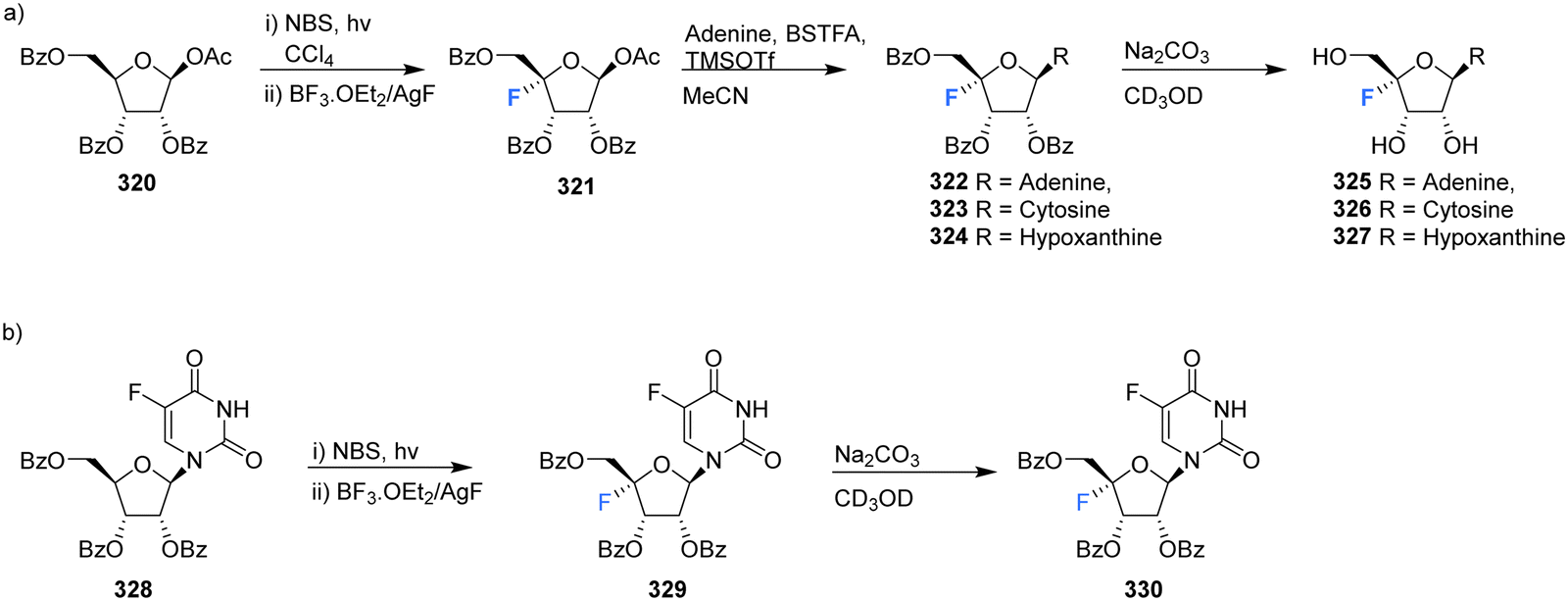 | ||
| Scheme 22 Synthesis of 4′-fluoro-nucleosides by a bromination-fluorination protocol developed by Lee et al.70 | ||
In another effort to circumvent generating 4′-fluoro epimers, Lee et al., developed a strategy to access 4′-fluoro-nucleosides 325–327 involving a successive bromination-fluorination protocol using 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribose 320 as a substrate.70 Reaction with N-bromosuccinimide (NBS) followed by treatment with boron trifluoride-etherate and silver fluoride (Scheme 22a) gave 4-fluoro-β-D-ribofuranose 321 which was then utilised in a modified Hilbert-Johnson N-glycosylation with adenine, cytosine or hypoxanthine to generate benzoyl protected derivatives 322–324. Finally, deprotection with methanolic sodium carbonate gave 4′-fluoro-nucleosides 325–327 as illustrated in Scheme 22a. An alternative approach involving the NBS mediated bromination of 5-fluorouracil 328, followed by fluorination gave 329 as illustrated in Scheme 22b. As before, deprotection with methanolic sodium carbonate was used to generate 4′-fluoro-5-fluorouracil 330.70
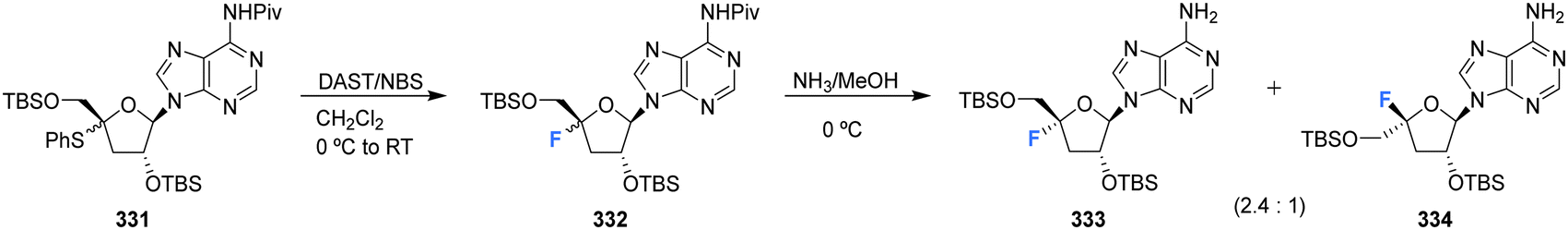
Kubota et al., developed a method for the synthesis of 4′-substituted cordycepin analogues. This involved the substitution of 4′-phenylsulfanyl as a leaving group from N6-pivaloylated 331.91 After reacting 331 with DAST/NBS in DCM, followed by deprotection with NH3/MeOH, 4′-fluoro-cordycepin analogues 333 and 334 could be acquired as a mixture of diastereomers as illustrated in Scheme 23.
 | ||
| Scheme 23 Synthesis of 4′-fluoro-nucleosides by substitution of 4′-phenylsulfanyl developed by Kubota et al.91 | ||
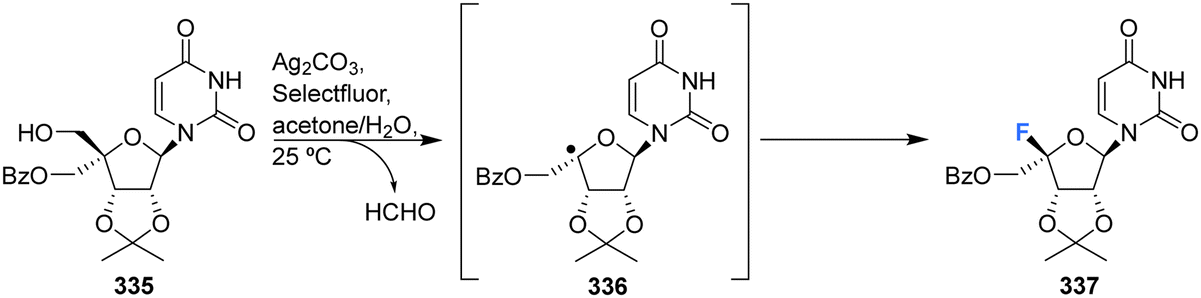
More recently Zhou et al., developed a protocol for the synthesis of glycosyl fluorides, involving the silver promoted radical dehydroxymethylative fluorination of carbohydrates under mild conditions.92 Mechanistically the reaction involves a radical fluorination via β-fragmentation of sugar-derived 5′-alkoxyl radical, and is conducted with Ag2CO3 and Selectfluor in aqueous acetone. This method was used to access protected 4′-fluoro-uridine 337 in good yield, as illustrated in Scheme 24.
 | ||
| Scheme 24 Synthesis of 4′-fluoro-nucleosides by radical dehydroxymethylative fluorination developed by Zhou et al.92 | ||
Concluding remarks
It is perhaps surprising that 4′-fluoro-nucelosides have not had a more prominent focus in bioactives discovery, particularly so given that the natural product nucleocidin has been known for over 50 years. There has been a rich and diverse chemistry exploring 2′-fluoro and 3′-fluoro nucleosides, entities which have been ubiquitous in drug discovery and some have reaped large rewards as effective commercial products. The chemistry required to introduce fluorine into the 4′-position of nucleosides is certainly more challenging, and perhaps the replacement of F for H in 4′-nucleosides has been viewed as a rather neutral modification relative to a much more proactive deoxyfluorination (F for OH) in 2′-fluoro and 3′-fluoro nucleotides. That said, the published and patented literature reviewed here indicate a recent and intensive interest in exploring 4′-fluoro-nucleosides, particularly in antiviral programmes, and we anticipate continued interest in this structural class. Elucidation of the enzymology involved in nucleocidin biosynthesis would further invigorate research into 4′-fluoro-nucleosides as it would offer a unique biotechnology, not available to other fluoro-nuceloside classes.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC for a grant and the EU Horizon 2020 Sinfonia consortia for financial support.References
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2017, 73, 1053–1066 CrossRef CAS.
- C. Isanbor and D. O'Hagan, J. Fluor. Chem., 2006, 127, 303–319 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- T. Fujiwara and D. O'Hagan, J. Fluor. Chem., 2014, 167, 16–29 CrossRef CAS.
- S. O. Thomas, V. L. Singleton, J. A. Lowery, R. W. Sharpe, L. M. Pruess, J. N. Porter, J. H. Mowat and N. Bohonos, Ant. Ann., 1956, 716–721 Search PubMed.
- A. Olszewska, R. Pohl and M. Hocek, J. Org. Chem., 2017, 82, 11431–11439 CrossRef CAS PubMed.
- M. Chrominski, M. R. Baranowski, S. Chmielinski, J. Kowalska and J. Jemielity, J. Org. Chem., 2020, 85, 3440–3453 CrossRef CAS PubMed.
- V. Kumar and E. Rozners, ChemBioChem, 2022, 23, e202100560 CAS.
- F. Levi-Acobas, P. Rothlisberger, I. Sarac, P. Marliere, P. Herdewijn and M. Hollenstein, ChemBioChem, 2019, 20, 3032–3040 CrossRef CAS PubMed.
- A. E. Klopffer and J. W. Engels, ChemBioChem, 2004, 5, 707–716 CrossRef.
- A. Istrate, M. Medvecky and C. J. Leumann, Org. Lett., 2015, 17, 1950–1953 CrossRef CAS PubMed.
- S. Frei, A. Istrate and C. J. Leumann, Beilstein J. Org. Chem., 2018, 14, 3088–3097 CrossRef CAS PubMed.
- R. El-Khoury and M. J. Damha, Acc. Chem. Res., 2021, 54, 2287–2297 CrossRef CAS.
- M. Hollenstein and C. J. Leumann, Org. Lett., 2003, 5, 1987–1990 CrossRef CAS.
- S. Ellipilli and K. N. Ganesh, J. Org. Chem., 2015, 80, 9185–9191 CrossRef CAS PubMed.
- M. Luo, E. Groaz, G. Andrei, R. Snoeck, R. Kalkeri, R. G. Ptak, T. Hartman, R. W. Buckheit, D. Schols, S. De Jonghe and P. Herdewijn, J. Med. Chem., 2017, 60, 6220–6238 CrossRef CAS.
- M. Luo, E. Groaz, R. Snoeck, G. Andrei and P. Herdewijn, ACS Med. Chem. Lett., 2020, 11, 1410–1415 CrossRef CAS PubMed.
- P. Liu, A. Sharon and C. K. Chu, J. Fluor. Chem., 2008, 129, 743–766 CrossRef CAS.
- K. W. Pankiewicz, Carbohydr. Res., 2000, 327, 87–105 CrossRef CAS.
- L. Eyer, R. Nencka, E. de Clercq, K. Seley-Radtke and D. Růžek, Antivir. Chem. Chemother., 2018, 26, 2040206618761299 CAS.
- S. Pal, G. Chandra, S. Patel and S. Singh, Chem. Rec., 2022, 22, e2021003 CrossRef.
- D. R. Nelson, J. N. Cooper, J. P. Lalezari, E. Lawitz, P. J. Pockros, N. Gitlin, B. F. Freilich, Z. H. Younes, W. Harlan, R. Ghalib, G. Oguchi, P. J. Thuluvath, G. Ortiz-Lasanta, M. Rabinovitz, D. Berastein, M. Bennett, T. Hawkins, N. Ravendhran, A. M. Sheikh, P. Varunok, K. V. Kowdley, D. Hennicken, F. McPhee, I. Rana, E. A. Hughes and A.-S. Team, Hepatology, 2015, 61, 1127–1135 CrossRef CAS PubMed.
- J. S. Roth, H. Ford, M. Tanaka, H. Mitsuya and J. A. Kelley, J. Chromatogr. B, 1998, 712, 199–210 CrossRef CAS.
- L. W. Hertel, G. B. Boder, J. S. Kroin, S. M. Rinzel, G. A. Poore, G. C. Todd and G. B. Grindey, Cancer Res., 1990, 50, 4417–4422 CAS.
- P. Kozuch, N. Ibrahim, F. Khuri, P. Hoff, E. Estey, V. Gandhi, M. Du, M. B. Rios, W. Plunkett, M. J. Keating and H. Kantarjian, Blood, 1999, 94, 127A–127A CrossRef.
- N. Bhuma, S. S. Burade, A. V. Bagade, N. M. Kumbhar, K. M. Kodam and D. D. Dhavale, Tetrahedron, 2017, 73, 6157–6163 CrossRef CAS.
- J. B. Chang, Acc. Chem. Res., 2022, 55, 565–578 CrossRef CAS.
- K. Detmer, D. Summerer and A. Marx, Eur. J. Org. Chem., 2003, 1837–1846 CrossRef.
- D. Summerer and A. Marx, J. Am. Chem. Soc., 2002, 124, 910–911 CrossRef CAS PubMed.
- M. Betson, N. Allanson and P. Wainwright, Org. Biomol. Chem., 2014, 12, 9291–9306 RSC.
- D. B. Smith, J. A. Martin, K. Klumpp, S. J. Baker, P. A. Blomgren, R. Devos, C. Granycome, J. Hang, C. J. Hobbs, W. R. Jiang, C. Laxton, S. Le Pogam, V. Leveque, H. Ma, G. Maile, J. H. Merrett, A. Pichota, K. Sarma, M. Smith, S. Swallow, J. Symons, D. Vesey, I. Najera and N. Cammack, Bioorg. Med. Chem. Lett., 2007, 17, 2570–2576 CrossRef CAS PubMed.
- G. Y. Wang, N. Dyatkina, M. Prhavc, C. Williams, V. Serebryany, Y. J. Hu, Y. F. Huang, J. Q. Wan, X. Y. Wu, J. Deval, A. Fung, Z. N. Jin, H. Fan, K. Shaw, H. Kang, Q. L. Zhang, Y. Tam, A. Stoycheva, A. Jekle, D. B. Smith and L. Beigelman, J. Med. Chem., 2019, 62, 4555–4570 CrossRef CAS PubMed.
- T. Xu, C. Xu, D. Liu, J. Fan, Y. Pan, R. T. Li and X. Chen; Int Patent Appl, 2nd May, WO2019/080898A1, 2019 Search PubMed.
- G. O. Morton, J. E. Lancaster, G. E. Vanlear, W. Fulmor and W. E. Meyer, J. Am. Chem. Soc., 1969, 91, 1535 CrossRef CAS.
- C. W. Waller, J. B. Patrick, W. Fulmor and W. E. Meyer, J. Am. Chem. Soc., 1957, 79, 1011–1012 CrossRef CAS.
- A. R. O. Pasternak, A. Bechthold and D. L. Zechel, ChemBioChem, 2022, 23, e202200140 CAS.
- T. Awakawa, L. Barra and I. Abe, J. Ind. Microbiol. Biotechnol., 2021, 48 DOI:10.1093/jimb/kuab001.
- D. A. Shuman, M. J. Robins and R. K. Robins, J. Am. Chem. Soc., 1970, 92, 3434–3440 CrossRef CAS.
- I. D. Jenkins, J. P. H. Verheyden and J. G. Moffatt, J. Am. Chem. Soc., 1976, 98, 3346–3357 CrossRef CAS.
- J. R. Florini, Nucleocidin, in D. Gottlieb and P. D. Shaw ed., Antibiotics, Mechanism of Action, Springer-Verlag, New York, vol. 1, 1967, pp. 427–433 Search PubMed.
- C. W. Waller, P. W. Fryth, B. L. Hutchings and J. H. Williams, J. Am. Chem. Soc., 1953, 75, 2025 CrossRef CAS.
- E. J. Tobie, J. Parasitol., 1957, 43, 291–293 CrossRef CAS.
- L. E. Stephen and A. R. Gray, J. Parasitol., 1960, 46, 509–514 CrossRef CAS.
- C. J. Bacchi, C. Lambros, B. Goldberg, S. H. Hutner and G. D. Carvalho, Antimicrob. Agents Chemother., 1974, 6, 785–790 CrossRef CAS PubMed.
- J. R. Florini, H. H. Bird and P. H. Bell, J. Biol. Chem., 1966, 241, 1091–1098 CrossRef CAS.
- I. W. Sherman, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1976, 53, 447–450 CrossRef CAS.
- J. Williamson and R. F. Macadam, Trans. R. Soc. Trop. Med. Hyg., 1976, 70, 130–137 CrossRef CAS PubMed.
- X. M. Zhu, S. Hackl, M. N. Thaker, L. Kalan, C. Weber, D. S. Urgast, E. M. Krupp, A. Brewer, S. Vanner, A. Szawiola, G. Yim, J. Feldmann, A. Bechthold, G. D. Wright and D. L. Zechel, ChemBioChem, 2015, 16, 2498–2506 CrossRef CAS PubMed.
- S. Zhang, D. Klementz, J. Zhu, R. Makitrynskyy, A. R. O. Pasternak, S. Gunther, D. L. Zechel and A. Bechtholda, J. Biotechnol., 2019, 292, 23–31 CrossRef CAS.
- X. Feng, N. Al Maharik, A. Bartholome, J. E. Janso, U. Reilly and D. O'Hagan, Org. Biomol. Chem., 2017, 15, 8006–8008 RSC.
- U. Ngivprom, S. Kluaiphanngam, W. J. Ji, S. Siriwibool, A. Kamkaew, J. R. K. Cairns, Q. Zhang and R. Y. Lai, RSC Adv., 2021, 11, 3510–3515 RSC.
- X. Feng, D. Bello, P. T. Lowe, J. Clark and D. O'Hagan, Chem. Sci., 2019, 10, 9501–9505 RSC.
- Y. W. Chen, Q. Z. Zhang, X. Feng, M. Wojnowska and D. O'Hagan, Org. Biomol. Chem., 2021, 19, 10081–10084 RSC.
- X. Feng, D. Bello and D. O'Hagan, RSC Adv., 2021, 11, 5291–5294 RSC.
- F. Deleeuw and C. Altona, Perkin Trans. 2, 1982, 375–384 RSC.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333–2344 CrossRef CAS.
- T. Hakoshima, H. Omori, K. Tomita, H. Miki and M. Ikehara, Nucleic Acids Res., 1981, 9, 711–729 CrossRef CAS PubMed.
- M. N. Aher, N. D. Erande, V. A. Kumar, M. Fernandes and R. G. Gonnade, Acta Crystallogr., 2020, 76, 346 CAS.
- T. Iimori, Y. Murai, Y. Wakizaka, Y. Ohtsuka, S. Ohuchi, Y. Kodama and T. Oishi, Chem. Pharm. Bull., 1993, 41, 775–777 CrossRef CAS.
- A. R. Maguire, W. D. Meng, S. M. Roberts and A. J. Willetts, J. Chem. Soc., Perkin Trans. 1, 1993, 1795–1808 RSC.
- S. Martinez-Montero, G. F. Deleavey, A. Kulkarni, N. Martin-Pintado, P. Lindovska, M. Thomson, C. Gonzalez, M. Gotte and M. J. Damha, J. Org. Chem., 2014, 79, 5627–5635 CrossRef CAS PubMed.
- Y. F. Zhou, K. Lu, Q. Li, C. C. Fan and C. Z. Zhou, Chem. – Eur. J., 2021, 27, 14738–14746 CrossRef CAS PubMed.
- H. Shimada, K. Haraguchi, K. Hotta, T. Miyaike, Y. Kitagawa, H. Tanaka, R. Kaneda, H. Abe, S. Shuto, K. Mori, Y. Ueda, N. Kato, R. Snoeck, G. Andrei and J. Balzarini, Bioorg. Med. Chem., 2014, 22, 6174–6182 CrossRef CAS PubMed.
- I. D. Jenkins, J. P. Verheyden and J. G. Moffiatt, J. Am. Chem. Soc., 1971, 93, 4323 CrossRef CAS.
- D. Guillerm, M. Muzard, B. Allart and G. Guillerm, Bioorg. Med. Chem. Lett., 1995, 5, 1455–1460 CrossRef CAS.
- B. A. Mayes, A. M. Moussa, A. J. Stewart and G. Gosselin, Int Patent, Appl, 26th June, WO2014/099941A1, 2014 Search PubMed.
- S. Lee, C. Uttamapinant and G. L. Verdine, Org. Lett., 2007, 9, 5007–5009 CrossRef CAS PubMed.
- M. A. Ivanov, G. S. Ludva, A. V. Mukovnya, S. N. Kochetkov, V. L. Tunitskaya and L. A. Alexandrova, Russ. J. Bioorg. Chem., 2010, 36, 488–496 CrossRef CAS PubMed.
- T. Xu, C. Xu, D. Liu, J. Fan, Y. Pan, T. R. Li and X. Chen, Int Patent Appl., 26th June, WO2019/080898Al, 2014 Search PubMed.
- G. R. Owen, J. P. H. Verheyden and J. G. Moffatt, J. Org. Chem., 1976, 41, 3010–3017 CrossRef CAS.
- Q. Li, J. L. Chen, M. Trajkovski, Y. F. Zhou, C. C. Fan, K. Lu, P. P. Tang, X. C. Su, J. Plavec, Z. Xi and C. Z. Zhou, J. Am. Chem. Soc., 2020, 142, 4739–4748 CrossRef CAS.
- J. Sourimant, C. M. Lieber, M. Aggarwal, R. M. Cox, J. D. Wolf, J. J. Yoon, M. Toots, C. Ye, Z. Sticher, A. A. Kolykhalov, L. Martinez-Sobrido, G. R. Bluemling, M. G. Natchus, G. R. Painter and R. K. Plemper, Science, 2022, 375, 161–167 CrossRef CAS PubMed.
- E. Malek-Adamian, M. B. Patrascu, S. K. Jana, S. Martinez-Montero, N. Moitessier and M. J. Damha, J. Org. Chem., 2018, 83, 9839–9849 CrossRef CAS PubMed.
- E. Malek-Adamian, J. Fakhoury, A. E. Arnold, S. Martinez-Montero, M. S. Shoichet and M. J. Damha, Nucleic Acid Ther., 2019, 29, 187–194 CrossRef CAS PubMed.
- S. Ajmera, A. R. Bapat, E. Stephanian and P. V. Danenberg, J. Med. Chem., 1988, 31, 1094–1098 CrossRef CAS PubMed.
- R. Abele, P. Alberto, S. Kaplan, P. Siegenthaler, V. Hofmann, H. J. Ryssel, D. Hartmann, E. E. Holdener and F. Cavalli, J. Clin. Oncol., 1983, 1, 750–754 CrossRef CAS PubMed.
- R. Abele, E. Kaplan, R. Grossenbacher, H. J. Schmid and F. Cavalli, Eur. J. Cancer Clin. Oncol., 1984, 20, 333–336 CrossRef CAS.
- T. Ishikawa, F. Sekiguchi, Y. Fukase, N. Sawada and H. Ishitsuka, Cancer Res., 1998, 58, 685–690 CAS.
- S. Martinez-Montero, G. F. Deleavey, N. Martin-Pintado, J. F. Fakhoury, C. Gonzalez and M. J. Damha, ACS Chem. Biol., 2015, 10, 2016–2023 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- G. Wang, S. P. Lim, Y. L. Chen, J. Hunziker, R. Rao, F. Gu, C. C. Seh, N. A. Ghafar, H. Y. Xu, K. Chan, X. D. Lin, O. L. Saunders, M. Fenaux, W. D. Zhong, P. Y. Shi and F. Yokokawa, Bioorg. Med. Chem. Lett., 2018, 28, 2324–2327 CrossRef CAS PubMed.
- Z. H. Zheng, E. Groaz, R. Snoeck, S. De Jonghe, P. Herdewijn and G. Andrei, ACS Med. Chem. Lett., 2021, 12, 88–92 CrossRef CAS.
- S. Martinez-Montero, G. F. Deleavey, A. Dierker-Viik, P. Lindovska, T. Ilina, G. Portella, M. Orozco, M. A. Parniak, C. Gonzalez and M. J. Damha, J. Org. Chem., 2015, 80, 3083–3091 CrossRef CAS.
- G. Y. Wang, N. Dyatkina, M. Prhavc, C. Williams, V. Serebryany, Y. J. Hu, Y. F. Huang, X. Y. Wu, T. Q. Chen, W. S. Huang, V. K. Rajwanshi, J. Deval, A. Fung, Z. N. Jin, A. Stoycheva, K. Shaw, K. Gupta, Y. Tam, A. Jekle, D. B. Smith and L. Beigelman, J. Med. Chem., 2020, 63, 10380–10395 CrossRef CAS PubMed.
- G. Y. Wang, J. G. Wan, Y. J. Hu, X. Y. Wu, M. Prhavc, N. Dyatkina, V. K. Rajwanshi, D. B. Smith, A. Jekle, A. Kinkade, J. A. Symons, Z. N. Jin, J. Deval, Q. L. Zhang, Y. Tam, S. Chanda, L. Blatt and L. Beigelman, J. Med. Chem., 2016, 59, 4611–4624 CrossRef CAS PubMed.
- L. Beigleman, J. Deval and M. Prhavc; Int Patent Appl, 21st March, WO2019/053696Al, 2019 Search PubMed.
- R. J. Ferrier and S. R. Haines, J. Chem. Soc., Perkin Trans. 1, 1984, 1675–1681 RSC.
- Y. Kubota, M. Ehara, K. Haraguchi and H. Tanaka, J. Org. Chem., 2011, 76, 8710–8717 CrossRef CAS.
- X. Zhou, H. Ding, P. W. Chen, L. Liu, Q. K. Sun, X. Y. Wang, P. Wang, Z. H. Lv and M. Li, Angew. Chem., Int. Ed., 2020, 59, 4138–4144 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |