
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The design of small-molecule prodrugs and activatable phototherapeutics for cancer therapy
Hai-Hao
Han
 aceh,
Han-Min
Wang
ce,
Paramesh
Jangili
d,
Mingle
Li
d,
Luling
Wu
*b,
Yi
Zang
ck,
Adam C.
Sedgwick
*f,
Jia
Li
*ceh,
Xiao-Peng
He
*aij,
Tony D.
James
*bg and
Jong Seung
Kim
*d
aceh,
Han-Min
Wang
ce,
Paramesh
Jangili
d,
Mingle
Li
d,
Luling
Wu
*b,
Yi
Zang
ck,
Adam C.
Sedgwick
*f,
Jia
Li
*ceh,
Xiao-Peng
He
*aij,
Tony D.
James
*bg and
Jong Seung
Kim
*d
aKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, P. R. China. E-mail: xphe@ecust.edu.cn
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk; wllcyl@126.com
cState Key Laboratory of Drug Research, Molecular Imaging Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: jli@simm.ac.cn
dDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: jongskim@korea.ac.kr
eUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, P. R. China
fChemistry Research Laboratory, University of Oxford, Mansfield Road, OX1 3TA, UK. E-mail: adam.sedgwick@chem.ox.ac.uk
gSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
hShandong Laboratory of Yantai Drug Discovery, Bohai Rim Advanced Research Institute for Drug Discovery, Yantai, Shandong 264117, P. R. China
iThe International Cooperation Laboratory on Signal Transduction, Eastern Hepatobiliary Surgery Hospital, Shanghai 200438, China
jNational Center for Liver Cancer, Shanghai 200438, China
kLingang laboratory, Shanghai 201203, China
First published on 13th January 2023
Abstract
Cancer remains as one of the most significant health problems, with approximately 19 million people diagnosed worldwide each year. Chemotherapy is a routinely used method to treat cancer patients. However, current treatment options lack the appropriate selectivity for cancer cells, are prone to resistance mechanisms, and are plagued with dose-limiting toxicities. As such, researchers have devoted their attention to developing prodrug-based strategies that have the potential to overcome these limitations. This tutorial review highlights recently developed prodrug strategies for cancer therapy. Prodrug examples that provide an integrated diagnostic (fluorescent, photoacoustic, and magnetic resonance imaging) response, which are referred to as theranostics, are also discussed. Owing to the non-invasive nature of light (and X-rays), we have discussed external excitation prodrug strategies as well as examples of activatable photosensitizers that enhance the precision of photodynamic therapy/photothermal therapy. Activatable photosensitizers/photothermal agents can be seen as analogous to prodrugs, with their phototherapeutic properties at a specific wavelength activated in the presence of disease-related biomarkers. We discuss each design strategy and illustrate the importance of targeting biomarkers specific to the tumour microenvironment and biomarkers that are known to be overexpressed within cancer cells. Moreover, we discuss the advantages of each approach and highlight their inherent limitations. We hope in doing so, the reader will appreciate the current challenges and available opportunities in the field and inspire subsequent generations to pursue this crucial area of cancer research.
 Hai-Hao Han | Hai-Hao Han is an Associate Professor at SIMM (CAS). He received his PhD in 2020 from ECUST under the supervision of Prof. Xiao-Peng He. His research interests include fluorescent probes for disease theranostic applications and targeted drug development. |
 Han-Min Wang | Han-Min Wang is currently pursuing a PhD degree at SIMM (CAS) with Prof. Yi Zang. Her research focuses on the biological research of new target for NASH treatment and development of new chemical probe tools for monitoring the development of NASH. |
 Paramesh Jangili | Paramesh Jangili received his PhD in 2016 from Jawaharlal Nehru Technological University-Hyderabad, India. He subsequently joined in Ewha Womans University, Korea and in 2017 joined Prof. Jong Seung Kim lab at Korea University. |
 Jong Seung Kim (Left) and Mingle Li (Right) | Jong Seung Kim (Left) received his PhD from the Department of Chemistry and Biochemistry at Texas Tech University in 1993. Currently he is a full professor in the Department of Chemistry at Korea University in Seoul. Mingle Li (Right) obtained his PhD degree in 2019 from the Dalian University of Technology under the supervision of Prof. Xiaojun Peng. Currently he is a Research Professor in Prof. Jong Seung Kim's lab at Korea University in Seoul. |
 Yi Zang | Yi Zang obtained her PhD from SIMM (CAS) in 2008 and is currently a professor at Lingang Laboratory. Her research mainly focuses on the biological research of AMPK, drug discovery for organ fibrosis and development of new chemical biology probe tools. |
 Adam C. Sedgwick (Left), Tony D. James (Centre) and Luling Wu (Right) | Adam C. Sedgwick (Left) is a Glasstone Research Fellow at the University of Oxford and is a Junior Research Fellow at Jesus College, Oxford. His research focus is on developing new chemical tools for molecular imaging, sensing, and theranostic applications. His h-index is 29 (Google Scholar). Tony D. James (Centre) is a Professor at The University of Bath and Fellow of the Royal Society of Chemistry. His research interests include many aspects of Supramolecular chemistry, including probes for redox imbalance and theranostic systems. His h-index is 80 (Google Scholar). Luling Wu (Right) was awarded scholarships by the China Scholarship Council and University of Bath to carry out a PhD at the University of Bath. His research focuses on fluorescent probes/prodrugs and imaging. His h-index is 17. |
 Jia Li | Jia Li received his PhD from SIMM (CAS) in 2000 and was promoted to professor in 2005. He has been the director of SIMM (CAS) since 2019. His research interests are centered on the investigation of mechanisms of metabolic diseases and medicinal chemical biology. |
 Xiao-Peng He | Xiao-Peng He is a professor at Feringa Nobel Prize Scientists Research Center, ECUST. He obtained his BSc (2006) and PhD (2011) from ECUST. He conducted postdoctoral research with Kaixian Chen (SIMM, CAS) from 2011 to 2013 at ECUST. His research interests are chemical probes for glycobiology and sugar-based drug discovery. |
Key learning points1. The importance of developing new and effective anticancer agents with different mechanism of action2. The design strategies used for the development of prodrugs 3. The importance of introducing a diagnostic component during cancer therapy 4. The advantages and disadvantages of each therapeutic approach (chemotherapy, photodynamic therapy, and photothermal therapy). 5. Future perspectives in the area of prodrug development. |
1. Introduction
Cancer is defined as a disease caused by the abnormal proliferation of cells. This uncontrollable cell growth not only accumulates to form solid tumours but leads to subsequent invasion to adjacent and distal tissues (and organs), a phenomenon which is known as cancer metastasis.1,2 Metastasis is the leading cause of cancer morbidity and mortality and responsible for ∼90% of cancer-related deaths.3 According to the International Agency for Research on Cancer, 19.29 million new cancer cases and 9.96 million cancer-related deaths were reported worldwide in 2020; unfortunately, these numbers are only expected to continue to increase.1 Thus, research efforts are extensively being devoted to developing new and effective treatment protocols. A primary focus is on overcoming the inherent limitations of current chemotherapeutics, such as poor aqueous solubility, poor selectivity toward cancer cells, the intrinsic or acquired resistance of tumours (multidrug resistance), and the dose-limiting toxicities (low maximum tolerated doses, MTD).4–6 Select examples of routinely used FDA-approved chemotherapeutics (Fig. 1) include doxorubicin (DOX, 1), camptothecin [CPT (2) CPT-11 (3) and its active metabolite, SN-38, (4)], gemcitabine (5), 5-fluorodeoxyuridine (FDU, 6), pazopanib (7), panobinostat (8), cisplatin (9), carboplatin (10), and oxaliplatin (11), all of which are discussed in this review. These therapeutics follow a similar mode of action that involves the inhibition of DNA replication, interference with RNA transcription, blockage of cell division, and inhibition of topoisomerases, which prevent cell division and tumour growth.7–9 Unfortunately, these therapies cannot distinguish between the uncontrollable growth of cancer cells and fast replicating healthy cells. Therefore, therapeutics that can target and treat cancers via new modes of action are particularly sought after. Crucial to their effective construction, aqueous solubility, cell uptake and tumour specificity are important factors that need to be considered.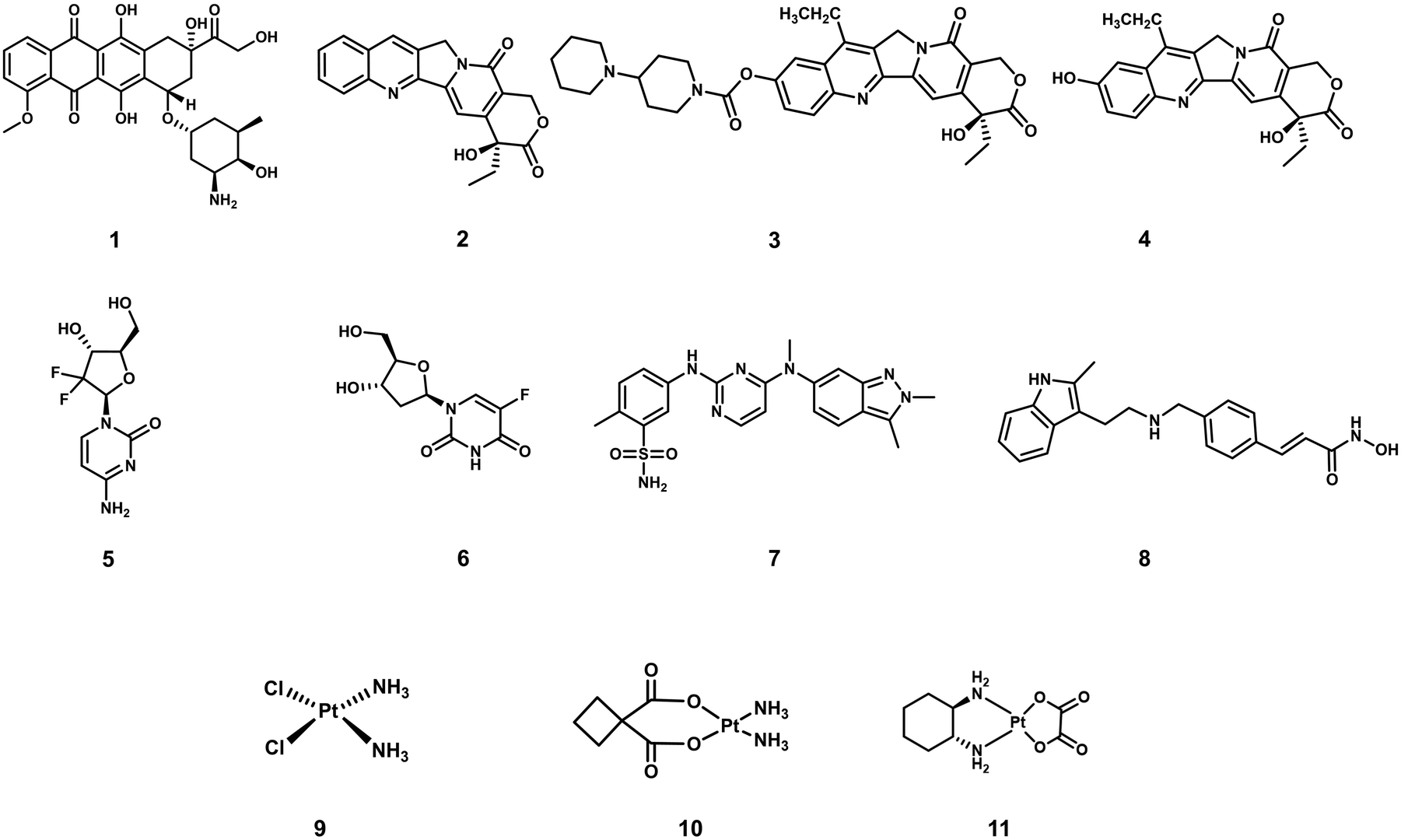 | ||
| Fig. 1 Chemical structures of select examples of FDA-approved chemotherapeutics. | ||
In recent years, a significant focus has been directed towards developing prodrug-based strategies for cancer therapy. Anticancer prodrugs are “masked” inactive chemotherapeutics designed to have little to no pharmacological activity. Beneficially, in the presence of a cancer-specific biomarker, the “masked to unmasked” conversion takes place to produce a therapeutic effect at the desired location. This enhanced specificity is designed to minimize off-target toxicities and improve therapeutic efficacies. Several prodrug examples have been reported with high tumour specificities, minimal off-target toxicities, and higher maximum tolerated doses.10–13 Prodrugs have also been developed to improve aqueous solubility and cell permeability of a therapeutic.14 Select examples of prodrug-based cancer treatments include FDA-approved Zytiga (hormone-refractory prostate cancer) and Ixazomib Citrate (multiple myeloma). Over recent decades, design strategies for small-molecule prodrugs with improved therapeutic efficacy and minimal side effects during disease treatments have been reviewed.15–17 The aforementioned review articles focus on the design and application strategies of prodrugs for individual chemotherapy or phototherapy, with minimal elaboration on the commonalities in the design of these therapeutic modalities. This tutorial review focuses on prodrugs that can be activated by specific biomarkers found in the tumour microenvironment or by external stimuli, as well as theranostics for precision-enhanced chemo- and phototherapy. In this review, we discuss recently reported prodrug strategies for chemo- and phototherapeutic applications. Each prodrug design and its advantages are compared to conventional chemotherapeutics and by describing each design strategy, we believe the importance of understanding factors that support the tumour microenvironment (TME) and aid cancer cell profileration will become apparent to the reader. Moreover, several reported prodrug examples that provide an integrated diagnostic response and the advantages of these theranostic systems are also discussed. The current limitations associated with each therapeutic strategy are discussed, and in addition, we will provide our own perspectives on the future directions for this important research area. This article provides a general overview on the design of prodrugs and activatable phototherapeutics which we believe will facilitate the development of improved therapies.
2. The design of small-molecule prodrugs and activatable phototherapeutics
Early prodrug examples were designed to improve solubility, cell permeability, and chemical stability. In recent years, the design of prodrugs has been focused towards overcoming the toxicity issues surrounding chemotherapeutics.18,19 Developing prodrugs usually requires synthetic modification of a therapeutic to “mask” the cytotoxic properties; this is often achieved by introducing a biomarker responsive “protecting group” (Scheme 1). Other prodrugs strategies include the use of protecting groups that are responsive to external stimuli such as light or ultrasound irradiation (Scheme 1).20,21 In some cases, the drugs themselves are fluorescent (e.g., doxorubicin,22 camptothecin23 and deferasirox24); as a result, a fluorescence “OFF-ON” switching can be used to highlight disease specific regions (i.e., diagnosis) while providing the real-time visualization of drug activation (Scheme 1). This is often referred to as theranostics (theragnostics).25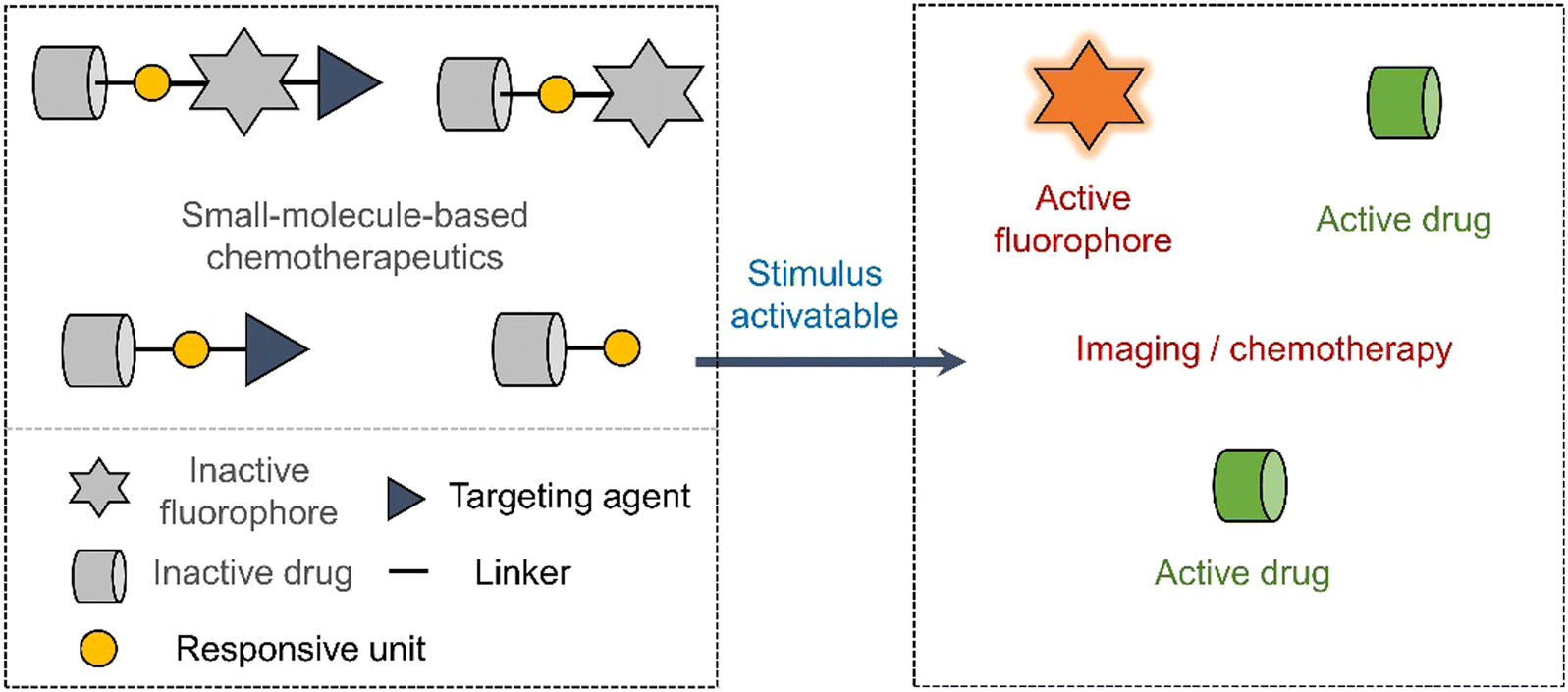 | ||
| Scheme 1 Basic schematic diagram of the various design strategies used to create small-molecule prodrug-based anticancer agents. | ||
Most reported prodrug strategies employ protecting groups that respond to specific biomarkers found within the tumour microenvironment (TME). TME is the environment around the tumour consisting of the blood vessels, rapidly proliferating cancer cells, endothelial cells, immune cells, fibroblasts, and an extracellular matrix.26,27 Several physiological processes are upregulated to sustain this microenvironment, which can be exploited for the design of prodrugs. A notable example is the enhanced cellular metabolism referred to as the Warburg effect, which leads to a lower extracellular pH being observed in tumour tissues (between 6.5 to 7.2) compared to that of normal tissues (pH around 7.4).28 Other examples include inflammatory biomarkers, such as reactive oxygen species (ROS) and enzymes, matrix metalloproteinases (MMPs), and hyaluronidase (HAdase). Hypoxia is a particular trait of the TME, resulting from increased oxygen consumption by cancer cells combined with an inadequate blood supply throughout the tumour (Scheme 2).29 Intracellular biomarkers such as glutathione (GSH), γ-glutamyltranspeptidase (GGT), and β-galactosidase (β-Gal)30,31 are upregulated in cancers; for example, the concentration of glutathione (GSH) in cancer cells is ∼20 mM, much higher than that in healthy cells (∼5 mM).32
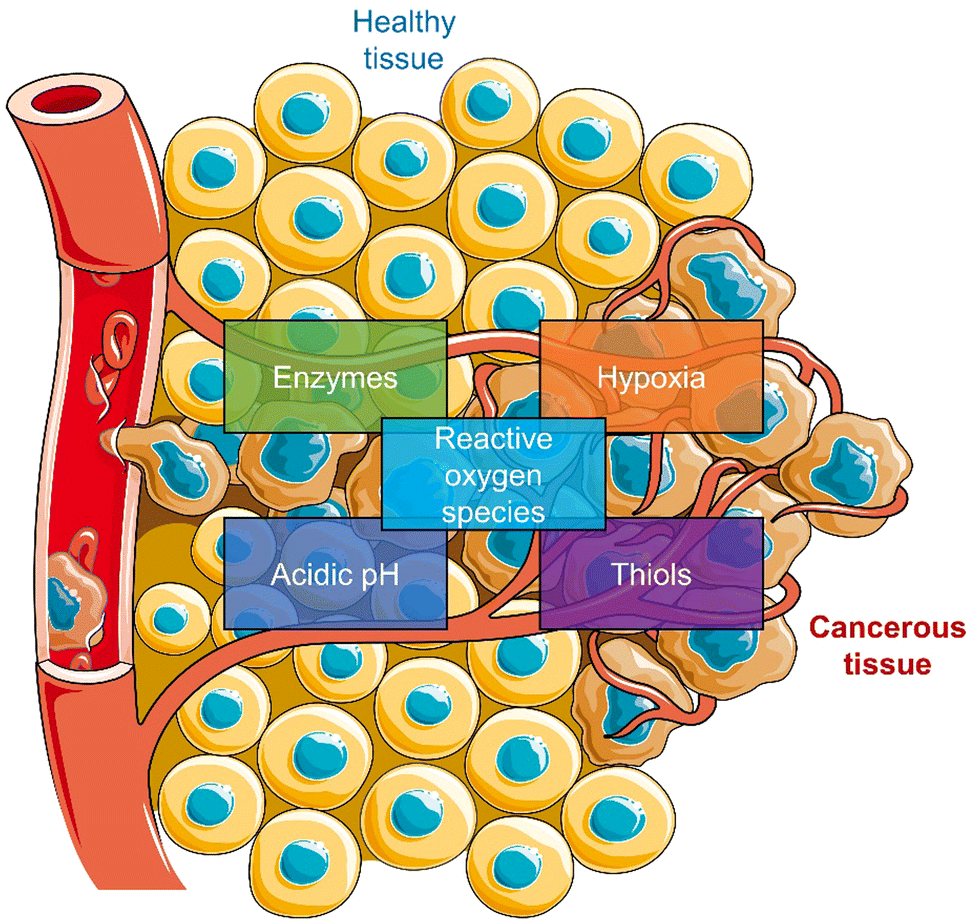 | ||
| Scheme 2 Schematic illustration of the characteristics of TME used to develop TME-responsive small-molecule prodrugs and activatable “smart” phototherapeutics (parts of the scheme were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/)).33 | ||
Despite the promise shown by currently reported prodrug strategies,11 the highly cytotoxic nature often remains and therefore impedes clinical translation. Low maximum tolerated doses are often seen due to their inability to target the tumour site effectively. Light-based treatments such as photodynamic therapy (PDT) offer a high precision, non-genotoxic and non-invasive approach to eradicate cancerous tissues.34,35 PDT is an FDA-approved therapeutic modality with examples including Photofrin® (porfimer sodium), Visudyne (Verteporfin or BPD-MA), and 5-aminolevulinic acid (ALA).36 In brief, PDT relies on the activation of photosensitizers by irradiation with a specific wavelength of light to produce cytotoxic ROS. The light-mediated production of ROS eradicates the surrounding diseased tissue. There are two types of photosensitized reactions, i.e., type I and type II. For type I, the light-activated photosensitizer undergoes electron transfer with surrounding biomolecules to afford free radicals, such as superoxide radicals and hydroxyl radicals, which can induce a cytotoxic effect.37,38 For type II, the triplet excited state of the light-activated photosensitizer undergoes direct energy transfer with ground-state oxygen (3O2) to generate singlet oxygen (1O2), thus directly leading to apoptosis or necrosis of cancer cells.39 Therefore, the hypoxic environment of a solid tumour often leads to poor efficacies being observed for Type II PDT agents.40–42 New photosensitizers that exhibit excellent tumour localising properties, high singlet oxygen quantum yields, and high therapeutic efficacies are continuously being reported.43 With the advancement in imaging technologies, photosensitisers are viewed as “all in one” in phototheranostics.43 However, light irradiation of the diseased tissue often results in damaging the surrounding healthy tissue. Efforts to improve the precision of PDT have led to the development of activatable photosensitizers (Scheme 3), in which the PDT excitation wavelength only exists when it is activated by a disease specific biomarker. This strategy exploits the changes in photophysical properties of a photosensitizer,44 therefore they can be seen as analogous to prodrugs or as fluorescent probes.45 Their simultaneous use as fluorescence probes creates the additional ability to define tumour margins between cancerous and healthy regions via imaging-guided PDT.46
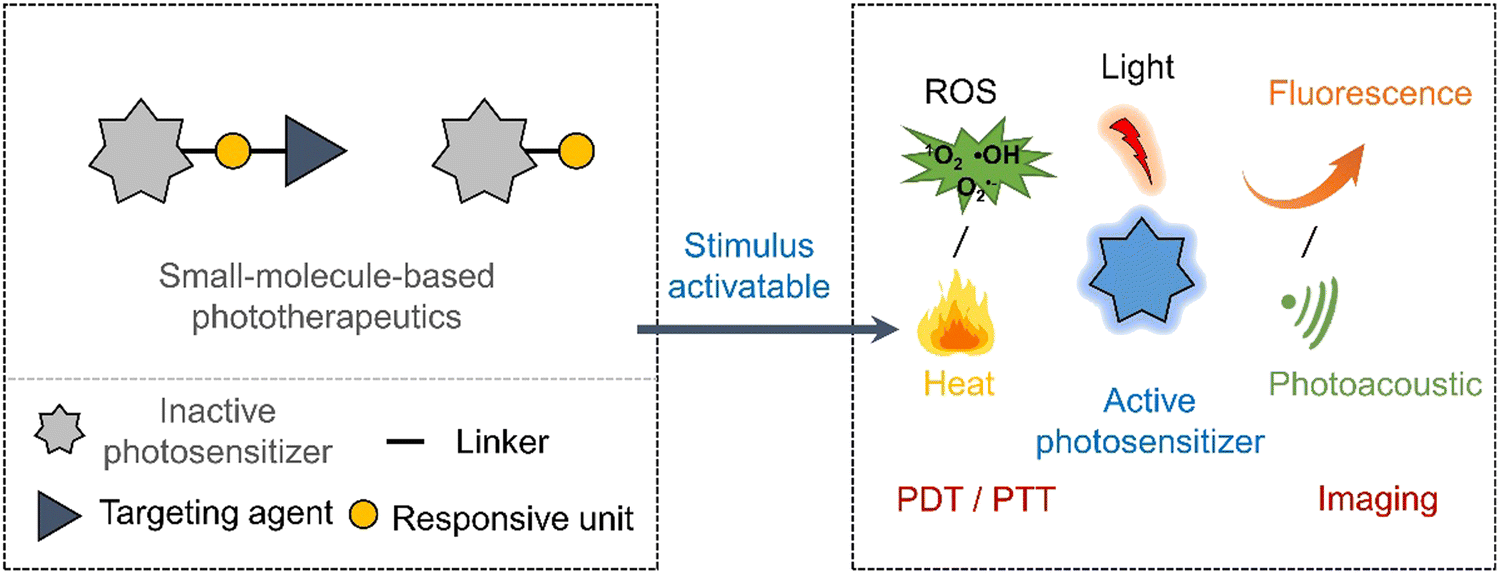 | ||
| Scheme 3 Basic schematic diagram of the various design strategies used to create small-molecule activatable “smart” phototherapeutics. | ||
In recent years, photothermal therapy (PTT) has been of great interest to researchers because of its minimal invasiveness, ability to overcome the oxygen requirements of PDT, and capacity to eradicate malignant tumours.47 PTT, which relies on the production of heat, is an attractive alternative approach to PDT.48 The photothermal agents generate heat upon light irradiation and induces cell death by the apoptosis or necrosis pathways.49 Reported PTT agents range from inorganic materials (e.g., silver, gold, transition-metal, and platinum nanoparticles and rare earth ions doped nanocrystals) to small organic molecules (e.g., cyanines, croconaines, porphyrins, and diketopyrrolopyrroles).50 However, compared to inorganic-based PTT agents, organic-based PTT agents are more promising due to their excellent biocompatibilities and ease of modification.48,51 An effective PTT agent is needed to accumulate at the solid tumour, and upon the light irradiation, the tumour temperatures exceeds >48 °C. Since PTT exploits the non-radiative decay of molecules, high molar absorptivity and low fluorescent quantum yield are desirable for PTT agents. To avoid the off-target phototoxicities for PTT agents, researchers have focused on developing activatable systems that undergo changes to photophysical properties at the region of interest (Scheme 3). This approach provides the potential to achieve differential cytotoxicity between cancer and nearby-normal cells. For the design of an activatable PTT agent, the presence of disease-related biomarkers needs to induce a change in UV absorption. These are similar design requirements to photoacoustic (PA) probes,52 PTT is therefore often used in combination with PA to facilitate real-time tracking in vivo.53–56 For a more extensive overview on the area of PTT and PA, the reader can be directed to an excellent review by Liu et al.51
Unfortunately, even though an experimental therapeutic can show promise in vitro, it is often not translated into small animal studies. A common issue mainly due to the factors such as limited tumour specificity, poor aqueous solubilities, rapid metabolism/excretion, and poor diffusion of prodrugs into the tumour mass. Drug permeation throughout the tumour is crucial since residual cancer cell survival can promote tumour regrowth and drug resistance. To overcome this hurdle, researchers have focused on introducing targeting units onto known chemotherapeutics.57 Such targeting functionalities exploit known interactions between small molecules and protein receptors overexpressed on the membrane surface of tumour cells or active transport mechanisms.57 This review will be broken down into sections by method of activation of a chemotherapeutic or phototherapeutic with cancer-related biomarkers, including enzymes, ROS, thiols, and other biomarkers specific to the TME and external stimulants, such as light and X-rays.
3. Small-molecule prodrugs and activatable phototherapeutics
3.1 Enzyme-responsive prodrugs and enzyme-mediated activation of phototherapeutics
Enzymes are critical to a number of metabolic processes and are highly specific with regards to the type of substrate, and their catalytic function.58 It is now well-understood that several enzymes are overexpressed in a number of types of cancer cell lines. Elevated enzymatic activities are believed to facilitate several pathological processes, ranging from tumour angiogenesis, cell invasion to metastasis.59,60 With the help of biologists, the biological substrates of these enzymes have been disclosed. This knowledge has then been used to functionalise chemotherapeutics, fluorescent probes and photosensitisers with enzyme-cleavable/activatable motifs and achieve controlled intracellular release of the active molecules. In this section, we discuss select examples of enzyme-based activatable chemotherapeutics and activatable “smart” phototherapeutic strategies.Most previous enzyme-based research has focused on the substrate specific properties of hydrolases (such as ester bonds that can be broken by esterases or short peptide substrates that can be broken by proteases) to construct prodrugs or “smart” phototherapeutic agents that enable controlled release and accumulation of drugs at specific biological targets. Most enzymatic reactions are fast and efficient, specific and mild compared to other stimulation conditions, and enzymatic-based therapeutic agents are able to exhibit higher reaction efficiencies (Table 1).
| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: CE: carboxylesterase, GGT: γ-glutamyltranspeptidase, β-Gal: β-galactosidase, DOX: doxorubicin, MMAE: monomethyl auristatin E, CAM: chick chorioallantoic membrane, PDT: photodynamic therapy, PTT: photothermal therapy, —: not mentioned. | ||||||
| 12 | CE | DOX | Chemotherapy | A549, HepG2, MCF7 and MCF7/DOX cells | DOX-resistant MCF7/DOX-derived tumour-bearing mice | 66 |
| 13 | Esterase and caspase-3 | DOX | Chemotherapy | U-87 MG cells | U-87 MG tumour-bearing mice | 74 |
| 14 | Cathepsin B | TPECM | PDT | MDA-MB-231 cells | — | 77 |
| 15 | GGT | Selenium-based rhodamine derivative | PDT | SHIN3 cells | Tumour-bearing CAM model | 84 |
| 16 | β-Gal | Hemicyanine dye | PTT | SKOV3 cells | SKOV3 tumour-bearing mice | 89 |
| 17 | β-Gal | MMAE | Chemotherapy | KB and HeLa cells | KB tumour-bearing mice | 30 |
Carboxylesterase (CE) enzyme is known to hydrolyse several carboxyl esters and amides catalytically, and it is seen as a useful tumour biomarker for patient staging.61,62 In light of this, a number of CE-responsive prodrugs have been developed for theranostic applications.63–65 Sharma et al. developed the prodrug 12 (Fig. 2a).6612 consists of a DOX that possessed both fluorescence and anti-cancer properties, a dichloroacetic acid (DCA) and a lipophilic cationic triphenylphosphonium (TPP) mitochondrial targeting unit. DOX is an FDA-approved chemotherapeutic with the nickname “the Red Devil” attributed to its fluorescent red appearance in solution. It is routinely used in the clinic to treat a range of cancers due to its broad anti-tumour activity; however, debilitating side effects, ranging from hair loss, bone marrow suppression, vomiting, and cardiotoxicity, are observed from its use. In addition, DOX-induced tumour cell resistance has limited its use as a first-line agent.67,68 DCA is a mitochondrial PDK inhibitor that promotes glucose oxidation rather than glycolysis by inhibiting pyruvate dehydrogenase kinase and increasing the flux of pyruvate into the mitochondria, helping overcome drug resistance.6912 was therefore designed to rationalise the fight against drug resistance by combining subcellular organelle (mitochondria) targeting with the modulation of tumour cell metabolism via DCA. As shown in Fig. 2a, the CE-mediated amide-bond hydrolysis of 12 resulted in the simultaneous release of DCA and subsequent release of DOX, which was confirmed by the enhanced fluorescence emission of free DOX characteristic fluorescence at 560 nm. Such CE-mediated transformation to release free DOX was further proved by high-performance liquid chromatography (HPLC) and liquid chromatography-mass spectroscopy (LC-MS). This change in fluorescence permitted the use of fluorescence microscopy to visualise the CE-mediated release of DOX in live cells. Cellular imaging and cytotoxicity studies were performed in a series of different cancer cell lines (i.e., A549-human lung adenocarcinoma epithelial cell; HepG2-human hepatic cancer cell) and normal cell lines (i.e., NHDFs-normal human dermal fibroblasts; IMR90-normal lung fibroblast cell line). In accordance with the design expectations, 12 was shown to target the mitochondria of cancer cells rather than normal cells because of the higher mitochondrial membrane potentials of cancer cells (Δψm = ∼−220 mV) compared with normal cells (∼−140 mV). The cytotoxicity was dependent on the CE activity, which was confirmed using a known CE inhibitor bis-(4-nitrophenyl)phosphate (BNPP). As a result the cytotoxicity of 12 towards healthy cells was mitigated. Interestingly, DOX was translocated to the nucleus from its initial mitochondrial localisation, thus reducing the efflux of DOX from cancer cells. Generally, ATP-binding cassette (ABC) transporter-mediated enhanced drug efflux (a term describing the expression of a transport protein that moves a drug from the intracellular to the extracellular space, thereby reducing the intracellular drug concentration) is known to contribute to drug resistance.70 Tumour cells normally produce ATP by conventing pyruvate to lactate through aerobic glycolysis, which contributes to tumour cell proliferation and promotes tumour metastasis.71 However, the released DCA fragment from 12 was shown to modulate the pyruvate metabolic pathway in cancer cells and trigger mitochondrial dysfunction by reducing lactate accumulation, glucose uptake and intracellular ATP levels. The above properties were believed to enable 12 to overcome drug resistance in MCF7 and DOX-resistant MCF7/DOX cell lines. The intraperitoneal administration of 12 in DOX-resistant MCF7/DOX-derived tumour-bearing mice models significantly inhibited tumour growth compared to the tumour volumes of control groups (DMSO and a simple mixture of DOX + DCA) (Fig. 2b). This work provides a new approach to overcome drug resistance by reversing cancer cell metabolism prior to drug activation, thereby increasing cancer cell apoptosis and inhibiting the regeneration of drug-resistant tumours.
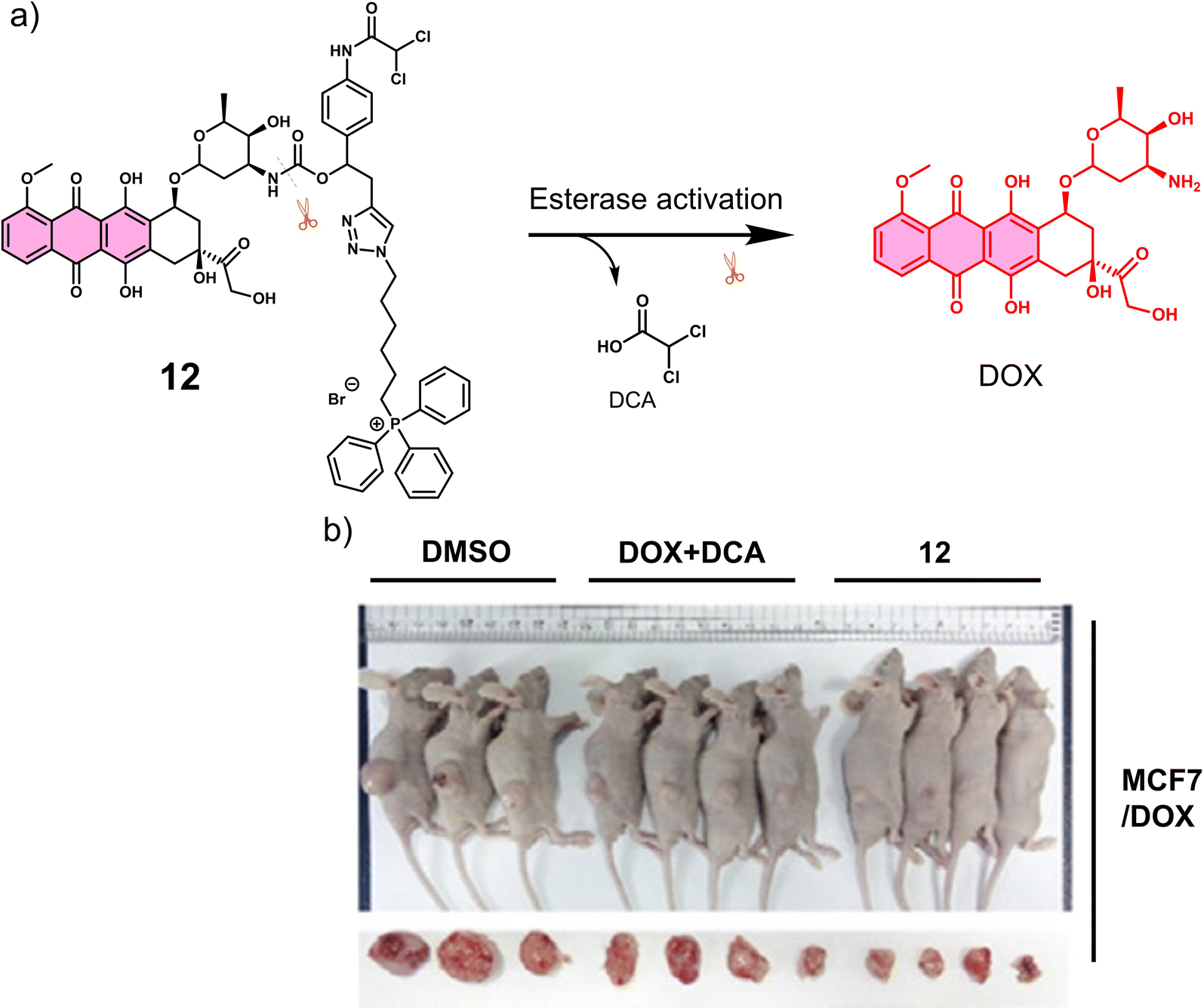 | ||
| Fig. 2 (a) Chemical structure of 12 and its CE-mediated release of DOX in MCF7/DOX-resistant cell lines. (b) Representative images of DOX-resistant MCF7/DOX-derived tumour-bearing mice treated with DMSO control, DOX + DCA, and 12. Reproduced with permission from ref. 66. Copyright (2018) Elsevier Inc. | ||
Proteases are enzymes that hydrolyse peptide bonds in proteins or peptides and play an important role in many diseases and biological processes, such as fetal and postnatal development, reproduction, signal transduction, immune response, many autoimmune and degenerative diseases, and cancer. Most proteases are specific in their mode of action, with hydrolysis usually occurring at specific amino acid residues, sequences or peptide bonds in the vicinity of the substrate protein or peptide.72 Caspase-3 is a cysteine-aspartic acid protease that cleaves various cellular targets (e.g., DNA) to mediate programmed cell death (apoptosis).7313, referred to as a “self-triggered apoptosis enzyme prodrug,” is a caspase-3 responsive prodrug (Fig. 3a).7413 consists of the cancer cell targeting integrin αvβ3-RGD (Arg-Gly-Asp) tripeptide, caspase-3 cleavable linker (DEVD tetrapeptide, Asp-Glu-Val-Asp), cellular esterase cleaved ester linkage and DOX. Notably, DOX is known to induce apoptosis-related markers, such as caspase-3.75 Therefore, it can activate more DOX, leading to the release of more caspase-3, and the cycle repeats itself leading to enhanced and widespread anti-cancer activity. As shown in Fig. 3a, the presence of caspase-3 results in the cleavage of the DEVD linker, followed by subsequent ester hydrolysis affords free DOX. Therefore, in this strategy, the release of DOX is designed to increase the intracellular expression of caspase-3 and enhance the enzymatic turnover of 13 (self-triggered). Esterase- and caspase-3-mediated hydrolysis was confirmed by HPLC (in solution) and confocal laser scanning microscopy (CLSM) images (in vitro). The selective cytotoxicity of 13 was found only in U-87 MG (human glioma cells, integrin αvβ3-positive) cell line rather than HT-29 (human colon cancer cells, integrin αvβ3-negative) cell lines. This selectivity was attributed to integrin-mediated endocytosis by the RGD targeting peptide, confirmed using a fluorescence imaging study. Corroborating the “self-triggered” properties, the release of DOX from 13 treated cells was found to induce a 154-fold increase in caspase-3 activity. Using in vivo models, compared to controls (RDEVD-DOX (RGD-deficient) and RGDDEV-DOX (DVED-deficient)), 13-administered U-87 MG tumour-bearing mice were found to have significantly reduced tumour volumes (near 0 mm3vs. greater than 200 mm3 (control)) after 7 days of treatment (3 mg kg−1, once a day for seven days) with 14 days of monitoring (Fig. 3b and c). This report demonstrates both the importance of cancer-targeting units and a prodrug strategy to achieve ideal antitumour activity in vivo.
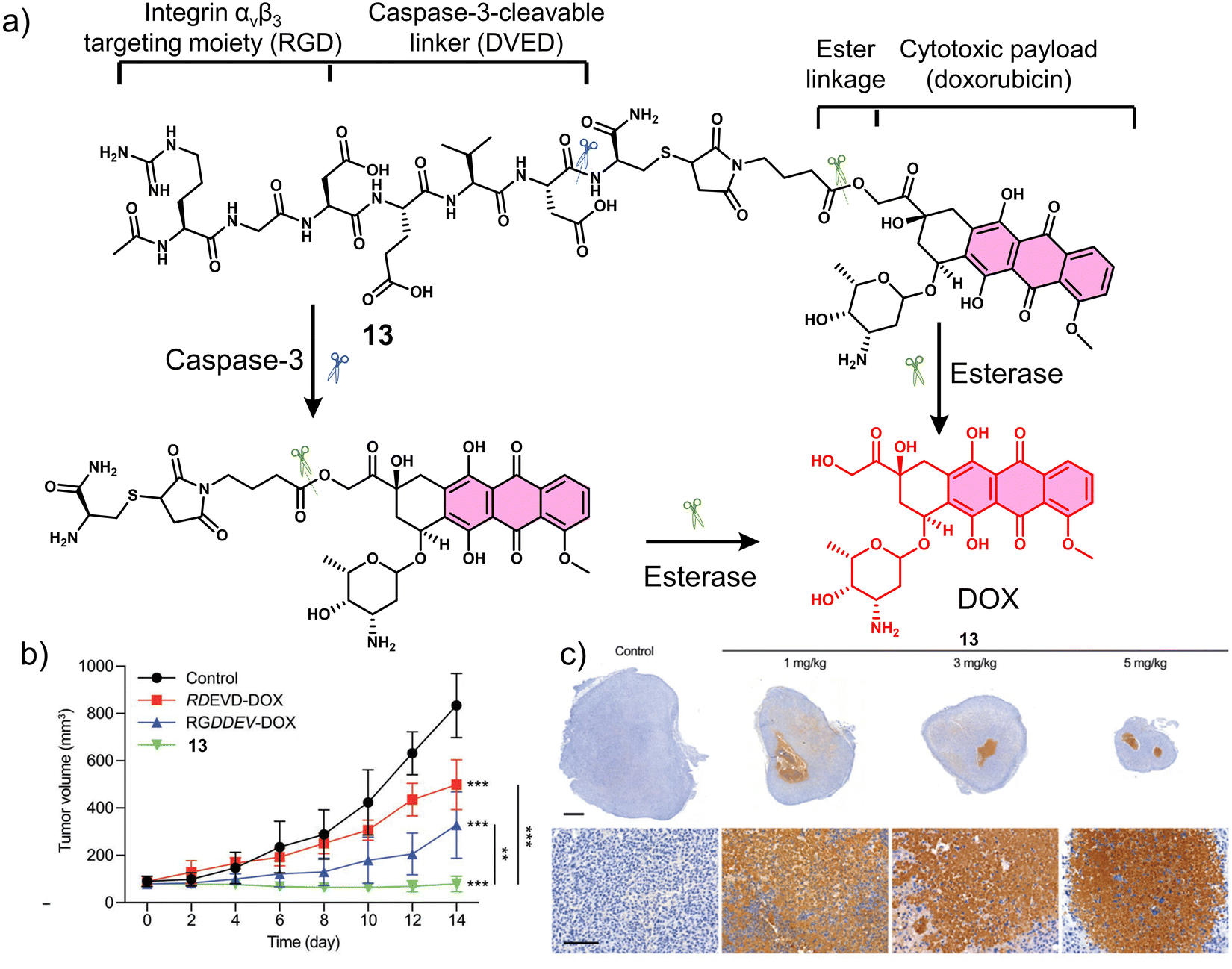 | ||
| Fig. 3 (a) Chemical structure of 13 and its enzymatic products (caspase-3 and esterase). (b) Tumour growth profiles of U-87 MG tumour-bearing mice that received saline as control, RDEVD-DOX (3 mg kg−1), RGDDEV-DOX (3 mg kg−1), or 13 (3 mg kg−1); n = 6. Data are mean ± s.d. **P < 0.01, ***P < 0.001. (c) Immunostaining of caspase-3 in the tumour sections from U-87 MG tumour-bearing mice treated with control and 13 (1 mg kg−1, 3 mg kg−1, and 5 mg kg−1); scale bar: 1 mm (upper panels) and 100 μm (lower panels). This is an open access article distributed under the terms of the Creative Commons CC BY license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. | ||
Cathepsin B is a lysosomal protease overexpressed in a range of cancers. It is responsible for promoting the spatial expansion of tumours, angiogenesis, and the metastasis of tumour cells within and outside the blood vessels through protein hydrolysis. Since the discovery of its specificity for the peptide sequence Gly-Phe-Leu-Gly (GFLG), numerous groups have developed cathepsin B-responsive systems.76 Liu and co-workers developed the multifunctional PDT agent, 14. This system consisted of an AIEgen (TPECM) that possessed both fluorescence and PDT properties, GFLG as the cathepsin B-responsive linker, cRGD for actively targeting αvβ3 integrin, and a hydrophilic peptide sequence (Fig. 4).77 AIEgens are molecules that emit minimal fluorescence in a mono-dispersed state; however, the fluorescence output is significantly enhanced when the molecules are aggregated (or rigidified).78,79 The initial good hydrophilicity of 14 meant that the AIEgen was unable to form fluorescent aggregates and produce ROS under light irradiation. However, the cathepsin B-mediated cleavage of the GFLG linker resulted in a concomitant increase in fluorescent intensity at 615 nm and a significant increase in ROS generated in the solution. This was further confirmed through fluorescence microscopy in MDA-MB-231 cells with high signal-to-noise ratio. Dose-dependent cytotoxicity was seen for 14 upon white light irradiation (0.25 W cm−2, 2 min) in MDA-MB-231 cells overexpressing αvβ3 integrin when compared to MCF-7 and 293T cell lines (αvβ3 integrin-negative controls). The cathepsin-B-mediated activation and phototoxicity of 14 were confirmed through the pre-treatment of MDA-MB-231 cells with cRGD, CA-074-Me (a cathepsin B inhibitor), and vitamin C (a ROS scavenger). All were shown to suppress its light-mediated cytotoxicity. Unique to this strategy is that this method does not require the use of any protecting groups or FRET pairs to deactivate the phototoxicity of the photosensitizer. Thus, it illustrates the unique advantages of AIE for the development of activatable photosensitizers. Although limited by the short emission wavelength, this study sets the basis for the use of AIE-active, enzyme-responsive PDT agents for cancer therapy.
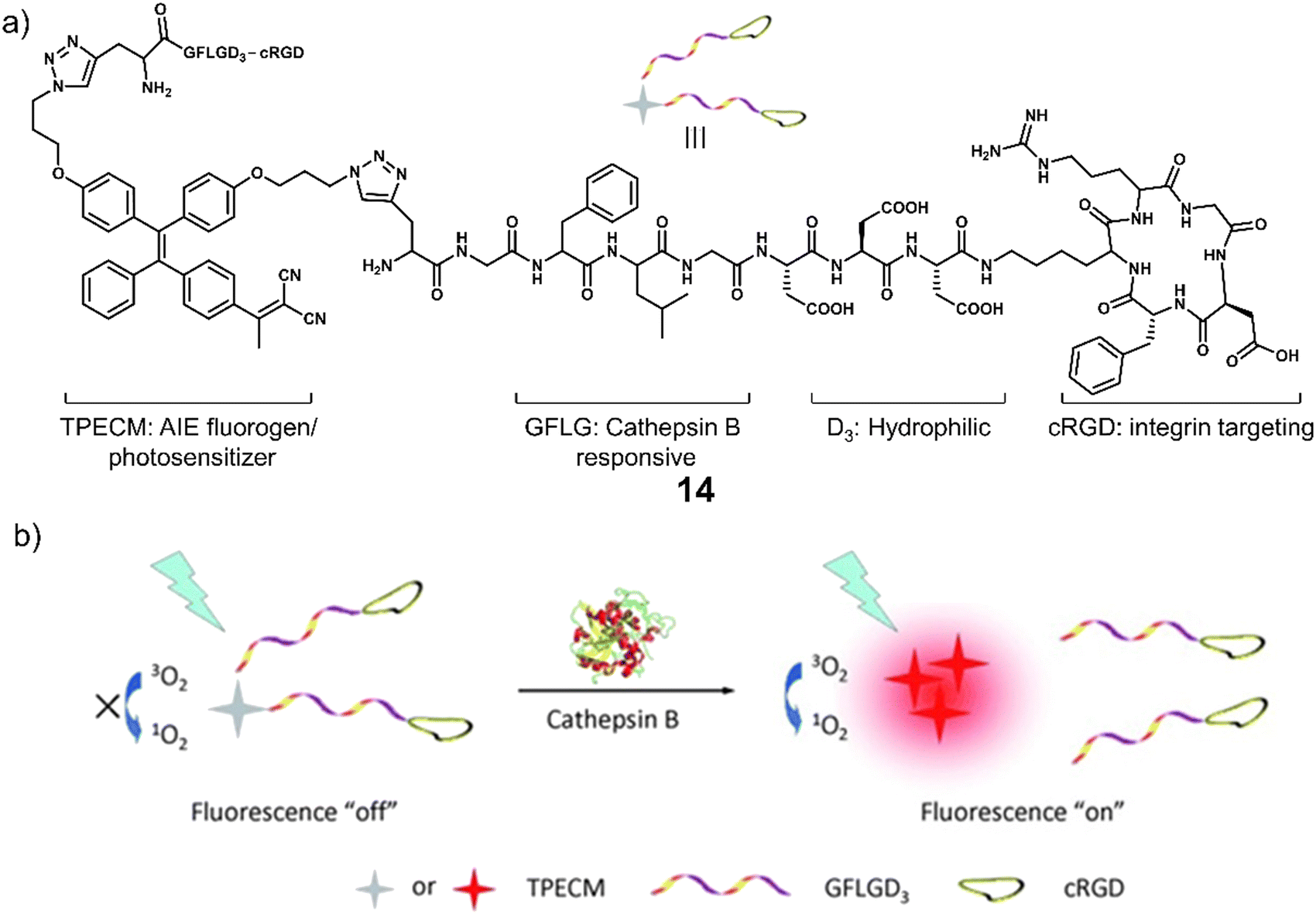 | ||
| Fig. 4 (a) Chemical structure of 14 and (b) the schematic illustration of 14 being activated by cathepsin B with a fluorescence “turn-on” signal and the ability to generate ROS upon white light irradiation. Reproduced with permission from ref. 77. Copyright (2014) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
γ-Glutamyltranspeptidase (GGT) is a cell surface N-terminal nucleophilic hydrolase that is associated with GSH metabolism. GGT is known to be overexpressed during oxidative stress and is believed to be associated with tumour progression, invasion, and the development of chemotherapeutic resistance.80,81 For these reasons, researchers have focused on the development of GGT-responsive systems for fluorescence imaging and therapeutic applications.82 Using the knowledge of prior GGT-responsive systems,83 Urano and co-workers developed a selenium-based rhodamine derivative, 15, for GGT-activated PDT (Fig. 5).84 The replacement of the oxygen atom on rhodamine with a selenium atom was found to increase the singlet oxygen quantum yield by 10.8 fold.85 Initially, 15 was non-fluorescent and displayed minimal phototoxicity under 532 nm light excitation. Which was attributed to the rhodamine derivative HMSeR being “masked’ with a glutamate functionality. In the presence of GGT, the glutamate protecting group was hydrolysed and released photoactive HMSeR, which is confirmed via increased fluorescence emission at 562 nm and the change in HPLC retention times. Light irradiation (510–550 nm, 50 mW cm−2, 1 min) of a SHIN3 cell line (human ovarian cancer cells with high expression of GGT) incubated with 15 (10 μM) resulted in a remarkable photocytotoxicity being observed (20% cell viability). The intracellular GGT-activation of 15 was confirmed using the pre-treatment of SHIN3 cells with GGT inhibitor, GGsTop. The in vivo antitumour studies in a tumour-bearing chick chorioallantoic membrane (CAM) model revealed that 15 was able to selectively eradicate GGT-positive tumours without causing unwanted phototoxicity to the surrounding healthy tissues. This study represents one of the first aminopeptidases-activatable PDT agents, which offers scope for the subsequent development of a variety of peptidase (overexpressed in a variety of different tumours)-activated PDT agents and long excitation wavelength GGT-responsive analogues.
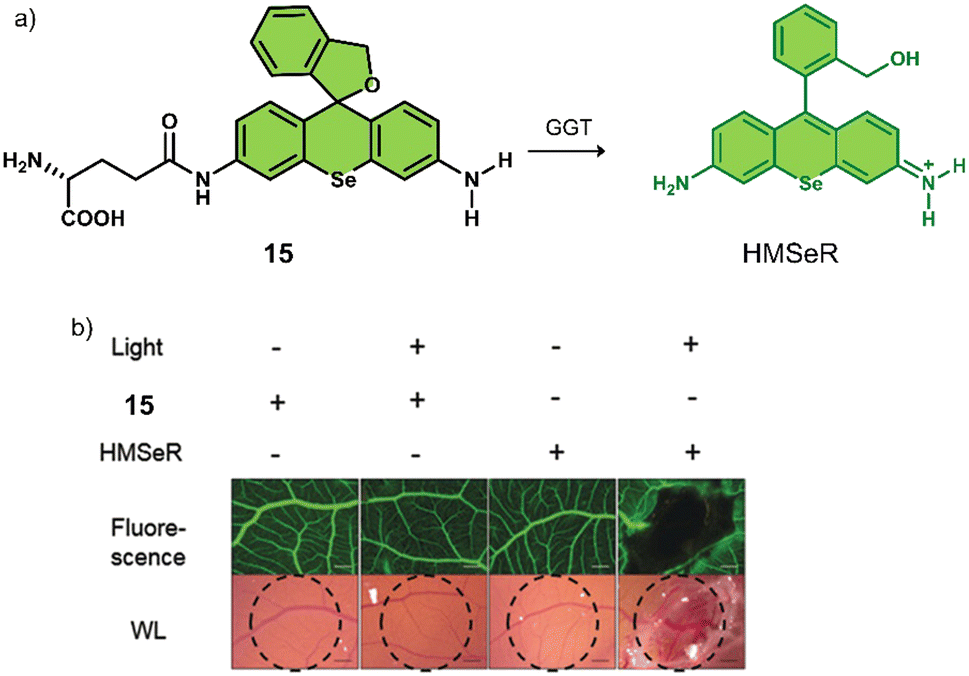 | ||
| Fig. 5 (a) Chemical structure of 15 and the GGT-activatable release of HMSeR. (b) Evaluation of vessel occlusion on CAM after photoirradiation (510–550 nm, 50 mW cm−2, 15 min) in the presence of 15 (100 μM, 25 μL) and HMSeR (100 μM, 25 μL). Reproduced with permission from ref. 84. Copyright (2017) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Glycosidases are involved in a variety of intracellular catabolic processes, such as endocytosis of glycoproteins and deglycosylation modifications following transport to lysosomes.86 Deficiency of glycosidases creates cellular storage disorders and their abnormalities can also cause severe multisystem disorders, therefore, glycosidases have become biomarkers used in the diagnosis of cancer and in monitoring its malignant progression, recurrence and prognosis.87 Glycosidase-responsive therapeutic agents are primarily activated by enzyme catalysis of substrates to release active therapeutic agents. β-Gal is a glycoside hydrolase that catalyses the hydrolysis of galactoside residues from glycoconjugates. The overexpression of β-Gal is associated with primary ovarian cancer and cell senescence.88 Zhen et al. developed a NIR fluorescent and PA-imaging PTT agent, β-Gal-responsive phototheranostic 16 used for the image-guided PTT treatment of ovarian cancer SKOV3 tumour-bearing mice models (Fig. 6).8916 consists of a galactose-functionalised NIR hemicyanine dye (CyOH) that possesses both imaging and PTT properties. A poly(ethylene glycol) (PEG) chain was added to provide good aqueous solubility. 16 initially exhibits minimal fluorescence and photoacoustic properties; however, the presence of β-Gal removes the galactosyl group to afford free CyOH-P, leading to a simultaneous enhancement in optical (fluorescent intensity and PA signal) and photothermal properties. A change in absorption from 600 nm to 688 nm and an increase in fluorescence intensity at 720 nm was observed. The co-incubation of 16 with SKOV3 (β-Gal-overexpressed ovarian cancer cells) and NIH-3T3 cells (mouse embryonic fibroblast cells, control) led to an enhanced fluorescent intensity being observed only in SKOV3 cells. Suggesting good selectivity for β-Gal in biological environments. 16 displayed significant phototoxicity towards SKOV3 cells following 680 nm laser irradiation (0.6 W cm−2, 5 min). The NIR fluorescence and the photoacoustic (PA) signals of the β-Gal-response to 16 were found to reach a maximum value at the tumour site 1 h post-injection in SKOV3 tumour-bearing mice. This confirmed overexpression of β-Gal at the tumour and identified an optimal time point for PTT treatment. A significant reduction in tumour volume was observed compared to the control (saline) when 16 (300 μM) was administered via tail vein, and the tumour region was subjected to light irradiation (680 nm laser, 0.6 W cm−2, 5 min). 16 represents the first example of combining both optical imaging and PTT.
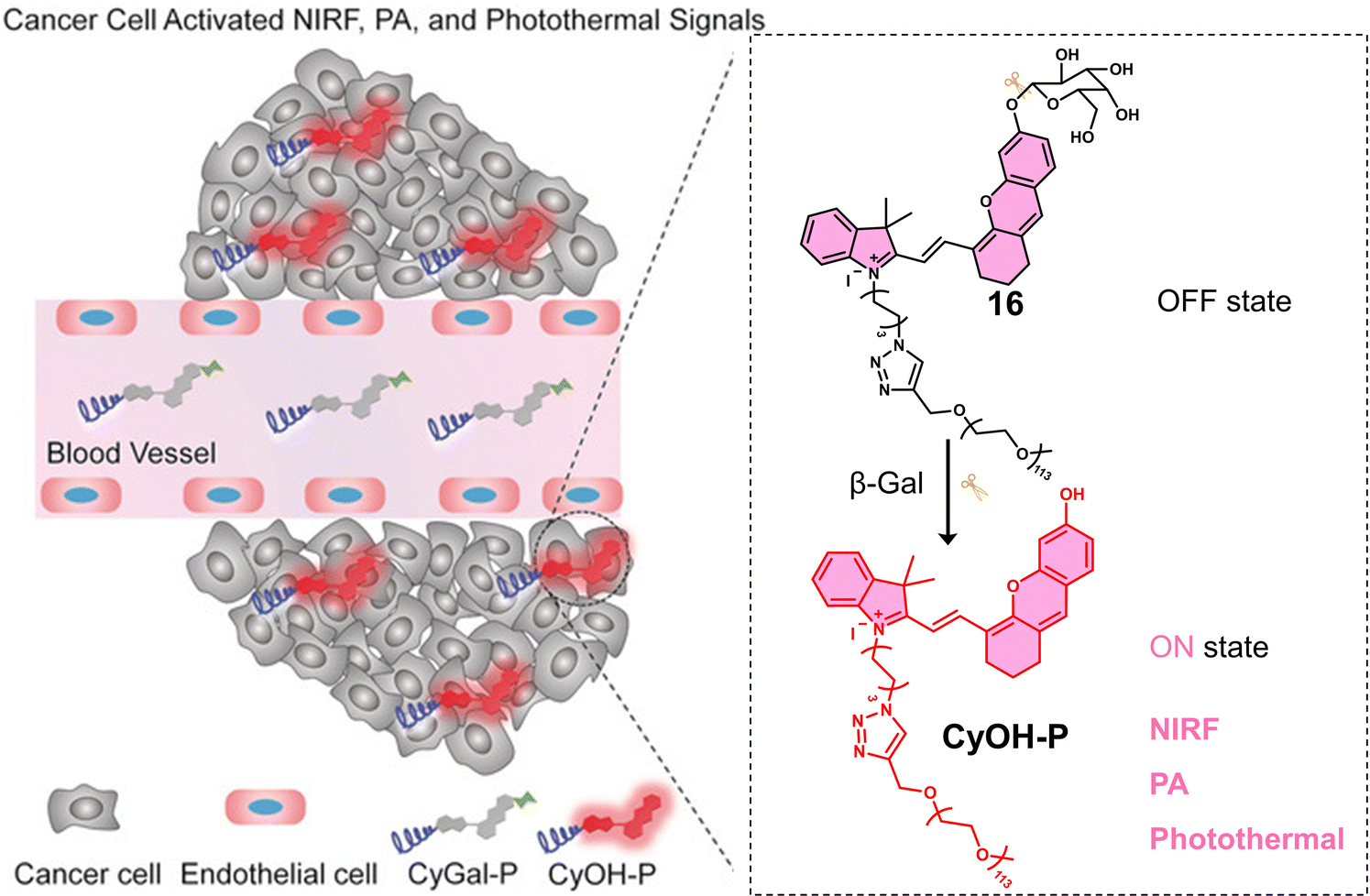 | ||
| Fig. 6 Chemical structure of 16 and schematic illustration of its β-Gal activated theranostic properties. Reproduced with permission from ref. 89. Copyright (2018) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
To improve the selectivity of β-Gal-activated prodrugs, Legigan et al. developed a folic acid-functionalised β-Gal responsive prodrug, 17 (Fig. 7a) for MMAE-based therapy for the treatment of KB-derived xenograft mice models.30 Monomethyl auristatin E (MMAE) is a synthetic anti-tumour agent derived from a peptide-based natural product, dolastatin-10. MMAE inhibits cell division by suppressing microtubule protein polymerization. Unfortunately, due to its potent toxicity, it cannot be used as a drug on its own. MMAE has therefore been approved for use as an antibody-drug conjugate (ADCs), including Tivdak and Adcetris.90 Due to the high production costs of ADCs, 17 was designed to offer an alternative therapeutic approach. 17 consisted of a self-immolative linker, which was connected to a folate subunit (targets folate receptors (FR) expressed on the surface of cancer cells), a galactose unit, and the antineoplastic monomethyl auristatin E (MMAE). In the presence of β-Gal, the glycosidic bond of 17 was hydrolysed, and the bioactive MMAE was released (confirmed by HPLC). As β-Gal is present in the lysosomes of both healthy and malignant cells, the introduction of the folate moiety affords specificity towards FR-positive tumour cells, thus avoiding non-selective drug release in non-malignant tissues. High cytotoxicity was seen in FR-positive KB (human oral epidermoid carcinoma cell line) and HeLa (human cervical carcinoma cell line) cells when compared with FR-negative A549 cells and a positive correlation with the expression of FR on the cancer cells was observed. Interestingly, it was shown that by using a co-culture study that the release of MMAE in FR-positive KB cells could spread to surrounding FR-negative A549 cancer cells and induce cell death. These results highlight the potential of 17 to overcome the significant heterogeneity of tumours, consisting of a wide range of tumour cells that may not express FR on the cancer cells. A significant reduction in the tumour volume with a high survival rate was observed with 17 in luciferase-transfected KB tumour-bearing mice models (Fig. 7b and c). This work represents one of the first generation β-Gal prodrugs that exhibits several advantages over the alternative and expensive antibody-directed enzyme prodrug therapy (ADEPT).
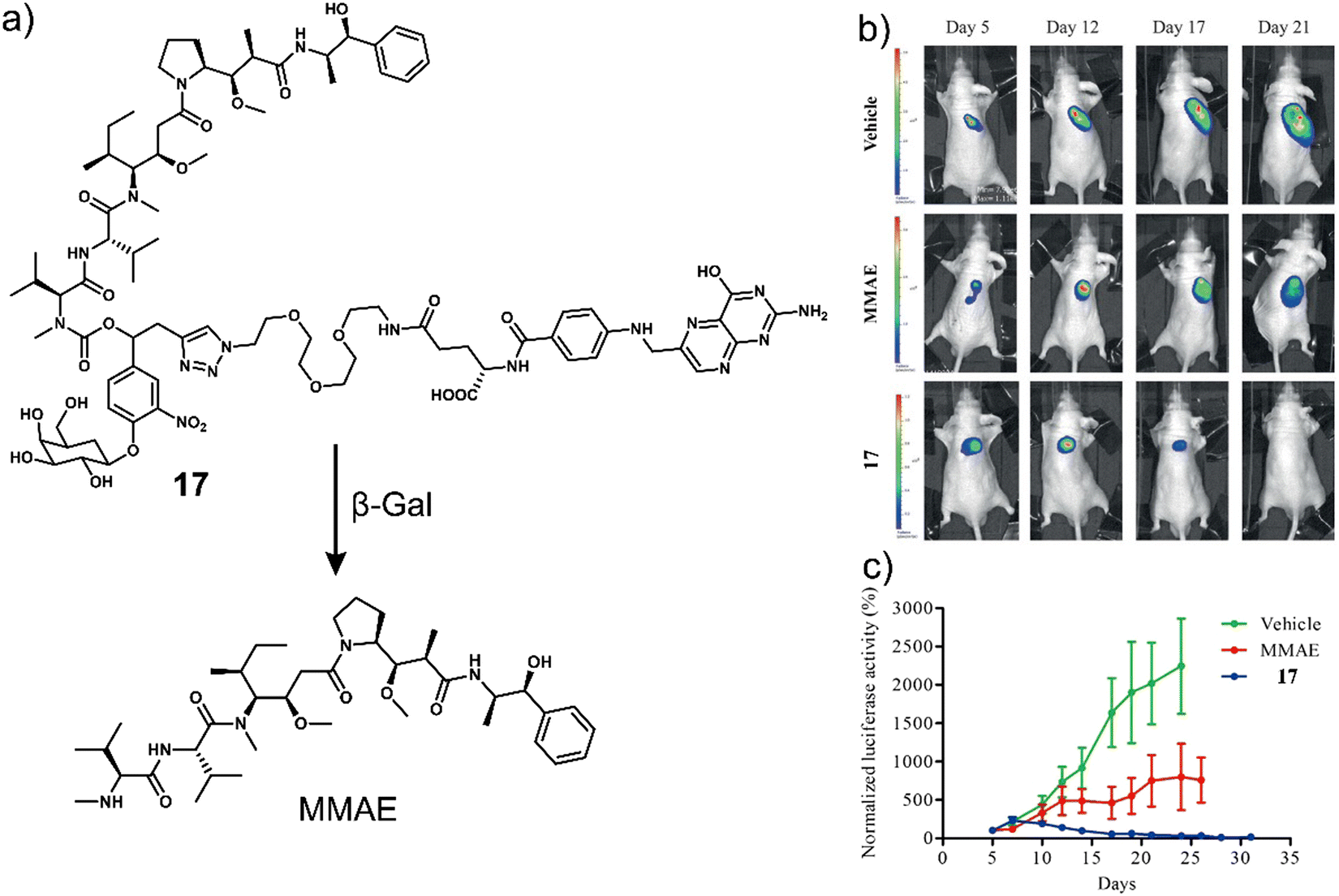 | ||
| Fig. 7 (a) Chemical structure of 17 and the resultant MMAE product with β-Gal. (b) Representative bioluminescence imaging of luciferase-transfected KB tumour-bearing mice at days 5, 12, 17, and 21 post-implantation when treated with vehicle (5% DMSO in PBS buffer), MMAE (intravenous administration at days 4, 7, 10, 14, and 17) or 17 (intravenous administration at days 5, 7, 10, 12, 14, 17, 19, 21, 23, and 26). (c) Tumour growth inhibition over time (days) with the vehicle, MMAE (1 mg kg−1) or 17 (5 mg kg−1). Reproduced with permission from ref. 30. Copyright (2012) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.2 Prodrugs and activatable phototherapeutics responsive to either oxidative or reducing environments
The dynamic balance between redox states is essential for maintaining the physiological function of cells.91 Redox homeostasis is achieved by regulating both ROS and reductive species. An imbalance in redox states is associated with cancer, with excessive ROS production and somewhat contradictory, excessive GSH concentrations being observed in cancer cells.92 To date, the consensus is that elevated reductive and oxidative stress may exist in different tumours (intertumoural) and even coexist at different progression stages within the same tumour (intratumoural). This includes the suborganelle level within the same cancer cell.93 For these reasons, researchers continue to explore prodrugs that respond to oxidative and reducive environments. In this section, we discuss prodrug examples that are designed to be activated in either reductive or oxidative environments (Table 2).| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: H2O2: hydrogen peroxide, GSH: glutathione, CPT: camptothecin, PPa: pheophorbide a, BTD: benzothiadiazole, PDT: photodynamic therapy, PTT: photothermal therapy, —: not mentioned. | ||||||
| 18 | H2O2 | SN-38 | Chemotherapy | B16F10 and HeLa cells | B16F10-induced metastatic lung tumour mice | 96 |
| 19 | H2O2 | Nitrogen mustard BrM | Chemotherapy | HL-60 cells | — | 97 |
| 21 | GSH | CPT | Chemotherapy | HepG2 cells | H22 tumour-bearing mice | 101 |
| 22 | GSH | PPa | PDT | U-87 MG cells | U-87 MG tumour-bearing mice | 102 |
| 23 | GSH | BODIPY fluorophore | PTT | HeLa cells | U14 tumour-bearing mice | 103 |
| 24 | GSH | Gemcitabine | Chemotherapy | A549, U87 and HEK 293 cells | A549 and U87 tumour-bearing mice | 104 |
| 25 | GSH | SN-38 | Chemotherapy | LoVo and SW620 cells | SW620 tumour-bearing mice | 31 |
| 26 | H2O2 and GSH sequentially | Benzothiadiazole | PDT | HeLa cells | — | 108 |
| 27–29 | GSH | Oxaliplatin | Chemotherapy | A549, A2780, HCT116, CT26 and EMT6 cells | A549, A2780, HCT116, CT26 and EMT6 tumour-bearing mice | 115 |
| 30 | GSH | Cisplatin | Chemotherapy | A2780, HeLa and MCF-7 cells | — | 116 |
| 31 | GSH | Carboplatin | Chemotherapy | A2780, HeLa and MCF-7 cells | — | 116 |
Several research groups have focused on the development of H2O2-responsive prodrugs. Examples include the use of boronate ester and α-ketoamide to construct ROS-activated therapeutic reagents. Kim et al. reported boronate ester-functionalized coumarin fluorophore-SN-38 conjugate (18) as a hydrogen peroxide (H2O2)-responsive theranostic system. H2O2 is well-known to be elevated in cancer cells and is believed to promote their differentiation, growth, and survival.94,9518 used a boronate ester H2O2-responsive moiety to mask the fluorescent properties of the coumarin fluorophore, which was used as a self-immolative linker for the chemotherapeutic SN-38 (Fig. 8a).96 The diagnostic boronate-functionalised coumarin allowed the direct monitoring of SN38 release. The addition of H2O2 (300 μM) to an aqueous solution of 18 (5 μM) was shown to oxidise the boronate moiety and release of SN-38 and fluorescent coumarin within 1 h, which was confirmed using fluorescence spectroscopy and fast atom bombardment mass spectroscopy (FAB-MS). The incubation of B16F10 and HeLa cell lines with 18 exhibited dose-dependent cytotoxicity in the presence of H2O2 (100 μM) for 48 h. Whereas, in the absence of H2O2, the cytotoxicity of 18 was found to be lower than SN-38. More importantly, 18 exhibited greater antitumour efficacy than SN-38 and extended the survival period by 6 days when compared with the control group (saline) (Fig. 8b–d). Although moderate antitumour effects were observed, this report has inspired others to develop systems for the fluorescence imaging of drug release in vitro and in vivo.
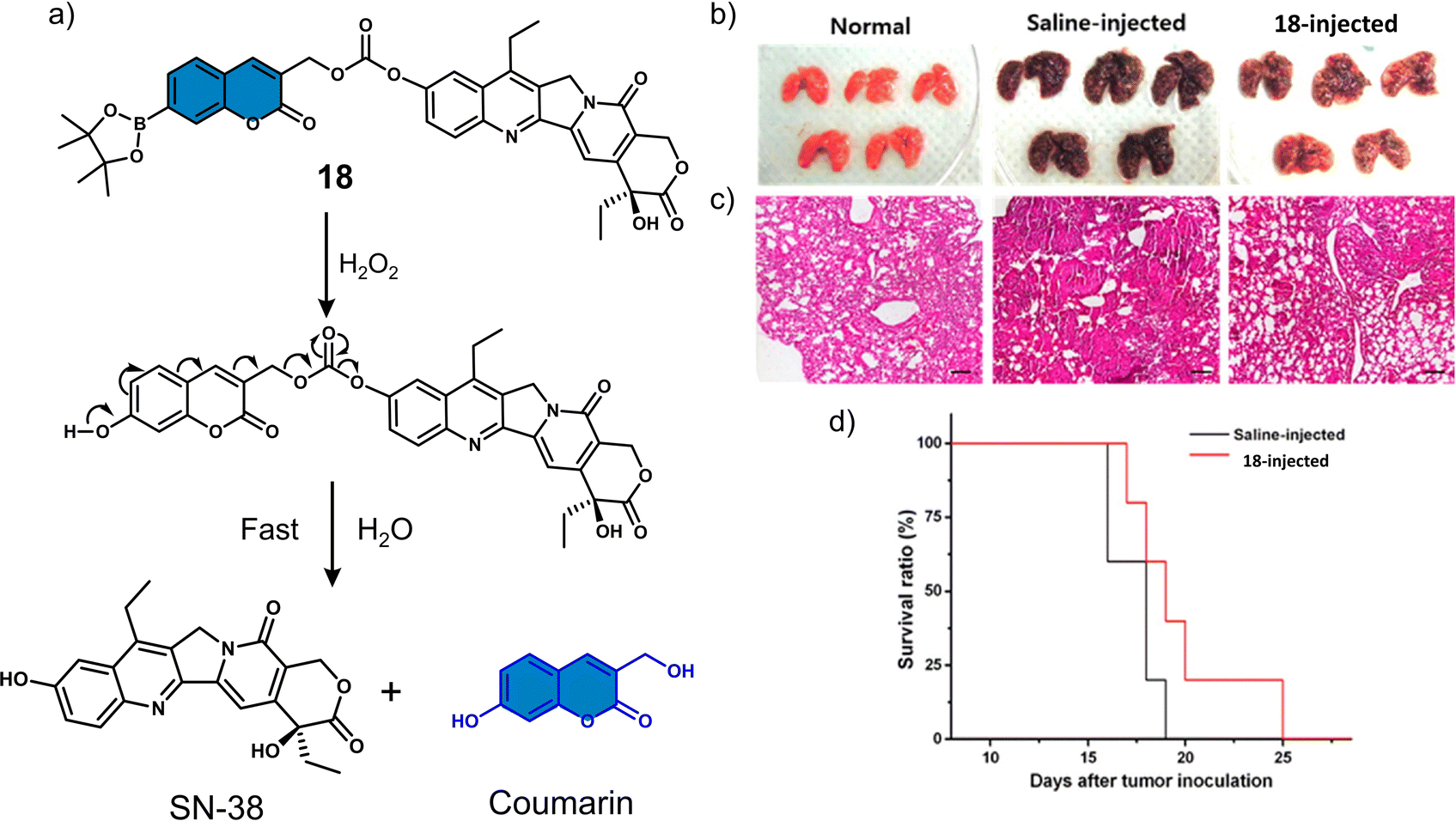 | ||
| Fig. 8 (a) Chemical structure of 18 and mechanism for the H2O2-mediated release of SN-38. (b) Representative images of lungs isolated from normal mice and B16F10-induced metastatic lung tumour mice that were administered intratracheally with saline or 18 (0.25 mg kg−1). (c) Histological sections of lung tissues with H&E staining. Scale bars: 100 μm. (d) Kaplan–Meier survival analysis of mice administered with saline or 18. Reproduced with permission from ref. 96. Copyright (2014) American Chemical Society. | ||
The vast majority (>90%) of reported H2O2-responsive prodrugs are based on phenylboronic acid/phenylboronic acid ester fragments, but deficiencies such as the high sensitivity of boronic acid esters to hydrolysis, off-target reactions between boronic acid and bio-diols, and the moderate selectivity of boronic acid esters for H2O2 limit their clinical translation. The Yin group developed a novel H2O2-responsive prodrug, 19, by functionalising anti-tumour active nitrogen mustard analogues with α-ketoamide unit (Fig. 9a).97 The strong electron-withdrawing nitro group of 19 enhance the electrophilicity of the adjacent carbonyl group, allowing a significantly faster reaction with H2O2.98 Considering the important role of the nitro group of 19 in H2O2-induced nucleophilic attack, the authors also synthesized a non-nitro analogue of 20 that is resistant to H2O2 as a negative control. 19 exhibited reasonable stability in the absence of H2O2 and displayed little cytotoxicity against cancer cells. HPLC, 1H NMR and mass spectroscopic analysis confirmed the release of nitrogen mustard N,N-bis(2-bromoethyl)benzene-1,4-diamine (BrM) when 19 (100 μM) was exposed to H2O2 (2 eq.). 19 exhibited a high antiproliferative effect in HL-60 promyelocytic cell line and pretreatment with the H2O2 scavenger N-acetyl-cysteine (NAC) reduced its anti-proliferative capacity. 20, which is inert to H2O2, exhibited relatively low cytotoxicity to HL-60 cells, and pretreatment with NAC did not reduce the toxicity of 20 to HL-60 cells (Fig. 9b). Mechanistic investigations indicated that 19 selectively releases nitrogen mustard by H2O2 activation, which effectively inhibits the proliferation of HL-60 cells through DNA cross-linking and mitochondria-dependent apoptotic pathways.
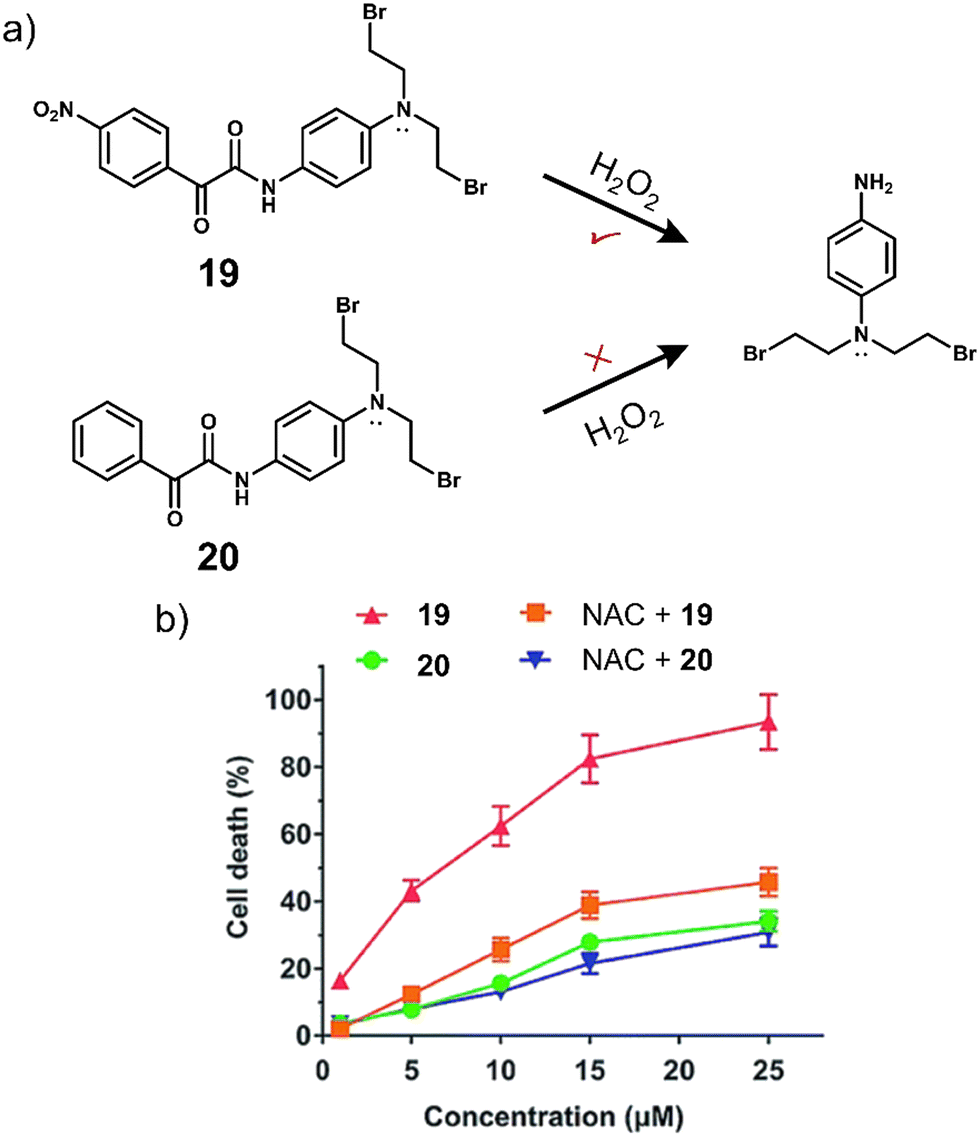 | ||
| Fig. 9 (a) Chemical structure of 19, 20 and the H2O2-mediated release mechanism of nitrogen mustard BrM. (b) The effects of NAC on the cytotoxicity of 19 and 20 in HL-60 cells after 72 h incubation. Reproduced with permission from ref. 97. Copyright (2015) The Royal Society of Chemistry. | ||
Several research groups have focused on the development of prodrugs that release a therapeutic in reducing environments. Examples include the use of disulfide bonds and dinitrobenzenesulfonyl groups to construct GSH-activated therapeutic systems. Glutathione (GSH) is a biological tripeptide (glutamic acid, cysteine, and glycine) that contains a free sulfhydryl group. It is the most abundant biological thiol in cells, regulating cellular redox by acting as a key antioxidant. As mentioned in brief earlier, elevated GSH concentrations are found in tumour cells (2–10 mmol L−1), which is believed to contribute to tumour progression and drug resistance.99 For these reasons, researchers have focused on developing GSH-responsive prodrugs and strategies that can reduce intracellular GSH concentrations to overcome potential drug resistance mechanisms.100
Kong et al. developed a fluorescence-based theranostic 21 for monitoring the GSH-mediated release of chemotherapeutic camptothecin (CPT) in a H22 tumour-bearing mice model. CPT is a natural product isolated from the Chinese endemic medicinal plant, Camptotheca acuminata, and has been found to inhibit the proliferation of cancer cells by selectively inhibiting DNA topoisomerase I (Topo I). 21 was constructed by conjugating a merocyanine-based dye with the anticancer agent, camptothecin (CPT), via a GSH-responsive disulfide linker (Fig. 10).10121 displayed an initial weak fluorescence emission intensity due to the amino unit of the merocyanine unit being capped. The addition of GSH induced disulfide cleavage and resulted in a significant increase in fluorescence signal at 702 nm, which was ascribed to the release of free merocyanine and CPT. This was further confirmed using high-resolution mass spectroscopy (HRMS). In vitro studies revealed that 21 exhibited higher antiproliferative activity in the hepatoma HepG2 cell line compared to a normal liver HL-7702 cell line. These results were attributed to the HepG2 cell lines having elevated intracellular GSH concentrations, which was confirmed via fluorescence microscopy by monitoring the increase in emission at 702 nm. Using the NIR-emissive properties of merocyanine, it was confirmed that 21 ehibited good tumour localisation properties in H22 tumour-bearing mice after intravenous injection (visualised using an IVIS LuminaIII in vivo imaging system).
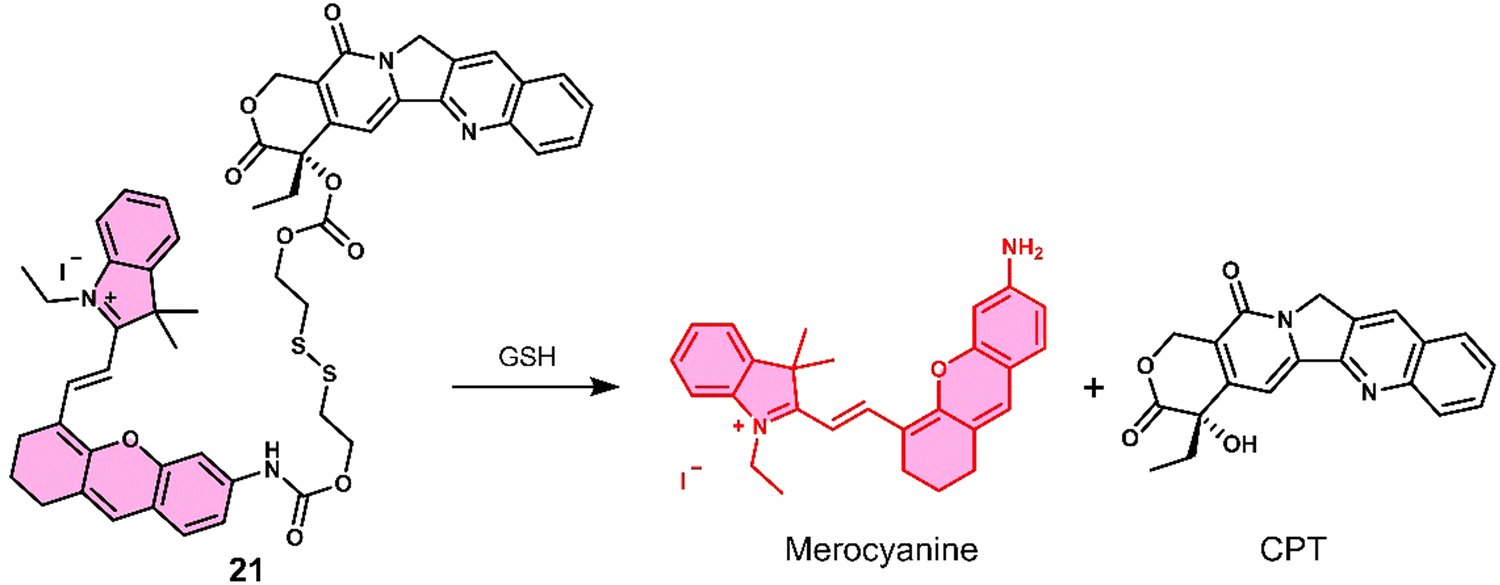 | ||
| Fig. 10 Chemical structure of 21 and the GSH-mediated release mechanism of CPT and fluorescent merocyanine dye. | ||
Ye and co-workers constructed the tumour-targeting GSH-activated PDT agent, 22, that could be visualised by dual-modality NIR fluorescence/MR imaging to allow image-guided PDT (Fig. 11).102 Combining the ability of MRI to image the whole body and the high sensitivity of fluorescence for functional imaging of tumour cells, the diagnostic accuracy and reliability can be substantially improved. 22 consisted of the photosensitizer pheophorbide a (PPa), the thiol-responsive disulfide linker, DOTA-Gd complex, and cyclic peptide cRGD, all of which were linked via an amino oxyluciferin fluorophore. Owing to hydrophobic PPa and hydrophilic RGD, 22 was found to self-assemble in an aqueous solution to form nanoparticles with a hydrodynamic size of ∼60 nm. This self-assembly resulted in high r1 relaxivity (20.0 ± 1.7 mm−1 s−1), quenched fluorescence at 677 nm, and a low 1O2 quantum yield. In the presence of GSH in a PBS-HSA mixture (PBS buffer containing 5% HSA), 22 was cleaved to 2-RGD and released photoactive PPa-SH. This structural transformation was confirmed by HPLC and matrix-assisted laser desorption/ionization mass spectroscopy (MALDI-MS). Hydrophobic PPa-SH was effectively bound to HSA, allowing it to disperse in water and exhibit a strong NIR fluorescence emission at 677 nm as well as enabling 1O2 generation under laser irradiation (690 nm, 40 mW cm−2, 5 min). Fluorescence imaging studies confirmed the cellular uptake of 22 naonoparticles in U-87 MG cells, followed by an increase in fluorescence emission at 690–750 nm when activated by intracellular GSH and HSA. An increase in T1-weighted MR images was observed after incubation of U-87 MG cells with 22 nanoparticles, which matched those observed by the fluorescence imaging. The light irradiation (690 nm, 40 mW cm−2, 180 s) of U-87 MG cells incubated with 22 nanoparticles (20 μM), displayed a significant photocytotoxicity (<10% cell viability); however, minimal cytotoxicity was observed for GES-1 cells under the same conditions. 22 nanoparticles using dual-modal fluorescence and MRI imaging exhibited good tumour localising ability in a U-87 MG tumour-bearing mice model. 22 nanoparticles (200 μL, 200 μM) significantly reduced tumour volume compared to control groups (PBS) under laser irradiation (690 nm, 800 mW cm−2, two consecutive exposures of 10 min each at an interval of 10 min). Good biosafety of 22 nanoparticles was confirmed since body weight losses were comparable to the PBS group.
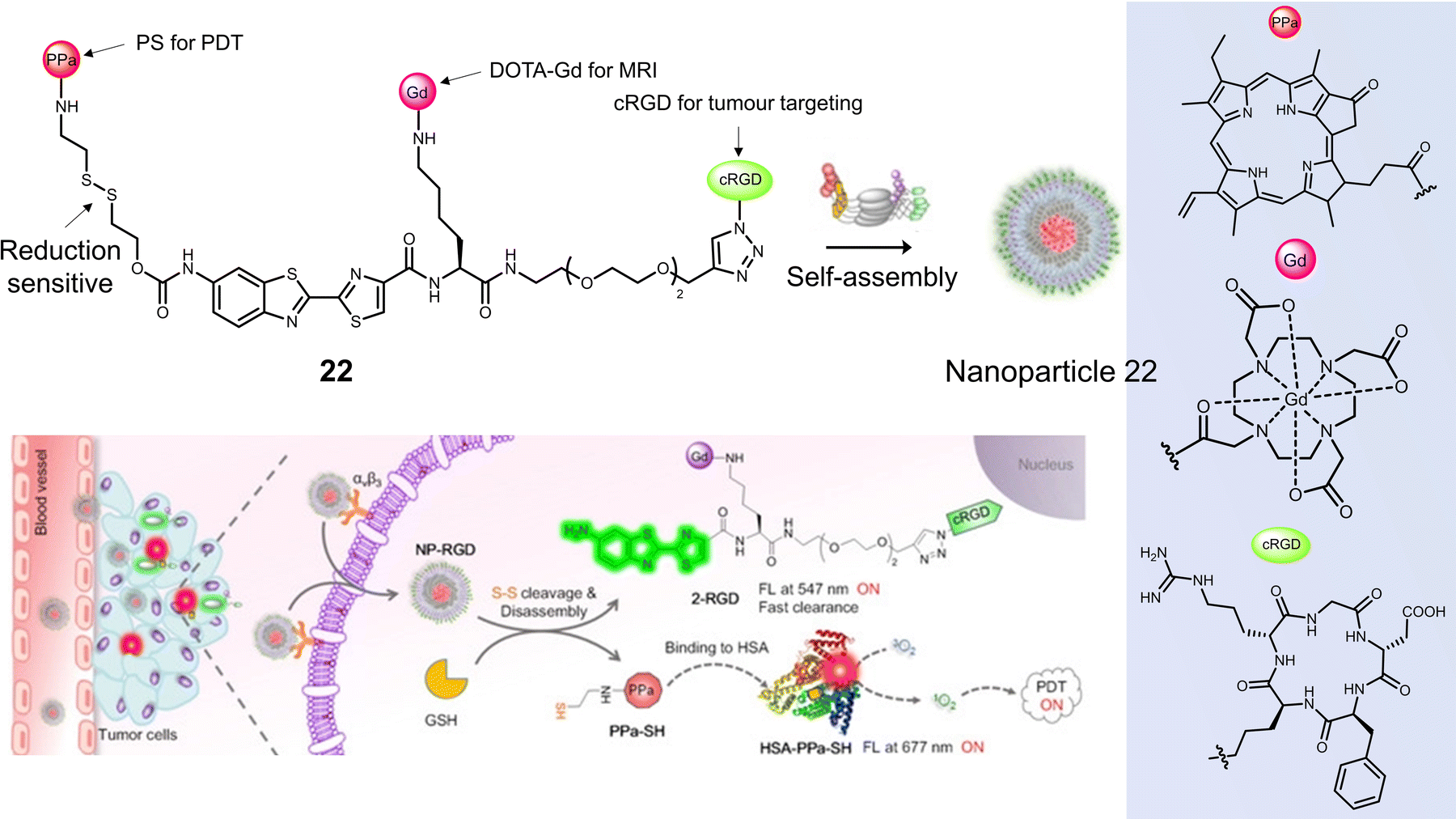 | ||
| Fig. 11 The chemical structure of 22 and schematic illustration of 22 targeting tumour cells by binding αvβ3 integrin with subsequent fluorescence turn-on and light-mediated therapeutic effect on cancer cells after activation by GSH and binding to HSA. Reproduced with permission from ref. 102. Copyright (2020) Wiley-VCH Verlag GmbH. | ||
Disulfide-based BODIPY scaffolds have also been used to construct promising PTT theranostic agents. Wang et al. reported the disulfide-based PTT agent 23 that uses a BODIPY fluorophore for both imaging and PTT. The amino unit on the phenyl group of the fluorophore was conjugated to PEG-based disulfide chain to afford good aqueous solubility and enable GSH detection (Fig. 12).10323 formed stable nanoparticles in an aqueous solution via self-assembly and was weakly emissive due to the aggregation-caused quenching (ACQ) effect. The GSH-mediated cleavage of the disulfide bond afforded soluble products, leading to an enhancement in fluorescent intensity and an increase in the diameter of the nanoparticles from 89 nm to 350 nm. Incubation of 23 nanoparticles with HeLa cells resulted in a low fluorescence emission being initially observed. However, treatment with GSH resulted in a significant increase in fluorescence emission. The NIR signal of 23 nanoparticles was found to reach its maximum value at 36 h post-injection in uterine cervical cancer (U14) tumour-bearing mice. The maximum PA signal at the tumour region was identified at 12 h post-injection of 23 nanoparticles. 23 nanoparticles exhibited excellent photothermal conversion efficiency upon NIR laser irradiation (808 nm, 0.5 W cm−2, 5 min), resulting in an increase of up to 47.7 °C in temperature (Fig. 12b). This resulted in successful tumour ablation with over a 90% decrease in tumour volume compared to controls (light alone and 23 nanoparticles without light) (Fig. 12c). Excellent biosafety of the agent was confirmed using haematoxylin and eosin (H&E) staining.
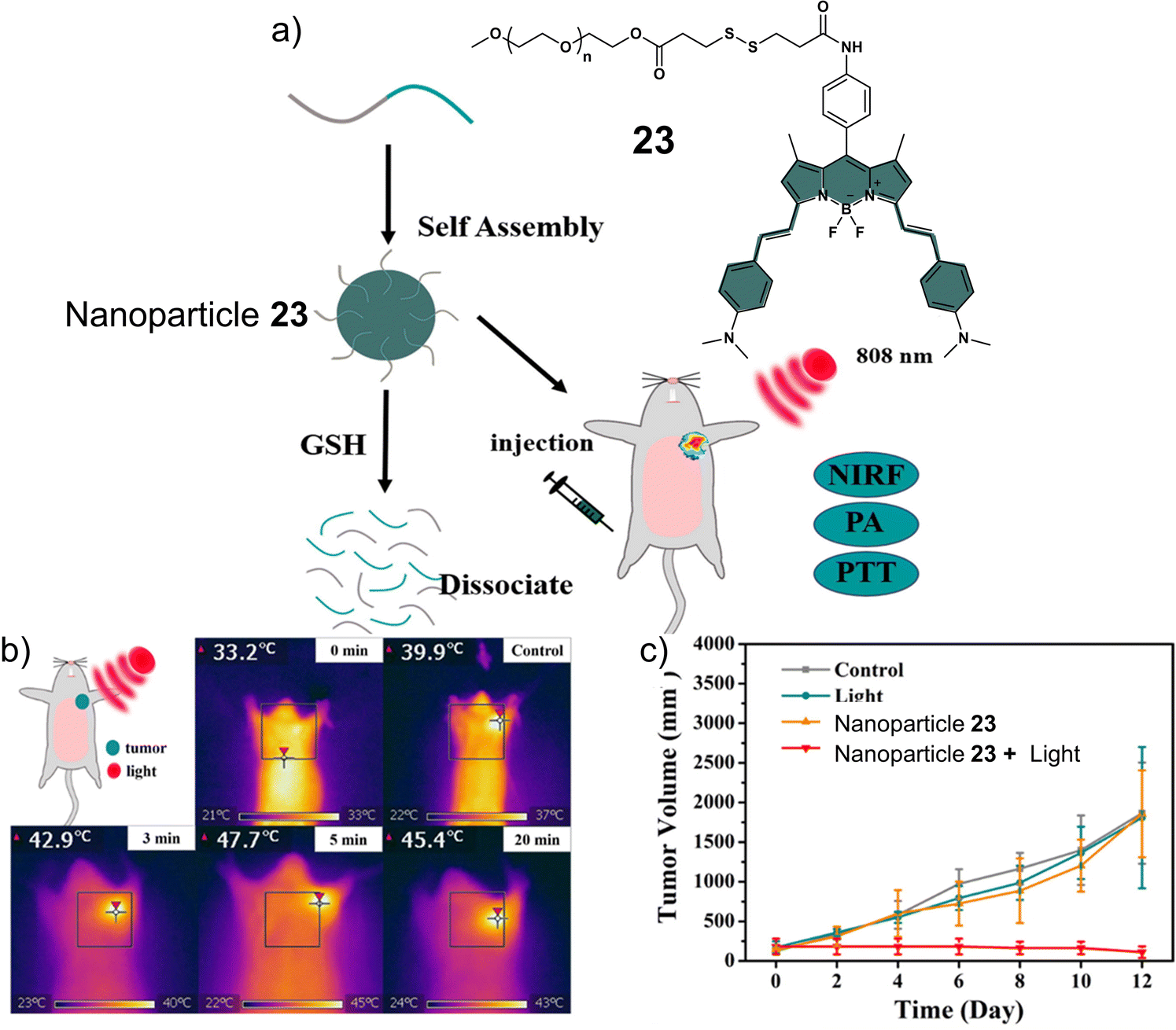 | ||
| Fig. 12 (a) Chemical structure of 23 and the activation of its theranostic properties by GSH. (b) Infrared thermal imaging of the uterine cervical cancer (U14) tumour-bearing mice after treatment with 23 nanoparticles under 808 nm irradiation (0.5 W cm−2, 5 min.). (c) The changing curve of real-time tumour volume of the uterine cervical cancer (U14) tumour-bearing mice 12 days after the treatment with 23 nanoparticles with or without 808 nm irradiation (0.5 W cm−2, 5 min) or control. Reproduced with permission from ref. 103. Copyright (2018) Elsevier B.V. | ||
Lucero et al. developed a GSH-responsive fluorescent and photoacoustic theranostic (24), and evaluated its properties in lung cancer A549 cells and an A549 tumour-bearing mice model.104 In this study, the authors initially focused on tuning the chemical reactivity of the traditional GSH-mediated nucleophilic aromatic substitution (SNAr) reaction of 2,4-dinitrobenzenesulfonyl.105 Since, it was believed that the high sensitivity of 24 towards GSH (micromolar) might lead to premature release in vivo and minimise selectivity between cancerous and healthy cells. Through extensive synthesis and evaluation, a 2-fluoro-4-nitrobenzenesulfonyl group was identified as an ideal candidate because its millilmolar reactivity towards GSH was believed to provide appropriate selectivity in vivo. 24 was constructed using 2-fluoro-4-nitrobenzenesulfonyl, a self-immolative hemicyanine chemical probe (HD-CH2OH), and the FDA-approved chemotherapeutic, gemcitabine (Fig. 13a). Gemcitabine is used for the treatment of a wide range of cancers, including breast, testicular, ovarian, and non-small cell lung cancer. Its mode of action is intracellular metabolism by nucleoside kinases to active nucleotides gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP), which lead to apoptosis by inhibiting the synthesis of DNA.106 In accordance with design expectations, minimal fluorescence and photoacoustic signals were observed for 24 alone in solution (pH 7.4, with 70% PBS/MeCN). The addition of GSH (10 mM) to an aqueous solution of 24 (5 μM) was shown to afford HD-CH2OH and result in the release of gemcitabine (confirmed by fluorescence, PA, MS, and nuclear magnetic resonance (NMR) analysis). For this system, a change in absorption from 570 nm to 680 nm and a significant increase in PA emission intensity at 690 nm was observed. 24 was then assessed in vitro using lung cancer A549, glioblastoma U87, and HEK 293 cells. 24 could readily distinguish GSH concentrations in lung cancer A549, glioblastoma U87, and HEK 293 using fluorescence microscopy, with the signal intensity correlating to antiproliferative activity; A549 cells displayed a greater fluorescence response and lower IC50 than U87 cells. A blind study was performed to demonstrate the importance of companion diagnostics,10724 was also able to differentiate between A549 tumour-bearing mice and U87 tumour-bearing mice by the GSH-activatable PA signal with 95% accuracy (Fig. 13b). Moreover, 24 exhibited a significantly better anti-tumour effect in A549 tumour-bearing mice than in U87 tumour-bearing mice ascribed to differences in endogenous GSH concentrations between the mice models (Fig. 13c).
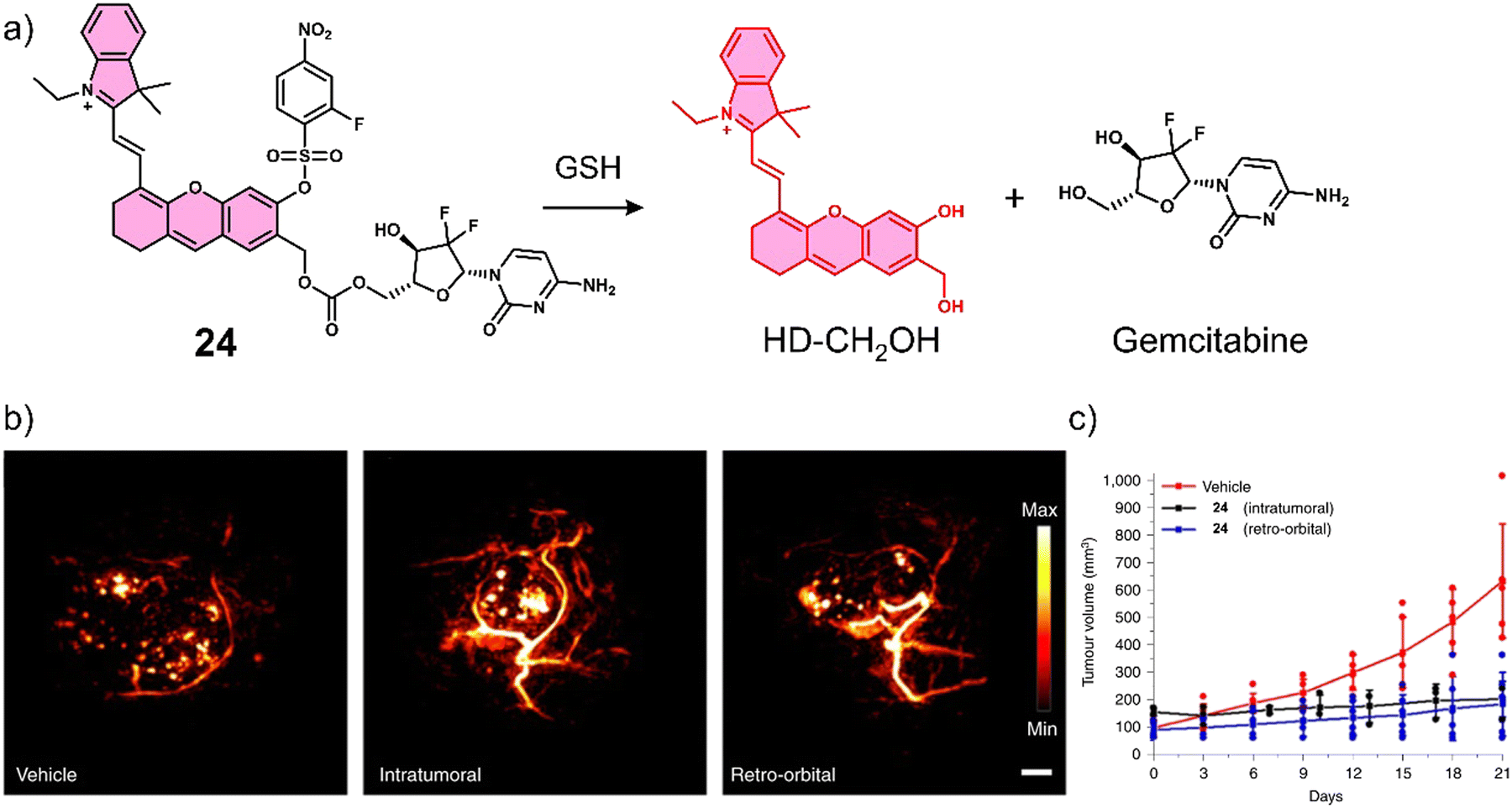 | ||
| Fig. 13 (a) Chemical structure of 24 and the GSH-responsive release of Gemcitabine and HD-CH2OH. (b) PA images of tumours after treatment with vehicle (10% DMSO/PBS), intratumoural injection of 24 (100 μM, 10% DMSO/PBS), and retro-orbital injection of PARx (400 μM, 10% DMSO/PBS). Samples were irradiated at 680 nm. Scale bar: 2 mm. (c) Average tumour volume after treatment with vehicle (n = 6 independent animals), intratumoural injection of 24 (n = 3 independent animals) and retro-orbital injection (n = 6 independent animals) of 24 (100 μM, 50 μL) once every 7 days for 21 days. Reproduced with permission from ref. 104. Copyright (2021) The Author (M. Y. Lucero and J. Chan). Published by Springer Nature. | ||
Considering that tumours can exist in a reductive and an oxidative state, Sharma et al. focused on identifying a suitable protecting group that could respond to either GSH or H2O2. As can be seen in Fig. 14a, a thioether linker was used to conjugate cancer-targeting cyclooxygenase-2 (COX-2) inhibitor, indomethacin with SN-38 to afford a theranostic 25.31 The inherent fluorescent properties of SN-38 were used to determine drug release in solution-based studies, in which the thioether linker was identified to undergo thiol-mediated hydrolysis (thiolysis) by GSH to release SN-38. Moreover, H2O2 was shown to oxidise the thioether linker to its sulfone analogue and undergo hydrolysis to release SN-38. SN-38 release by either GSH or H2O2 was studied by fluorescence spectroscopy with increased fluorescence emission intensity at 540 nm. In vitro analysis found greater antiproliferative activities in cell lines that overexpressed COX-2 (LoVo and SW620) than in those with a lower COX-2 expression (NHDFs, normal human dermal fibroblasts; MCF10A, human breast epithelial cell lines), which illustrated the importance of the indomethacin targeting unit. Moreover, the treatment of LoVo cells with either H2O2 or N-acetylcysteine (a GSH precursor) was shown to induce the intracellular release of SN-38 via monitoring changes in fluorescence emission intensity. Subsequent animal-based studies were performed using a colon cancer (SW620) tumour-bearing mice model via intraperitoneal injection (25 (5 mg kg−1 d−1), SN-38 (5 mg k−1g d−1), or vehicle (DMSO)) once every five days for three weeks. The tumour volume of 25-treated mice decreased significantly relative to what was seen for the controls about 17 mm3 and 12 mm3 (DMSO and SN-38, respectively) (Fig. 14b and c). Moreover, using immunoblotting assays, 25 was shown to significantly inhibit the expression of pro-inflammatory factors (e.g., TNF-α, IL-6, and VEGF). These results were ascribed to the anti-inflammatory properties of indomethacin. Minimal toxicity was observed for 25 with normal blood parameters (e.g., AST, ALT, and serum creatine); this indicates a good safety profile using this strategy. We anticipate this approach will inspire others to develop other multi-biomarker responsive prodrugs or even exploit the GSH or H2O2-responsive thioether linker for use in other applications.
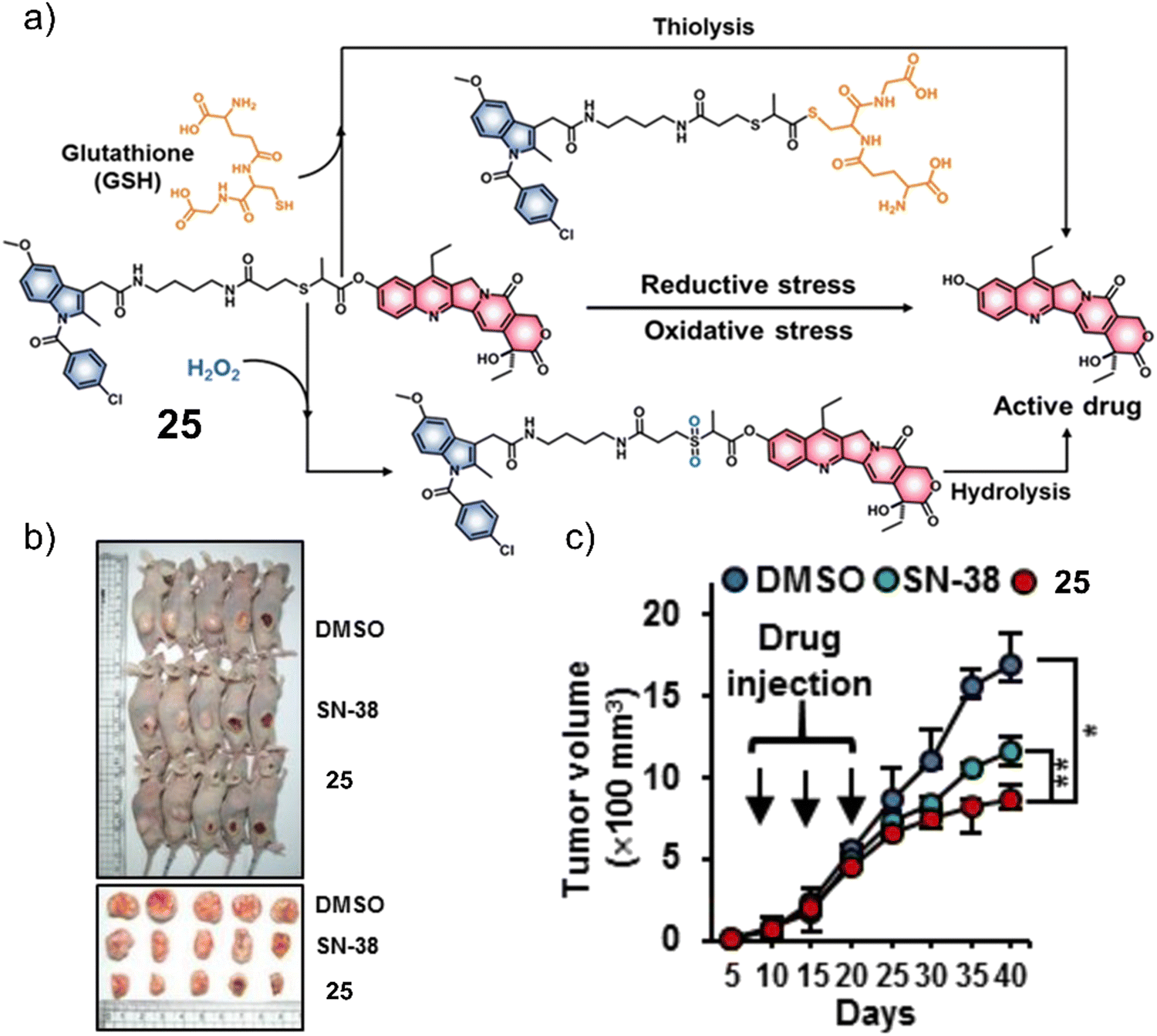 | ||
| Fig. 14 (a) Chemical structure of 25 and the products of the GSH or H2O2-mediated release of SN-38. (b) Representative images of SW620 tumour-bearing mice treated with control (DMSO), SN-38, and 25. (c) Tumour growth inhibition of SW620 tumour-bearing mice over 40 days with DMSO, SN-38 (5 mg kg−1 d−1), and 25 (5 mg kg−1 d−1) once every five days for three weeks. n = 5, mean ± SD, *P < 0.05, **P < 0.01. Reproduced with permission from ref. 31. Copyright (2019) American Chemical Society. | ||
A theranostic system co-activated by H2O2 and GSH is also used in the PDT treatment of tumour cells. Wang and co-workers reported the mitochondria-targeted PDT agent, 26, that was shown to be activated by H2O2 and GSH sequentially (Fig. 15a).10826 consisted of the PS benzothiadiazole (BTD) functionalised with a TPP mitochondrial targeting unit and a boronic ester protecting group that was covalently attached to a thiol unit.109,11026 initially exhibited a low fluorescence emission at 510 nm in PBS buffer (pH 7.4), and the sequential addition of H2O2 and GSH was shown to afford TPP-NH2ArSO3H, a process initiated by H2O2-mediated removal of the boronic ester and subsequently completed by a series of redox reactions mediated by H2O2 and GSH. This sequential activation resulted in a concomitant increase in fluorescence intensity at 510 nm and increased ROS production (both 1O2 and O2˙−). TPP-NH2ArSO3H exhibits a high two-photon (TP) absorption cross-section (δ = 41 ± 4 GM in methanol) at 800 nm, allowing its NIR TP activation for greater tissue penetration. Fluorescence microscopic evaluation confirmed the mitochondrial localisation of 26 in HeLa cells. The PDT efficacy of 26 was evaluated in HeLa cells, pretreated with H2O2 (50 μM). Significant cell death was observed after irradiating the cells with a two-photon laser at 800 nm (120 s) (Fig. 15b). Remarkably, TPP-NH2ArSO3H exhibited mixed type 1 and type 2 mechanisms; therefore, minimal changes to its phototoxicity were observed under hypoxic conditions. 26 represents the first H2O2/GSH sequentially activated mitochondria-targeted PDT agent with mixed type I and type II mechanisms.
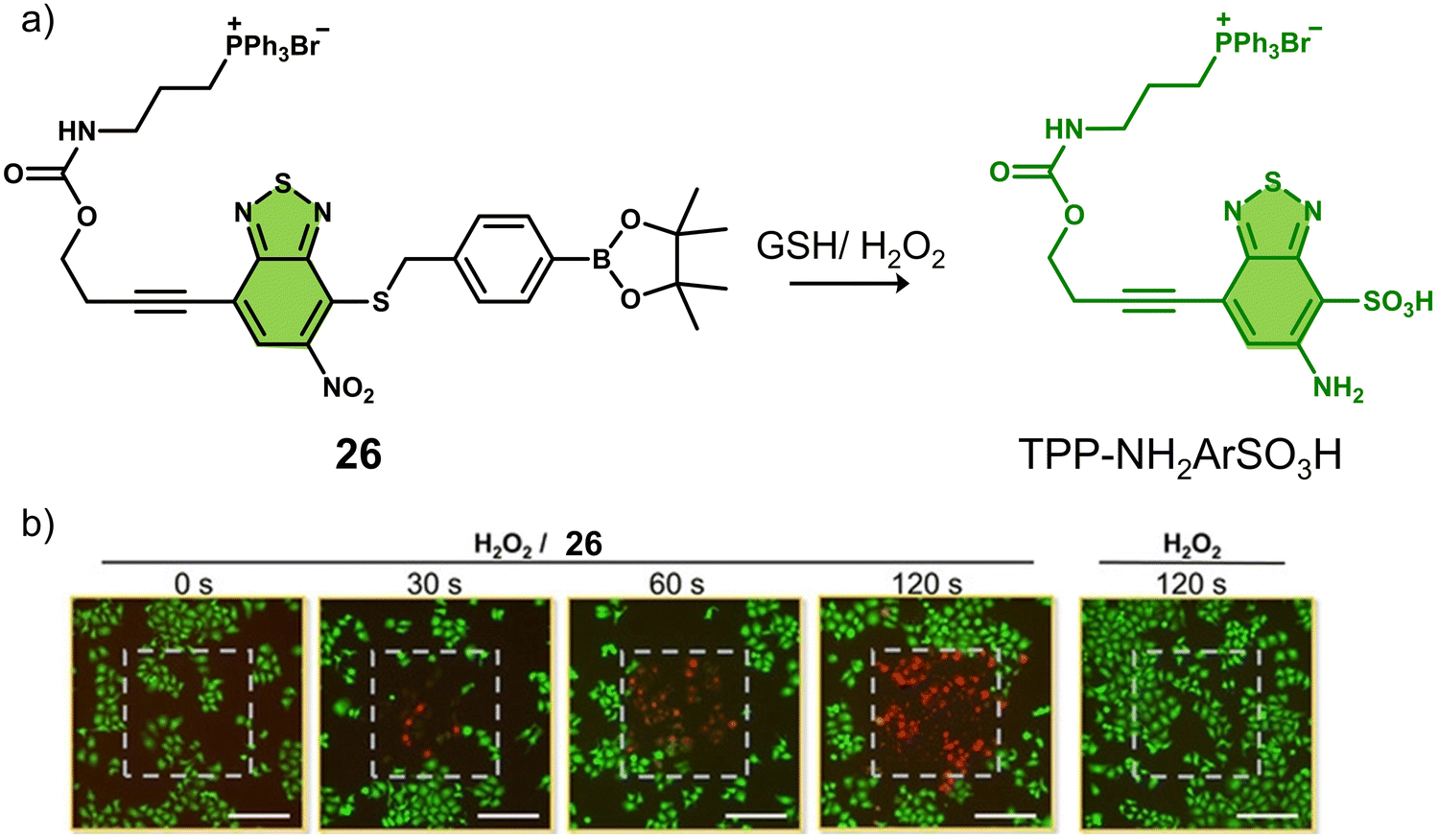 | ||
| Fig. 15 (a) The chemical structure of 26 and the H2O2/GSH -activatable TPP-NH2ArSO3H release mechanism. (b) Live/dead staining of HeLa cells that were pretreated with H2O2 (50 μM) followed by treated with 26 (0 or 10 μM). The cells were irradiated by a two-photon laser (λ = 800 nm) for 0, 30, 60, and 120 s. The laser irradiation area (400 μm × 400 μm) in each image was labeled with a gray dashed square. Scale bars: 200 μm. Reproduced with permission from ref. 108. Copyright (2020) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Nearly 50% of cancer patients with solid tumours will receive platinum (Pt)-based therapy.111 The Pt(II)-based therapeutics that have been approved to treat cancers include cisplatin, carboplatin, nedaplatin, oxaliplatin, and lobaplatin. Unfortunately, their poor tumour specificity leads to dose-limiting toxicities and a narrow therapeutic index, severely limiting their clinical efficacy. In recent years, researchers have focused on overcoming these limitations by developing Pt(IV)-based prodrugs. The key advantage of using Pt(IV)-based prodrugs is their relative non-toxic nature until reduced intracellularly to their cytotoxic Pt(II) form (see Fig. 16). In addition, the ease of modification of the axial ligands allows the ability to tailor the pharmacological properties of these molecules. For an extensive overview on platinum-based therapies, the reader can turn to an excellent review by Lippard and co-workers.112
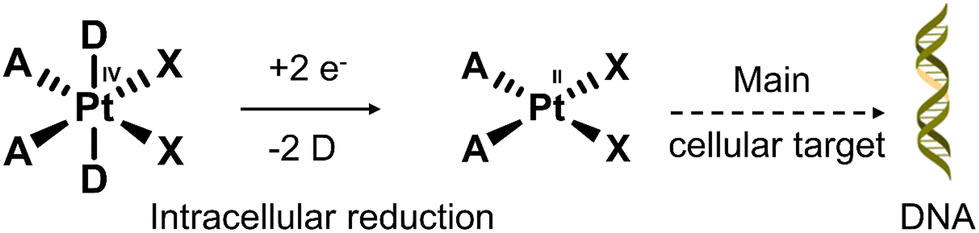 | ||
| Fig. 16 The conversion mechanism of the prodrug Pt(IV) complex to the Pt(II) complex under a reducing environment. D = axial ligands that modulate the lipophilicity and the optimal redox properties; X = leaving groups that control the rate of aquation and stability in the biological medium; A = non-leaving groups that are closely related to the antiproliferative activity. | ||
In 1988, the Sessler group introduced the world to “texaphyrins” which are penta-aza Schiff base expanded porphyrins, that form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with an extensive array of functional metal cations.52,113 Notably, the paramagnetic gadolinium(III) metallotexaphyrin (MGd) was found to be more reducible than porphyrins, endowing its use as a redox mediator. Furthermore, in recent studies, the ability of MGd to catalyse the intracellular reduction of oxaliplatin-based Pt(IV) prodrugs to cytotoxic Pt(II) has been confirmed, i.e., oxaliplatin.114 This led to the construction of a series of Pt(IV)-based texaphyrin conjugates, namely 27, 28, 29.115 It was rationalised that the tumour-localising properties of MGd would enable the effective delivery of Pt(IV) and selective reduction of Pt(IV) to Pt(II) in solid tumours. These candidates consisted of an MGd, which is conjugated to oxaliplatin Pt(IV)-derivatives via a succinate linker (Fig. 17a). Each compound differed by the axial ligand (L = Cl, 27; OAc, 28; OH, 29), which plays a key role in the rate of reduction and hydrolysis of the complexes. 28 was identified as the most stable candidate, and the reduction of Pt(IV) was shown by the reverse-phase (RP)-HPLC. In vitro analysis indicated that 28 exhibited a broad antiproliferative activity compared to oxaliplatin across several cell lines, including platinum-resistant ones. This broad anticancer effect translated to A549, A2780 (ovarian cancer), and HCT116 (human colon cancer cells) tumour-bearing mice and syngeneic tumour (CT26, murine colorectal carcinoma cells; EMT6, murine mammary carcinoma cells) mice models. 28 (70 mg kg−1 per dose on days 1, 5, 9, 13) via tail vein injection of mice with patient-derived xenografts (PDXs) was shown to reduce the tumour volume significantly when compared to vehicle and oxaliplatin (4 mg kg−1 per dose on days 1, 5, 9, 13) (Fig. 17b). Remarkably, these results confirmed that 28 is more effective in retarding and inhibiting tumour growth than the most closely related platinum-based treatments currently available. Which clearly illustrates the promise of Pt(IV)-based prodrugs for potential clinical translation.
:1 complexes with an extensive array of functional metal cations.52,113 Notably, the paramagnetic gadolinium(III) metallotexaphyrin (MGd) was found to be more reducible than porphyrins, endowing its use as a redox mediator. Furthermore, in recent studies, the ability of MGd to catalyse the intracellular reduction of oxaliplatin-based Pt(IV) prodrugs to cytotoxic Pt(II) has been confirmed, i.e., oxaliplatin.114 This led to the construction of a series of Pt(IV)-based texaphyrin conjugates, namely 27, 28, 29.115 It was rationalised that the tumour-localising properties of MGd would enable the effective delivery of Pt(IV) and selective reduction of Pt(IV) to Pt(II) in solid tumours. These candidates consisted of an MGd, which is conjugated to oxaliplatin Pt(IV)-derivatives via a succinate linker (Fig. 17a). Each compound differed by the axial ligand (L = Cl, 27; OAc, 28; OH, 29), which plays a key role in the rate of reduction and hydrolysis of the complexes. 28 was identified as the most stable candidate, and the reduction of Pt(IV) was shown by the reverse-phase (RP)-HPLC. In vitro analysis indicated that 28 exhibited a broad antiproliferative activity compared to oxaliplatin across several cell lines, including platinum-resistant ones. This broad anticancer effect translated to A549, A2780 (ovarian cancer), and HCT116 (human colon cancer cells) tumour-bearing mice and syngeneic tumour (CT26, murine colorectal carcinoma cells; EMT6, murine mammary carcinoma cells) mice models. 28 (70 mg kg−1 per dose on days 1, 5, 9, 13) via tail vein injection of mice with patient-derived xenografts (PDXs) was shown to reduce the tumour volume significantly when compared to vehicle and oxaliplatin (4 mg kg−1 per dose on days 1, 5, 9, 13) (Fig. 17b). Remarkably, these results confirmed that 28 is more effective in retarding and inhibiting tumour growth than the most closely related platinum-based treatments currently available. Which clearly illustrates the promise of Pt(IV)-based prodrugs for potential clinical translation.
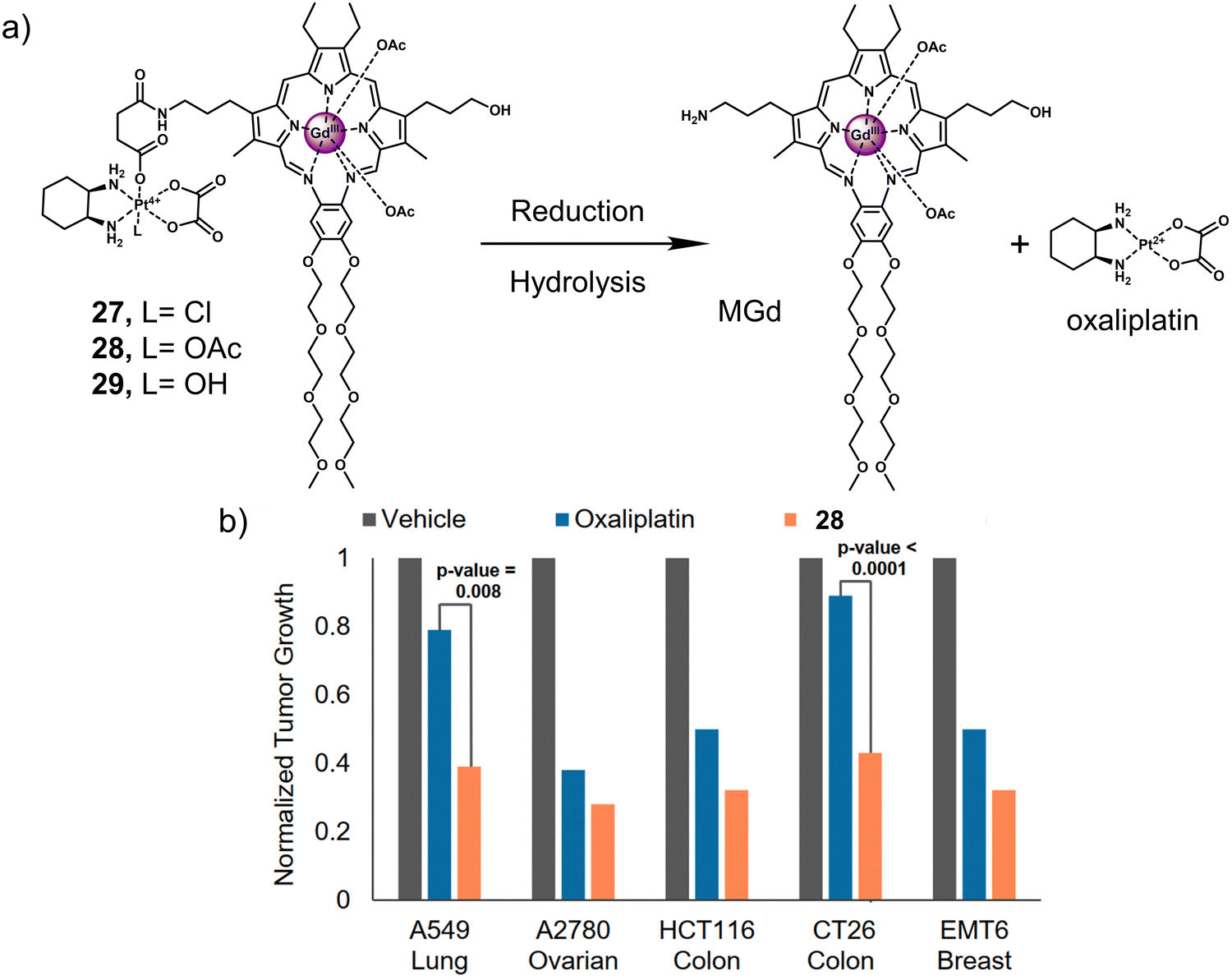 | ||
| Fig. 17 (a) Chemical structure of 27, 28, 29 and the release of therapeutic oxaliplatin and MGd under the sequential action of reduction and hydrolysis. 27, L= Cl; 28, L= OAc; 29, L= OH. (b) In vivo efficacy of 28 (70 mg kg−1 per dose on days 1, 5, 9, 13) vs. oxaliplatin (4 mg kg−1 per dose on days 1, 5, 9, 13) in A549, A2780 and HCT116 tumour-bearing mice models and syngeneic tumour (CT26, EMT6) mice models. The study endpoint for the A549 model was 30 d. The study endpoint for all other models was the day at which the vehicle-treated mice reached maximum tumour burden. Reproduced with permission from ref. 115. Copyright (2020) Published by National Academy of Sciences. | ||
Another two gadolinium-based theranostics, 30 and 31, were reported for tandem MR imaging of Pt(IV) reduction in A2780 and HeLa cells (Fig. 18).116 The Gd(III)-DOTA MR contrast agent was conjugated to Pt(IV) cisplatin and carboplatin derivatives by the axial ligand to afford 30 and 31, respectively. The relaxivity (r1 and r2) of both 30 and 31 was greater than that of the Gd(III) MR contrast agent, Gd-1, due to an increase in rotation-related time (τR) and change in the inner sphere hydration number (q) of Gd(III). Excellent stability was observed in the absence of reductants. However, the relaxivity (r1) of both 30 and 31 decreased significantly and converged with that of the parent scaffold Gd(III) MR contrast agent Gd-1 when GSH (5 mM) was added. This resulted in the conversion of both 30 and 31 to carboxylic-derived monomeric Gd(III) complexes Gd-1 and the active Pt(II) analogues, confirmed by HPLC analysis. This apparent change in r1 before and after the reduction of 30 and 31 enabled the monitoring of intracellular reduction of the Pt(IV) derivatives. Higher IC50 values were observed for 30 and 31 compared to cisplatin and carboplatin, respectively. This was ascribed to poor cell permeability. However, it is believed that higher concentrations of 30 and 31 can be used to address the lower cytotoxicity. In addition, the cell impermeable Gd(III) complex Gd-1 released by intracellular degradation of 30 and 31 can be used as a contrast agent for MR imaging. An increased r1 was observed in 30-treated A2780 cells and in HeLa cells compared to the untreated control group. This example demonstrates the ability to use MRI to visualise and monitor the release of Pt(IV) derivatives in cells.
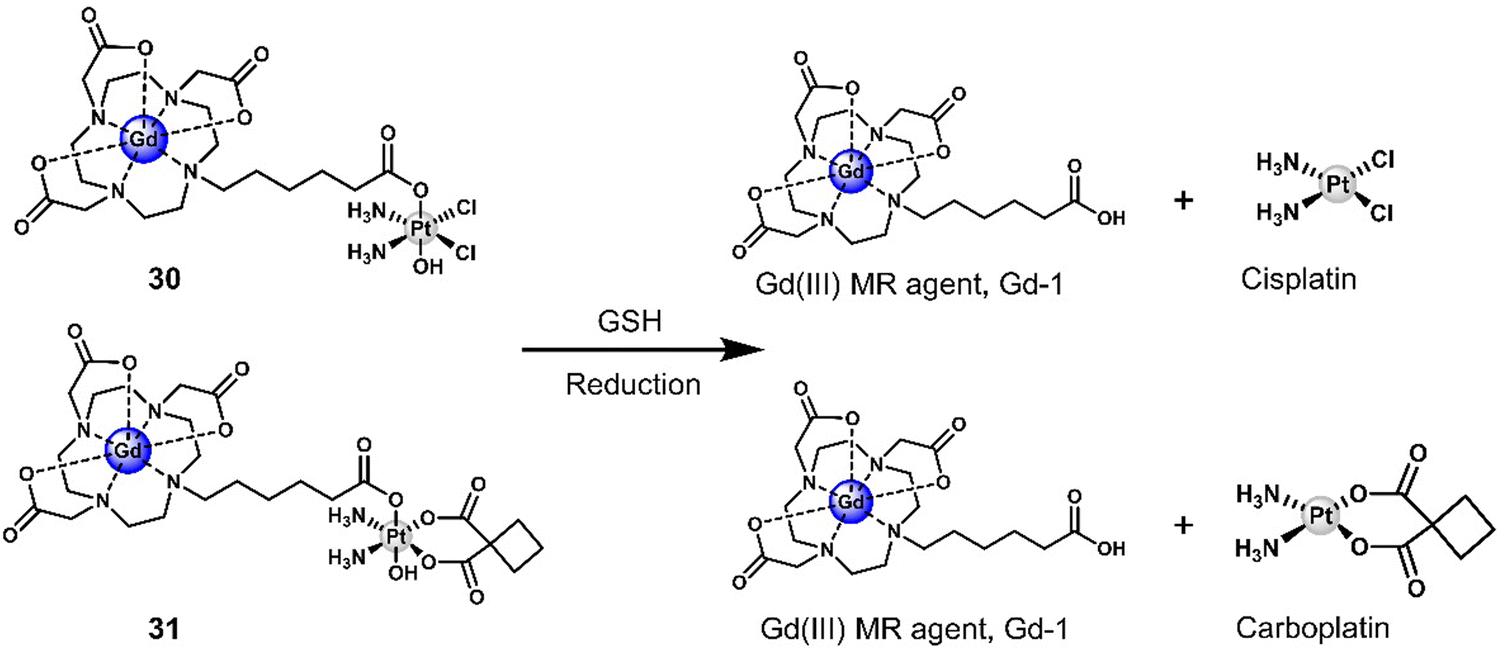 | ||
| Fig. 18 Chemical structure of 30, 31, and the GSH-responsive release of therapeutic Pt(II) drug (cisplatin and carboplatin) and a Gd(III) MR contrast agent, Gd-1. DOTA: 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetic acid. | ||
3.3 Hypoxia-responsive prodrugs and phototherapeutics
Hypoxia is a common feature of all solid tumours, which arises from an inadequate blood supply within tumour tissue affording low oxygen environments. This observation is exacerbated with tumour growth as oxygen supply is unable to meet the metabolic demands of tumour cells, thus causing the intratumoural microenvironment to exhibit a significant hypoxic phenotype.117,118 It is well-established that the degree of hypoxia is correlated with the local concentration of reducing enzymes, such as nitroreductase (NTR) and azoreductase. These enzymes are known for reducing nitro-aromatic and azoaromatic-derivatives, respectively. Researchers have therefore exploited nitroaromatics and azoaromatic units as hypoxia-based protecting groups to develop prodrugs that are activated in the hypoxic environment of tumours (Scheme 4) (Table 3).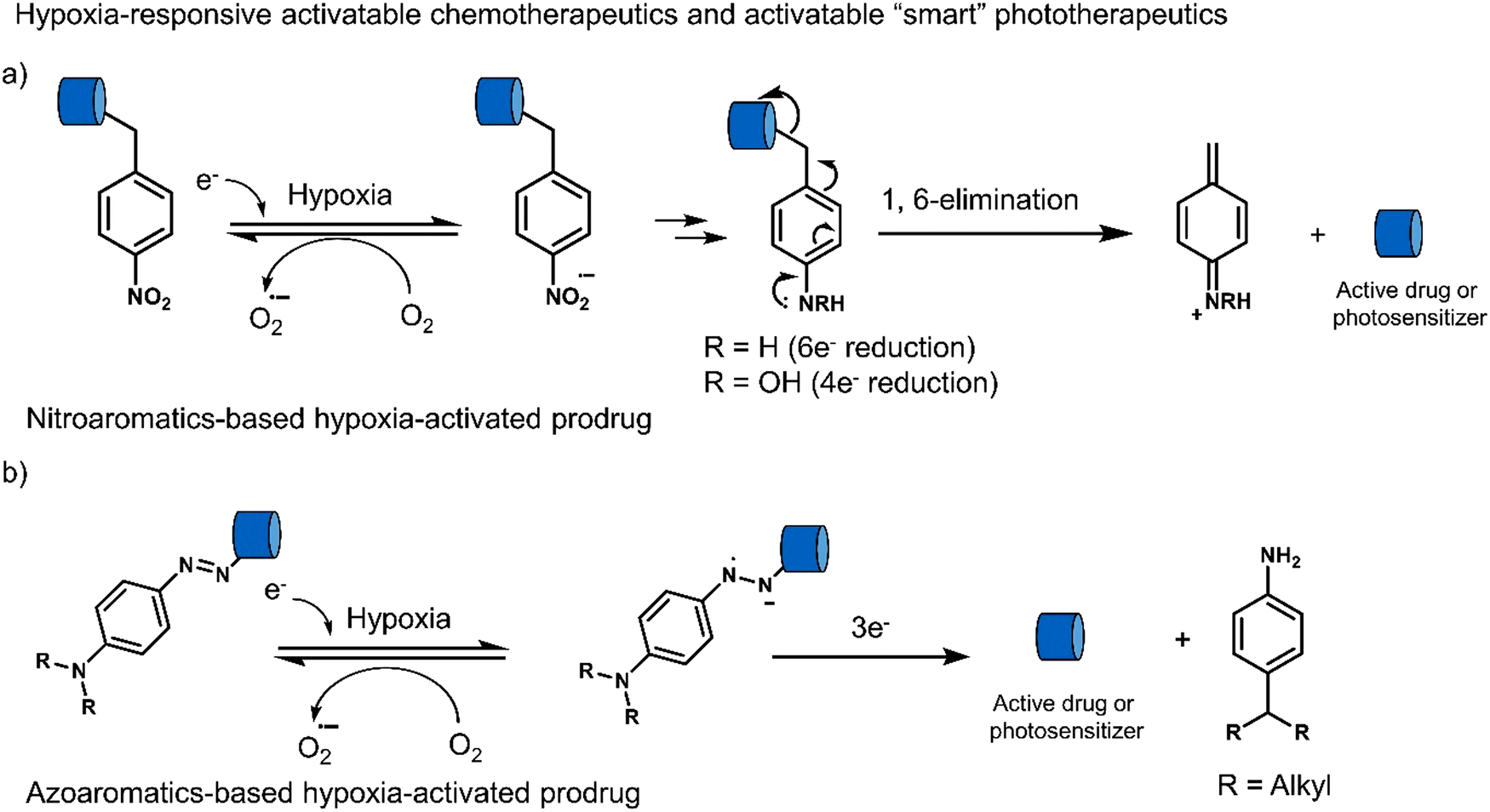 | ||
| Scheme 4 Basic mechanism of nitroaromatic-based and azoaromatic-based prodrugs activation. | ||
| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: FDU: 5-fluorodeoxyuridine, PPA: pyropheophorbide α, SN-38: the active metabolite of camptothecin, PDT: photodynamic therapy, PTT: photothermal therapy, —: not mentioned. | ||||||
| 32 | Hypoxia | FDU | Chemotherapy | MGC-803 and MCF-7 cells | MCF-7 tumour-bearing mice | 119 |
| 33 | Hypoxia | Iodinated heptamethine cyanine | PTT | HeLa, HepG2, and A549 cells | HeLa tumour-bearing mice | 120 |
| 34 | Hypoxia | 3,4-Difluorobenzyl curcumin agent | Chemotherapy | CD133+ MDA-MB-231 cells and tumour spheroids | MDA-MB-231 tumour-bearing mice | 122 |
| 36 | Hypoxia | Panobinostat | Chemotherapy | OE21 and HCT116 cells | OE21 tumour-bearing mice | 124 |
| 37 | Hypoxia | IR-1048 | PTT | — | A549 tumour-bearing mice | 129 |
| 38 | Hypoxia | SN-38 | Chemotherapy | A549, HeLa, HepG2, MCF-7, and MDA-MB-231 cells | 4T1 tumour-bearing mice | 130 |
| 39 | Hypoxia | Seleno-rosamine scaffold | PDT | A549 cells | — | 131 |
| 40 | Hypoxia | PPA | PDT | BEL-7402 cells | — | 132 |
A particularly illustrative example is the chemotherapeutic 32 developed by Liu et al.,119 which contains a hypoxia-responsive 4-nitrobenzyl protecting group, 4′-(diethylamino)-1,1′-biphenyl-2-carboxylate linker (and fluorescent reporter), and the chemotherapeutic 5-fluorodeoxyuridine (FDU). FDU is commonly used to treat colorectal cancer as well as kidney and stomach cancers through the inhibition of thymidylate synthase, and ultimately the synthesis of the DNA. As shown in Fig. 19a, the nitroaromatic of 32 was reduced by the nitroreductase to release FDU and 4′-(diethylamino)-1,1′-biphenyl-2-carboxylate, which isomerises to afford fluorescent 7-(diethylamino)coumarin (CM). The reaction progress was confirmed via an increased fluorescence emission at 530 nm and HPLC studies. In vitro studies of 32 were carried out in MGC-803 (human gastric carcinoma) and MCF-7 (human breast cancer cell line) cancer cell lines under normoxic and hypoxic conditions, in which the antiproliferative effect of 32 was found to be higher in both cell lines under hypoxic conditions. Compared to FDU, the prodrug 32 exhibited minimal cytotoxicity to a normal cell line (BRL-3A, a rat liver-derived cell line), suggesting the potential to overcome the known off-target toxicities of FDU. The treatment of MCF-7 tumour-bearing mice with 32 (10 mg kg−1 once every four days for 12 days) was shown to reduce the tumour size compared to controls (Fig. 19b and c). The tumour reduction volume induced by 32 was 86% greater than that of the control group (saline).
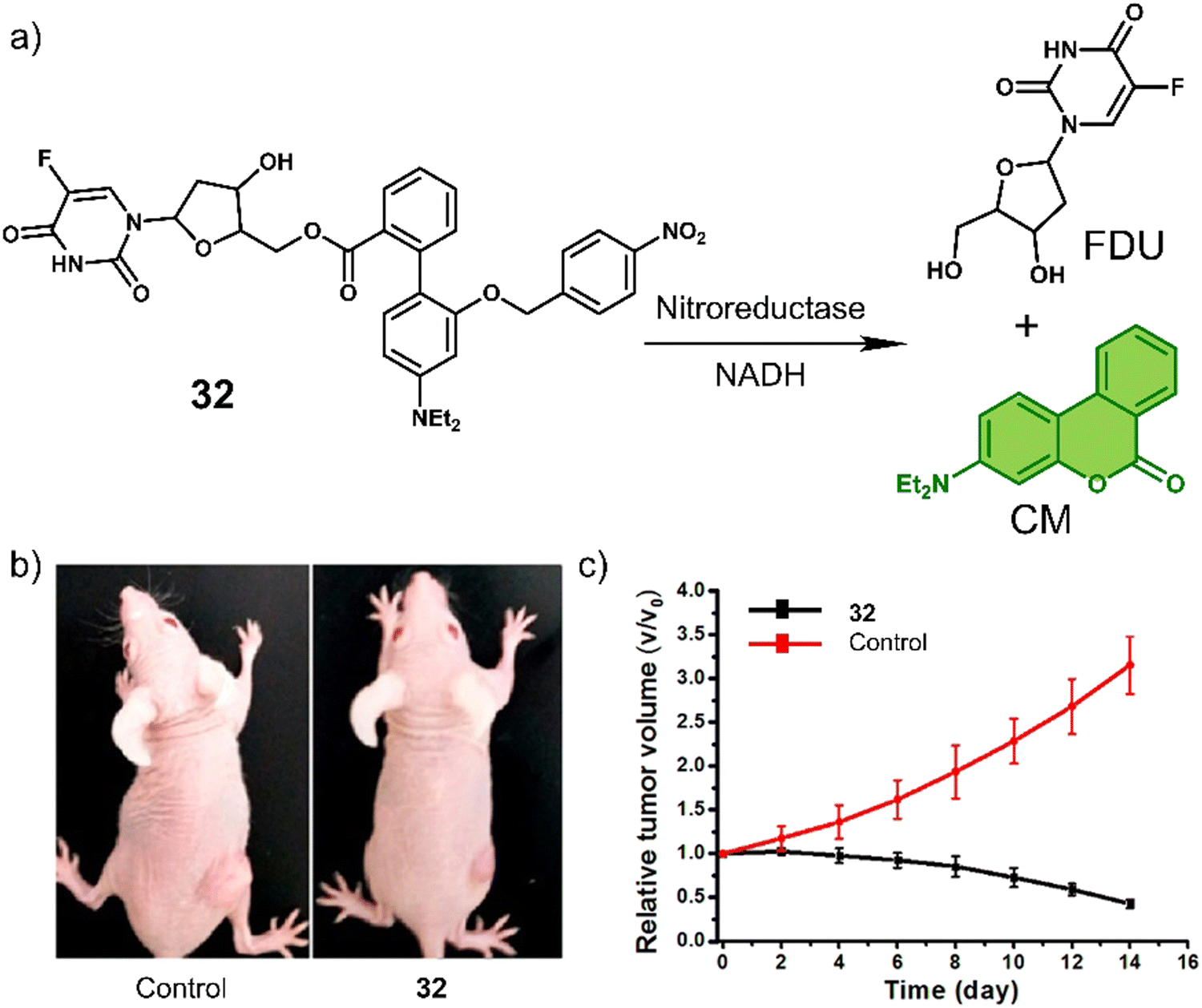 | ||
| Fig. 19 (a) Chemical structure of 32 and the hypoxia-induced release of FDU and 7-(diethylamino)coumarin (CM). (b) Representative images of MCF-7 tumour-bearing mice after treatment with the control group (saline) and 32. (c) Tumour growth inhibition over the time with the control group (saline) and 32 (10 mg kg−1). Reproduced with permission from ref. 119. Copyright (2018) American Chemical Society. | ||
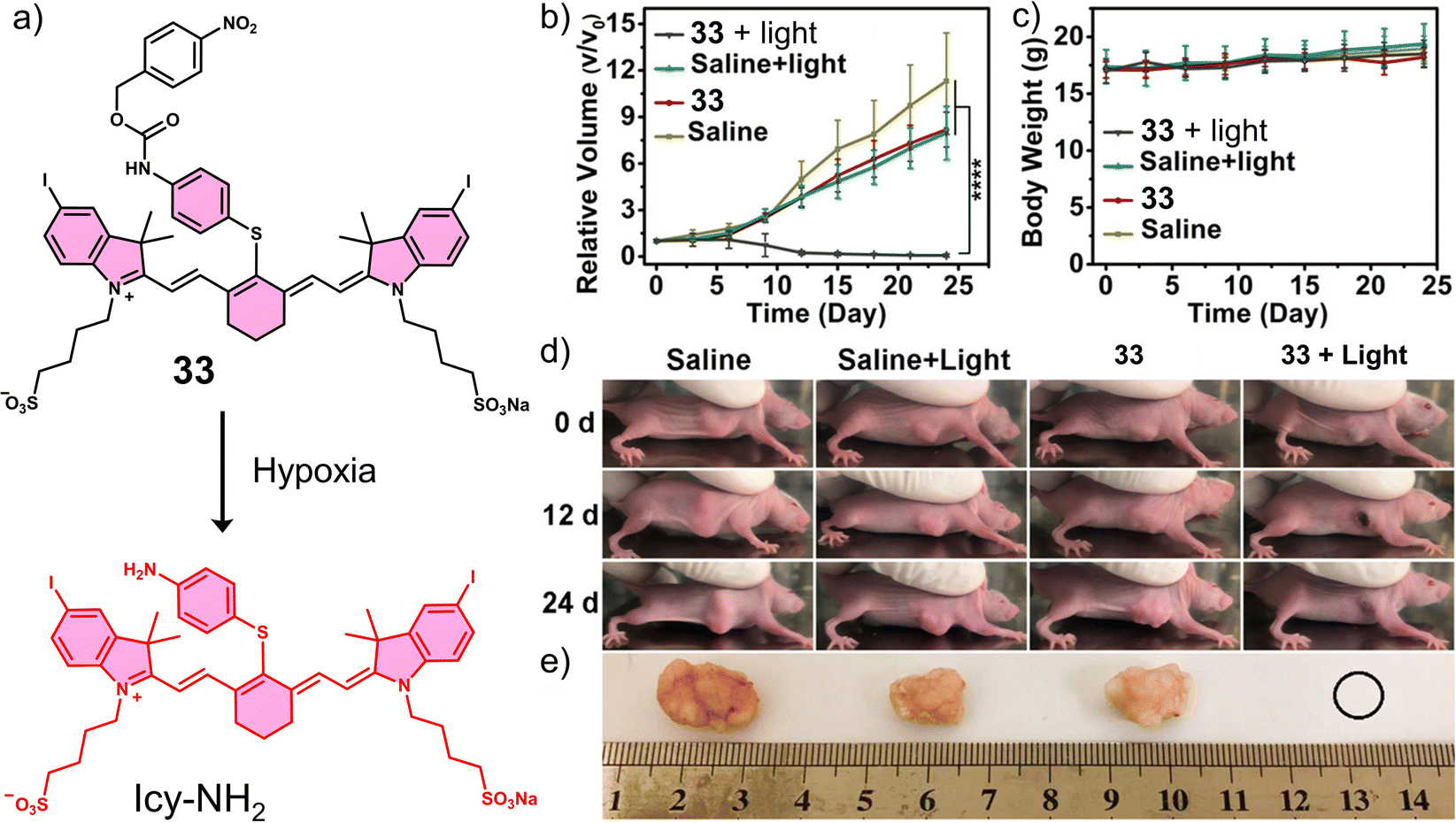
In another study, the nitrobenzyl hypoxia release trigger was used in the construction of an activatable phototherapeutic. Peng and co-workers developed a hypoxia-activatable PTT agent, 33 (Fig. 20a), for the PTT treatment of HeLa tumour-bearing mice models.120 An interesting aspect to the design of this system is that it can convert from a PDT agent to a PTT agent under hypoxic conditions, thus overcoming the oxygen requirement of PDT. 33 exploited the hypoxia-responsive 4-nitrobenzyl group and an iodinated heptamethine cyanine dye as the PS. In the presence of NADH and NTR, the nitroaromatic unit was reduced, followed by the release of Icy-NH2, which was confirmed via fluorescence and HRMS studies. Both 33 and Icy-NH2 exhibited high molar extinction coefficients (2.35 × 105 M−1 cm−1 and 2.21 × 105 M−1 cm−1, respectively) at 837 and 835 nm, respectively. Initially, 33 displayed a high 1O2 quantum yield and high fluorescence emission at 837 nm. However, upon being converted to Icy-NH2, the 1O2 quantum yield and fluorescence emission decreased significantly, and the non-radiative relaxation pathway was enhanced for efficient heat production. Therefore, as the surrounding oxygen environment changed, the therapeutic function of 33 was switched from PDT to PTT under 808 nm light irradiation. 33 exhibited high cytotoxicity upon 808 nm light (240 J cm−2) under normoxic conditions via PDT and under hypoxic conditions via PTT. In vivo experiments using HeLa tumour-bearing mice models revealed the fluorescence intensity of 33 was inversely proportional to the level of NTR activity and temperature. The 808 nm light irradiation (0.6 W cm−2, 7 min) of 33 was shown to increase the temperature of the tumour to 60 °C, which reduced the tumour size significantly compared to controls (Fig. 20b–d). This work demonstrates a unique strategy that exploits the advantageous properties of both PDT and PTT and serves as a new approach for the development of intelligent phototherapeutic agents that maximise photon efficiency.
 | ||
| Fig. 20 (a) Chemical structure of 33 and its reduction mechanism under hypoxic environment to afford Icy-NH2. (b) Relative tumour volume and (c) Body weights of HeLa tumour-bearing mice after the treatment of saline or 33 (15 nM, 100 μL) with or without 808 nm irradiation (252 J cm−2, 7 min) once. ****P < 0.0001. (d) Images of HeLa tumour-bearing mice of (b) at 0, 12, and 24 days post-treatment. (e) Images of tumours harvested from (d) at 24 days post-treatment. Reproduced with permission from ref. 120. Copyright (2020) American Chemical Society. | ||
Cancer stem cells (CSCs) contribute to tumour regeneration, metastasis, and recurrence. CSCs have therefore been a focal point of cancer research over the last decade, with several reports on their detection and treatment.121 Breast CSCs have been associated with tumourigenesis, chemoresistance, and poor patient prognosis. For these reasons, the hypoxia-responsive prodrug 34 was developed for the treatment of breast cancer stem cells (CSCs) (Fig. 21a).12234 was functionalised with the carbonic anhydrase IX (CAIX) inhibitor acetazolamide (Az), the hypoxia-responsive dimethylnitrothiophene unit, and the experimental anti-CSC drug 3,4-difluorobenzyl curcumin (CDF). Dimethylnitrothiophene has a lower reduction potential than the more widely used nitrobenzyl hypoxia release trigger. Furthermore, the presence of dimethyl results in favourable reaction kinetics. Additionally, by replacing the CDF group with a naphthalimide fluorophore, the corresponding fluorescent analogue 35 was constructed, which could release the fluorophore in a similar manner to 34 for the targeted imaging of CSCs. Az was used as a targeting unit for CSCs (MDA-MB-231, human breast cancer cell line) and an inhibitor for CSC proliferation.123 In solution studies, UV/vis spectroscopy and HPLC analysis, CDF was shown to be released from 35 in the presence of E. coli NTR and NADH. Under the same conditions, the fluorescence intensity of 35 increased at 550 nm using fluorescence microscopy. The fluorescence of 35 could be activated in CD133+ (a biomarker of cancer stemness) MDA-MB-231 cells but not in BJ cells (CAIX negative) under mild hypoxic conditions (3% O2), and the fluorescence was suppressed in CAIX knock-down CD133+ MDA-MB-231 cells or under normoxic conditions. The dimethylnitrothiophene-based hypoxia-sensitive group was readily activated under 3% O2, with significant cytotoxicity observed for CD133+ MDA-MB-231 cells under hypoxic conditions. Also, similar cytotoxicity was observed using a tumour spheroid in vitro assay (CD133+ MDA-MB-231 tumour spheroids). Single tail-vein injection of 35 in MDA-MB-231 tumour-bearing mice was shown via fluorescence imaging (Maestro™ In Vivo Fluorescence Imaging System) to predominantly accumulate at the tumour site over 144 hours. Besides, the tail vein administration of 34 (once a week for 3 weeks at a dose of 50.0 nM in 100 μL of PBS) resulted in a significant reduction in tumour volume compared to control groups (R-Az, without drug CDF; R-CDF, without targeting group Az) (Fig. 21b). Confirming the effectiveness of 34 to target CSCs, CD133+ MDA-MB-231 cells, and nude mice injected with 34-treated CD133+ MDA-MB-231 cells under hypoxic conditions exhibited suppressed tumourigenesis. However, for mice injected with DMSO-treated cells rapid tumour growth was observed, which demonstrated that 34 could be used to overcome tumourigenesis associated with CSCs.
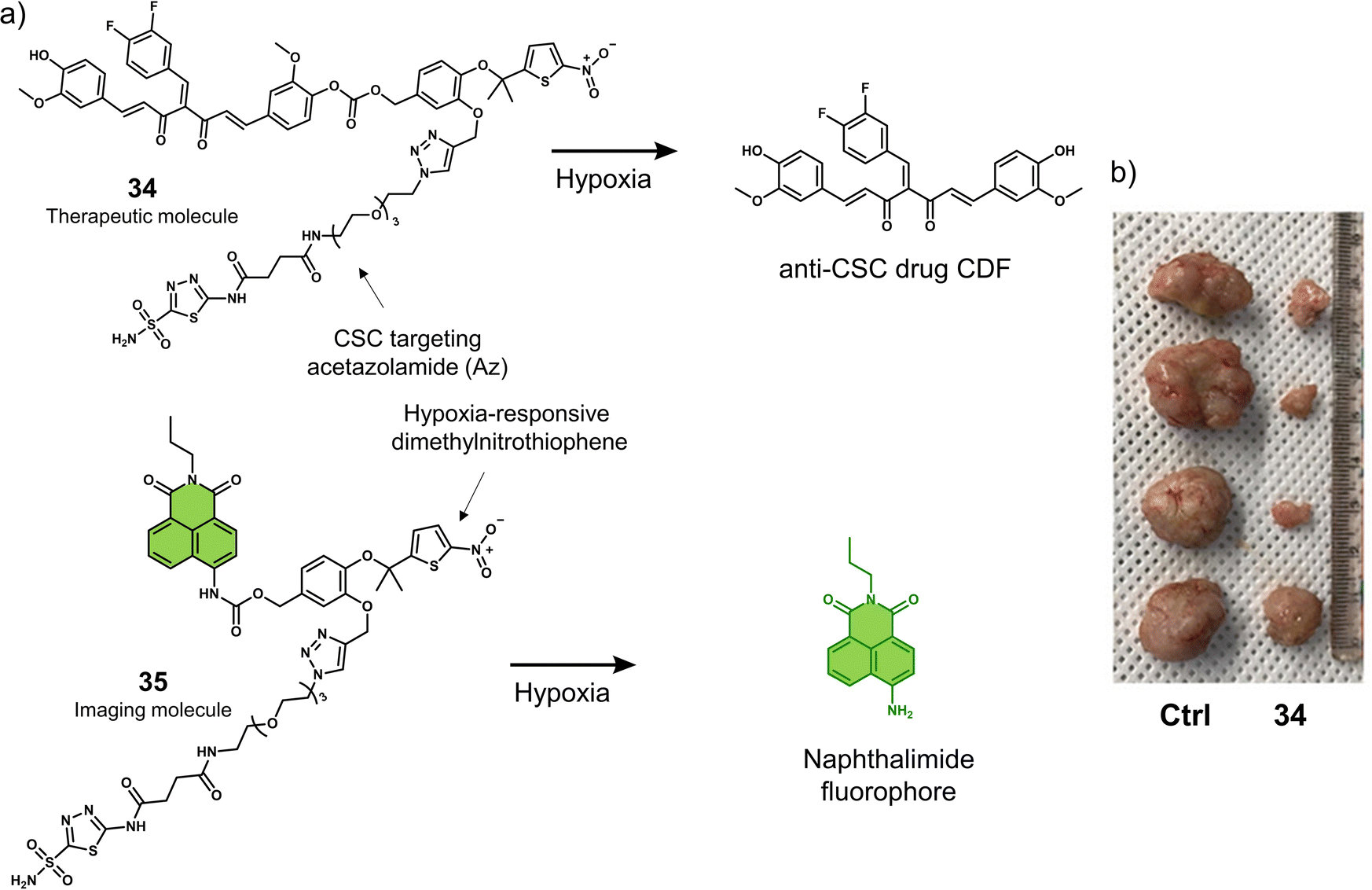 | ||
| Fig. 21 (a) Chemical structures of 34 and 35 and the reaction mechanism under hypoxic conditions. (b) Representative images of excised tumours from MDA-MB-231 tumour-bearing mice treated with control (DMSO), and 34 (50.0 nM in 100 μL of PBS) once per week for 3 weeks. Reproduced with permission from ref. 122. Copyright (2021) American Chemical Society. | ||
Nitroimidazole groups have also been incorporated into activatable chemotherapeutics. Skwarska et al. functionalised the lysine deacetylase (KDAC) inhibitor panobinostat with 1-methyl-2-nitroimidazole-unit to develop a hypoxia-responsive prodrug, 36 (Fig. 22a).124 KDACs are enzymes that promote tumour malignancy by deacetylating lysine residues found on histones or non-histones. Overexpressed class I KDACs are found in various cancer types (e.g., lung, breast, pancreatic, and gastric cancer) and are associated with poor prognosis.125–127 Several KDAC inhibitors have been approved for the treatment of hematological malignancies, such as panobinostat.12836 was found stable under normoxic conditions and displayed little inhibitory activity against KDAC. LC-MS analysis revealed the release of panobinostat when 36 was exposed to NADPH-CYP reductase (CYP004) under hypoxic conditions (<0.1% O2). 36 exhibited a higher antiproliferative effect in OE21 and HCT116 cancer cell lines under hypoxic conditions (<0.1% O2) than under normoxic (21% O2) conditions (Fig. 22b). In addition it exhibited excellent growth inhibition of HCT116 tumour spheroids. The treatment of OE21 tumour-bearing mice with 36 (50 mg kg−1, three doses on days 1, 3 and 5) was shown to reduce the tumour size compared to the control (vehicle) and significantly increased the survival (range 8–28 days) of the mice evaluated.
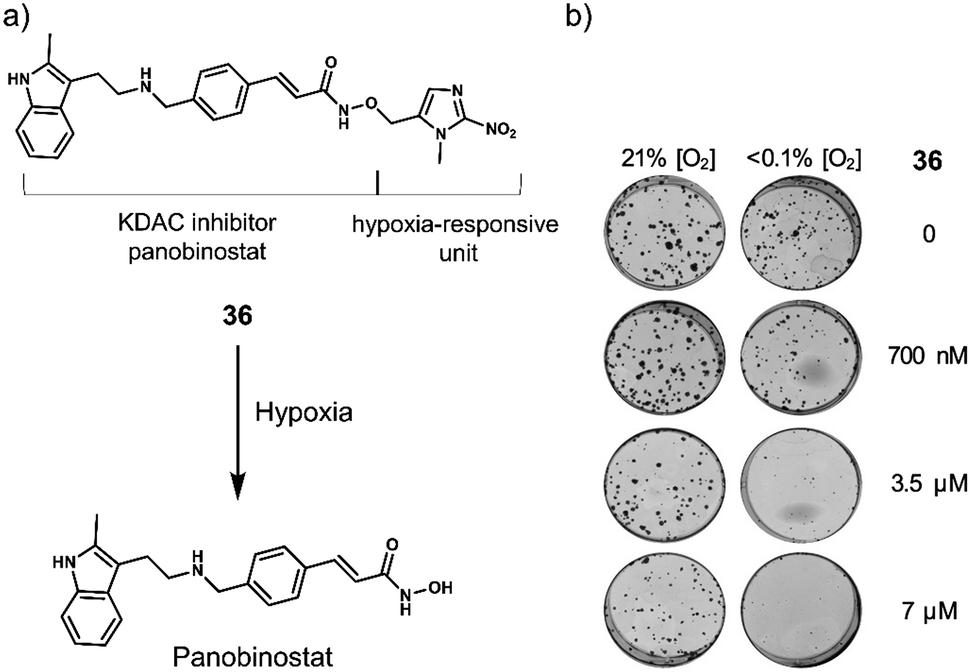 | ||
| Fig. 22 (a) Chemical structure of 36 and the hypoxia-mediated release of the KDAC inhibitor panobinostat. (b) Images of OE21 cells treated with 36 (0–7 μM) under hypoxic (<0.1% O2) and normoxic (21% O2) conditions for 24 hours, after which 36 was removed, and the cells were allowed to form colonies for 7 days under normoxic conditions. Reproduced with permission from ref. 124. Copyright (2021) Elsevier Inc. | ||
Meng et al. reported a hypoxia-responsive PTT agent, 37, that contains an nitroimidazolyl scaffold, for the near-infrared II (NIR-II) fluorescence/PA imaging and PTT treatment of A549 tumour-bearing mice models (Fig. 23).12937 was synthesised through a simple one-step conjugation reaction between 2-(2-nitroimidazolyl)ethylamine and commercially available NIR-II dye, IR-1048. It displayed minimal NIR-II fluorescence and had an absorption in the range of 900–1060 nm. Upon activation by nitroreductase (NTR) and NADH, 37 produced a NIR-II fluorescence emission at 1046 nm, an increase in photoacoustic intensity, and a high photothermal conversion efficiency (20.2%). In vivo studies using an A549 tumour-bearing mice model revealed strong NIR-II fluorescence emission and PA signal at the tumour site. This was indicative of hypoxia-mediated activation of IR1048-MZ. Remarkably, a rapid temperature increase from 30 °C to 58 °C and complete tumour ablation was observed with no tumour recurrence being seen after 30 days.
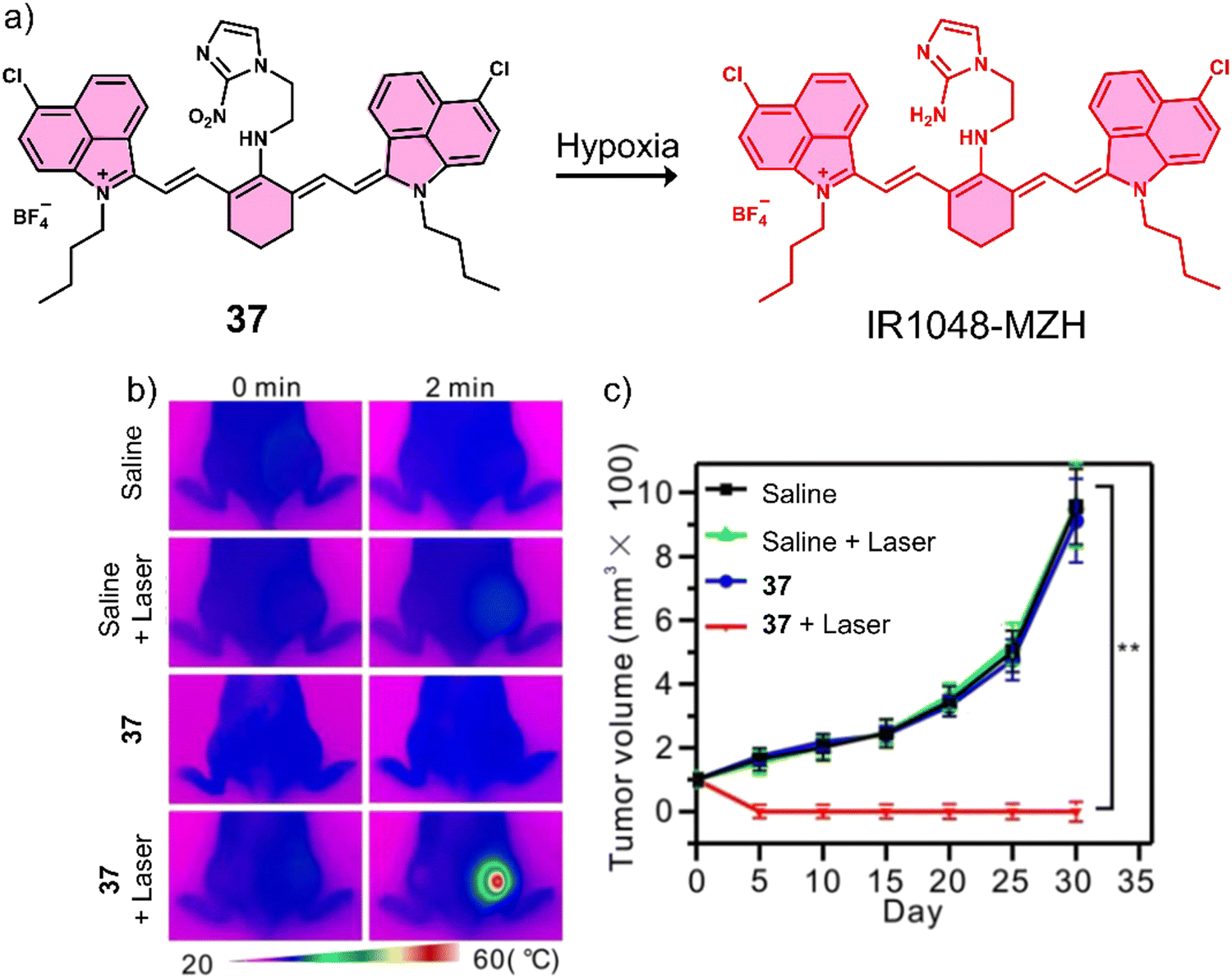 | ||
| Fig. 23 (a) Chemical structure of 37 and the phototheranostic properties under hypoxic conditions in tumour cells. (b) Infrared thermal imaging of the A549 tumour-bearing mice injected with saline or 37 with or without 980 nm NIR laser irradiation (0.1 W cm−2, 2 min). (c) The A549 tumour growth curves over 30 days after treatment with saline or 37 (200 μL, 40 μg mL−1) with or without NIR laser irradiation (980 nm, 0.1 W cm−2, 2 min). n = 5, **P < 0.01. Reproduced with permission from ref. 129. Copyright (2018) Published by Ivyspring International Publisher. | ||
Motivated to exploit the hypoxic environment of solid tumours, Zhou et al. developed the theranostic 38 that employs an aryl-azo benzyl alcohol motif to mask both the pharmacological activity and fluorescence emission of SN-38 (Fig. 24a).130 Under hypoxic conditions, the addition of rat liver microsomes (0–240 μg mL−1) to an aqueous solution of 38 (0.1 mM NADH as a coenzyme) resulted in an increase in fluorescence intensity at 560 nm, which was ascribed to the release of SN-38 (confirmed by MS-spectroscopic analysis). Fluorescence microscopy was used to visualise the release of SN-38 in HeLa cells under glutathione ethyl ester-induced hypoxic conditions and in living C. elegans. 38 exhibited selective toxicity for several cancer cell lines (A549, HeLa, HepG2, MCF-7, and MDA-MB-231) over normal cells (WI-38, human embryonic fibroblasts and BJ, human skin fibroblasts) under hypoxic conditions. Good stability was observed for 38 under normoxic conditions. The tail vein injection of 38 (10 μM, 20 μM in 100 μL PBS, respectively, twice a week for three weeks) in the murine mammary carcinoma 4T1 tumour-bearing mice model exhibited a dose-dependent reduction in tumour growth compared to the control group (Fig. 24b and c). This work demonstrated the utility of the aryl-azo benzyl alcohol motif for the development of hypoxia-responsive prodrugs.
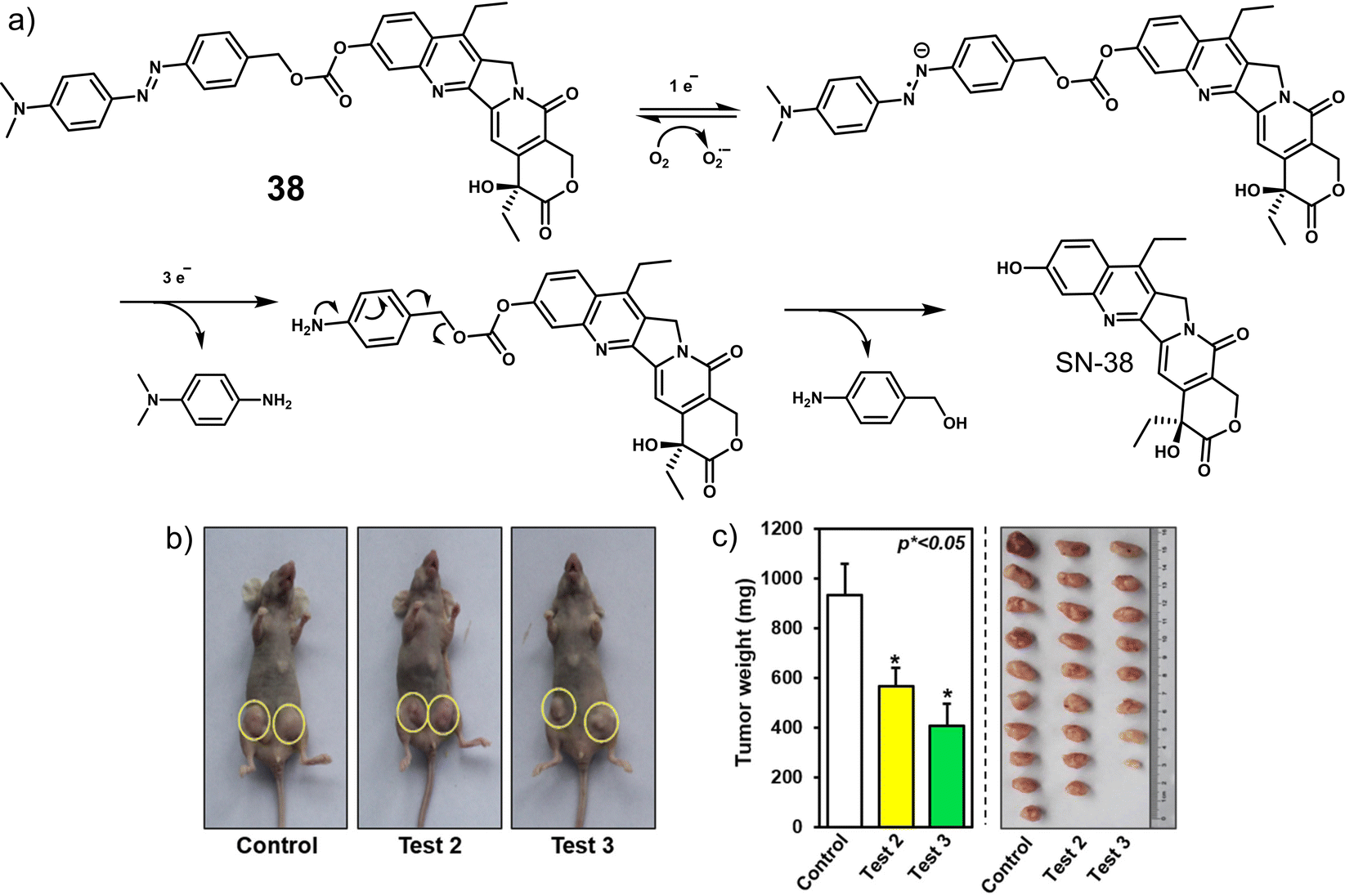 | ||
| Fig. 24 (a) Chemical structure of 38 and the mechanism for the hypoxia-activated release of SN-38. (b) Photographic images of tumour-bearing xenograft mice (n = 11 per test group) and (c) Tumours weight with dissected tumour tissue treated with PBS, test 2 (38, 10 μM, 100 μL PBS) or test 3 (38, 20 μM, 100 μL PBS) via tail vein twice weekly for three weeks. Reproduced with permission from ref. 130. Copyright (2018) Elsevier B.V. | ||
Piao et al. reported the hypoxia-responsive PDT agent, 39 (Fig. 25a), for the selective PDT treatment of A549 cancer cells under hypoxia.131 A seleno-rosamine scaffold (SeR) was used as the fluorophore as well as the photosensitizer with a high 1O2 quantum yield (Φ1O2= 0.56). The amino unit was functionalised with an azo group to afford hypoxia-responsive 39. This azo unit served to quench the fluorescence emission at 557 nm and 1O2 production at 532 nm. Under hypoxic conditions, 39 was shown to be readily reduced by NADPH (50 μM) rat liver microsomes to afford fluorescent SeR. The phototoxicity of 39 was measured using a live/dead cell viability assay with ethidium homodimer (EthD, staining dead cells) and Calcein AM (staining live cells). The phototherapeutic effect of 39 in A549 cells was shown to be significantly higher under hypoxia (from extreme hypoxia 0.1% to mild hypoxia 8% oxygen concentrations) than normoxic conditions (Fig. 25b). Although this work exhibits great promise, the excitation wavelength is not optimal for in vivo applications as such further development is needed to afford long-wavelength derivatives.
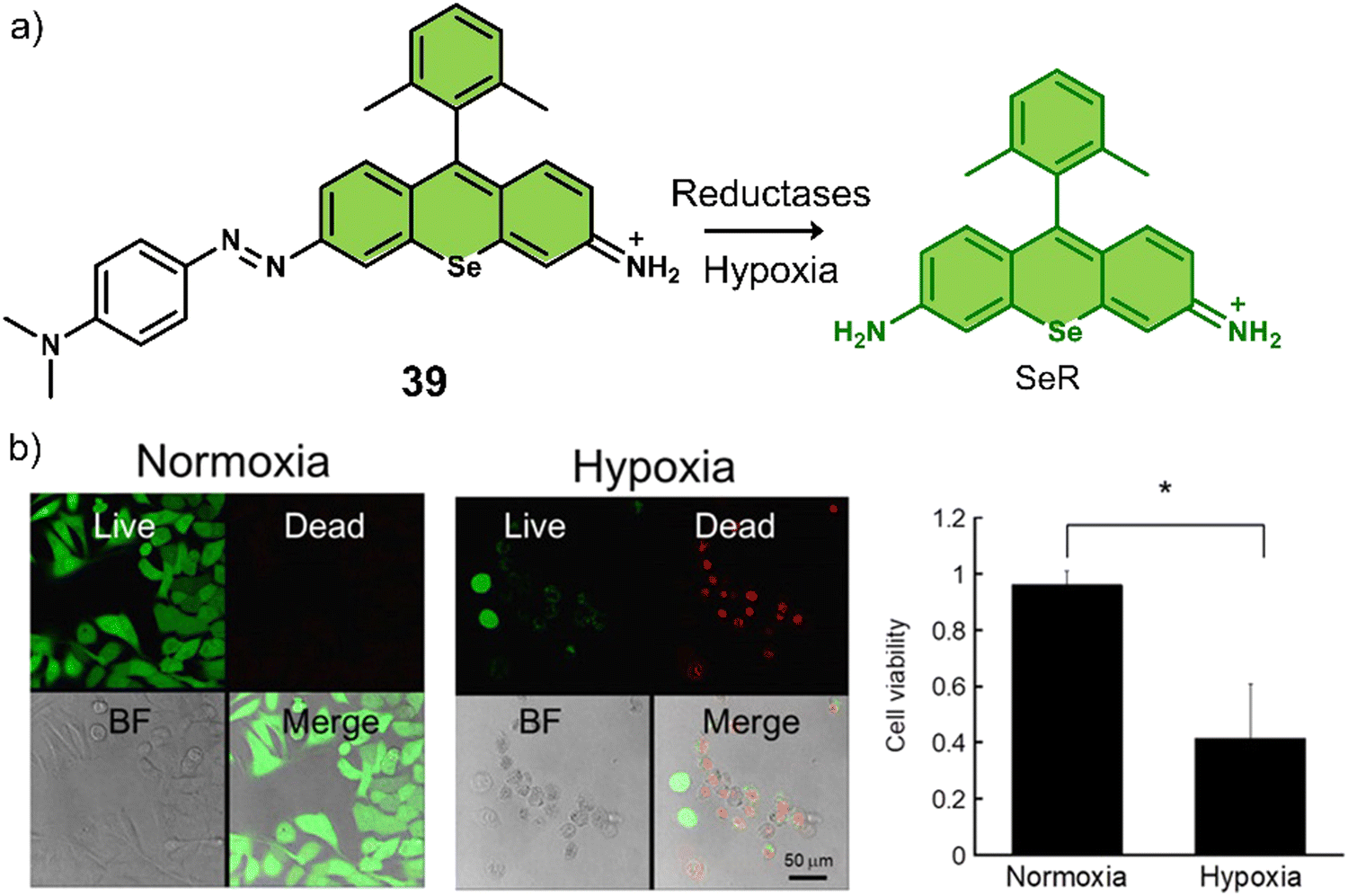 | ||
| Fig. 25 (a) Chemical structure of 39 and the hypoxia-activatable release of SeR. (b) Live/dead staining images of A549 cells that were photoirradiated (535 nm, 22 mW cm−2, 2 min) under normoxia and hypoxia after treatment with 39 (1 μM). Values are represented as mean ± SD (n = 3). *P < 0.05. Reproduced with permission from ref. 131. Copyright (2017) American Chemical Society. | ||
Another hypoxia-responsive PDT agent (40) exploits FRET to achieve simultaneous tumour imaging and PDT (Fig. 26).132 With 40, pyropheophorbide α (PPA, FRET donor) was covalently linked to fluorescent NIR dye SiR-665 (FRET acceptor) via an azo-benzene linker. Due to FRET and the presence of the azo group, 40 exhibited low fluorescence emission and singlet oxygen yield. However, in a hypoxic environment (NADPH and liver microsomes), an increased fluorescence emission at 680 nm and an enhanced 1O2-generation were observed. With these promising results, the ability of 40 for hypoxia-specific fluorescence imaging and photodynamic ablation of cancer cells in vitro was demonstrated in a BEL-7402 cell line (human hepatoma cells).
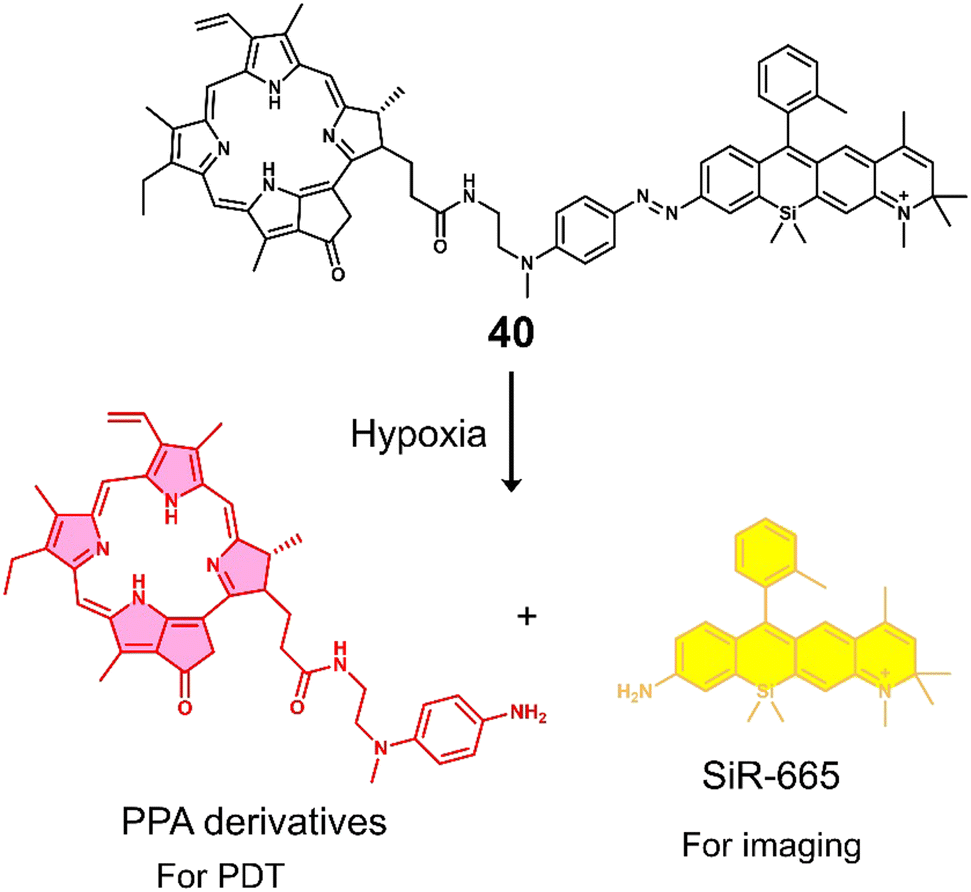 | ||
| Fig. 26 Chemical structure of 40 and the hypoxia-activated release of the PS PPA derivatives and NIR-dye SIR-665. | ||
3.4 Acidic pH-responsive activatable chemotherapeutics and activatable “smart” phototherapeutics
As stated briefly in the introduction, the TME is known to be acidic (pH 6.5 to 7.2) due to the increased glycolytic metabolism of cancer cells (Warburg effect). The acidic TME is viewed as a hallmark trait of cancers.133,134 The acidic intra- and extracellular pH in solid tumour tissues is crucial for cancer cell proliferation, migration, invasion, metastasis, evasion of immune surveillance, and drug resistance.135 This well-known property of the TME has resulted in a vast number of reports directed towards the development of acidic responsive prodrug systems.136 In this section, we discuss some selected examples of acidic responsive prodrugs that have been proven successful for chemotherapeutic and phototherapeutic applications (Table 4).| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: DOX: doxorubicin, PDT: photodynamic therapy, PTT: photothermal therapy, —: not mentioned. | ||||||
| 41 | Acid pH | DOX | Chemotherapy | A549 and HepG2 cells | — | 137 |
| 42 | Acid pH | DOX | Chemotherapy | U87 cells | — | 139 |
| 43 | Acid pH | DOX | Chemotherapy | A549 and CT26 cells | — | 140 |
| 44 | Acid pH | Iodinated-IR783 | PDT | HepG2 cells | — | 141 |
| 45 | Acid pH | p-Phenyleneethynylene-based derivative in zwitterionic form | PDT | HeLa cells | — | 142 |
| 46 | Acid pH | Heptamethine cyanine dye, IR-822 | PTT | MCF-7 cells | MCF-7 tumour-bearing mice | 143 |
| 47 | Acid pH | Rhodamine lactam analogue | PTT | HeLa, U-87 MG, and RAW 264.7 cells | H22 tumour-bearing mice | 145 |
Several research groups have focused on the development of prodrugs that release a therapeutic in acidic environments. Examples include the use of acid-sensitive breakable bonds (e.g., hydrazone) and 'ionisable' chemical groups (e.g., tertiary amines) to construct acid-sensitive therapeutic reagents (Scheme 5).
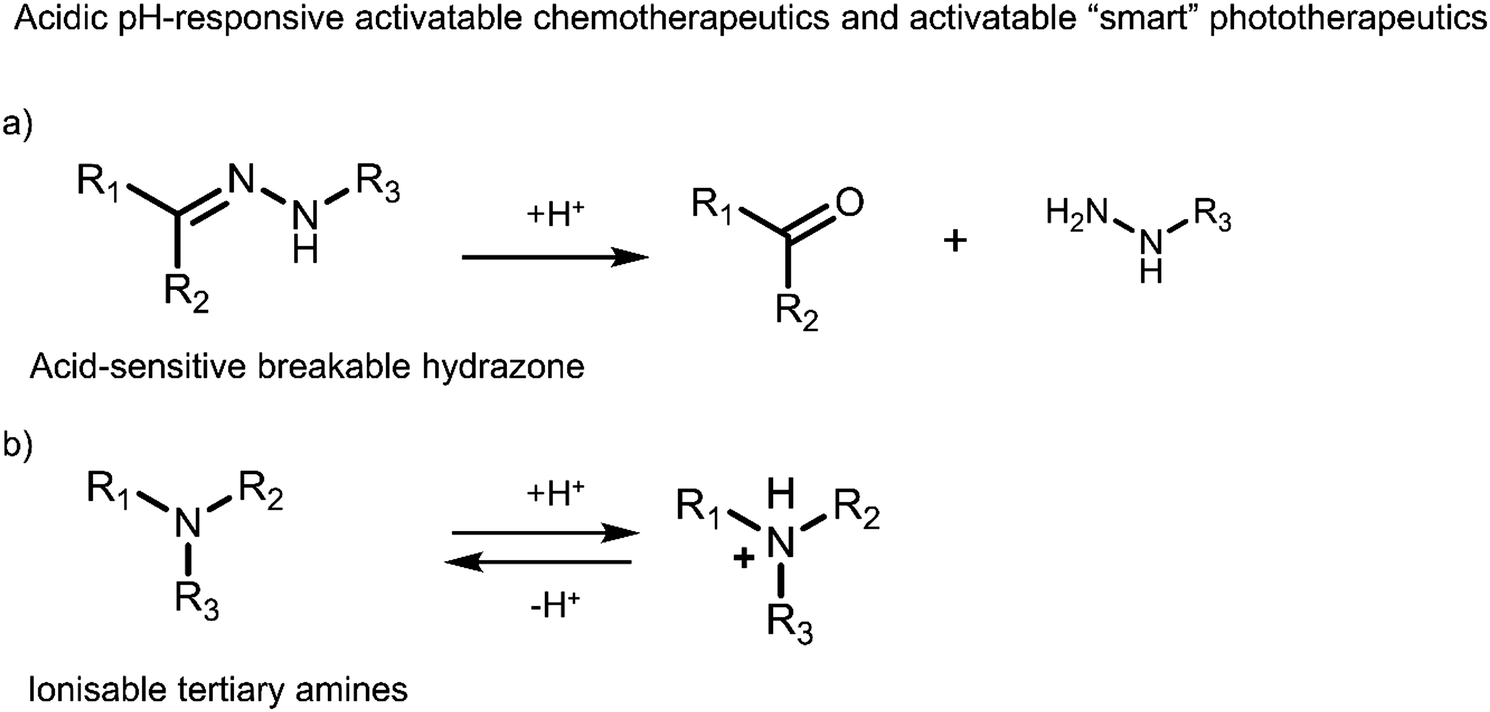 | ||
| Scheme 5 Basic mechanism of hydrazone-based and tertiary amines-based prodrugs activation. | ||
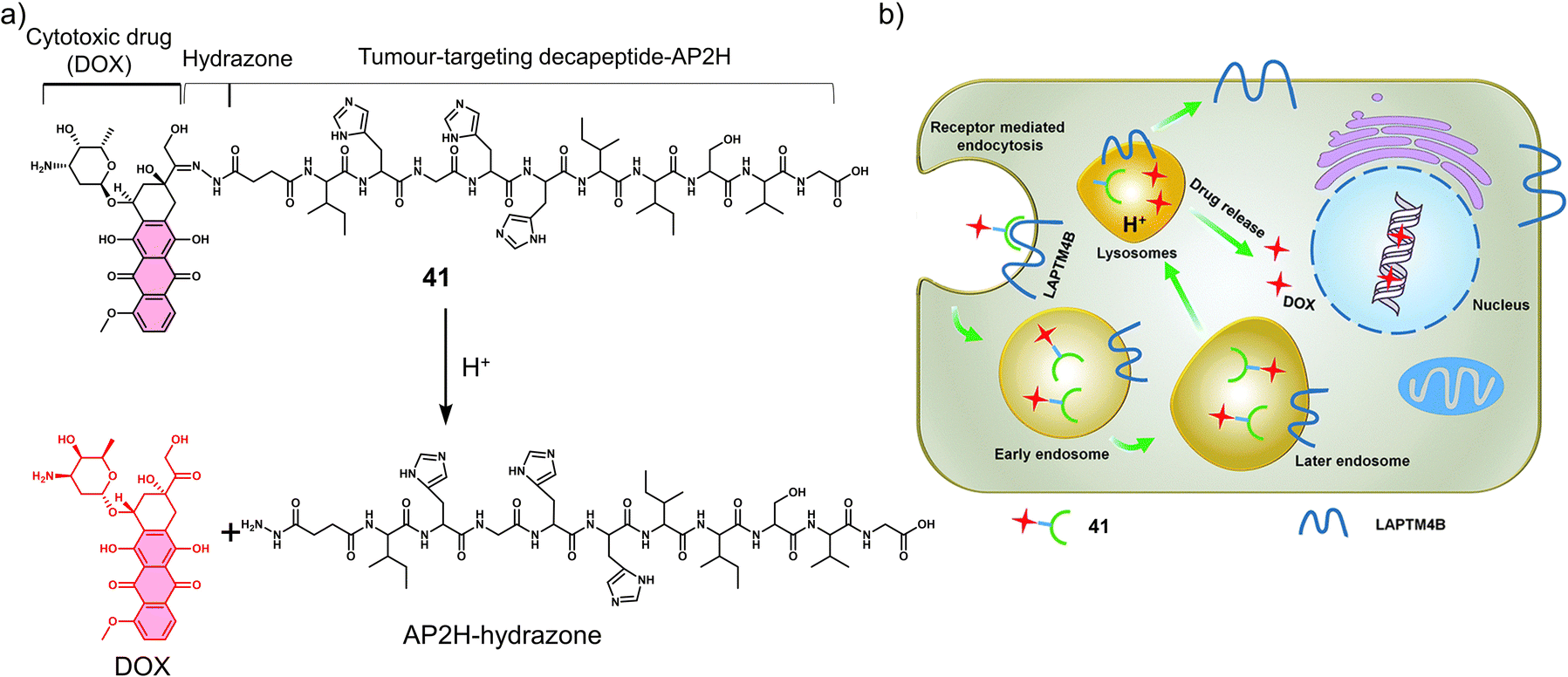 | ||
| Fig. 27 (a) Chemical structure of 41 and the acidic pH-activatable release of DOX. (b) The schematic illustration of LAPTM4B guided 41 internalization and acidic pH-mediated drug release process. Reproduced with permission from ref. 137. Copyright (2015) The Royal Society of Chemistry. | ||
Zhao and co-workers reported the acid-responsive theranostic 41 for the targeted delivery of DOX to cancer cells. In this theranostic, the tumour-targeting decapeptide AP2H (IHGHHIISVG) was linked to DOX via an acid-sensitive hydrazone bond (Fig. 27).137 AP2H is specific for the tumour-associated protein, lysosomal protein transmembrane 4 beta (LAPTM4B), that is expressed in various malignancies, including breast cancer, ovarian cancer, hepatocellular carcinoma (HCC), gastric cancer, and cervical cancer.138 HPLC studies revealed that over a period of 48 h at pH 5.0, 89% of DOX was released from 41, whereas at pH 6.0 and 7.4, only 31% and 10% were released under identical conditions, respectively. Encouraged by the excellent release profile at low pH values, 41 was shown to exhibit dose-dependent toxicity in LAPTM4B-positive A549 and HepG2 cell lines with IC50 of 1.14 μM and 4.0 μM, respectively. At the same time, minimal cytotoxicity was observed in a human embryonic kidney HEK 293 cell line (healthy cells), which illustrated the effectiveness of the AP2H targeted strategy for cancer cells. Fluorescence co-localisation experiments revealed that 41 was first localised in endosomes and lysosomes, which was ascribed to the targeting ability of AP2H for the LAPTM4B protein. Since endosomes and lysosomes are known to have a pH range of 4.5–6.5, DOX was released efficiently, which led to its translocation to the cell nucleus to activate the anticancer effect (DNA damage).
Another acid-responsive hydrazone-based DOX prodrug (42) was developed by Li et al. for in vitro evaluation in U87 cells. 42 consisted of an αvβ3 integrin targeting moiety (GRGDS), the coumarin fluorophore, and DOX. The fluorescent peptide backbone was conjugated to DOX through a hydrazone maleimide linker (Fig. 28).139 Under aqueous physiological conditions (pH 7.4), 42 showed a low emission intensity; however, as the pH value decreased to 5.0, a rapid fluorescence enhancement was observed at 460 nm. These results were ascribed to the hydrolysis of the hydrazone unit and the release of DOX (confirmed by HPLC analysis). Fluorescence spectroscopy results revealed that 93.8% of DOX from 42 was released at pH 5.0 after 11 h, while only about 40.7% was released at pH 7.4. Furthermore, in vitro studies indicated that upon incubation with αvβ3 integrin-positive human glioblastoma U87 cells, 42 exhibited concentration-dependent anticancer effects (IC50 = 0.19 μg mL−1). The result of time-dependent fluorescence imaging of 42 in U87 cells confirmed the suitability of the 42 to monitor DOX release in vitro in real-time.
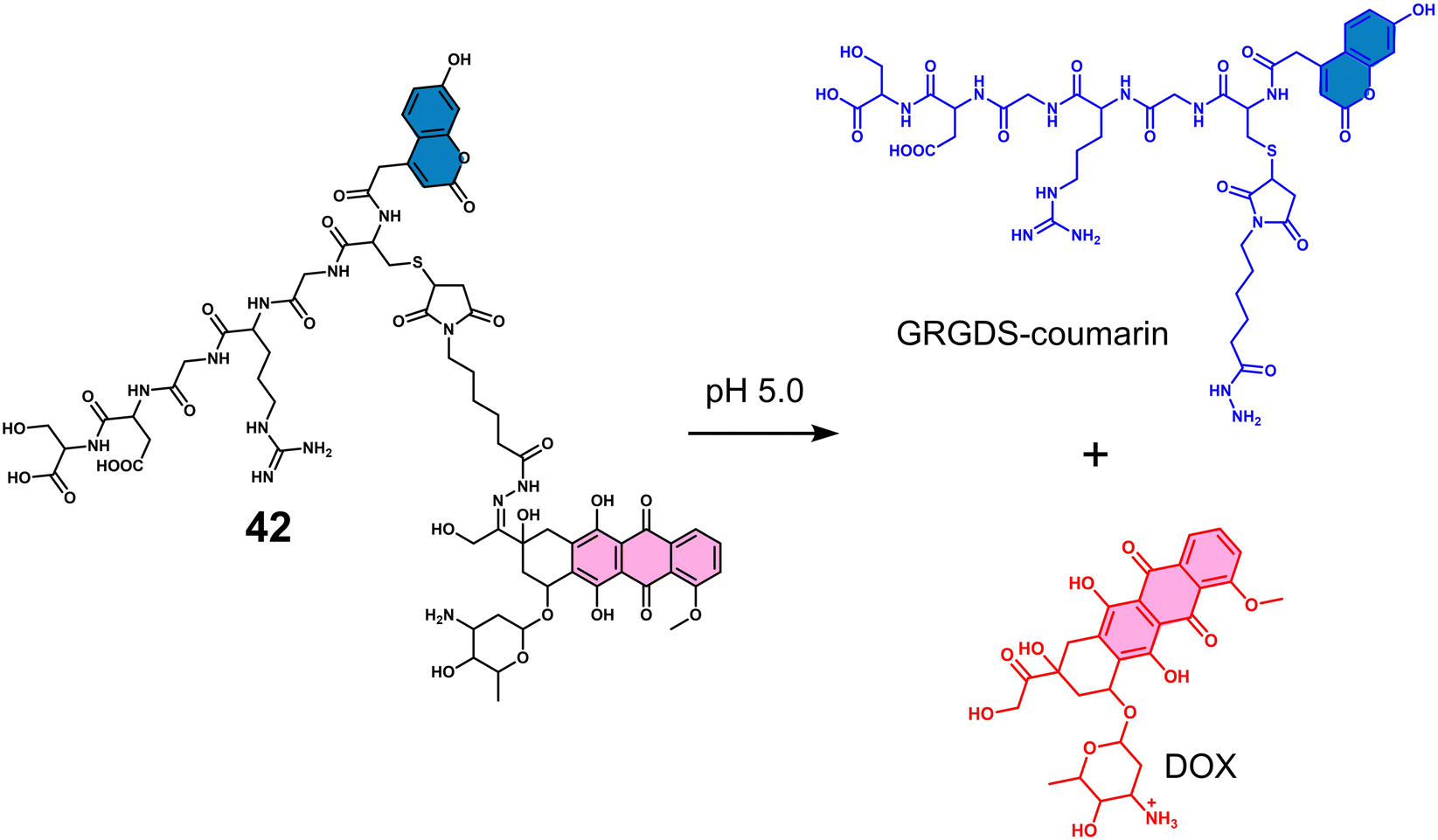 | ||
| Fig. 28 Chemical structure of 42 and the acid-mediated release of DOX. | ||
Sessler and co-workers reported the multimodal theranostic hydrazone-based MGd and DOX conjugate (43, Fig. 29a) for the acid-triggered release of DOX in A549 and CT26 live cells.14043 consisted of MGd conjugated to two molecules of DOX by an acid-responsive hydrazone linker. Each component in this strategy can be monitored by two complementary imaging methods, fluorescence spectroscopy and MR imaging. Initially, the fluorescence emission of DOX was quenched due to the paramagnetic nature of MGd. In acidic environments (pH 5.0), the hydrazone bonds of 43 underwent acid hydrolysis, resulting in the release of free DOX, which consequently showed a significant increase in the fluorescence signal at 593 nm. Relatively small changes in fluorescence emission intensity were observed at pH 7.4. This acid-triggered transformation was further confirmed by reverse-phase high-performance liquid chromatography (RP-HPLC) and electrospray ionization mass spectroscopy (ESI-MS). 43 exhibited higher antiproliferative activity in cancer cell lines (A549 and CT26) than in a normal fibroblast cell line NIH3T3 (Fig. 29b). These results were attributed to the fast uptake of 43 by cancer cells compared to normal cells, and once internalised, 43 undergoes acid-mediated hydrazone cleavage, releasing free DOX. The paramagnetic Gd(III) center of MGd was used for MR imaging of A549 and CT26 cancer cells, which revealed a strong MR signal when either cell line was treated with 4 μM of 43. More importantly, the MRI signal of 43 was generated without the need for hydrolysis of hydrazone to generate DOX and generate a fluorescence signal.
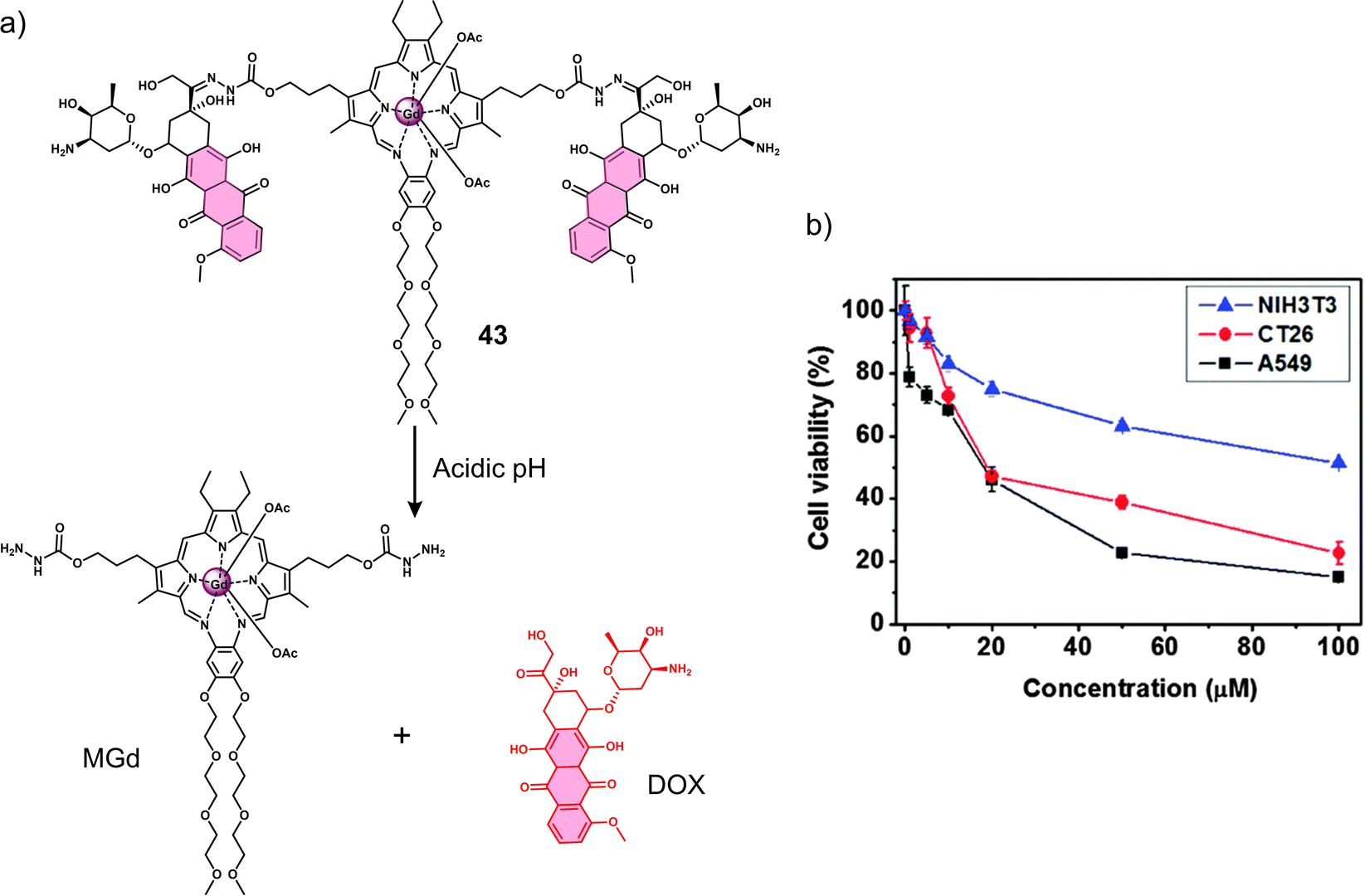 | ||
| Fig. 29 (a) Chemical structure of 43 and the acid-responsive release of the Gd(III) texaphyrin and DOX. (b) Antiproliferative activity of 43 incubated at various concentrations for 48 h with A549, CT26, and NIH3T3 using a standard MTT assay. Reproduced with permission from ref. 140. Copyright (2016) The Royal Society of Chemistry. | ||
In contrast, “ionisable” groups are often introduced in the construction of phototherapeutic agents, enabling the photophysical properties of the phototherapeutic agent to be reversibly switched under different pH environments. Kamkaew and co-workers reported the synthesis of the pH-responsive PDT agent, 44, for the treatment of hepatocellular carcinoma. The introduction of the pH-sensitive N-methylpiperazine unit onto iodinated-IR783 afforded an attractive system (44) that exhibits high 1O2 quantum yield and enabled the NIR image-guided PDT of acidic environments (Fig. 30).141 The addition of iodine atoms to IR783 was used to enhance the PDT properties through the heavy atom effect. At neutral pH, 44 was non-fluorescent with a low 1O2 production. However, in acidic environments (pH 3.0–5.0), the two nitrogen atoms of N-methylpiperazine unit become protonated, blocking the PeT (photoinduced electron transfer) process, thereby facilitating the fluorescence enhancement and increase in 1O2 generation under NIR light irradiation (850 nm, 30 mW cm−2). A live/dead cytotoxicity assay (Calcein AM and propidium iodide co-staining) of 44 indicated that the phototoxicity to HepG2 cells under 850 nm light irradiation at pH 5.0 was significantly higher than at pH 7.4. No dark toxicity was observed. Excellent penetration depth was obtained when a 5 mm piece of pork tissue was placed between the cells and the light source, attributed to the long excitation wavelengths. The above results confirm the potential of 44 for the PDT of solid tumours.
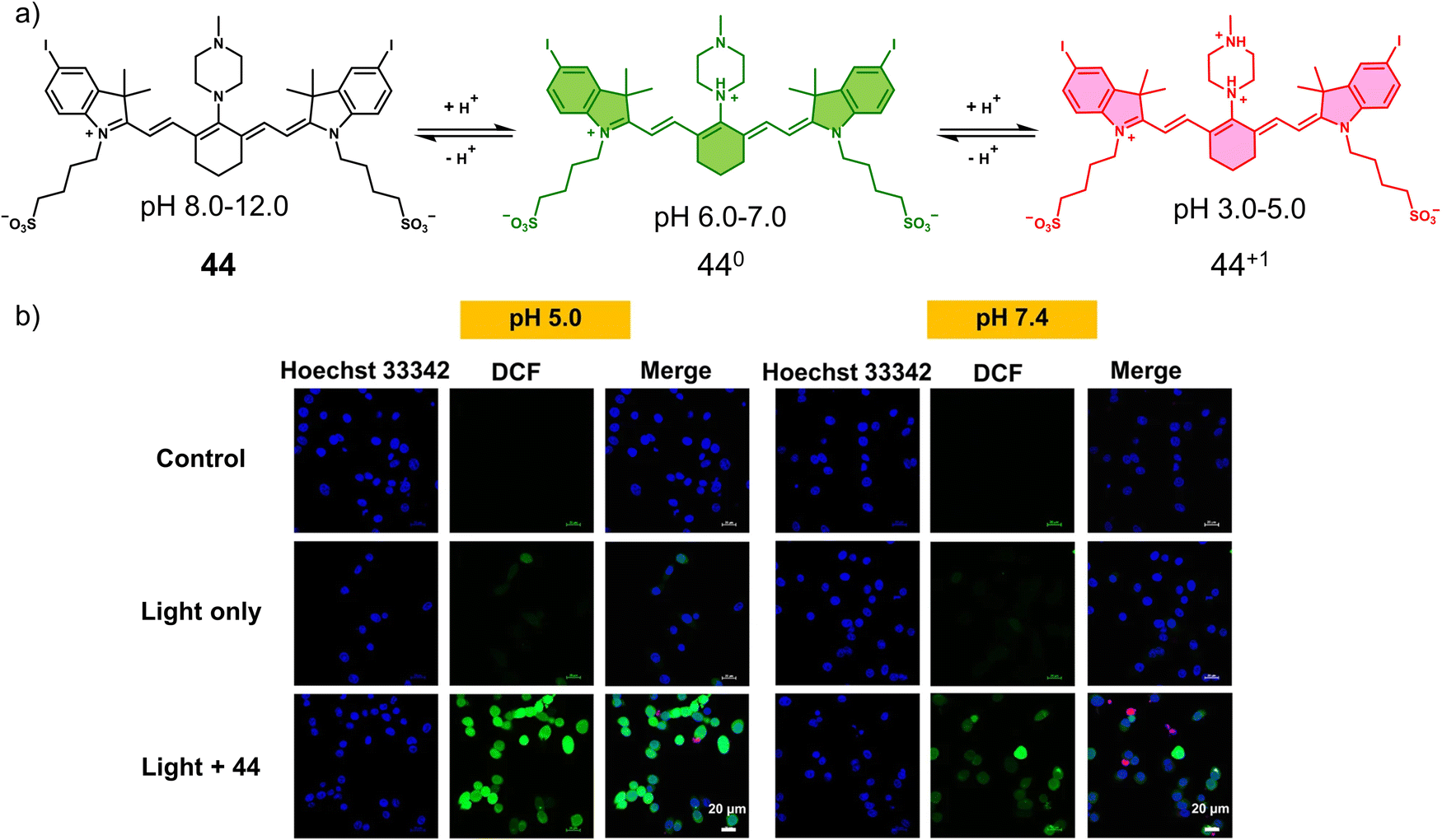 | ||
| Fig. 30 (a) Chemical structure of 44 and its pH-activated mechanism at values 6.0–7.0 and 3.0–5.0. (b) Detection of intracellular ROS generated by 44 (20 μM) at pH 5.0 and 7.4 in HepG2 cells using 2,7-dichloro-dihydro-fluorescein diacetate (DCFH-DA) cellular ROS detection assay. Scale bars: 20 μm. Reproduced with permission from ref. 141. Copyright (2020). The Author (S. Siriwibool, N. Kaekratoke, K. Chansaenpak, K. Siwawannapong, P. Panajapo, K. Sagarik, P. Noisa, R.-Y. Lai and A. Kamkaew). | ||
Hu et al. developed a unique PDT agent, 45, where the intersystem crossing (ISC) process (singlet oxygen activation) was shown to be reversibly activated via a pH-induced conjugated backbone twist (CBT) (Fig. 31a).142 The twisting was achieved through changes in intramolecular electrostatic interactions between positively charged quaternary ammonium functional groups and negatively charged terminal carboxyl groups. At neutral pH, the anionic form of 45 was not twisted, whereas in acidic pH, electrostatic interactions induced twisting. Techniques, including femtosecond transient absorption (fs-TA) spectroscopy and quantum chemical calculations, indicated that the ISC efficiency (ΦISC) was improved by increasing the degree of twisting of the molecule. Thus, in acidic pH (∼6.0) (compound's isoelectric pH), 45 exhibited a high singlet oxygen quantum yield. White light irradiation (10 mW cm−2, 5 min) of 45 treated HeLa cells exhibited a good acidic-pH selectivity at pH 6.0 compared to cells at pH 7.4 (Fig. 31b). Since previously discussed activatable PDT agents are irreversible, this work provides an alternative and reversible strategy for the activation of a PS. To the best of our knowledge, this is the first time that a pathological factor (i.e., pH) has been stimulated to modulate the reversible switching of ISC and thus improve the precision of photodynamic therapy. However, this concept has not yet been applied for in vivo studies and is still some way from practical application, so we believe that this system required further development and optimisation.
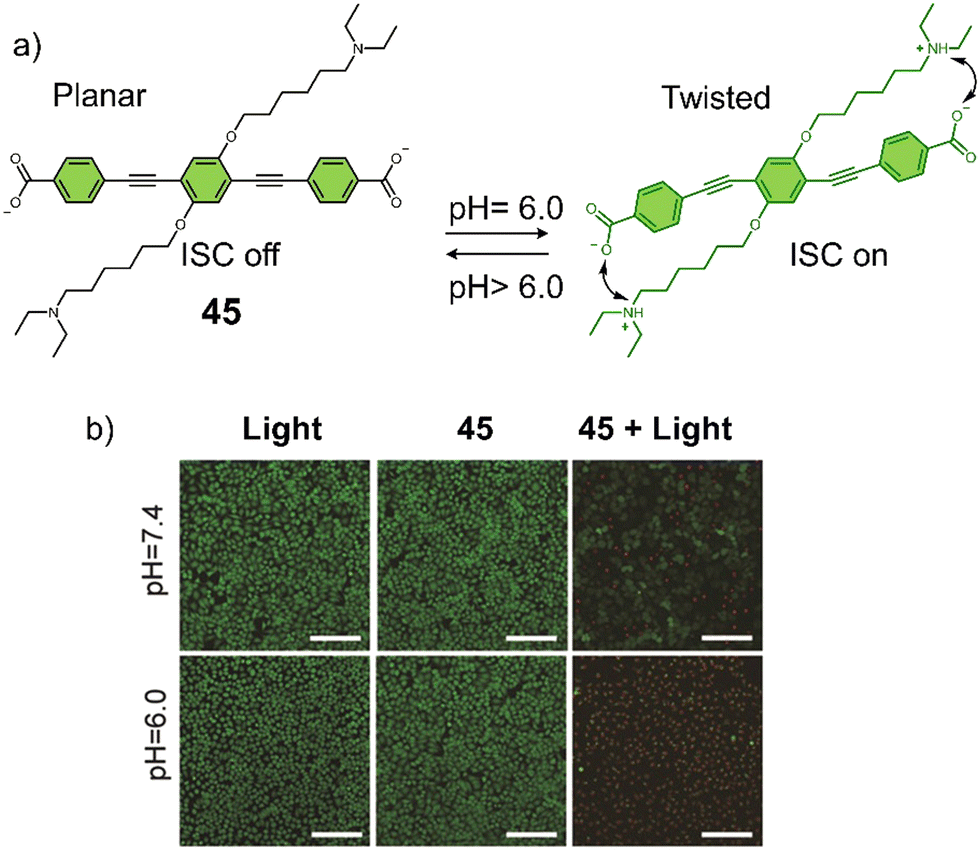 | ||
| Fig. 31 (a) The chemical structure and design of 45 with pH-reversible conjugated backbone twisting (CBT) for the reversible switching of ISC. (b) Live/dead staining images of HeLa cells in media pH 6.0 and pH 7.4 upon the light irradiation after treatment with 45. Scale bar: 160 μm. The green color represents live cells, and the red color represents dead cells. Reproduced with permission from ref. 142. Copyright (2019) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Cai and co-workers reported the acid-responsive PTT agent 46 (Fig. 32a) consisting of NIR-fluorescent heptamethine cyanine dye, IR-822, with N1-(pyridin-4-ylmethyl)ethane-1,2-diamine (PY) as the pH-responsive moiety.14346 was shown to be non-fluorescent at neutral pH. However, in acidic environments, the “N” of pyridine became protonated and resulted in an increase in fluorescence emission at 792 nm. This was accompanied by a change in absorption, which led to an increase in the PA signal at 637 nm. 46 exhibited good photothermal-conversion efficiency under NIR 808 nm laser irradiation (0.4 W cm−2, 3 min), with a temperature rise from 25.7 °C to 52.4 °C. In MCF-7 cells, 46 exhibited a gradual increase in fluorescence intensity as the pH of the microenvironment decreased. The phototheranostic properties of 46 were evaluated using a MCF-7 tumour-bearing mice model (Fig. 32b and c). The NIR fluorescence and PA imaging indicated that the maximum imaging intensity corresponding to 46 was seen 14 h post-injection. Under low-powered laser irradiation (808 nm, 0.4 W cm−2, 3 min), 46 was found to exhibit significant tumour ablation, and no tumour recurrence was seen after 30 days. The mice treated with 46 and light irradiation similarly maintained a high survival rate when compared to control groups (those treated with PBS or 46 without light irradiation).
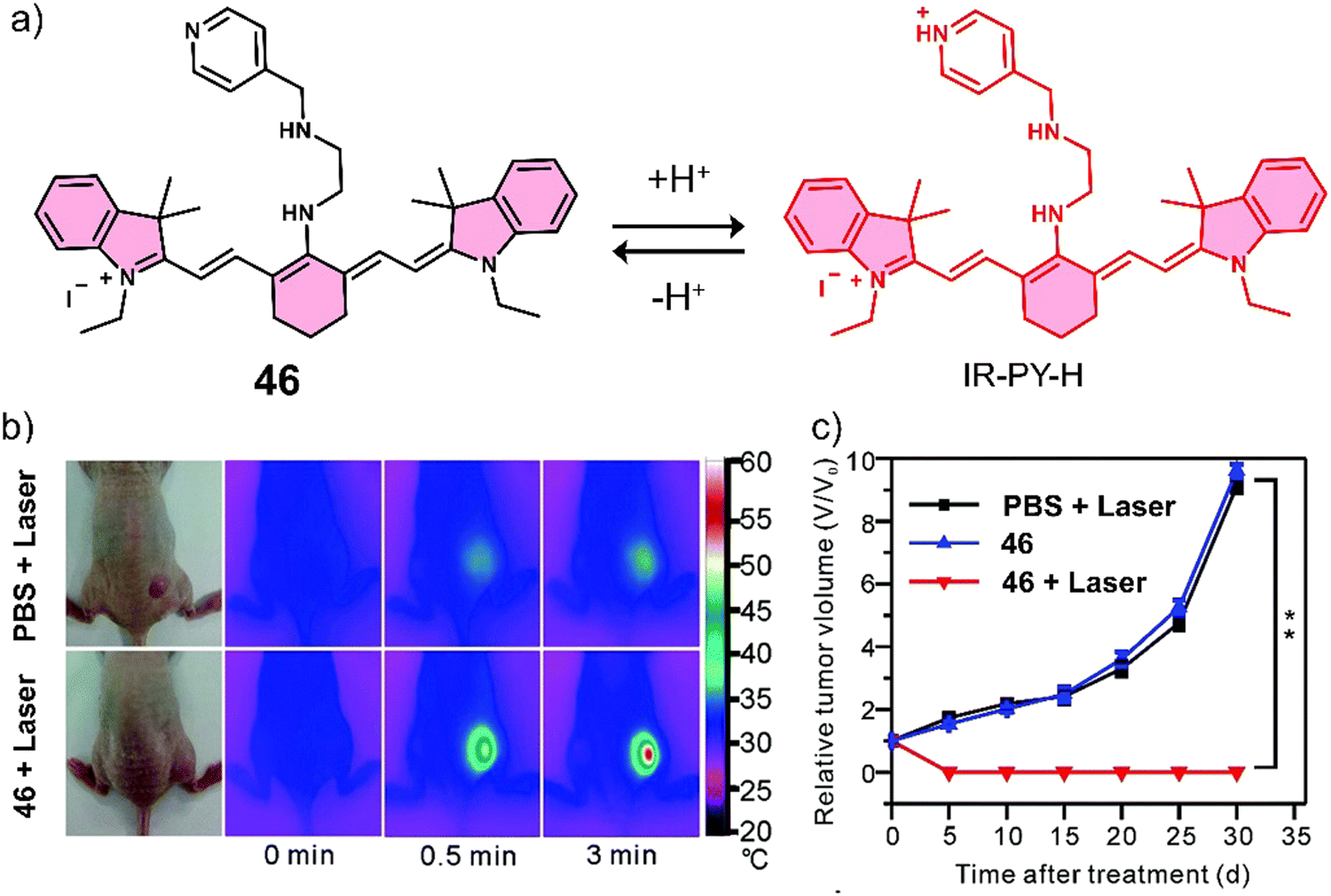 | ||
| Fig. 32 (a) Chemical structure of 46 and activation at acidic pH. (b) Infrared thermal images of MCF-7 tumour-bearing mice after tail-vein injection of PBS (100 μL), or 46 (100 μL, 40 μg mL−1) upon the NIR laser irradiation (808 nm, 0.4 W cm−2, 3 min) once. (c) The tumour growth curve for 30 days post-treatment for the different groups. The data are shown as mean ± SD (n = 10), **P < 0.01. Reproduced with permission from ref. 143. Copyright (2017) The Royal Society of Chemistry. | ||
Han and co-workers developed the PTT agent 47 that uses a sialic acid (SA) targeting unit for selective imaging and cancer treatment in vitro and in vivo. Tumour cells are well-known to have an increased metabolic demand for SA, which has led researchers to exploit this functionality as a targeting strategy.144 The SA unit was linked to a pH-activatable NIR PTT agent (a rhodamine lactam analogue) (Fig. 33a).145 Under physiological conditions (pH 7.2), 47 exhibited a relatively weak fluorescence emission intensity at 740 nm. However, in acidic environments (pH 4.5–6.0), 47 can be readily isomerised into highly fluorescent fluorone by proton-mediated ring opening of the intramolecular lactam,146 and an enhanced NIR fluorescence emission was observed, accompanied by a strong increase in absorption at 715 nm and good photothermal conversion efficiency upon 660 nm laser illumination (0.5 W cm−2, 600 s). The acid-responsiveness of 47 was confirmed in HeLa, U-87 MG, and macrophage RAW 264.7 cell lines with an increased fluorescence intensity being observed in lysosomes. The fluorescence response was confirmed through the pre-incubation of HeLa, U-87 MG, and RAW 264.7 cell lines with a V-ATPase inhibitor, bafilomycin A1 (BFA), which causes an increase in lysosomal pH.14747 exhibited the strongest NIR fluorescence emission at 750 nm at the tumour site 48 h post-injection in a H22 tumour-bearing mice model. Remarkably, the signal corresponding to 47 was found to last for 144 hours. Demonstrating its utility as a phototheranostic agent, significant phototoxicity was observed for HeLa, U-87 MG, and RAW 264.7 cells after NIR laser irradiation at 660 nm (0.5 W cm−2, 10 min) (Fig. 33b).
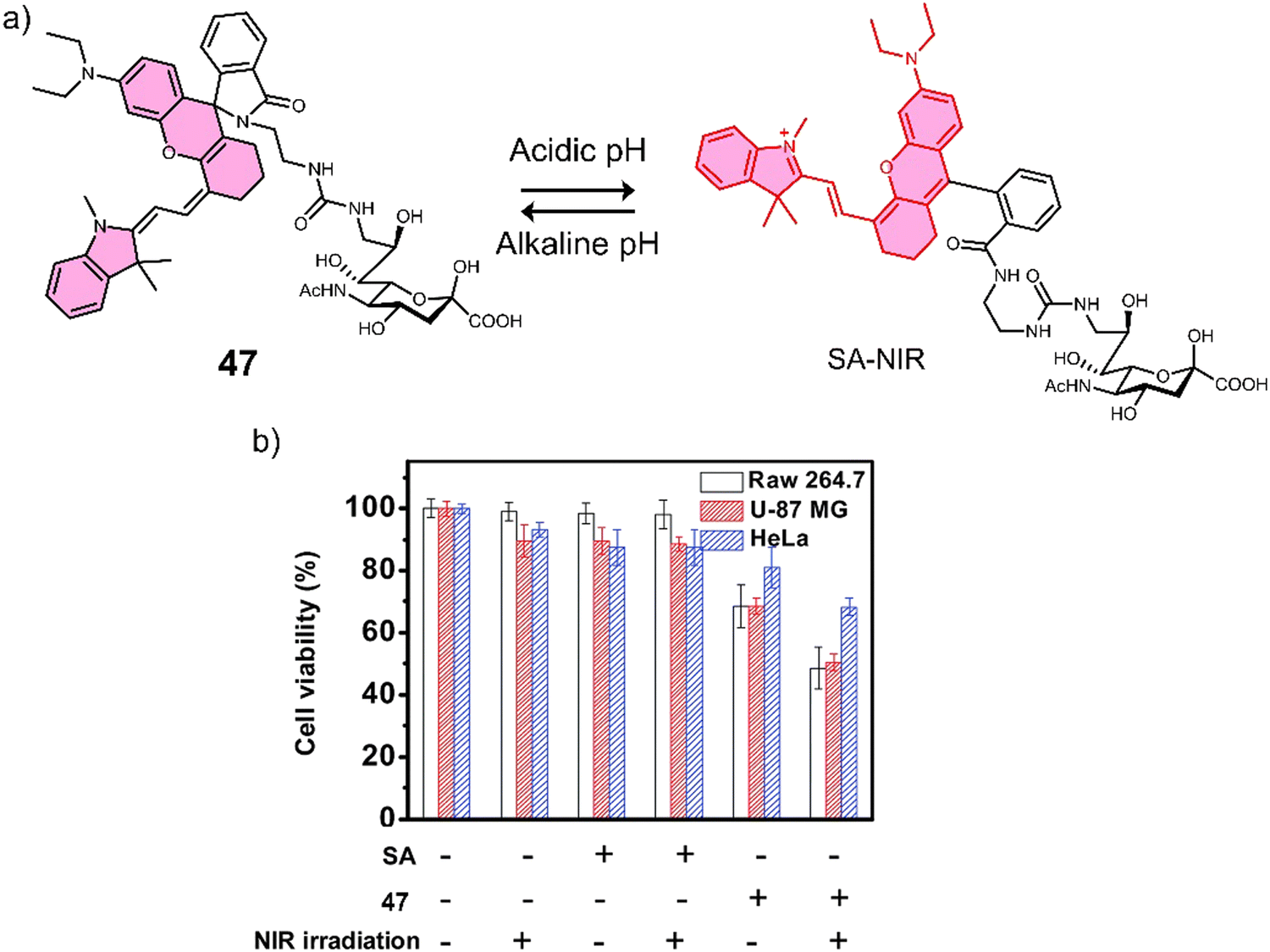 | ||
| Fig. 33 (a) Chemical structure of 47 for targeted imaging of cancerous HeLa, U-87 MG, and macrophage RAW 264.7 cell lines that overexpress SA receptors and activation at acidic pH. (b) Phototoxicity study with HeLa, U87-MG, and RAW 264.7 cell lines. Cell viability was determined by MTT assay. Reproduced with permission from ref. 145. Copyright (2014) The Royal Society of Chemistry. | ||
3.5 The use of external stimuli for the release of therapeutics
Photocages are a class of functional groups that are designed to release “cargo” under the irradiation of light. The reader can be directed to several excellent reviews for an extensive overview of photocages.148–150 Owing to the non-invasive, high spatial resolution, and spatio-temporal control of light, several photocages have been developed for chemotherapeutic applications.151–153 Initial reports for these light-responsive strategies relied on the use of a nitrobenzyl photoresponsive group. Unfortunately, this unit requires activation using short excitation wavelengths (<500 nm), which are associated with poor tissue penetration and potential phototoxicity.154 In recent years, a significant focus has been directed towards light-activatable systems that require long excitation wavelengths and that exhibit enhanced photoconversion efficiencies (Table 5). For an extensive overview on the photoremovable protecting units, the reader can be directed to the following excellent reviews.155,156| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: DOX: doxorubicin, PDT: photodynamic therapy, —: not mentioned. | ||||||
| 56 | Visible light | CA-074 and WinterGreen | Chemotherapy and PDT | MDA-MB-231 cells | — | 158 |
| 57 | Red-light | Oxaliplatin | Chemotherapy | Platinum-sensitive A2780, platinum-resistant A2780cis and MCF-7 cells | 4T1 tumour-bearing mice | 159 |
| 58 | X-ray | Pazopanib | Chemotherapy | HUVEC cells | HT29 tumour-bearing mice | 161 |
| 59 | X-ray | DOX | Chemotherapy | HeLa cells | HeLa tumour-bearing mice | 161 |
Recently, the work of the Winter group has led to the identification of a series of BODIPY-based photocages (e.g., 48–55) that are activated under visible or NIR light irradiation (Fig. 34).157 Some of these photocages are now commercially available from Sigma Aldrich under the trade names of WinterGreen carbamoyl imidazole photocage (51) and WinterRed Photocage (52) (Fig. 34). A recent example from the Winter group for these systems was with the covalent attachment of the cell impermeable CTSB inhibitor CA-074 with WinterGreen to form 56 (Fig. 35).158 It was rationalized that cell uptake could be achieved via the use of a photocage i.e., 56. 56 was found to be cell-permeable, and irradiation using blue light (460–470 nm) was shown to release CA-074 and BODIPY (WinterGreen). Exhibiting cytotoxicity toward MDA-MB-231 cells at 72 h incubation with the EC50 (half maximal effective concentration) of 0.78 μM under light irradiation (460–470 nm, 15 min). Also, minimal toxicity was observed towards normal MCF-10A cell lines with/without light irradiation (EC50 = 3.5 μM and 27 μM, respectively). Mechanistic studies indicated that CTSB inhibition was enhanced by the production of 1O2 due to light irradiation of the BODIPY fragment. Flow cytometry confirmed that 10 μM of 56 combined with light irradiation (460–470 nm, 15 min) was sufficient to achieve similar toxicities to 4 h of treatment using 500 mM H2O2 (known to mediate cell necrosis).
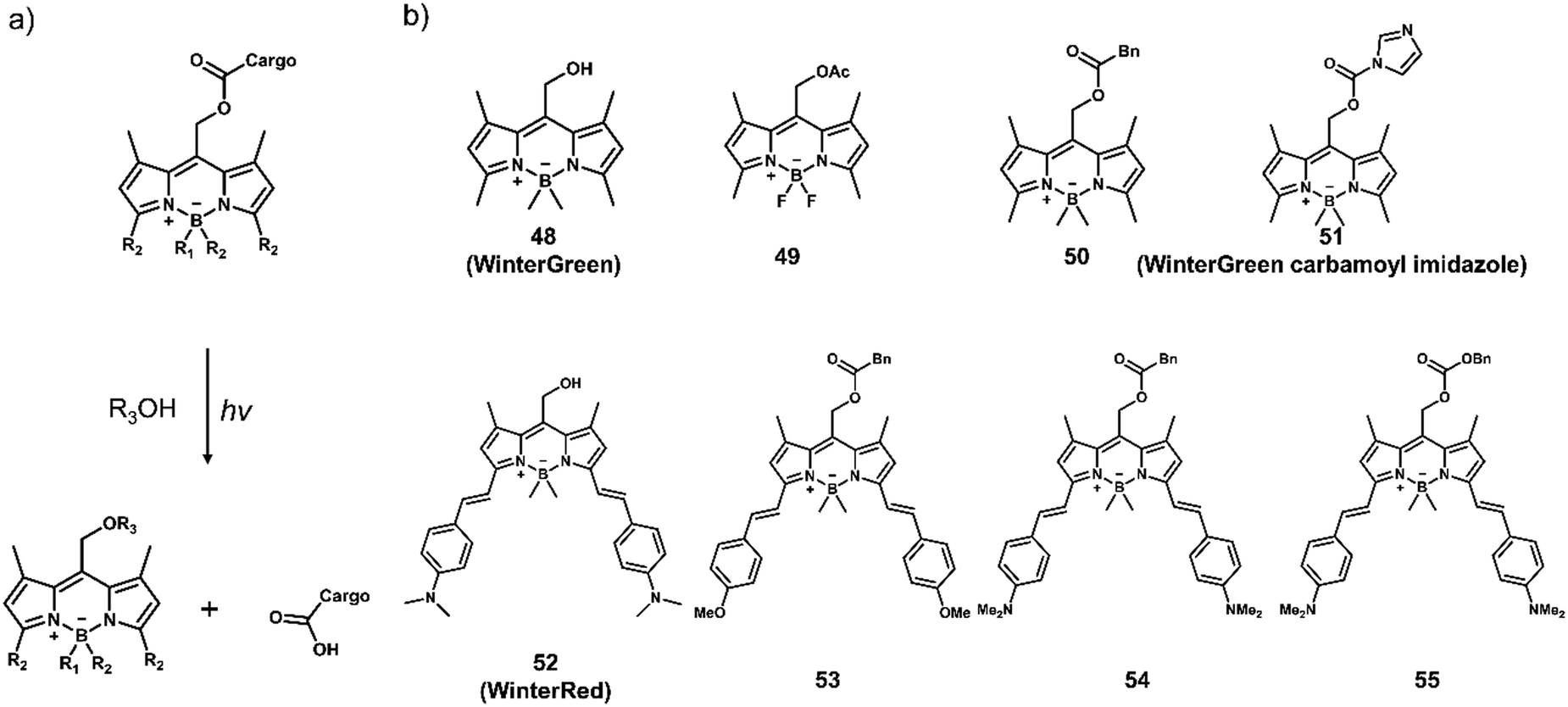 | ||
| Fig. 34 (a) Light-activated release mechanism and (b) representative chemical structures of BODIPY-based photocages (48–55) reported by the Winter group. | ||
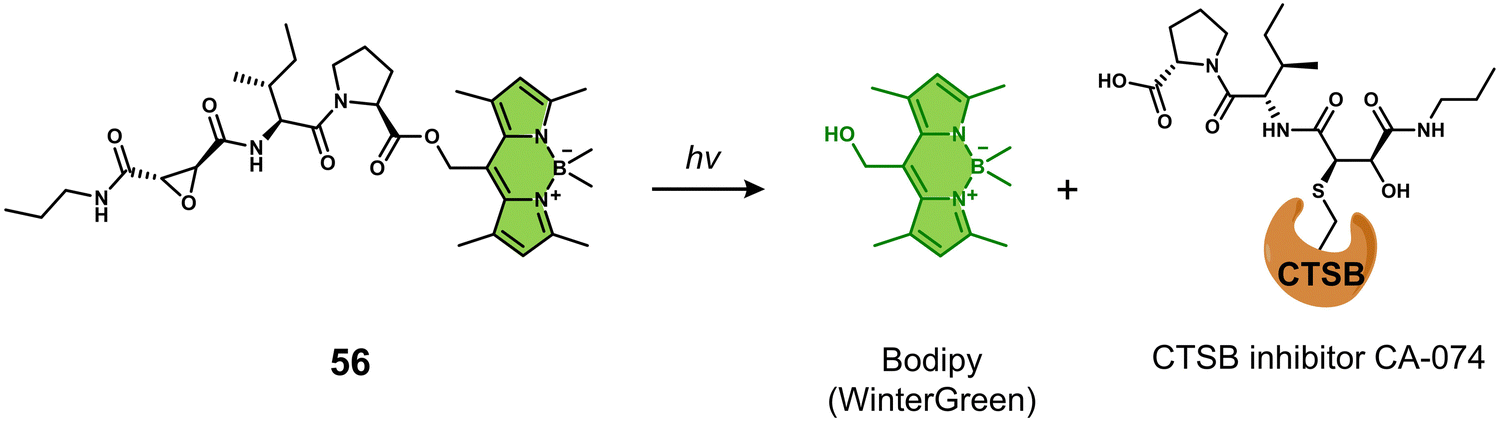 | ||
| Fig. 35 Chemical structure of 56 and the light-responsive (460–470 nm) release of the CTSB inhibitor CA-074. | ||
Recently, Zhu and co-workers developed a light-activatable (650/660 nm) oxaliplatin Pt(IV) conjugate photosensitizer (57, Fig. 36a).159 In this photosensitizer, Pyropheophorbide α (PPA) was linked to the axial position of the oxaliplatin Pt(IV) prodrug, and it was rationalised that the light activation of ROS could prove synergistic for Pt(IV) prodrugs.160 Remarkably, the light irradiation of 57 using a low-intensity red light excitation (650 nm, 7 mW cm−2) was shown via MS and HPLC to reduce Pt(IV) to Pt(II) and release PPA. 57 displayed enhanced cytotoxicity towards cancer cells [(platinum-sensitive A2780 and platinum-resistant A2780cis, human ovarian cancer cell; MCF-7, human breast cancer cell; 4T1, murine breast cancer cell line) (IC50 < 0.2 μM)] with light irradiation (650 nm, 7 mW cm−2, 15 min) than without light (IC50 > 10 μM). Minimal dark toxicity was observed towards MRC-5 cells [(MRC-5, normal human lung fibroblast) (IC50 > 10 μM)]. 57 combined with light irradiation (660 nm, 100 mW cm−2, 10 min) was found to have greater antitumour activity in vivo than oxaliplatin and a mixture of oxaliplatin with PPA in a murine mammary adenocarcinoma 4T1 tumour-bearing mice model (Fig. 36b and c). This study represents the first example where red light-controlled activation of platinum prodrugs has been used in vivo. We anticipate that this work will motivate others to develop new strategies for the light-controlled activation of platinum (Pt(IV)) prodrugs.
 | ||
| Fig. 36 (a) Chemical structure of 57 and the light-activated (650/660 nm) release mechanism of oxaliplatin. (b) Tumour growth inhibition over time with control groups (solvent + light, oxaliplatin + light, PPA + oxaliplatin + light, 57) and 57 that i.v. injected at a Pt dose of 3.5 μmol kg−1 every two days for 10 days upon the light (660 nm, 100 mW cm−2, 10 min). i.v.; intravenous injection. Arrows represent the time of administration and irradiation. **P < 0.01, ***P < 0.001, mean ± SD; n = 5. (c) Representative images of excised tumours at the study endpoint. Reproduced with permission from ref. 159. Copyright (2019) Elsevier Inc. | ||
Radiotherapy is often used in combination with chemotherapy to enhance the inhibition of tumour growth. However, the use of radiotherapy (X-rays) to activate prodrugs has remained elusive. The development of X-ray-activatable prodrugs provides the potential to activate therapeutics specific to the tumour area during conventional X-ray radiotherapy, potentially reducing patient side effects. In 2021, Bradley and co-workers reported the use of a medical radiotherapy X-ray source to selectively activate prodrugs to elicit their antiproliferative effects.161 An extensive screening process was used to identify molecules that have the potential to undergo a chemical transformation during X-ray radiation (60 Gy). As a result 4-acetamidobenzenesulfonyl azide and 4-(hydroxymethyl)-2,3,5,6-tetrafluoroaryl azide were identified that undergo transformation into 4-acetamidobenzenesulfonamide and 4-(hydroxymethyl)-2,3,5,6-tetrafluoroaniline, respectively. To illustrate the clinical potential of this strategy, the authors synthesised a pazopanib-based prodrug (58, Fig. 37a) functionalised with the X-ray activatable sulphonyl azide moiety. Pazopanib is a small molecule inhibitor of the vascular endothelial growth factor receptor (VEGFR) approved for the treatment of advanced kidney cancer.162 HPLC analysis demonstrated that 90% of 58 was transformed under X-ray irradiation (60 Gy). Moreover, activation of 58 was shown to be radiation dose-dependent and was effective on HUVEC (human umbilical vein endothelial) cell lines in vitro. A similar antiproliferative effect was observed to that of pazopanib when cells incubated with 58 (0, 5, and 10 μM) were subjected to X-ray irradiation (0, 6, 12, and 24 Gy). Intratumoural injection of 58 (100 mg kg−1) in conjunction with X-ray irradiation (6 Gy) in HT29 tumour-bearing mice afforded comparable tumour inhibition as pazopanib (100 mg kg−1), with prolonged survival times compared to the control groups (pazopanib or X-ray only) (Fig. 37b). To demonstrate the general nature of this strategy, the authors further developed 59 through the X-ray activatable 4-(hydroxymethyl)-2,3,5,6-tetrafluoroaryl azide moiety conjugated to DOX via a carbamate linker. 59 exhibited promising results both in vitro and in vivo (0, 5, and 10 μM in vitro; 10 mg kg−1in vivo) upon X-ray radiation (6, 12, 24, 36, 48, and 60 Gy in vitro; 6 Gy in vivo) in HeLa cell lines and HeLa tumour-bearing mice models (Fig. 38c). This pioneering work illustrated a unique prodrug design that has potential for clinical translation where the selective activation of antitumour agents can overcome off-target toxicities of currently available chemotherapeutics.
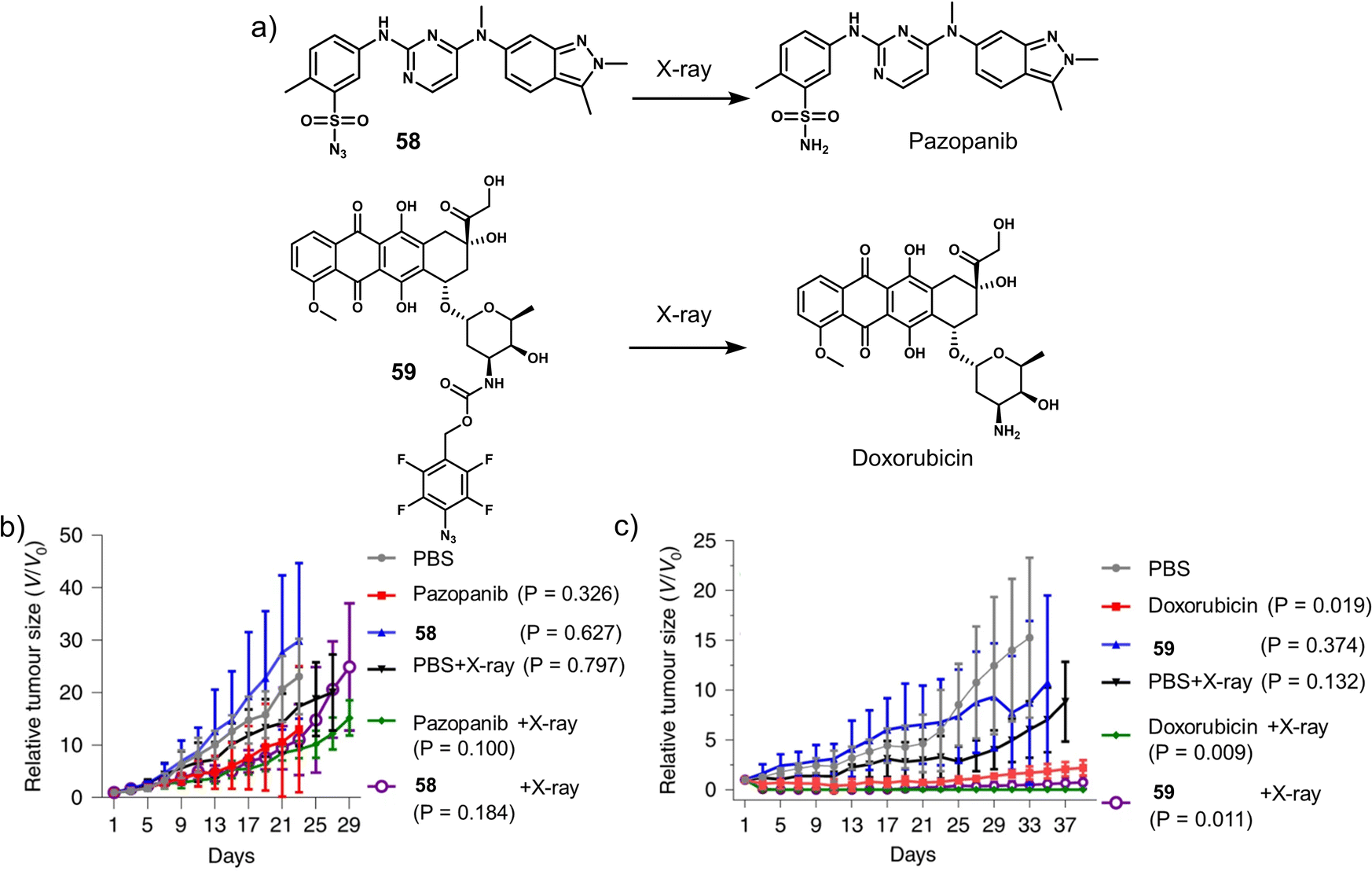 | ||
| Fig. 37 (a) Chemical structures of 58, 59, and the X-ray-activated release of pazopanib and doxorubicin (DOX). (b) Tumour growth inhibition of HT29 tumour-bearing mice models over the time with PBS, pazopanib (100 mg kg−1), 58 (100 mg kg−1) with or without X-ray irradiation (6 Gy) after 4 h of post intratumoural injection. (c) Tumour growth inhibition of HeLa tumour-bearing mice models over the time with PBS, doxorubicin (10 mg kg−1), 59 (10 mg kg−1) with or without X-ray irradiation (6 Gy) after 4 h of post intratumoural injection. Mean ± SD; n = 5. Reproduced with permission from ref. 161. Copyright (2021) The Author (J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley), under exclusive licence to Springer Nature Limited. | ||
3.6 Prodrugs and phototherapeutics that respond to more than one stimuli
Due to the complexity and high heterogeneity of the human body, it is difficult to selectively target tumours and avoid unwanted toxicity issues with just one method of activation. Therefore, researchers have been actively developing dual-responsive prodrugs and phototherapeutics to substantially enhance the precision of their activation in vivo (Table 6).| Stimuli | Active anti-cancer therapeutic agent | Treatment | In vitro models | Ex vivo/in vivo models | Ref. | |
|---|---|---|---|---|---|---|
| Note: CPT: camptothecin, DOX: doxorubicin, GSH: glutathione, PDT: photodynamic therapy, —: not mentioned. | ||||||
| 60 | Acid pH and GSH | CPT | Chemotherapy | A549 cells | A549 tumour-bearing mice | 163 |
| 61 | Acid pH and caspase-3 | DOX | Chemotherapy | U87 cells | — | 164 |
| 62 | Acid pH and thiol | Heptamethine cyanine IR765 | PDT | A549 and MCF-7 cells | MCF-7 tumour-bearing mice | 166 |
| 63 | Acid pH and GSH | Cy7 | PTT | — | 4T1 tumour-bearing mice | 167 |
Zhu and co-workers reported sequentially activatable prodrug, 60 (Fig. 38a), for the programmable drug release of CPT in A549 lung cancer cells and tumours.16360 consisted of an acid-sensitive tertiary amine-containing diblock copolymer conjugated to a cyanine fluorophore (Cy). The Cy fluorophore was functionalised with a disulfide linker to allow its conjugation to CPT and to enable the GSH-mediated release of CPT. In neutral aqueous conditions, 60 was shown to self-assemble to form nanomicelles and quench the fluorescence emission of Cy at 830 nm. In an acidic environment (pH < 6.0), the tertiary amine group of 60 became protonated, and subsequently, the nanomicelles disassembled, resulting in an increase in fluorescence intensity at 830 nm. Moreover, the presence of GSH was shown to hydrolyse the disulfide bond and release CPT, which resulted in a simultaneous change in fluorescence emission intensity to 650 nm. 60 exhibited a dose-dependent cytotoxicity in A549 cells, while no remarkable cytotoxicity was observed in normal QSG-7701 cells, which are typically known to have low intracellular concentrations of GSH. A549 cells, which were pre-incubated with NEM (N-ethylmaleimide, GSH inhibitor) and NaHCO3, separately, showed a significant attenuation of the antiproliferative activity of 60. These results revealed that the release of CPT could only be achieved at low pH and in the presence of a high concentration of GSH, both of which are present in cancer lesions. Furthermore, an in vivo fluorescence imaging (IVIS Spectrum CT imaging system) study demonstrated that 60 predominantly accumulated at the tumour site in A549 tumour-bearing mice. Moreover, 60 significantly reduced the tumour volume compared to control groups (PBS, CPT, and Cy-S-CPT) (Fig. 38b and c), with no loss in the body weight. This work highlights the attractive nature of employing a sequential activation strategy to enhance antitumour activity and alleviate any potential adverse effects in vivo.
 | ||
| Fig. 38 (a) Chemical structures of 60 and the sequentially-activatable release mechanism of CPT. (b) Tumour volumes over 15 days and treatment with PBS, CPT, Cy-S-CPT, and 60 administered at a CPT-equivalent dose of 10 mg kg−1 every 3 days via intravenous injections and (c) representative images of excised tumours of the A549 tumour-bearing mice exposed to PBS, CPT, Cy-S-CPT, and 60. Note: Cy-S-CPT is not functionalised with the diblock copolymer and serves as the control to 60. Reproduced with permission from ref. 163. Copyright (2018) The Royal Society of Chemistry. | ||
Owing to the low pH of the TME and the presence of caspase-3 during cytotoxic drug-induced apoptosis, Li et al. developed 61 designed to release DOX and confirm the induction of apoptosis (Fig. 39).16461 was functionalised with αvβ3 integrin (overexpressed in cancer cells)-targeting group Arg-Gly-Asp (RGD) peptide, a fluorophore 5(6)-carboxylfluorescein (FAM), a fluorescent quencher 4-(dimethylaminoazo)benzene-4-carboxylic acid (Dabcyl), and a caspase-3-cleavable peptide Asp-Glu-Val-Asp (DEVD), all of which were linked to DOX via an acid-labile hydrazone bond. Owing to the targeting nature of RGD tripeptide, 61 was shown to successfully target human glioblastoma U87 cells that overexpress αvβ3 integrin when compared to African green monkey kidney COS-7 cells. 61 exhibited significant antiproliferative activity with IC50 values of 4.3 μM in αvβ3 integrin-positive U87 cells. Initially, the fluorescence of both DOX and FAM were quenched by Dabcyl via Förster resonance energy transfer (FRET).165 However, in acidic environments, the hydrazone bond was cleaved, and the fluorescence emission of DOX at 590 nm was restored. Confocal laser scanning microscopy (CLSM) was used to monitor the release of DOX in U87 cells in real-time. The release of DOX-induced apoptosis resulted in the overexpression of apoptotic biomarker caspase-3 and the hydrolysis of DEVD sequence. Which restored the fluorescence emission of FAM in U87 cells monitored using CLSM. Overall, this strategy enabled monitoring of both DOX release and the induction of apoptosis.
 | ||
| Fig. 39 Chemical structure of 61 and the low pH/caspase-3 sequentially-activated release of DOX. | ||
Cai and co-workers reported the dual-responsive (acid- and thiol-responsive) activatable phototheranostic, 62, for tumour-targeted NIR/PA imaging and PDT in vitro and in vivo (Fig. 40).16662 was developed by covalently linking fluorescent dye 5′-carboxyrhodamine (Rho) with NIR fluorescent photosensitizer heptamethine cyanine IR765 (Cy) using a disulfide linker. With increasing concentrations of GSH (500 μM), the fluorescence emission intensity at 580 nm increased, which was ascribed to the cleavage of the disulfide linker and release of Rho. Decreasing the pH of the solution led to the protonation of the amino unit on Cy and an increase in fluorescence emission intensity at 765 nm. Fluorescence imaging indicated the preferential cellular uptake of 62 in cancerous A549 and MCF-7 cells rather than in normal 293-T (human embryonic kidney) cells. Concentration-dependent phototoxicity was observed for MCF-7 cells under 660 nm laser irradiation (30 mW cm−2, for 5 min), and minimal dark toxicity was observed. NIR fluorescence and PA imaging exhibited good tumour specificity as well as activation by thiols and acidic pH. Maximum imaging intensity values were seen at 15 h after tail vein injection, and this identified time point was used for light irradiation. 62 (150 μL, 150 μM, tail vein) with laser irradiation (660 nm, 100 mW cm−2, 30 min) resulted in a significant reduction in tumour volume, with no obvious side effects being observed by haematoxylin and eosin (H&E) staining. This confirmed a good biosafety profile with no off-target dark toxicities. As such, this work represents the first thiol/pH dual-responsive OFF-ON PDT agent with multimodal imaging and tumour-targeting properties.
 | ||
| Fig. 40 (a) The chemical structure of 62 and its dual low pH and GSH-mediated release of Rho and Cy-H. (b) Illustrated action of the mechanism of 62 in tumour of the mice. Reproduced with permission from ref. 166. Copyright (2017) Published by Ivyspring International Publisher. | ||
Given that a low pH and high GSH concentration are two of the main biomarkers observed in tumour regions, Wang et al. developed a dual-activatable PTT agent, 63, that was activated in the presence of both to provide precise NIR/PA imaging and PTT (Fig. 41).16763 used a citraconic group as the acidic pH-responsive moiety, thiol-protected cysteine (Cys) was used as the GSH-responsive unit and Cy7 as the fluorescent/PA reporter and PTT agent. In an acidic environment (pH 6.5) and the presence of GSH conditions, the citraconic group in 63 was hydrolysed, and a GSH-induced disulfide reduction occurred simultaneously. The free thiol intermediate underwent an intermolecular condensation to form a cyclic dimer, which then self-assembled into homogeneous nanoparticles. 63 exhibited enhanced NIR fluorescence and PA signals in the tumour site of 4T1 tumour-bearing mice after intravenous injection, attributed to the enhanced permeability and retention (EPR) effect168 of the nanoparticles, and achieved maximum values at 4 h and 12 h post-injection, respectively. 63 exhibited excellent photothermal efficacy when activated by low intratumoural pH and high levels of GSH upon 808 nm NIR laser irradiation (1 W cm−2, 10 min), resulting in a rapid local temperature increase of 17 °C. This then led to tumour ablation in more than 60% of mice after 30 days of treatment without causing any acute or systemic toxicity as determined by measuring the body weights and by H&E staining.
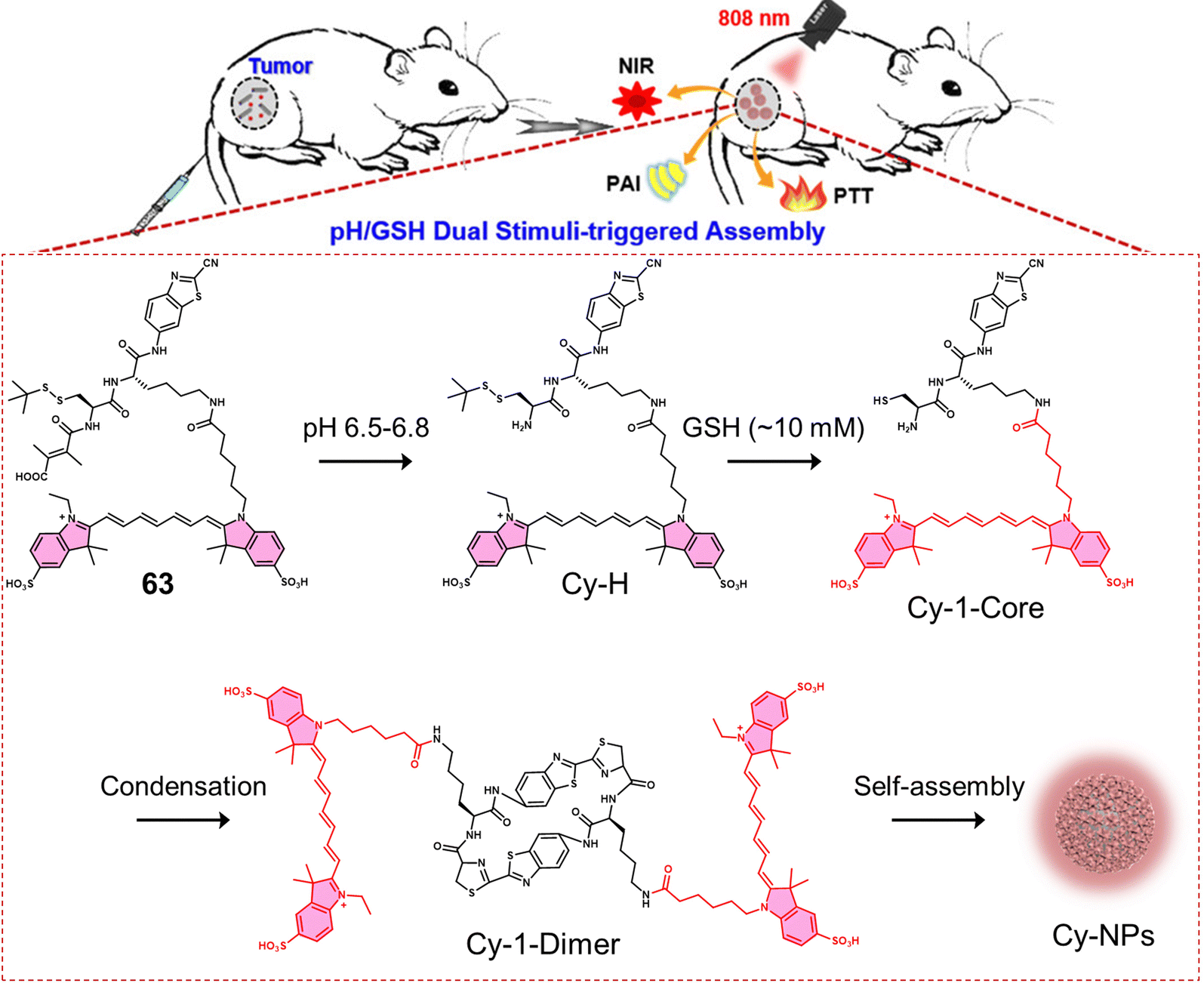 | ||
| Fig. 41 Chemical structures of 63 and the low pH/high GSH sequential activated molecular assembly for enrichment in and eradication of tumour cells. Reproduced with permission from ref. 167. Copyright (2020) American Chemical Society. | ||
4. Conclusion & perspective
In this tutorial review, we focused our attention on prodrugs that can be activated by specific biomarkers found in the tumour microenvironment or by external stimuli, as well as theranostics for the precision-enhanced chemo- and phototherapy of cancer. Tumour microenvironment biomarkers are typically acidic pH, hypoxia, an oxidative and reductive environment, and different enzymes that could be more abundant in cancer cells and readily activate responsive moieties present in prodrugs. The direct chemical modification of clinically approved drugs helps significantly reduce the excessive time burden involved in the drug development process and provides the potential to improve drug efficacy and overcome any adverse side effects. As seen throughout the review, activatable chemotherapeutics as well as phototherapeutic systems (i.e., PDT and PTT) provide improved selectivity for cancer cells and enable the ability to define tumour margins.Although significant progress has been made, the clinical translation of these agents remains challenging. For example, in terms of pH activation, current systems require relatively acidic environments (pH ≤ 5) to be effective in vivo. Therefore, since the tumour microenvironment is a mildly acidic environment (e.g., pH 6.5–7.2), prodrugs with improved pH sensitivity are required. Furthermore, the refinement of the chemical structures is essential to identify optimal pharmacokinetics and pharmacodynamics. This optimization is needed to minimise off-target toxicity and improve tumour specificity. As such, we anticipate that dual or multi-activatable prodrugs are prime targets to provide enhanced tumour specificity for targeted cancer therapy. We are confident that throught sustained research effort that in the near future improved activatable chemotherapeutics and activatable “smart” phototherapeutics will be developed for clinical use as effective agents for cancer treatment.
Abbreviations
| ADC | Antibody-drug conjugate |
| ADEPT | Antibody-directed enzyme prodrug therapy |
| Az | Acetazolamide |
| BFA | Bafilomycin A1 |
| BODIPY | Boron-dipyrrome-thenes |
| BTD | Benzothiadiazole |
| CAIX | Carbonic anhydrase IX |
| CAM | Chorioallantoic membrane |
| CBT | Conjugated backbone twist |
| CBT | 2-Cyanobenzothiazole |
| CDF | 3,4-Difluorobenzyl curcumin |
| CE | Carboxylesterase enzymes |
| CLSM | Confocal laser scanning microscopy |
| CM | 7-(Diethylamino)coumarin |
| CPT | Camptothecin |
| CSCs | Cancer stem cells |
| Cy | Cyanine |
| Cys | Cysteine |
| Dabcyl | 4-(Dimethylaminoazo)benzene-4-carboxylic acid |
| DCA | Dichloroacetic acid |
| DDS | Drug delivery systems |
| DEVD | Asp-Glu-Val-Asp |
| DOX | Doxorubicin |
| ESI-MS | Electrospray ionization mass spectroscopy |
| FAM | 5(6)-Carboxylfluorescein |
| FDU | 5-Fluorodeoxyuridine |
| FRET | Förster resonance energy transfer |
| FRs | Folate receptors |
| fs-TA | Femtosecond transient absorption |
| GFLG | Gly-Phe-Leu-Gly |
| GGT | γ-Glutamyltranspeptidase |
| GSH | Glutathione |
| H&E | Haematoxylin and eosin |
| H+ | Hydrogen ion |
| H2O2 | Hydrogen peroxide |
| HAdase | Hyaluronidase |
| HCC | Hepatocellular carcinoma |
| HPLC | High-performance liquid chromatography |
| HRMS | High-resolution mass spectroscopy |
| ISC | Intersystem crossing |
| LAPTM4B | Lysosomal protein transmembrane 4 beta |
| LC-MS | Liquid chromatography-mass spectrometry |
| MMAE | Monomethyl auristatin E |
| MMPs | Matrix metalloproteinases |
| MRI | Magnetic resonance imaging |
| MTD | Maximum tolerated doses |
| NADH | Nicotinamide adenine dinucleotide |
| NAFLD | Non-alcoholic fatty liver disease |
| NEM | N-Ethylmaleimide |
| NHDFs | Normal human dermal fibroblasts |
| NIR | Near-infrared fluorescence |
| NTR | Nitroreductase |
| O2˙− | Superoxide anion radicals |
| PA | Photoacoustic |
| PDT | Photodynamic therapy |
| PDT | Photodynamic therapy |
| PEG | Poly(ethylene glycol) |
| PeT | Photoinduced electron transfer |
| PPA | Pyropheophorbide α |
| PPa | Pheophorbide a |
| PS | Photosensitiser |
| PTT | Photothermal therapy |
| Pyro | Pyropheophorbide α |
| RGD | Arg-Gly-Asp |
| Rho | 5′-Carboxyrhodamines |
| RP-HPLC | Reverse-phase high-performance liquid chromatography |
| SA | Sialic acid |
| SAHA | Suberoylanilide hydroxamic acid |
| TME | Tumour microenvironment |
| TPP | Triphenylphosphine |
| VEGFR | Vascular endothelial growth factor receptor |
| XIAP | X-linked inhibitor of apoptosis protein |
| β-Gal | β-Galactosidase |
| Φ 1O2 | 1O2 quantum yields |
| Φ ISC | Intersystem crossing efficiency |
| 1O2 | Singlet oxygen |
| 3O2 | Ground state oxygen |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
X.-P. H. thanks the National Natural Science Foundation of China (No. 21788102, 91853201, 82130099 and 9185920077), the Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX03), the National Science Foundation of Shanghai (No. 21XD1404600, 21JC1406600, and 22140901000), the International Cooperation Program of Shanghai Science and Technology Committee (No. 17520750100), the Fundamental Research Funds for the Central Universities (222201717003) and the Programme of Introducing Talents of Discipline to Universities (B16017) for financial support. J. L. and Y. Z. thank the National Natural Science Foundation of China (No. 82130099, 82151219, 31871414 and 81971265) and the Shanghai Municipal Science and Technology Major Project (No. 22ZR1415200). H.-H. H. thanks the National Natural Science Foundation of China (No. 22107029) and Project funded by the China Postdoctoral Science Foundation (No. 2020M681196). T. D. J. wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01). L. W. wishes to thank the China Scholarship Council and the University of Bath for supporting his PhD in the UK. J. S. K. thank the financial support received from the National Research Foundation of Korea (CRI project no. 2018R1A3B1052702, 2019M3E5D1A01068998). M. L. wishes to thank the support of the Brain Pool Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant No. 2020H1D3A1A02080172, M. L.). A. C. S. would like to thank the Glasstone Research fellowship (University of Oxford) and Jesus College, Oxford for support.References
- J. Fares, M. Y. Fares, H. H. Khachfe, H. A. Salhab and Y. Fares, Signal Transduction Targeted Ther., 2020, 5, 28 CrossRef PubMed.
- D. Hanahan, Cancer Discov., 2022, 12, 31–46 CrossRef CAS PubMed.
- T. N. Seyfried and L. C. Huysentruyt, Crit. Rev. Oncog., 2013, 18, 43–73 CrossRef PubMed.
- R. Haddad, N. Alrabadi, B. Altaani and T. Li, Polymers, 2022, 14, 658 CrossRef CAS PubMed.
- K. Wu, N. A. Yee, S. Srinivasan, A. Mahmoodi, M. Zakharian, J. M. Mejia Oneto and M. Royzen, Chem. Sci., 2021, 12, 1259–1271 RSC.
- K. E. Arnst, S. Banerjee, H. Chen, S. Deng, D.-J. Hwang, W. Li and D. D. Miller, Med. Res. Rev., 2019, 39, 1398–1426 CrossRef PubMed.
- D. S. Shewach and R. D. Kuchta, Chem. Rev., 2009, 109, 2859–2861 CrossRef CAS PubMed.
- H. Kitao, M. Iimori, Y. Kataoka, T. Wakasa, E. Tokunaga, H. Saeki, E. Oki and Y. Maehara, Cancer Sci., 2018, 109, 264–271 CrossRef CAS PubMed.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS PubMed.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS PubMed.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- V. Abet, F. Filace, J. Recio, J. Alvarez-Builla and C. Burgos, Eur. J. Med. Chem., 2017, 127, 810–827 CrossRef CAS PubMed.
- X. Zhang, X. Li, Q. You and X. Zhang, Eur. J. Med. Chem., 2017, 139, 542–563 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- Y. Chen, L. Li, W. Chen, H. Chen and J. Yin, Chin. Chem. Lett., 2019, 30, 1353–1360 CrossRef CAS.
- M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- J. B. Zawilska, J. Wojcieszak and A. B. Olejniczak, Pharmacol. Rep., 2013, 65, 1–14 CrossRef CAS PubMed.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS PubMed.
- J. Guo, X. Pan, C. Wang and H. Liu, Bioconjugate Chem., 2022, 33, 993–1010 CrossRef CAS PubMed.
- J. M. Silva, E. Silva and R. L. Reis, J. Controlled Release, 2019, 298, 154–176 CrossRef CAS PubMed.
- N. S. H. Motlagh, P. Parvin, F. Ghasemi and F. Atyabi, Biomed. Opt. Express, 2016, 7, 2400–2406 CrossRef CAS PubMed.
- J. Dey and I. M. Warner, J. Lumin., 1997, 71, 105–114 CrossRef CAS.
- A. Steinbrueck, A. C. Sedgwick, H.-H. Han, M. Y. Zhao, S. Sen, D.-Y. Huang, Y. Zang, J. Li, X.-P. He and J. L. Sessler, Chem. Commun., 2021, 57, 5678–5681 RSC.
- L. H. Goetz and N. J. Schork, Fertil. Steril., 2018, 109, 952–963 CrossRef PubMed.
- M. Wang, J. Zhao, L. Zhang, F. Wei, Y. Lian, Y. Wu, Z. Gong, S. Zhang, J. Zhou, K. Cao, X. Li, W. Xiong, G. Li, Z. Zeng and C. Guo, J. Cancer, 2017, 8, 761–773 CrossRef CAS PubMed.
- Y. Yuan, Y.-C. Jiang, C.-K. Sun and Q.-M. Chen, Oncol. Rep., 2016, 35, 2499–2515 CrossRef CAS PubMed.
- G. Hao, Z. P. Xu and L. Li, RSC Adv., 2018, 8, 22182–22192 RSC.
- H. Alimoradi, S. S. Matikonda, A. B. Gamble, G. I. Giles and K. Greish, Curr. Pharm. Des., 2016, 22, 2808–2820 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed.
- A. Sharma, M.-G. Lee, M. Won, S. Koo, J. F. Arambula, J. L. Sessler, S.-G. Chi and J. S. Kim, J. Am. Chem. Soc., 2019, 141, 15611–15618 CrossRef CAS PubMed.
- C. Lin, H. He, Y. Zhang, M. Xu, F. Tian, L. Li and Y. Wang, RSC Adv., 2020, 10, 3084–3091 RSC.
- Servier Medical Art, https://smart.servier.com/, accessed February 2022.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820–15826 CrossRef CAS PubMed.
- J. Xie, Y. Wang, W. Choi, P. Jangili, Y. Ge, Y. Xu, J. Kang, L. Liu, B. Zhang, Z. Xie, J. He, N. Xie, G. Nie, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2021, 50, 9152–9201 RSC.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS PubMed.
- M. Li, Y. Shao, J. H. Kim, Z. Pu, X. Zhao, H. Huang, T. Xiong, Y. Kang, G. Li, K. Shao, J. Fan, J. W. Foley, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2020, 142, 5380–5388 CrossRef CAS PubMed.
- Z.-S. Yang, Y. Yao, A. C. Sedgwick, C. Li, Y. Xia, Y. Wang, L. Kang, H. Su, B.-W. Wang, S. Gao, J. L. Sessler and J.-L. Zhang, Chem. Sci., 2020, 11, 8204–8213 RSC.
- H.-B. Cheng, B. Qiao, H. Li, J. Cao, Y. Luo, K. M. K. Swamy, J. Zhao, Z. Wang, J. Y. Lee, X.-J. Liang and J. Yoon, J. Am. Chem. Soc., 2021, 143, 2413–2422 CrossRef CAS PubMed.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- P. Sarbadhikary, B. P. George and H. Abrahamse, Theranostics, 2021, 11, 9054–9088 CrossRef CAS PubMed.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- M. R. K. Ali, M. A. Rahman, Y. Wu, T. Han, X. Peng, M. A. Mackey, D. Wang, H. J. Shin, G. Chen Zhuo, H. Xiao, R. Wu, Y. Tang, D. M. Shin and M. A. EI-Sayed, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3110–E3118 CAS.
- Y. Xu, Y. Wang, J. An, A. C. Sedgwick, M. Li, J. Xie, W. Hu, J. Kang, S. Sen, A. Steinbrueck, B. Zhang, L. Qiao, S. Wageh, J. F. Arambula, L. Liu, H. Zhang, J. L. Sessler and J. S. Kim, Bioact. Mater., 2022, 14, 76–85 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- Y. Ren, A. C. Sedgwick, J. Chen, G. Thiabaud, C. V. Chau, J. An, J. F. Arambula, X.-P. He, J. S. Kim, J. L. Sessler and C. Liu, J. Am. Chem. Soc., 2020, 142, 16156–16160 CrossRef CAS PubMed.
- J. Chen, A. C. Sedgwick, S. Sen, Y. Ren, Q. Sun, C. Chau, J. F. Arambula, T. Sarma, L. Song, J. L. Sessler and C. Liu, Chem. Sci., 2021, 12, 9916–9921 RSC.
- H.-H. Han, H. Tian, Y. Zang, A. C. Sedgwick, J. Li, J. L. Sessler, X.-P. He and T. D. James, Chem. Soc. Rev., 2021, 50, 9391–9429 RSC.
- P. S. Low, S. Singhal and M. Srinivasarao, Curr. Opin. Chem. Biol., 2018, 45, 64–72 CrossRef CAS PubMed.
- W.-T. Dou, H.-H. Han, A. C. Sedgwick, G.-B. Zhu, Y. Zang, X.-R. Yang, J. Yoon, T. D. James, J. Li and X.-P. He, Sci. Bull., 2022, 67, 853–878 CrossRef CAS PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- M. Vellard, Curr. Opin. Biotechnol, 2003, 14, 444–450 CrossRef CAS PubMed.
- M. H. Baig, M. Adil, R. Khan, S. Dhadi, K. Ahmad, G. Rabbani, T. Bashir, M. A. Imran, F. M. Husain, E. J. Lee, M. A. Kamal and I. Choi, Semin. Cancer Biol., 2019, 56, 1–11 CrossRef CAS PubMed.
- A. Sreedhar and Y. Zhao, Biomed. Rep., 2018, 8, 3–10 CAS.
- S. E. Pratt, S. Durland-Busbice, R. L. Shepard, K. Heinz-Taheny, P. W. Iversen and A. H. Dantzig, Clin. Cancer Res., 2013, 19, 1159–1168 CrossRef CAS PubMed.
- L. Her and H.-J. Zhu, Drug Metab. Dispos., 2020, 48, 230–244 CrossRef CAS PubMed.
- H. Dong, L. Pang, H. Cong, Y. Shen and B. Yu, Drug Delivery, 2019, 26, 416–432 CrossRef CAS PubMed.
- J. Zhuang, N. Li, Y. Zhang, B. Li, H. Wen, X. Zhang, T. Zhang, N. Zhao and B. Z. Tang, CCS Chem., 2021, 4, 1028–1043 CrossRef.
- C. Perez, K. B. Daniel and S. M. Cohen, ChemMedChem, 2013, 8, 1662–1667 CrossRef CAS PubMed.
- A. Sharma, M.-G. Lee, H. Shi, M. Won, J. F. Arambula, J. L. Sessler, J. Y. Lee, S.-G. Chi and J. S. Kim, Chem, 2018, 4, 2370–2383 CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- C. Carvalho, R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS PubMed.
- E. D. Michelakis, L. Webster and J. R. Mackey, Br. J. Cancer, 2008, 99, 989–994 CrossRef CAS PubMed.
- W. Li, H. Zhang, Y. G. Assaraf, K. Zhao, X. Xu, J. Xie, D.-H. Yang and Z.-S. Chen, Drug Resistance Updates, 2016, 27, 14–29 CrossRef PubMed.
- X. Lin, Z. Xiao, T. Chen, S. H. Liang and H. Guo, Front. Oncol., 2020, 10, 317 CrossRef PubMed.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283, 30433–30437 CrossRef PubMed.
- K. G. Ponder and L. H. Boise, Cell Death Discovery, 2019, 5, 56 CrossRef PubMed.
- S. W. Chung, J. U. Choi, Y. S. Cho, H. R. Kim, T. H. Won, P. Dimitrion, O.-C. Jeon, S. W. Kim, I.-S. Kim, S. Y. Kim and Y. Byun, Adv. Sci., 2018, 5, 1800368 CrossRef PubMed.
- S. Bae, P. M. Siu, S. Choudhury, Q. Ke, J. H. Choi, Y. Y. Koh and P. M. Kang, Am. J. Physiol. Heart Circ. Physiol., 2010, 299, H1374–H1381 CrossRef CAS PubMed.
- Y. Li, T. Mei, S. Han, T. Han, Y. Sun, H. Zhang and F. An, Chin. Chem. Lett., 2020, 31, 3027–3040 CrossRef CAS.
- Y. Yuan, C.-J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- A. C. Sedgwick, K.-C. Yan, D. N. Mangel, Y. Shang, A. Steinbrueck, H.-H. Han, J. T. Brewster, X.-L. Hu, D. W. Snelson, V. M. Lynch, H. Tian, X.-P. He and J. L. Sessler, J. Am. Chem. Soc., 2021, 143, 1278–1283 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS PubMed.
- A. Corti, M. Franzini, A. Paolicchi and A. Pompella, Anticancer Res., 2010, 30, 1169–1181 CAS.
- M. H. Hanigan, H. F. Frierson, P. E. Swanson and B. R. De Young, Hum. Pathol., 1999, 30, 300–305 CrossRef CAS PubMed.
- B. He, X. Sui, B. Yu, S. Wang, Y. Shen and H. Cong, Drug Delivery, 2020, 27, 1474–1490 CrossRef CAS PubMed.
- F. Zhou, S. Yang, C. Zhao, W. Liu, X. Yao, H. Yu, X. Sun and Y. Liu, Theranostics, 2021, 11, 7045–7056 CrossRef CAS PubMed.
- M. Chiba, Y. Ichikawa, M. Kamiya, T. Komatsu, T. Ueno, K. Hanaoka, T. Nagano, N. Lange and Y. Urano, Angew. Chem., Int. Ed., 2017, 56, 10418–10422 CrossRef CAS PubMed.
- M. W. Kryman, G. A. Schamerhorn, J. E. Hill, B. D. Calitree, K. S. Davies, M. K. Linder, T. Y. Ohulchanskyy and M. R. Detty, Organometallics, 2014, 33, 2628–2640 CrossRef CAS PubMed.
- J. Garbe and M. Collin, J. Innate Immun., 2012, 4, 121–131 CrossRef CAS PubMed.
- E. Waidely, A.-R. O. Al-Yuobi, A. S. Bashammakh, M. S. El-Shahawi and R. M. Leblanc, Analyst, 2016, 141, 36–44 RSC.
- X. Chai, H.-H. Han, A. C. Sedgwick, N. Li, Y. Zang, T. D. James, J. Zhang, X.-L. Hu, Y. Yu, Y. Li, Y. Wang, J. Li, X.-P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 18005–18013 CrossRef CAS PubMed.
- X. Zhen, J. Zhang, J. Huang, C. Xie, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 7804–7808 CrossRef CAS PubMed.
- A. Abawi, X. Wang, J. Bompard, A. Bérot, V. Andretto, L. Gudimard, C. Devillard, E. Petiot, B. Joseph, G. Lollo, T. Granjon, A. Girard-Egrot and O. Maniti, Int. J. Mol. Sci., 2021, 22, 4103 CrossRef CAS PubMed.
- D. Trachootham, W. Lu, M. A. Ogasawara, N. R.-D. Valle and P. Huang, Antioxid. Redox Signaling, 2008, 10, 1343–1374 CrossRef CAS PubMed.
- X. Guo, Y. Cheng, X. Zhao, Y. Luo, J. Chen and W.-E. Yuan, J. Nanobiotechnol., 2018, 16, 74 CrossRef PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- C. Lennicke, J. Rahn, R. Lichtenfels, L. A. Wessjohann and B. Seliger, Cell Commun. Signaling, 2015, 13, 39 CrossRef PubMed.
- H. Sies, Redox Biol., 2017, 11, 613–619 CrossRef CAS PubMed.
- E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- T. Meng, J. Han, P. Zhang, J. Hu, J. Fu and J. Yin, Chem. Sci., 2019, 10, 7156–7162 RSC.
- X. Xie, X. Yang, T. Wu, Y. Li, M. Li, Q. Tan, X. Wang and B. Tang, Anal. Chem., 2016, 88, 8019–8025 CrossRef CAS PubMed.
- A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS PubMed.
- L. Kennedy, J. K. Sandhu, M.-E. Harper and M. Cuperlovic-Culf, Biomolecules, 2020, 10, 1429 CrossRef CAS PubMed.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450–6456 CrossRef CAS PubMed.
- R. An, X. Cheng, S. Wei, Y. Hu, Y. Sun, Z. Huang, H.-Y. Chen and D. Ye, Angew. Chem., Int. Ed., 2020, 59, 20636–20644 CrossRef CAS PubMed.
- X. Wang, W. Lin, W. Zhang, C. Li, T. Sun, G. Chen and Z. Xie, J. Colloid Interface Sci., 2019, 536, 208–214 CrossRef CAS PubMed.
- M. Y. Lucero and J. Chan, Nat. Chem., 2021, 13, 1248–1256 CrossRef CAS PubMed.
- A. C. Sedgwick, J. E. Gardiner, G. Kim, M. Yevglevskis, M. D. Lloyd, A. T. A. Jenkins, S. D. Bull, J. Yoon and T. D. James, Chem. Commun., 2018, 54, 4786–4789 RSC.
- W. Plunkett, P. Huang, Y. Z. Xu, V. Heinemann, R. Grunewald and V. Gandhi, Semin. Oncol., 1995, 22, 3–10 CAS.
- N. Papadopoulos, K. W. Kinzler and B. Vogelstein, Nat. Biotechnol., 2006, 24, 985–995 CrossRef CAS PubMed.
- J. Sun, K. Du, J. Diao, X. Cai, F. Feng and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 12122–12128 CrossRef CAS PubMed.
- M. Weber, H.-H. Han, B.-H. Li, M. L. Odyniec, C. E. F. Jarman, Y. Zang, S. D. Bull, A. B. Mackenzie, A. C. Sedgwick, J. Li, X.-P. He and T. D. James, Chem. Sci., 2020, 11, 8567–8571 RSC.
- H.-H. Han, A. C. Sedgwick, Y. Shang, N. Li, T. Liu, B.-H. Li, K. Yu, Y. Zang, J. T. Brewster, M. L. Odyniec, M. Weber, S. D. Bull, J. Li, J. L. Sessler, T. D. James, X.-P. He and H. Tian, Chem. Sci., 2020, 11, 1107–1113 RSC.
- M. Miller, A. Mellul, M. Braun, D. Sherill-Rofe, E. Cohen, Z. Shpilt, I. Unterman, O. Braitbard, J. Hochman, E. Y. Tshuva and Y. Tabach, iScience, 2020, 23, 101262 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- J. L. Sessler, T. Murai, V. Lynch and M. Cyr, J. Am. Chem. Soc., 1988, 110, 5586–5588 CrossRef CAS.
- G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef CAS PubMed.
- G. Thiabaud, G. He, S. Sen, K. A. Shelton, W. B. Baze, L. Segura, J. Alaniz, R. M. Macias, G. Lyness, A. B. Watts, H. M. Kim, H. Lee, M. Y. Cho, K. S. Hong, R. Finch, Z. H. Siddik, J. F. Arambula and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7021–7029 CrossRef CAS PubMed.
- C. J. Adams and T. J. Meade, Chem. Sci., 2020, 11, 2524–2530 RSC.
- A. E. Nejad, S. Najafgholian, A. Rostami, A. Sistani, S. Shojaeifar, M. Esparvarinha, R. Nedaeinia, S. H. Javanmard, M. Taherian, M. Ahmadlou, R. Salehi, B. Sadeghi and M. Manian, Cancer Cell Int., 2021, 21, 62 CrossRef PubMed.
- J.-N. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160–6224 CrossRef CAS PubMed.
- W. Liu, H. Liu, X. Peng, G. Zhou, D. Liu, S. Li, J. Zhang and S. Wang, Bioconjugate Chem., 2018, 29, 3332–3343 CrossRef CAS PubMed.
- X. Zhao, S. Long, M. Li, J. Cao, Y. Li, L. Guo, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2020, 142, 1510–1517 CrossRef CAS PubMed.
- L. Yang, P. Shi, G. Zhao, J. Xu, W. Peng, J. Zhang, G. Zhang, X. Wang, Z. Dong, F. Chen and H. Cui, Signal Transduction Targeted Ther., 2020, 5, 8 CrossRef PubMed.
- J. H. Kim, P. Verwilst, M. Won, J. Lee, J. L. Sessler, J. Han and J. S. Kim, J. Am. Chem. Soc., 2021, 143, 14115–14124 CrossRef CAS PubMed.
- F. E. Lock, P. C. McDonald, Y. Lou, I. Serrano, S. C. Chafe, C. Ostlund, S. Aparicio, J.-Y. Winum, C. T. Supuran and S. Dedhar, Oncogene, 2013, 32, 5210–5219 CrossRef CAS PubMed.
- A. Skwarska, E. D. D. Calder, D. Sneddon, H. Bolland, M. L. Odyniec, I. N. Mistry, J. Martin, L. K. Folkes, S. J. Conway and E. M. Hammond, Cell Chem. Biol., 2021, 28, 1258–1270.E13 CrossRef CAS PubMed.
- Z. Zhang, H. Yamashita, T. Toyama, H. Sugiura, Y. Ando, K. Mita, M. Hamaguchi, Y. Hara, S. Kobayashi and H. Iwase, Breast Cancer Res. Treat., 2005, 94, 11–16 CrossRef CAS PubMed.
- T. Sudo, K. Mimori, N. Nishida, R. Kogo, T. Iwaya, F. Tanaka, K. Shibata, H. Fujita, K. Shirouzu and M. Mori, Oncol. Rep., 2011, 26, 777–782 CAS.
- W. Feng, B. Zhang, D. Cai and X. Zou, Cancer Lett., 2014, 347, 183–190 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- X. Meng, J. Zhang, Z. Sun, L. Zhou, G. Deng, S. Li, W. Li, P. Gong and L. Cai, Theranostics, 2018, 8, 6025–6034 CrossRef CAS PubMed.
- Y. Zhou, M. Maiti, A. Sharma, M. Won, L. Yu, L. X. Miao, J. Shin, A. Podder, K. N. Bobba, J. Han, S. Bhuniya and J. S. Kim, J. Controlled Release, 2018, 288, 14–22 CrossRef CAS PubMed.
- W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713–13719 CrossRef CAS PubMed.
- C. Wang, S. Wang, Y. Wang, H. Wu, K. Bao, R. Sheng and X. Li, Sci. Rep., 2020, 10, 12127 CrossRef CAS PubMed.
- N. Piasentin, E. Milotti and R. Chignola, Sci. Rep., 2020, 10, 13613 CrossRef CAS PubMed.
- B. Lin, H. Chen, D. Liang, W. Lin, X. Qi, H. Liu and X. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 11157–11166 CrossRef CAS PubMed.
- E. Boedtkjer and S. F. Pedersen, Annu. Rev. Physiol., 2020, 82, 103–126 CrossRef CAS PubMed.
- J.-T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X.-Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- Y. Jin, Y. Huang, H. Yang, G. Liu and R. Zhao, Chem. Commun., 2015, 51, 14454–14457 RSC.
- L. Wang, Y. Meng and Q.-Y. Zhang, BMC Cancer, 2019, 19, 293 CrossRef PubMed.
- S.-Y. Li, L.-H. Liu, H.-Z. Jia, W.-X. Qiu, L. Rong, H. Cheng and X.-Z. Zhang, Chem. Commun., 2014, 50, 11852–11855 RSC.
- M. H. Lee, E.-J. Kim, H. Lee, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, Chem. Commun., 2016, 52, 10551–10554 RSC.
- S. Siriwibool, N. Kaekratoke, K. Chansaenpak, K. Siwawannapong, P. Panajapo, K. Sagarik, P. Noisa, R.-Y. Lai and A. Kamkaew, Sci. Rep., 2020, 10, 1283 CrossRef CAS PubMed.
- W. Hu, T. He, H. Zhao, H. Tao, R. Chen, L. Jin, J. Li, Q. Fan, W. Huang, A. Baev and P. N. Prasad, Angew. Chem., Int. Ed., 2019, 58, 11105–11111 CrossRef CAS PubMed.
- X. Meng, W. Li, Z. Sun, J. Zhang, L. Zhou, G. Deng, P. Gong and L. Cai, J. Mater. Chem. B, 2017, 5, 9405–9411 RSC.
- S. T. Teoh, M. P. Ogrodzinski, C. Ross, K. W. Hunter and S. Y. Lunt, Front. Oncol., 2018, 8, 174 CrossRef PubMed.
- X. Wu, M. Yu, B. Lin, H. Xing, J. Han and S. Han, Chem. Sci., 2015, 6, 798–803 RSC.
- Z. Li, Y. Song, Y. Yang, L. Yang, X. Huang, J. Han and S. Han, Chem. Sci., 2012, 3, 2941–2948 RSC.
- T. Yoshimori, A. Yamamoto, Y. Moriyama, M. Futai and Y. Tashiro, J. Biol. Chem., 1991, 266, 17707–17712 CrossRef CAS PubMed.
- L. Josa-Culleré and A. Llebaria, ChemPhotoChem, 2021, 5, 296–314 CrossRef.
- D. C. F. Monteiro, E. Amoah, C. Rogers and A. R. Pearson, Acta Crystallogr., Sect. D: Struct. Biol., 2021, 77, 1218–1232 CrossRef CAS PubMed.
- P. Dunkel and J. Ilaš, Cancers, 2021, 13, 3237 CrossRef CAS PubMed.
- S. Bonnet, Dalton Trans., 2018, 47, 10330–10343 RSC.
- M. M. Dcona, K. Mitra and M. C. T. Hartman, RSC Med. Chem., 2020, 11, 982–1002 RSC.
- B. M. Vickerman, E. M. Zywot, T. K. Tarrant and D. S. Lawrence, Nat. Rev. Chem., 2021, 5, 816–834 CrossRef PubMed.
- Y. Hou, Z. Zhou, K. Huang, H. Yang and G. Han, ChemPhotoChem, 2018, 2, 1005–1011 CrossRef CAS.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- N. P. Toupin, K. Arora, P. Shrestha, J. A. Peterson, L. J. Fischer, E. Rajagurubandara, I. Podgorski, A. H. Winter and J. J. Kodanko, ACS Chem. Biol., 2019, 14, 2833–2840 CrossRef CAS PubMed.
- Z. Wang, N. Wang, S.-C. Cheng, K. Xu, Z. Deng, S. Chen, Z. Xu, K. Xie, M.-K. Tse, P. Shi, H. Hirao, C.-C. Ko and G. Zhu, Chem, 2019, 5, 3151–3165 CAS.
- X. Feng, D. Jiang, T. Kang, J. Yao, Y. Jing, T. Jiang, J. Feng, Q. Zhu, Q. Song, N. Dong, X. Gao and J. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17817–17832 CrossRef CAS PubMed.
- J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley, Nat. Chem., 2021, 13, 805–810 CrossRef CAS PubMed.
- B. Rini and M. Y. Al-Marrawi, Expert Opin. Pharmacother., 2011, 12, 1171–1189 CrossRef CAS PubMed.
- C. Yan, Z. Guo, Y. Liu, P. Shi, H. Tian and W.-H. Zhu, Chem. Sci., 2018, 9, 6176–6182 RSC.
- S.-Y. Li, L.-H. Liu, L. Rong, W.-X. Qiu, H.-Z. Jia, B. Li, F. Li and X.-Z. Zhang, Adv. Funct. Mater., 2015, 25, 7317–7326 CrossRef CAS.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- X. Meng, Y. Yang, L. Zhou, L. Zhang, Y. Lv, S. Li, Y. Wu, M. Zheng, W. Li, G. Gao, G. Deng, T. Jiang, D. Ni, P. Gong and L. Cai, Theranostics, 2017, 7, 1781–1794 CrossRef CAS PubMed.
- A. Wang, Q. Mao, M. Zhao, S. Ye, J. Fang, C. Cui, Y. Zhao, Y. Zhang, Y. Zhang, F. Zhou and H. Shi, Anal. Chem., 2020, 92, 16113–16121 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |