
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable cycloaliphatic polyurethanes: from synthesis to applications
Agathe
Mouren
 and
Luc
Avérous
*
and
Luc
Avérous
*
BioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: luc.averous@unistra.fr
First published on 15th December 2022
Abstract
Polyurethanes (PUs) are a versatile and major polymer family, mainly produced via polyaddition between polyols and polyisocyanates. A large variety of fossil-based building blocks is commonly used to develop a wide range of macromolecular architectures with specific properties. Due to environmental concerns, legislation, rarefaction of some petrol fractions and price fluctuation, sustainable feedstocks are attracting significant attention, e.g., plastic waste and biobased resources from biomass. Consequently, various sustainable building blocks are available to develop new renewable macromolecular architectures such as aromatics, linear aliphatics and cycloaliphatics. Meanwhile, the relationship between the chemical structures of these building blocks and properties of the final PUs can be determined. For instance, aromatic building blocks are remarkable to endow materials with rigidity, hydrophobicity, fire resistance, chemical and thermal stability, whereas acyclic aliphatics endow them with oxidation and UV light resistance, flexibility and transparency. Cycloaliphatics are very interesting as they combine most of the advantages of linear aliphatic and aromatic compounds. This original and unique review presents a comprehensive overview of the synthesis of sustainable cycloaliphatic PUs using various renewable products such as biobased terpenes, carbohydrates, fatty acids and cholesterol and/or plastic waste. Herein, we summarize the chemical modification of the main sustainable cycloaliphatic feedstocks, synthesis of PUs using these building blocks and their corresponding properties and subsequently present their major applications in hot-topic fields, including building, transportation, packaging and biomedicine.
 Agathe Mouren | Agathe Mouren obtained her Master's Degree in Engineering in Paper, Print Media and Biomaterials at Grenoble INP – Pagora, UGA in Grenoble, France. She started her PhD in Polymer Science in 2020 at the Institute of Chemistry and Processes for Energy, Environment and Health, University of Strasbourg/CNRS in Strasbourg, France, under the supervision of Prof. Luc Avérous. Her interests cover several aspects of renewable polymer materials. During her doctoral studies, her research focuses on the synthesis of new renewable polymers from building blocks obtained by biological recycling. |
 Luc Avérous | Prof. Luc Avérous is a Group Leader (BioTeam) and Head of the Polymer Research Department at ICPEES (UMR CNRS – University of Strasbourg), in France. In 2003, he became a Full Professor at ECPM (Strasbourg-France), where he teaches polymer science and engineering. Over the past 25 years, his major research projects have dealt with biobased and renewable polymers. As a leading international expert in these fields, he has developed strong international collaborations with several labs and major companies. He has co-edited 4 books and published hundreds of scientific communications including 20 review articles, with around 20 000 citations. |
1. Introduction
Currently, polymer-based materials, also known as “plastics”, are essential with wide range of applications and can be found in all aspects of daily life due to their amazing and incomparable properties such as light weight, processability, versatility, durability, adaptability, energy saving and low cost.1 Since 1950, the annual production of plastics has increased, reaching around 370 million tons in 2020. However, this has resulted in a corresponding increase in plastic pollution globally due to the huge and continuous production of microplastics, which affects the environment including oceans, rivers, deserts, mountaintops, and tropical forests. Plastic pollution irreversibly changes the functioning of ecosystems and affects human health.2–4 Moreover, the extraction of fossil resources and production of plastic contribute to greenhouse gas (GHG) emissions. Consequently, different strategies have been explored to develop sustainable and renewable materials from the cradle to grave, with a controlled end of life. To date, most plastics are produced from fossil resources, but new resources are being increasingly explored and developed such as CO2, plastic waste and biomass for their synthesis to reduce (i) the use of the limited fossil resources and (ii) the global environmental impact.5,6 To improve the global life cycle analysis (LCA), at the end of life of plastics, currently the focus is biodegradability/compostability7–9 and recycling (mechanical recycling, chemical/biological recycling, and upcycling).10,11 Accordingly, more and more materials are initially tailored to make their recycling easier.12Among the different available polymers, PUs are the sixth most produced polymer family. The PU market size was valued at 71 billion USD in 2020, which is expected to reach 73 billion USD in 2028.13 The PU market is divided into various parts comprising foams, coatings, elastomers and adhesives.14 PUs are mainly characterized by the presence of urethane groups in their polymer chains. Urethane groups can be synthesized via several pathways, but the most common to date is the addition between hydroxyl group (OH) and isocyanate (NCO) groups. The corresponding polyisocyanates and polyols are mainly fossil-based and present different architectures.
IUPAC defines aliphatic compounds as acyclic, cyclic, saturated or unsaturated carbon compounds, excluding aromatic compounds.15 It is well known that aromatic and aliphatic building blocks can bring specific advantages and drawbacks to the final polymer properties. For instance, acyclic aliphatic compounds result in good hydrophobicity, high UV stability, and hydrolysis and oxidation resistance. They also present chain mobility in the polymer structure, reducing the glass transition temperature (Tg) and rigidity. This can lead to amorphous structures, which are preferred for certain applications requiring high transparency. Acyclic aliphatic building blocks result in low viscosity, easy synthesis process and avoid the use of solvents. However, linear aliphatic building blocks result in also low thermal stability, and their active functional groups are in general less reactive than aromatic ones. Aromatic building blocks give interesting polymer properties after synthesis such as stiffness, thermal resistance and barrier properties. The π–π interactions between aromatic rings increase the rigidity, Tg and mechanical behavior. However, their drawbacks include low resistance to photo-oxidation, leading to yellowing due to UV degradation and absorption. Consequently, the viscosity increases and the processability and solubility decrease.16–21 Cycloaliphatic compounds, which represent cyclic aliphatics, are a large class of chemicals, with the advantages of linear aliphatic and aromatic building blocks in terms of thermal properties, thermal stability and processability. Furthermore, the final materials properties such as flexibility, Tg, transparency and mechanical properties are intermediate between aromatic and linear aliphatic.22–27
Currently, research and production are focused on the development of new sustainable PU systems. The corresponding building blocks or/and polymers can be obtained from plastic waste or/and from biomass using chemical and biochemical (biotech) processes, which can be coupled, leading to a “chem-biotech” process.
Intensive efforts are devoted to chemical recycling, where plastics are depolymerized into intermediates and building blocks. They can be repolymerized to produce the same polymer with virgin-like properties or can be converted to obtain another polymer. If the final product is of higher value, this recycling type is called “upcycling”.28 Three main chemical processes are available for recycling plastics depending on their chemical nature, which have different advantages and drawbacks, as follows: (i) chemical reactions such as solvolysis can be applied to polymers with ether, ester and acid amide bonds. (ii) Dissolution/precipitation is applicable to mixed plastics, which may also contain additives and impurities. (iii) Thermomechanical routes such as pyrolysis are mostly applied to polyolefins.29
Due to recent biotechnological approaches, it is possible to use plastic waste as an interesting carbon source by microbial depolymerization30 or enzymatic plastic degradation31 into oligomers and monomers, which can be recovered to finally obtain polymers via a chemical or biochemical pathway. In the case of “biodegradation”, biotic degradation leads to mineralization in the presence of microorganisms, with the production of CO2 or CH4 in aerobic or anaerobic systems, respectively.32 These different short molecules can be used as alternative carbon-based substrates for microbes by fermentation to produce (i) bacterial polymers, as in the case of polyhydroxyalkanoates (PHAs), and (ii) a wide variety of valuable products such as glycolipids, aromatics, organic acids and alcohols.33
Also, biomass can be employed to obtain biobased polymers.34–37 Biobased polymers are defined by IUPAC as materials composed or derived in whole or in part of biological products obtained from biomass, with a positive C14 content.38 Biomass is based on all living biological organisms, e.g., plants, mushrooms, animals and microorganisms. According to the initial biobased resource, a wide range of polymer structures can be directly or indirectly obtained, e.g., from polysaccharides, vegetable oils, proteins, bacterial polyesters, lipids, terpenes and lignins. However, an additional synthesis step may be required.
Acyclic aliphatic feedstock such as triglycerides, e.g., from vegetable oils, animal fats and micro-algae, are advantageous to give final aliphatic architectures using oleo-chemistry. Triglycerides can be employed to easily obtain fatty acids, with numerous structural variations such as (i) chain length of up to 24 carbons, (ii) different number of double bonds (saturation, mono and poly-unsaturation), (iii) different types and numbers of other active groups (carboxylic, OH, and epoxy groups) and (iv) different stereochemistry of the double bonds. Thus, triglycerides, fatty acids and their derivatives have been intensively studied and developed for the synthesis of various aliphatic polymers in the frame of the oleochemistry.39–42
Aromatic architectures can also be obtained. The most studied sustainable aromatic resources include lignins, tannins and their derivatives. Besides their abundance and low cost, various chemical modifications are possible to develop rigid and aromatic biobased polymers due to their high OH contents.16,17,43
Based on acyclic aliphatic and aromatic compounds, a large range of sustainable cycloaliphatic resources is available to obtain different cycloaliphatic PU architectures with specific properties. This latter point is the core of the topic of this review, which is structured into different parts. The first section of this original review provides some generalities on PUs, their chemistry, and the main chemicals and the different strategies employed to increase their sustainability. Secondly, we focus on the synthesis of biobased cycloaliphatic PUs and their main properties, also presenting their main sustainable building blocks and final materials. Finally, in the last section, we introduce the main applications of these biobased cycloaliphatic PUs, e.g., foams, coatings, adhesives and membranes, including some biomedical applications.
2. Generalities
2.1. Introduction to PU chemistry
Isocyanates were discovered in 1848 by Wurtz. Subsequently, in the 19th century, Curtius and Hofmann developed isocyanate chemistry. In 1937, Otto Bayer and co-workers synthesized the first PU via the reaction of a polyester diol with a diisocyanate.44 Then, during World War II, the first PUs were commercialized as elastomers, coatings, and adhesives.45 Subsequently, the first PU foams and structured thermosets appeared in the 1950s.46Nowadays, the most common route to synthesize PUs is the fast and exothermic polyaddition reaction between polyols to introduce H-labile groups and polyisocyanates, with or without catalysts.47 The two main PU categories are thermoplastics and thermosets. Thermoplastic PUs (TPUs) are obtained via a reaction between compounds with a functionality of 2 (f = 2). The final polymer is soluble in different solvents and can be thermo-reprocessed. Thermoset PUs are synthesized with at least one polyfunctional reagent with f > 2. A third category is often added with covalent adaptable networks (CANs), which can be under an external stimulus, dissociative or associative, such as vitrimers without the loss of their global architectural integrity. CANs present the advantages of both systems, i.e., the recyclability of thermoplastics and the high mechanical properties of thermosets.48
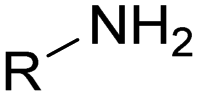
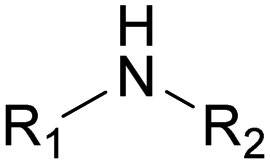
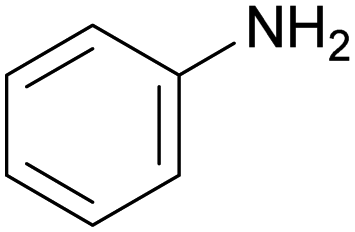
The nature of the H-labile group is related to the reactivity towards isocyanates, as shown in Table 1. For instance, the different amines (from primary, secondary, aliphatic or aromatic groups) are more reactive than OH groups. Water and the primary OH group have similar reactivity. Urea, tertiary OH, aromatic OH and urethane are 2 to 300 times less reactive than secondary OH.49,50
A large variety of polyols, polyisocyanates, chain extenders and cross-linkers allows high variability in the final polymer architectures. For instance, long chain polyol brings softness and elasticity to the final PUs, whereas a high level of cross-linking results in rigidity.50
| Hydrogen active compound | Chemical structure | Relative reaction rate (non-catalyzed, 25 °C) |
|---|---|---|
| Primary aliphatic amine |
|
2500 |
| Secondary aliphatic amine |
|
500–1250 |
| Primary aromatic amine |
|
5–7.5 |
| Primary OH |
|
2.5 |
| Water |

|
2.5 |
| Carboxylic acid |
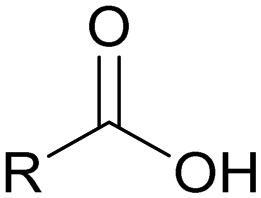
|
1 |
| Secondary OH |

|
0.75 |
| Urea |

|
0.375 |
| Tertiary OH |

|
0.0125 |
| Phenolic OH |
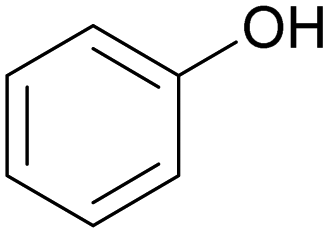
|
0.0025–0.0125 |
| Urethane |
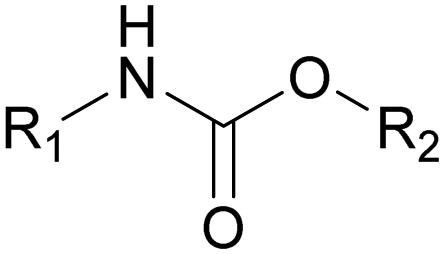
|
0.0025 |
It is also possible to tailor the final properties of PUs by the incorporation of additives or modifying their processing conditions. These different variabilities result in high versatility for a wide range of applications.14
2.2. Main PU chemicals for synthesis
Isocyanate chemistry. The high reactivity of the isocyanate group with hydrogen-labile groups explains why the PU chemistry is so widespread. Isocyanates react easily with alcohols, thiols, amine, water, urethanes, urea groups, carboxylic acids, cyclic anhydrides, and epoxide compounds or self-react with other isocyanates.51 The main reactions of isocyanates with H-labile groups are summarized in Fig. 1.
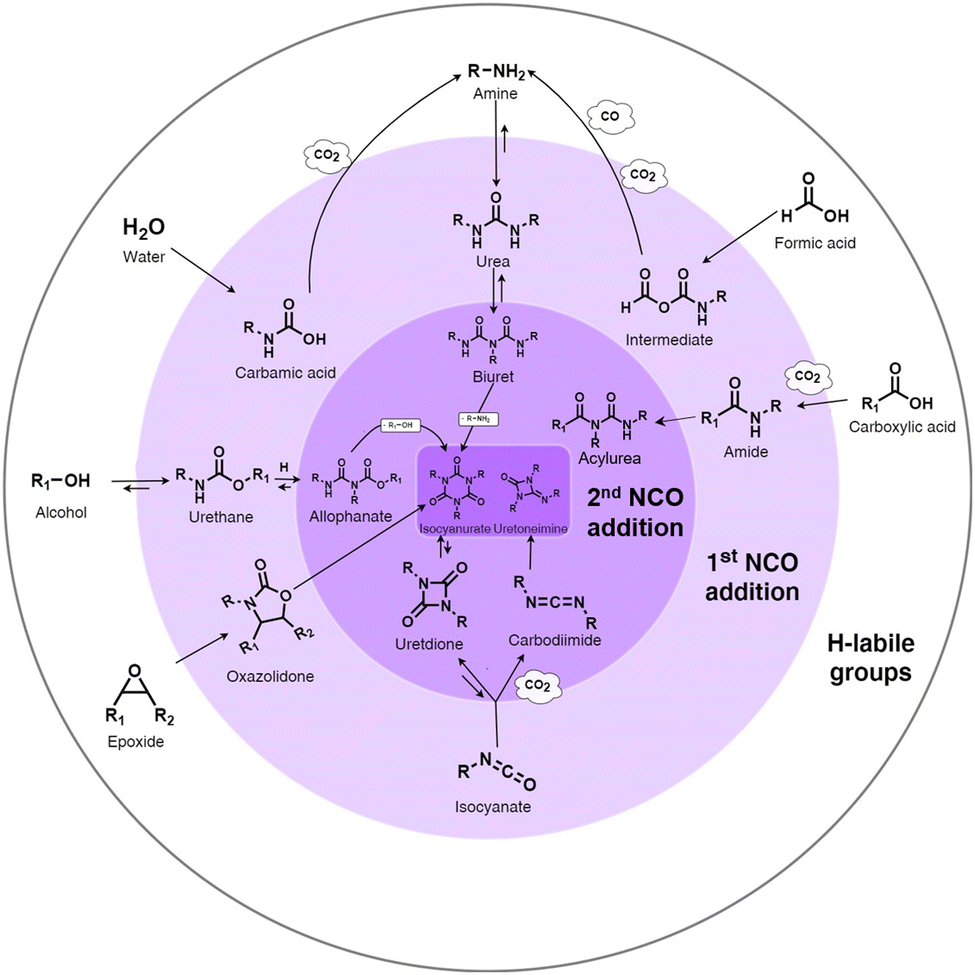 | ||
| Fig. 1 Summary of the main chemical reactions between isocyanate and H-labile groups, adapted from ref. 49. | ||
The reaction between isocyanate and alcohols is the main route for the synthesis of PUs. This reaction is exothermic with an enthalpy release of 100 kJ mol−1.50 Isocyanate can react with water to form an unstable carbamic acid in the first step. The decomposition of carbamic acid leads to the formation of an amine and CO2 release with an enthalpy release of 196 kJ mol−1. In the third step, the amine group reacts with isocyanate to form urea. The use of CO2 as a blowing agent and the heat produced during this reaction can be the basis for the synthesis of some PU foam, starting from room temperature.52 Isocyanates can also react with urethane to form allophanate at temperatures higher than 110 °C.53 In a similar route, disubstituted urea and isocyanate react to form biuret at temperatures higher than 120 °C. The formation of allophanate and biuret formation is reversible. Amide and CO2 are produced by the reaction between carboxylic acid and isocyanate. The further addition of isocyanate to amide leads to the formation of acylurea.54 The dimerization of isocyanates often occurs with aromatic isocyanates due to their high reactivity.55 Depending on the condition and catalysts, the reaction products can be uretidinedione and carbodiimide. Carbodiimide is produced by the thermal degradation of isocyanate at temperatures higher than 160 °C.56 In the presence of special catalysts such as potassium acetate, the trimerization of isocyanate can occur to form heterocyclic isocyanurate, finally forming polyisocyanurate (PIR), which is increasingly used to produce specific foams with higher fire resistance.50,57 In the case of the reaction between isocyanate and cyclic anhydride, cyclic imide is produced. Oxazolidones are cyclic urethanes formed by the reaction of isocyanates with epoxide compounds, under specific catalyst conditions and at temperatures between 50 °C and 60 °C.58
Polyisocyanates for PU synthesis. Isocyanates can be synthesized through four main routes, as presented in Fig. 2. Curtius, Hoffman or Lossen rearrangement involves azide intermediary reaction products that are unstable, and thus not suitable for large-scale production. Primary amine phosgenation is used to produce isocyanate at the industrial level, despite the toxicity of phosgene.59,60
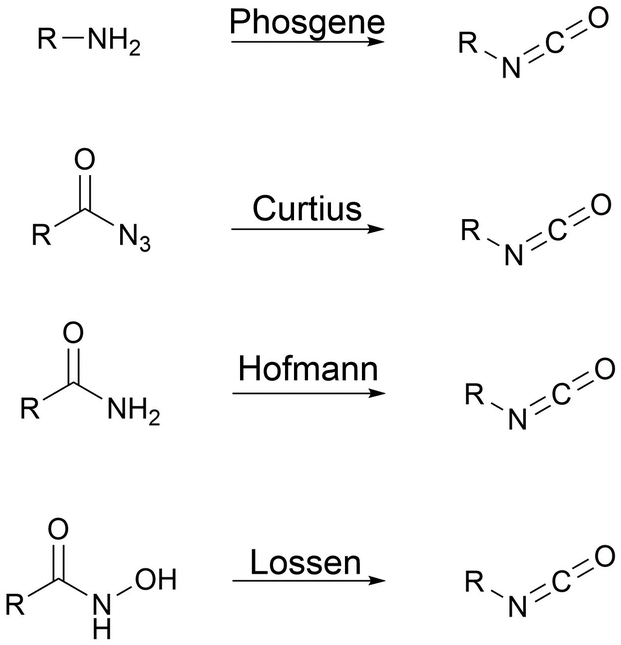 | ||
| Fig. 2 Isocyanate synthesis by phosgene route and rearrangements. | ||
Polyisocyanates can be aromatic, aliphatic, cycloaliphatic or polycyclic. The main diisocyanates used in PU chemistry are as follows: (i) toluene diisocyanate (TDI), which is commercialized as a mixture of 2,4 and 2,6 isomers, (ii) 4,4′-methylenediphenyl diisocyanate (MDI) and polymeric 4,4′-MDI (pMDI), (iii) 1,6-hexamethylene diisocyanate (HDI), (iv) 4,4′-dicyclohexyl diisocyanate (HMDI) and (v) isophorone diisocyanate (IPDI). Their chemical structures are presented in Table 2. All these polyisocyanates are bifunctional except pMDI, which has an average f of 2.7. Also, they are all fossil-based and synthesized from phosgene chemistry.61,62 Given that aromatic diamines are more available and cheaper than aliphatic diamines, aromatic isocyanates such as TDI and MDI represent around 95% of the diisocyanate market for the production of PUs.63 Aromatic isocyanates are efficient given that they have higher reactivity due to the negative charge delocalization on their aromatic ring.64 The stability of aromatic isocyanates results in enhanced thermal and flame retardant properties. Aliphatic isocyanates are also useful for UV and oxidation resistance.20
| Isocyanate | Chemical structure | k 1 | k 1/k2 |
|---|---|---|---|
| 2,4-TDI |
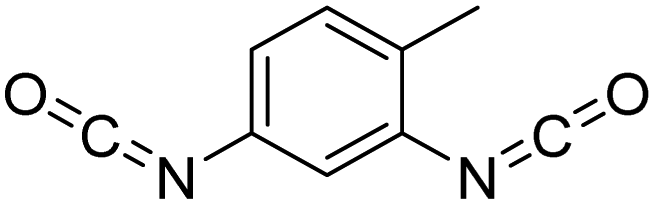
|
400 | 12.121 |
| MDI |

|
320 | 2.909 |
| HDI |
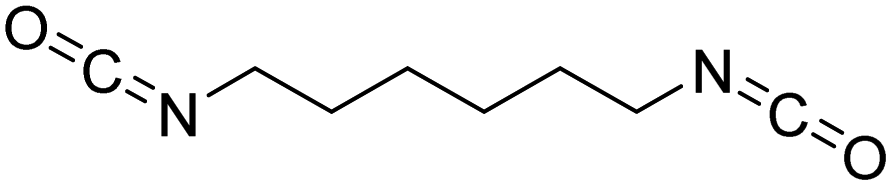
|
1 | 2.000 |
| HMDI |

|
0.57 | 1.425 |
| IPDI |
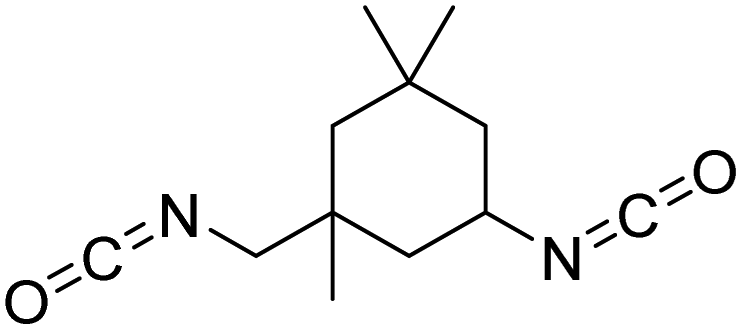
|
0.62 | 2.695 |
In a symmetric diisocyanates, both NCO groups have the same reactivity. However, once a nucleophilic compound is added to an isocyanate group to form the first urethane group, the second unreacted NCO group reactivity is lowered. This is due to the electron-releasing effect of PUs.50,59,63 For example, the first isocyanate groups of MDI and HDI are 2.9 and 2.0 times more reactive than the second one, respectively. This reactivity gap is accentuated in the case of asymmetric diisocyanates. For example, the addition of the first OH group to 2,4-TDI is 12 times faster than the second one. Temperature and catalysts decrease the selectivity of the OH groups for isocyanates.63
Polyols characteristics. Polyols are liquid and viscous monomeric, oligomeric or polymeric compounds based on OH groups with an f of at least equal to 2. The final properties of PUs are strongly impacted by the characteristics polyols such as f, molar mass, hydroxyl number, chemical structure and reactivity.
Specifically, f is directly correlated with the morphology (thermoplastic and thermoset) of the final PUs and the corresponding mechanical behavior. The density of OH groups (IOH) is expressed as milligrams of KOH per gram of molecule (mg KOH g−1) and the molar mass of polyols is usually in the range of 0.5 to 10 kDa.
Types of polyols. For the synthesis of PUs, different types of polyols are used, ranging from short (chain extender, cross-linkers) to long polyols, which are based on polyether, polyester, polycarbonate (PC) or polyacrylic architectures. Some basic chemical structures are presented in Fig. 3.
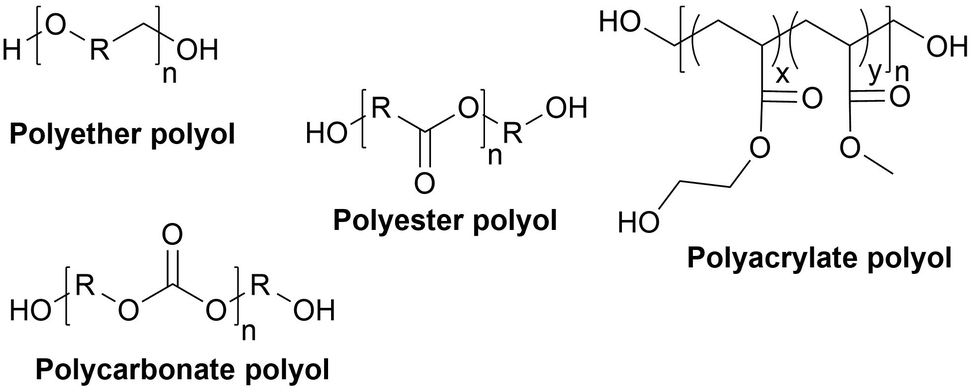 | ||
| Fig. 3 General chemical structures of main polyols for the synthesis of PUs. | ||
Polyether polyols are often produced via the ring-opening polymerization (ROP) from epoxides such as propylene oxide (PO), ethylene oxide (EO) or butylene oxide with a polyol as the initiator to obtain secondary OH. One conventional polyether polyol is polytetramethylene ether glycol (PTMEG), which has been commercialized by BASF (Germany) as PolyTHF® (PTHF) and obtained from tetrahydrofuran (THF). This soft polyol is commonly used for the synthesis of elastomeric PUs. Poly(ethylene glycol) (PEG) and poly(propylene glycol) (PPG) are also broadly used. Polyether polyols have many advantages such as low cost and low viscosity, and they can endow the final PUs hydrolytic stability and flexibility. However, they present very weak oxidative and thermal stability.59,65
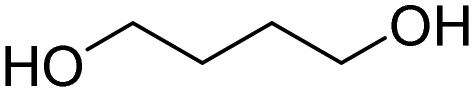
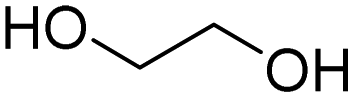
The production of polyester polyols is based on the polycondensation between polyhydroxyls and polyacid building blocks or from hydroxyl-acids (such as lactic acid), by ROP from cyclic lactone (lactide and caprolactone), or/and by transesterification from polyesters with a short polyol (glycolysis). There are numerous inexpensive and commercially available building blocks, but the most common for polyacids are adipic acid, succinic acid, phthalic anhydride, and polycaprolactone (PCL). The most common short polyols with f = 2 are ethylene glycol, 1,3-propylene glycol, 1,4-butanediol (BDO), and 1,6-hexane diol (HDO), such as Realkyd® produced by Arkema (France). Glycerol is largely used when f > 2 is desired to obtain the final thermosets.66 Different polyols can be combined to tailor their functionality, properties, and cost.67 The synthesis of polyester polyols is relatively easy to control to obtain primary OH end groups. Ester bonds lead to sensitivity to hydrolysis. Consequently, the final materials can be degraded under long-term exposure to moisture. PUs with polyester polyols generally have higher mechanical properties (rigidity) compared to polyether polyols due to the molecular rigidity of the ester bonds. However, these PUs need to be synthesized at high temperature due to their higher viscosity. Aromatic polyester polyols are remarkable due to their fire resistance. Aliphatic polyester polyols are used when UV and abrasion resistance are required, which allow higher deformation at low temperatures.20,65
PC polyols were initially prepared via the reaction of a diol and a phosgene. The most prevalent process is the transesterification of glycol and dialkyl or diaryl carbonates at temperatures of up to 200 °C. Another method is the ROP of cyclic carbonate with a polyol as the initiator. Poly(1,6-hexanediol) carbonate, obtained by the polycondensation of 1,6-hexanediol and diphenyl carbonate, is a PC polyol widely employed for the synthesis of PUs. Covestro commercialized it as Desmophen®.68 PUs with PC polyols are suitable for long-life applications because they are extremely hydrolysis and oxidation resistant.50,65
Acrylic polyols contain both vinyl and OH groups. They are obtained via the free radical copolymerization of acrylic or methacrylic acid esters, including methyl, ethyl, and butyl methacrylate. They can be copolymerized with functional groups such as styrene, vinyl ester and maleates. The free OH groups are usually added by ester functionalization in a second step. The properties of the final PUs depend on the nature of their monomers. For example, methyl methacrylate results in hardness and water and UV resistance compared to styrene, which is UV sensitive. Some monomers can provide flexibility, adhesion or solvent/grease resistance. However, due to their high costs, these polyols are only used for high-performance PU coating applications such as automotive refinishing.69 These polyols can be found on the market, for instance BASF (Germany) proposes a wide range of acrylic polyols named Joncryl®.70



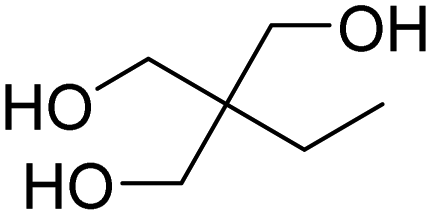
Polyamines can also replace polyols (short or long) to react with isocyanate to synthesize polyureas. Their advantage is that aliphatic or aromatic amines react faster than OH with NCO (see Table 2) and the urea bonds present higher rigidity and thermal stability. For instance, elastane fibers are poly(urethane-urea) polymers. They are prepared from diamine chain extenders, MDI and either polyether or polyester soft segments. EDA is the most common diamine chain extender.65
2.3. Main PU domains
PUs are a versatile family with various properties because of the diversity of the chemical structures of their building block. Based on these different macromolecular architectures, different PU domains exist with the corresponding structures, shapes, morphologies, and applications, such as foams, coatings, thermoplastics, and adhesives.49The foam morphology drives the final properties and behavior. Generally, F-PUFs exhibit a low density, low compressive strength, high recovery properties and high water absorption. They are commonly used for a wide range of comfort and cushioning applications such as transportation, furniture, and bedding. They have the ability to be foamed through the slabstock process or molding.76,77 R-PUFs are mainly designed for thermal insulation and energy recovery due to their high density of closed cells. These cells contain the vaporized blowing agent with low thermal conductivity. These elements provide high compressive strength, good heat stability and good barrier properties. R-PUFs are convenient for building application because of their ability to be on-site sprayed. It is also possible to manufacture R-PUF board stocks or sandwich panels.78 Between F-PUF and R-PUF, intermediate systems can be created with intermediate behaviors.
The main PU coating technology is based on a two-component system with the reaction between polyisocyanate and polyol (usually with f > 2) at ambient temperature, with an adequate catalyst in a solvent. The coating is generally sprayed as a clear coat in automotive and aviation applications.80 To reduce the emission of volatile organic compounds (VOC), the solvent can be replaced with water for amphiphilic systems. Water-borne systems are based on dispersible polyols and aliphatic isocyanates because MDI and TDI have high reactivity with water.81 Also, a solvent-free two-component PU is available for floor coating. Low viscosity is required and fillers are added to achieve the final required properties.82 There is also an oven curing system for metal coating. A blocked isocyanate is unblocked when heating and react rapidly with a polyol to form the final coating. These systems can be solvent- or water-borne, or in powder form.83 Moisture-cured systems are interesting as anti-corrosion coatings without mixing and metering. Curing reaction occurs between an isocyanate-terminated prepolymer and water to form urea, which results in hardness and resistance to water and chemicals. However, this reaction is generally long and difficult to control because it is dependent on the room temperature and humidity.84 Urethane acrylate coatings are another one-component solvent-free system. The fast prepolymer curing is induced by the UV cross-linking of free acrylates at room temperature. This reaction is often activated by a photo initiator.85
2.4. Strategies to increase PU sustainability
Sustainable development is a significant concept due to growing concerns about environmental and socio-economic issues, which aims to create a healthy and safe future for humanity.91Accordingly, PUs can also be considered as a sustainable solution to address economic and environmental concerns. For instance, in building insulation, R-PUFs are used for energy saving, which have a lightweight structure, reducing the transport and energy costs. Another example is the highly durable PU coatings, which protect and increase the shelf-life of products.92
However, the sustainability of PUs need to be increased by reducing the health issues and GHG emissions linked to their production and by considering their end-of-life. Thus, to achieve these objectives, we can find different sustainable strategies. The first global strategy is based on the valorization of plastic waste (PU and others) at the end of life, as follows: (i) conventionally by different types of recycling and (ii) by bioproduction using biotechnology strategies; in this case, the waste is used as a carbon source.93–95 The design of the polymers can be managed to ease the recycling at the end of their life such as in the case of CANs. The second popular global approach is to develop biobased PUs from biomass resources. The last approach is based on the ban of the toxic polyisocyanates to develop PUs without isocyanates, also called non-Isocyanate PUs (NIPUs).
PU waste generally comes from post-consumer products and production waste from various markets such as construction, furniture, bedding, automotive and clothing. Thus, the valorization of these products at the end of their life cycle is now essential in the context of sustainable global development. However, this valorization is limited due to the lack of efficient collection and separation processes of the different PU resources, leading to landfilling and mismanaged plastic waste, which are the main cause of plastic pollution from the land to ocean with the production of microplastics.96
Energy recovery processes such as incineration are a well-known technique, but the toxic gas and GHG emissions, such as nitrogen dioxide and CO2, and low value of the recovered products are not satisfactory, as in the case of the gasification routes for syngas production (CO and H2).97 Thus, recycling is much more desirable as a sustainable approach for dealing with PU waste in terms of the circular economy and can generate an important profit-pool according to a recent McKinsey report.98 Given that conventional mechanical recycling can be only used on some TPUs by re-melting with the loss of some thermo-mechanical properties, several additional and sustainable recycling methods have been developed. For example, chemical and biological recycling can produce monomers and oligomers from this type of plastic waste. These building blocks can be used for the (re-)polymerization of a second generation (2G) of polymers with equivalent or other applications compared to the initial polymer.29 These processes are increasingly implemented in industries, but are still not fully attractive due to the increase in cost. Thus, to overcome this economic drawback, upcycling processes can convert plastic waste into more valuable chemicals through different processes from chemistry and/or biotechnologies.99
Chemical recycling. Chemical recycling allows waste to be treated chemically to recover some monomers and building blocks, which can be reused in the synthesis of polymers of 2G in a sustainable approach. Also, mixed polymers and composites can be converted into higher quality materials compared to mechanical recycling.29
Different techniques have been developed such as hydrolysis, aminolysis, phosphorolysis, glycolysis, pyrolysis, and hydrogenation.100,101 Hydrolysis was the first chemical recycling method developed for flexible foams. PU waste is placed at high temperature and high pressure with water to form polyols, amine intermediates and CO2. After treatment, the initial isocyanates can be regenerated from the recovered amine intermediates.101 Aminolysis involves a transesterification reaction between the urethane ester group of PU foams and ammonia or amine to produce bi or polyfunctional amines and alcohols.102 Mixing PU waste with phosphoric and phosphonic acid ester leads to the formation of a liquid product containing PU oligomers with flame retardancy, adhesive properties and UV resistance.103 Based on short polyols, glycolysis has largely been developed for the recycling of R- and F-PUFs. It is based on transesterification reaction and can take place on the urethane groups or on the ester groups in the case of PUs based on polyester polyols, at high temperature in the presence of a catalyst. In this process, it is possible to recover polyol from PUFs at 190 °C and reuse them as building blocks for the synthesis of polymers.104 Pyrolysis involves the thermal decomposition of long-chain polymers into ash, gas and light hydrocarbons such as ethylene, benzene, and aniline under high pressure, which can be used for the synthesis of chemicals. Moreover, isocyanate can be recovered after first degradation stage (100–300 °C) where the urethane bonds are broken.105 Hydrogenation is similar to the pyrolysis process, but degradation takes place under high-pressure hydrogen instead of inert gas.101
Depending on the chemical recycling process, end-of-life products and production scraps can be transformed into oligo-urethanes, polyols, amine, gas, ash, and short hydrocarbons. Among these processes, only glycolysis and gasification are conducted on a large scale. However, their major drawbacks are the initial product separation, high energy consumption (high temperature and pressure), and the need for chemicals such as organic solvents and catalysts.101
Biological recycling. An emerging recycling route catalyzed by biological entities such as bacteria, fungi, and enzymes has been reported in several reviews.31,106–109 Biological recycling is greener than the conventional chemical recycling and does not require high temperature and chemicals compared to chemical recycling. Efficient poly(ethylene terephthalate) (PET) depolymerization has been achieved by Carbios (France) on an industrial scale.110 However, the case of urethane groups is more challenging, given that these chemical groups are much more robust than ester bonds. Consequently, PUs from polyester polyols are much more susceptible to biological degradation than that from polyester polyols.
Most researchers use microbial communities to recycle PU foams and enzymes for the depolymerization of coatings and TPUs. It was observed that PU foams from polyether polyols were easily degraded by fungi due to the easy penetration of their mycelium into the pores of PUs. Alternatively, the capacity of bacteria to form biofilms was used to degrade PU foams from polyester polyols.101 Enzymatic degradation is also interesting due to the possibility to control bond cleavage and generate building blocks for re-polymerization. Some recent studies reported that urethane bonds can be bio-lysed by combining esterases and amidases. This approach showed promising results.111–113 Furthermore, different sustainable approaches have been recently developed, e.g., involving an initial biochemical step to obtain building blocks by enzymatic depolymerization, followed by a chemical step to obtain a new generation of polymers in a perpetual and sustainable “chem-biotech” cycle.111
Environmental impact of plastic waste treatment. Particularly, in terms of CO2 emissions (carbon footprint), the environmental impact of each recycling method varies. This can be studied according to plastic production, product manufacturing, waste collection, transport, sorting and recycling steps. The environmental impact depends on the energy in the process, the quality, and the quantity of the output products and emissions. The plastic waste processing methods that produce the most CO2-equivalent emissions correspond to incineration, incineration with energy recovery, landfill, pyrolysis, mechanical recycling, solvolysis and dissolution/precipitation, in decreasing order.29,114 Results show that breaking chemical structure or bonds and obtaining low-quality end products have a higher environmental impact. Moreover, if the CO2 costs were factored into the product price, circular options will be more cost-effective than producing neat polymers from fossil resources.29
Upcycling. To date, the low cost of producing virgin monomers from fossil carbon feedstocks makes chemical or biological recycling industrially very unattractive. Thus, creating value-added materials and chemicals from plastic waste can be an answer to this economic drawback. The upcycling of unsorted mixed plastic wasted into value-added sustainable materials has been developed using a chem-biotech approach.111 In the first step, engineered mixtures of enzymes are used to depolymerize polymers. Mixed microbial cultures convert the plastic waste into value-added products and building-blocks, before (re-) polymerization or formulation of a 2G polymer, with or without a new architecture.115
Due to the large variety of PU complex structures, chemical and biological degradation lead to a wide variety of end products including organic acids, alcohols, aromatics, and amines such as EG, BDO, adipic acid, 4,4′-methylenedianiline (MDA) and 2,4′-toluene diamine (TDA).111 These monomers can be polymerized into PUs, polyesters, polyamides.116 MDA and TDA can be extracted and converted into MDI and TDI via the phosgene route for the synthesis of PUs.106
They can also serve as a carbon substrate for microbial communities to be metabolized into other monomers (e.g., BDO into succinic acid).117 EG can be used as a substrate by microorganisms for the production of PHAs and glyoxylic acid.118 EG has also been converted by modified bacteria into engineered extracellular building block hydroxyalkanoyloxy-alkanoate (HAA), which has been used as polyol in the synthesis of TPUs.119
These upcycling strategies for mixed plastic waste can improve the profits from cheap carbon sources and improve the carbon yield compared to other recycling processes. However, the limitations of this highly attractive and eco-friendly chem-biotech approach are the low degradation activities of microbes and enzymes. Secondly, the separation of the PU degradation product has low efficiency and is expensive. Thirdly, the toxicity of diamines such as MDA and TDA reduces the biocatalytic activity and growth of mixed cultures.116,120
Reprocessable thermosets. The specific design of new macromolecular architectures considering the end of life of these materials can be the answer to ease the recycling of waste and to increase the sustainability, such as in the case of the CANs. CANs present outstanding behavior and interest as sustainable thermoset alternative materials.121 These polymers are based on dynamic covalent bonds to form reversible networks, for instance, with a thermal input.122 These CANs can be dissociative or associative. In the case of dissociative CANs, their bonds are completely broken in the first step before being reformed, such as in the case of Diels Alder chemistry.123 Associative CANs also exist, where their bonds are simultaneously broken and formed in exchange reactions, such as for vitrimers.124 This chemistry confers self-healing properties and high recyclability to reuse them for several cycles. Various recent reviews show that these properties can be introduced to PUs.125–127 Besides, it also provides access to shape-memory materials, especially in the biomedical field.128,129
In the particular case of PUs, the urethane group can show dynamic behavior depending on its nature and the urethane/free alcohol ratio via tanscarbamoylation. Moreover, it has been shown that the appropriate catalyst can enhance the associative and/or dissociative carbamate exchange mechanisms depending on its nature.130 Recent studies have proven the possibility of the reuse and recycling of PU foams by compression molding or extrusion due to this dynamic behavior.131,132
Due to environmental concerns and limited petroleum resources, there has been interest in replacing fossil raw materials with renewable alternatives, e.g., biomass. The majority of renewable carbon is embedded in plant-derived macromolecules such as cellulose, lignin, and starch or in smaller molecules such as terpenes, vegetable oils and carbohydrates or even in biogenic CO2. There are two strategies for moving towards polymers using renewable resources.5
The first pathway is based on a type of “copy and paste” strategy, developing conventional chemical bricks, but in this case from biomass, with a rather easy and fast implementation. This strategy is often based on the sugar fermentation process as in the case of the bioproduction of ethanol in high yield by alcoholic fermentation, for the synthesis of conventional polymers. As an example, ethylene is obtained from bioethanol by dehydration and can be used as a monomer for the production of biobased low-density polyethylene (LDPE) as done by Braskem (Brazil). Ethylene glycol can be obtained from biobased ethylene and acts as a diol for PET, as in the case of the “plant bottle” developed by the Coca-Cola Company. However, the major roadblocks are biomass conversion efficiency and competitiveness.133 However, this competitiveness can vary with the emergence of strong challengers, e.g., new cheap fossil resources, as in the case of the recent strong development of shale gas production (mix of C1 to C4) in North America. This fast development is often unpredictable.
The second strategy is based on the conversion (with or without chemical modifications) of renewable feedstock into novel polymers with “new” macromolecular architectures and with sometimes “new” properties and applications, compared to conventional fossil-based polymers. The diversity of biomass-derived molecules or biomacromolecules is an advantage, but it is a challenge to achieve a controlled molar mass and specific microstructure architecture to finally develop specific properties to find a market. Thus, to solve these issues, efficient catalysis, controlled polymerization process and polymer processing are required. Given that biobased building blocks or biopolymers present interesting chemical structures and are widely available, a wide range of new materials and additives with specific properties and application is now produced. For example, different biobased polylactides (PLAs) with controlled structures and properties are now produced on an industrial scale starting from biomass-derived lactic acid.5
According to the different resources, processes and approaches, numerous and an increasing quantity of biobased compounds are produced and commercialized in the last few decades, such as biobased polyols and polyisocyanates.
Biobased polyols. Polyols are the main component for the synthesis of PUs, which is biobased. In 2019, they represented a global production of around 22 Mt. The demand for biobased polyols is increasing, and the different strategies currently employed for their production has recently been summarized.134
The growing interest in carbohydrate-containing biomass has led to the large-scale production of various biobased monomers such as EG, 1,3-propanediol (PDO), BDO, isosorbide, succinic acid, azelaic acid, sebacic acid, terephthalic acid, 1,5-pentamethylenediamine (PDA), and 1,6-hexamethylene diamine.
In the current approach to increase the renewable content in polymers, the first step is to replace the initiator polyol for the synthesis of polyethers with biobased compounds such as sorbitol or sucrose. This leads to the formation of partially renewable polyols and PUs. Fully biobased polyether polyols can be synthesized via the polycondensation of PDO from glucose (Susterra®, DuPont Tate & Lyle BioProducts, US) to produce PDO (Velvetol®, WeylChem-Germany).135,136 BASF used the biobased BDO from Genomatica (US), also known as “BioBDO”, to synthesize polytetrahydrofurane polyester polyol by polycondensation (PolyTHF®).137,138 These short diols can also be used to produce fully biobased polyester polyol via condensation with biobased dicarboxylic acids such as adipic and succinic acid such as Biosuccinium® commercialized by Roquette (France) in association with DSM (The Netherlands).139
The interest in vegetable oils, and generally on oil-chemistry producing biobased PUs has particularly increased in the last two decades. Several reviews reported the strong evolution of the synthesis of PUs from vegetable oil.49,87,140–146 The use of vegetable oil for the synthesis of polyols or polyols based on fatty acids is in full expansion because this raw material is currently abundant, cheap, and renewable with a growing production. This market was estimated to around US$7 billion in 2020 with an annual growth of 7%.147 A large range of fatty acids can be obtained via the hydrolysis of vegetable oils. They have several active and functional chemical groups such as double bonds, which can be used to develop very rich chemistry. For instance, the epoxidation of double bonds by H2O2 and the opening of oxiranes under acidic condition are the most common route for the synthesis of polyols.148 Several grades of polyols based on vegetable oils are already commercially available, such as Radia (Oléon-France) or BiOH® (Cargill-US).149,150 They can be used to improve the hydrophobicity of PUs. Dimer fatty acids are interesting because of their high molar mass. Dimerization is mainly based on the Diels Alder reaction between unsaturated fatty acids, followed by the separation of the different obtained chemicals. Mainly aliphatic and cycloaliphatic C36-dicarboxylic acids are formed. Depending on the feedstock and reaction conditions, this reaction also leads to the formation of bicyclic, aromatic dimer acids or mixture of saturated and unsaturated branched C18-carboxylic acids with pending chains to bring softness.151,152 Dimerization of fatty acid can take place to lead to the production of cyclo-polyols (Priplast™, Croda Germany) or through polycondensation with glycol to form polyester polyols (Sovermol®, BASF Germany).153,154 These polyols are also used to improve the hydrolytic stability and aging properties in PUs.155,156
Biobased polyisocyanates. During the last few decades, different biobased isocyanates have emerged at the industrial level to develop fully biobased polymers. Several polyisocyanates are commercially available such as dimer fatty acid-based diisocyanate (DDI), lysine derivative diisocyanate (EELDI) and sugar-based diisocyanate (PDI). Their renewable contents are 91%, 75% and 71%, respectively,140 and their chemical structures are presented in Table 4. Other diisocyanates from available biomass such as different fractioned lignins or isosorbide (bicyclic diol) have also been studied at an academic level in recent years.157 Recently, Covestro (Germany) achieved fully biobased aniline production from sugar using the chem-biotech approach.158,159 This precursor is involved in the synthesis of MDI. GHG neutral pMDI has been recently developed by BASF (Germany) under the brand Lupranat® ZERO from renewable raw materials.160
| Name | Chemical structure | Precursor | Biocontent | Commercial name and supplier | Applications |
|---|---|---|---|---|---|
| Pentamethylene diisocyanate (PDI) |
|
Glucose | 70% | STABiO™ D-370 N (Mitsui) | TPUs, flexible foams and coatings |
| PDI-trimer |
|
Glucose | 71% | DESMODUR® Eco 7300 (Covestro) | Hardener for PU coatings |
| Dimeryl diisocyanate (DDI) |

|
Dimer fatty acids | 91% | DDI 1410 (BASF) | Outdoor |
| Lysine ethyl ester diisocyanate (EELDI) |

|
L-Lysine | 75% | LDI (Alfa-Aesar™) | Biomedical |
| Polymeric 4,4′-methylenediphenyl diisocyanate (pMDI) |

|
Unknown | Unknown | Lupranat® ZERO | R-PUFs (thermal insulation) |
Fatty acid-based isocyanates have been studied because of their attractive chemical structures based on long aliphatic chains. Soybean oil was brominated and converted by silver cyanate (AgNCO) to diisocyanate.161,162 Aliphatic diisocyanate was synthesized from oleic acid via Curtius arrangement.163,164 Castor oil was converted to aliphatic diisocyanate via a non-phosgene route.165 Diisocyanates can be produced from dimer fatty acids via the formation of dimer diamine. Dimer fatty acid can react with ammonia to be converted into dinitrile, and then be hydrogenated in dimer diamine with a catalyst.166 There are two routes to convert dimer diamine into dimer diisocyanate, as follows: (i) phosgenation167 and (ii) reacting dimer acid chloride with sodium azide.168 The most commercialized diisocyanate form dimer fatty acid is DDI from BASF for the synthesis of coatings and elastomers.140 DDI has low toxicity, low vapor pressure and high resistance to moisture and UV light.151
Polyisocyanates based on the lysine amino acid have been studied because of the non-toxic products from their degradation (amino acid), which is advantage for biomedical applications. They also have low vapor pressure, which is a key point, with these toxic compounds, for handling and processing.169–171 Polyisocyanates such as lysine methyl ester diisocyanate, EELDI and lysine triisocyanate are products from amine-terminated lysine ester phosgenation.172–174
PDI was the first commercialized biobased diisocyanate. Mitsui Chemicals (Japan) commercialized it as STABiO™.175 In the synthesis of PDI, starch is converted into PDA through biotechnological and chemical processes before being phosgenated.176–178 The trimeric PDI has been commercialized since 2015 by Covestro (Germany) as DESMODUR® Eco 7300.179 This hardener shows good resistance to aging, scratching and chemical treatment.180 Isosorbide and isomannide have been recently studied for the synthesis of diisocyanates. Also, 1,4:3,6 dianhydrohexitols are converted into the corresponding 2,5-diamino compounds before phosgenation into diisocyanate.181,182 Another route to synthesize isosorbide-based isocyanates is via Curtius rearrangement after esterification with succinic anhydride.183
Furan-based diisocyanates have been synthesized since 1962 starting from methyl furoate and hydroxymethylfurfural.184–189 Lignin is the second most abundant biopolymer and largely studied for its derived aromatic components. Lignin-derived phenolic acids such as vanillic acid and syringic acid have been used as precursors for the synthesis of diisocyanates via Curtius rearrangement.190,191
Environmental impact of biobased PUs from renewable resources. Beginning-of-life options are as important as the end-of-life scenarios. Indeed, feedstock extraction, processing energies and transports have a strong environmental impact. Consequently, the interest in non-food, renewable resources for the production of PUs is increasing due to their enhanced sustainability compared to fossil fuel extraction and refining.192 Various renewable feedstocks can be used for the synthesis of polyols, where the choice of feedstock is important. A life cycle assessment (LCA) following the cradle-to-factory gate approach compared different PU coatings from fossil-based conventional polyester polyols and biobased polyester polyols obtained with renewable monomers such as succinic acid, glycerol and PDO. It was found that the biobased PU had the lowest environmental impact. The GHG emissions and non-renewable energy use were reduced by 75% and 35%, respectively.193 However, this approach did not take consider the side effects of feedstock farming such as acidification and eutrophication due to fertilizer and pesticide use.192 Another LCA following the cradle-to-gate scenario on the production of polyols for PUs from rapeseed oil showed that although biobased polyols have better environmental impact compared to fossil-based polyols, their effects can be negative on land use, marine eutrophication and ecotoxicity.194 Recently, an LCA on biobased R-PUFs from azelaic acid and lignin proved that the final properties of the materials related to their application are the key factor for higher environmental performances. In the case of thermal insulation, the lowest thermal conductivity and density present the lowest environmental impact.195
Sustainability is not only based on the origin of the feedstocks, but also about the process, use and end of life. Biobased polymers contribute to a more sustainable economy, but some drawbacks remain, including the cost and efficiency of biorefineries and the negative agricultural impacts. Thus, a combination of bioeconomy and circularity (circular bioeconomy) is needed together with emerging recycling technologies such as biological, chemical and upcyling.192
R&D on the synthesis of safer PUs without isocyanate has been strengthened over the two last decades and companies around the world are now under pressure to minimize their isocyanate consumption and develop non-toxic compounds. The most promising approach to synthesize NIPUs seems thus far to be the polyaddition of cyclic carbonates and amines, as described in numerous reviews.197–202 Given that the primary and secondary OH groups are formed by opening cyclic carbonates, NIPUs are called polyhydroxyurethanes (PHUs). One of the great advantages of this approach is the low toxicity of cyclo-carbonates, which are based on CO2 consumption during their synthesis. The cyclo-carbonate and amine components can be obtained from biobased resources, which can lead to the formation of cross-linked materials such as fully biobased PUFs.203,204 However, there are other routes to synthesize NIPUs, such as polycondensation, ring-opening polymerization and rearrangement.205 Polycondensation (or transurethanization) involves the use of a bifunctional carbamate, a diamine and a polyol. NIPUs can also be synthesized via the ring-opening polymerization of 6–7 membered cyclic carbamates, but this involves phosgene, which is harmful. Several rearrangement reactions can be employed for the synthesis of NIPUs including Curtius, Hofmann, and Lossen rearrangements, but isocyanate intermediates are formed, which is not a significant improvement over the conventional route.206
3. Sustainable cycloaliphatic building blocks for the synthesis of PUs
The use of sustainable raw materials for the synthesis of polymer is the result of a long trend over several decades. Today, this approach is increasingly evident, as shown herein previously. Thus, various cycloaliphatic monomers or building blocks from biomass such as terpenes, vegetable oils and carbohydrates have been intensively studied for the synthesis of PUs over the past two decades. Examples of cycloaliphatic molecules from these families are limonene, pinene, abietic acid, isosorbide, glucose, and dimers of fatty acids.207,208 They are often chemically modified to become polyols, diisocyanates, and diamines, which are suitable for the synthesis of PUs. Certain mono-functional compounds such as cholesterol, which is a lipid of animal origin, and its derivatives are also used in the synthesis of PUs.209 The synthesis of sustainable cycloaliphatic PUs and NIPUs is presented in this section, depending on the type of sustainable feedstock, which is mainly, but not only, biobased, and their corresponding chemical modifications. However, because the production of building blocks from plant metabolism often has low productivity,210 microbial production can be a strong economic and sustainable alternative. Employing an upcycling strategy, it will soon be possible to widely bioproduce these cycloaliphatic building blocks from different enzymatic systems and the combination of metabolic engineering of mixed microbial cultures from plastic waste as a substrate.115 However, thus far, the meaningful bioproduction of cycloaliphatics is only possible by extraction from biomass.211,2123.1. From terpenes
Terpenes (or terpenoids or isoprenoids) are a part of the hydrocarbon-rich group of molecular biomass, representing over 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds. Terpenes are composed of a hydrocarbon backbone and isoprene (2-methyl-1,4-butadiene) units. They can also have different active groups such as double bonds, OH and carboxyl groups. They can be acyclic or cyclic, consisting of 1 to 14 aliphatic rings.213 Terpenes are present in different types of biomass such as plants, bacteria, animals, and fungi. Terpenes are biosynthesized by many plants and trees, and are found in essential oils, resins, pigments, and latexes, among others.
000 compounds. Terpenes are composed of a hydrocarbon backbone and isoprene (2-methyl-1,4-butadiene) units. They can also have different active groups such as double bonds, OH and carboxyl groups. They can be acyclic or cyclic, consisting of 1 to 14 aliphatic rings.213 Terpenes are present in different types of biomass such as plants, bacteria, animals, and fungi. Terpenes are biosynthesized by many plants and trees, and are found in essential oils, resins, pigments, and latexes, among others.
The easiest approach to produce a mixture of terpenes is extraction from turpentine. This is the volatile fraction of pine resin, with a production of 350 kt year−1.214 Rosin is the non-volatile part of resin. Terpenes are composed of a functionalized cycloaliphatic structure, which can undergo polymerization with or without prior chemical modification.215
Bacteria such as Escherichia coli possess a native metabolic pathway for isoprene, the terpene elementary unit. The bioproduction of high-value terpenes has been achieved using recombinant terpene biosynthesis modules, metabolic engineering and site-directed mutagenesis. New terpene architectures have been extended through this approach.211
Terpenes are abundant, inexpensive, biodegradable, easy to isolate and do not compete with food or feed. They have been used for various applications such as fragrances, flavors, perfumes, pharmaceuticals, drugs, dyes, varnishes, and biofuels.216 The use of terpenes as building blocks for the synthesis of polymers has become attractive, as shown by several recent reviews on this topic.217–221 The most commonly studied terpenes for polymerization are α- and β-pinene, limonene, and myrcene because they can be easily isolated from the turpentine in orange peel in reasonable amounts. Besides, their double bonds are interesting for the synthesis of polymers because they can react directly by ionic or radical polymerizations. They can also be functionalized to develop different polymers by polycondensation or ROP.220
Production/extraction. Limonene is a chiral molecule produced by over 300 plants. The (R)-enantiomer represents over 90% of orange peel oil, which is a waste product from the citrus industry. It can also be obtained by the isomerization of pinene.217 Recently, the bioproduction of limonene from recombinant Escherichia coli has been achieved using glucose and glycerol as carbon sources.222,223
Up to 85 kt of limonene is produced annually worldwide, which is commonly used in the fragrance, flavor and pharmaceutical industries. Limonene possesses one double bond in its ring and another one as a side group. Because limonene is an abundant and inexpensive monomer, these alkyl groups are attractive for cationic and condensation polymerization and for chemical modifications before polymerization.217
Chemical modifications. Several chemical modifications have been developed on limonene for the synthesis of cycloaliphatic PUs and PHUs. The global pathways are presented in Fig. 4 with different parts from 1 to 8.
 | ||
| Fig. 4 Review of the chemical modifications of limonene for the synthesis of PUs (from 1 to 8). | ||
Click chemistry can be considered as a green approach using the principles of green chemistry.18 Click reactions exhibit high yields, short reaction times, high chemoselectivity and simple reaction conditions.224 Photochemical click chemistry has been carried out to synthesize limonene-based polyols (1) and (4), dithioester (2) and diamine (5) for the synthesis of PUs.
In detail, various mercaptans such as 1-thio-glycerol react via thiol-ene addition with limonene under UV radiation in the presence of a photo initiator catalyst to form polyol (1).225 R-PUFs were prepared by mixing limonene-based polyol (1) with another commercial one, and pMDI and water. High thermal stability higher than 250 °C was achieved with a Tg above 200 °C due to the rigidity of the limonene ring.226
Limonene dimercaptan (3) was synthesized as a reaction intermediate for the synthesis of polyol (4). Firstly, limonene reacted by thiol-ene addition with thioacetic acid to form dithioester (2) before being transesterified to obtain dimercaptan (3). The triazabicyclodecene (TBD) catalyst was used to reduce the reaction time compared to the original base-catalyzed reaction in a green chemistry approach.227 Limonene dimercaptan (3) has often been transformed into polyol (4) for the synthesis of R-PUFs via thiol-ene addition with alkoxylated compounds such as 2-allylphenol, eugenol and glycerol-1-allylether.226,228
PHUs have also been synthesized from dithiol (3) and carbamate monomers from hydroxamic fatty acid derivative with two double-bonded end groups by thiol-ene addition. It has been shown that the bulky structure of limonene reduced the crystallinity of NIPU, leading to amorphous polymers with a low Tg of around −13 °C.229
Thiol-ene addition has also been reported for the synthesis of diamines (5). It was used as a reaction intermediate for the synthesis dicarbamate (6) to synthesize NIPUs. Cysteamine hydrochloride reacted with limonene to from diamine (5) under UV radiation with dimethylpropylamine as the photo-initiator. Then, diamine (5) reacted with dimethyl carbonate to form dicarbamate (6) with TBD catalyst. NIPUs were synthesized from dicarbamate (6) with polyol (1) or TBD-catalyzed fatty acid-derived diols. The bulky cycloaliphatic units of limonene dicarbamate (6) and diol (1) led to the formation of short polymers with low molar masses (6150–7900 Da), crystalline domains and Tm of up to 69 °C.230
Another way to produce limonene-based PHUs is the reaction between biscyclocarbonate (8) with acyclic or cycloaliphatic polyamines.231,232 This monomer (8) was produced from limonene dioxide (7), which can be obtained by various catalytic routes.233–236 Several pathways for the production of biscyclocarbonate (8) have been studied, but the most attractive one is the catalytic carbonization of epoxide (7) with CO2. Low molar masses were obtained (from 600 to 1840 Da) for NIPUs obtained by reaction between (H) and diamines (f = 2).237–241
For cross-linked PHUs synthesized from limonene biscyclocarbonate (H) and polyamines with f > 2, the Young's modulus and Tg increased with amine functionality and flexibility. The purification of limonene biscyclocarbonate (8) by recrystallization significantly increased the thermoset properties of the NIPUs. Very rigid and brittle polymers were obtained with an increase in the biscyclocarbonate (8) content due to the rigidity of the cycloaliphatic structure of limonene.232
Some other modifications of limonene into polyol, diamine or diisocyanate can be found in the literature. However, to the best of our knowledge, the synthesis of PUs using these modified building blocks has not been reported to date. For instance, short and long diol syntheses have been described through the hydroformylation of limonene. In the second step, two routes were developed to form a limonene-based diamine. This building block can be used as a precursor for the synthesis of diisocyanates or NIPUs.242 Besides, a patent describes the modification of limonene to form diisocyanate.243 However, the production of PUs has not been reported using these different methods.
Production/extraction. There are three main types of rosins, i.e., gum rosin, wood rosin and tall oil rosin. Gum rosin is the non-volatile fraction of pine resin. Wood rosin is recovered from aged pine stumps. Tall oil is an abundant and inexpensive by-product of Kraft processing of softwoods in the paper industry, which is composed of rosin and fatty acids.217
The annual global rosin production is over 1000 kt. It is composed of rosin acids (about 90%) and about 10% of by-products. Rosin acids consist mainly of isomeric abietic-type acid and its isomers (40–60%) and pimaric-type acids (9–27%), as shown in Fig. 5. Abietic acid is characterized by a rigid aliphatic tricyclic structure with one or more unsaturations.244 Its hydrophobicity allows it to be used as a water-resistant agent for paper coating, or in antifouling, adhesive and printing inks. In the last two decades, rosin chemical modifications have been studied for the synthesis of sustainable cycloaliphatic polymers.245
 | ||
| Fig. 5 Chemical structures of main rosin acids. | ||
Chemical modifications. The main chemical pathways are presented in Fig. 6 with different parts, from 9 to 29. Early studies on the modification of rosin acids for the synthesis of PUs are based on Diels Alder reaction to produce diacids or acid anhydrides. Firstly, abietic acid, which is the main component of rosin acids, is isomerized at high temperature to form levopimaric acid (9). Then, Diels Alder reaction at high temperature with excess maleic anhydride, fumaric acid or acrylic acid leads to the formation of maleopimaric acid (MPA) (10), fumaropimaric acid (FPA) (11) and acrylopimaric acid (APA) (12), respectively.246,247
 | ||
| Fig. 6 Review of abietic acid chemical modifications for the synthesis of PUs (from 9 to 29). | ||
In the synthesis of rosin-based Pus, the MPA (10) monomer has been widely used to produce various building blocks for the production of PUs such as polyester polyols (13) and (17), or cyclocarbonates (19).
Different polyester polyols (13) have been synthesized via the esterification of MPA (10) with various diols, with different molar masses. R-PUFs were designed from pMDI, polyester polyols (13), polyether polyols, blowing agent, surfactant and catalysts. It appeared that rosin-based polyols (13) and low molar mass diols resulted in the formation of very thermally stable foams.248 Also, it seems that R-PUFs from rosin-based polyester polyols (13) were more resistant to compression and exhibited lower thermal conductivity than commercial foam due to the rigid cycloaliphatic structure of rosin.249
MPA (B) was also the starting point to design a specific rosin-based diol (17) as a chain extender for the synthesis of TPUs. Firstly, MPA (10) reacted with 4-aminobenzoic acid by imidization to form a diacid (15). Acids were chlorinated (16) with thionyl dichloride and DMF at 85 °C for 5 h. Then, EG reacted with acyl chloride (16) by esterification to form a diol (17). Prepolymers were synthesized via the reaction between IPDI and PTHF (2 kDa). Rosin-based diol (17) was used as a chain extender. Atomic force microscopy (AFM) studies showed that the HSs domains increased with an increase in the content of rosin-based diol (17) due to the rigidity of the aliphatic rings. Also, imide groups participated in inter- and intra-molecular interactions, increasing the thermodynamic incompatibility between the HSs and SSs, thus developing microphase separation.250
MPA (10) was further described as the origin of a specific rosin-based cyclic carbonate (19). MPA (10) was esterified with epichlorohydrin, and then reacted with aqueous sodium hydroxide and tetrabutyl ammonium bromide as a phase transfer catalyst to form triglycidyl ester of maleopimaric acid (18).251 This monomer (18) has been used in cycloaddition with carbon dioxide to form a rosin-based cyclic carbonate (19). PHUs were synthesized via the ROP between this monomer (19) and various aliphatic diamines or triamines. The Tg is correlated with the chemical structures of selected amines. A higher Tg (73 °C) was obtained with cycloaliphatic rigid amine (isophorone diamine) compared to the lowest (18 °C) value obtained for linear flexible aliphatic amines (triethylenetetramine).252
MPA (10) was also studied for polymer grafting to increase the thermal stability of R-PUFs due to the rigidity of rosin. MPA (10) was grafted by imidization on the amine groups of amino-functionalized polydimethylsiloxane. R-PUF was produced from pMDI, modified polysiloxane, polyether polyol, blowing agent, surfactant and catalysts. The free amino groups of modified polysiloxane reacted with isocyanates to form urea links. SSs based on polysiloxane reduced the R-PUF compression resistance, whereas rosin-grafted polysiloxane enhanced it. The improvement in mechanical properties was attributed to the rigidity of the cycloaliphatic MPA structure and imide bonds.253
APA (12) and MPA (10) were produced from levopimaric acid by Diels Alder reaction. The esterification of APA (12) with diethylene glycol gave trifunctional polyester polyol (14) for the production of waterborne PUs (WPUs). Then, rosin-based polyester polyol (14) reacted with TDI and a polyether polyol to form a prepolymer. Chain extenders were added to complete the reaction before neutralizing and dispersing the mixture in water, in a green chemistry approach. It appeared that the corresponding PU films with high rosin polyol content (14) presented improved mechanical and thermal properties, water resistance and antibacterial properties. These results are attributed to the rigid structure of rosin and cross-link density of PU network.254
Diacid dimer (20) was largely studied as a platform molecule from abietic acid for the synthesis of PUs and design of other building blocks. Diacid dimer (20) was produced via abietic acid cationic dimerization.244 Using this molecule, abietic acid-based diisocyanate (AADI) (21) was synthesized in two steps via Curtius rearrangement. Subsequently, it was used to synthesize thermoset PUs with PCL diol and N,N,N′,N′-tetrakis(2-hydroxypropyl)ethylenediamine (TKED) as a chain extender. In this case, the HSs were composed of TKED and rigid cyclic aliphatic structure of AADI (21). The impact of the HS content in PU has been studied. It has been shown that increasing the HS content favored microphase separation. By increasing the AADI (21) and TKED content, the cross-linking density also increased. Also, the chain mobility decreased and the steric hindrance increased, leading to higher mechanical and thermal properties.255
Rosin dimer (20) was also modified into a short diol (23) for the synthesis of TPU as a rigid chain extender. Dimer (20) was chlorinated with oxalyl chloride (22) before being esterified with EG. Then, the prepolymer of MDI and polybutylene adipate (PBA) was extended with rosin-based diol (23) at different contents. Due to the high molar masses and high chain entanglements of the bulky cycloaliphatic structure of the new diol (23), the thermal stability and the mechanical properties of the final TPUs increased.256
Other polyols, (24) and (25) from abietic acid, have also been described in recent studies. Tall oil with different rosin contents ranging from 2% to 20% reacted with diethanolamine and triethanolamine secondary amines or OH groups to form polyols, (24) and (25), respectively. They have been used to extend the TPU prepolymer of MDI and polyadipate. The degree of phase separation was higher for TPU with the rosin-based chain extenders (24 and 25) compared to EG due to its rigidity.257,258
The addition of abietic mono acid during the preparation of PUs was also intensively studied. The carboxylic acid and isocyanate groups reacted to produce an amide linkage (26) for the synthesis of R-PUF with significant compression strength improvement due to the rigid cycloaliphatic structure.259 In the formulation of TPUs, the addition of rosin resin in the mixture with BDO, as the chain extender, increased the HS content and HS/SS phase separation because of the rigidity of the urethane and amide bonds, and the cycloaliphatic structure of rosin. The molar mass, crystallization time, viscosity, rheological, mechanical and adhesion properties increased with an increase in rosin content.260–262 It was shown that after a certain rosin amount (50 eq.%), the storage modulus decreased because of the rosin bulky structure, which increased the separation of the linear PU chains.263 Monomer with imide-ureas (27) were produced via a second amide reaction with isocyanate groups. These linkages were described as HSs due to their rigidity and ability to form hydrogen bonds. Similar results to that with rosin acid as the chain extender were observed, where an increasing rosin content led to a higher molar mass and viscosity.264
Recently, hydrogenated abietic acid (28) was recently used in the acrylate formulation of PUs with IPDI and 2-hydroxyethyl acrylate. The bulky and rigid cycloaliphatic structure of rosin decreased the volume shrinkage properties compared to the linear structures. Also, the imide-urea linkages (29) formed strong hydrogen bonds, increasing the adhesion to glass, PET, PVC and PC by up to 50% compared to the commercial ref. 265.
Production/extraction. Pinene is an abundant bicyclic monoterpene with two structural isomers, i.e., α-pinene and β-pinene, which represent between 75% to 90% of softwood essential oils.266 Turpentine is a volatile by-product from the papermaking kraft process. This is the largest and cheapest source of terpenoids from biomass, with a production of 360 kt/year.267 Turpentine is composed of different terpenoids, such as α-pinene (75–85%), β-pinene (up to 3%) camphene and limonene (5–15%). Pinene can also be found in other plants or can be biosynthesized from engineered microbial communities from glycerol, which can be obtained after plastic waste recycling (chemical or biological).268 Various applications have been developed from neat pinene in medicine, fuel production and flavor or fragrance.269
Chemical modifications. Fig. 7 presents the chemical modification of pinene for the synthesis of PUs with different parts, from 30 to 44. Pinene-based polyols (36, 35), diamines (31) and diisocyanates (42, 43, 44) have been developed as particular PU components.
 | ||
| Fig. 7 Review of the chemical modification of pinene for the synthesis of PUs (from 30 to 44). | ||
Two polyols (36, 37) have been synthesized from α-pinene-based epoxide (E) opened with diamine or polyol, respectively. Firstly, the acid-catalyzed isomerization of α-pinene led to the formation of terpinene (32).216 Then, it reacted with maleic anhydride to form terpinene anhydride (33) by Diels Alder reaction.270 ROP with epichlorohydrin led to the formation of pinene-based epoxy resin (34).271 Then, the secondary amine of diethanolamine reacted with epoxy groups of compound (34) to form a polyol (35) for the synthesis of cross-linked PUs. Flexible transparent films were obtained with Tg in the range of 5 °C to 37 °C.272 These films showed good chemical resistance and mechanical properties due to their cross-linked structure after curing. Another pinene-based polyol has been synthesized from epoxy resin (34). Epoxy groups were opened with a diol or triol to form a polyol (36) under acidic conditions.273 WPUs have been synthesized from this polyol (36) with HDI trimer. Flexible transparent films were obtained with a Tg in the range of 35 °C to 40 °C and high water-resistance and adhesion properties.274
Pinene-based diamines (31) have been designed to synthesize fully biobased NIPUs. Firstly, N,N′-diacetyl-p-menthane-1,8-diamine (30) was synthesized from turpentine with acetonitrile under acid conditions. Then, amide bonds were hydrolyzed under the catalysis of sodium hydroxide solution in EG to form p-menthane-1,8-diamine (MDA) (31).275,276 Biobased PHUs were developed by the reaction of cyclic carbonated soybean oil with MDA (31) at 150 °C for a long time (60 h), similar to the synthesis of NIPUs, and without solvent, following a green chemistry approach. Good thermal stability was obtained. Dynamic mechanical analysis and gel content measurement confirmed that the curing was uniform and the cross-linking density was higher at the used stoichiometry.277
The synthesis of pinene-based diisocyanates has been recently patented. Several routes have been studied, starting from α-pinene or β-pinene. Firstly, pinene was oxidized to pinene-derived ketone (37, 38). After a subsequent reaction step, pinene-derived diamines were formed (39, 40, 41). They were converted via a phosgene route into diisocyanates (42, 43, 44). These diisocyanates can react to form PUs.278
Production from biomass and waste. Betulin is a natural pentacyclic lupene-type triterpene, which can be found in more than 200 plant species, especially in the Betulaceae family. Chiefly, large quantities of betulin can be extracted (from 10 to 45 wt%) from the outer bark of white birch. Given that bark extractives are mainly hydrophobic, debarking is necessary to avoid issues in pulp industries. Bark is usually burned for energy recovery, but its extract can be highly valuable.279,280 The bioproduction of betulin has recently been developed by metabolic engineering with glycerol and ethanol as the substrate, which can be obtained from plastic waste biorecycling.210
Neat betulin has been widely studied for pharmacological applications such as anti-cancer, anti-HIV, and cosmetic applications.281 For the extraction of biomass, several methods of sublimation with organic solvent have been studied.282 The interest in the synthesis of polymers using betulin is increasing. Indeed, its high melting point (256 °C) is due to its rigid aliphatic pentacyclic structure based on 30 carbon atoms. Besides, betulin possesses two OH groups (potential bifunctionality) and an isopropenyl group, which can be interesting for different chemical modifications.283
Chemical modifications. The kinetics of the polyaddition reaction between betulin OH groups and polymeric MDI isocyanates groups were studied for the synthesis of PUs. Different solvents and temperatures have been tested using dibutyltin dilaurate (DBTL) as a catalyst. Consequently, molar masses in the range of 4150 to 7200 Da were obtained. The tensile strength and elongation at break increased with an increase in the molar mass. However, the reaction between the isocyanate and OH groups was not complete, which was explained by the high steric hindrance of betulin, limiting the obtained molar mass.284
Betulin-based PUs have been synthesized through a two-pot route. Firstly, anionic ROP with EO occurred. After the isolation and purification of betulin with polyethylene oxide chains, step-growth polymerization with IPDI occurred in the presence of a catalyst. A one-pot synthesis route has also been developed. The catalyst switch strategy has been employed with base-catalyzed ROP and acid-catalyzed step-growth polymerization. To avoid the high crystallinity linked to long polyethylene oxide grafted chains, 1,4-benzenedimethanol has been added in different ratios with betulin. Subsequently, a molar mass in the range of 16.9 to 83.3 kDa was obtained. When the gap between two betulin units increased, hydrophobic crystalline nano-domains were formed. Thus, homogeneous hydrophobicity increased with an increase in the betulin content because of the restricted phase separation.285
3.2. From cholesterol
Cholesterol is a lipid from the sterol family, which is involved in different biochemical mechanisms in living organisms such as animals and human beings and is a derivate of terpenes. Specifically, it is an amphiphilic molecule found in the plasma of cellular membranes, and its chemical structure is presented in Fig. 8 (left). It possesses a polar secondary alcohol, non-polar rigid tetracyclic ring structure and a flexible branched carbon chain. Cholesterol is obviously biocompatible, which is a key factor in its use as a building block for the synthesis of polymers. Given that it possesses one OH group, it has been used for the synthesis of biocompatible polymers.286 Polymers with cholesterol in their main chain have been synthesized using it as an initiator for free radical polymerization, ROP, esterification. The synthesis of polymers bearing cholesterol side chains has also been reported, which are usually grafted via polymerization or radical polymerization.209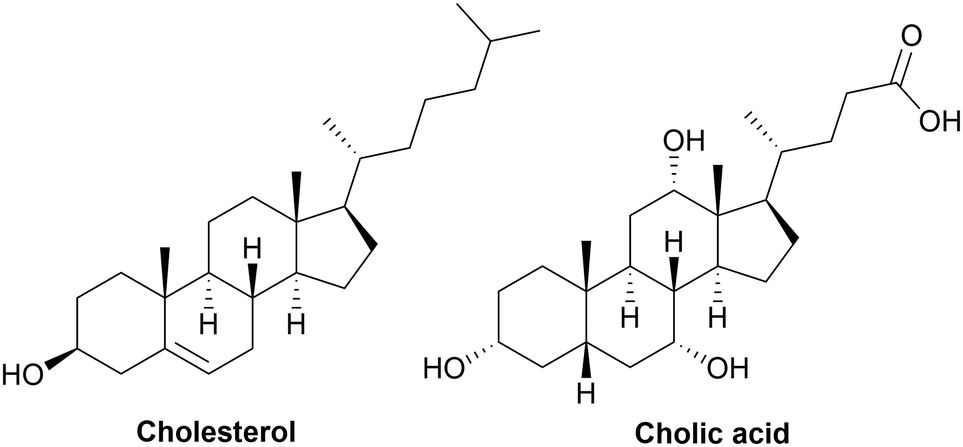 | ||
| Fig. 8 Chemical structure of cholesterol (left) and cholic acid (right). | ||
TPU with cholesterol-based side-chains have been studied as a biocompatible adhesive. Firstly, a TPU was synthesized from MDI and a polyol such as PCL or PTHF. Then, a second addition of MDI was performed, allowing the grafting of cholesterol on the urethane linkage by the formation of allophanate. Consequently, this created a high-affinity surface for the attachment and adhesion of cells.287,288
As an example, the synthesis of grafted cholesterol in PU from MDI and PTHF has been reported. Cholesterol was grafted by the second addition of MDI. Cross-linked networks were obtained when MDI did not react with cholesterol, but with two urethane bonds. The pendant chains of the bulky tetracyclic cholesterol prevented the crystallization of the HSs, leading to fully amorphous materials. Moreover, the Tg increased from −63 C to −55 °C with an increase in cholesterol content due to its rigidity.289
Cholesterol is also known as a precursor of bile acids, which help to solubilize fats for vitamin metabolism. Bile acids, such as cholic acid, chenodeoxy cholic acid, deoxycholic acid and lithocholic acid are formed in the liver of mammals and birds after the metabolism of cholesterol.290 The chemical structure of cholic acid is presented in Fig. 8 (right). Cholic acids can be employed for the synthesis of fully biobased thermoset PUs. Firstly, coatings with hydrophobic properties were synthesized using EELDI as the diisocyanate and cholic acid as the polyol. Their Tg was high, ranging from 106 °C to 118 °C.290 Then, cholic acid was used as the initiator for the ROP of ε-caprolactone on its primary OH group. This polyol has been mixed with EELDI to produce biobased and biocompatible coatings. Semi-crystalline networks were obtained with Tm in the range of 45 °C to 65 °C. Furthermore, an elastic modulus in the range of 121 to 171 MPa was obtained, with an increase in elongation at break from 11% without cholic acid to 327% with the longest cholic acid-based polyol.291
3.3. From carbohydrates
Carbohydrates are the largest worldwide and biobased feedstock. Based on the degree of polymerization (DP) or number of monomeric sugar units, a well-known classification has been reported. In this frame, polysaccharides were defined as a group of substances with a DP above 10, comprising starch, which is composed of amylose and amylopectin. Non-starch polysaccharides are composed of cellulose, hemicellulose, pectin, and others. Oligosaccharides represent short-chain carbohydrates with a DP in the range of 3 to 9, which are mainly composed of malto-oligosaccharides from maltodextrin. Finally, sugars with a DP below 2 are composed of monosaccharides, disaccharides and sugar alcohols. Monosaccharides can be glucose, fructose or galactose. Disaccharides can be sucrose, lactose, or maltose. Sugar alcohols are composed of compounds such as sorbitol, mannitol, lactitol, xylitol, erythritol, isomalt, inositol and maltitol.292Given that carbohydrates possess at least two available OH groups (f ≥ 2), they can be used as polyols or to produce polyisocyanates, diamines, biscyclocarbonates and other components for the synthesis of PUs. Among then, isohexides such as isosorbide are commercially available sugar-based diols, which have been intensively studied for the synthesis of polymers. Due to their two secondary OH groups, various functionalization and chemical modifications are possible to create renewable building blocks for the synthesis of PUs. Other carbohydrates have been used with or without chemical modifications for the synthesis of PUs such as glucose, sucrose, starch, fructose, and others.293–298
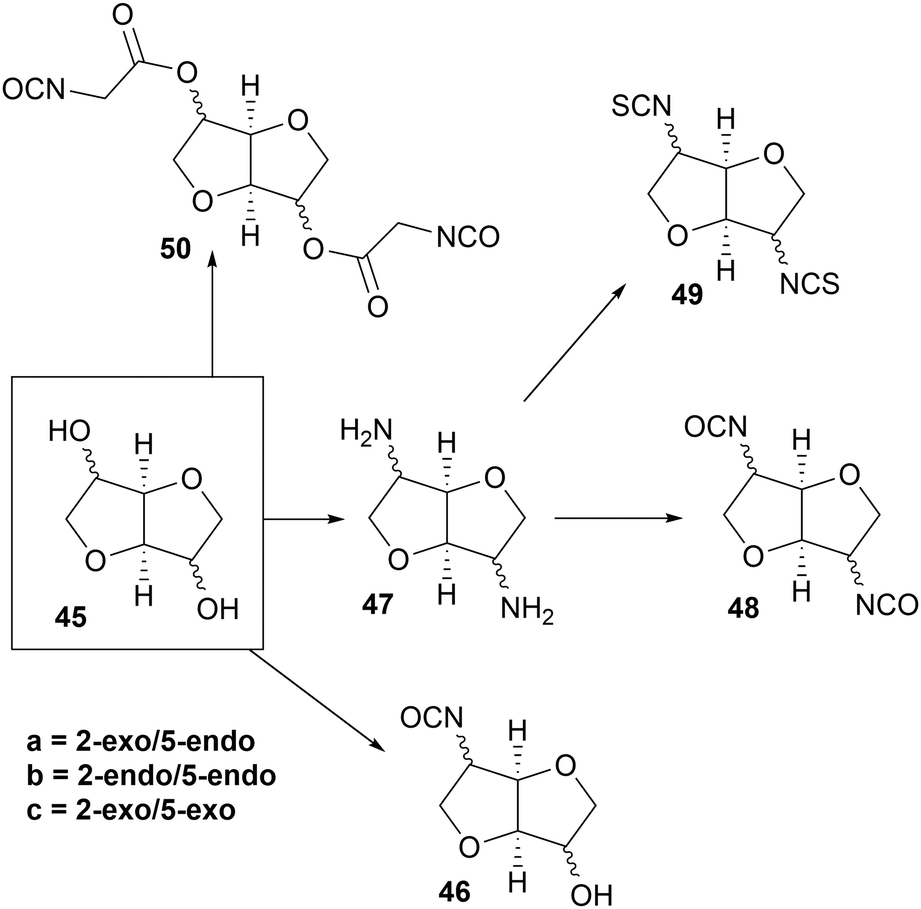 | ||
| Fig. 9 Isohexide-derived diamine, isocyanates and isothiocyanates (from 45 to 50). | ||
Production. The large-scale production of isosorbide is relatively easy, given that it can be obtained from polysaccharides such as cellulose or starch in some steps. Firstly, the enzymatic depolymerization of starch or cellulose leads to the production of D-glucose. The hydrogenation of this compound gives D-sorbitol. Double dehydration is the final step in the formation of isosorbide. However, several side products are formed, decreasing the yield. Accordingly, various synthetic strategies and catalysts have been reviewed to overcome this technical challenge.301 However, the starting materials for the synthesis of isomannide are fructose and mannitol, which are less available than glucose. Isoidide is very interesting due to its reactivity. However, its precursor L-idose cannot be produced from plant biomass.302 The industrial production of isosorbide on a large scale has already been achieved by Roquette (France) using different derivates.303 This recent commercial and now largely available biobased chemical is employed as a “catalyst” for numerous global projects, e.g., renewable polymer synthesis. In 2018, the isosorbide market size was valued at 396 million USD, which is projected to reach 430 million USD by 2025.304 Recently, the sustainable biosynthesis of limonene was achieved through combinatorial metabolic engineering. The main advantage of this is that the used engineered cyanobacteria only required CO2 and light for growth.305
The two furan units of isosorbide endow the corresponding polymers with a rigid structure and mechanical strength, leading to a high Tg, tensile modulus and UV stability. Isosorbide is also biodegradable, soluble in water, and generally recognized as safe and non-toxic, and thus used in food packaging and medical applications.306
Isosorbide has high stability and two functional OH groups, making it an ideal and versatile platform molecule. Easy conversion reactions are possible for various applications such as green solvents, additives (plasticizers and surfactants), and renewable polymers.307
PU synthesis from neat isohexide. Isosorbide and its isomers have been intensively used without chemical modification for the synthesis of Pus during these last two decades. The synthesis and corresponding properties of PUs are reported in Table 5.
| Isohexide | Diisocyanate | Other diol/diamine | Reaction conditions | T g of SSs (°C) | T m (°C) | M n (kDa) | Strength at break (MPa) | Elongation at break (%) | Modulus (MPa) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 45a | HDI | Castor oil | 160 °C, 20 min, | / | / | / | 0.8 to 12.9 | 42 to 363 | 5 to 19 | 320 |
| 45a | MDI | / | DBTL, 60 °C, 6 h | / | / | / | / | / | / | 370 |
| 45a | MDI | Radia 7282 | 80 °C, DBTL | −48 to −41 | 194 to 219 | 12 to 39 | / | / | / | 371 |
| 45a | DDI | PBS diol | 80 °C, 15 h | −41 to −38 | 57 to 75 | 21 to 35 | 7.0 to 13.0 | 87 to 715 | 107 to 226 | 322 |
| 45a | DDI | BDO | 80 °C, 24h, DBTL | −15 to −1 | 50 to 84 | 4 to 18 | / | 30 to 700 | 45 to 40 | 323 |
| 45a | HDI | Castor oil, aminodisulfide | 160 °C, 20 min | 20 to 29 | / | / | 25.0 | 140 to 146 | 53 to 255 | 372 |
| 45a | MDI | PTHF, Castor oil, BDO | DMF, 40/60/80 °C, 16 h, DBTL | −33 to −30 | 24 to 29 | 10 to 22 | 1.3 to 14.1 | 28 to 1358 | / | 319 |
| 45a | HDI | PCL diol | DMF, 100 °C, Cat. | / | / | / | / | / | / | 373 |
| 45a | IPDI, HMDI | PPG | 80 °C, 4 h, DMDEE | 54 to 68 | / | 7 to 15 | / | / | / | 309 |
| 45a | EELDI | PCL diol | 70 °C, 6 h, DBTL | / | / | 36 | / | / | / | 324 |
| 45a | HDI | PCL diol | 120 °C, 12 h | / | / | / | / | / | 374 | |
| 45a | HDI | PCL diol, castor oil | DMF, 80 °C, 8 h, DBTL | −43 to −33 | 30 to 36 | 59 to 80 | 9.5 to 13.9 | 596 to 1319 | / | 375 |
| 45a | MDI | PTHF, BDO | 75 °C, 3h then 120 °C 16 h | −64 to −56 | 168 to 206 | 19 to 57 | 10.9 to 36.4 | 264 to 668 | / | 308 |
| 45a | HDI | PCL diol | 120 °C, 12 h | / | / | / | / | / | / | 376 |
| 45a | HDI | PCL diol | 120 °C, 12 h | −48 to −35 | 32 to 56 | / | 12.8 to 37.5 | 900 to 1520 | 377 | |
| 45a | HDI | PTHF | 120 °C, 12 h | −47 to −38 | / | 16 to 153 | 5.9 to 19.1 | 153 to 1695 | 16 to 41 | 318 |
| 45a,b | MDI | PTHF, BDO | / | −60 to −50 | 147 to 199 | 16 to 27 | 24.0 to 48.0 | 462 to 570 | 8 to 39 | 378 |
| 45a | HDI, MDI, TDI, | Bisphenol A | NMP, 100 °C, 3 h, n-butyltin oxide hydroxide | 74 to 178 | / | 4 to 7 | / | / | / | 316 |
| 45a | DDI, EELDI | Dimethylol propionic acid (DMPA) | 2-Butanone, 70 °C, 4–6 h, DBTL | 18 to 58 | / | 9 to 29 | / | / | / | 317 |
| 45a | DDI | DMPA | 2-Butanone, 70 °C, 4–6 h, DBTL | 15 to 19 | / | 7 to 27 | / | / | / | 379 |
| 45a | DDI, EELDI | DMPA | 2-Butanone, 40 °C, 2–5 h, DBTL | / | 10 to 16 | / | / | / | 380 | |
| 45a | DDI, EELDI, HDI, IPDI | DMPA | 2-Butanone, 70 °C, 4–6 h, DBTL | 28 to 60 | / | 9 to 17 | / | / | / | 381 |
| 45a | HDI, MDI | BDO | DMF, RT, 24 h, DBTL | 15 to 115 | 162 to 233 | 9 to 14 | / | / | / | 310 |
| 45a | HDI | PC diol | 120 °C, 12 h | −37 to −33 | 63 to 70 | 56 to 126 | 13.0 to 54.0 | 955 to 1795 | 43 to 549 | 313 |
| 45a | HDI | PTHF, glycerin, castor oil, | DMF, 80 °C, 24 h, DBTL | 3 to 7 | / | / | / | / | / | 311 |
| 45a | HDI | PCL diol | 150 °C, 12 h | 31 to 42 | / | 16 to 50 | 5.4 to 9.8 | 97 to 1245 | 8.2 to 42 | 321 |
| 45a | HDI, IPDI | PPG | THF, 60 °C, 2 h | −63 to −24 | / | 30 to 75 | / | / | / | 312 |
| 45a | TDI | PPG | Foam | / | / | / | / | / | / | 382 |
| 45a,b | MDI, PPDI | / | Cyclohexanone, 70 °C, 24 h | 122 to 143 | / | / | / | / | / | 314 |
| 45a | HDI | Castor oil | / | 65 | 164 to 170 | / | 48.3 to 55.8 | 305 to 409 | 1248 to 1549 | 383 |
| 45b,c | Acrylate isocyanate | 1,8-Octanedithiol | / | 15 to 19 | 40 to 158 | 8 to 22 | 37.0 to 56.0 | 462 to 1658 | 3 to 350 | 326 |
| 45a | TDI | Chromophore | DMAc, 70 °C, 24 h, DBTL | 60 to 120 | / | / | / | / | / | 315 |
| 45a | HDI | PCL diol | DMF, 100 °C, Cat. | / | 78 to 183 | 224 | 0.2 to 3.9 | 49 to 306 | 1 to 28 | 384 |
| 45a | DDI | Oligo-PHB diol | 70 °C, 24 h | −14 to 4 | 66 to 141 | / | 0.3 to 7.5 | 50 to 843 | 0.2 to 138 | 325 |
| 45a | HDI | Soybean oil-based amide diol, DMPA | 78 °C, 3 h | −1 to 9 | / | / | 0.7 to 8.2 | 406 to 794 | 4 to 63 | 385 |
| 45a | HDI | D,L-Lactide, PEG | Toluene/DMF, 75 °C, 24 h | 38 to 43 | / | 31 to 49 | 32.3 to 52.9 | 4 to 16 | 3062 to 3588 | 386 |
| 45a,b,c | HDI, MDI | / | DMAc, 80 °C, 24 h, DBTL | 65 to 183 | / | / | / | / | / | 330 |
Neat isosorbide, BDO and common polyether polyols such as PTHF and PPG react with general diisocyanates such as MDI and HDI.308–310 The synthesis of PUs can be carried out in one or two steps. DBTL, dimorpholinodiethyl ether (DMDEE), and n-butyltin oxide hydroxide hydrate can be used as catalysts. Several green or common solvents such as DMF, THF, 2-butanone, cyclohexanone, NMP, and DMAc are employed.309,311–317
As an example, TPU was synthesized from HDI with different PTHF/isosorbide ratios without a catalyst, in a green chemistry approach. Isosorbide as a cycloaliphatic compound endowed the TPU with rigidity, which was translated by an increase in Tg and elastic modulus from −48 °C to −39 °C and 16 to 42 MPa, respectively, with a high elongation (around 500%) decrease.318
More recent research has been focused on using biobased polyols such as castor oil, poly(butylene succinate) diol (PBS diol) and PCL diol.319–321 Some research groups have also used biobased diisocyanates such as DDI and EELDI to synthesize fully biobased PUs.322–325 Oligo-poly(β-hydroxybutyrate) (PHB) diols have been recently developed with different molar masses and associated with isosorbide at various ratios. These polyol mixtures react with DDI as diisocyanate without solvent and without catalyst to synthesize fully biobased TPU. A wide range of thermo-mechanical properties was obtained. The crystallinity, Tg, strain at break and elastic modulus increased with an increase in isosorbide content due to its rigid cycloaliphatic structure.325
The impact of the stereochemistry of isomannide and isoidide integrated in linear PUs was studied. Isohexide-based acrylate was synthesized via the reaction between isohexide and 2-isocyanatethyl acrylate in THF with DBTL as the catalyst. This monomer reacted in a second step by thiol-Michael addition with 1,8-octanedithiol in with dimethylphenylphosphine as the catalyst. This reaction was conducted using the green chemistry concept, i.e., click chemistry at low temperature. Isohexide-based PUs showed a wide range of behaviors, for instance, isoidide-based PUs, which present tough semi-crystalline plastic behavior compared to isomannide-based PUs, which presents amorphous elastomer properties. Rheological studies proved that isoidide stereochemistry led to more linear polymer chains with less entanglement compared to isomannide.326
PU synthesis from isohexide derivates. The difference in reactivity between both secondary OH groups can lead to low molar mass polymers, for instance, in the case of isosorbide. Various chemical modifications have been studied to lead to a broad spectrum of functional building blocks for the synthesis of PUs such as diisocyanate, polyols and also diamine and biscyclocarbonate for the synthesis of NIPU. The synthesis reaction conditions and final properties of these isohexide-based PUs are summarized in Table 6.
| Diol/diamine | Diisocyanate, cyclocarbonate | Other diol/diamine | Reaction conditions | T g of SSs (°C) | T m (°C) | M n (KDa) | Ref. |
|---|---|---|---|---|---|---|---|
| 46c | / | / | DMAc, DBTL | 118 | 145 to 194 | 8 to 12 | 328 |
| 47a, BDO, BDA, BDT | 48a, 48b, 48c, 49c | / | DMAc, 80 °C, 24 h, DBTL | 48 to 112 | 90 to 305 | 9 to 20 | 182 |
| 2,5-Bis(hydroxymethyl)furan | 50a, 50b | / | DMF, 30 °C, 24 h, DBTL | 34 to 52 | 240 to 248 | 8 to 12 | 327 |
| 45a, 45b | 50a, 50b | / | DMF, 120 °C, DBTL | 78 to 81 | 116 to 117 | 14 to 15 | 183 |
| Commercial diamines | 51 | / | DMF, 25 °C, 12 h, cat. | −8 to 59 | / | / | 343 |
| 47c | Diglycerol carbonate | / | 80–100 °C, 5 h | 65.5 | / | 5 | 344 |
| 52a | MDI, HDI | / | DMAc, 80 °C, 24 h, DBTL | 52 to 183 | 200 to 283 | / | 329 |
| 45a, 45b, 45c, 52a | MDI | / | DMAc, 80 °C, 24 h, DBTL | / | / | / | 330 |
| 53a | pMDI | Jeffol SG | Foam | / | / | / | 228 |
| 54a, 54b, 54c | MDI, HDI | / | NMP/DMF/DMAc, 60 °C, 18h | 52 to 153 | 225 to 330 | / | 387 |
| 55a | HDI | / | 150 °C, 12 h | 7 to 23 | 33 to 40 | / | 335 |
| 56a | MDI | / | Foam | / | / | / | 331 |
| 57a | IPDI | / | Toluene, DBTL, 120 °C, 30 min | / | / | / | 332 |
| 57a | MDI | / | Xylene, RT, 48 h, DBTL | / | / | / | 333 |
| 58a | CN981 | / | 65 °C, UV curing | 58 to 62 | / | / | 342 |
| 59a | HDI | 3-(4-Hydroxy-3-methoxyphenyl)acrylate | Acetone, 60 °C, 5 h, DBTL | 39 to 56 | / | / | 336 |
| 59a | MDI | / | RT, 48 h, DBTL | / | / | 2 | 337 |
| 59a | MDI | PDO | RT | / | / | 30 to 47 | 338 |
| 60a | TDI, MDI, IPDI, HDI | Terephthaloyl chloride | Chlorobenzene, 160 °C, 4 h, cat. | 78 to 159 | / | 4 to 24 | 339 |
| 45a, 60a | HDI | / | Toluene, 50 °C, 2 h, Sn(Oct)2 | 56 to 59 | / | 64 to 84 | 340 |
| 45b, 61b | PPDI, TDI | / | DMF, 80 °C, 6 h | / | / | 4 to 17 | 341 |
| 62, 63 | HMDI | PEG 200 | THF, 90 °C, 5 h | −38 to −8 | / | / | 345 |
| 63, 64, 65 | HMDI | PPG | / | / | / | / | 346 |
| 63, 64 | TDI | PCL diol, BDO | / | / | / | / | 347 |
| 65 | MDI | PCL diol, BDO | 120 °C, 4h, DBTL | / | / | 22 to 52 | 388 |
| 65 | Polypropylene oxide toluene diisocyanate | / | DMSO, RT, 10 to 24 h, DBTL | −60 | / | / | 389 |
| 65 | HDI | PCL diol | DMSO, 80 °C, 72 h | / | / | / | 390 |
| 65 | HDI | Methanol, butanol, butylamine | NMP, 115 °C, 6 h | / | 99 to 129 | / | 391 |
| 66 | IPDI | / | THF or DMF, 60 °C, 22 h, DBTL | 111 | / | 10 to 34 | 348 |
| 67 | IPDI | / | THF or DMF, 60 °C, 22 h, DBTL | −8 to 149 | / | 6 to 11 | 348 |
| 71, 72, 73 | HDI, MDI | / | DMF, 40 or 60 °C, 48 h, DBTL | 50 to 161 | 126 to 185 | 10 to 56 | 351 |
| 68, 69 | HDI | / | THF, 70 °C, 6 to 60 h, DABCO | / | / | 1 to 16 | 392 |
| 74 | / | / | THF, 60 °C, 48 h, Zr(acac)4 | 64 | 212 | 11 | 393 |
| 69 | HDI, EELDI | / | DMF, 25 °C/50 °C, 24 h/72 h, DBTL | / | / | 5 to 14 | 350 |
| 70 | HDI, EELDI | / | DMF, 25 °C/50 °C, 24 h/72h, DBTL | / | / | 5 to 13 | 350 |
| 45a, 71 | HDI, MDI | PCL diol, BDO | DMF, RT, 24 h, DBTL | −60 to −3 | 40 to 54 | 20 to 56 | 394 |
| 75, 76 | HDI, MDI | BDO | DMF, 40 °C, 24 h, DBTL | 35 to 126 | 137 to 232 | 40 to 65 | 395 |
| 77 | BICMC, MDI, IDPI, HDI | 1,4-Cyclohexanediol | DMF, 60 °C, 24 h, DBTL | 119 to 250 | 199 to 265 | 21 to 61 | 396 |
| 78 | HDI, IPDI, BICMC, MDI | / | 1,4-Dioxane, 80 °C, 24 h, DBTL | 143 to 248 | / | 8 to 22 | 397 |
| Neopentyl glycol | 39 to 162 | / | 10 to 13 | ||||
| 79 | pMDI | / | Foam | / | / | / | 349 |
Isohexide-based amines, isocyanates and isothiocyanates for the synthesis of PUs are reported in Fig. 9 with different ways, from 45 to 50. Using a phosgene route, diisocyanates (48a, 48b, 48c) and diisothiocyanate (49c) have been designed from diamine (47) derived from isosorbide (45a), isomannide (45b) and isoidide (45c), respectively. Poly(thio)urethanes and poly(thio)ureas were synthesized by reacting these diiso(thio)cyanates with various diols and diamines in DMAc with DBTL as the catalyst.182 Other isosorbide- and isomannide-based diisocyanates have been obtained in several synthesis steps with succinic acid and phosgene (50a, 50b). Biobased PUs have been obtained by mixing these diisocyanates (50a, 50b) with isosorbide, isomannide and bis-hydroxymethylfuran in DMF with DBTL as the catalyst to obtain biobased PUs.183,327 Also, isoidide-based monomer (46c) bearing one OH and one isocyanate group has been synthesized to produce [AB]-type homo-PUs.328
Moreover, various isohexide-based polyols have been employed in the literature for the synthesis of PUs, such as polyether polyols, polyester polyols and polyacrylates. The final building block structures are reported in Fig. 10 with different ways (from 52 to 60).
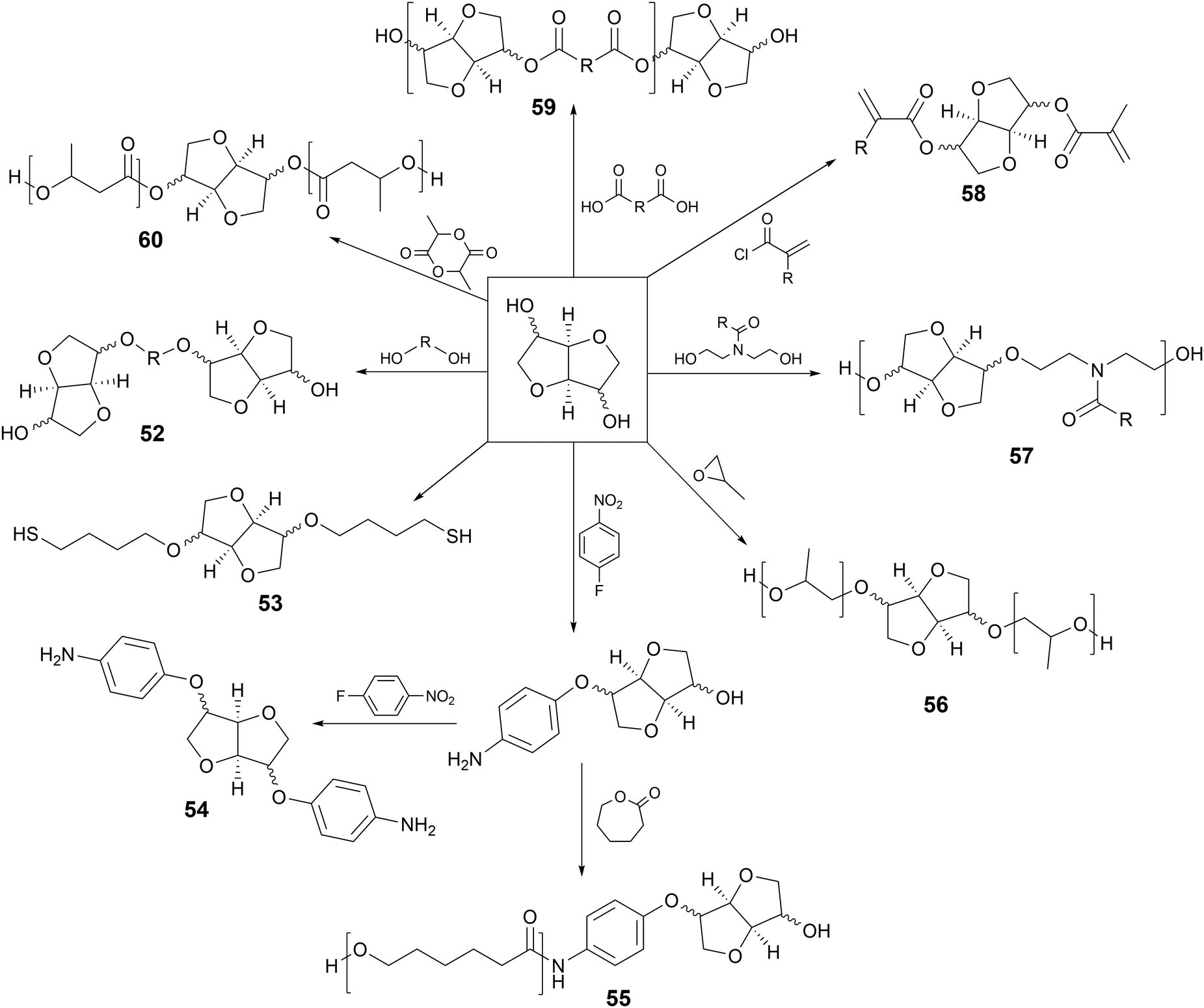 | ||
| Fig. 10 Isohexide-based polyols, polyamines and dimercaptan for the synthesis of PUs (from 52 to 60). | ||
Two polyether polyols were designed from isohexides for the development of various types of PUs. Firstly, an isosorbide-based ether-diol (52a) was produced in several steps, including the protection of the exo OH group by alkylation, followed by a deprotection step. Microwave irradiation was used to improve the regioselectivity and reduce the reaction time during the alkylation step. Various aromatic and linear diols were used for this alkylation step such as bisphenol A, thiodiphenol, BDO, HDO, and PEG. These isosorbide-based ether-diols (52a) were successfully polymerized with MDI or HDI.329,330 Secondly, the oxypropylation of isosorbide led to another polyether polyol (56a) with various OH indexes. R-PUFs with improved thermal and dimensional stabilities were obtained compared to PPG as the polyol.331
Polyetheramides were also synthesized for the preparation of PUs for efficient applications. Polyetheramide polyols (57a) were designed from fatty acid and isosorbide. Firstly, fatty acid was converted to fatty amide by amidation with diethanolamine. It reacted in a second step with isosorbide to form polyetheramide polyols (57a). These products were used for the efficient synthesis of coatings.332,333 Dietheramides (54a, 54b, 54c) were developed from the reaction between three isohexides isomers with 4-fluoronitrobenzene, which were used for the synthesis of polyurea with high thermal resistance.334
Also, four biobased isohexide-based polyester polyols were produced via esterification reaction or ROP. Firstly, the reaction of 4-fluoronitrobenzene with the endo group of isosorbide led to the formation of amino-alcohol, which was used as an initiator for the ROP of ε-caprolactone on its primary amine group. These polyester polyols (55a) were used for the synthesis of PUs with HDI.335 Secondly, a condensation reaction was performed for polyester polyols (59a) between isosorbide and various dicarboxylic acids such as α-ketoglutaric acid, dimer fatty acid and azelaic acid. TPUs and coatings were prepared using these polyester polyols and MDI or HDI.336–338 Then, isosorbide was also used by several groups as an initiator for the ROP of D,L-lactide to obtain polyester polyols (60a). Various diisocyanates have been employed for the synthesis of TPUs with increased molar masses and mechanical properties.339,340 Recently, an isomannide-based polyol (61) with oxide ending groups was synthesized via the pathway described in Fig. 11. This polyol was mixed with p-phenylene diisocyanate (PPDI) or TDI to form biobased cycloPUs. It has been showed that the diisocyanate source has a strong impact on the morphology of the final material.341
 | ||
| Fig. 11 Isomannide-based polyol (61) synthesis route. | ||
Recently, isosorbide di(meth)acrylates (58a) were designed from (meth)acryloyl chloride and isosorbide. They were mixed with acrylic PUs to prepare UV curable coatings with a high storage modulus and good impact properties.342
In a sustainable approach, biobased polythioether was prepared by click chemistry from isosorbide dithiol (53a) for the preparation of PUF. This new polyol structure was developed via photochemical or thermal thiol-ene reaction with alkoxylated aromatic phenols containing double bonds such as ortho allyl phenol and eugenol.228 Also, to avoid the handling of the harmful phosgene and isocyanate, isosorbide-based PHUs have been developed via the addition of isosorbide-based biscyclocarbonate (51) with various available linear or cycloaliphatic diamines.343 The synthesis route is reported in Fig. 12. Another NIPU was synthesized from isoidide diamine (47c) with diglycerol dicarbonate.344
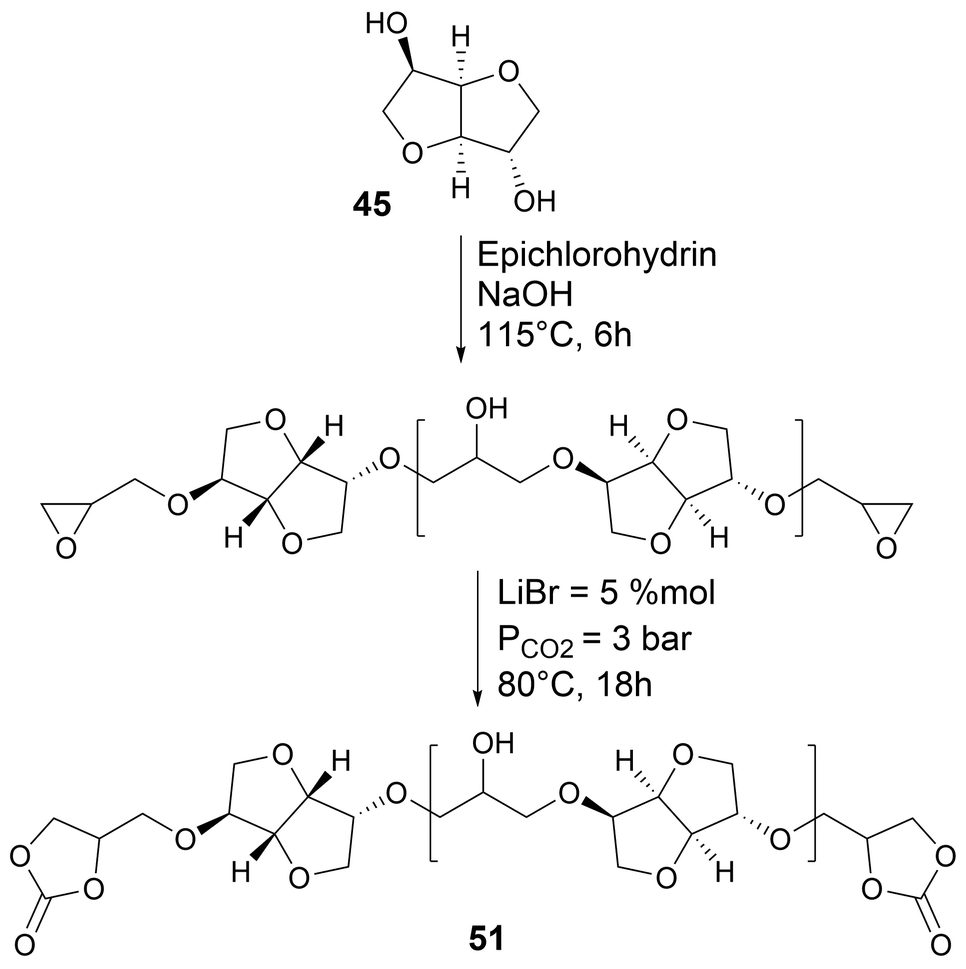 | ||
| Fig. 12 Isosorbide-derived biscyclocarbonate (51) synthesis route for the synthesis of NIPU. | ||
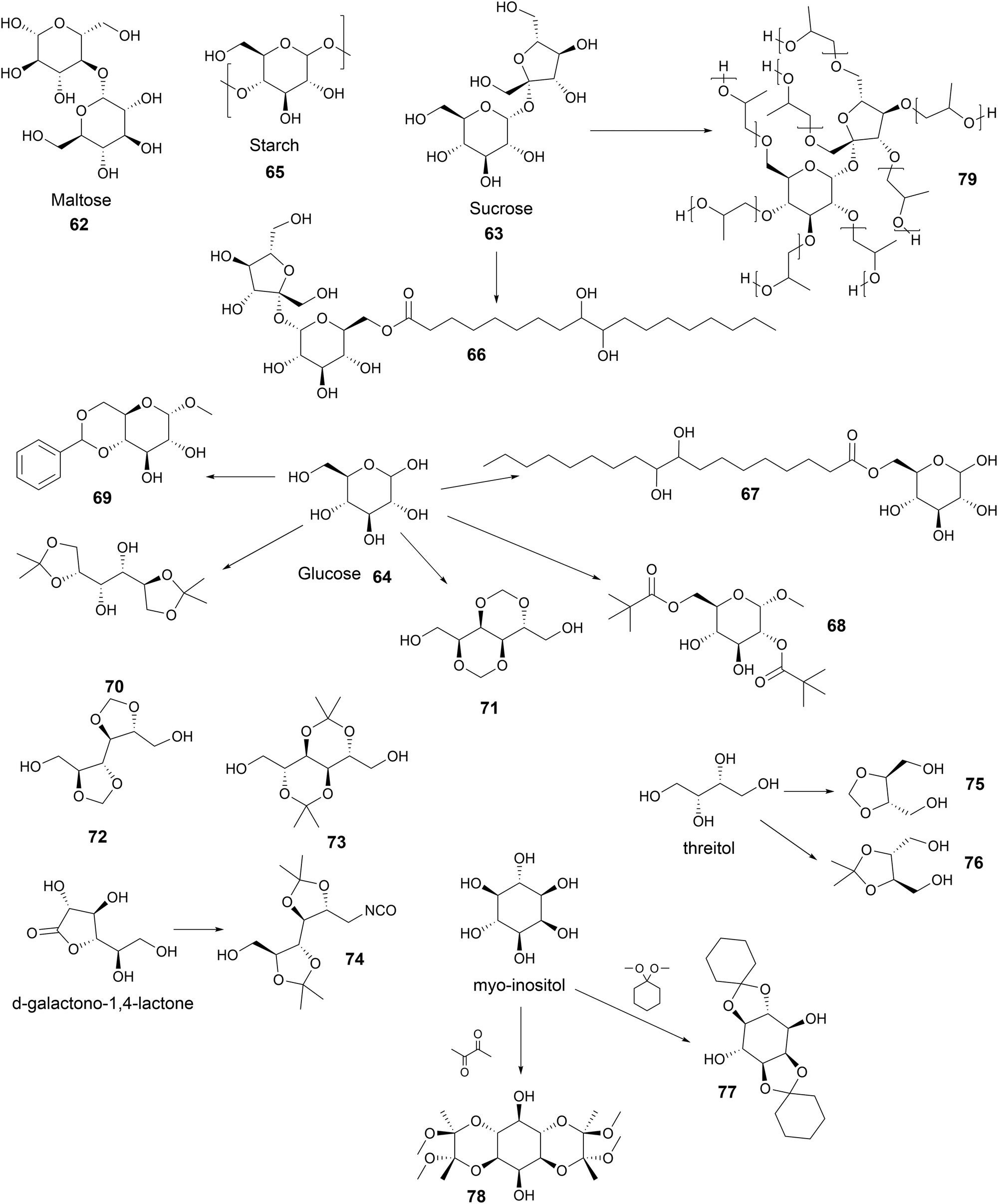 | ||
| Fig. 13 Summary of carbohydrate (un)modified structures for PUs synthesis, from 62 to 79. | ||
Due to their different OH groups, maltose (62), sucrose (63), glucose (64) and starch (65) have been intensively used as renewable cross-linkers in PUs.345–348
Modified carbohydrates have also been designed to be used as biobased cross-linkers for the synthesis of thermoset PUs. Firstly, the esterification of the primary OH group of sucrose (63) and glucose (64) has been performed with fatty acid to form new biobased cross-linkers (66 and 67), respectively. Also, a highly functional polyether polyol (79) has been designed via the oxypropylation of sucrose (63). R-PUFs were prepared using these polyols and polymeric MDI. High physical and mechanical properties were obtained.349
New OH-bearing PUs with high hydrophilicity and degradability have been designed from carbohydrates due to their hydrolysable acetals. The OH group protection of various carbohydrates by acetalization can occur, leading to the creation of cycloaliphatic structures, as presented in Fig. 13 (from 69 to 78). The starting carbohydrates were glucose, theritol, myo-inositol and d-galacto-1,4-lactone, a galactose derivative. It appeared that the thermal stability was higher for PUs with free OH groups than for that with protective acetals.350 It has also been observed that PUs from D-gluco, galacto, and D-manno building blocks (71, 72, 73) with protected secondary OH groups had similar properties to that of PU from isosorbide, respectively.351
3.4. From vegetable oil and fatty acids
Vegetable oils are present in many plants, but are especially abundant in soybean, palm, rapeseed, coconut, corn, sunflower, castor, cottonseed, and linseed. Each resource presents a particular lipid profile with strong variations in its fatty acid content.352,353 Metabolic engineering enables the design oils with customizable lipid profiles and properties. Some microorganisms have been engineered to accumulate a high amount of certain lipids with easy extraction, using industrial waste materials as a carbon source.354The synthesis of PUs via oleochemistry has become a hot topic in the last two decades to obtain thermosets or TPUs due to the abundance, very low cost, non-toxicity and attractive chemical structures of vegetable oils.355–358 Their chemistry is rich. For instance, their double bonds can be functionalized into OH groups and form polyols using different approaches. Ozonolysis, epoxidation, hydroformylation, transesterification, amidation and thiol-ene coupling are the most developed methods for the functionalization of double bonds for the synthesis of polyols using vegetable oils.359
Dimers of diisocyanate, polyol, diamine, and dicyclocarbonate with cyclohexene ring can be produced via the dimerization of fatty acids, e.g., Diels Alder cycloaddition [4+2] of linoleic and linolenic acids at high temperature.360 The pending aliphatic chains of these dimers can bring mobility to the final polymers. It is also possible to eliminate the residual double bonds by hydrogenation to increase the resistance of polymers to UV light, temperature, and oxidation, consequently increasing their shelf life.323
The condensation of a dimer fatty acid with excess linear diols has been conducted to prepare new biobased polyester polyols from vegetable oils. This building block was used in addition to MDI and BDO to produce TPU. Molar masses in the range of 15 to 38 kDa were obtained depending on the HS content. Organized segregation in microdomains was observed, with the highest interaction between HSs and SSs at the highest HS content.361,362 Polyester polyol was synthesized from dimer fatty acid and 2,2-dimethylpropane-1,3-diol by esterification. WPUs have been produced from this polyol and MDI.363
The synthesis of NIPU from vegetable oil derivatives has also been studied to avoid the handling of the harmful isocyanate. Cyclocarbonates and diamines from dimer fatty acids have been designed and reported, which will be presented in the following section.
Dimer-based diamines can be obtained from dimer fatty acids via two different routes, i.e., direct amination and amidification with diamine excess. However, the second route requires the removal of a huge excess of diamine. Croda (Germany) developed several Priamine©, dimer diamines synthesized from dimer fatty acids, called Pripol©. This hydrophobic diamine exhibited low viscosity, which is interesting for solvent-free formulation and synthesis.364 The amine ratio and functionality strongly impact the properties of the final polymer. Indeed, the quantity of trimer fatty acid side products directly impacted the cross-linking density and resulted in specific structuration.365
Thermoplastics and thermoset PHUs were synthesized using sebacic biscyclocarbonate and dimer-based diamine with various functionalities and contents. Amorphous polymers were obtained with a Tg in the range of −23 °C to −14 °C.366 Sustainable PHUs were designed via green chemistry approaches, without solvent and catalyst. In this case, a fatty acid dimer was chlorinated before being esterified with glycerol carbonate to form a biobased biscyclocarbonate, with trimethylamine as the catalyst. The properties and molar masses of the final NIPUs were consistently lower than that of conventional PUs. Aminolysis reaction has lower reactivity than urethanisation between isocyanate and primary alcohol, leading to longer reaction time with lower molar masses.365
The esterification strategy was used with glycerol carbonate without the halogenation step to produce dimer-based biscyclocarbonate. PHUs and water-borne NIPUs were prepared from this building block and dimer diamines through bulk polymerization and mini-emulsion process, respectively. However, the hydrolysis of cyclocarbonate occurred during the mini-emulsion process, leading to the formation of low molar mass polymers. All the NIPUs exhibited a low Tg due to the long linear fatty chains presented in both components.367
Dimer-based diamines can be converted into diisocyanate via the phosgene or hydrazine routes.165 The Cognis-BASF (Germany) Company developed heptyl-3,4-bis(9-isocyanatononyl)-1-pentylcyclohexane (abbreviated as DDI) under the trade name DDI1410® from linoleic acid. Its purity is higher than 90% with a non-negligible amount of mono-functional isocyanates.317 It is also possible to obtain and isolate trimers as co-products from Diels Alder cycloaddition.368 The synthesis of several PUs has been realized using DDI as a flexible diisocyanate to create polymers with a high biobased content.
DDI was used as a soft building block for the synthesis of biobased TPU. Isosorbide and BDO acted as HSs. All the polymers showed HS/SS segregation by SAXS with separate isosorbide and BDO domains.323 The same research group studied the preparation of biobased TPUs from DDI, HDI, oligo-PHB diol and various chain extenders. The final biobased contents were in the range of 78% to 87%. DDI has been used as long and flexible hydrophobic parts, whereas HDI was used to increase the crystallinity. It is interesting to note that DDI was considered as 86% participating in the SSs due to its flexible C36 chain. The remaining 14% was considered as HSs, mainly based on the urethane group parts. The mechanical results showed higher Young's moduli and lower elongation at break for the TPU with longer prepolymers and highly reactive chain extenders.369
4. Applications of sustainable cycloaliphatic PUs in relation to their properties
Sustainable cycloaliphatic building blocks enable the synthesis of PUs with novel macromolecular architectures to bring specific properties such as particular mechanical properties, high thermal stability, and sometimes biocompatibility, satisfying the requirements of various applications. In this part, the applications of renewable cycloaliphatic compounds of these functional materials in the fields of foams, adhesives, coatings, TPUs, shape-memory PUs (SMPUs) and microspheres are reviewed in connection with the properties of the materials, as summarized in Fig. 14. | ||
| Fig. 14 Summary of cycloaliphatic PUs applications. | ||
4.1. Foams
Due their particular rigidity, cycloaliphatics are mainly used in R-PUF applications, e.g., thermal insulation. Various cycloaliphatic polyols have been used in the synthesis of R-PUFs, such as limonene, isosorbide, and sucrose. As examples, R-PUFs from limonene-based polyol (A) showed a compressive strength of 195 kPa and closed cell content of 90%.226 Limonene polyol (4) was used to produce R-PUFs with a closed cell content of above 90% and a high compression strength between 177 and 412 kPa.228 R-PUFs from oxypropylated isosorbide polyols (56a) with various OH indices were prepared with improved thermal stability, compressive strength, dimensional stability and reduced water absorption compared to 1,2-propanediol-based polymers.331 Highly functional sucrose-based polyols (79) were used for the synthesis of R-PUFs with high dimensional stability, low friability and compression strength between 126 and 152 kPa.349Flame retardancy has become a hot topic in the last decade, and particularly for PU foams.398 Many strategies have been employed, including the use of additives and halogen-, nitrogen- or phosphorous-based compounds for the synthesis of polymers, especially R-PUFs. For instance, rosin has been intensively studied as a flame retardant agent in R-PUFs. It was shown that rosin-based foam was less flammable and required less than half the amount of flame retardant than that with conventional polyol.399
Various other rosin-based polyols have been developed for the synthesis of R-PUFs with improved thermal stability, flame retardation and compression resistance compared to conventional flame-retardants for R-PUFs.400–402 Grafting or combining MPA or FPA with phosphorus, polysiloxanes or other halogen-free compounds further improved the flame retardancy of R-PUFs.253,403–405
4.2. Coatings
Coatings can be applied to a material to protect it from corrosion, radiation, moisture, and biological degradation for a large range of final applications. Coatings are the second most important market of PUs after foams due to their excellent abrasion resistance, toughness, low-temperature flexibility, corrosion and chemical resistance.37 Accordingly, due to the hydrophobicity of most cycloaliphatic structures, they have been largely studied as coating polymers with low surface energy.Rosin has been intensively developed in WPUs for biomedical application due to its antibacterial activity.406–408 WPUs from rosin-based polyester polyol (14) were developed with improved mechanical, thermal, water resistance and antibacterial properties. The activity of the antibacterial films increased with an increase in content polyol (14) because it degraded the outer membrane of the bacteria, eventually leading to cell death. Furthermore, water absorption was reduced from 80% to 15% due to the increased cross-linked density and presence of unsaturated double bonds, providing a greater barrier to water migration.254
Due to the hydrophobicity of rosin, various WPU coatings with low water absorption and high mechanical properties have been designed using rosin. For example, the addition of 30 wt% of maleopimaric acid polyester polyol (13) to the WPU formulation, improved its thermal degradation from 170 °C to 237 °C and tensile strength from 7 to 23 MPa and decreased the water absorption from 79% to 15%.409 Two different smooth and flexible WPU transparent films were also designed using rosin-based polyols (13 or 15). It appeared that the impact strength, hardness, water-resistance and thermal resistance of these films increased with an increase in NCO/OH ratio.274,410 Two-component WPU (2K-WPU) films with low water absorption have been prepared from rosin-based polyol with f = 3. The impact of rosin-based polyester polyol content on the properties of the WPUs were investigated. The water absorption decreased with an increase in rosin polyol content due to the hydrophobicity of the rosin cycloaliphatic structure. Consequently, the tensile strength improved from 7 to 16 MPa and the soluble fraction decreased due to the increase in cross-linking density caused by the trifunctionality of the rosin-based polyol. Also, gloss and hardness were improved with rosin polyol content.411,412
Recent works have investigated isosorbide modification for the preparation of PU coatings. It has been shown that the isosorbide cycloaliphatic structure increased the thermal stability, Tg and antimicrobial properties.333,337,342 Then, isosorbide di(meth)acrylate has been designed as a photocurable reactive diluent for UV-curable coatings. It was mixed with PU acrylic to test its diluting ability. The results were similar to that obtained with a commercial diluent. Moreover, the Tg, hardness, storage modulus, and impact properties increased. Nevertheless, the bulky structure of isosorbide allowed segmented fluidity in the network, leading to the formation of flexible high-performance photosensitive resins.342
Biobased PU coatings were developed from IPDI, PLA-based diol, BDO and antifouling additives such as rosin and non-toxic butenolide. The release of butenolide was responsible for the antifouling properties. The addition of rosin delayed the release of butenolide by increasing the polymer self-polishing. Strong antifouling properties were maintained for at least two months.419
Multiblock-like amphiphilic PUs have been synthesized using betulin and ethylene oxide. PUs with different betulin contents have been spin-coated for protein-resistance tests. The results showed that an increase in betulin content decreased the phase separation and increased the homogeneity of hydrophobicity. It improved the broad-spectrum protein resistance. Indeed, when the betulin content was too low, hydrophobic crystalline domains were formed due to phase separation, providing enough sites for protein adsorption. This type of hydrophobic and protein-resistant polymer can be interesting for antifouling applications.285
4.3. Adhesives
Adhesives are one of the major applications of PUs, which are mainly used in the automotive, footwear and furniture industries. PU adhesives can be cured with heat for rigid adhesives or with moisture for elastic adhesives containing urea bonds. Two-component adhesives have also been developed. PU adhesives have many advantages such as the ability to form strong covalent bonds quickly, compatibility with most substrates, and easy handling. Increasing efforts have been devoted to the solventless preparation of renewable PU adhesives to reduce the environmental impact and toxic VOC emissions.420 Vegetable oils and rosin systems have been intensively studied for the development of biobased adhesives.TPU sealants containing rosin as a chain extender were developed as sealants for defects in disc regeneration surgery. In the case of a mixture with BDO as the chain extender, when the rosin content increased, the urethane and urethane-amide HS content increased as well as HS/SS segregation. The storage modulus decreased with an increase in the rosin content due to the bulky chemical structure of rosin compared to the linear BDO.263,421
Biomedical TPU tissue adhesives were designed from oligo-PHB diol, DDI, HDI and various chain extenders. A mixture of flexible DDI and hard HDI resulted in mid-range properties. TPU adhesive systems were applied on different substrates for lap-shear adhesion tests. Bovine muscle, liver tissues and porcine skin tissues have been tested for biomedical application. Higher reactive chain extenders showed improved adhesion on muscle tissues. Highest adhesions were obtained on skin substrate.369
Hydrogenated rosin has been studied in PUA systems, and their adhesion to different substrates tested, such as glass, PET, PC and PVC. The photopolymerization rate was impacted by a larger stereoelectronic effect and higher Tg due to the bulky cycloaliphatic chemical structure of rosin. The adhesion on these different substrates increased with an increase in the rosin-based content. This was due to the small volume shrinkage during UV-curing, leading to less internal stress. Also, imide-urea bonds were created after the second addition of isocyanate. These bonds formed strong hydrogen bonds with the different substrates, especially with polar ones such as glass, increasing the adhesion.265
4.4. TPUs
TPUs exhibit many useful properties such as flexibility, elasticity, strength, good abrasion resistance, and transparency. In a sustainable development approach and to reduce the carbon footprint and improve LCA, particular TPUs have been designed using renewable building blocks. In general, cycloaliphatics enable the creation of TPU architectures with high rigidity and thermal stability.Rosin-based polyols (24, 25) have been compared to EG as chain extenders in the TPU formulation with MDI and polyadipate. The degree of phase separation was higher for TPU with rosin-based chain extenders (24, 25) compared to EG. The TPU with polyols (24, 25) were stiffer due to the rigidity of rosin, leading to a higher storage modulus and thermal stability and lower relaxation potential.257,258
In the case of isosorbide-based polyester polyols, TPUs (60a) were synthesized with different diisocyanates such as TDI, MDI, IPDI, and HDI. Isosorbide was used to obtain a high Tg in the range of 78 °C to 159 °C. SnCl2 catalyst combined with aromatic solvent such as chloro-benzene or xylene was the best effective combination in this case. Indeed, it is known that metal chloride catalysts are the most efficient for condensation reactions. With a too high excess of diisocyanate compared to the conventional stoichiometry, the Tg increased and allophanate formation was favored, leading to the formation of 3D cross-linked structures. Due to the high content of H-bonds, the thermal stability decreased with excess isocyanate and the adhesion to polar surfaces and solubility in polar nucleophilic solvent increased.339
Electrospinning has also been used to prepare highly elastic scaffolds composed of HDI, isosorbide and polycarbonate diol. High thermal stability and mechanical properties were obtained due to the rigid cycloaliphatic structure of isosorbide with high elongation at break between 365% and 953%. Moreover, the hydrophilicity of isosorbide increased the cell adhesion and proliferation. However, these TPUs were too stiff for soft tissue application.374,376 In one case, it has been shown that cycloaliphatic building blocks in TPU scaffolds slowed down their hydrolytic degradation. Indeed, DDI-based urethane groups are less hydrolysable than esters groups due to the hydrophobic nature of the dimer.369
4.5. SMPUs
As described earlier, dynamic chemistry has been intensively studied in the last few decades to increase the shelf life of materials. This chemistry enables the repair of damaged polymer materials by reprocessing or by self-healing, even if they are thermosets.125 Given that it is a relatively new topic, the main issues are to create dynamic materials with high strength and stiffness and with scalable and affordable chemistry. Then, shape-memory effects have also been studied to facilitate self-repair and reprocessing in a sustainable development approach.129 Renewable cycloaliphatic building blocks have been used in these fields to improve the mechanical properties due to their rigidity.SMPUs with improved properties have been synthesized from highly functional rosin-based polyester polyols with different amounts of HDI and DBTL as the catalyst. The mechanical properties of the PU network improved with an increase in isocyanate content by increasing the crosslinking density and HS content. With the HDI content increased from 30% to 80%, the tensile strength and toughness increased significantly and the elongation at break decreased from 119% to 61%. These PU networks showed improved mechanical properties compared to previously reported ones due to their aliphatic rings and the combined effect of physical and chemical crosslinking. Concerning self-healing properties, network rearrangement took place by transcarbamoylation at high temperature. After 4 h at 160 °C, PU with 70% HDI exhibited good self-healing and welding capabilities. For the thermal-responsive shape memory tests, the initial state was reached within 1 min. Pictures of the shape-recovery performance of PU containing 70 mol% of HDI and 30 mol% of rosin-based polyol are presented in Fig. 15. These material properties were reached because of the topological rearrangement due to transcarbamoylation exchange reactions, catalyzed by DBTL, chemical and physical crosslinking networks, and a suitable content of HSs and SSs.425
 | ||
| Fig. 15 Shape memory performance of PUs with 30 mol% of rosin-based polyol and 70 mol% of HDI after different treatments. Reproduced from ref. 417. | ||
Other renewable dynamic isosorbide-based poly(urethane-urea) networks with high mechanical properties have been designed using isosorbide, castor oil, and 4-aminophenyl disulfide. Isosorbide was incorporated to reduce the crosslinking density with the same mechanical behavior due to its rigidity brought by its cycloaliphatic structure. Good thermal resistance and high mechanical properties were obtained with a Young's modulus of 255 MPa, tensile strength of 25 MPa and elongation at break of 140%. With associative disulfide exchange reaction, these materials were could be reprocessed at high temperature. Fully recovered mechanical properties were obtained after three thermal remolding cycles at 180 °C.372
Dynamic biobased PHUs have been synthesized from pinene-derived diamine (31) and cyclic-carbonated soybean oil. Pinene-derived amino groups catalyzed the synthesis reaction and flexible soybean oil chains and pinene-derived carbamate bonds catalyzed transesterification reactions, which are responsible for dynamic bond exchange. No solvent or catalyst was added in the green chemistry approach. SMPUs with a stoichiometric ratio between pinene-based diamines (31) and cyclocarbonates showed the best thermo-mechanical properties due to the high cross-linking density. The tensile strength increased to 2.2 MPa and the elongation at break decreased to 106%. SMPUs with a high content of pinene-derived diamines (31) had longer relaxation time and lower relaxation rate due to the rigidity of the cycloaliphatic structure, which limited the chain mobility. Transesterification and transcarbonation reactions led to materials with self-healing, reprocessability, shape-memory properties and chemical recyclability. Chemical recycling in ethanol was possible due to the exchange reaction with OH groups from ethanol. Triple-shape memory behavior was possible because of the additional reversible phase transition. At 140 °C, a topology-freezing transition (or dynamic bond exchange) occurred and the material could be reshaped to its initial form. After several reprocessing cycles, the mechanical and thermal properties were almost completely recovered at more than 85% of the original value, as shown in Fig. 16.277
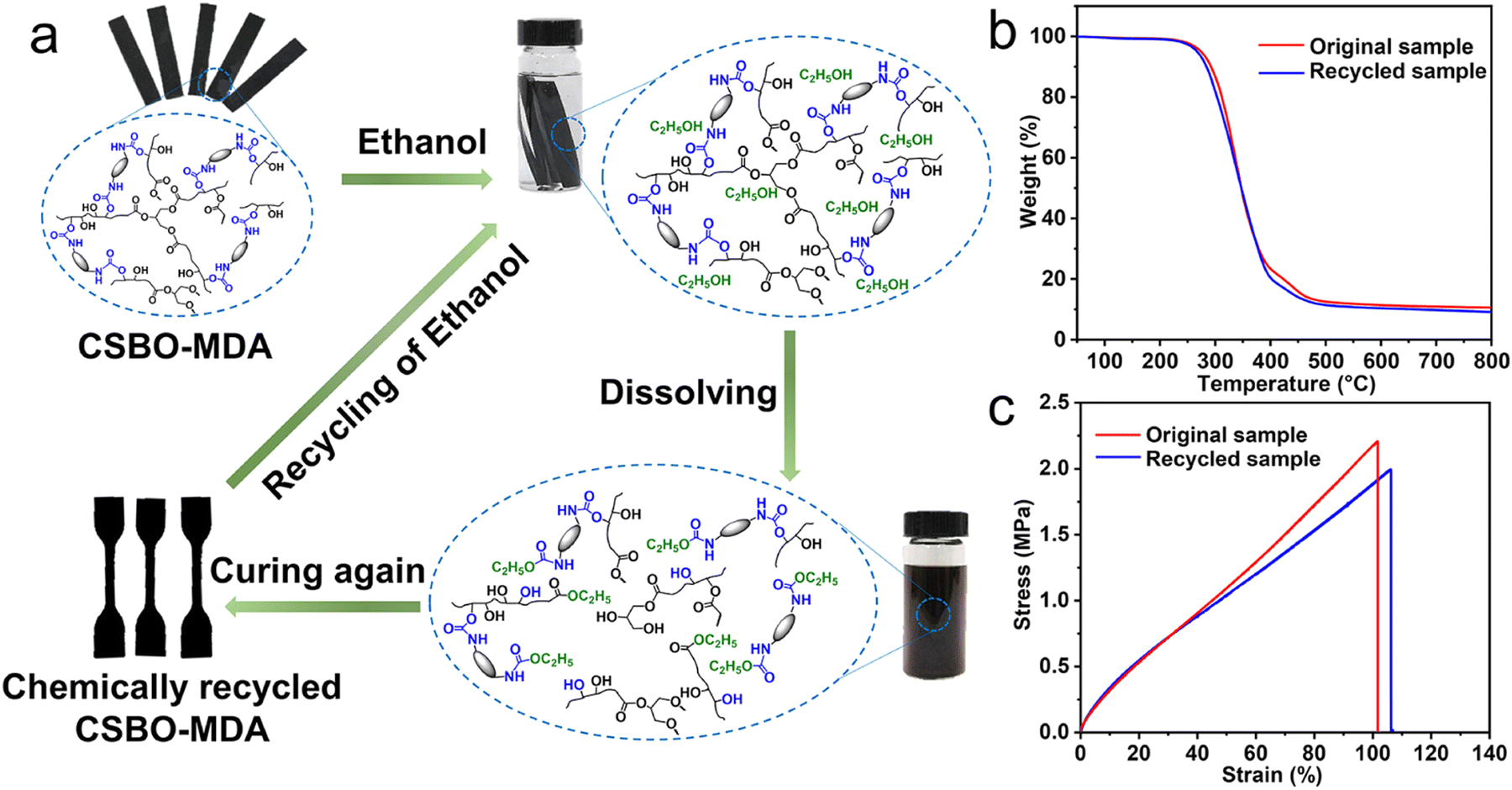 | ||
| Fig. 16 (a) Chemical recycling of cured CSBO-MDA1.0 PHUs. CSBO corresponds to cyclic-carbonated soybean oil. Ratio between CSBO and MDA is 1.0. (b) TGA curves and (c) stress–strain curves of original and recycled CSBO–MDA samples. Reproduced from ref. 272. | ||
PUs containing grafted and free cholesterol have been synthesized from MDI and PTHF. Cholesterol was grafted by the second addition of MDI, with the formation of allophanate. Cross-linked networks were obtained when MDI did not react with cholesterol, forming two urethane bonds. The tensile stress increased with an increase in cholesterol and cross-linking density from 18 to 47 MPa. The elongation at break decreased from 2013% to 1609% when the cholesterol content of increased because cholesterol hindered the stretching of the PU. Shape-recovery tests were conducted at 0 °C and 45 °C. When the amount of grafted cholesterol increased, the shape recovery at 45 °C and 0 °C increased, due to physical and chemical cross-linking. However, when free cholesterol increased, the shape recovery at 45 °C and 0 °C decreased, with premature breakage of PUs with a higher cholesterol content. The absence of chemical and physical cross-linking of PUs with free cholesterol led to a decrease in shape recovery.289
Rosin-based diisocyanate (21) has been used as a building block for the synthesis of thermo-responsive biobased SMPU. It has been shown that the microphase segregation increased with an increase in rosin-based diisocyanate (21) content. Also, increasing the cross-linking density resulted in improved thermal and mechanical properties and shape-recovery performance. The elastic modulus and Tα (relaxation temperature associated with Tg) increased from 2.2 to 3.3 GPa and from 61 °C to 78 °C, respectively. The shape fixity ratio and shape recovery ratio increased from 98.3% to 99.4% and from 96.7% to 98.8%, respectively. Also, these SMPUs showed higher hydrolytic degradability in buffer solution without enzymatic catalyst, reaching up to 71% weight loss within 8 weeks due to their numerous hydrolysable groups.255
Three different isosorbide-based SMPUs were developed for biomedical application. Firstly, SMPUs with enhanced mechanical properties were designed from two diisocyanates composed of HDI and isosorbide units. Poly(DL-lactic acid) diol was selected as the soft chain polyol and isosorbide as the chain extender. Excellent mechanical properties were obtained due to the increase in HS content. The strength at break, elongation at break and Young's modulus ranged from 32 to 53 MPa, 4% to 16% and 3062 to 3588 MPa, respectively. Moreover, good shape-memory characteristics were achieved with a shape fixity ratio of up to 99.8% and recovery ratio of up to 90.2%. In vitro degradation tests were successful with almost total degradation within 120 days. Thus, high-value bone repair application can be envisaged.386 Secondly, SMPUs based on isosorbide, castor oil, PCL diol and HDI have been designed for biomedical application. Isosorbide and castor oil were used to form net points, and PCL diol acted as the switching segment. High shape-memory properties were obtained with 95% shape recovery ratio and 90% shape fixity ratio. Moreover, shape recovery tests in 37 °C water bath were successful with shape recovery within 20 s, as shown in Fig. 17. Due to their high cell viability and proliferation, these materials can be envisaged for stent with self-expansion. The proof of concept test showed the self-expansion of the stent within 18 s.375 Thirdly, thermally responsive SMPUs from isosorbide, MDI and different amounts of PCL diol have been designed. The PU containing 30% of PCL exhibited good shape-memory properties. 95% shape-fixing ratio was obtained due to the PCL crystalline domains. The 71% shape-recovery ratio was due to the amorphous PU domains due to the bulky structure of cycloaliphatic isosorbide. This material could be knotted by itself in a water bath at 40 °C, illustrating its potential for smart suture application. Bioresorbability characteristics, cell adhesion and proliferation were proven, confirming its high potential for application in the biomedical field.377
 | ||
| Fig. 17 Self-expansion of stent made with PUs from isosorbide, PCL diol, castor oil and HDI in 37 °C water bath. Reproduced with authorization from ref. 347. | ||
4.6. Microspheres or microcapsules for adsorption
Microcapsules or microspheres are interesting due to their controlled surface permeability, high specific area, high adsorption capacity and stability in aqueous media. They have been used in various high-value fields such as water treatment and as drug carriers. Due to their relatively high biocompatibility and versatility, good flexibility and elasticity, PUs can be remarkable in this field.426 Several renewable cycloaliphatic building blocks previously presented in this review are biocompatible. Then, some of them have been studied for microcapsule development for the adsorption of drugs, gases, and fragrances.The synthesis of bioresorbable PU microspheres has been investigated with rosin as the raw material. The degradability and compatibility of rosin in the human body make it an excellent candidate for adsorption and drug release applications. Rosin-based polyester diol was used for the synthesis of WPUs with pendant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. The addition of styrene led to the formation of microspheres. The effects of temperature and pH on their absorption properties were investigated, and it was found that they can be used for pH-sensitive drug-release application.427
C. The addition of styrene led to the formation of microspheres. The effects of temperature and pH on their absorption properties were investigated, and it was found that they can be used for pH-sensitive drug-release application.427
Hollow-porous rosin-based PU microspheres have been developed for drug release applications. Also, a rosin-based polyester polyol was used for the synthesis of PUs. The effect of the chain extender type and amount and impact of solvent were investigated. The morphology, particle size, particle distribution and buffer volume of the microspheres were impacted by these parameters.428
PU networks from betulin, using triphenylmethane triisocyanate as a cross-linker have also been prepared. Three rigid aliphatic cycles of the betulin introduced frustrated packing. This high free volume allowed the synthesis of microporous PU to be envisaged. Specifically, 10 μm microglobules were formed by precipitation during synthesis. This morphology is suitable for gas adsorption membrane applications. The pore size was between 0.5 and 1 nm, which is interesting for the gas separation kinetics. These nanosizes can be reduced by hydrogen bonds due to the urethane linkages. Promising CO2/N2 gas selectivity was observed by adsorption studies.429
Isosorbide-based microcapsules have been prepared via interfacial polycondensation. Naphthol ether fragrance was encapsulated in a shell composed of isosorbide and MDI. Impregnation was chosen to apply these microcapsules on textile fibers. Isosorbide was chosen to replace the conventional and toxic bisphenol A. Due to the hydrophobicity of MDI and the hydrophilic character of isosorbide, microcapsules were successfully synthesized. They presented a unimodal volume distribution, with the average particle size of 27 μm. Impregnation of the textile fibers was achieved with the absence of aggregates and good resistance to washing.370
5. Conclusion and outlook
PUs are a very important polymer family and most of their building blocks/monomers come from fossil resources to date. However, due to environmental concerns, legislation and oil rarefaction and its price fluctuation, attention has been given to renewable and sustainable feedstock and recycling. Among the wide range of chemical structures, cycloaliphatic building blocks are attractive given that they combine the main advantages of linear aliphatic and aromatic compounds. In this frame, different sustainable compounds such as terpenes, cholesterol, carbohydrates, and fatty acids derivate have been reported as cycloaliphatic structures for the synthesis of PUs.These building blocks possess different types of reactive and modifiable groups such as carbon–carbon double bonds, OH groups and carboxylic acids. Many strategies have been developed to transform these chemical structures into functional building blocks for the preparation of sustainable PUs. In the case of carbon–carbon double bonds, a large and rich chemistry can be developed such as click chemistry with Diels Alder or thiol-ene addition, considering the principle of green chemistry. In the case of OH groups and acids, various methods of esterification and etherification have been employed to synthesize new cycloaliphatic polyesters or polyether polyols. They partially or totally replaced conventional linear polyols, chain extenders and cross-linkers. The rigidity brought by the cycloaliphatic structure results in an increase in Tg and higher thermal stability of novel PUs compared to linear aliphatic polyols. In the frame of the synthesis of conventional PUs and contrary to polyols, only few renewable and sustainable cycloaliphatic diisocyanates have been developed, which are rarely described in the literature. Their synthesis require many steps, including the handling of the harmful phosgene. Some non-phosgene routes have also been described with low yield. However, in a sustainable development strategy, renewable cycloaliphatic polyamines and polycyclocarbonates were also developed for the synthesis of NIPUs. Mostly low molar mass PUs have been obtained because it is difficult to get the right stoichiometric conditions. The second reason is that aminolysis has lower reactivity than urethanisation.
These sustainable and renewable cycloaliphatic building blocks bring interesting properties to the final Pus, as follows:
• Stiffness
• High thermal stability
• Multiphasic structure associated with HS/SS micro segregation
• Low surface energy
The stiffness and thermal stability caused by the cycloaliphatic structure enable the production of PUs with enhanced mechanical and thermal properties for various applications such as R-PUFs, where these properties are particularly required and associated with flame-retardant properties, e.g., building insulation and high-performance coatings with resistance to hydrolysis and oxidation.
Novel multiphasic structures can be created due to the H-bonding ability of the functional groups of these building blocks, enabling the creation of TPUs with hydrophobic crystalline HS domains, with increased HS/SS phase segregation. This physical cross-linking improved the mechanical properties of TPUs and H-bonding ability also intensively used in the field of adhesives.
These HS crystalline domains combined with bulky apolar structure provide PUs with low surface energy. They present hydrophobicity and low adhesion of bacterial cells. Materials with high cross-linking density have enhanced transparency, gloss, and hardness.
Cycloaliphatics combine the main advantages of aromatic and linear aliphatic compounds, but some drawbacks still remain. Due to their specific structure, the high steric hindrance of the functional groups reduces their reactivities and often requires specific reaction conditions with chemical modification to obtain higher reactive groups before polymerization. Moreover, moderate barrier properties were obtained in the case of inhomogeneous structures with segregation. These morphologies result in different diffusion pathways, decreasing the permeability properties. Also, due to their bulky and rigid structure, the viscosity may increase, causing difficulties in processing, for instance, during the fabrication of the foams or coatings. Similarly, a higher Tg can make PUs brittle.
Given that the environmental impact is increasingly considered, PUs from cycloaliphatics with improved environmental impact have been designed such as renewable SMPUs, WPU coatings, antifouling coatings and UV-curing adhesives and coatings. The trend and perspectives in this field are also based on green chemistry to produce materials with high sustainable carbon content, extended shelf life and controlled end of life. Biomass is a well-known renewable feedstock with increasing control, but other sustainable carbon sources have been investigated, such as plastic waste. Several processes for chemical and biological recycling have been developed, but they are still less commercially attractive compared to low-cost virgin compounds. Thus, to overcome this key point, research on upcycling is increasing to convert plastic waste into value-added chemicals through biotechnology, which is associated with chemistry or not.
Author contributions
Agathe Mouren: Conceptualization, data curation, methodology, writing – orginal draft, Writing – review & editing. Luc Avérous: conceptualization, methodology, writing – review & editing, supervision, funding acquisition, validation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement No. 870294.References
- Plastics – the Facts 2020, https://plasticseurope.org/fr/knowledge-hub/plastics-the-facts-2020-2/, (accessed April 13, 2022).
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. D. Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- A. D. Vethaak and J. Legler, Science, 2021, 371, 672–674 CrossRef CAS PubMed.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed.
- A.-C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- T. Narancic, S. Verstichel, S. Reddy Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. Babu Padamati and K. E. O’Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed.
- A. Morinval and L. Averous, Polym. Rev., 2021, 0, 1–69 Search PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- Polyurethane Market Size, Share & Trends Report, 2021-2028, https://www.grandviewresearch.com/industry-analysis/polyurethane-pu-market, (accessed April 13, 2022).
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- G. P. Moss, P. a S. Smith and D. Tavernier, Pure Appl. Chem., 1995, 67, 1307–1375 Search PubMed.
- C. Zhang, J. Xue, X. Yang, Y. Ke, R. Ou, Y. Wang, S. A. Madbouly and Q. Wang, Prog. Polym. Sci., 2022, 125, 101473 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. M. Paraskar, M. S. Prabhudesai, V. M. Hatkar and R. D. Kulkarni, Prog. Org. Coat., 2021, 156, 106267 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- S. Awasthi and D. Agarwal, J. Coat. Technol. Res., 2007, 4, 67–73 CrossRef CAS.
- H. Ni, J. L. Daum, P. R. Thiltgen, M. D. Soucek, W. J. Simonsick, W. Zhong and A. D. Skaja, Prog. Org. Coat., 2002, 45, 49–58 CrossRef CAS.
- E. Schab-Balcerzak, D. Sęk, A. Volozhin, T. Chamenko and B. Jarząbek, Eur. Polym. J., 2002, 38, 423–430 CrossRef CAS.
- M. Hasegawa, M. Fujii and Y. Wada, Polym. Adv. Technol., 2018, 29, 921–933 CrossRef CAS.
- Y.-H. Wu, C.-C. Wang and C.-Y. Chen, Polym. J., 2021, 53, 695–702 CrossRef CAS.
- Y. Zhuang, J. G. Seong and Y. M. Lee, Prog. Polym. Sci., 2019, 92, 35–88 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS.
- A. Magnin, E. Pollet, V. Phalip and L. Avérous, Biotechnol. Adv., 2020, 39, 107457 CrossRef CAS PubMed.
- J. A. Glaser, in Plastics in the Environment, ed. A. Gomiero, IntechOpen, 2019 Search PubMed.
- T. Tiso, B. Winter, R. Wei, J. Hee, J. de Witt, N. Wierckx, P. Quicker, U. T. Bornscheuer, A. Bardow, J. Nogales and L. M. Blank, Metab. Eng., 2022, 71, 77–98 CrossRef CAS PubMed.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC.
- A. Llevot and M. Meier, Polym. Int., 2019, 68, 826–831 CrossRef CAS.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- A. Noreen, K. M. Zia, M. Zuber, S. Tabasum and A. F. Zahoor, Prog. Org. Coat., 2016, 91, 25–32 CrossRef CAS.
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CrossRef CAS.
- C. Zhang, T. F. Garrison, S. A. Madbouly and M. R. Kessler, Prog. Polym. Sci., 2017, 71, 91–143 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- M. R. Islam, M. D. H. Beg and S. S. Jamari, J. Appl. Polym. Sci., 2014, 131(18) DOI:10.1002/app.40787.
- S. Miao, P. Wang, Z. Su and S. Zhang, Acta Biomater., 2014, 10, 1692–1704 CrossRef CAS PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- J. M. Buist, ed., Developments in polyurethane, Applied Science Publishers, London, 1978 Search PubMed.
- G. M. Kline, J. Eng. Mech. Div., 1956, 82, 1071–22 CAS.
- O. Bayer, Angew. Chem., 1947, 59, 257–272 CrossRef.
- D. Randall and S. Lee, eds., The polyurethanes book, Distributed by John Wiley & Sons, Huntsman Polyurethanes; New York, Everberg, Belgium, 2002 Search PubMed.
- M. A. Lucherelli, A. Duval and L. Avérous, Prog. Polym. Sci., 2022, 101515 CrossRef CAS.
- J. Peyrton and L. Avérous, Mater. Sci. Eng., R, 2021, 145, 100608 CrossRef.
- M. Ionescu, Chemistry and technology of polyols for polyurethanes, Rapra Technology, Shawbury, Shrewsbury, Shropshire, UK, 2005 Search PubMed.
- R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47–76 CrossRef CAS.
- J. Beckerdite, R. Herrington, K. Hock and D. C. Company, Flexible Polyurethane Foams, Dow Chemical Company, Dow Plastics, 1991 Search PubMed.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J.-P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- P. F. Wiley, J. Am. Chem. Soc., 1949, 71, 3746–3748 CrossRef CAS.
- L. C. Raiford and H. B. Freyermuth, J. Org. Chem., 1943, 08, 230–238 CrossRef CAS.
- T. W. Campbell and K. C. Smeltz, J. Org. Chem., 1963, 28, 2069–2075 CrossRef CAS.
- K. H. Slotta and H. Dressler, Berichte Dtsch. Chem. Ges. B Ser., 1930, 63, 888–898 CrossRef.
- J. S. Senger, I. Yilgor, J. E. McGrath and R. A. Patsiga, J. Appl. Polym. Sci., 1989, 38, 373–382 CrossRef CAS.
- M. Szycher, Szycher's handbook of polyurethanes, CRC Press, Boca Raton, 1999 Search PubMed.
- H.-J. Knölker, T. Braxmeier and G. Schlechtingen, Angew. Chem., Int. Ed. Engl., 1995, 34, 2497–2500 CrossRef.
- J. H. Saunders and R. J. Slocombe, Chem. Rev., 1948, 43, 203–218 CrossRef CAS PubMed.
- S. Ozaki, Chem. Rev., 1972, 72, 457–496 CrossRef.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- D. C. Allport, D. S. Gilbert and S. M. Outterside, Eds., MDI and TDI: a safety, health and the environment: a source book and practical guide, J. Wiley, New York, 2003 Search PubMed.
- M. F. Sonnenschein, Polyurethanes: science, technology, markets, and trends, Wiley, Hoboken, Second edition., 2020 Search PubMed.
- Polyester polyols by Arkema, https://www.arkema.com/global/en/products/product-finder/product-range/coatingresins/polyesterpolyols/, (accessed November 10, 2021).
- P. Furtwengler, R. Perrin, A. Redl and L. Avérous, Eur. Polym. J., 2017, 97, 319–327 CrossRef CAS.
- Desmophen® | Polyols by Covestro, https://solutions.covestro.com/en/brands/desmophen, (accessed November 10, 2021).
- U. Meier-Westhues, Polyurethanes: coatings, adhesives and sealants, Vincentz Network, Hannover, 2007 Search PubMed.
- Joncryl® acrylic resins & emulsions, https://www.basf.com/us/en/products/General-Business-Topics/dispersions/Products/joncryl0.html, (accessed November 10, 2021).
- Y. Zhang, Y. Qi and Z. Zhang, Prog. Org. Coat., 2016, 97, 115–121 CrossRef CAS.
- M. S. Sanchez-Adsuar and J. M. Martín-Martínez, J. Adhes. Sci. Technol., 1997, 11, 1077–1087 CrossRef CAS.
- X. Liu, T. Wang, J. Li, J. Cheng and J. Zhang, J. Polym. Res., 2015, 22, 149 CrossRef.
- M. Möller and H.-U. Moritz, J. Appl. Polym. Sci., 2006, 101, 4090–4097 CrossRef.
- K. Ashida, Polyurethane and related foams chemistry and technology, CRC Taylor & Francis, Boca Raton, 2007 Search PubMed.
- G. Woods, Flexible polyurethane foams: chemistry and technology, Applied Science Publishers, London; Englewood, NJ, 1982 Search PubMed.
- N. Mills, in Polymer Foams Handbook, ed. N. Mills, Butterworth-Heinemann, Oxford, 2007, pp. 19–37 Search PubMed.
- N. Mahmood, Z. Yuan, J. Schmidt and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- M. Melchiors, M. Sonntag, C. Kobusch and E. Jürgens, Prog. Org. Coat., 2000, 40, 99–109 CrossRef CAS.
- T. Engbert, M. Bock and O. Kirihara, Shikizai Kyokaishi, 1999, 72, 102–107 CAS.
- M. J. Dvorchak, J. Coat. Technol., 1997, 69, 47–52 CrossRef CAS.
- P. Zhang, H. Fan, K. Hu, Y. Gu, Y. Chen, J. Yan, S. Tian and Y. He, Prog. Org. Coat., 2018, 120, 88–99 CrossRef CAS.
- I. Acar and M. Orbay, Polym. Eng. Sci., 2011, 51, 746–754 CrossRef CAS.
- D. K. Chattopadhyay, B. Sreedhar and K. V. S. N. Raju, J. Appl. Polym. Sci., 2005, 95, 1509–1518 CrossRef CAS.
- J. Xu, Y. Jiang, T. Zhang, Y. Dai, D. Yang, F. Qiu, Z. Yu and P. Yang, Prog. Org. Coat., 2018, 122, 10–18 CrossRef CAS.
- A. Baldan, Int. J. Adhes. Adhes., 2012, 38, 95–116 CrossRef CAS.
- A. Tenorio-Alfonso, M. C. Sánchez and J. M. Franco, J. Polym. Environ., 2020, 28, 749–774 CrossRef CAS.
- E. K. Leitsch, W. H. Heath and J. M. Torkelson, Int. J. Adhes. Adhes., 2016, 64, 1–8 CrossRef CAS.
- J. Datta and P. Kasprzyk, Polym. Eng. Sci., 2018, 58, E14–E35 CrossRef CAS.
- Z. S. Petrović and J. Ferguson, Prog. Polym. Sci., 1991, 16, 695–836 CrossRef.
- B. Hopwood, M. Mellor and G. O’Brien, Sustainable Dev., 2005, 13, 38–52 CrossRef.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- C. G. Schirmeister and R. Mülhaupt, Macromol. Rapid Commun., 2022, 43, 2200247 CrossRef CAS PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- Recycling and the future of the plastics industry | McKinsey, https://www.mckinsey.com/industries/chemicals/our-insights/how-plastics-waste-recycling-could-transform-the-chemical-industry, (accessed July 26, 2022).
- B. M. Weckhuysen, Science, 2020, 370, 400–401 CrossRef PubMed.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, J. Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- A. Kemona and M. Piotrowska, Polymers, 2020, 12, 1752 CrossRef CAS PubMed.
- S. Bhandari and P. Gupta, in Recycling of Polyurethane Foams, ed. T. Sabu, R. Ajay Vasuedo, K. Krishnan, V. K. Abitha, and T.Martin George, William Andrew Publishing, 2018, pp. 77–87 Search PubMed.
- K. Troev, G. Grancharov, R. Tsevi and A. Tsekova, Polymer, 2000, 41, 7017–7022 CrossRef CAS.
- D. Simón, A. de Lucas, J. F. Rodríguez and A. M. Borreguero, Polym. Degrad. Stab., 2016, 133, 119–130 CrossRef.
- R. Font, A. Fullana, J. A. Caballero, J. Candela and A. García, J. Anal. Appl. Pyrolysis, 2001, 58–59, 63–77 CrossRef CAS.
- M. Cregut, M. Bedas, M.-J. Durand and G. Thouand, Biotechnol. Adv., 2013, 31, 1634–1647 CrossRef CAS PubMed.
- F. Valerio, J. Waste Manage., 2010, 30, 2354–2361 CrossRef CAS PubMed.
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed.
- J.-G. Gu and J.-D. Gu, J. Polym. Environ., 2005, 13, 65–74 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- A. Magnin, E. Pollet, R. Perrin, C. Ullmann, C. Persillon, V. Phalip and L. Avérous, J. Waste Manage., 2019, 85, 141–150 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, E. Pollet and L. Avérous, J. Waste Manage., 2021, 132, 23–30 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, A. Bazin, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 4234–4241 CrossRef CAS PubMed.
- A. E. Schwarz, T. N. Ligthart, D. Godoi Bizarro, P. De Wild, B. Vreugdenhil and T. van Harmelen, J. Waste Manage., 2021, 121, 331–342 CrossRef CAS PubMed.
- H. Ballerstedt, T. Tiso, N. Wierckx, R. Wei, L. Averous, U. Bornscheuer, K. O’Connor, T. Floehr, A. Jupke, J. Klankermayer, L. Liu, V. de Lorenzo, T. Narancic, J. Nogales, R. Perrin, E. Pollet, A. Prieto, W. Casey, T. Haarmann, A. Sarbu, U. Schwaneberg, F. Xin, W. Dong, J. Xing, G.-Q. Chen, T. Tan, M. Jiang and L. M. Blank, Environ. Sci. Eur., 2021, 33, 99 CrossRef CAS PubMed.
- J. Liu, J. He, R. Xue, B. Xu, X. Qian, F. Xin, L. M. Blank, J. Zhou, R. Wei, W. Dong and M. Jiang, Biotechnol. Adv., 2021, 48, 107730 CrossRef CAS PubMed.
- W.-J. Li, T. Narancic, S. T. Kenny, P.-J. Niehoff, K. O’Connor, L. M. Blank and N. Wierckx, Front. Microbiol., 2020, 11, 382 CrossRef PubMed.
- B. Mückschel, O. Simon, J. Klebensberger, N. Graf, B. Rosche, J. Altenbuchner, J. Pfannstiel, A. Huber and B. Hauer, Appl. Environ. Microbiol., 2012, 78, 8531–8539 CrossRef PubMed.
- T. Tiso, T. Narancic, R. Wei, E. Pollet, N. Beagan, K. Schröder, A. Honak, M. Jiang, S. T. Kenny, N. Wierckx, R. Perrin, L. Avérous, W. Zimmermann, K. O’Connor and L. M. Blank, Metab. Eng., 2021, 66, 167–178 CrossRef CAS PubMed.
- R. Wei, T. Tiso, J. Bertling, K. O’Connor, L. M. Blank and U. T. Bornscheuer, Nat. Catal., 2020, 3, 867–871 CrossRef CAS.
- A. Khan, N. Ahmed and M. Rabnawaz, Polymers, 2020, 12, 2027 CrossRef CAS PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- K.-K. Tremblay-Parrado, C. Bordin, S. Nicholls, B. Heinrich, B. Donnio and L. Avérous, Macromolecules, 2020, 53, 5869–5880 CrossRef CAS.
- R. Hajj, A. Duval, S. Dhers and L. Avérous, Macromolecules, 2020, 53, 3796–3805 CrossRef CAS.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- K. R. Gajbhiye, B. P. Chaudhari, V. B. Pokharkar, A. Pawar and V. Gajbhiye, Int. J. Pharm., 2020, 588, 119781 CrossRef CAS PubMed.
- P. Nellepalli, T. Patel and J. K. Oh, Macromol. Rapid Commun., 2021, 42, 2100391 CrossRef CAS PubMed.
- S. Wendels and L. Avérous, Bioact. Mater., 2021, 6, 1083–1106 CrossRef CAS PubMed.
- C. C. Hornat and M. W. Urban, Prog. Polym. Sci., 2020, 102, 101208 CrossRef CAS.
- F. Elizalde, R. H. Aguirresarobe, A. Gonzalez and H. Sardon, Polym. Chem., 2020, 11, 5386–5396 RSC.
- B. Quienne, F. Cuminet, J. Pinaud, M. Semsarilar, D. Cot, V. Ladmiral and S. Caillol, ACS Sustainable Chem. Eng., 2022, 10(21), 7041–7049 CrossRef CAS.
- D. T. Sheppard, K. Jin, L. S. Hamachi, W. Dean, D. J. Fortman, C. J. Ellison and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 921–927 CrossRef CAS PubMed.
- Y.-S. Jang, B. Kim, J. H. Shin, Y. J. Choi, S. Choi, C. W. Song, J. Lee, H. G. Park and S. Y. Lee, Biotechnol. Bioeng., 2012, 109, 2437–2459 CrossRef CAS PubMed.
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, ACS Sustainable Chem. Eng., 2021, 9, 10664–10677 CrossRef CAS.
- Propanediol | Susterra® renewably sourced propanediol, https://duponttateandlyle.com/susterra/, (accessed November 10, 2021).
- Velvetol® | High Performance Polyether Polyols | WeylChem – WeylChem, https://www.weylchem.com/products/velvetolr, (accessed November 10, 2021).
- Genomatica Products, https://www.genomatica.com/products/, (accessed November 10, 2021).
- BASF now offers bio-based PolyTHF, https://www.basf.com/global/en/media/news-releases/2015/03/p-15-163.html, (accessed November 10, 2021).
- Acide succinique BIOSUCCINIUM®, https://fr.roquette.com/industries/produits-phares/industry-performance-materials-biosuccinum, (accessed November 10, 2021).
- P. Furtwengler and L. Avérous, Polym. Chem., 2018, 9, 4258–4287 RSC.
- M. Desroches, M. Escouvois, R. Auvergne, S. Caillol and B. Boutevin, Polym. Rev., 2012, 52, 38–79 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Biomacromolecules, 2010, 11, 2825–2835 CrossRef CAS PubMed.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- Z. S. Petrović, Polym. Rev., 2008, 48, 109–155 CrossRef.
- D. P. Pfister, Y. Xia and R. C. Larock, ChemSusChem, 2011, 4, 703–717 CrossRef CAS PubMed.
- J. C. Ronda, G. Lligadas, M. Galià and V. Cádiz, Eur. J. Lipid Sci. Technol., 2011, 113, 46–58 CrossRef CAS.
- Natural Oil Polyols Market Size Report, 2021–2028, https://www.grandviewresearch.com/industry-analysis/natural-oil-polyols-nop-market, (accessed November 10, 2021).
- A. Arbenz, R. Perrin and L. Avérous, J. Polym. Environ., 2018, 26, 254–262 CrossRef CAS.
- Polyester Polyols | OLEON, https://www.oleon.com/product/derivatives/polyester-polyols, (accessed November 10, 2021).
- What are BiOH® polyols? https://www.cargill.com/bioindustrial/what-are-bioh-polyols, (accessed November 10, 2021).
- R. Höfer, Industrial Biorefineries & White Biotechnology, Elsevier, 2015, pp. 127–155 Search PubMed.
- E. Hablot, B. Donnio, M. Bouquey and L. Avérous, Polymer, 2010, 51, 5895–5902 CrossRef CAS.
- Solutions from the Nature – Sovermol®, https://www.basf.com/vn/en/products/paint-coating-industry/sovermol.html, (accessed November 10, 2021).
- PriplastTM polyester polyols for enhanced flexibility | Croda Smart Materials | Smart Materials, https://www.crodasmartmaterials.com/en-gb/brands/priplast, (accessed November 10, 2021).
- M. A. Sawpan, J. Polym. Res., 2018, 25, 184 CrossRef.
- M. S. Gaikwad, V. V. Gite, P. P. Mahulikar, D. G. Hundiwale and O. S. Yemul, Prog. Org. Coat., 2015, Complete, 164–172 CrossRef.
- B. V. Tawade, R. D. Shingte, S. S. Kuhire, N. V. Sadavarte, K. Garg, D. M. Maher, A. B. Ichake, A. S. More and P. P. Wadgaonkar, Polyurethanes Today, 2017, 11, 1202 Search PubMed.
- Bio-aniline – Basic chemicals from biomass | Covestro, https://www.covestro.com/en/sustainability/flagship-solutions/bio-based-aniline, (accessed July 25, 2022).
- United States, US10173969B2, 2019.
- BASF expands portfolio of climate friendly products introducing the first isocyanate not carrying a CO2 backpack, https://www.basf.com/global/en/media/news-releases/2022/02/p-22-132.html, (accessed July 28, 2022).
- G. Çaylı and S. Küsefoğlu, J. Appl. Polym. Sci., 2008, 109, 2948–2955 CrossRef.
- E. Taylan and S. H. Küsefoğlu, J. Appl. Polym. Sci., 2011, 119, 1102–1110 CrossRef CAS.
- L. Hojabri, X. Kong and S. S. Narine, J. Polym. Sci. Part Polym. Chem., 2010, 48, 3302–3310 CrossRef CAS.
- L. Hojabri, X. Kong and S. S. Narine, Biomacromolecules, 2009, 10, 884–891 CrossRef CAS PubMed.
- A. S. More, T. Lebarbé, L. Maisonneuve, B. Gadenne, C. Alfos and H. Cramail, Eur. Polym. J., 2013, 49, 823–833 CrossRef CAS.
- United States, US3217028A, 1965.
- United States, US3465023A, 1969.
- United States, US3725450A, 1973.
- J. Han, B. Chen, L. Ye, A. Zhang, J. Zhang and Z. Feng, Front. Mater. Sci. China, 2009, 3, 25–32 CrossRef.
- S. A. Guelcher, A. Srinivasan, J. E. Dumas, J. E. Didier, S. McBride and J. O. Hollinger, Biomaterials, 2008, 29, 1762–1775 CrossRef CAS PubMed.
- A. Pavelková, P. Kucharczyk, Z. Kuceková, J. Zedník and V. Sedlařík, J. Bioact. Compat. Polym., 2017, 32, 225–241 CrossRef.
- J. S. Nowick, N. A. Powell, T. M. Nguyen and G. Noronha, J. Org. Chem., 1992, 57, 7364–7366 CrossRef CAS.
- European Union, EP1020435A4, 2000.
- United States, US20060167303A1, 2006.
- STABiOTM, Bio-based 1,5-pentamethylene diisocyanate (PDI) based polyisocyanate| Business and Products | MITSUI CHEMICALS, INC., https://jp.mitsuichemicals.com/en/service/packaging/coatings/stabio/index.htm, (accessed November 10, 2021).
- European Union, EP2543736B1, 2019.
- United States, US9376404B2, 2016.
- United States, US10173970B2, 2019.
- Desmodur® eco N 7300, https://solutions.covestro.com/en/products/desmodur/desmodur-eco-n-7300_84603813-18871289, (accessed November 10, 2021).
- W. Li, H. Li, C. Wu, B. Han, P. Ouyang and K. Chen, Chem. Eng. J., 2021, 425, 131527 CrossRef CAS.
- J. Thiem and F. Bachmann, Makromol. Chem., 1991, 192, 2163–2182 CrossRef CAS.
- F. Bachmann, J. Reimer, M. Ruppenstein and J. Thiem, Macromol. Chem. Phys., 2001, 202, 3410–3419 CrossRef CAS.
- M. D. Zenner, Y. Xia, J. S. Chen and M. R. Kessler, ChemSusChem, 2013, 6, 1182–1185 CrossRef CAS PubMed.
- United States, US3049552A, 1962.
- J. L. Cawse, J. L. Stanford and R. H. Still, Makromol. Chem., 1984, 185, 697–707 CrossRef CAS.
- J. L. Cawse, J. L. Stanford and R. H. Still, Br. Polym. J., 1985, 17, 233–238 CrossRef CAS.
- M. N. Belgacem, J. Quillerou and A. Gandini, Eur. Polym. J., 1993, 29, 1217–1224 CrossRef CAS.
- A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203–1379 CrossRef CAS.
- C. N. D. Neumann, W. D. Bulach, M. Rehahn and R. Klein, Macromol. Rapid Commun., 2011, 32, 1373–1378 CrossRef CAS PubMed.
- S. S. Kuhire, S. S. Nagane and P. P. Wadgaonkar, Polym. Int., 2017, 66, 892–899 CrossRef CAS.
- World Intellectual Property Organization, WO2016103283A1, 2016.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- M. N. Garcia Gonzalez, M. Levi and S. Turri, J. Cleaner Prod., 2017, 164, 171–178 CrossRef CAS.
- A. Fridrihsone, F. Romagnoli, V. Kirsanovs and U. Cabulis, J. Cleaner Prod., 2020, 266, 121403 CrossRef CAS.
- A. Manzardo, A. Marson, M. Roso, C. Boaretti, M. Modesti, A. Scipioni and A. Lorenzetti, ACS Omega, 2019, 4, 14114–14123 CrossRef CAS PubMed.
- J. E. Lockey, C. A. Redlich, R. Streicher, A. Pfahles-Hutchens, P. (Bert), J. Hakkinen, G. L. Ellison, P. Harber, M. Utell, J. Holland, A. Comai and M. White, J. Occup. Environ. Med., 2015, 57, 44–51 CrossRef CAS PubMed.
- K. Błażek and J. Datta, Crit. Rev. Environ. Sci. Technol., 2019, 49, 173–211 CrossRef.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- Y. Suryawanshi, P. Sanap and V. Wani, Polym. Bull., 2019, 76, 3233–3246 CrossRef CAS.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- H. Khatoon, S. Iqbal, M. Irfan, A. Darda and N. K. Rawat, Prog. Org. Coat., 2021, 154, 106124 CrossRef CAS.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- C. Carré, H. Zoccheddu, S. Delalande, P. Pichon and L. Avérous, Eur. Polym. J., 2016, 84, 759–769 CrossRef.
- H. Blattmann, M. Lauth and R. Mülhaupt, Macromol. Mater. Eng., 2016, 301, 944–952 CrossRef CAS.
- P. Stachak, I. Łukaszewska, E. Hebda and K. Pielichowski, Materials, 2021, 14, 3497 CrossRef CAS PubMed.
- O. Kreye, S. Wald and M. A. R. Meier, Adv. Synth. Catal., 2013, 355, 81–86 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- K. Y. Sen and S. Baidurah, Curr. Opin. Green Sustainable Chem., 2021, 27, 100412 CrossRef CAS.
- Y. Zhou, V. A. Briand, N. Sharma, S. Ahn and R. M. Kasi, Materials, 2009, 2, 636–660 CrossRef CAS.
- E. Czarnotta, M. Dianat, M. Korf, F. Granica, J. Merz, J. Maury, S. A. B. Jacobsen, J. Förster, B. E. Ebert and L. M. Blank, Biotechnol. Bioeng., 2017, 114, 2528–2538 CrossRef CAS PubMed.
- K. Kemper, M. Hirte, M. Reinbold, M. Fuchs and T. Brück, Beilstein J. Org. Chem., 2017, 13, 845–854 CrossRef CAS PubMed.
- M. Li, F. Hou, T. Wu, X. Jiang, F. Li, H. Liu, M. Xian and H. Zhang, Nat. Prod. Rep., 2020, 37, 80–99 RSC.
- F. Zhou and E. Pichersky, Curr. Opin. Plant Biol., 2020, 55, 1–10 CrossRef CAS PubMed.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- V. Mahendra, Appl. Mech. Mater., 2019, 890, 77–91 Search PubMed.
- M. Eggersdorfer, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2000.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- J. Zhao and H. Schlaad, in Bio-synthetic Polymer Conjugates, ed. H. Schlaad, Springer, Berlin, Heidelberg, 2013, pp. 151–190 Search PubMed.
- J. Alonso-Gutierrez, R. Chan, T. S. Batth, P. D. Adams, J. D. Keasling, C. J. Petzold and T. S. Lee, Metab. Eng., 2013, 19, 33–41 CrossRef CAS PubMed.
- C. Willrodt, C. David, S. Cornelissen, B. Bühler, M. K. Julsing and A. Schmid, Biotechnol. J., 2014, 9, 1000–1012 CrossRef CAS PubMed.
- S. P. Shirame and R. B. Bhosale, in Green Chemistry, eds. H. E.-D. M. Saleh and M. Koller, InTech, 2018 Search PubMed.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- R. K. Gupta, M. Ionescu, D. Radojcic, X. Wan and Z. S. Petrovic, J. Polym. Environ., 2014, 22, 304–309 CrossRef CAS.
- C. S. Marvel and L. E. Olson, J. Polym. Sci., 1957, 26, 23–28 CrossRef CAS.
- M. L. Shrestha and M. Ionescu, J. Polym. Environ., 2018, 26, 2257–2267 CrossRef CAS.
- L. Filippi and M. A. R. Meier, Macromol. Rapid Commun., 2021, 42, 2000440 CrossRef CAS PubMed.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- Y. Mahamat Ahmat, S. Madadi, L. Charbonneau and S. Kaliaguine, Catalysts, 2021, 11, 847 CrossRef CAS.
- A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- A. Rehman, E. Russell, F. Saleem, F. Javed, S. Ahmad, V. C. Eze and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 104680 CrossRef CAS.
- L. Charbonneau, X. Foster and S. Kaliaguine, ACS Sustainable Chem. Eng., 2018, 6, 12224–12231 CrossRef CAS.
- K. A. Maltby, M. Hutchby, P. Plucinski, M. G. Davidson and U. Hintermair, Chem. – Eur. J., 2020, 26, 7405–7415 CrossRef CAS PubMed.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- A. J. Kamphuis, F. Milocco, L. Koiter, P. P. Pescarmona and E. Otten, ChemSusChem, 2019, 12, 3635–3641 CrossRef CAS PubMed.
- G. Fiorani, M. Stuck, C. Martín, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2016, 9, 1304–1311 CrossRef CAS PubMed.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- A. Causero, C. Troll and B. Rieger, Ind. Eng. Chem. Res., 2020, 59, 15464–15477 CrossRef CAS.
- European Union, EP0266661A2, 1988.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Eur. Polym. J., 2015, 67, 409–417 CrossRef CAS.
- L. Vevere, A. Fridrihsone, M. Kirpluks and U. Cabulis, Polymers, 2020, 12, 2706 CrossRef CAS PubMed.
- G.-F. Chen, Prog. Org. Coat., 1992, 20, 139–167 CrossRef CAS.
- A. M. Atta, R. Mansour, M. I. Abdou and A. M. Sayed, Polym. Adv. Technol., 2004, 15, 514–522 CrossRef CAS.
- Y. Zhang, S. Shang, X. Zhang, D. Wang and D. J. Hourston, J. Appl. Polym. Sci., 1995, 58, 1803–1809 CrossRef CAS.
- J. F. Jin, Y. L. Chen, D. N. Wang, C. P. Hu, S. Zhu, L. Vanoverloop and D. Randall, J. Appl. Polym. Sci., 2002, 84, 598–604 CrossRef CAS.
- L. Zhang, Y. Jiang, Z. Xiong, X. Liu, H. Na, R. Zhang and J. Zhu, J. Mater. Chem. A, 2013, 1, 3263–3267 RSC.
- X. Q. Liu, W. Huang, Y. H. Jiang, J. Zhu and C. Z. Zhang, eXPRESS Polym. Lett., 2012, 6, 293–298 CrossRef CAS.
- G. Liu, G. Wu, J. Chen and Z. Kong, Prog. Org. Coat., 2016, 101, 461–467 CrossRef CAS.
- S. Wang, X. Yang, Z. Li, X. Xu, H. Liu, D. Wang, H. Min and S. Shang, Compos. Sci. Technol., 2020, 198, 108272 CrossRef CAS.
- X. Xu, Z. Song, S. Shang, S. Cui and X. Rao, Polym. Int., 2011, 60, 1521–1526 CrossRef CAS.
- P. Gnanasekar, J. Chen, S. R. Goswami, H. Chen and N. Yan, ChemSusChem, 2020, 13, 5749–5761 CrossRef CAS PubMed.
- W. Xie, Q. Yan and H. Fu, Polym. Adv. Technol., 2021, 32, 4415–4423 CrossRef CAS.
- K. Pietrzak, M. Kirpluks, U. Cabulis and J. Ryszkowska, Polym. Degrad. Stab., 2014, 108, 201–211 CrossRef CAS.
- K. Mizera, M. Kirpluks, U. Cabulis, M. Leszczyńska, M. Półka and J. Ryszkowska, Ind. Crops Prod., 2018, 113, 98–110 CrossRef CAS.
- C.-C. Hsieh and Y.-C. Chen, J. Cleaner Prod., 2020, 276, 124203 CrossRef CAS.
- F. Arán-Aís, A. M. Torró-Palau, A. C. Orgilés-Barceló and J. M. Martín-Martínez, J. Adhes. Sci. Technol., 2000, 14, 1557–1573 CrossRef.
- F. Arán-Aís, A. M. Torró-Palau, A. C. Orgilés-Barceló and J. M. Martín-Martínez, J. Adhes. Sci. Technol., 2002, 16, 1431–1448 CrossRef.
- M. S. Sánchez–Adsuar, E. Papon and J.-J. Villenave, J. Appl. Polym. Sci., 2001, 82, 3402–3408 CrossRef.
- P. Carbonell-Blasco, J. M. Martín-Martínez and I. V. Antoniac, Int. J. Adhes. Adhes., 2013, 42, 11–20 CrossRef CAS.
- F. Arán-Aís, A. M. Torró-Palau, A. César Orgilés-Barceló and J. M. Martín-Martínez, Int. J. Adhes. Adhes., 2005, 25, 31–38 CrossRef.
- B. Liu, J. Nie and Y. He, Int. J. Adhes. Adhes., 2016, 66, 99–103 CrossRef CAS.
- K. A. C. Vespermann, B. N. Paulino, M. C. S. Barcelos, M. G. Pessôa, G. M. Pastore and G. Molina, Appl. Microbiol. Biotechnol., 2017, 101, 1805–1817 CrossRef CAS PubMed.
- J. D. Tibbetts and S. D. Bull, Green Chem., 2021, 23, 597–610 RSC.
- S. H. Helalat, C. Jers, M. Bebahani, H. Mohabatkar and I. Mijakovic, Microb. Cell Fact., 2021, 20, 187 CrossRef CAS PubMed.
- D. Risner, M. L. Marco, S. A. Pace and E. S. Spang, Processes, 2020, 8, 263 CrossRef CAS.
- M. C. Kloetzel, Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 1–59 Search PubMed.
- G. M. Wu, Z. W. Kong and F. Q. Chu, Chem. Ind. For. Prod., 2007, 27, 57–62 CAS.
- G.-M. Wu, Z.-W. Kong, H. Huang, J. Chen and F.-X. Chu, J. Appl. Polym. Sci., 2009, 113, 2894–2901 CrossRef CAS.
- G.-M. Wu, Z.-W. Kong, J. Chen and J.-C. Jiang, J. Appl. Polym. Sci., 2011, 120, 579–585 CrossRef CAS.
- G. Wu, Z. Kong, C. Chen, J. Chen, S. Huo and J. Jiang, J. Appl. Polym. Sci., 2013, 128, 132–138 CrossRef CAS.
- S. S. Kovals'skaya, N. G. Kozlov and T. S. Tikhonova, Chem. Nat. Compd., 1989, 25, 552–557 CrossRef.
- S.-C. Xu, S.-J. Zhu, J. Wang, L.-W. Bi, Y.-X. Chen, Y.-J. Lu, Y. Gu and Z.-D. Zhao, Chin. Chem. Lett., 2017, 28, 1509–1513 CrossRef CAS.
- X. Liu, X. Yang, S. Wang, S. Wang, Z. Wang, S. Liu, X. Xu, H. Liu and Z. Song, ACS Sustainable Chem. Eng., 2021, 9, 4175–4184 CrossRef CAS.
- United States, US10590069B2, 2020.
- A. I. Smeds, L. Vähäsalo, J. Rahkila, P. C. Eklund and S. M. Willför, Holzforschung, 2019, 73, 1017–1033 CrossRef CAS.
- J. Rizhikovs, J. Zandersons, G. Dobele and A. Paze, Ind. Crops Prod., 2015, 76, 209–214 CrossRef CAS.
- S. Amiri, S. Dastghaib, M. Ahmadi, P. Mehrbod, F. Khadem, H. Behrouj, M.-R. Aghanoori, F. Machaj, M. Ghamsari, J. Rosik, A. Hudecki, A. Afkhami, M. Hashemi, M. J. Los, P. Mokarram, T. Madrakian and S. Ghavami, Biotechnol. Adv., 2020, 38, 107409 CrossRef CAS PubMed.
- P. A. Krasutsky, Nat. Prod. Rep., 2006, 23, 919–942 RSC.
- M. Okada, K. Suzuki, Y. Mawatari and M. Tabata, Eur. Polym. J., 2019, 113, 12–17 CrossRef CAS.
- V. A. Erä, P. Jääskeläinen and K. Ukkonen, Angew. Makromol. Chem., 1980, 88, 79–88 CrossRef.
- Y. Chen, Q. Song, J. Zhao, X. Gong, H. Schlaad and G. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 6593–6600 CrossRef CAS PubMed.
- L. Hosta-Rigau, Y. Zhang, B. M. Teo, A. Postma and B. Städler, Nanoscale, 2013, 5, 89–109 RSC.
- S. J. Stachelek, I. Alferiev, H. Choi, A. Kronsteiner, P. Uttayarat, K. J. Gooch, R. J. Composto, I.-W. Chen, R. P. Hebbel and R. J. Levy, J. Biomed. Mater. Res., Part A, 2005, 72A, 200–212 CrossRef CAS PubMed.
- R. N. Jana and J. W. Cho, Fibers Polym., 2009, 10, 569–575 CrossRef CAS.
- Y.-C. Chung, H. Y. Kim, J.-H. Yu and B. C. Chun, Macromol. Res., 2015, 23, 350–359 CrossRef CAS.
- G. Acik, H. R. F. Karabulut, C. Altinkok and A. O. Karatavuk, Polym. Degrad. Stab., 2019, 165, 43–48 CrossRef CAS.
- B. Acik, G. Acik and H. Erdemi, Eur. Polym. J., 2021, 146, 110247 CrossRef CAS.
- J. H. Cummings and A. M. Stephen, Eur. J. Clin. Nutr., 2007, 61, S5–S18 CrossRef CAS PubMed.
- J. A. Galbis, M. de, G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- S. K. Bobade, N. R. Paluvai, S. Mohanty and S. K. Nayak, Polym.-Plast. Technol. Eng., 2016, 55, 1863–1896 CrossRef CAS.
- P. D. Raytchev, C. Besset, E. Fleury, J.-P. Pascault, J. Bernard and E. Drockenmuller, Pure Appl. Chem., 2012, 85, 511–520 CrossRef.
- J. Xiang, S. Yang, J. Zhang, J. Wu, Y. Shao, Z. Wang and M. Yang, Polym. Bull., 2022, 79, 2667–2684 CrossRef CAS.
- J. A. Galbis and M. G. García-Martín, in Carbohydrates in Sustainable Development II, ed. A. P. Rauter, P. Vogel and Y. Queneau, Springer, Berlin, Heidelberg, 2010, pp. 147–176 Search PubMed.
- R. S. Malani, V. C. Malshe and B. N. Thorat, J. Coat. Technol. Res., 2022, 19, 361–375 CrossRef CAS.
- D. J. Saxon, A. M. Luke, H. Sajjad, W. B. Tolman and T. M. Reineke, Prog. Polym. Sci., 2020, 101, 101196 CrossRef CAS.
- G. Flèche and M. Huchette, Starch - Stärke, 1986, 38, 26–30 CrossRef.
- C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- Addit. Polym., 2015, 2015, 8–9.
- Global Isosorbide Market Size | Industry Report, 2019–2025, https://www.grandviewresearch.com/industry-analysis/isosorbide-industry, (accessed April 11, 2022).
- P.-C. Lin, F. Zhang and H. B. Pakrasi, Metab. Eng. Commun., 2021, 12, e00164 CrossRef CAS PubMed.
- O. Gómez-de-Miranda-Jiménez-de-Aberasturi, A. Centeno-Pedrazo, S. Prieto Fernández, R. Rodriguez Alonso, S. Medel, J. María Cuevas, L. G. Monsegue, S. De Wildeman, E. Benedetti, D. Klein, H. Henneken and J. R. Ochoa-Gómez, Green Chem. Lett. Rev., 2021, 14, 533–543 Search PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- I. Javni, O. Bilić, N. Bilić, Z. S. Petrović, E. A. Eastwood, F. Zhang and J. Ilavský, J. Appl. Polym. Sci., 2015, 132(47) DOI:10.1002/app.42830.
- B. S. Gregorí Valdés, C. S. B. Gomes, P. T. Gomes, J. R. Ascenso, H. P. Diogo, L. M. Gonçalves, R. Galhano dos Santos, H. M. Ribeiro and J. C. Bordado, Polymers, 2018, 10, 1170 CrossRef PubMed.
- R. Marín, A. Alla, A. M. de Ilarduya and S. Muñoz-Guerra, J. Appl. Polym. Sci., 2012, 123, 986–994 CrossRef.
- S. Oprea, V.-O. Potolinca and V. Oprea, Eur. Polym. J., 2016, 83, 161–172 CrossRef CAS.
- I. Ristić, I. Krakovsky, T. Janić, S. Cakić, A. Miletić, M. Jotanović and T. Radusin, J. Therm. Anal. Calorim., 2018, 134, 895–901 CrossRef.
- S.-Y. Oh, M.-S. Kang, J. C. Knowles and M.-S. Gong, J. Biomater. Appl., 2015, 30, 327–337 CrossRef CAS PubMed.
- Q. Shou, R. Yuan, G. Ma, X. Liang, J. Wan, M. Xian and Q. Wang, Polymer, 2019, 164, 118–125 CrossRef CAS.
- E. C. Varkey and K. Sreekumar, J. Mater. Sci., 2010, 45, 1912–1920 CrossRef.
- C.-H. Lee, H. Takagi, H. Okamoto, M. Kato and A. Usuki, J. Polym. Sci. Part -Polym. Chem., 2009, 47, 6025–6031 CrossRef CAS.
- Y. Li, B. A. J. Noordover, R. A. T. M. van Benthem and C. E. Koning, Eur. Polym. J., 2014, 59, 8–18 CrossRef CAS.
- H.-J. Kim, M.-S. Kang, J. C. Knowles and M.-S. Gong, J. Biomater. Appl., 2014, 29, 454–464 CrossRef CAS PubMed.
- S. T. Cho, J. I. So, J.-Y. Jung, S. Hwang, S.-H. Baeck and S. E. Shim, Macromol. Res., 2019, 27, 153–163 CrossRef CAS.
- X.-P. An, J.-H. Chen, Y.-D. Li, J. Zhu and J.-B. Zeng, Sci. China: Mater., 2018, 61, 993–1000 CAS.
- H. Park, M.-S. Gong and J. C. Knowles, J. Mater. Sci.: Mater. Med., 2013, 24, 281–294 CrossRef CAS PubMed.
- T. Calvo-Correas, M. D. Martin, A. Retegi, N. Gabilondo, M. A. Corcuera and A. Eceiza, ACS Sustainable Chem. Eng., 2016, 4, 5684–5692 CrossRef CAS.
- M. Charlon, B. Heinrich, Y. Matter, E. Couzigné, B. Donnio and L. Avérous, Eur. Polym. J., 2014, 61, 197–205 CrossRef CAS.
- X. He, Z. Zhai, Y. Wang, G. Wu, Z. Zheng, Q. Wang and Y. Liu, J. Appl. Polym. Sci., 2012, 126, E354–E361 CrossRef.
- S. Wendels, B. Heinrich, B. Donnio and L. Avérous, Eur. Polym. J., 2021, 153, 110531 CrossRef CAS.
- C. J. Stubbs, J. C. Worch, H. Prydderch, Z. Wang, R. T. Mathers, A. V. Dobrynin, M. L. Becker and A. P. Dove, J. Am. Chem. Soc., 2022, 144, 1243–1250 CrossRef CAS PubMed.
- R. J. Kieber, S. A. Silver and J. G. Kennemur, Polym. Chem., 2017, 8, 4822–4829 RSC.
- F. Bachmann, J. Reimer, M. Ruppenstein and J. Thiem, Macromol. Rapid Commun., 1998, 19, 21–26 CrossRef CAS.
- N. Kasmi, M. Roso, N. Hammami, M. Majdoub, C. Boaretti, P. Sgarbossa, C. Vianello, G. Maschio, M. Modesti and A. Lorenzetti, Des. Monomers Polym., 2017, 20, 547–563 CrossRef CAS PubMed.
- M. Beldi, R. Medimagh, S. Chatti, S. Marque, D. Prim, A. Loupy and F. Delolme, Eur. Polym. J., 2007, 43, 3415–3433 CrossRef CAS.
- T. Jiang, W. Wang, D. Yu, D. Huang, N. Wei, Y. Hu and H. Huang, J. Polym. Res., 2018, 25, 140 CrossRef.
- M. Alam, Int. J. Polym. Anal. Charact., 2019, 24, 533–547 CrossRef CAS.
- C. K. Patil, H. D. Jirimali, J. S. Paradeshi, B. L. Chaudhari, P. K. Alagi, P. P. Mahulikar, S. C. Hong and V. V. Gite, Prog. Org. Coat., 2021, 151, 106084 CrossRef CAS.
- R. Medimagh, M. M. Smaidia, H. Bennour and S. Chatti, Polym. Int., 2015, 64, 513–520 CrossRef CAS.
- A. El Mahdi, M. M'sahel and R. Medimagh, Macromol. Chem. Phys., 2017, 218, 1700077 CrossRef.
- A. A. Gadgeel and S. T. Mhaske, Prog. Org. Coat., 2021, 150, 105983 CrossRef CAS.
- J. C. Khanderay and V. V. Gite, J. Appl. Polym. Sci., 2019, 136, 47558 CrossRef.
- X. X. Li, M. H. Sohn and U. R. Cho, Elastomers Compos., 2019, 54, 225–231 CAS.
- H. R. Kricheldorf, R. Mix and S. M. Weidner, J. Polym. Sci. Part Polym. Chem., 2014, 52, 867–875 CrossRef CAS.
- Y. Ma, J. Liu, M. Luo, J. Xing, J. Wu, H. Pan, C. Ruan and Y. Luo, RSC Adv., 2017, 7, 13886–13895 RSC.
- F. Bayrak, E. Ay, A. Oral, T. Karayıldırım and K. Ay, Iran. Polym. J., 2022, 31, 413–423 CrossRef CAS.
- G. Wei, H. Xu, L. Chen, Z. Li and R. Liu, Prog. Org. Coat., 2019, 126, 162–167 CrossRef CAS.
- V. Besse, R. Auvergne, S. Carlotti, G. Boutevin, B. Otazaghine, S. Caillol, J.-P. Pascault and B. Boutevin, React. Funct. Polym., 2013, 73, 588–594 CrossRef CAS.
- J. L. J. van Velthoven, L. Gootjes, D. S. van Es, B. A. J. Noordover and J. Meuldijk, Eur. Polym. J., 2015, 70, 125–135 CrossRef CAS.
- B. Ates, S. Koytepe, M. G. Karaaslan, S. Balcioglu and S. Gulgen, Int. J. Adhes. Adhes., 2014, 49, 90–96 CrossRef CAS.
- R. Lalwani and S. Desai, J. Appl. Polym. Sci., 2010, 115, 1296–1305 CrossRef CAS.
- J. Kloss, F. S. M. de Souza, E. R. da Silva, J. A. Dionísio, L. Akcelrud and S. F. Zawadzki, Macromol. Symp., 2006, 245–246, 651–656 CrossRef.
- A. Boyer, C. E. Lingome, O. Condassamy, M. Schappacher, S. Moebs-Sanchez, Y. Queneau, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2012, 4, 296–306 RSC.
- M. Ionescu and Z. S. Petrović, J. Cell. Plast., 2010, 46, 223–237 CrossRef CAS.
- K. Hashimoto, K. Yaginuma, S. Nara and H. Okawa, Polym. J., 2005, 37, 384–390 CrossRef CAS.
- B. Begines, F. Zamora, E. Benito, M. de, G. García-Martín and J. A. Galbis, J. Polym. Sci. Part Polym. Chem., 2012, 50, 4638–4646 CrossRef CAS.
- L. Maisonneuve, G. Chollet, E. Grau and H. Cramail, OCL, 2016, 23, D508 CrossRef.
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1800524 CrossRef PubMed.
- R. Ledesma-Amaro, Eur. J. Lipid Sci. Technol., 2015, 117, 141–144 CrossRef CAS.
- E. Hablot, D. Zheng, M. Bouquey and L. Avérous, Macromol. Mater. Eng., 2008, 293, 922–929 CrossRef CAS.
- K.-K. Tremblay-Parrado, C. García-Astrain and L. Avérous, Green Chem., 2021, 23, 4296–4327 RSC.
- J. Peyrton, C. Chambaretaud, A. Sarbu and L. Avérous, ACS Sustainable Chem. Eng., 2020, 8, 12187–12196 CrossRef CAS.
- A. E. Coman, J. Peyrton, G. Hubca, A. Sarbu, A. R. Gabor, C. A. Nicolae, T. V. Iordache and L. Averous, Eur. Polym. J., 2021, 149, 110363 CrossRef CAS.
- R. S. Malani, V. C. Malshe and B. N. Thorat, J. Coat. Technol. Res., 2022, 19, 201–222 CrossRef CAS.
- B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC.
- C. Bueno-Ferrer, E. Hablot, F. Perrin-Sarazin, M. C. Garrigós, A. Jiménez and L. Averous, Macromol. Mater. Eng., 2012, 297, 777–784 CrossRef CAS.
- C. Bueno-Ferrer, E. Hablot, M. del, C. Garrigós, S. Bocchini, L. Averous and A. Jiménez, Polym. Degrad. Stab., 2012, 97, 1964–1969 CrossRef CAS.
- X. Liu, K. Xu, H. Liu, H. Cai, J. Su, Z. Fu, Y. Guo and M. Chen, Prog. Org. Coat., 2011, 72, 612–620 CrossRef CAS.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- C. Carré, L. Bonnet and L. Avérous, RSC Adv., 2015, 5, 100390–100400 RSC.
- C. Carré, L. Bonnet and L. Avérous, RSC Adv., 2014, 4, 54018–54025 RSC.
- E. Rix, E. Grau, G. Chollet and H. Cramail, Eur. Polym. J., 2016, 84, 863–872 CrossRef CAS.
- S. K. Green, R. E. Patet, N. Nikbin, C. L. Williams, C.-C. Chang, J. Yu, R. J. Gorte, S. Caratzoulas, W. Fan, D. G. Vlachos and P. J. Dauenhauer, Appl. Catal., B, 2016, 180, 487–496 CrossRef CAS.
- S. Wendels, R. Balahura, S. Dinescu, S. Ignat, M. Costache and L. Avérous, Eur. Polym. J., 2022, 164, 110968 CrossRef CAS.
- N. Azizi, Y. Chevalier and M. Majdoub, Ind. Crops Prod., 2014, 52, 150–157 CrossRef CAS.
- H. Blache, F. Méchin, A. Rousseau, É. Fleury, J.-P. Pascault, P. Alcouffe, N. Jacquel and R. Saint-Loup, Ind. Crops Prod., 2018, 121, 303–312 CrossRef CAS.
- J.-H. Chen, D.-D. Hu, Y.-D. Li, J. Zhu, A.-K. Du and J.-B. Zeng, Polym. Test., 2018, 70, 174–179 CrossRef CAS.
- K. Gorna and S. Gogolewski, J. Biomed. Mater. Res., Part A, 2006, 79A, 128–138 CrossRef CAS PubMed.
- S.-M. Hong, J.-W. Kim, J. C. Knowles and M.-S. Gong, J. Biomater. Appl., 2017, 31, 1026–1038 CrossRef CAS PubMed.
- S.-M. Hong, J.-R. Cha and J.-G. Kim, Polym. Test., 2020, 91, 106852 CrossRef CAS.
- S. B. Jo, U. Erdenebileg, K. Dashnyam, G.-Z. Jin, J.-R. Cha, A. El-Fiqi, J. C. Knowles, K. D. Patel, H.-H. Lee, J.-H. Lee and H.-W. Kim, J. Tissue Eng., 2020, 11, 2041731419900424 Search PubMed.
- Y.-S. Joo, J.-R. Cha and M.-S. Gong, Mater. Sci. Eng., C, 2018, 91, 426–435 CrossRef CAS PubMed.
- H.-N. Kim, D.-W. Lee, H. Ryu, G.-S. Song and D.-S. Lee, Molecules, 2019, 24, 1061 CrossRef PubMed.
- Y. Li, B. A. J. Noordover, R. A. T. M. van Benthem and C. E. Koning, Eur. Polym. J., 2014, 52, 12–22 CrossRef CAS.
- Y. Li, B. A. J. Noordover, R. A. T. M. van Benthem and C. E. Koning, ACS Sustainable Chem. Eng., 2014, 2, 788–797 CrossRef CAS.
- Y. Li, B. A. J. Noordover, R. A. T. M. van Benthem and C. E. Koning, Prog. Org. Coat., 2015, 86, 134–142 CrossRef CAS.
- S.-R. Shin, J.-Y. Liang, H. Ryu, G.-S. Song and D.-S. Lee, Molecules, 2019, 24, 1347 CrossRef PubMed.
- W.-J. Si, L. Yang, Y.-X. Weng, J. Zhu and J.-B. Zeng, Polym. Test., 2018, 69, 9–15 CrossRef CAS.
- Y. K. Tsui and S. Gogolewski, J. Mater. Sci.: Mater. Med., 2009, 20, 1729–1741 CrossRef CAS PubMed.
- Y. Xia and R. C. Larock, ChemSusChem, 2011, 4, 386–391 CrossRef CAS PubMed.
- W. Yang, D. Guan, J. Liu, Y. Luo and Y. Wang, New J. Chem., 2020, 44, 3493–3503 RSC.
- R. Medimagh, A. Saadaoui, S. Mghirbi, S. Marque, D. Prim, A. Fildier, A. Bulete, G. Raffin and S. Chatti, J. Polym. Res., 2014, 21, 486 CrossRef.
- S.-K. Ha and H.-C. Broecker, Macromol. Mater. Eng., 2003, 288, 569–577 CrossRef CAS.
- A. L. Da Róz, A. A. S. Curvelo and A. Gandini, Carbohydr. Polym., 2009, 77, 526–529 CrossRef.
- M. Barikani and M. Mohammadi, Carbohydr. Polym., 2007, 68, 773–780 CrossRef CAS.
- K. Wilpiszewska and T. Spychaj, Carbohydr. Polym., 2007, 70, 334–340 CrossRef CAS.
- R. Garçon, C. Clerk, J.-P. Gesson, J. Bordado, T. Nunes, S. Caroço, P. T. Gomes, M. E. Minas da Piedade and A. P. Rauter, Carbohydr. Polym., 2001, 45, 123–127 CrossRef.
- R. V. Gómez and O. Varela, Macromolecules, 2009, 42, 8112–8117 CrossRef.
- R. Marín and S. Muñoz-Guerra, J. Appl. Polym. Sci., 2009, 114, 3723–3736 CrossRef.
- R. Marín and S. Muñoz-Guerra, J. Polym. Sci. Part Polym. Chem., 2008, 46, 7996–8012 CrossRef.
- A. Sudo, Y. Shibata and A. Miyamoto, J. Polym. Sci. Part Polym. Chem., 2013, 51, 3956–3963 CrossRef CAS.
- A. Yoshida and A. Sudo, J. Polym. Sci. Part Polym. Chem., 2017, 55, 3798–3803 CrossRef CAS.
- D. A. Purser and J. L. McAllister, in SFPE Handbook of Fire Protection Engineering, ed. M. J. Hurley, D. Gottuk, J. R. Hall, K. Harada, E. Kuligowski, M. Puchovsky, J. Torero, J. M. Watts and C. Wieczorek, Springer, New York, NY, 2016, pp. 2308–2428 Search PubMed.
- G. W. Hedrick and A. J. Green, Prod. RD, 1973, 12, 319–321 CAS.
- L. Gao, G. Zheng, Y. Zhou, L. Hu, G. Feng and M. Zhang, Polym. Degrad. Stab., 2014, 101, 92–101 CrossRef CAS.
- M. Zhang, Z. Luo, J. Zhang, S. Chen and Y. Zhou, Polym. Degrad. Stab., 2015, 120, 427–434 CrossRef CAS.
- China, CN101891883B, 2012.
- China, CN102352026A, 2012.
- L. Gao, G. Zheng, Y. Zhou, L. Hu, G. Feng and Y. Xie, Ind. Crops Prod., 2013, 50, 638–647 CrossRef CAS.
- S. Wang, P. Sun, X. Xu, H. Liu, D. Wang and X. Huang, Adv. Eng. Mater., 2021, 2101044 Search PubMed.
- C. Yu, C. Yan, J. Shao and F. Zhang, Colloid Polym. Sci., 2021, 299, 1489–1498 CrossRef CAS.
- H. Moustafa, N. A. Darwish and A. M. Youssef, Food Chem., 2022, 371, 131193 CrossRef CAS PubMed.
- L. Huo, D. Wang, H. Liu, P. Jia and J. Gao, J. Biomater. Sci., Polym. Ed., 2016, 27, 1100–1114 CrossRef CAS PubMed.
- X. Xu, S. Shang, Z. Song, S. Cui, H. Wang and D. Wang, BioResources, 2011, 6, 2460–2470 CAS.
- G. Wu, Z. Kong, J. Chen, S. Huo and G. Liu, Prog. Org. Coat., 2014, 77, 315–321 CrossRef CAS.
- H. Si, H. Liu, S. Shang, J. Song, S. Liao, D. Wang and Z. Song, Prog. Org. Coat., 2016, 90, 309–316 CrossRef CAS.
- H. Si, H. Liu, S. Shang, J. Song, S. Liao, D. Wang and Z. Song, J. Appl. Polym. Sci., 2016, 133(15) DOI:10.1002/app.43292.
- S. A. Madbouly and J. U. Otaigbe, Prog. Polym. Sci., 2009, 34, 1283–1332 CrossRef CAS.
- R. D. Toker, N. Kayaman-Apohan and M. V. Kahraman, Prog. Org. Coat., 2013, 76, 1243–1250 CrossRef CAS.
- C. Zhang, K.-C. Huang, H. Wang and Q. Zhou, Prog. Org. Coat., 2020, 148, 105855 CrossRef CAS.
- L. Dall Agnol, F. T. G. Dias, H. L. Ornaghi, M. Sangermano and O. Bianchi, Prog. Org. Coat., 2021, 154, 106156 CrossRef CAS.
- M. Lejars, A. Margaillan and C. Bressy, Chem. Rev., 2012, 112, 4347–4390 CrossRef CAS PubMed.
- J. Pan, Q. Xie, H. Chiang, Q. Peng, P.-Y. Qian, C. Ma and G. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 1671–1678 CrossRef CAS.
- H. Y. Chiang, J. Pan, C. Ma and P.-Y. Qian, Biofouling, 2020, 36, 200–209 CrossRef CAS PubMed.
- S. Sahoo, S. Mohanty and S. K. Nayak, J. Macromol. Sci., Part A, 2018, 55, 36–48 CrossRef CAS.
- P. Carbonell-Blasco, I. Antoniac and J. M. Martin-Martinez, Key Eng. Mater., 2014, 583, 67–79 Search PubMed.
- J. Fu, L. Wang, H. Yu, M. Haroon, F. Haq, W. Shi, B. Wu and L. Wang, Prog. Org. Coat., 2019, 131, 82–99 CrossRef CAS.
- C.-A. Xu, Z. Qu, M. Lu, H. Meng, Y. Zhan, B. Chen, K. Wu and J. Shi, Colloids Surf., A, 2021, 614, 126146 CrossRef CAS.
- S. Gogolewski, K. Gorna, E. Zaczynska and A. Czarny, J. Biomed. Mater. Res., Part A, 2008, 85A, 456–465 CrossRef CAS PubMed.
- J. Li, W. Yang, Z. Ning, B. Yang and Y. Zeng, Polymers, 2021, 13, 3538 CrossRef CAS PubMed.
- X. Liu, S. She, W. Tong and C. Gao, RSC Adv., 2014, 5, 5775–5780 RSC.
- J. Shao, C. Yu, F. Bian, Y. Zeng and F. Zhang, ACS Omega, 2019, 4, 2493–2499 CrossRef CAS PubMed.
- C. Yu, H. Liu, J. Shao and F. Zhang, J. Renewable Mater., 2022, 10, 1049–1061 CAS.
- J. Jeromenok, W. Böhlmann, C. Jäger and J. Weber, ChemistryOpen, 2013, 2, 17–20 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |