
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocontrolled RAFT polymerization: past, present, and future
Yungyeong
Lee
a,
Cyrille
Boyer
 *c and
Min Sang
Kwon
*ab
*c and
Min Sang
Kwon
*ab
aDepartment of Materials Science and Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: minsang@snu.ac.kr
bResearch Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea
cCluster for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, and Australian Centre for NanoMedicine, UNSW Sydney, Kensington, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au
First published on 11th April 2023
Abstract
In this review, we provide a brief history, progress, and applications, and discuss the remaining challenges of photocontrolled reversible addition–fragmentation chain transfer (RAFT) polymerization (i.e., photoinduced electron/energy transfer-RAFT (PET-RAFT), photoiniferter, and photomediated cationic RAFT polymerization). Among these, visible-light-driven RAFT polymerization has attracted particular attention in recent years due to its benefits, including low energy consumption and the safe reaction procedure. Moreover, the incorporation of visible-light photocatalysis in the polymerization has conferred attractive features, such as spatiotemporal control and oxygen tolerance; however, a clear understanding of the reaction mechanism has not been completely provided. We also present recent research efforts to elucidate the polymerization mechanisms with the aid of quantum chemical calculations combined with experimental evidence. This review offers an insight into the better design of polymerization systems for desired applications and helps realize the full potential of photocontrolled RAFT polymerization in both academic- and industrial-scale applications.
 Yungyeong Lee | Yungyeong Lee received her B.S. and M.S. degrees in Chemistry from Ulsan National Institute of Science and Technology (UNIST) in 2017 and 2019, respectively. Since 2021, she has been pursuing a PhD in Materials Science and Engineering (MSE) at Seoul National University (SNU) under the supervision of Prof. Min Sang Kwon. Her research interest focuses on the development of organic photocatalysts, investigation of their photophysical and electrochemical properties, and application to visible-light-mediated polymerization reactions particularly in aqueous systems. |
 Cyrille Boyer | Prof Cyrille Boyer is a Full Professor in the School of Chemical Engineering, Deputy Head of School, and Co-Director of Australian Centre for Nanomedicine. In 2022, he was awarded an Australian Laureate Fellowship. Cyrille's research interests mainly cover the preparation of functional macromolecules using photocatalysts, which find applications in various areas, including in nanomedicine and in energy storage. The research of his group has been recognized by several research awards, including 2018 IUPAC-Polymer International Young Researcher award, 2016 ACS Biomacromolecules/Macromolecules Young Researcher Award, Le Fevre Memorial Prize for Chemistry; and 2015 Malcolm McIntosh Prize for Physical Science. |
 Min Sang Kwon | Min Sang Kwon received his B.S. degrees in Chemistry and MSE from SNU. Then, he did his PhD in Chemistry at SNU under the guidance of Prof. Eun Lee. After finishing his postdoctoral studies at SNU with Prof. Soo Young Park, he moved to University of Michigan to work with Prof. Jinsang Kim. In 2016, he began his independent career at UNIST as an Assistant Professor. In 2020, he moved back to his alma mater, SNU, where he is currently an Associate Professor of MSE. He has a broad interest in organic chemistry, photochemistry, and polymer chemistry. |
1. Introduction
1.1. Visible light as an energy source for photomediated reactions
Visible light refers to electromagnetic waves with wavelengths in the range of approximately 400–750 nm that are detectable by the human eye.1 As visible light constitutes approximately half of the solar energy reaching the surface of earth, tremendous efforts have been devoted to utilize visible light as an energy source in the field of solar energy utilization including photovoltaics and solar fuels. Owing to Ciamician's pioneering insight, influenced by photosynthesis in nature,2 light has been consistently used as an energy source for organic reactions.3 Because most organic molecules do not absorb light in the visible range, ultraviolet (UV) light has long been employed as the primary energy source for photochemical reactions. However, due to its high energy, UV light often causes substantial side reactions and thereby reduces the reaction efficiency, which consequently limits the scope of photochemical reactions. Therefore, compared to conventional thermal reactions, photochemical reactions have been considerably underexplored.In 2008, MacMillan,4 Yoon,5 and Stephenson6 and coworkers reported that complex organic reactions proceed very efficiently in the presence of photoredox catalysts (PCs) under visible light irradiation. From these pioneering studies, organic chemists recognized that low-energy visible light could be a suitable energy source for various organic reactions, which led to the origin of a new field called “visible-light photocatalysis”.7–9 Thereafter, for more than a decade, visible-light photocatalysis has indeed been used as a mild and efficient method to activate molecules for a wide range of organic transformations that cannot be realized by other methodologies of chemical catalysis.10–13 Under irradiation, excited-state PCs engage in electron or energy transfer and consequently generate highly reactive radical species from the corresponding stable substrates. These previously inaccessible reaction scaffolds have enabled significant development of radical chemistry in organic synthesis.
1.2. Visible-light-driven reversible deactivation radical polymerization
Reversible deactivation radical polymerization (RDRP) is a powerful technique to prepare polymers with controlled molecular weights, narrow molecular weight distributions (MWDs), compositions, functionalities, and architectures that impact the properties of polymers.14,15 As this method provides appropriate controllability and compatibility with various monomers under mild conditions, it has been employed in numerous applications such as coatings, adhesives, cosmetics, ink materials for printing, detergents, paints, and surfactants.16 In RDRP, heat is typically utilized as an energy source for polymerization in the presence of an initiator or a catalyst.17,18Consistent efforts have been made to utilize light as an energy source for RDRP because of the potential benefits, including spatiotemporal control over polymerization and fast kinetics at ambient temperature as compared to conventional thermal processes.19–21 Early systems for photomediated RDRP, which utilized high-energy UV light, had mostly relied on (i) a traditional photoinitiating system based on Norrish type I/II reactions,22 (ii) a catalytic species with absorption only in the UV range,23,24 or (iii) a chain transfer agent (CTA) (i.e., RAFT agent) that undergoes photolysis under UV light irradiation.25–27 However, these processes typically exhibited insufficient controllability of polymerization, side reactions and/or too fast kinetics due to the high energy of UV light. Nevertheless, it should be noted that in the field of polymer chemistry, a variety of UV-light-induced photoinitiating systems had been actively investigated before the emergence of visible-light photocatalysis owing to their utility in industrial applications such as photoresists and/or photocuring systems.
With regard to the milder reaction conditions of polymerization, visible light has been introduced as an energy source for RDRP since the early 2010. Inspired by visible-light photocatalysis, in 2012 Hawker and Fors demonstrated atom transfer radical polymerization (ATRP) of methacrylates using tris(2-phenylpyridine)iridium (Ir(ppy)3) as a PC and a 50 W fluorescent lamp.28 Matyjaszewski and coworkers reported another photomediated ATRP using a copper (Cu) complex under violet and blue light irradiation (392 and 450 nm, respectively),29 where photoinduced reduction of the Cu2+ complex and subsequent activation of the alkyl halide initiator were achieved by irradiation rather than heating. By the addition of visible-light-absorbing dyes to indirectly reduce the Cu2+ complex and generate initiating radicals, Yagci and coworkers further expanded the polymerization wavelength to 400–500 nm.30 In the case of RAFT polymerization, in 2014, Boyer and coworkers employed Ir(ppy)3 to activate the reversible addition–fragmentation chain transfer (RAFT) polymerization of various vinylic monomers (including (meth)acrylamides, (meth)acrylates, styrene, and vinyl esters) under blue light irradiation (435 nm).31 In 2015, Boyer,32 Qiao,33 and coworkers separately reported the successful RAFT polymerization of (meth)acrylates under blue or green light irradiation (435 and 530 nm, respectively) in the absence of PCs. These visible-light-driven RDRPs retained excellent controllability of polymerization similar to conventional thermal RDRPs. Since these initial studies, various types of visible-light-driven RDRPs have been extensively studied from different perspectives such as (i) reaction optimization, (ii) mechanistic studies, (iii) understanding the unique features of each polymerization, (iv) catalyst/reagent developments, (v) syntheses of polymers with a novel structure, and (vi) new applications.19,20,34–36
1.3. Visible-light-driven RAFT polymerization
In conventional RAFT polymerization, an initiator generates radicals typically by thermal decomposition and a CTA ensures the controllability of polymerization via RAFT equilibrium.37 In contrast, in photomediated RAFT polymerization, a photoinitiating system mostly driven by UV light has replaced the conventional thermal initiator. Despite the simplicity and convenience of photomediated polymerization, which proceeds by irradiation rather than heating, the high energy of UV light caused undesired side reactions and lowered the controllability of polymerization.38 Boyer and coworkers pioneered the use of low-energy visible light for photomediated RAFT polymerization in 2014 by the incorporation of PCs, named photoinduced electron/energy transfer-RAFT (PET-RAFT) polymerization.31 In 2015, Boyer,32 Qiao,33 and coworkers separately reported visible-light-driven photoiniferter polymerization of specific (meth)acrylates. The proposed PET-RAFT polymerization was based on electron/energy transfer between a visible-light-absorbing PC and a CTA,31 whereas photoiniferter polymerization was based on visible-light-driven photolysis of a CTA in the absence of PCs and had been previously demonstrated only under UV light.39,40 More recently, cationic RAFT polymerization, which involves the formation of carbocations as propagating species,41,42 was realized under visible light in the presence of a PC, as reported by Fors and coworkers.43 Since then, numerous studies have improved the utility of reaction systems, for example, by investigating PCs and/or CTAs to enhance the rate of polymerization under a broad range of irradiation wavelengths. It has also been revealed that these visible-light-driven RAFT polymerizations exhibit decent controllability of polymerization as well as attractive features including spatiotemporal control, oxygen tolerance, and unique selectivity of polymerization, which are dependent on reaction conditions (i.e., irradiation wavelengths and the combination of a CTA, monomer, and PC if employed), and high compatibility with other polymerization methods, which was not observed in conventional RDRP.44 Therefore, visible-light-driven RAFT polymerization has enabled the preparation of polymers with high levels of structural complexities and functionalities and reached a wide range of advanced applications, such as advanced manufacturing, nanomedicine, and energy. Fig. 1 depicts the timeline of milestones toward the development of visible-light-driven RAFT polymerization.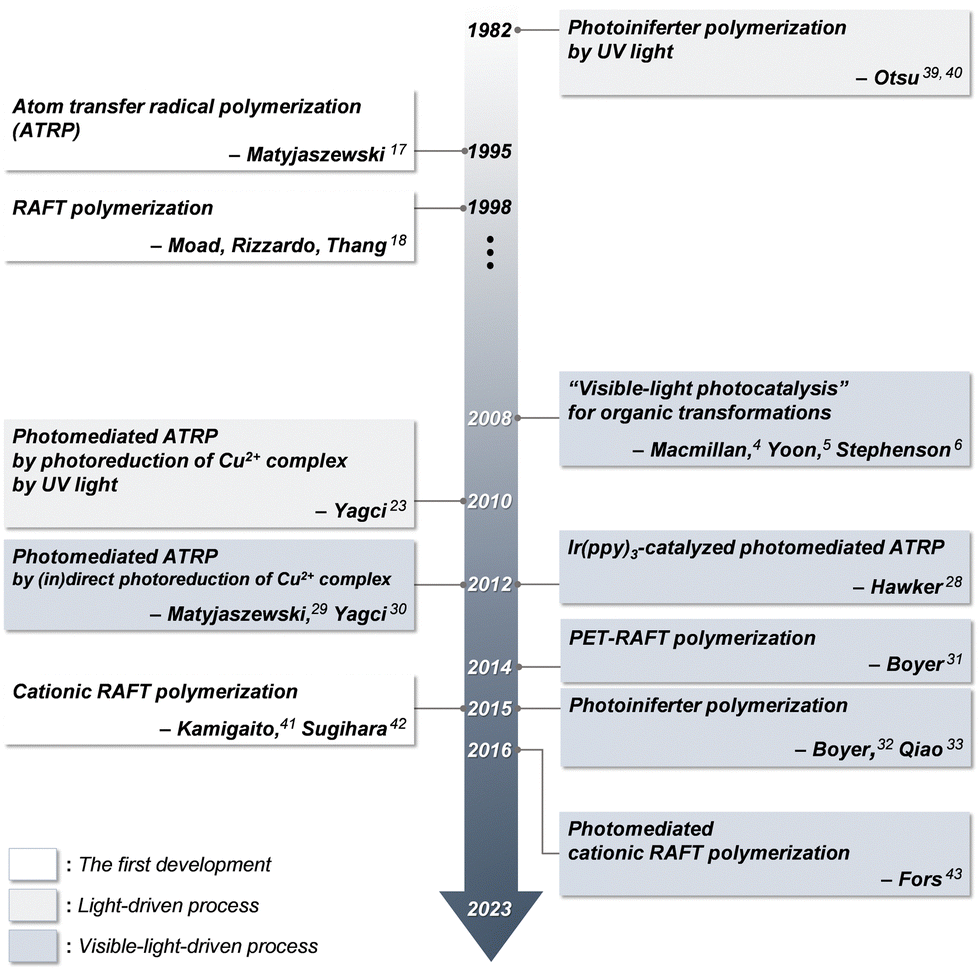 | ||
| Fig. 1 Timeline of milestones toward the development of photocontrolled RAFT polymerization. | ||
1.4. Scope of this review
Given these attractive and unique features, several highly cited review articles have already discussed photocontrolled RAFT polymerization either as a part of photomediated RDRP or photoredox catalysis,19,20,45–47 or as an independent topic with a particular emphasis on mechanistic studies,48 oxygen tolerance,49 spatiotemporal regulation of the reaction system by light,50,51 fabrication of advanced materials,52 and environmental compatibility of polymerization.44 The recent review articles have provided an insight into the remaining challenges and underscored the promising future of photomediated RAFT polymerization for its wide applicability in the industry.34,53 However, as compared to PET-RAFT polymerization, only a few articles54,55 have paid considerable attention to photoiniferter polymerization until Hartlieb in 2022 outlined the progress of photoiniferter polymerization from early systems to the current state.56 It was because photoiniferter polymerization has mostly utilized UV light thus far, in contrast to PET-RAFT polymerization which utilized the milder visible light.Visible light is more than an energetically mild and environmentally benign energy source. Convergence of visible-light photocatalysis and photomediated RDRP has also brought decent controllability of polymerization and distinct features such as oxygen tolerance. Furthermore, visible and near-infrared (NIR) light allows utilizing wavelength-orthogonal chemistry for activating wavelength-selective photoinduced reactions in an orthogonal manner. In this regard, it is timely to provide a detailed review on photocontrolled RAFT polymerization reactions (i.e., PET-RAFT, photoiniferter, and photomediated cationic RAFT polymerization) with a particular emphasis on visible-light-driven systems, and summarize their concept, method, mechanism, and applications. In addition to the unprecedently wide versatility of the polymerization ranging from precision polymer synthesis in academic fields to 3D/4D printing technologies in industrial fields, we describe the recent increasing efforts to uncover and elucidate the underlying mechanistic backgrounds from a photophysical point of view with the help of computational studies. As the broad applicability of the polymerization originates from the mechanism, this review provides an overview of the current status and the remaining challenges in photocontrolled RAFT polymerization.
2. Mechanism
In this section, the basic mechanisms and current studies related to the mechanistic understanding of various visible-light-driven RAFT polymerizations are described. The methods mostly do not require exogenous initiators and share a RAFT process, which underpins the controllability similar to that in conventional thermal RAFT polymerization. Based on the initiation mechanism, the methods can be categorized as follows.In PET-RAFT polymerization, an initiating radical is generated by the interaction between an excited-state PC and a CTA followed by (i) fragmentation of the reduced CTA via either an oxidative31 or a reductive quenching cycle,57 and/or (ii) homolysis of the CTA via energy transfer.46 The CTA can be oxidized as in the case of photomediated cationic RAFT polymerization.43 Photoiniferter polymerization includes direct homolysis of the CTA under irradiation in the absence of a PC.40,58 Very recently, the RAFT process has been combined with inner-sphere electron transfer (ISET)59 and hydrogen atom transfer (HAT).60
Despite the burgeoning applications of visible-light-driven RAFT polymerization in numerous fields, several questions, such as whether it is electron or energy transferred from the excited-state PC to the CTA, still remain unsolved. It is also noteworthy that the electron/energy transfer processes may also depend on the type of PC and CTA. Herein, the importance of enhancing fundamental understanding of the mechanism and thereby the recent efforts utilizing quantum chemical (QC) calculations, kinetic modeling, laser flash photolysis, or transient absorption spectroscopy to answer these unsolved questions are summarized.
2.1. Conventional RAFT polymerization (RAFT process)
RAFT polymerization, which was firstly developed by Rizzardo, Moad, Thang, and coworkers in 1998,18 relies on degenerative chain transfer to reversibly deactivate active propagating radicals. By the addition of a CTA possessing a thiocarbonylthio (TCT) group to conventional radical polymerization, the RAFT process successfully realized RAFT polymerization with controlled and living behavior (Scheme 1). Firstly, an external initiator is fragmented, typically by heat or light, to generate initiating radicals. After several additions of monomers, the propagating radical is added to the CTA. The resulting TCT intermediate radical undergoes fragmentation to liberate the R group as another propagating radical. These two propagating radicals are reversibly added and fragmented to become dormant and active species for polymerization, respectively, which is called RAFT main equilibrium. Via this RAFT main equilibrium, the propagating radicals share equal probabilities for chain growth. It is noted that addition and fragmentation occur faster than propagation, which prevents bimolecular termination between two radical species. Therefore, polymers possessing TCT groups exhibit low dispersity and can be reactivated by adding new monomers. As controlled and living radical polymerization is realized through an efficient RAFT process, for each monomer family, a CTA with appropriate R and Z groups must be chosen to ensure energetically favorable addition, fragmentation, and propagation. Dithiobenzoates (DTBs), dithiocarbamates (DTCs), trithiocarbonates (TTCs), and xanthates are representative classes of CTAs categorized based on the Z groups. Since the development of RAFT polymerization, the influence of R and Z groups on polymerization control has been thoroughly studied. The well-established general guidelines for the selection of CTAs for a certain monomer are well described in the review articles by the inventors of RAFT polymerization in the Commonwealth Scientific and Industrial Research Organization (CSIRO)61 and Perrier.37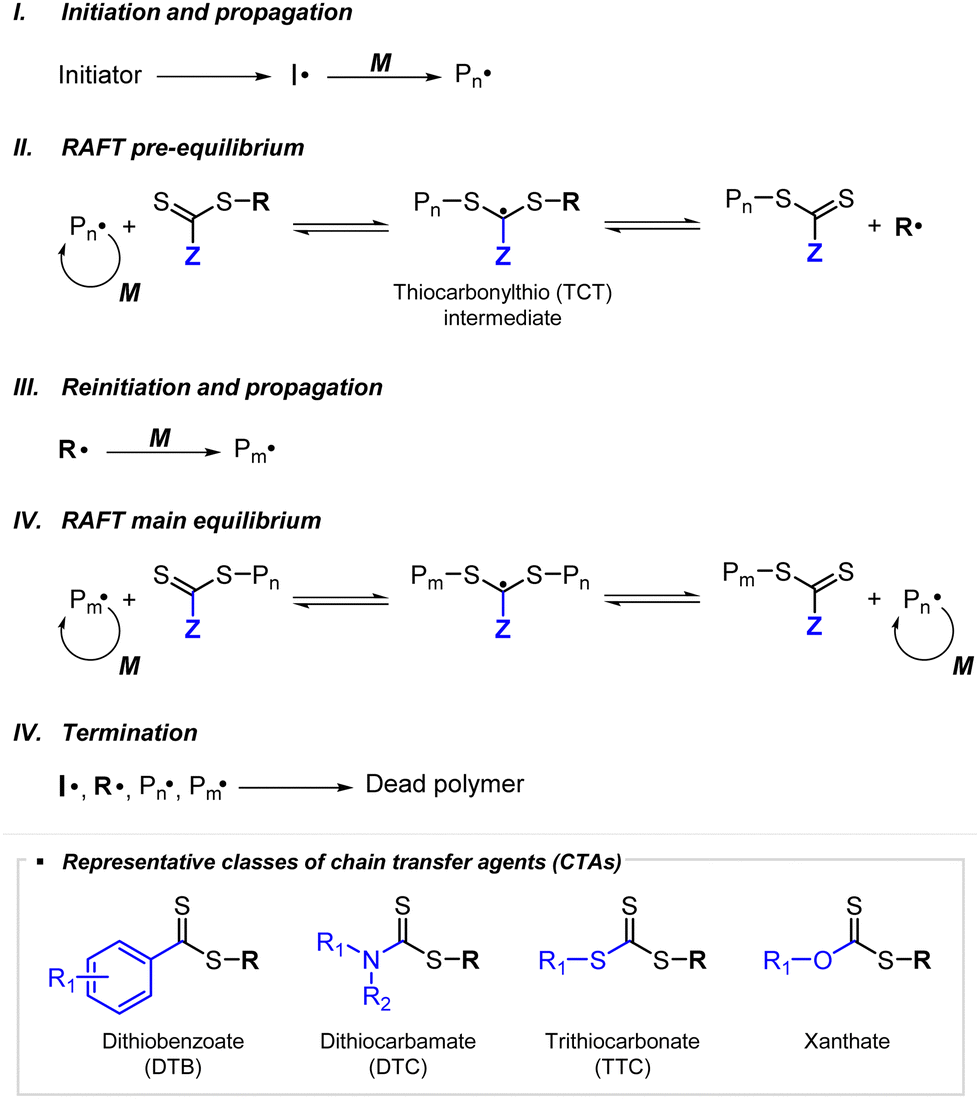 | ||
| Scheme 1 Mechanism of conventional RAFT polymerization. | ||
2.2. Visible-light-driven RAFT polymerization
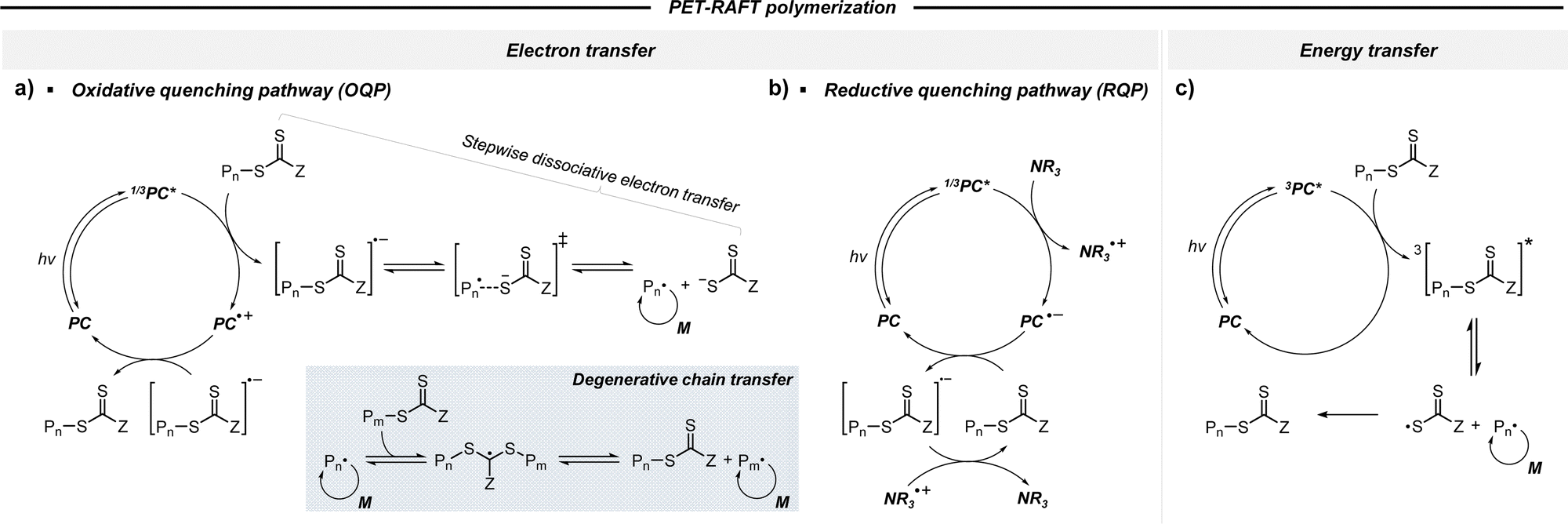 | ||
| Scheme 2 Mechanism of PET-RAFT polymerization. Electron transfer via (a) oxidative quenching pathway (OQP) and (b) reductive quenching pathway (RQP) in the presence of a reducing agent (e.g., tertiary amine). (c) Energy transfer. | ||
2.2.1.1. Electron transfer: oxidative quenching pathway (OQP). The OQP involves oxidation of an excited-state PC. Electron transfer from an excited-state PC to a ground-state CTA leads to the formation of a one-electron-oxidized PC (PC˙+) and an anionic intermediate of the CTA, which then undergoes fragmentation to afford a propagating radical and a TCT anion. Stepwise dissociative electron transfer (DET) is considered to occur during the activation step (Scheme 2a). Based on the transfer coefficient (α) measured by cyclic voltammetry (CV), Moad, Strover, and coworkers claimed that electron transfer from the excited-state PC to the CTA and dissociation of the C–S bond occur in a stepwise manner.62 More recently, Denifl, Moad, Coote, and coworkers thoroughly investigated the reduction of the selected CTAs (cyanomethyl benzodithioate and dimethyl trithiocarbonate) and their subsequent chemical reactions by performing low-energy electron attachment experiments in the gas phase (Fig. 2).63 Interestingly, in these experiments, the authors observed that anion intermediates were (i) generated and (ii) selectively dissociated into TCT anions and carbon radicals through C–S bond cleavage. These observations strongly supported the involvement of stepwise DET in the electron-transfer-driven RAFT process.
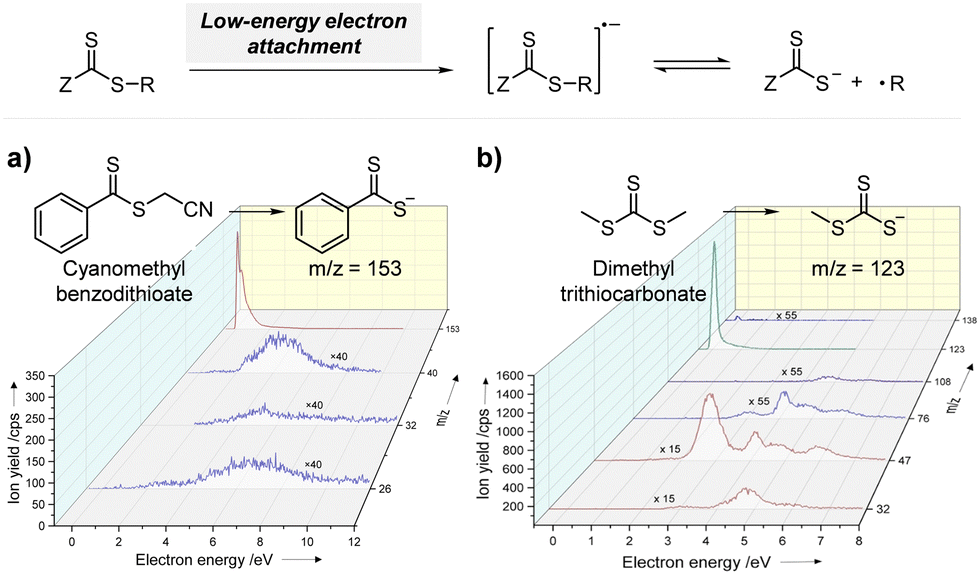 | ||
| Fig. 2 Electrochemical activation of the CTA by low-energy electron attachment and intensity map of TCT anions generated from subsequent dissociation of (a) cyanomethyl benzodithioate and (b) dimethyl trithiocarbonate. Adapted with permission from ref. 63 (Copyright 2021 John Wiley & Sons, Inc.). | ||
Recently, the research groups of Boyer and Liu,64 and Kwon65 separately reported that the selectivity for the CTA in PET-RAFT polymerization is closely related to the stepwise DET mechanism. Boyer and coworkers observed that metal naphthalocyanine (MNC)-catalyzed PET-RAFT polymerization of methyl acrylate (MA) under NIR light irradiation (780 nm) showed substantial monomer conversion and decent control for TTCs with a tertiary R group, whereas no polymerization occurred for TTCs with a secondary R group.64 QC calculations combined with experiments revealed that an activation barrier exists for the dissociation of the C–S bond of a CTA anion intermediate, depending on the R group. Similar results were also reported by Kwon and coworkers.65 Using Ag2S nanocrystals (NCs) as a PC under red light irradiation (635 nm), PET-RAFT polymerization of methacrylates was achieved for TTCs (CDTPA), whereas no polymers were obtained for DTBs (CPADB). According to QC calculations, a significantly higher activation energy was obtained for the stepwise dissociation of the anion intermediate of CPADB compared to that of CDTPA (45.47 vs. 21.86 kJ mol−1), implying that anion intermediates generated by photoinduced electron transfer play an essential role in the observed Z-group selectivity (Fig. 3).
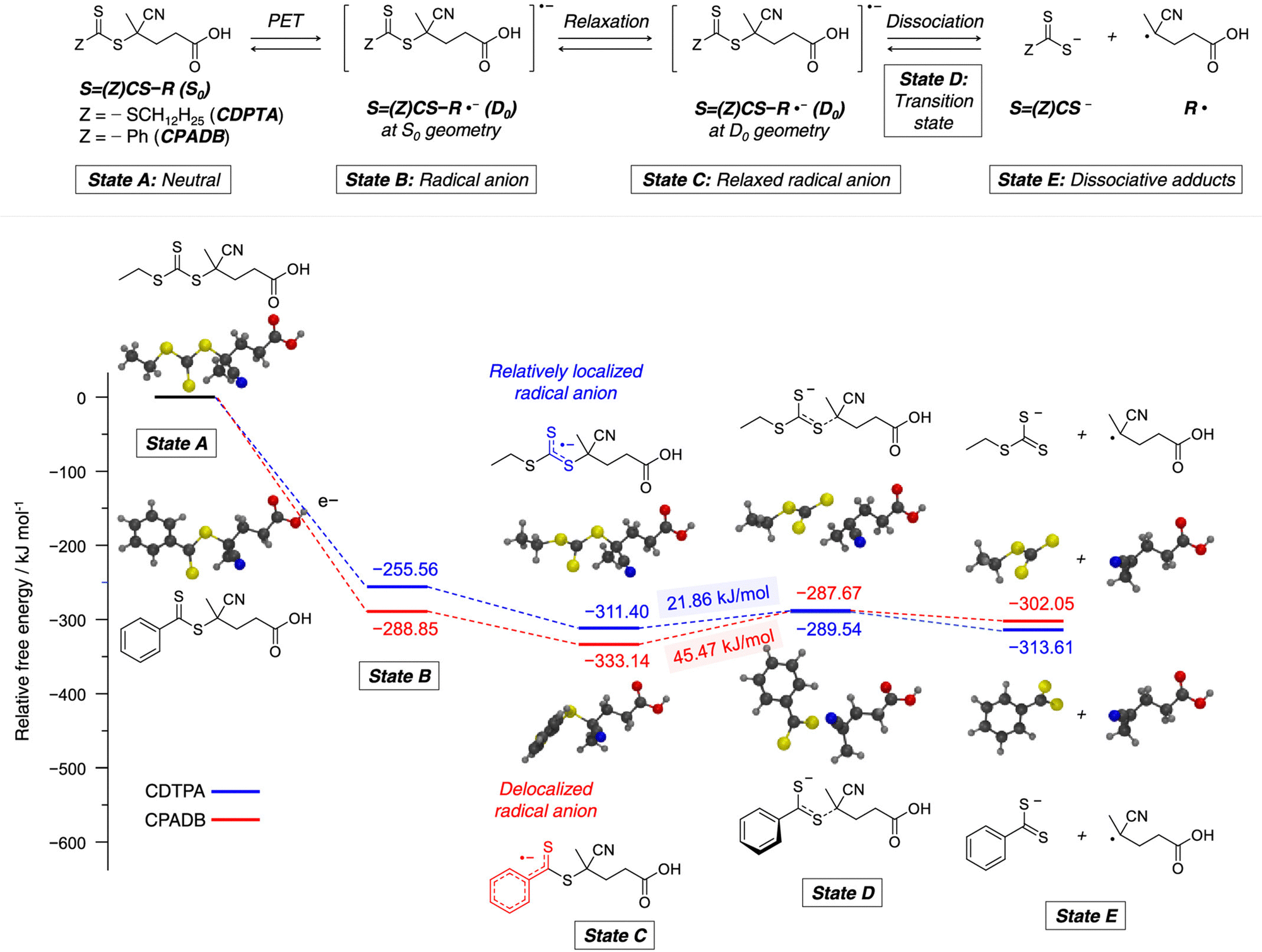 | ||
| Fig. 3 Calculated energy profiles for stepwise dissociation of anion intermediates of CDTPA (blue) and CPADB (red). Adapted with permission from ref. 65 (Copyright 2023 American Chemical Society). | ||
Although OQPs mostly rely on photoinduced outer-sphere electron transfer (OSET) between the PC and CTA, Pan and coworkers recently reported PET-RAFT polymerization based on photoinduced ion-pair ISET (IP-ISET) (Scheme 3).59 The authors demonstrated successful PET-RAFT polymerization of various vinylic monomers under visible-light irradiation in the presence of a zwitterionic borane catalyst ([L2B]+X−) and CTA. Based on QC calculations combined with experiments, [L2B]+[ZCS2]− generated in situ from [L2B]+X− and CTA was considered to be an active catalytic species. The stability of [L2B]˙ formed by ion-pair electron transfer and the very long excited-state lifetime of [L2B]+* allowed the reaction to proceed well even at extremely low catalyst concentrations as low as 1 ppb, which is approximately three orders of magnitude lower than those of OSET-based systems. Unlike the reported PET-RAFT polymerization, this method showed excellent tolerance to phenolic radical inhibitors, owing to the strong hydrogen bond between the phenolic group of the inhibitor and X− of [L2B]+X−.
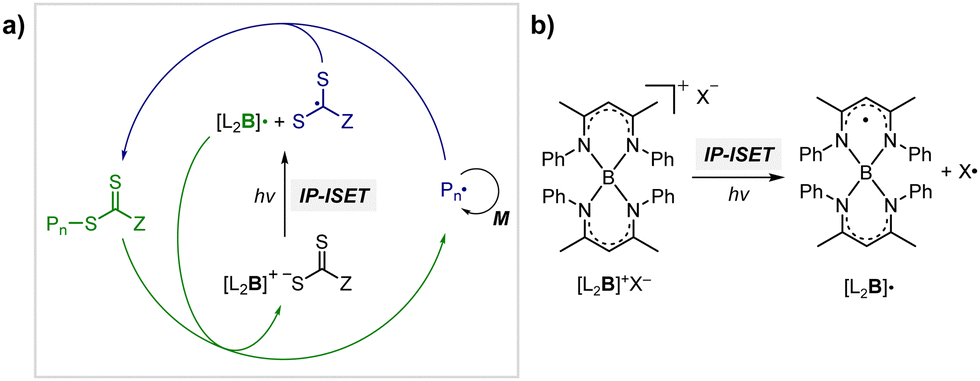 | ||
| Scheme 3 (a) Mechanism of PET-RAFT polymerization based on photoinduced inner-sphere electron transfer (ISET). (b) Chemical structures of [L2B]+ and [L2B]˙.59 | ||
2.2.1.2. Electron transfer: reductive quenching pathway (RQP). The RQP involves reduction of an excited-state PC by adding a sacrificial reducing agent (Scheme 2b). A one-electron-reduced PC (PC˙−) is initially formed through electron transfer from a sacrificial reducing agent to an excited-state PC. Subsequently, one electron was transmitted from PC˙− to the CTA, generating propagating radical species and a TCT anion through a TCT anionic intermediate. In 2015, Boyer and coworkers proposed PET-RAFT polymerization via RQP using a tertiary amine as a sacrificial reducing agent.66 This protocol provided faster kinetics and enhanced oxygen tolerance without severely sacrificing controllability compared to PET-RAFT polymerization via OQP (i.e., in the absence of tertiary amines). Since this report, PET-RAFT polymerization via RQP has been widely used when fast polymerization kinetics under ambient conditions, such as polymerization under biorelevant conditions, are required.67–70
At approximately the same time in 2015, the use of tertiary amines in photocontrolled RAFT polymerization in the absence of PCs was proposed by Qiao and coworkers (Scheme 4).71 Herein, the CTA directly absorbed light and tertiary amines, as a catalyst, reduced the excited-state CTA. This electron transfer from the tertiary amine to the excited-state CTA generated the anionic intermediate of the CTA, which then underwent fragmentation to provide a propagating radical and a TCT anion. Qiao and coworkers used 365 nm UV LEDs as the light source, and Konkolewicz and coworkers later demonstrated that the polymerization could proceed properly under visible light (<530 nm).72 It is noted that this mechanism of tertiary amine-catalyzed generation of the TCT anion in the absence of a PC cannot be excluded in PET-RAFT polymerization via RQP, which complicates a complete understanding of the polymerization results.
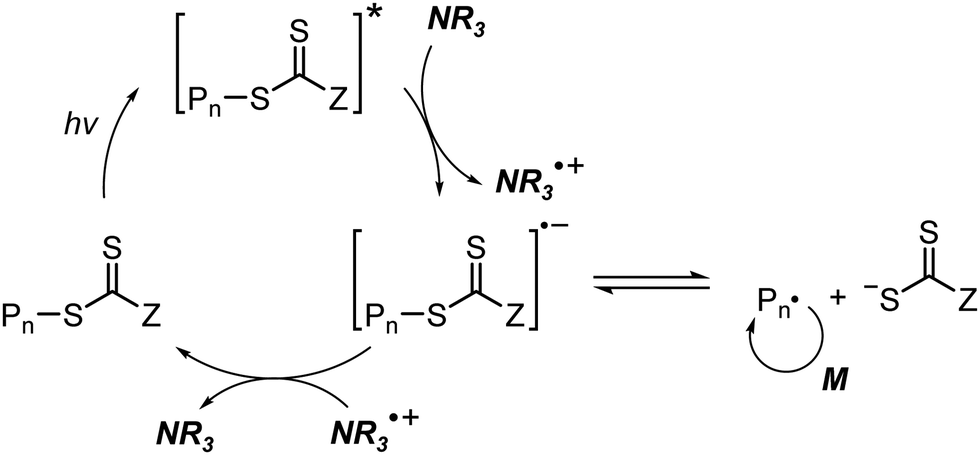 | ||
| Scheme 4 Mechanism of tertiary amine-catalyzed generation of the TCT anion in the absence of a PC. | ||
In PET-RAFT polymerization via RQP, the polymerization features, such as the rate of polymerization, oxygen tolerance, and controllability, were found to be sensitive to the type73 and amount of tertiary amines,71,72 as well as the irradiation wavelength.57 This sensitivity likely arises because the rate of electron transfer from a tertiary amine to an excited-state PC (or an excited-state CTA) relies on the amount and redox potentials of the amine. Furthermore, one-electron-oxidized tertiary amines may cause various side reactions by providing hydrogen and/or generating reactive α-amino radical species,74,75 leading to poor controllability. However, the effects of tertiary amine intermediates on the polymerization have not been thoroughly analyzed and hence, combined efforts involving QC calculations, kinetics simulations, and chromatographic analyses are required.
2.2.1.3. Energy transfer. An alternative pathway involves photoinduced energy transfer, in which energy is transferred from an excited-state PC to a CTA, promoting the CTA to an electronically excited state. Then, active propagating radicals are generated via homolytic C–S bond cleavage of the excited-state CTA (Scheme 2c). Energy transfer is likely to occur from the T1 state of the PC to the triplet excited states of the CTA.
Energy transfer is a photophysical process by which energy is transferred from one molecular entity (donor, D) in an excited state to another molecular entity (acceptor, A) to be raised to a higher energy state. The process can be distinguished into three types:76,77 primitive energy transfer by emission-reabsorption, Förster resonance energy transfer, and Dexter energy transfer. In photocatalysis of organic molecules, Dexter energy transfer is of predominant relevance.77 As Dexter energy transfer is based on simultaneous electron exchange between D and A, which requires the overlap of the wavefunctions, the energy transfer occurs over very short distances within approximately 10 Å, which approaches the collisional diameter.78,79 In addition, the electron exchange is governed by the Wigner spin conservation rule and hence, the spin-allowed processes are single–singlet and triplet–triplet energy transfer. Among these, triplet–triplet energy transfer is preferred owing to the typically longer lifetime of triplet states compared to singlet states. The longer lifetime increases the probability of collisions. Because this is essentially an electron transfer process, the kinetics can be described using Marcus theory.80 Therefore, the rate of Dexter energy transfer is strongly influenced by the thermodynamic driving force, reorganization energy, and electronic coupling between D and A; nevertheless, in the synthetic community, the thermodynamic driving force (i.e., the energy gap between the lowest triplet state of D and A) is mostly utilized to estimate the energy transfer efficiency because the reorganization energy and electronic coupling are rather difficult to measure. When the thermodynamic driving force is large as in exergonic energy transfer, the energy transfer gets faster close to the diffusion limit. The studies on the energy transfer pathway in PET-RAFT polymerization are discussed in the next section.
2.2.1.4. Mechanistic understanding of PET-RAFT polymerization. The important debate with respect to the mechanism of PET-RAFT polymerization is how an excited-state PC interacts with a CTA: via either electron transfer or energy transfer. Substantial research efforts to answer this difficult question have been continuously made until recently, and these efforts are well summarized in a recent review article by Konkolowicz and coworkers.48 Smith and coworkers supported an electron transfer mechanism based on calculations of the energetics of selected PCs (ZnTPP and pheophorbide A (PheoA)), various CTAs, and charge-transfer complexes that can be formed between them.81 Falvey and coworkers also supported an electron transfer mechanism based on studies using laser flash photolysis for (i) triplet–triplet energy transfer between a triplet sensitizer (e.g., anthraquinone, Rose Bengal, and benzophenone) and a CTA, and (ii) subsequent decomposition of an excited-state CTA.82 Although energy transfer from the sensitizer to the CTA was efficient, decomposition of the CTA was rarely observed, suggesting that radical generation by energy transfer is rather limited compared to radical generation by electron transfer. Very recently, Kwon and coworkers successfully resolved the mechanistic complexity of PET-RAFT polymerization by using appropriately designed Ag2S NCs as a PC.65 Owing to the small bandgap and moderate
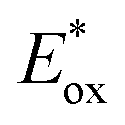 of the Ag2S NCs, PET-RAFT polymerization proceeded solely through electron transfer. Successful polymerization of methyl methacrylate (MMA) in the presence of Ag2S NCs confirmed that the electron transfer readily occurs in PET-RAFT polymerization. However, it is noteworthy that the mechanism depends on the type of PCs. In the case of [Ru(bpy)3]2+ and Ir(ppy)3, the research groups of Allonas and Boyer83,84 and Konkolewicz85 provided evidence for an energy transfer process. Allonas, Boyer, and coworkers calculated the Gibbs free energy change during electron transfer (ΔGET) and triplet–triplet energy transfer (ΔGEnT) based on the redox potentials and triplet energies of PCs and a series of CTAs obtained from QC calculations and CV measurements.83,84 ΔGET and ΔGEnT were then plotted against the rate constant for quenching (kq) of an excited-state PC obtained from laser flash photolysis experiments. A substantial correlation was observed between kq and ΔGEnT, whereas no relationship was found between kq and ΔGET, suggesting that energy transfer likely occurs between an excited-state PC and a CTA. Konkolewicz and coworkers also supported an energy transfer mechanism based on the simulation results of polymerization kinetics.85 Using PET-RAFT polymerization of MA using Ir(ppy)3 and TTC as a model system, the kinetics data obtained by experiment were compared with the data simulated using kinetic models of possible PET-RAFT mechanisms. Among them, the energy transfer mechanism was found to be the best fit.
of the Ag2S NCs, PET-RAFT polymerization proceeded solely through electron transfer. Successful polymerization of methyl methacrylate (MMA) in the presence of Ag2S NCs confirmed that the electron transfer readily occurs in PET-RAFT polymerization. However, it is noteworthy that the mechanism depends on the type of PCs. In the case of [Ru(bpy)3]2+ and Ir(ppy)3, the research groups of Allonas and Boyer83,84 and Konkolewicz85 provided evidence for an energy transfer process. Allonas, Boyer, and coworkers calculated the Gibbs free energy change during electron transfer (ΔGET) and triplet–triplet energy transfer (ΔGEnT) based on the redox potentials and triplet energies of PCs and a series of CTAs obtained from QC calculations and CV measurements.83,84 ΔGET and ΔGEnT were then plotted against the rate constant for quenching (kq) of an excited-state PC obtained from laser flash photolysis experiments. A substantial correlation was observed between kq and ΔGEnT, whereas no relationship was found between kq and ΔGET, suggesting that energy transfer likely occurs between an excited-state PC and a CTA. Konkolewicz and coworkers also supported an energy transfer mechanism based on the simulation results of polymerization kinetics.85 Using PET-RAFT polymerization of MA using Ir(ppy)3 and TTC as a model system, the kinetics data obtained by experiment were compared with the data simulated using kinetic models of possible PET-RAFT mechanisms. Among them, the energy transfer mechanism was found to be the best fit.
In addition to the abovementioned issue, several issues remain unsolved, as introduced by Konkolewicz and coworkers.48 First, the mechanism of electron transfer during OQP needs to be reconsidered. According to the current mechanism where the TCT anion is mainly involved in the regeneration of the ground-state PC, PET-RAFT polymerization could be significantly retarded or even stop at very low catalytic loading of the PC, as demonstrated by Konkolewicz and coworkers.85 This finding is inconsistent with the previously reported experimental results. As such, in-depth studies are required to determine which pathways are involved in the reduction of oxidized cationic PC intermediates. Second, more in-depth understanding of the overall picture of what drives electron or energy transfer is required. The experimental results thus far suggest that the mechanism is dependent on the reaction system composition (i.e., combination of PC, CTA, and monomer), whereas the exact mechanistic background remains unknown. Third, the fate of TCT radicals, which can be generated in PET-RAFT polymerization via energy transfer or photoiniferter polymerization, needs to be understood better. Herein, excessive TCT radicals could be accumulated by the termination of propagating radical species, as observed for TCT radicals generated by electron transfer, consequently leading to significant rate retardation. However, this retardation was not observed in the experimental studies and could be related to the formation of disulfide compounds that have not been much investigated. Fourth, the process of C–S bond dissociation in the energy transfer process remains unclear. As studied by Falvey and coworkers, C–S bond dissociation of the CTA in the triplet excited state was not sufficiently efficient to drive polymerization.82 However, the study was performed in the absence of a monomer and thus, additional experiments in the presence of a monomer are necessary to draw a clear conclusion. Last, the direct spectroscopic observation of essential intermediates such as one-electron-oxidized/reduced PC, TCT anions, and TCT radicals would help to unambiguously identify the underlying mechanisms.
2.2.2.1. Basic mechanism. Photoiniferter polymerization under UV light irradiation, firstly proposed by Otsu and coworkers in 1982 as a type of RDRP,39,86 has garnered renewed interest since Boyer,32 Qiao,33 and coworkers separately expanded the energy source of polymerization to visible light. Herein, a TCT-based CTA is a key agent that simultaneously acts as an initiator, a transfer agent, and a reversible terminator (ini-fer-ter), thereby eliminating the need for a PC or an exogenous initiator. As depicted in Scheme 5, the CTA directly absorbs light to become an excited state, which is followed by the homolysis of the C–S bond to produce an active initiating/propagating radical and a persistent TCT radical.56 The propagating radicals can participate in degenerative chain transfer, a key mechanistic aspect of RAFT polymerization, and the TCT radical can deactivate active radical species in a reversible fashion as seen in ATRP. As this process requires the absorption of light by the CTA, the irradiation wavelength for photoiniferter polymerization is quite limited in contrast to PET-RAFT polymerization.
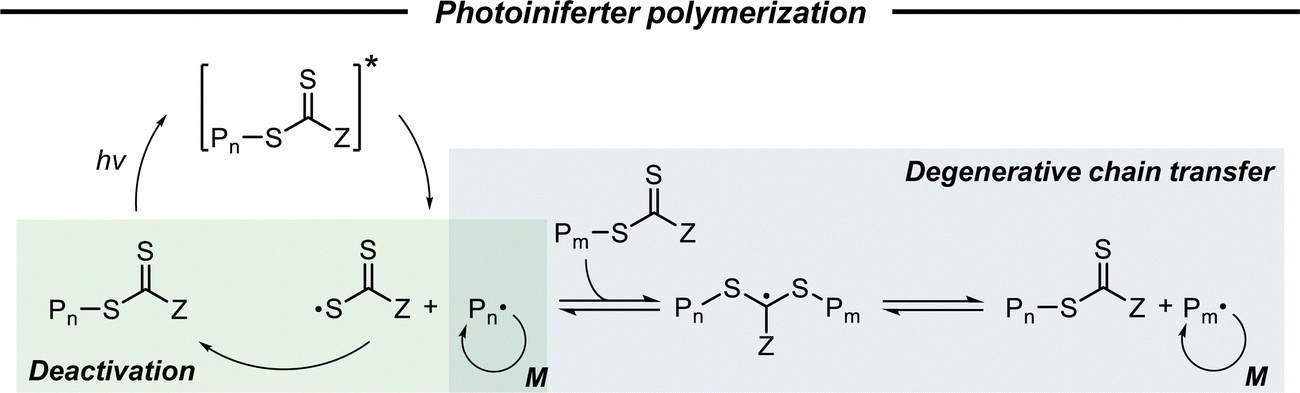 | ||
| Scheme 5 Mechanism of photoiniferter polymerization. | ||
2.2.2.2. Mechanistic understanding of photoiniferter polymerization. The most essential issue in photoiniferter polymerization is to clearly understand the mechanism of photolysis of the C–S bond, an important process that determines the concentrations of radicals and CTAs during polymerization. Recently, Konkolewicz and coworkers reported that the rate of photoiniferter polymerization of MMA was significantly affected by the Z group of DTBs.87 The rate of polymerization was nicely correlated with the redox potentials of CTAs, implying that the homolysis of the C–S bond is closely related to the electronic properties of CTAs. Falvey and coworkers suggested that higher singlet excited states such as S2 states are involved more in the photolysis than S1 or T1 states based on energetics considerations.82 Although these studies provided useful mechanistic insights into photolysis of the C–S bond, a precise understanding of the entire process of photoiniferter polymerization is still lacking and is expected to be provided through advanced QC calculations combined with well-designed experiments. For example, Kwon, Min, and coworkers very recently proposed that S1/S0 conical intersection (CI) acted as an activation barrier for photolysis of the C–S bond.88 CDTPA under 515 nm irradiation (10 mW cm−2) led to controlled and quantitative polymerization of MMA, whereas polymerization of MA provided adducts of (MA)n–CDTPA rather than polymers. QC calculations of reaction intermediates proposed that relative energies at the adiabatic S1 state, CI, and after dissociation were critical for the observed monomer–CTA selectivity (Fig. 4). Energy profiles for the process from Frank–Condon excitation, structural relaxation, and CI were similar for adducts of both monomers, whereas energy profiles for the process from CI to bond dissociation were more favorable in MMA–CDTPA due to the large stabilization energy of generated radicals. Other possible processes from CI include nonradiative decay channels, hindering the C–S bond photolysis and subsequent polymerization.
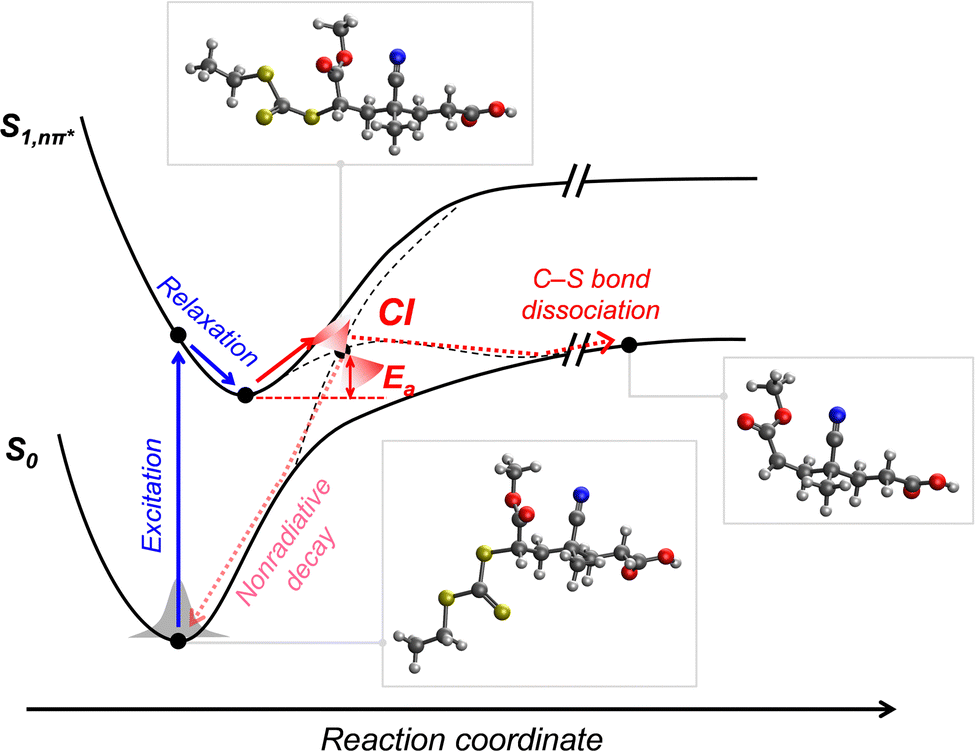 | ||
| Fig. 4 Pathway for photolysis of the C–S bond in MA–CDTPA proposed by QC calculations.88 | ||
Next, two different mechanisms (i.e., degenerative chain transfer and reversible deactivation) can affect the control of RAFT polymerization, depending on the type of CTA employed in the reaction. These two mechanisms could jointly operate in the case of xanthate (with a low transfer constant).56 In contrast, in the other study using TTC (with a higher transfer constant), preliminary calculations suggested that the reversible deactivation did not contribute primarily to the control over polymerization.89 Quantitative analysis of the contribution of two mechanisms needs to be provided by further investigation using experimental and theoretical studies.
Lastly, as in PET-RAFT polymerization, the fates of the CTA and the corresponding TCT radical need to be clarified. Although the photodegradation of TCT groups under UV light irradiation with high energy has been reported,19,26 the case under visible-light irradiation with low energy has not been observed.
2.2.3.1. Photomediated cationic RAFT polymerization. Cationic RAFT polymerization was firstly reported by the research group of Kamigato41 and Sugihara.42 Herein, in the presence of protonic acid initiators to protonate a CTA, the fragmentation of the CTA cation generated carbocations as propagating species, which were polymerized in a controlled fashion via degenerative chain transfer. Fors and coworkers then developed a photomediated version of cationic RAFT polymerization by introducing highly oxidizing 2,4,6-tris(p-methoxyphenyl)pyrylium tetrafluoroborate as a PC to oxidize a CTA and generate a carbocation (Scheme 6).43 One-electron oxidation of the CTA by the excited-state PC upon irradiation was followed by mesolytic cleavage of the resulting CTA radical cation to yield the CTA radical and carbocation as propagating intermediates. Similar to conventional cationic RAFT polymerization, the polymerization was controlled via degenerative chain transfer. The use of a PC provided additional regulation over the propagation step by photo-reversible generation of the carbocation. Reduction of the CTA radical by the one-electron-reduced PC generated the CTA anion and PC, which capped the carbocation chain-end and closed the catalytic cycle. Therefore, the photocontrolled behavior of polymerization (i.e., the generation of the propagating carbocation and thus polymerization only proceed under light irradiation) could only be provided by a PC that possessed proper
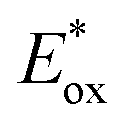 and E0red for favored one-electron oxidation of the CTA and reduction of the CTA radical, respectively.90 Based on the investigation of the detailed mechanism using selected PCs and CTAs, Fors and coworkers concluded that in the case of pyrylium derivatives, a singlet excited state might involve electron transfer as the low fluorescence quantum yield (ΦFL) of the PC led to a lower polymerization rate. The concern over the direct oxidation of the monomer instead of the CTA by the excited-state PC was also resolved by Stern–Volmer analysis and CV measurements. Both the CTA and isobutyl vinyl ether (iBVE) monomer could be oxidized, but it was revealed that the oxidation of the CTA was favored by lower E0ox. The bimolecular quenching constant between the excited-state PC and CTA determined by Stern–Volmer analysis was two orders of magnitude higher than that between the PC and iBVE. Lastly, E0red of the PC was important for efficient capping of the propagating chain-end and deactivation of polymerization, providing perfect temporal control.
and E0red for favored one-electron oxidation of the CTA and reduction of the CTA radical, respectively.90 Based on the investigation of the detailed mechanism using selected PCs and CTAs, Fors and coworkers concluded that in the case of pyrylium derivatives, a singlet excited state might involve electron transfer as the low fluorescence quantum yield (ΦFL) of the PC led to a lower polymerization rate. The concern over the direct oxidation of the monomer instead of the CTA by the excited-state PC was also resolved by Stern–Volmer analysis and CV measurements. Both the CTA and isobutyl vinyl ether (iBVE) monomer could be oxidized, but it was revealed that the oxidation of the CTA was favored by lower E0ox. The bimolecular quenching constant between the excited-state PC and CTA determined by Stern–Volmer analysis was two orders of magnitude higher than that between the PC and iBVE. Lastly, E0red of the PC was important for efficient capping of the propagating chain-end and deactivation of polymerization, providing perfect temporal control.
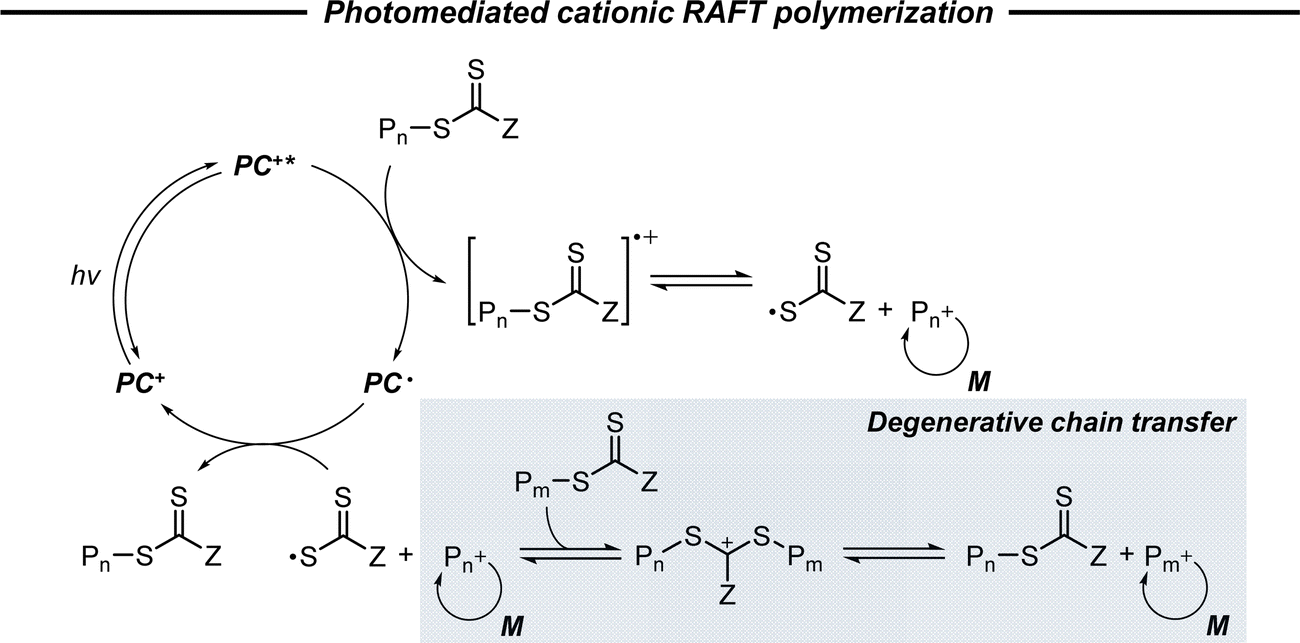 | ||
| Scheme 6 Mechanism of photomediated cationic RAFT polymerization. | ||
2.2.3.2. Hydrogen atom transfer (HAT)-mediated photomediated RAFT polymerization. Fors and coworkers introduced a HAT manifold into photomediated RAFT polymerization.60 Herein, a benzophenone analogue capable of Norrish type II reaction was used as a PC and TTC-derived disulfide was used as a CTA precursor, unlike in conventional PET-RAFT polymerization. Under irradiation using a compact fluorescent lamp (CFL), the PC absorbed light to generate a PC intermediate with diradical character (Scheme 7). This PC intermediate abstracted the C–H bond of the substrate through hydrogen abstraction, and the resulting radical intermediates of the substrate initiated the polymerization. On the other hand, the disulfide generated two TCT radicals through S–S bond homolysis. One TCT radical was combined with the propagating radical to generate a macro-CTA, whereas the other TCT radical reacted with the PC intermediate, ketyl radical derived from benzophenone, to generate trithiocarbonic acid and regenerate the PC. As stated by the authors, this method will be useful to graft CTA moieties into hydridic C–H bonds of substrates (e.g., biomolecules and backbone of commodity polymers) for subsequent polymerization.
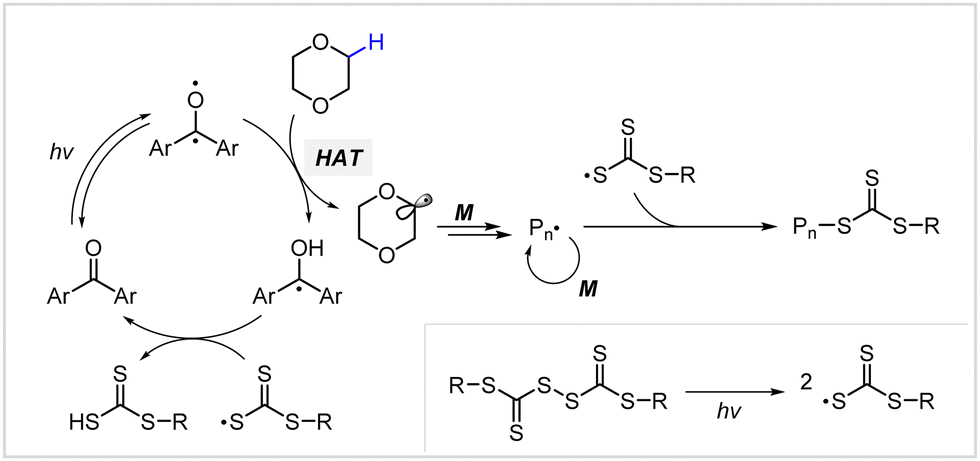 | ||
| Scheme 7 Mechanism of hydrogen atom transfer (HAT)-mediated photomediated cationic RAFT polymerization.60 | ||
3. Distinct features
In addition to the use of green and abundant visible light as an energy source, the distinct features of visible-light-driven RAFT polymerization endow the reaction system with controllability and the synthesized polymers with structural diversity, functionality, and excellent livingness, which otherwise are not realizable via conventional thermally initiated reactions. These features are attributed to the abovementioned mechanisms and can be primarily categorized into (i) spatiotemporal control, (ii) selectivity, (iii) orthogonality, and (iv) oxygen tolerance. It is noted that PET-RAFT polymerization distinctly demonstrates all four features. As active species are generated by light irradiation, spatiotemporal control is commonly achieved regardless of the polymerization mechanism (Section 3.1). Except for photomediated cationic RAFT polymerization, in PET-RAFT and photoiniferter polymerizations, active species are radicals so that the reactions can simultaneously occur with other reactions possessing different (such as ionic) intermediates and can be orthogonally controlled by turning the light on/off (Section 3.3). Meanwhile, the other two features (i.e., selectivity and oxygen tolerance) have mainly been examined for PET-RAFT polymerization and still remain elusive for other methods.3.1. Spatiotemporal control
Every photomediated RAFT polymerization operates by the absorption of light by either a PC or a CTA, and is therefore inherently a spatiotemporally controlled process that only occurs where and when the light is irradiated. Perfect temporal control directs the mechanism of the reaction toward a photocontrolled process necessitating the use of light and compounds that appropriately absorb light and play the role. For example, imperfect control or increase in monomer conversion even without light irradiation, which has been observed in some photomediated cationic RAFT polymerizations, has been ascribed to the decomposition of the PC or slow deactivation of the CTA owing to improper balance between ground- and excited-state redox potentials of the PC and CTA.43,90–92 Slow deactivation of the CTA results in longer lifetime of active propagating species such that these species remain after the cessation of irradiation and consume monomers. This loss of control caused by the remaining radicals, however, can be utilized to induce latent RAFT polymerization in the dark using the energy stored during precedent irradiation.93 Here, eosin Y (EY)-mediated photoinduced conversion of triplet oxygen into singlet oxygen which was then reduced by ascorbic acid in the dark generated the initiating hydroxyl radical.On the other hand, PET-RAFT polymerization can be temporally switched by other stimuli (e.g., temperature, pH, atmosphere, external magnetic field, and chemical reagents) in combination with light under which the photocatalytic activity of the PC is reversibly controllable (Fig. 5). The listed examples of dual-gated polymerizations have significant prospects for industrial applications as they offer additional means of control other than light and/or easy separation of PCs from the polymerization mixture for reuse.
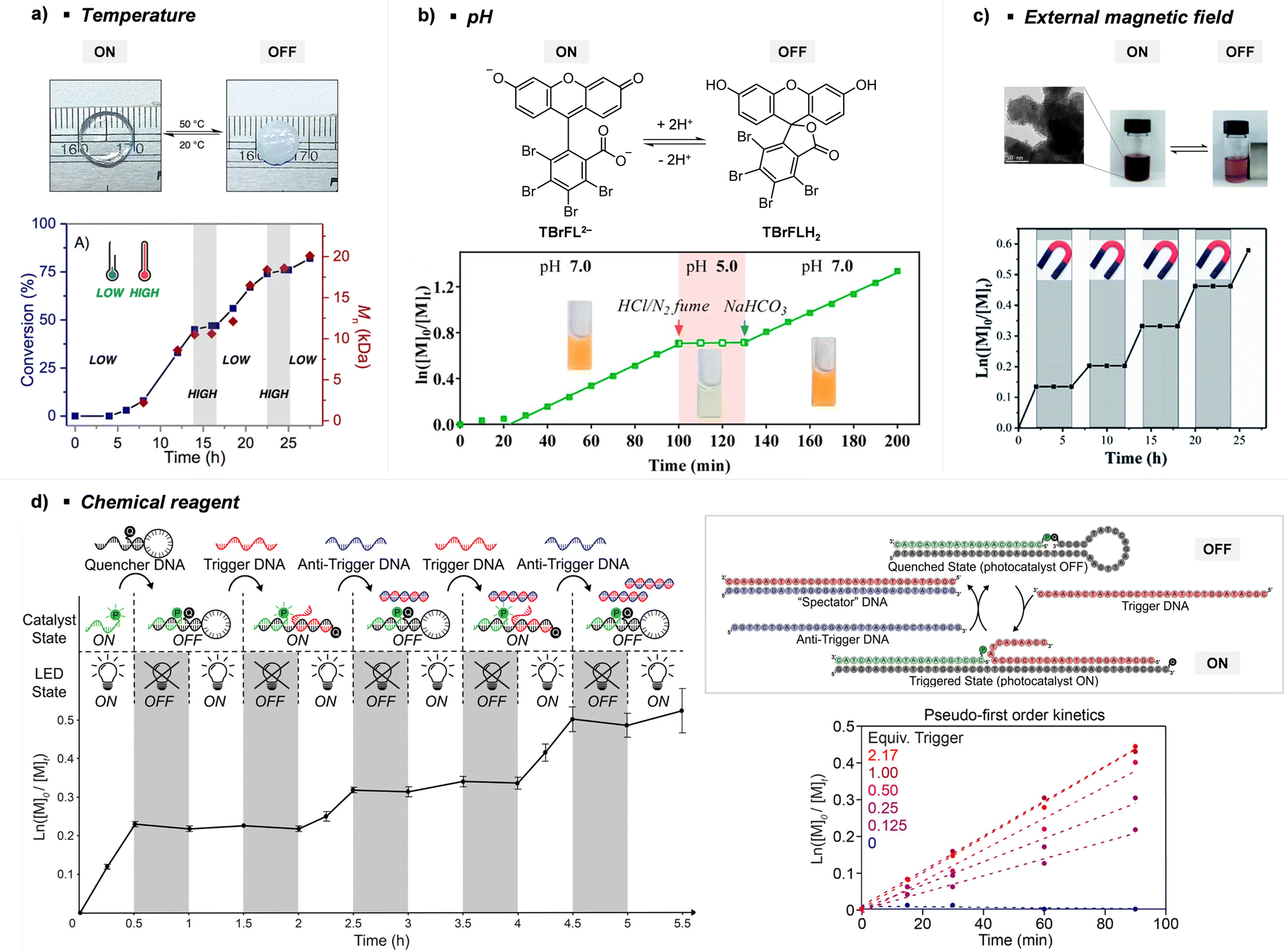 | ||
| Fig. 5 Temporal control of PET-RAFT polymerization by various stimuli other than light: (a) heat, (b) pH, (c) external magnetic field, and (d) chemical reagents. Adapted with permission from (a) ref. 94 (Copyright 2017 American Chemical Society), (b) ref. 96 (Copyright 2019 American Chemical Society), (c) ref. 103 (Copyright 2020 The Royal Society of Chemistry), and (d) ref. 104 (Copyright 2023 American Chemical Society). | ||
Johnson and coworkers embedded 10-phenylphenothiazine (PTH) in a poly(N-isopropylacrylamide (NIPAm))-based gel in which at temperatures above the lower critical solution temperature (LCST), the PC could not be accessed by light and other reagents, and consequently the polymerization could not proceed (Fig. 5a).94 Polymerization only proceeded at temperatures below the LCST and under light irradiation. In contrast, in nanocomposites composed of cross-linked hyperbranched polyglycerol, polyfluorene backbones decorated with poly(NIPAm) brushes, and Ru(bpy)3Cl2, heat-driven shrinkage of poly(NIPAm) chains generated open pores for reagents to enter and polymerization proceeded.95
Boyer and coworkers discovered a pH-switchable xanthene-based PC TBrFL (Fig. 5b).96 Because of the pH-responsiveness of xanthene dyes, at pH = 5, TBrFL became colorless and inactive to catalyze PET-RAFT polymerization, whereas at pH = 7, the original visible-light absorption property was restored to turn on the reaction. pH was lowered by adding gaseous HCl/N2 or CO2 and was recovered by NaHCO3 or N2, respectively. This pH-responsive photocatalytic performance of xanthene dyes was retained in heterogeneous PCs where Erythrosin B (EB) and Rose Bengal were conjugated to porous polymers.97 ZnTPPS4− demonstrated pH-dependent polymerization kinetics.98 The polymerization kinetics were retarded by lowering the pH of the reaction mixture from 8.6 to 3.5. This was presumably because higher concentration of the hydroxide ion at higher pH weakened the coordination of the zinc core of ZnTPPS4− with the CTA or monomer and promoted the absorption of light by ZnTPPS4− and subsequent polymerization, as evidenced by the higher molar absorption coefficient of the reaction mixture at irradiation wavelength. In contrast, ZnOETPP exceptionally utilized oxygen for the polymerization process so that polymerization did not occur in the presence of CO2 or N2.99 This role of oxygen contradicted its conventionally recognized role of inhibiting polymerization by scavenging the propagating radicals (for a detailed explanation, please see Section 3.4). Copolymerization of macrocyclic allylic sulfones with acrylate and acrylamide monomers was reversibly switched by alternate introduction of Ar and SO2.100 During radical ring-opening cascade polymerization, macrocyclic allylic sulfones release SO2 and the resulting secondary alkyl radicals participate in propagation. However, the gradually accumulated SO2 in the reaction system recombined with the alkyl propagating radicals and inhibited polymerization in the late stages particularly at low reaction temperature. Density functional theory (DFT) calculations and electron paramagnetic resonance (EPR) spectroscopy evidenced that Gibbs free energy change upon extrusion and recombination of SO2 from the monomer was relatively small such that SO2 could negatively affect the polymerization behavior. Thus, the introduction of Ar re-initiated the SO2-inhibited polymerization, whereas the introduction of exogenous SO2 switched off the polymerization.
The hybrid nanocomposites of magnetic Fe3O4, silica, and PCs (Ir(ppy)3101 and ZnTPP102) reported by Cai, Zhang, and coworkers or γ-Fe2O3 nanoparticles (NPs) doped with carbon dots reported by Qiao, Pang, and coworkers103 were responsive to an external magnetic field. Upon applying a magnetic field, these nanohybrid PCs exhibited agglomeration and could not catalyze polymerization. This was attributed to the reduced visible-light absorption and limited access and electron transfer between PCs (bound to the surface of the nanocomposites) and CTA (Fig. 5c).
Lastly, Martell and coworkers conjugated DNA aptamers and a quencher dye with EY to switch on the photocatalytic activity of EY by adding chemical reagents that could be hybridized with the DNA (Fig. 5d).104 As absorption of the quencher dye overlaps with emission of EY, fluorescence and photocatalytic activity of EY were switched off when the quencher dye and EY were in proximity through static quenching and Förster resonance energy transfer (FRET). The added chemical reagent non-covalently hybridized with the DNA to induce a conformational change, remove the proximity between the quencher dye and EY, and switch on the photocatalytic activity of EY. In particular, when trigger DNA was used as the chemical reagent, anti-trigger DNA with complementary sequences could inactivate the trigger DNA resulting in double-stranded spectator-DNA and a photocatalytically inactivate PC. Therefore, the activity of the PC could be reversibly switched, and the polymerization rate could be modulated by the amount of trigger-DNA relative to that of quencher-DNA. As sequences and secondary structures of DNA aptamers can be designed to specifically fit diverse trigger molecules such as glucose, hydrocortisone, and zinc ions, these switchable DNA–PC conjugates were expected to be expanded to various PCs and chemical reagents, only if appropriate pair of PC and quencher dye possessing overlapping emission and absorption spectra could be identified.
3.2. Selectivity
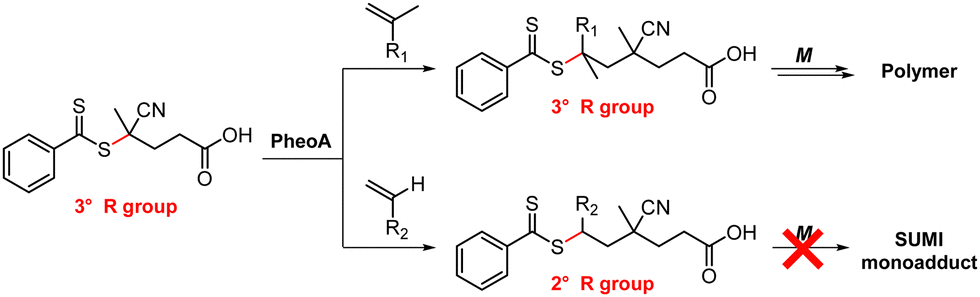
For successful photocontrolled RAFT polymerization, effective photolysis of the CTA (activation), insertion of the monomer, and deactivation of the CTA are required. It has been shown that each step is highly selective toward reagents and reaction conditions. First of all, the irradiation wavelength should match the absorption profile of either PC or CTA (depending on the type of polymerization) for their excitation. In PET-RAFT polymerization the (de)activation of the CTA by the PC at the initiation stage and that of the CTA with monomers inserted at the propagation stage are mainly governed by thermodynamic constraints where the reactivity of the excited-state PC is determined by the energy absorbed at the irradiation wavelength. In addition to irradiation at the appropriate wavelength, selectivity of the PC toward the CTA and selective insertion of the monomer into the CTA have been observed. These selectivity issues can be leveraged to prepare polymers with desired sequences and complex architectures.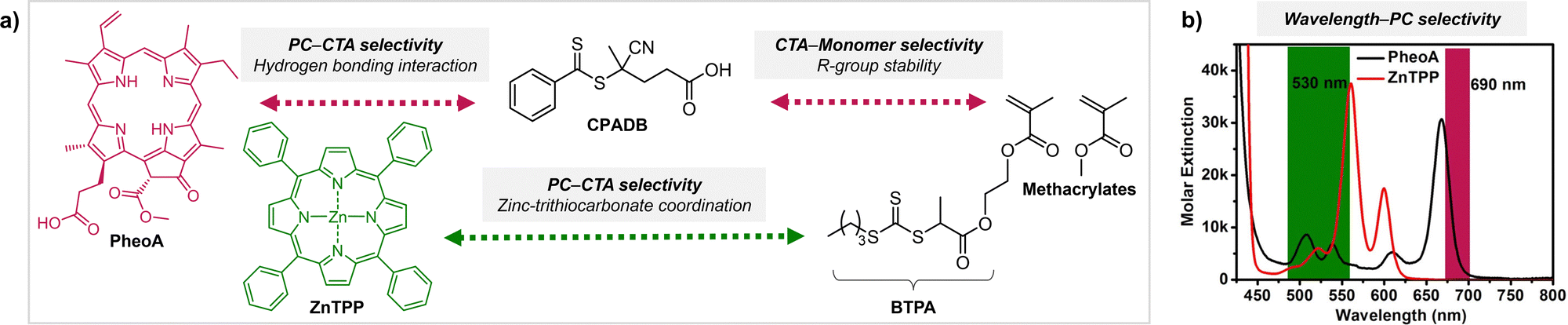 | ||
| Scheme 8 (a) Selectivity issues in PET-RAFT polymerization from a reaction mixture of two PCs (PheoA and ZnTPP), two CTAs (CPADB and BTPA), and methacrylate monomers. The two PCs can be selectively activated at different wavelengths as per (b) UV-vis absorption profiles. Adapted with permission from (b) ref. 106 (Copyright 2016 American Chemical Society). | ||
 | ||
| Scheme 9 Monomer-dependent selective photoactivation of CPADB.106 | ||
CTAs possessing the same Z group but different R groups can also be selectively fragmented. Matyjaszewski and coworkers in collaboration with Boyer reported that during photoiniferter polymerization of MMA in the presence of CDTPA and BTPA, only CDTPA was consumed for polymerization.110 This was because the photolysis of CDTPA was favored owing to the higher stability and radical stabilization energy (RSE) of the tertiary R group, whereas BTPA with the secondary R group did not undergo fragmentation or participate in chain transfer with the poly(MMA) radical (Scheme 10a). The retarded polymerization rate upon the addition of an excess amount of BTPA or DTPA evidenced that both CTAs participated in the formation of the intermediate adduct radical, whereas only the adduct with CDTPA resulted in the fragmentation and release of the R group for propagation. By changing the irradiation wavelength from green (520 nm, 4.25 mW cm−2) to blue (465 nm, 6.5 mW cm−2), the remaining intact BTPA could be consumed for polymerization of MA or N,N-dimethyl acrylamide (DMAm) in which both the R group of the CTA and the monomer generated secondary R groups (Scheme 10b).
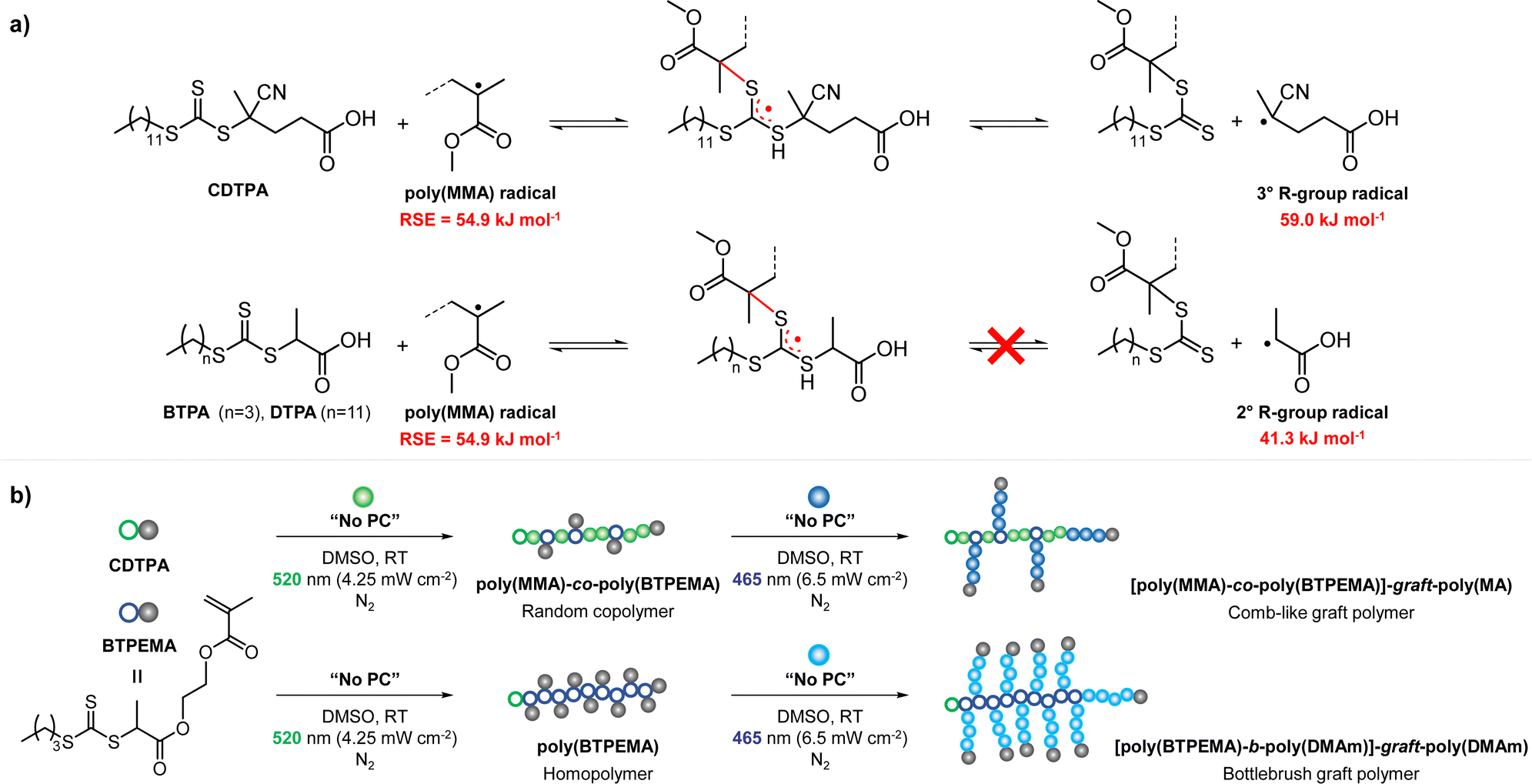 | ||
| Scheme 10 (a) Selective fragmentation of CTAs after the addition of the poly(MMA) radical. (b) Stepwise synthesis of comb-like and bottlebrush polymers via selective fragmentation of CTAs under irradiation at different wavelengths.110 | ||
As it is the change in C–S bond stability and balance among the various possible reactions (e.g., chain transfer, dissociation, propagation, and recombination) within the RAFT equilibrium that dictate the selectivity in the activation of the CTA to add a monomer, the existence of a PC is not necessary for the selective activation of the CTA and subsequent polymerization; nevertheless, the PC facilitates the reaction or allows upscale of the reaction system because of oxygen tolerance. For further discussion on SUMI and utilization of the unique selectivity of photocontrolled RAFT polymerization to confer polymers with complex architectures, please see Sections 4.3.1.2 and 4.3.2, respectively.
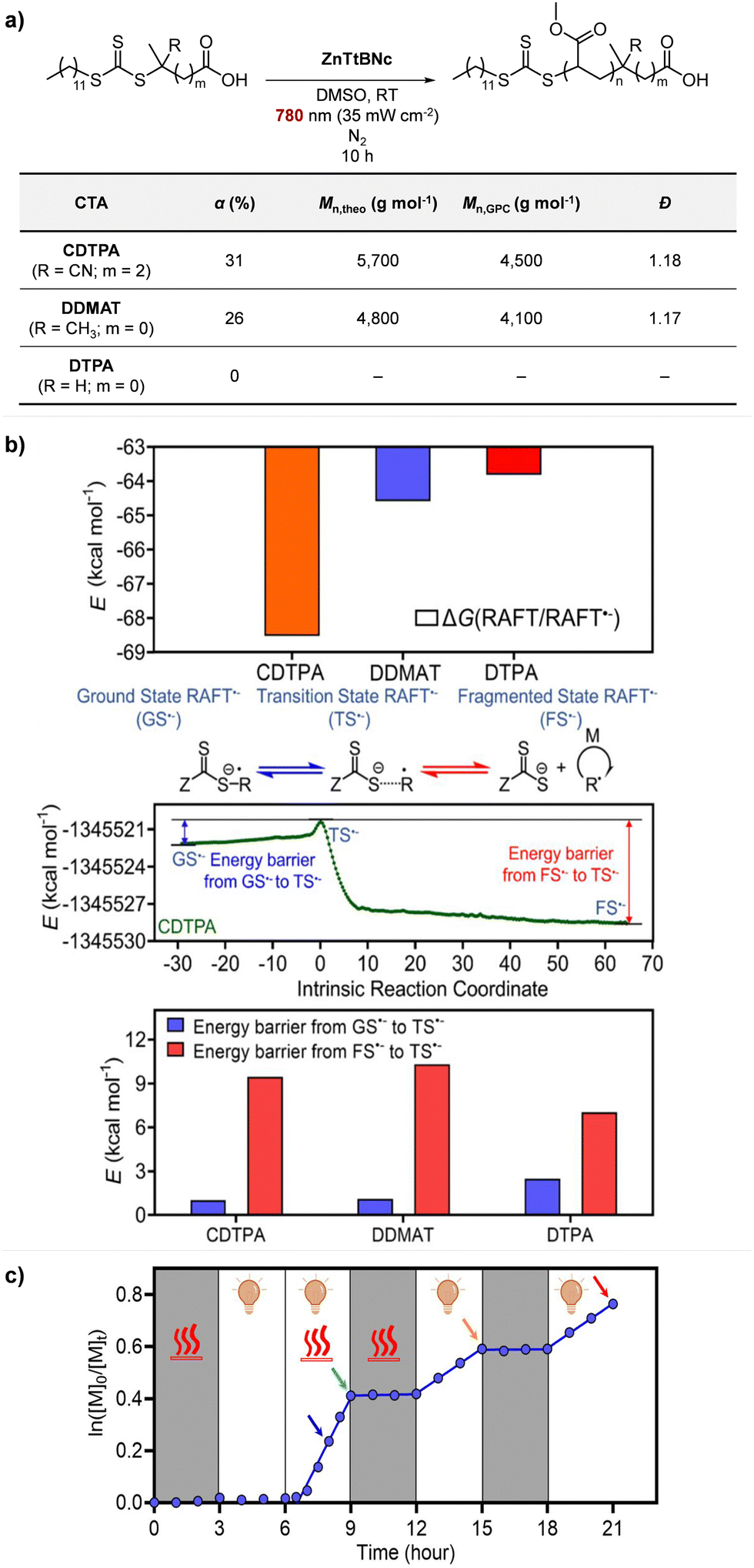 | ||
| Fig. 6 (a) PET-RAFT polymerization of MA. (b) Comparison of Gibbs free energy change, reaction path calculated by the IRC method, and energy barriers in CTA˙− fragmentation. (c) Heat-assisted photoactivation (at 52 °C) of DTPA for PET-RAFT polymerization of MA. Adapted with permission from ref. 64 (Copyright 2022 American Chemical Society). | ||
3.3. Orthogonality
Multiple reactions that can occur either individually or simultaneously in one pot without interfering with each other are said to be orthogonal. For example, in terms of the type of reactive species, PET-RAFT or photoiniferter polymerization with radical intermediates is orthogonal to the reactions, including (photomediated) cationic RAFT or anionic or cationic ring-opening polymerization (ROP), involving ionic intermediates. In terms of the energy source, light can be turned on and off without interrupting other stimuli. Moreover, light at certain wavelengths that can selectively activate one of the several combined organic photochemical reactions has paved the way for efficient synthesis of advanced functional materials through, for example, formation and post-modification of the polymer network and programmed patterning of given surfaces.112–114 Therefore, researchers either orthogonally turned on and off the photomediated reversible (de)activation of the radicals produced from the PC and/or CTA to ionic polymerizations mediated by other stimuli (e.g., heat,115 electricity,116,117 and the presence of specific reagents118–122) or selectively activated two photomediated polymerizations via irradiation at disparate wavelengths.123–126This orthogonality that allows on-demand control of each polymerization system has been applied to the one-pot synthesis of polymers with complex architectures and compositions as discussed in Sections 4.3.1.1 and 4.3.2. By combining two orthogonal polymerizations, the scope of the monomers incorporated along the single backbone has widened. The microstructure of multiblock copolymers (e.g., order, length, and number of blocks) could also be facilely manipulated by tuning the order and length of the applied external stimuli, and the number of switching between two polymerizations.
3.4. Oxygen tolerance
Due to its radical scavenging effect, oxygen is detrimental to radical polymerization and thus needs to be removed before the reaction. To conduct the reaction in the presence of oxygen, oxygen tolerance is of significant interest as the degassing process is time-consuming and limits the practicability of polymerization from both biological and industrial perspectives. Among the various photomediated RAFT polymerizations discussed herein, oxygen tolerance of PET-RAFT polymerization has been extensively recognized,49,127 as compared to the cases of photoiniferter and photomediated cationic RAFT polymerizations. It is because inactivation of oxygen occurs via a photoinduced electron or energy transfer mechanism in the presence of the PC and/or any additives that play a supporting role (Section 3.4.1.1). In contrast, photoiniferter polymerization involving direct photolysis of the CTA lacks the ability to consume oxygen, and thus is intrinsically sensitive to oxygen. Therefore, in the reported studies, alternative approaches such as not stirring the reaction mixture or use of solvent with high viscosity and low oxygen solubility, to physically block the diffusion of oxygen into the polymerization mixture, or additives were employed. Every reported photomediated cationic RAFT polymerization has been performed in deoxygenated media, providing no hint towards oxygen tolerance of the reaction system. As cationic reaction intermediates are not affected by radical-scavenging oxygen, cationic polymerization is known to be insensitive to oxygen;22,128 nevertheless, the impact of oxygen on photocatalytic systems needs to be investigated.Meanwhile, in some cases, oxygen could in turn be transformed into an initiating source or even facilitate the activation of the CTA, which contradicts its known inhibitory role. These examples of oxygen-initiated or -accelerated polymerizations are discussed in Section 3.4.2. This section describes the three categories of oxygen tolerance reported to date in various polymerization reactions (Scheme 11).
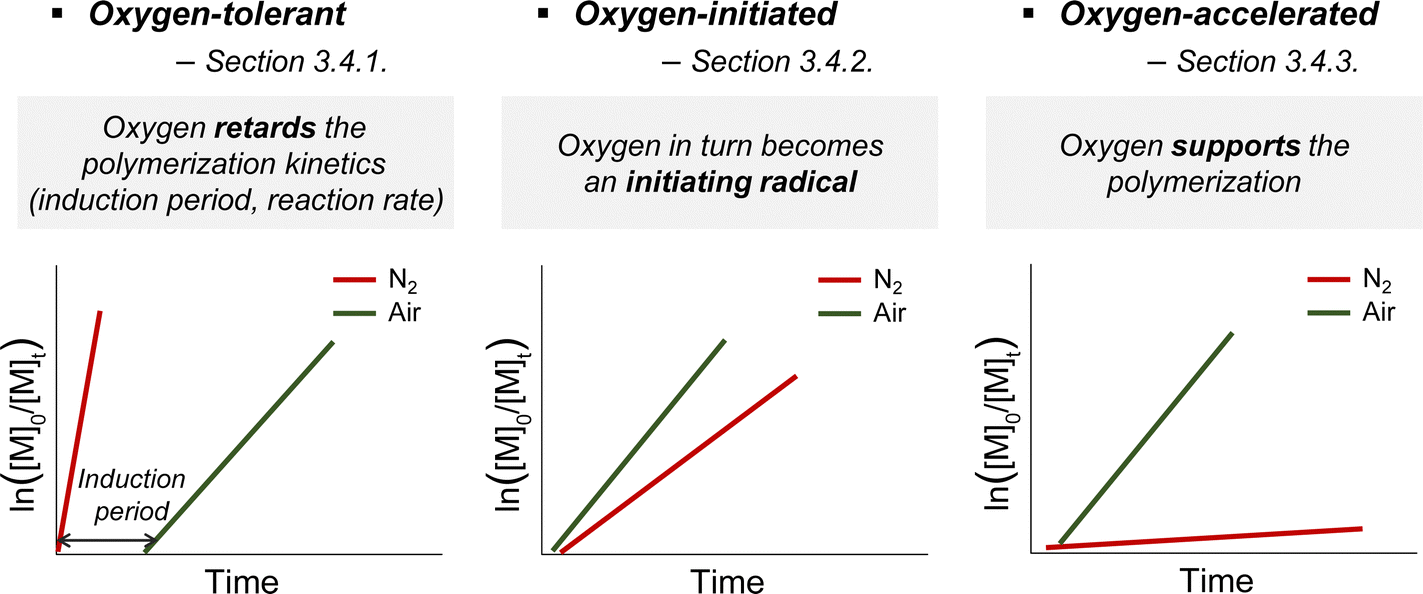 | ||
| Scheme 11 Three categories of oxygen tolerance. Schematic representation of the kinetic plot of ln([M]0/[M]t) vs. reaction time in (a) oxygen-tolerant, (b) oxygen-initiated, and (c) oxygen-accelerated PET-RAFT polymerizations. | ||
It should be noted that, regardless of the type of mechanism, ‘the presence of oxygen’ can refer to the reaction being performed in either a completely ‘open-to-air’, ‘sealed but not degassed’ container or other containers where the volume of the remaining head space substantially affects the amount of oxygen and controllability of polymerization. In any case, oxygen tolerance has extended the applications of photocontrolled RAFT polymerization to 3D/4D printing or surface functionalization of given surfaces or biomolecules in which the deoxygenation process can be time-consuming, cumbersome, or harsh to the substrate.
3.4.1.1. PET-RAFT polymerization. Although the consumption of oxygen accompanies the induction period or slower polymerization kinetics as compared to the case in the absence of oxygen, the polymerization reaction would eventually proceed. Thus, well-defined polymers with high end-group fidelity in the presence of oxygen could be realized. The mechanism to achieve oxygen tolerance depends on the reaction system composition and the concomitant role of the excited-state PC as a reductant (via OQP) or an oxidant (via RQP). In brief, oxygen is reduced by the excited-state PC in OQP and by the ground-state anion of the PC in RQP. Photoinduced energy transfer from the triplet excited-state PC to triplet oxygen is another route for oxygen tolerance (Scheme 12).
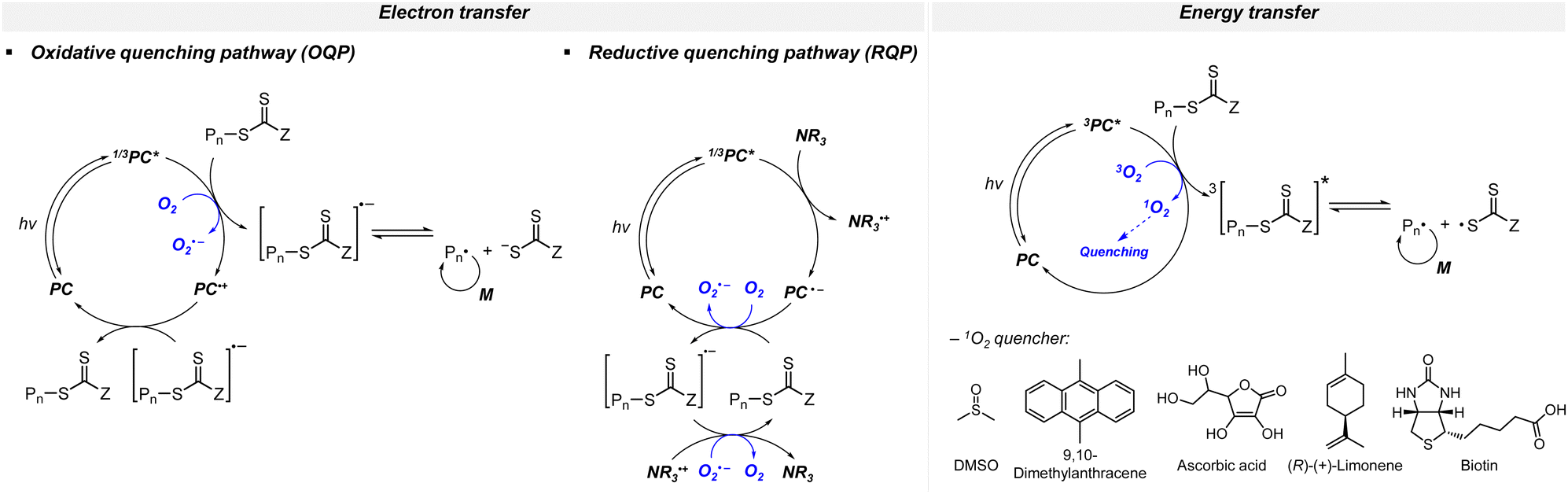 | ||
| Scheme 12 Mechanisms of the three categories of oxygen tolerance in PET-RAFT polymerization. | ||
Electron transfer: oxidative quenching pathway (OQP). Oxygen tolerance of PET-RAFT polymerization was discovered by Boyer and coworkers in 2014 in their first study of PET-RAFT polymerization using Ir(ppy)3 as a PC.31 Strongly reducing excited-state Ir(ppy)3 was expected to reduce oxygen into a superoxide radical anion, which allows the polymerization in a non-degassed sealed vessel. The kinetics of the polymerization of MMA and MA in DMSO were almost identical to that in a degassed vessel after the induction period to consume oxygen, indicating no degradation of the PC or CTA. With the preserved photocatalytic activity and chain ends, a di- or tri-block copolymer was successfully synthesized by simply adding the monomer and solvent to the reaction mixture. Ru(bpy)3Cl2 with reducing property (
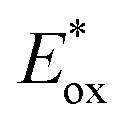 = −0.81 V vs. SCE) similar to that of Ir(ppy)3 (
= −0.81 V vs. SCE) similar to that of Ir(ppy)3 (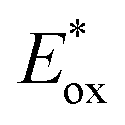 = −1.73 V vs. SCE) also exhibited oxygen tolerance for the polymerization of MA and DMAm in DMSO despite the retardation in polymerization kinetics in the presence of air.129 It was attributed to the sluggish consumption of oxygen by the PC. Whereas the induction period was decreased with an increase in the catalyst loading, the polymerization rate was not significantly affected.
= −1.73 V vs. SCE) also exhibited oxygen tolerance for the polymerization of MA and DMAm in DMSO despite the retardation in polymerization kinetics in the presence of air.129 It was attributed to the sluggish consumption of oxygen by the PC. Whereas the induction period was decreased with an increase in the catalyst loading, the polymerization rate was not significantly affected.
Electron transfer: reductive quenching pathway (RQP). In their first study of PET-RAFT polymerization using organic dyes, Boyer and coworkers observed that EY and fluorescein with lower
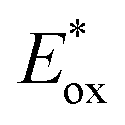 (PC*/PC˙+) than E0red(CTA/CTA˙−) and sufficiently low ΦFL allowed effective PET and polymerization of methacrylates in DMSO under blue light irradiation.66 The apparent propagation rate (kappp) which was lowered by oxygen was restored by adding TEA. The shortened inhibition period suggested that via RQP, the one-electron-reduced EY, generated by the reduction of excited-state EY by TEA, concurrently reduced oxygen to a superoxide radical anion and activated the CTA to allow polymerization without degassing. TEA of at least one equivalent to the CTA was required to completely reduce dissolved oxygen in the reaction vessel. The concentrations of TEA and EY were also important as the highly increased polymerization rate (i.e., high radical flux and high catalyst turnover) led to an increase in polymer dispersity (Đ = 1.4).130 In addition to the tertiary amine, ascorbic acid (or ascorbate)131–133 was added to the reaction system to induce reductive quenching of other PCs including ZnTPPS4− 131,133 and poly(boron dipyrromethene-alt-fluorene),132 and concomitant oxygen tolerance in the reaction system.
(PC*/PC˙+) than E0red(CTA/CTA˙−) and sufficiently low ΦFL allowed effective PET and polymerization of methacrylates in DMSO under blue light irradiation.66 The apparent propagation rate (kappp) which was lowered by oxygen was restored by adding TEA. The shortened inhibition period suggested that via RQP, the one-electron-reduced EY, generated by the reduction of excited-state EY by TEA, concurrently reduced oxygen to a superoxide radical anion and activated the CTA to allow polymerization without degassing. TEA of at least one equivalent to the CTA was required to completely reduce dissolved oxygen in the reaction vessel. The concentrations of TEA and EY were also important as the highly increased polymerization rate (i.e., high radical flux and high catalyst turnover) led to an increase in polymer dispersity (Đ = 1.4).130 In addition to the tertiary amine, ascorbic acid (or ascorbate)131–133 was added to the reaction system to induce reductive quenching of other PCs including ZnTPPS4− 131,133 and poly(boron dipyrromethene-alt-fluorene),132 and concomitant oxygen tolerance in the reaction system.
The study of the effect of tertiary amines on PET-RAFT polymerization using EY by Ferji and coworkers revealed that the stability of the amine radical cation impacted the efficiency of the reduction of excited-state EY, subsequent reduction of the CTA, and polymerization in DMSO in the presence of air.73 Herein, the authors concluded that (i) oxygen tolerance originated from photoinduced conversion of oxygen to inactive singlet oxygen rather than from the reduction of oxygen to the superoxide radical anion and (ii) the tertiary amine facilitated the reduction of the CTA by preferably generating the EY radical anion. Among five amines (TEA, triethanolamine (TEOA), tributylamine, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), and 4-(dimethylamino)pyridine (DMAP)), tributylamine and DMAP increased the amine radical stability and conferred oxygen tolerance because of the long alkyl chain and π-bond conjugation, respectively, whereas TEOA in combination with EY produced an initiating radical even in the absence of a CTA to provide polymers with high dispersity.
To date, a variety of amines such as TEA,134 TEOA,67,135–140N,N,N′,N′-tetramethylethylenediamine (TEMED)141 and PMDETA68–70 have been employed as cocatalysts with EY in various solvents for 3D printing resin, surface-initiated polymerization from proteins and DNA, and polymerization in continuous flow reactors, where oxygen tolerance and accelerated polymerization kinetics are highly advantageous. Another xanthene-based organic PC EB/TEOA system was also employed for 3D printing in aqueous solutions without prior deoxygenation.142 However, some reaction systems were still degassed despite the use of amines, and the reason for degassing and the results of negative controls such as polymerization without a PC, amine, or CTA were not provided, which hampered the elucidation of the exact role of an amine additive. Moreover, Boyer, Sumerlin and coworkers reported that the reaction mechanism became complicated depending on the reaction conditions (e.g., irradiation wavelength, reagents, concentration, and solution pH) and concomitantly generated different reaction intermediates.57
Amine-facilitated reduction of oxygen was also reported for graphitic carbon nitride (g-C3N4).143,144 Qiao and coworkers observed a significant decrease in the inhibition period (from 3 hours to 30 minutes) after the addition of TEOA into PET-RAFT polymerization of MA under UV light irradiation (365 nm, 3.5 mW cm−2).143 The efficient deoxygenation was ascribed to an additional pathway to reduce dissolved oxygen via g-C3N4-mediated electron transfer from TEA. Ferji and coworkers again examined the influence of the amount of amine on polymerization control.144 In g-C3N4-catalyzed PET-RAFT polymerization of MMA as a model monomer in the presence of tributylamine under irradiation at 405 nm (60 mW cm−2), loss of polymerization control was noticed when five equivalents of tributylamine relative to TTC were added. It was attributed to the production of species that could initiate free radical polymerization via the interaction of the oxidized amine with another amine. The authors thus set the appropriate molar ratio of amine to CTA as one.
Energy transfer. Oxygen can also be inactivated via the photoinduced energy transfer mechanism. Owing to the excitation of triplet oxygen to the singlet state via triplet–triplet annihilation with excited-state ZnTPP and subsequent consumption of singlet oxygen by DMSO to form dimethyl sulfone, the concentration of dissolved oxygen was kept sufficiently low to continue the polymerization of DMAm even under fully open conditions.145 However, in other solvents with properties (i.e., boiling point, molecular weight, and dielectric constant) similar to those of DMSO, polymerization became considerably slower with longer inhibition time, implying the intrinsic role of DMSO in the consumption of singlet oxygen. Therefore, to realize oxygen tolerance in other solvents, additives as singlet oxygen quenchers were necessary.146 Among 9,10-dimethylanthracene, ascorbic acid, and (R)-(+)-limonene, 9,10-dimethylanthracene was the most effective additive, demonstrating similar kinetics to the polymerization with deoxygenation, a very short inhibition period, and excellent temporal control. ZnTMPyP, another water-soluble analogue of ZnTPP, with biotin was employed in the oxygen-tolerant aqueous dispersion polymerization of hydroxypropyl methacrylate (HPMA) for photoinitiated polymerization-induced self-assembly (photo-PISA) in a 96-well plate without deoxygenation.147 Herein, the thioether group of biotin was oxidized to sulfoxide upon trapping of singlet oxygen. When the thioether group was a part of the monomer, polymerization of 2-(methylthio)ethyl methacrylate (MTEMA) bearing the thioether group using ZnTPP was oxygen-tolerant without any additives.148 This oxygen tolerance of ZnTPP was also retained when embedded in 2D metal–organic framework (MOF) nanosheets.149 Similarly, sodium ascorbate significantly reduced singlet oxygen in Ru(bpy)3Cl2-catalyzed rapid and controlled polymerization of acrylamides in water and enabled the facile preparation of triblock copolymers within an hour without deoxygenation.150
3.4.1.2. Photoiniferter polymerization. In the abovementioned mechanisms of conferring oxygen tolerance to PET-RAFT polymerization, oxygen is commonly consumed and inactivated via additional reaction pathways mediated by a PC with or without additives. In contrast, photoiniferter polymerization in which a reactive radical can only be generated by direct photolysis of the CTA innately suffers from rather slow reaction kinetics and is not tolerant to oxygen. Thus, oxygen tolerance has rarely been achieved.
For instance, TTC-based CTAs in the presence of tertiary amines could mediate polymerization under UV71 or blue light irradiation151,152 to prepare polymers in the absence of a PC. Energetically favored photoinduced electron transfer from the amine to the excited-state CTA and then from the resulting CTA anion to oxygen led to in situ removal of oxygen such that polymerization without prior deoxygenation proceeded after the induction period (Scheme 13).151 Regarding the mechanistic origin of oxygen tolerance, photoinduced energy transfer (i.e., generation of singlet oxygen) was excluded by the similar monomer conversions of MA in DMSO and in toluene despite the different abilities of the solvents to scavenge singlet oxygen. Very recently, Zhang, Weng, and coworkers reported an oxygen-tolerant polymerization system using CPADB under irradiation with a household lamp (14 W, 2.4 mW cm−2) where the amine was incorporated as a part of 2-(dimethylamino)-, 2-(diethylamino)-, or 1-(dipropylamino)ethyl methacrylate monomers.153 However, with a target degree of polymerization (DP) of 400, the relatively high dispersity (1.42–2.12), deviation of molecular weight from theoretical values, and/or the color change of solution from red to yellow (i.e., degradation of CPADB) suggested the need for further optimization of reaction conditions. Gibson and coworkers disregarded the high dispersities of polymers synthesized via amine-catalyzed photolysis of TTC and only focused on the quick preparation of large libraries of polymers via high-throughput (HTP) synthesis in a 96-well plate. Dioxane with degassing ability inferior to that of DMSO was thus used as a solvent owing to the ease of purification of polymers by simple evaporation of solvent under vacuum.152 It has to be noted that categorizing these reports which harnessed amine additives as photoiniferter polymerization might be controversial because herein amine-catalyzed electron transfer instead of the direct photolysis of the CTA initiated the polymerization, which was similar to PC-catalyzed electron transfer in PET-RAFT polymerization. Meanwhile, Chen and coworkers introduced Zn0.64Fe2.36O4 into DMSO154,155 or ZnO NPs into aqueous medium156 to reduce oxygen to a hydroxyl radical via photoinduced generation of electron–hole pairs under sunlight irradiation, and enabled open-to-air photoiniferter polymerization from various CTAs despite the poor controllability (Đ = 1.35–1.56).
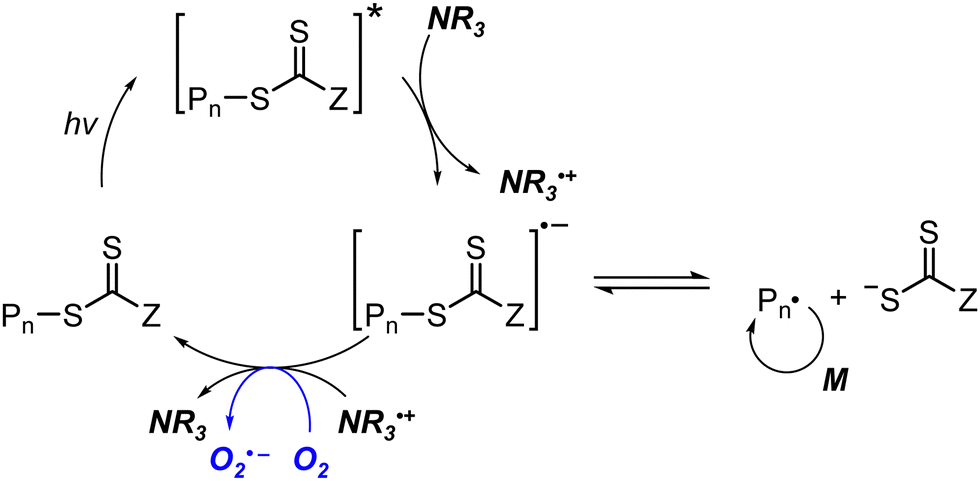 | ||
| Scheme 13 Tertiary amine-assisted oxygen-tolerant polymerization in the absence of a PC. | ||
Oxygen tolerance was also achieved without additives. The so-called “polymerization through oxygen” was reported by Johnson and coworkers even in a completely open vial, where the diffusion of oxygen into the polymerization site was substantially restricted by the high viscosity and low solubility of oxygen in DMSO and not stirring the reaction mixture.157 Polymerization of acrylates and acrylamides from TTCs was thus conducted at the bottom of the reaction vessel directly above the high-intensity irradiation source (450 nm, ≥503 mW cm−2) to ensure a high radical flux and a short inhibition period arising from high light intensity. Although this oxygen-tolerant and rapid polymerization retained the controlled and living nature, the molecular weight of the polymer was larger than the theoretical value due to partial degradation of the CTA located near the solution/air interface. The use of deep eutectic solvents (DES) instead of DMSO improved the “polymerization through oxygen” approach.158 DES, composed of hydrogen-bonding-accepting tetrabutylammonium chloride and hydrogen-bonding-donating ethylene glycol, provided an enhanced polymerization rate and higher stability of the end group of TTCs and DTBs against photodegradation under 465 nm irradiation (0.7 or 3.1 mW cm−2). In addition, oxygen tolerance behavior was more pronounced such that stirring the reaction mixture in an open vial did not affect the polymerization rate. Similar results were observed with PET-RAFT polymerization using Ir(ppy)3, EY, and ZnTPP as a PC, although the exact role of DES needs to be elucidated. The effects of composition of DES, solubility of monomers and polymers in DES, and propagation rate constants (kps) of monomers in DES should also be considered.
Meanwhile, oxidative decomposition of xanthates and concomitant molecular weight discrepancy were also observed during additive-free polymerization of n-butyl acrylate (n-BA) under violet light irradiation in a closed non-degassed reaction vessel.159 Subsequently, Konkolewicz and coworkers filled the remaining headspace in the reaction vessel with an inert solvent rather than air to block the diffusion of oxygen.160 The combination of two immiscible liquids (water and mineral oil) allowed the preparation of aqueous- or organic-soluble polymers after the induction period to consume the already dissolved oxygen.
Benefitting from the simple system composition, photoiniferter polymerization has recently been applied to 3D printing where rapidness and efficiency of polymerization may have higher significances than precise controllability of polymerization. Zhu, Li, and coworkers conducted open-to-air rapid 3D printing using a commercial digital light processing (DLP) technique-based 3D printer, where the oxygen was consumed by the radicals generated after the photolysis of xanthate EXEP under violet light irradiation (405 nm, 2.0 mW cm−2).161 Herein, xanthate was employed because of its favored photolysis due to lower stability of the intermediate radical and the weaker C–S bond as compared to those of TTCs. The radicals generated after photolysis consumed oxygen thereby affording oxygen tolerance to the reaction system. It is noted that in the preparation of a linear polymer under blue light irradiation (460–470 nm), deviation of molecular weight because of the oxidation of some portion of EXEP was observed with a long induction period.162
3.4.1.3. Photomediated cationic RAFT polymerization and others. Oxygen tolerance in PET-RAFT and photoiniferter polymerization is achieved by converting oxygen to reactive oxygen species which is then trapped or deactivating oxygen by coupling with excess radicals. For example, DMSO acts as both a solvent and a singlet oxygen quencher, and thus it is extensively used for oxygen-tolerant polymerization. In contrast, oxygen tolerance of photomediated cationic RAFT polymerization remains elusive presumably because cationic propagating species cannot capture oxygen and this polymerization method is not compatible with DMSO due to the high Lewis basicity of DMSO.121 Cationic reaction intermediates are insensitive to oxygen, but oxygen tolerance of photomediated cationic RAFT polymerization has not been studied.
In other polymerizations, enzymes are utilized. This enzymatic degassing strategy harnesses the intrinsic enzymatic activity to reduce oxygen, so the oxygen tolerance is achieved regardless of irradiation and decoupled from light. Glucose oxidase (GOx) or pyranose oxidase (P2Ox) in the presence of glucose reduces oxygen to hydrogen peroxide. Thus, in situ removal of oxygen was leveraged for EY-catalyzed surface-initiated PET-RAFT polymerization from CTA-tethered substrates under ambient conditions.163 Nanopure water had to be used as a solvent as the enzymatic stability of GOx and the polymerization rate could be influenced by residual minerals and salts in deionized water. Flavin adenine dinucleotide (FAD), a cofactor within GOx, was reduced to FADH− by glucose. Upon violet light irradiation (405 nm, 11.5 W), FADH− was excited, became a highly reducing state, and initiated polymerization by transferring an electron to a monomer or CTA. In addition to the deoxygenation capability by GOx, oxygen was consumed by FADH− regenerating FAD. Therefore, an excess of glucose was necessary initially for GOx and then for FAD to continuously generate FADH− that played a catalytic role. This photoenzymatic RAFT polymerization was employed for ultrahigh-molecular-weight (UHMW) polymer synthesis in an aqueous solution from N-vinylcaprolactam,164 oligo(ethylene glycol) methyl ether methacrylate (OEGMA), and other monomers.165
In conventional photomediated radical polymerization based on Type II visible-light-sensitive photoinitiating systems, oxygen tolerance is achieved by coinitiators and additives (e.g., amine, thiol, silane, germane, borane, phosphine, and phosphite).128,166,167 These hydrogen donors quench inactive peroxyl radicals by hydrogen abstraction and generate new initiating radicals. Page and coworkers recently reported rapid visible-light-driven thiol–ene reaction-based 3D printing of acrylic resin under ambient conditions using a three-component photoinitiating system (ZnTPP as a PC, and electron-donating and electron-accepting coinitiators) (Scheme 14).168,169 The thiol quenched a peroxyl radical which was formed by the addition of oxygen to the propagating radical. The resulting thiyl radical was substantially reactive than the peroxyl radical, so consequently decreased the oxygen inhibition period and continued the polymer network formation. Moreover, the thiol reduced excited-state ZnTPP to yield a thiyl radical which in turn was incorporated as a thioether within the network. The thioether also contributed to oxygen tolerance by scavenging singlet oxygen which was generated by TTA between excited-state ZnTPP and triplet oxygen. Only 1 wt% thiol was sufficient to achieve oxygen tolerance, and the mechanical properties of the printed objects were adjustable by the thiol content.
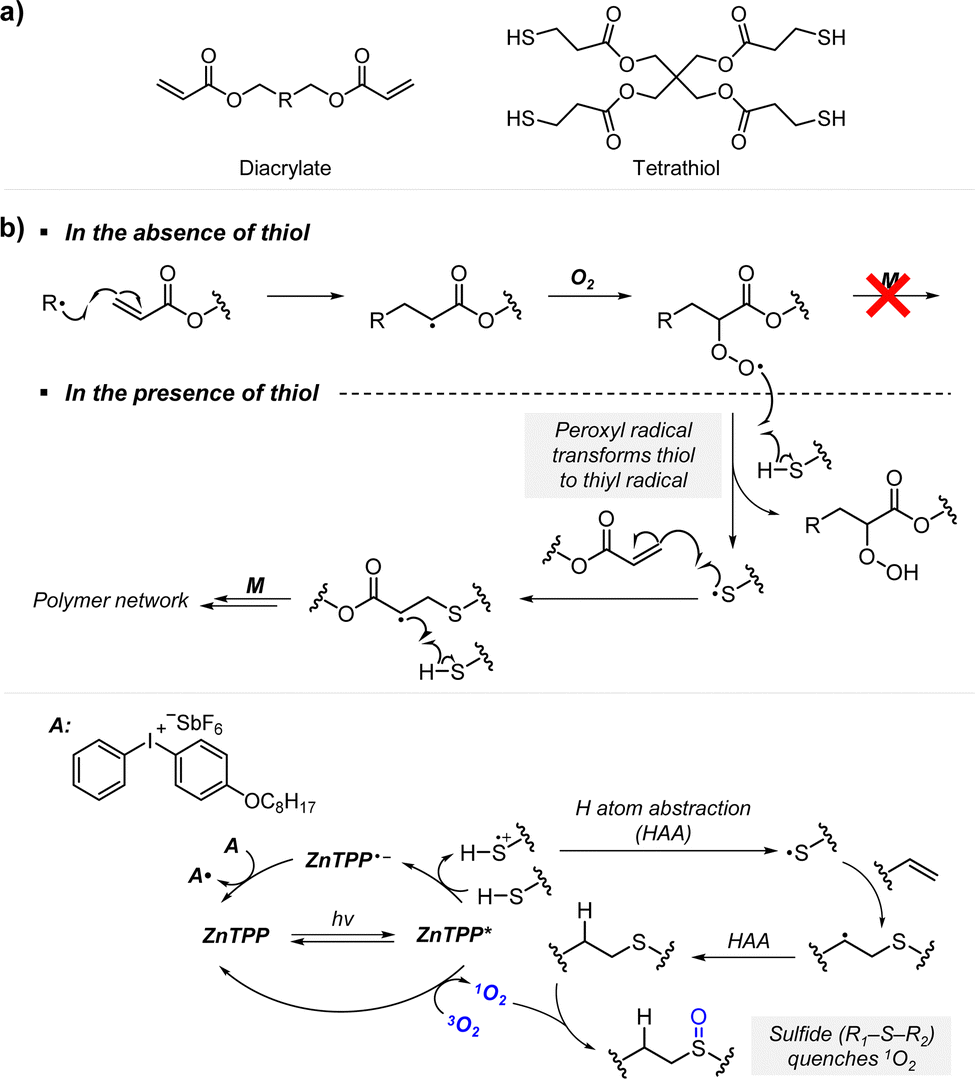 | ||
| Scheme 14 Oxygen-tolerant thiol–ene reaction-based visible-light-driven 3D printing of acrylic resin.169 (a) Structure of diacrylate and tetrathiol monomers. (b) Mechanism of oxygen tolerance in the presence of ZnTPP and thiol. | ||
Ascorbic acid converts singlet oxygen into hydrogen peroxide and then reduces hydrogen peroxide into hydroxyl radicals. Hydroxyl radicals enable polymerization in ultralow volumes (μL scale) where deoxygenation is difficult.170 On the other hand, hydroxyl radical-initiated polymerization could also proceed in the dark via the hydroxyl radicals after a brief irradiation period.93 Hydrogen peroxide, a precursor of hydroxyl radicals, was generated during irradiation. Under near neutral and basic conditions where hydrogen peroxide remained stable, the temporal control was restored as in the case of ZnTPPS4−/ascorbic acid-catalyzed PET-RAFT polymerization of DMAm in water.131 Inferior temporal control of polymerization was also observed for a fluorescein/ascorbic acid system.171 Only the prior deoxygenation prevented the generation of the latent radicals and stopped the reaction in the dark, but the polymerization rate accordingly decreased owing to lowered amount of radicals.
Oxygen in the presence of water can also generate hydroxyl radicals. Very recently, Hou, Xiao, Zhao, and coworkers reported that 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy)-azine based covalent organic frameworks (COFs) generated initiating hydroxyl radicals from water and atmospheric air under white light irradiation (15 mW cm−2).172 Photoinduced production of the superoxide radical anion followed by reaction with water generated hydroxyl radicals. Thus, in a completely open vial, the polymerization rate increased with an increase in the volumetric ratio of water to dioxane, whereas polymerization did not proceed in the absence of water. Nonpolar solvents were not appropriate reaction media due to inefficient stabilization of photogenerated carriers. Adding methylene blue or benzoquinone to scavenge the hydroxyl radical or superoxide radical anion, respectively, led to no polymerization, supporting the initiation mechanism. TEA promoted polymerization by enhancing charge separation in the COF and the capability to transform singlet oxygen into a superoxide radical anion.
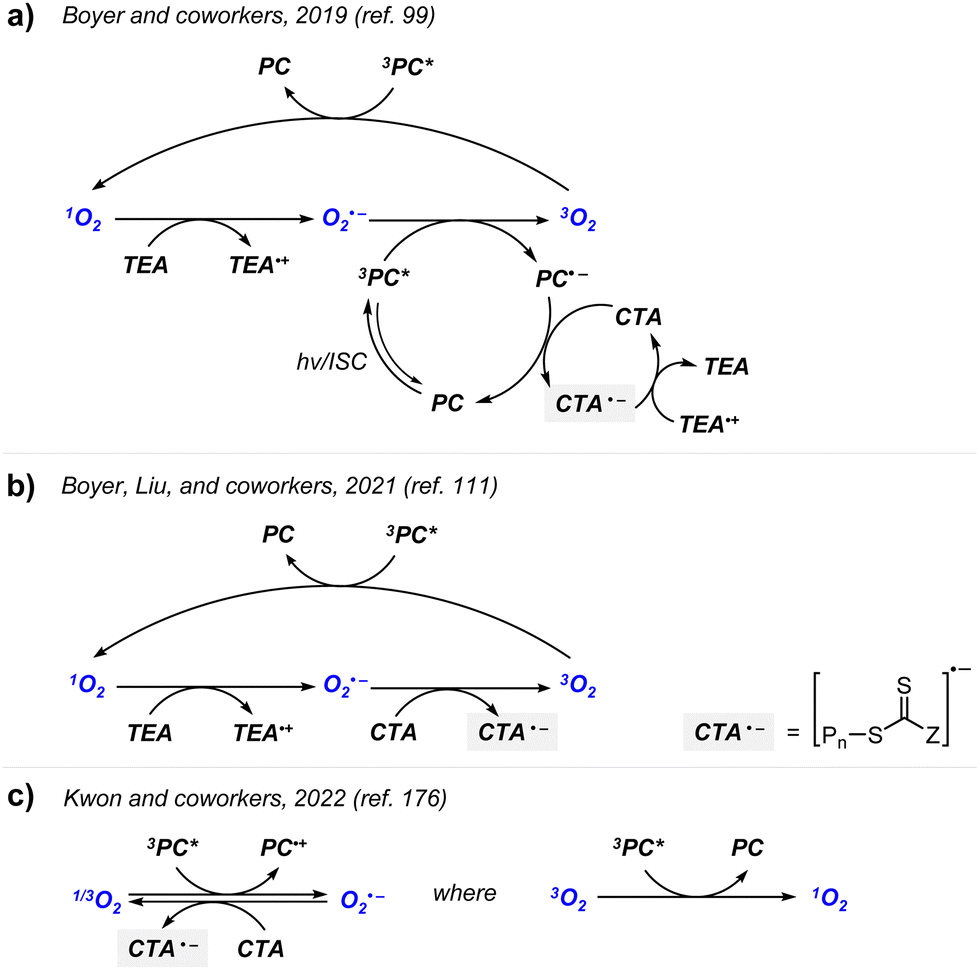 | ||
| Scheme 15 Three distinct mechanisms proposed for oxygen-accelerated PET-RAFT polymerization. Generation of the superoxide radical anion to reduce the CTA was achieved as described in (a) ref. 99 and (b) ref. 111 in the presence or (c) absence of TEA as an additive.176 It is noted that to emphasize the participation of oxygen, the complete catalytic cycles of reagents are not described for clarity. | ||
Boyer and coworkers firstly reported the surprisingly supportive role of oxygen in ZnOETPP/TEA-catalyzed PET-RAFT polymerization under far-red light irradiation (690 nm, 3.0 mW cm−2).99 DMAm and MA could be polymerized in the presence of PC, TEA, and oxygen. “On/off” switchability of the reaction by the introduction and removal of oxygen combined with a constant polymerization rate in the “on” state implied the essential role of oxygen as a catalyst. Moreover, pure oxygen, when compared to atmospheric air, further increased the polymerization kinetics. Based on the experimental investigation and DFT calculations of the formation of singlet oxygen and the superoxide radical anion, the authors proposed the mechanism of oxygen-catalyzed PET-RAFT polymerization. The superoxide radical anion, which was generated via the reduction of singlet oxygen by TEA, reduced excited-state ZnOETPP to proceed PET-RAFT polymerization via RQP (Scheme 15a and Fig. 7).
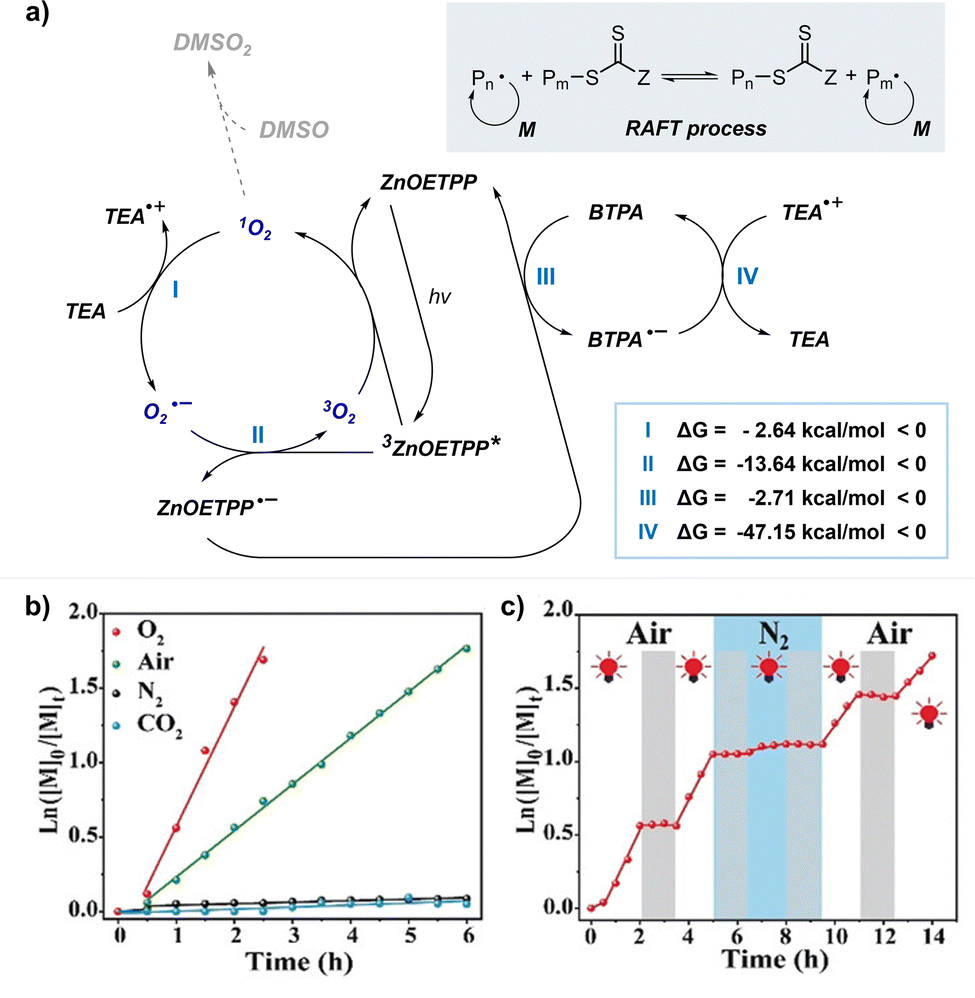 | ||
| Fig. 7 (a) The proposed mechanism of ZnOETPP/TEA-catalyzed PET-RAFT polymerization accelerated by oxygen. Kinetic plots of ln([M]0/[M]t) vs. reaction time (b) under different atmospheres and (c) in gas and light dual-gated polymerization. Monomer: MA; CTA: BTPA; additive: TEA (one equivalent relative to the CTA). Adapted with permission from ref. 99 (Copyright 2019 John Wiley & Sons, Inc.). | ||
The peculiar role of oxygen, namely the ‘oxygen-mediated reductive quenching pathway (O-RQP)’, was also reported by Boyer, Liu, and coworkers.111 Herein, oxygen allowed the activation of the CTA, which otherwise was restricted by the unfavorable thermodynamics of the PC exhibiting long wavelength absorption either via conventional OQP or RQP. The excited-state PC generates singlet oxygen, which is reduced to a superoxide radical anion by TEA. It was this superoxide radical anion that directly activated the CTA via single-electron transfer and activated the polymerization (Scheme 15b and Fig. 8). Combined experimental and theoretical investigations of the PET-RAFT polymerization of MA in DMSO using four model (metallo)porphyrin-based PCs revealed that the thermodynamic viability of O-RQP predominantly depended on the singlet oxygen generation ability of the PC, rather than the electron transfer ability. In particular, via O-RQP in the presence of oxygen, the fastest kinetics was achieved as compared to that via other pathways. Finally, as expected, four phthalocyanines that were previously considered inactive for PET-RAFT polymerization via the conventional mechanisms became successful PC candidates for well-controlled PET-RAFT polymerization under red to NIR light irradiation (590, 660, 730, and 780 nm depending on PCs, 10 mW cm−2) via the newly proposed O-RQP (Scheme 16a). This mechanism was partially valid for aqueous polymerization in water using a water-soluble phthalocyanine ZnPCS4− (Scheme 16b).173 Herein, reactive oxygen species did not directly activate the CTA, but participated in the generation of other initiating radicals. Water as a hydrogen donor transformed the superoxide radical anion into a hydrogen peroxyl radical, which is a precursor of hydrogen peroxide. Hydrogen peroxide was then activated by excited-state ZnPCS4− to form an initiating hydroxyl radical. Based on experimental and DFT calculations, this proposed pathway was more favored than OQP and RQP, leading to successful demonstration of polymerization in water under NIR irradiation (730 nm, 12 mW cm−2). Another water-soluble phthalocyanine possessing several ethylene glycol units could also catalyze PET-RAFT polymerization under NIR irradiation (730 nm, 75.7 mW cm−2) only in the presence of hydrogen peroxide under a N2 atmosphere,174 supporting the proposed mechanism for initiation. This newly proposed O-RQP is thus promising to widen the scope of PCs in catalyzing PET-RAFT polymerization at longer wavelengths; nevertheless, the stability of PC candidates in the presence of reactive oxygen species must be considered. Hou, Xiao, and coworkers reported a similar system for heterogeneous PCs involving porphyrins.175 As the head space or amount of oxygen in the sealed reaction vessel increased, the polymerization rate was accelerated albeit with the lengthened induction period. TEA-catalyzed depletion of singlet oxygen and the formation of the superoxide radical anion were experimentally confirmed by UV/Vis and electron spin resonance (ESR) spectroscopy.
 | ||
| Fig. 8 (a) Chemical structure of ZnTSP. (b) Kinetic plot of ln([M]0/[M]t) vs. reaction time and temporal control of model PET-RAFT polymerization via OQP (green), RQP (blue), and O-RQP (red). (c) Schematic representation of thermodynamic viability (denoted by Gibbs free energy change [kcal mol−1]) for each mechanism. Written in grey are threshold values for each step. Monomer: MA; CTA: DTPA; additive: TEA (0.5 equivalent relative to the CTA). Adapted with permission from ref. 111 (open access). | ||
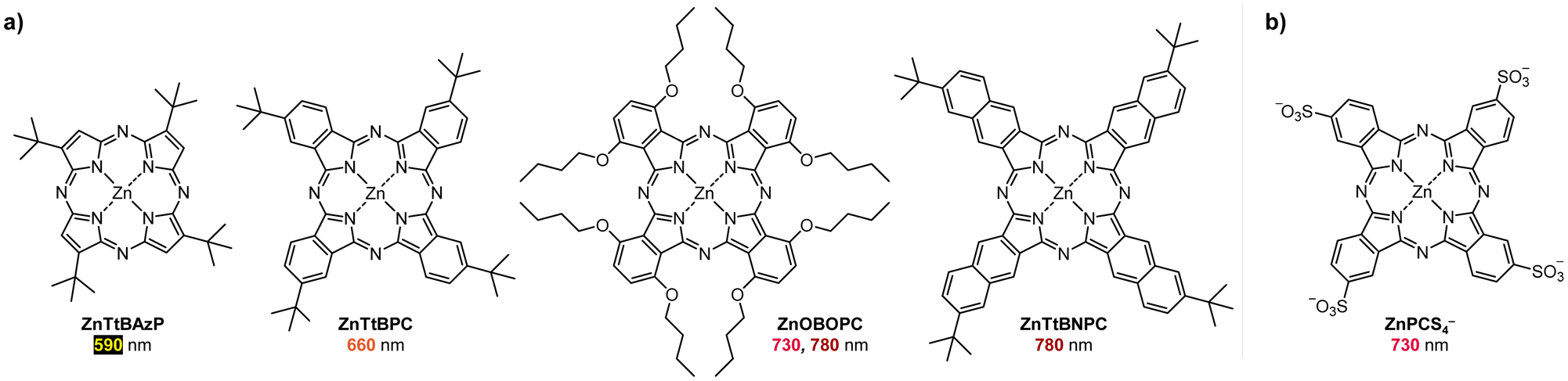 | ||
| Scheme 16 Chemical structures of the phthalocyanines discovered for PET-RAFT polymerization at longer wavelengths (>590 nm) via O-RQP.111 | ||
Very recently, Kwon and coworkers in collaboration with Gierschner and Koo reported 3DP-MSDP-IPN as a novel organic PC for additive-free oxygen-accelerated polymerization in ambient and aqueous environments.176 The mechanism was similar to O-RQP111 in terms of generation of singlet oxygen by the excited-state PC and activation of the CTA by the superoxide radical anion. However, Stern–Volmer analysis suggested that the excited-state PC was likely to be quenched by oxygen rather than by the CTA, which means that in the presence of a PC possessing a suitable redox potential, the superoxide radical anion could be generated without an external reductant (Scheme 15c). Ag2S NCs also exhibited a similar phenomenon.65 Using potassium superoxide as a source of the superoxide radical anion, the polymerization still proceeded in the absence of Ag2S NCs, confirming the proposed mechanism for oxygen-accelerated polymerization.
Nevertheless, it should be noted that the abovementioned oxygen-accelerated polymerization, such as via O-RQP, is mechanically distinct from the previously reported oxygen-demanding polymerization using alkylborane (complex) as a precursor of the initiating radical. For example, triethylborane reacts with oxygen to liberate an ethyl radical, which reacts with another oxygen to generate a peroxyl radical. The peroxyl radical then reacts with triethylborane to generate an ethyl radical as an initiating species (Scheme 17a). Therefore, polymerization can be initiated and externally regulated by the presence of oxygen.177 If alkylboranes are complexed, the complex first needs to be decomplexed for further processes.178 Wu and coworkers reported both oxygen-demanding and photocontrolled polymerization based on light-induced decomplexation of the alkylborane complex.179 Upon UV light irradiation, the photoacid in the triethylborane-amine/photoacid complex 1,3-diaminopropane-triethylborane (DAPTB)/Ph2I+ generated a proton which subsequently released ethylborane to consecutively initiate polymerization (Scheme 17b).
4. PET-RAFT polymerization
4.1. Overview
This section aims to summarize the research on PET-RAFT polymerization. First of all, the reported PCs are outlined in Section 4.2. The next sections summarize the applications of PET-RAFT polymerization such as for preparation of polymers with precise control over their characteristics (e.g., sequence, architecture, polymer tacticity, and dispersity). PET-RAFT polymerization has been widely applied to various applications owing to its uniquely distinct features in contrast to the conventional RAFT polymerization. In particular, surface-initiated polymerization to functionalize a given surface, 3D/4D printing technologies, and stereolithography along with the advent of advanced techniques, such as flow chemistry and high-throughput (HTP) synthesis, would pave the way to the industrial applications. If worth mentioning, the newly designed functional monomers are also introduced.4.2. PCs for polymerization
Since the first employment of Ir(ppy)3 as a PC in 2014,31 along with the increasing number of applications, the scope of PCs has been accordingly widened to realize PET-RAFT polymerization of various monomers under a wide range of wavelengths. In brief, the development of PCs has been directed as follows. Concerns regarding environmental issues of transition metals have led to development of metal-free (in)organic alternatives, which increased the environmental compatibility of PCs and eliminated the potential harm in applying the prepared polymeric materials in electronic and/or biomedical fields.180,181 In the case of biomedical applications, PCs that can be activated under irradiation at longer wavelengths are desired as deeper penetration length and lower energy would allow polymerization in the presence of biomolecules and eventually enable in vivo polymerization in the near future. Last but not least, heterogeneous PCs that can be easily purified and reused for the next polymerizations have also gained interest. In any case, highly efficient photocatalytic performance of a PC that realizes polymerization at low catalytic loading and/or endows the reaction system with oxygen tolerance is desired.During the search of PCs, researchers initially adapted PCs that were efficient for other types of visible-light-driven organic transformations including RDRPs, solar energy conversion, or water splitting, and successfully demonstrated PET-RAFT polymerization using a wide range of PCs. Apart from these trial-and-error strategies, recently emerging approaches are based on investigation of the structure–property/performance relationship of conventional PCs and computer-guided design of novel PCs.96,182 These general design strategies have been quite well established and allow efficient, systematic, and rational design of novel PCs. For details on the rational design of PCs, please refer to the review articles by Boyer, Miyake, Liu, and coworkers, who have summarized the effects of the structures of PCs on their photocatalytic activities (light absorption, quantum yield, and redox properties) in various photocontrolled polymerizations (including PET-RAFT and photomediated cationic RAFT polymerizations),183 and Kwon and coworkers, who have particularly focused on the recently emerged purely organic PCs since 2016.184
In the following sections, the reported PCs are categorized into metal-based, organic, inorganic, and modified PCs. The types of PCs are briefly described, while the detailed applications are mentioned throughout Sections 4.3 and 4.4. For representative PCs, structures of the PCs are provided with the used wavelengths and monomers reported for polymerization (Table 1). It is noted that PET-RAFT polymerization provided high tolerance of functional groups incorporated into monomers (Scheme 18). For example, by PET-RAFT polymerization using Ir(ppy)3100 or EY,185 macrocyclic allylic sulfones, which introduce degradable ester bonds along the polymer backbone, could be evenly distributed within a nearly ideal random copolymer of macrocyclic allylic sulfones and acrylic monomers, which otherwise is not achieved by thermally initiated RAFT polymerization. The even distribution of degradable moieties along the copolymers led to significantly improved degradation behaviors.
| PC | Irradiation wavelengths | CTA–monomer pair | ||
|---|---|---|---|---|
| Dithiobenzoates | Trithiocarbonates | Xanthates | ||
| Metal-based PCs – Section 4.2.1 | ||||
|
Blue (435–470 nm) | MMA, OEGMA, TFEMA | CoAEMA, MAEFe | ←(Methacrylates) |
| t-BA, BzA, MA, OEGA, TFEA | ←(Acrylates) | |||
| HPMAm | ←(Methacrylamides) | |||
| DEAm, DMAm, NAM, NIPAm, N-acryloyl amino acids | ←(Acrylamides) | |||
| (Others)→ | Isoprene, St, VAc, vinyl ketones, macrocyclic allylic sulfone | DVP, NVP, VAc, VBz, VCz, VP | ||
| White | Fluorinated monomers | VDF | ||
|
Blue (435–470 nm) | BzMA, MMA, OEGMA | HPMA, MAA | |
| DEGA, MA, NASS, OEGA | ||||
| DEAm, DMAm, NAM, NIPAm | ||||
| St | ||||
|
BzMA, DMAPS-MA, GMA, HEMA, HPMA, MMA, MPC, MTEMA, NBMA, NHSMA, OEGMA, TFEMA | |||
| Red (635 nm) | n-BA, t-BA, Boc-AEA, BzA, CHA, DEGA, DMAEA, EA, EHA, n-HA, HEA, HPA, IBOA, MA, MSEA, OEGA, TEGDA, TFEA | |||
| Yellow (560 nm) | ||||
| Green (530 nm) | AAm, AAPTAC, t-BAm, Boc-AEAm, Cp-DIBAC, DAAm, DEAm, DMAm, HEAm, NAM, NIPAm, PEAm | |||
| St, VBA | ||||
| White | DEAm, DMAm, HEAm, NAM | |||
| Organic PCs – Section 4.2.2 | ||||
|
BzMA, DMAEMA, GMA, HEMA, MAA, MMA, OEGMA, PFPMA | HFBMA, MMA | ||
| n-BA, t-BA, HEA, MA, OEGA, PEGDA | ||||
| Blue (450–483 nm) | CBMAm, HPMAm | |||
| Green (520–530 nm) | AMPS, DAAm, DEAm, DMAm, FSAm, HEAm, NAM, NIPAm, PEG-based monomers, saccharide- and peptide-based monomers | |||
| BTP, NaSS, St, vinyl ketones, macrocyclic allylic sulfone | ||||
 | ||
| Scheme 18 Chemical structures and abbreviations of monomers employed for PET-RAFT polymerization. The asterisk denotes that only a selected monomer is shown. | ||
Chlorophyll a, a naturally occurring visible-light-absorbing PC for photosynthesis in green plants, is considered the first non-transition metal-based PC and it allowed utilization of low-energy red light (635 nm) for PET-RAFT polymerization for the first time.194,195 Bacteriochlorophyll a, which is present in purple bacteria and absorbs at even longer wavelength (>900 nm), further expanded the wavelength range of PET-RAFT polymerization to the far-red (780 nm) and NIR region (850 nm).196 Metalloporphyrins were also effective under red light irradiation (635 nm).105 Among the metalloporphyrins embedded with different core metals (Zn2+, Ni2+, Co2+, and Fe3+), only ZnTPP catalyzed the polymerization as its high triplet state quantum yield (0.88) and low ΦFL (0.04) increased the probability of photoinduced electron transfer at the triplet excited state. Aluminum (Al3+) porphyrin complexes were recently discovered for polymerization under blue light irradiation (460 nm, 5 mW cm−2).197 On the other hand, free base porphyrin without the core metal still retained photocatalytic ability albeit with a slow polymerization rate, owing to ineffective collision between free base porphyrin and CTA.198 The polymerization rate was significantly enhanced by covalently tethering the CTA to free base porphyrin.
ZnTPP demonstrated a wide absorption profile, so PET-RAFT polymerization could be performed under blue, green, yellow, orange, and red light irradiation (460, 522, 565, 595, and 635 nm, respectively).105 Nevertheless, red light has been typically used in subsequent studies. Several ZnTPP derivatives were employed as well. Water-soluble analogues including ZnTPPS4−98,131,133,199 and ZnTMPyP147,200,201 or supramolecular ZnTPOR/cucurbit[7]uril which exhibited improved water-solubility compared to ZnTPOR202 catalyzed polymerization in aqueous systems. In the case of supramolecular ZnTPOR/cucurbit[7]uril, host–guest complexation between cucurbit[7]uril and TPOR also prevented self-aggregation-induced quenching of excited-state ZnTPOR in water and reduction of photocatalytic performance. Similarly, self-aggregation of ZnTPP at high concentration in DMSO was reduced by linking polyhedral oligomeric silsesquioxane (POSS) to ZnTPP.203 ZnOETPP could absorb far-red light (690 nm), but could catalyze polymerization only in the presence of oxygen and TEA, which is contradictory to the common role of oxygen in inhibiting the polymerization99
Very recently, phthalocyanines were discovered for PET-RAFT polymerization under red to NIR light irradiation (Scheme 16).111,173 These molecules exhibiting long wavelength absorption were inactive for polymerization via OQP and RQP owing to the unfavorable thermodynamics, but became active for polymerization via O-RQP in the presence of oxygen and tertiary amines. For a detailed explanation on the role of oxygen in the case of ZnOETPP and phthalocyanines, please see Section 3.4.2.2.
 | ||
| Scheme 19 Four representative scaffolds of organic PCs discovered for PET-RAFT polymerization. | ||
EY has been widely applied as a PC in various organic transformation reactions via photoredox catalysis.205 In this regard, researchers have employed other classes of previously reported organic PCs (e.g., phenothiazines, phenazines, and cyanoarenes) for PET-RAFT polymerization (Scheme 19). For example, PTH, which realized photoinduced organocatalyzed ATRP (O-ATRP) for the first time,206 promoted the PET-RAFT polymerization of acrylates and acrylamides under irradiation using a CFL207 or at 400 nm.208 Incorporation of electron withdrawing groups (i.e., n-butylphenyl and hydrofluorocarbons) into PTH caused a red-shift in absorption profiles, and thus enhanced visible-light-absorbing abilities in the case of PTH-2,209 PTH-3,210,211 and PTH-4.212 Moreover, fluorinated PTH-4 provided better control over the polymerization of fluorinated polymers as compared to non-fluorinated PTH-3, owing to the fluorine–fluorine interaction between the PC and the propagating chain-end.212 PTH-5 with the extended conjugation exhibited lower 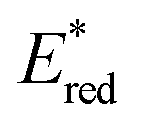 and higher E0ox than PTH-3, which led to favored initial electron transfer from the excited-state PC to xanthate and subsequent electron transfer from the xanthate anion to the one-electron-oxidized PC.213 A series of phenazine-based PCs developed by structural modification of 5,10-diphenyl-5,10-dihydrophenazine exhibited disparate photocatalytic activities in PET-RAFT polymerization under blue light irradiation (460 nm).125 Likewise, covalent structural modification of the known PC scaffolds, including extension of the conjugated structure or change of substituents, has been widely applied to modulate the performance of PCs in polymerization under desired reaction conditions (e.g., irradiation wavelengths and chemical properties of reagents).
and higher E0ox than PTH-3, which led to favored initial electron transfer from the excited-state PC to xanthate and subsequent electron transfer from the xanthate anion to the one-electron-oxidized PC.213 A series of phenazine-based PCs developed by structural modification of 5,10-diphenyl-5,10-dihydrophenazine exhibited disparate photocatalytic activities in PET-RAFT polymerization under blue light irradiation (460 nm).125 Likewise, covalent structural modification of the known PC scaffolds, including extension of the conjugated structure or change of substituents, has been widely applied to modulate the performance of PCs in polymerization under desired reaction conditions (e.g., irradiation wavelengths and chemical properties of reagents).
As the structures of PCs affect their catalytic abilities for PET-RAFT polymerization, a comprehensive investigation of commercial fluorescein derivatives guided the development of a novel PC Henry-1.214 Herein, the photophysical properties studied by DFT calculations were compared with the experimental results of the polymerization. On the other hand, the discovery of TBrFL was fully guided by DFT calculations.96 In a similar vein, via a synthetic platform proposed for the design of organic PCs based on strongly twisted donor–acceptor scaffolds,182 a combination of various donors and acceptors provided a large number of PC candidates with predictable photophysical and electrochemical characteristics. Thus, highly efficient cyanoarene derivatives 4DP-IPN215 and 3DP-MSDP-IPN176 were newly designed for PET-RAFT polymerization in an organic and aqueous system, respectively. The outstanding photocatalytic performances of all systematically designed PCs (i.e., Henry-1, TBrFL, 4DP-IPN, and 3DP-MSDP-IPN) were ascribed to efficient generation of a triplet excited state and appropriate excited-state redox potentials, proposing the key design principle of next-generation PCs for PET-RAFT polymerization.
Thus far, perylene,216 benzaldehyde,217–219 benzothiadiazole,220 PheoA,106 reduced free base porphyrin,221 fluorophenyl bacteriochlorin,222 self-assembled carboxylated porphyrin (SA-TCPP),223 g-C3N4,143,144,224,225 poly(1,4-diphenylbutadiyne)-nanofibers (PDNB-NF),226 imine-based COFs,227 and (heteroatom-doped) carbon dots228 have been reported as organic PCs. Among them, SA-TCPP, g-C3N4, imine-based COFs, and PDPB-NF were heterogeneous, so they can be separated after the polymerization and reused several times. Moreover, biomolecules such as flavin mononucleotide229 and FAD cofactor164,165 were employed. FAD is a cofactor of GOx and is reduced to FADH− in the presence of glucose. FADH− exhibits a high excited-state reducing power, so could catalyze photoenzymatic RAFT polymerization under violet light irradiation (405 nm).
| PC | Irradiation wavelengths | ||||
|---|---|---|---|---|---|
| Ag3PO4 | Blue (465 nm) | Green (525 nm) | Red (625 nm) | NIR (780, 940 nm) | Sunlight |
| Ag2S NCs | Blue (455 nm) | Green (515 nm) | Red (630 nm) | Sunlight | |
| Zn0.64Fe2.36O4 | Sunlight | ||||
| Bi2O3 | White | ||||
| CdSe QDs | Blue (465 nm) | Green (532 nm) | White, sunlight | ||
| Si QDs | Blue (460 nm) | White | |||
| CsPbBrxI3−x NCs | Blue (460 nm) | Green (535 nm) | Red (635 nm) | ||
| NaYF4:Yb3+/Tm3+ and NaYF4:Yb3+/Er3+ NPs | NIR (980 nm) | ||||
It is noted that Ag3PO4 could be activated in the NIR region (940 nm) owing to the localized surface plasmon resonance effect.230 NaYF4:Yb3+/Tm3+ and NaYF4:Yb3+/Er3+ NPs,238 and CsPbBrxI3−x perovskite NCs237 could also utilize NIR light owing to their upconversion properties and an extremely large two-photon absorption cross-section, respectively. NaYF4:Yb3+/Tm3+ and NaYF4:Yb3+/Er3+ NPs absorbed NIR light (980 nm) and emitted light at 325–380 and 425–500 nm via upconversion.238
Moreover, based on the heterogeneity of inorganic PCs, recovery and reusability of the used PCs for the next polymerizations were demonstrated for several PCs such as CdSe-based QDs,233 Zn0.64Fe2.36O4,154 and Bi2O3.232 These PCs were typically retrieved by simple centrifugation, whereas magnetic Zn0.64Fe2.36O4 could be recovered with a magnet.
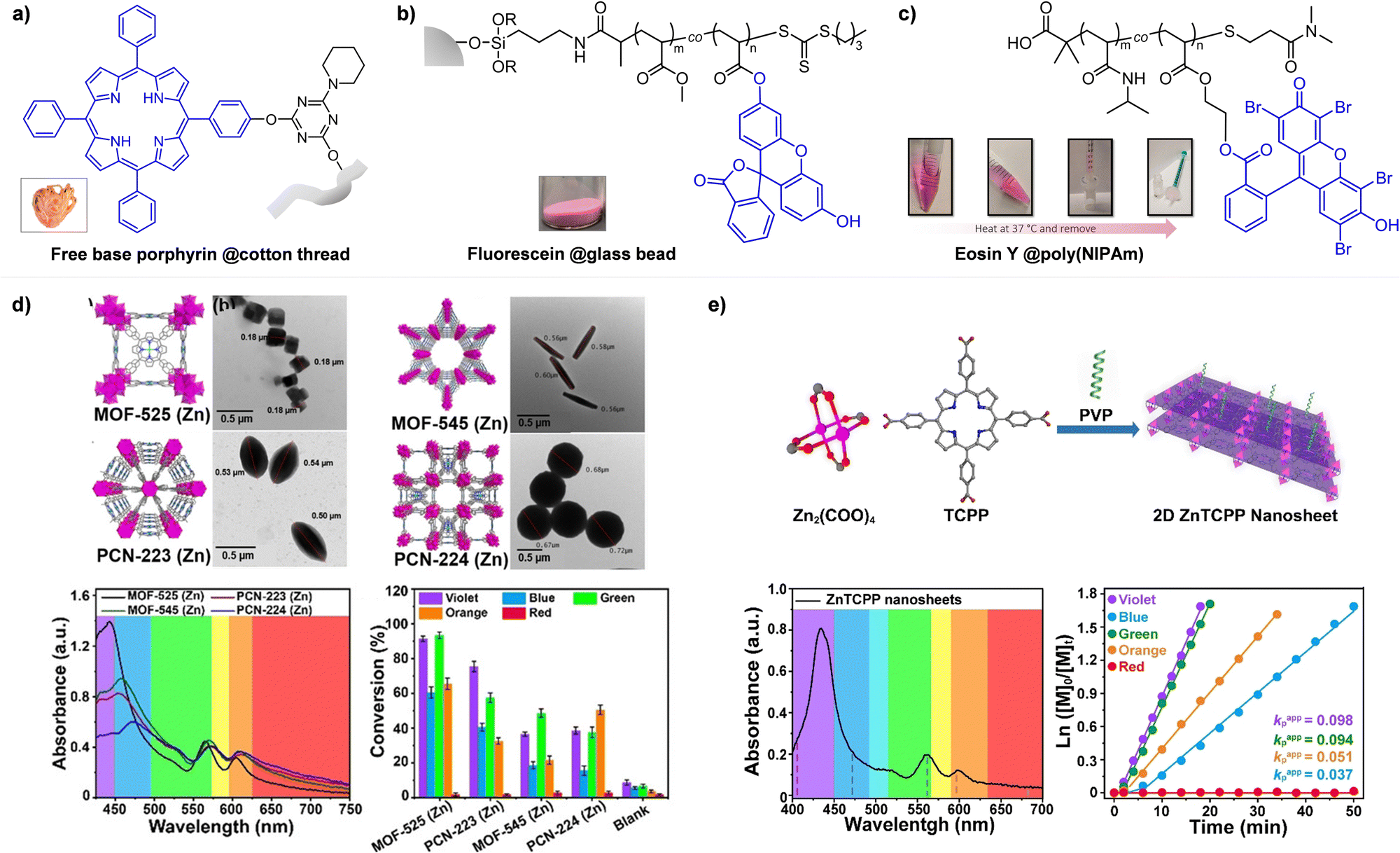 | ||
| Fig. 9 Chemical structures and photographs of (a) free base porphyrin, (b) fluorescein, and (c) EY immobilized on heterogeneous supports or polymers. (d) Crystal structures, TEM images (top) and UV/Vis absorption spectra of 3D zinc porphyrinic MOFs, and monomer conversions in the polymerization of MA from BTPA using various 3D zinc porphyrinic MOFs under irradiation (9 mW cm−2) (bottom). (e) Schematic representation and UV/Vis absorption spectrum of the 2D zinc porphyrinic MOF, and kinetic plots of ln([M]0/[M]t) vs. reaction time in the polymerization of MA from BTPA using the 2D zinc porphyrinic MOF under irradiation (10 mW cm−2). Adapted with permission from (a) ref. 240 (Copyright 2018 American Chemical Society), (b) ref. 245 (Copyright 2022 The Royal Society of Chemistry), (c) ref. 247 (Copyright 2021 American Chemical Society), (d) ref. 257 (Copyright 2020 John Wiley & Sons, Inc.), and (e) ref. 258 (Copyright 2021 John Wiley & Sons, Inc.). | ||
PCs can be covalently incorporated into a macromolecular network. Via the introduction of polymerizable vinyl moieties into PCs, followed by copolymerization with different monomers, macromolecular versions of PTH94 and EY247,248 were prepared. Copolymerization with NIPAm resulted in PC-embedded poly(NIPAm)s with thermoresponsive behavior, allowing the purification of macromolecular PCs by heating the reaction mixture at temperatures above the LCST (Fig. 9c).94,247 Amine-functionalized EY served as a monomer for ROP between diglycidyl and diamine monomers for the preparation of an interpenetrating polymer network.249 The prepared PC-based copolymers could be grafted onto the commercial ultrafiltration membranes.250,251 These PC-tethered membranes served as an integrated synthesis-separation system by catalyzing the polymerization and efficiently filtering unreacted monomers and other reagents after the polymerization. On the other hand, Sonogashira–Hagihara reactions between halogen substituents of xanthene dyes (i.e., EB and Rose Bengal) and dialkynes97 and Debus–Radziszewski reactions between amine-terminated porphyrins and imidazolium bromides252 generated PC-conjugated porous polymer frameworks. Cross-linking between hydroxy-terminated porphyrins and dichlorodimethylsilanes provided twisted 3D structures with open catalytic sites.253,254 The effective catalytic performance was ascribed to the large surface area of catalytically active sites, prevention of aggregation of porphyrins and concomitant reduction of quenching of excited-state porphyrins.
COFs or MOFs also served as heterogeneous platforms. 2D porphyrinic COFs were synthesized through polycondensation reactions between amine-terminated porphyrins and multivalent aldehydes.255,256 3D MOFs synthesized from zirconium-based clusters and porphyrins showed catalytic activities dependent on their crystal structures (Fig. 9d).257 During the synthesis of 3D MOFs from free base- or zinc-embedded porphyrins as organic ligands and zinc-based clusters as metal nodes, addition of polyvinylpyrrolidone (PVP) induced anisotropic growth of the frameworks and generation of ultrathin 2D nanosheets (Fig. 9e).258 Ultrathin 2D nanosheets demonstrated more rapid polymerization kinetics than 3D MOFs owing to the higher aspect ratio, larger surface area, and more accessible catalytically active sites.149,258,259
4.3. Applications
High end-group fidelity in PET-RAFT polymerization allows the one-pot synthesis of multiblock copolymers comprised of vinyl monomers with various side chains via chain extension with subsequently added monomers. Multiple extensions technically would lead to the preparation of (ultra)high-molecular-weight polymers (Section 4.3.5). Vinyl ethers and cyclic monomers (e.g., cyclic esters, thiiranes, and epoxides) which are ionically (not radically) polymerized can also be manually incorporated into the polymers at the designated positions with tunable lengths and numbers of blocks. This is owing to the orthogonality and compatibility of PET-RAFT polymerization with other polymerizations (Section 4.3.1.2). Moreover, by introducing specially designed monomers, CTA, or any additives, the control of architecture (Section 4.3.2), stereochemistry (Section 4.3.3), and dispersity (Section 4.3.4) has been achieved. It is noted that to fully demonstrate the versatility of PET-RAFT polymerization, strategies that include “click” reactions to synthesize structurally diverse polymers by linking multiple polymers possessing pre- or post-functionalized chain ends into a single chain are not discussed here. Becer and coworkers described various methodologies for multiblock copolymer synthesis.2604.3.1.1. Multiblock copolymer synthesis. Multiblock copolymers are synthesized by a number of successful chain extensions. After each step, end-group fidelity should remain high, and the reactivity of the PC should be retained to restart polymerization reaction upon addition of new monomers. In their first discovery of PET-RAFT polymerization using Ir(ppy)3 as a PC in 2014, Boyer and coworkers leveraged the effective retention of the DTB end group of the polymer to demonstrate whether chain extension could yield di-, tri-, or even deca-block copolymers.31 The decablock copolymer of MA ([poly(MA)]10, Mn,GPC = ∼82
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Đ = ∼1.40) was synthesized from BTPA by ten repeated quantitative polymerizations of MA (58–144 equivalents to the macroinitiator) dissolved in degassed DMSO as the viscosity of the reaction mixture increased with the number of polymerizations. Neither isolation of the macroinitiators nor an additional amount of PC was required. The tailing of GPC traces at the low molecular weight region was attributed to dead polymers and the gradual decrease in end-group fidelity (Fig. 10a). The final end-group fidelity after the tenth polymerization was calculated to be 68% from the deconvolution of the MWD. Similarly, the pentablock copolymer of five different monomers (methyl, ethyl, n-propyl, n-butyl, and n-pentyl acrylate) with the target DP of 50 for each monomer was synthesized from BSTP (Mn,GPC = 28390 g mol−1, Đ = 1.14, and end-group fidelity = ∼80%).261 By adding Ir(ppy)3 of 10 ppm relative to the monomer in the polymerization of the first monomer, >89% monomer conversion was achieved in 24 hours and 48 hours for the first three and rest two reactions, respectively. The authors decided to increase the reaction time instead of the catalytic loading to avoid the loss of the end group which appeared in the GPC traces. A functional triblock copolymer of the monomers possessing ketone and trimethylsilyl-protected alkynes could also be synthesized although the fast reactivity of the latter monomer was considered to increase the dispersity to 1.24.
000 g mol−1 and Đ = ∼1.40) was synthesized from BTPA by ten repeated quantitative polymerizations of MA (58–144 equivalents to the macroinitiator) dissolved in degassed DMSO as the viscosity of the reaction mixture increased with the number of polymerizations. Neither isolation of the macroinitiators nor an additional amount of PC was required. The tailing of GPC traces at the low molecular weight region was attributed to dead polymers and the gradual decrease in end-group fidelity (Fig. 10a). The final end-group fidelity after the tenth polymerization was calculated to be 68% from the deconvolution of the MWD. Similarly, the pentablock copolymer of five different monomers (methyl, ethyl, n-propyl, n-butyl, and n-pentyl acrylate) with the target DP of 50 for each monomer was synthesized from BSTP (Mn,GPC = 28390 g mol−1, Đ = 1.14, and end-group fidelity = ∼80%).261 By adding Ir(ppy)3 of 10 ppm relative to the monomer in the polymerization of the first monomer, >89% monomer conversion was achieved in 24 hours and 48 hours for the first three and rest two reactions, respectively. The authors decided to increase the reaction time instead of the catalytic loading to avoid the loss of the end group which appeared in the GPC traces. A functional triblock copolymer of the monomers possessing ketone and trimethylsilyl-protected alkynes could also be synthesized although the fast reactivity of the latter monomer was considered to increase the dispersity to 1.24.
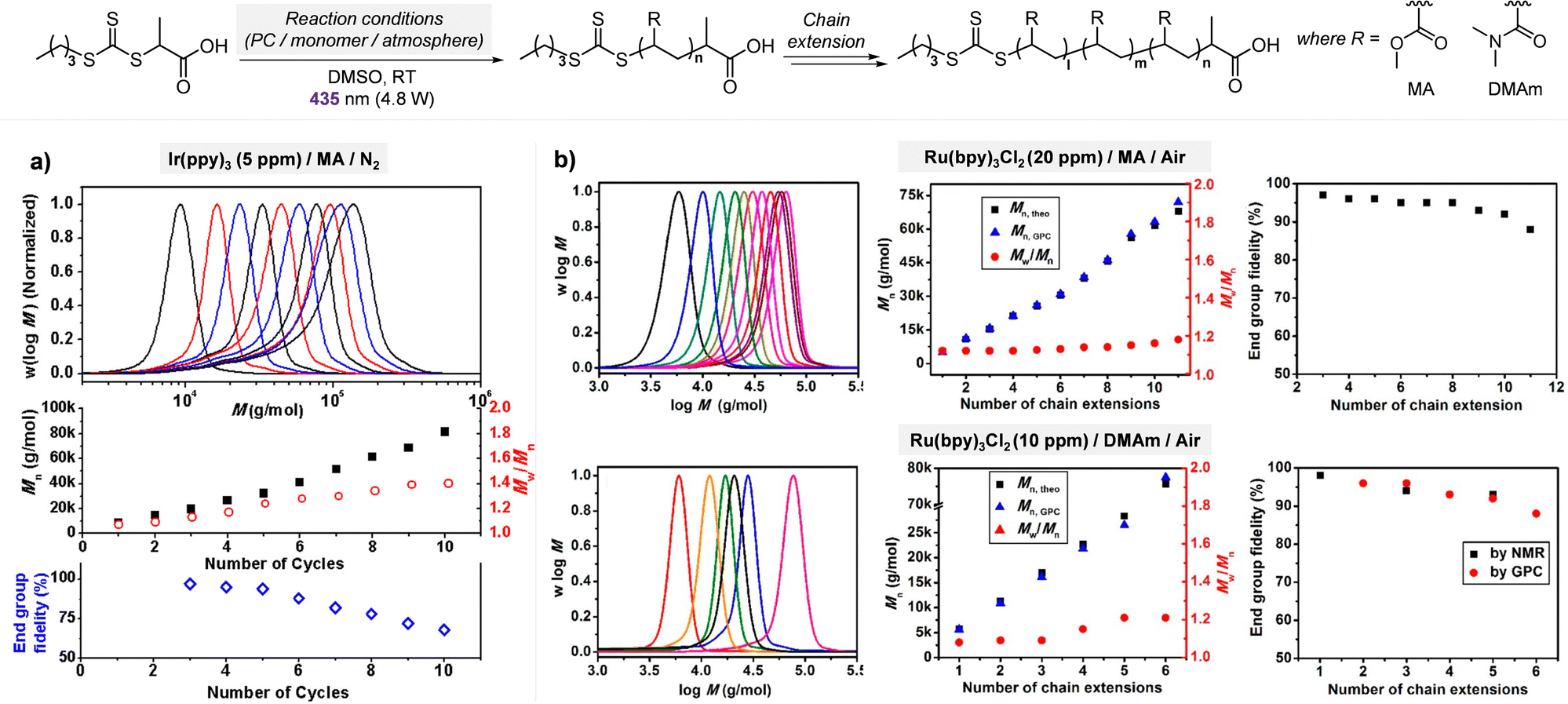 | ||
| Fig. 10 Multiblock polymers of MA and DMAm obtained by PET-RAFT polymerization. Evolution of MWDs, Mn, Mw/Mn, and end-group fidelity vs. number of cycles of chain extensions for (a) Ir(ppy)3- and (b) Ru(bpy)3Cl2-catalyzed reactions. Adapted with permission from (a) ref. 31 (Copyright 2014 American Chemical Society) and (b) ref. 129 (Copyright 2014 American Chemical Society). | ||
Ru(bpy)3Cl2 was the first reported PC that enabled the synthesis of [poly(MA)70]10-b-poly(MA)90 and [poly(DMAm)60]5-b-poly(DMAm)300 in DMSO in the presence of oxygen when used at 20 ppm and 10 ppm, respectively, under blue light irradiation (435 nm, 4.8 W) (Fig. 10b).129 Herein, neither the reaction vessel nor the monomer solution was degassed, but polymerization was controlled such that the end-group fidelity determined by GPC remained higher than 86% and dispersity did not exceed 1.2. Owing to its aqueous solubility, Ru(bpy)3Cl2 was also employed for the oxygen-tolerant and rapid synthesis of a water-soluble triblock copolymer.150 Ru(bpy)3Cl2 (25 ppm relative to the monomer) with ascorbate (two equivalents relative to BTPA) was employed to prepare poly(N-acryloylmorpholine (NAM))40-b-poly(N,N-diethylacrylamide (DEAm))40-b-poly(DMAm)40 in water within an hour under blue light irradiation (470 nm, 26 mW cm−2). Employment of a continuous flow reactor further enhanced the polymerization kinetics. EY disodium salt in combination with TEOA (0.01 and one equivalent relative to the CTA, respectively) allowed the rapid oxygen-tolerant synthesis of the triblock copolymer poly(DMAm)100-b-poly(DMAm)50-b-poly(DMAm)50 (Đ = 1.39) and [poly(DMAm)10]3 (Đ = 1.24) within 15 minutes without prior deoxygenation.136 Meanwhile, ZnTPP that could achieve oxygen tolerance in DMSO without any additives solely pushed the limit to HTP polymerization in a 96-well plate. At diluted monomer concentration (1.0 M), 400 ppm of ZnTPP under yellow light irradiation (560 nm, 9.7 mW cm−2) provided [poly(NAM)25]6 (Mn,GPC = 20900 g mol−1 and Đ = 1.19) with low molecular weight tailing.262 This simple and efficient ZnTPP-catalyzed HTP polymerization has enabled the simple and efficient synthesis of high-order block copolymers to construct a library of polymers with varying sequences, compositions, molar ratios between different monomer functionalities (e.g., cationic, hydrophobic, and hydrophilic), or structures, which revealed the relationship between the structures and properties of polymers in antimicrobial activities263,264 and polymer–protein binding interactions.265,266
Liao, Lalevée, and coworkers developed a novel organic PC oxygen-doped anthanthrene (ODA) for metal-free synthesis of [poly(n-BA)]10 (Mn,GPC = ∼143.2 kg mol−1 and Đ = ∼1.31) (Fig. 11).267 The polymerization of the first block with ODA (5 ppm), BTPA as the CTA, and n-BA as the monomer (100 equivalents relative to BTPA) under blue light irradiation (455 nm, 3 mW cm−2) was followed by multiple chain extensions with degassed n-BA (100 equivalents relative to the macroinitiator) for four hours per step.
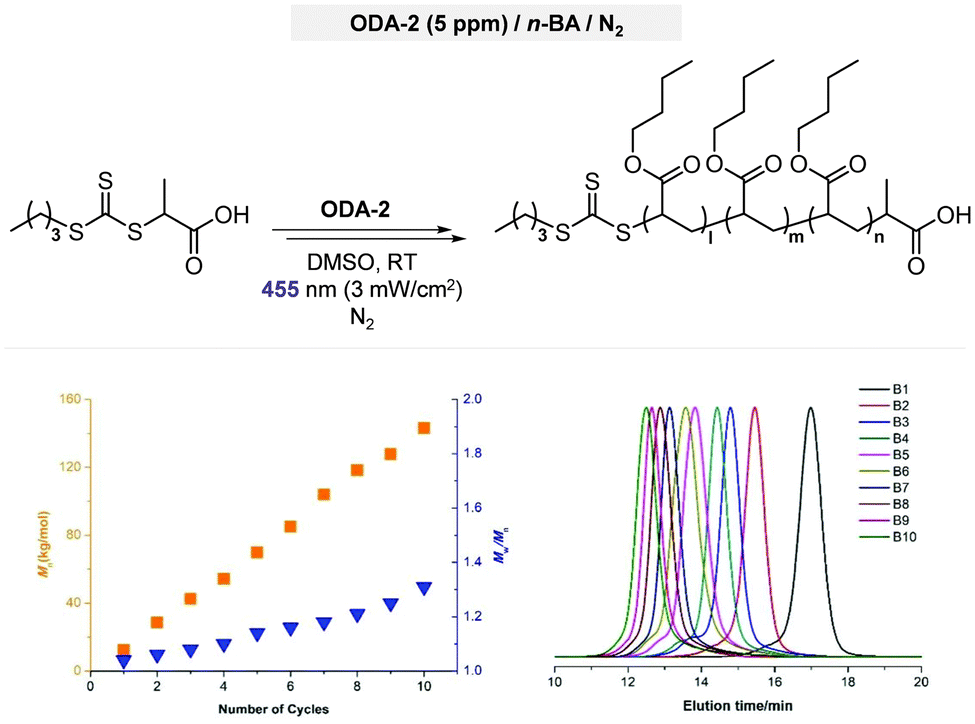 | ||
| Fig. 11 Multiblock polymers of n-BA obtained by ODA-2-catalyzed PET-RAFT polymerization. Evolution of Mn, Mw/Mn, and MWDs vs. number of cycles of chain extensions. Adapted with permission from ref. 267 (Copyright 2022 The Royal Society of Chemistry). | ||
4.3.1.2. Incorporation of various monomers into a single polymer chain. PET-RAFT polymerization with radical intermediates is orthogonal to other polymerizations with ionic intermediates. Moreover, the use of light as an energy source allows selective activation of PET-RAFT polymerization and consumption of radically polymerizable vinyl monomers (e.g., (meth)acrylates and (meth)acrylamides) from the reaction mixture in which ionically polymerizable monomers (e.g., vinyl ether and cyclic monomers) also exist. The remaining monomers can be independently polymerized in the presence of corresponding initiators. Therefore, using appropriately designed initiators, various monomers can be incorporated into a single polymer chain via dual polymerization processes.
First of all, the same dormant TTC can be activated to undergo either PET-RAFT or (photomediated) cationic RAFT polymerization. Fors and coworkers, for the first time, reported wavelength-dependent activation of Ir(ppy)3 at 450 nm for PET-RAFT polymerization of MA and of pyrylium tetrafluoroborate salt at 520 nm for photomediated cationic RAFT polymerization of iBVE (Table 3, entry 1).124 However, as the pyrylium salt also exhibited absorption at 450 nm, the catalyst loading had to be regulated to selectively activate PET-RAFT polymerization and avoid the synthesis of tapered block copolymers. On the other hand, cationic RAFT polymerization could be activated by (electro)chemical means which are completely orthogonal to light stimuli. The CTA was oxidized by adding ferrocenium tetrafluoroborate salt and deactivated by adding one equivalent of DTC anion into the reaction mixture to cap the propagating cations and reduce the ferrocenium salt (Table 3, entry 2).119 In the electrochemical activation, the CTA was oxidized by anodic oxidation of ferrocene (as a redox mediator) to ferrocenium and deactivated by applying cathodic current, and the concentration of ferrocene should be kept low to prevent the quenching of the triplet excited-state of Ir(ppy)3 and block the PET-RAFT polymerization (Table 3, entry 3).116 Instead, Alaniz, Sepunaru, and coworkers utilized PTH as the redox mediator (Table 3, entry 4).117 As PTH catalyzed the PET-RAFT polymerization of MA (380 nm, 3 mW cm−2), switching between two polymerizations by toggling two corresponding external stimuli successfully generated block copolymers of vinyl ether and acrylate up to tetrablocks. Meanwhile, Satoh and coworkers reported that the change of irradiation wavelength could affect the ratio of the two monomers separately incorporated by two orthogonal processes and the number of interconversions (Table 3, entry 5).121 Herein, cationic RAFT polymerization was kept constantly “on” by the presence of Lewis acid B(C6F5)3 and only ZnTPP-catalyzed PET-RAFT polymerization could be reversibly activated. Under red light irradiation (630 nm, 45 mW), both MA and iBVE participated in polymerization. The diad sequence of iBVE in 13C NMR spectra, which was not observed in the PET-RAFT polymerization of MA and iBVE, confirmed that the Lewis acid-catalyzed cationic RAFT and ZnTPP-catalyzed PET-RAFT polymerization from the common CTA were interconvertible. Under green light irradiation (525 nm, 20 mW), the contribution of PET-RAFT polymerization was higher and the consumption of iBVE was lower than that under red light irradiation owing to more active generation of the TTC anion.
| Entry | PET-RAFT polymerization | Initiator | Combined polymerization | Ref. | |||
|---|---|---|---|---|---|---|---|
| PC (stimuli) | Monomer | Mechanism | Reagents (stimuli) | Monomer | |||
| 1 | Ir(ppy)3 (on/off: light, 450 nm) | MA |
|
Cationic RAFT polymerization | Pyrylium salt (on/off: light, 520 nm) | iBVE | 124 |
| Cationic RAFT polymerization | |||||||
| 2 | Ir(ppy)3 (on/off: light, 450 nm) | MA |
|
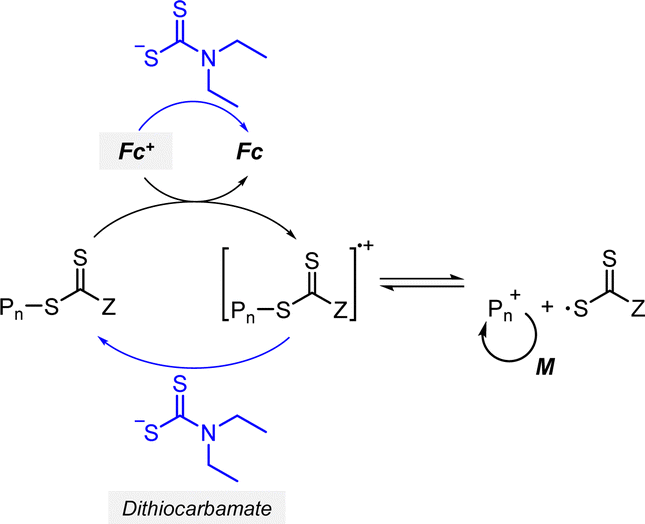
|
Ferrocenium tetrafluoroborate salt (FcBF4)/dithiocarbamate (on/off: chemical redox reaction) | iBVE | 119 |
| Cationic RAFT polymerization | |||||||
| 3 | Ir(ppy)3 (on/off: light, 455 nm) | MA |
|
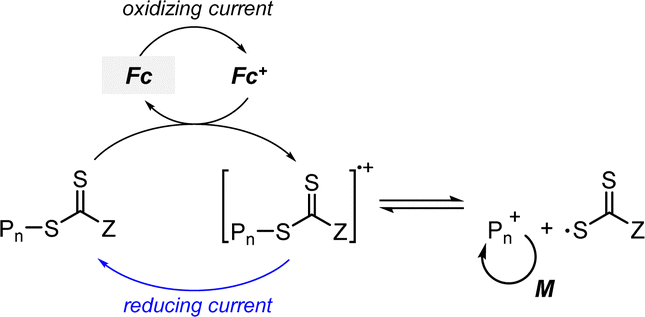
|
Ferrocene (Fc) (on/off: electrical redox reaction) | iBVE | 116 |
| Cationic RAFT polymerization | |||||||
| 4 | 10-phenyl phenothiazine (PTH) (on/off: light, 380 nm) | MA |
|

|
PTH (on/off: electrical redox reaction) | iBVE | 117 |
| Cationic RAFT polymerization | |||||||
| 5 | ZnTPP (on/off: light, 630 nm) | MA |
|
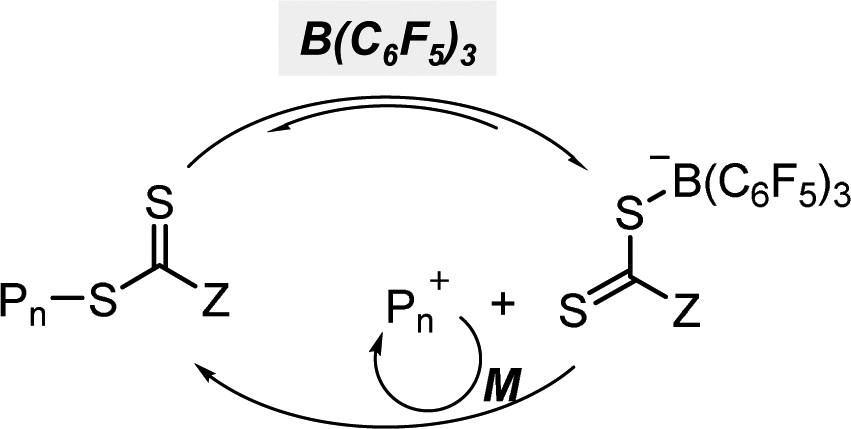
|
B(C6F5)3 (only on) | iBVE | 121 |
| 6 | Ir(ppy)3 (on/off: light, 460 nm) | MA |
|
Cationic ring-opening polymerization | Diphenyl phosphate (on) | ε-Caprolactone | 123 |
| Diphenyliodinium triflate (DPITF) + Ir(ppy)3 (on/off: light, blue) | |||||||
| 7 | Ir(ppy)3 (on/off: light, blue) | MA |
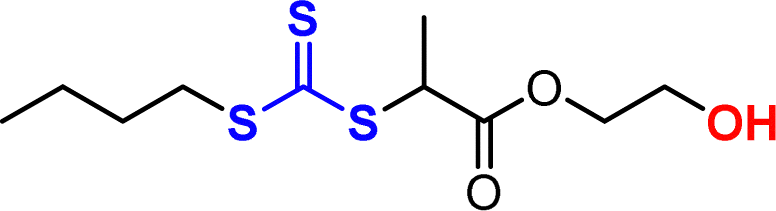
|
Cationic ring-opening polymerization |
|
δ-Valerolactone | 120 |
| Merocyanine ↔ spiropyran (on/off: light, 460 nm) | |||||||
| 8 | ZnTPP (on/off: light, 635 nm) | MA |
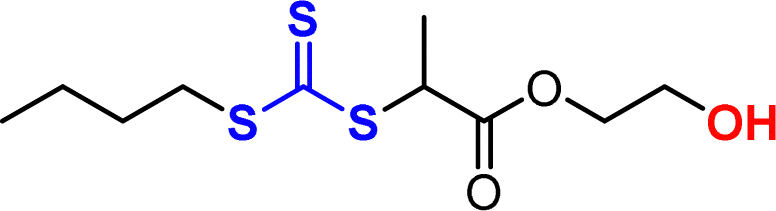
|
Cationic ring-opening polymerization |
|
δ-Valerolactone | 118 |
| Anionic ring-opening polymerization | |||||||
| 9 | Ir(ppy)3 (on/off: light, blue) | DMAm, NIPAm |
|
|
Tetraphenylphosphonium chloride (TPPCl) (on/off: heat, >50 °C) | Thiiranes | 115 |
Other monomers including cyclic monomers (e.g., cyclic esters and epoxides) can be incorporated into a single polymer chain using a bifunctional initiator prepared by the functionalization of the CTA with a hydroxyl group, for example, via the esterification of the carboxylic acid moiety in the R group. Boyer and coworkers firstly reported the one-pot synthesis of block copolymers by a combination of PET-RAFT polymerization and another polymerization method (Table 3, entry 6).123 The block copolymer of ε-caprolactone and MA was synthesized using diphenyl phosphate and Ir(ppy)3 and the ratio of the length of each block was simply controlled via the temporal control of PET-RAFT polymerization (460 nm, 0.7 mW cm−2). You and coworkers employed diphenyliodonium triflate that only generated photoacid in the presence of Ir(ppy)3 under blue light irradiation (10 W) and then catalyzed the cationic ROP of δ-valerolactone (Table 3, entry 7).120 Ir(ppy)3 could still catalyze PET-RAFT polymerization after the addition of MA to the reaction mixture. In this case, both processes required irradiation such that only diblock copolymers with defined orders could be obtained. Boyer and coworkers reported the generation of photoacid via intramolecular cyclization of merocyanine to spiropyran followed by dissociation of the proton under blue light irradiation (460 nm, 0.7 m cm−2) (Table 3, entry 8).118 ZnTPP that absorbs at 635 nm afforded orthogonality in wavelength and thus selective activation of two polymerizations. However, upon changing the light source from blue to red, the ROP of δ-valerolactone was not completely inactivated due to slow inactivation of the photoacid molecule. Effective switching between anionic ROP and PET-RAFT polymerization was realized using a combination of heat and light as two external stimuli (Table 3, entry 9).115 Herein, the anionic ROP of the cyclic thiirane monomer using quaternary onium salts (tetraphenylphosphonium chloride) and TTC only proceeded at >50 °C due to the energy barrier to open the ring such that PET-RAFT polymerization of acrylamides under blue light irradiation (5 W) was orthogonally conducted at room temperature. As the reaction temperature and irradiation wavelength were easily switched in an orthogonal manner, interconversion of two polymerizations five times from symmetric TTC provided an undecablock copolymer (Fig. 12). Programmed heating and/or irradiation, such as intermittent irradiation during heating, enabled precise insertion of the DMA block at any position within the homopolymer of thiiranes, which would otherwise require careful consideration of the reactivities of monomers.
 | ||
| Fig. 12 Synthesis of an undecablock copolymer by multiple cycles of heating and irradiation. Under heating, 2-(phenoxymethyl) thiirane (POMT) was selectively consumed by TTC-mediated anionic ROP whereas under irradiation DMAm was consumed by PET-RAFT polymerization. Adapted with permission from ref. 115 (open access). | ||
Lastly, in addition to ionic polymerization, radical-mediated O-ATRP was selectively activated with PET-RAFT polymerization. Boyer, Miyake, and coworkers reported that N,N-diaryl dihydrophenazine could catalyze not only O-ATRP but also PET-RAFT polymerization, based on the similarity between two reaction mechanisms and hence the required catalytic properties.125 Interestingly, PET-RAFT polymerization required less amount of PC as compared to O-ATRP of MMA. Therefore, using alkyl bromide-functionalized CTA and by increasing the loading of PC from 50 to 500 ppm, diblock copolymers could be obtained via PET-RAFT polymerization of MA followed by O-ATRP of MMA.
4.3.1.3. Single unit monomer insertion (SUMI). SUMI refers to selective insertion of a single unit monomer at a time.268 As the stability of the C–S bond changes after the insertion of a certain monomer, carefully designed reaction conditions (the combination of CTA, monomer, and PC) can only activate the initial CTA to which the monomer is added.109 The monoadduct can be isolated and its synthesis can be confirmed by NMR, mass spectrometry, and gel permeation chromatography. Sequential SUMI of a series of monomers leads to discrete oligomers with known monomer sequences, which would pave the way for synthetic polymer chemistry to reach DNA or proteins possessing a high precision in the monomer sequence and the resulting functionalities. For example, the well-defined oligomers can be the building blocks for higher-order polymers or can provide a hint at the stereochemical effect on the activation of the diastereomeric oligomeric CTA, which has been challenged by the difficulties in the preparation of appropriate materials.269–271 Visible-light-driven RAFT polymerization under mild reaction conditions in the absence of exogenous radical initiators is advantageous to prepare polymers with high end-group fidelity and prevent the formation of initiator-derived byproducts, when compared to conventional thermally initiated polymerizations.
For successful SUMI (i.e., to limit multiple additions of monomers), dissociation of the initial CTA should be preferred compared to that of the monoadduct via the increase in C–S bond dissociation energy along with the insertion of a monomer with a less stable R group (Scheme 20a). If the monoadduct decomposes, deactivation of the propagating radical instead of propagation by either recombination or chain transfer to the initial CTA is required (Scheme 20b). The use of non-homopolymerizable monomers, such as styrene, N-substituted maleimide, VAc, limonene, and indene, with low kp is also helpful. The blue shift of the UV absorption band of the monoadduct to lower wavelengths as compared to that of the initial CTA enabled selective and efficient activation of the initial CTA by light.272 However, in cases where the selected monomers were purposely inserted in desired sequences via success SUMI, PCs such as PheoA,106 Ir(ppy)3,89 and/or ZnTPP89,269,270,273,274 were necessary to achieve reasonably high reaction efficiencies under mild reaction conditions. Using ZnTPPS4−, a water-soluble analogue of ZnTPP, Moad and coworkers observed the significant enhancement of reaction kinetics in SUMI of DMAm as a model monomer in water.199
 | ||
| Scheme 20 (a) Mechanism and (b) requirements for SUMI, where kd (k−d) is the decomposition (recombination) rate constant of the initial CTA under irradiation, kadd is the addition rate constant of a monomer to the R group radical of the initial CTA, kp is the propagation rate constant of the second monomer to the R group radical of the SUMI monoadduct, ktr is the chain transfer rate constant of the R group radical of the initial CTA to the SUMI monoadduct, k−tr is the chain transfer rate constant of the R group radical of the SUMI monoadduct to the initial CTA, and kd′ (k−d′) is the decomposition (recombination) rate constant of the SUMI monoadduct under irradiation. | ||
For example, in the first report of the synthesis of trimers by Boyer, Moad, Hawker, Xu, and coworkers, the insertion of the first monomer styrene into CDTPA under green light irradiation (530 nm, 0.6 m cm−2) was followed by the Ir(ppy)3-catalyzed PET-RAFT polymerization of the second monomer N-substituted maleimide under blue light irradiation (460 nm, 0.8 mW cm−2) (Scheme 21a).89 The combination of styrene and maleimides is well-known for alternative copolymerization due to the formation of donor–acceptor pairs. The last third monomer VAc or limonene was inserted using ZnTPP under red light irradiation (635 nm, 0.4 mW cm−2) or Ir(ppy)3, respectively, because Ir(ppy)3 resulted in multiple addition of VAc. Further functionalization of the trimers provided a new methacrylate monomer possessing a precisely defined side chain of hexamer as a potential building block for periodic polymers. Later, Boyer and coworkers conducted sequential SUMI of up to five monomers in the gram scale while addressing scalability problems by the use of flow reactors for reaction and automated flash chromatography for purification.273 ZnTPP in DMSO imparted oxygen tolerance and non-homopolymerizable electron-donating indene and electron-accepting N-substituted maleimide derivatives were alternatively polymerized (Scheme 21b).
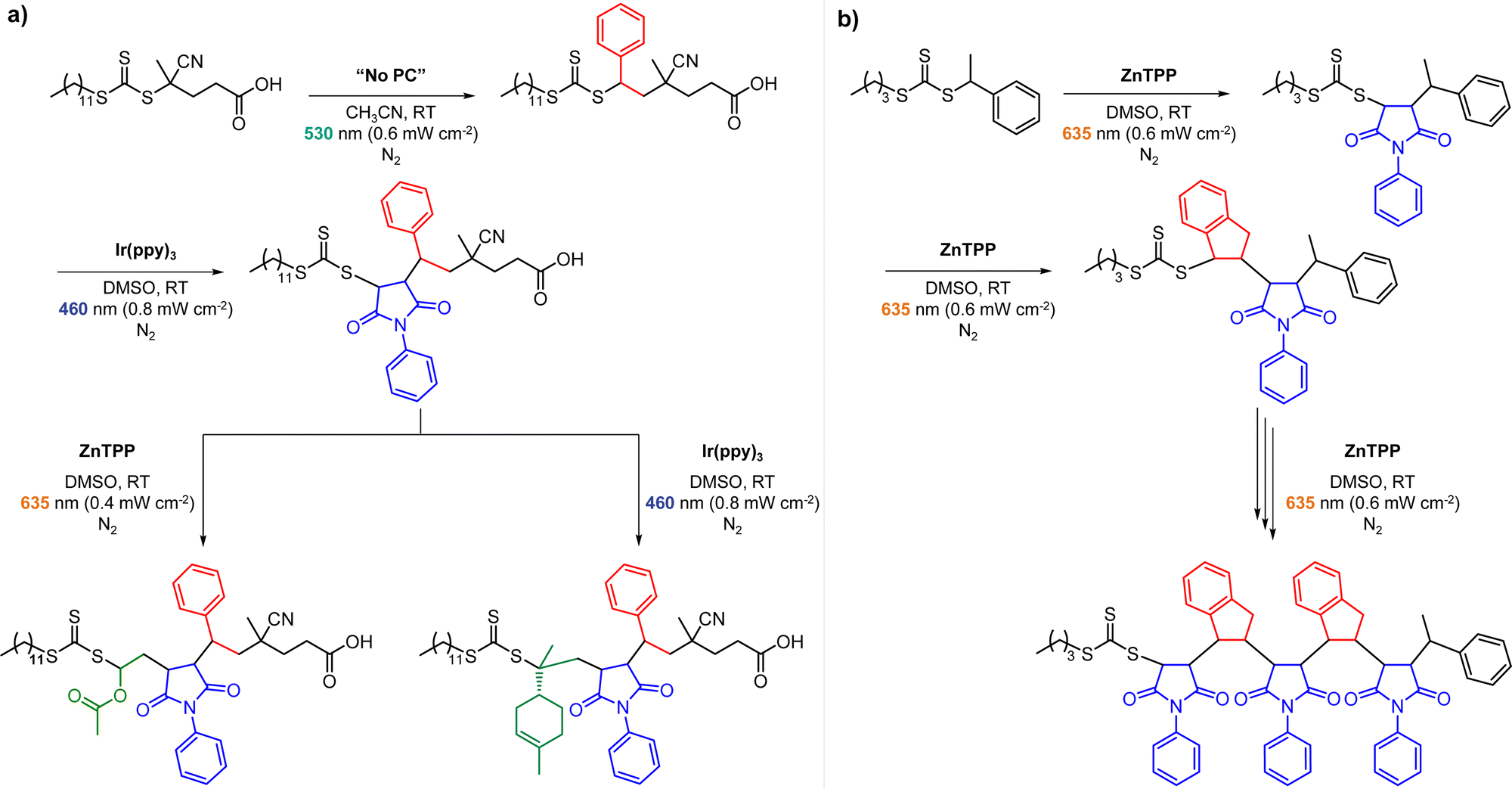
Meanwhile, discrete oligomeric CTAs have aided the investigation of the stereochemical effect on photoactivation, copolymerization kinetics of a certain monomer in the RAFT process, and the influence of precisely designed sequences on polymeric properties. Into the pool of pairs of electron-donating (e.g., indenes and (β-methyl)styrene derivatives) and electron-accepting monomers (e.g., N-substituted maleimides), various functionalities can be introduced as their substituents. Initially, SUMI of various α,β-disubstituted vinyl monomers led to the synthesis of a stereospecific discrete oligomeric CTA.269,270 Insertions of the examined cyclic N-substituted maleimides in trans-configurations were preferred owing to steric hindrances and restricted rotations of cyclic monomers, as combinedly evidenced by structural analysis by single-crystal X-ray diffraction and QC calculations.269
Xu and coworkers reported that for the successful SUMI of β-methylstyrene derivatives, careful selection of the CTA was required.275 When β-methylstyrene derivatives were inserted next to maleimide into TTCs with different R groups using ZnTPP, the dimer was formed in 50% yield despite the complete conversion of the starting monoadduct of maleimide due to the degradation of the desired dimer after a certain reaction time. The proposed degradation mechanism was based on the β-methylstyrenenic carbon radical, which when fragmented abstracts hydrogen from an excess of β-methylstyrene derivatives and can no longer participate in the RAFT process (i.e., chain transfer or recombination). Therefore, the authors postulated that to prevent the degradation of the product, (i) chain transfer of the fragmented β-methylstyrenenic carbon radical should occur faster than the hydrogen abstraction and (ii) the starting monoadduct should possess a better leaving R group than β-methylstyrenes. As expected, quantitative formation of the dimer was achieved when the first monomer was changed from maleimide to acrylonitrile (AN).
Xiao, Lai, and coworkers synthesized trimers consisting of two N-substituted maleimides and 5,6-benzo-2-mehtylene-1,3-dioxepane (BMDO) in between from n-butyl benzyl TTC (BBTC).274 BMDO is a cyclic ketene acetal monomer whose radical ROP affords a polymer with a degradable ester bond in the backbone. Contrary to the successful insertion of N-substituted maleimide into BBTC using ZnTPP in DMSO under red light irradiation (620 nm), the use of thermal initiator lauroyl peroxide in toluene was required for the insertion of BMDO due to hydrolysis of BMDO in hygroscopic DMSO and poor solubility of ZnTPP in toluene (Scheme 22a). Two model trimers, BBTC-N-phenyl maleimide (PMI)-BMDO-N-methyl maleimide (MMI) and BBTC-PMI-BMDO-PMI, were degradable under basic conditions and cytocompatible with the human normal cell line (HUVEC) at up to 600 μmol L−1 concentration (Scheme 22b), but only BBTC-PMI-BMDO-MMI was cytotoxic to the human cancer cell line (MCF-7) (Scheme 22c) implying the potential of sequence-defined oligomers for biological applications.
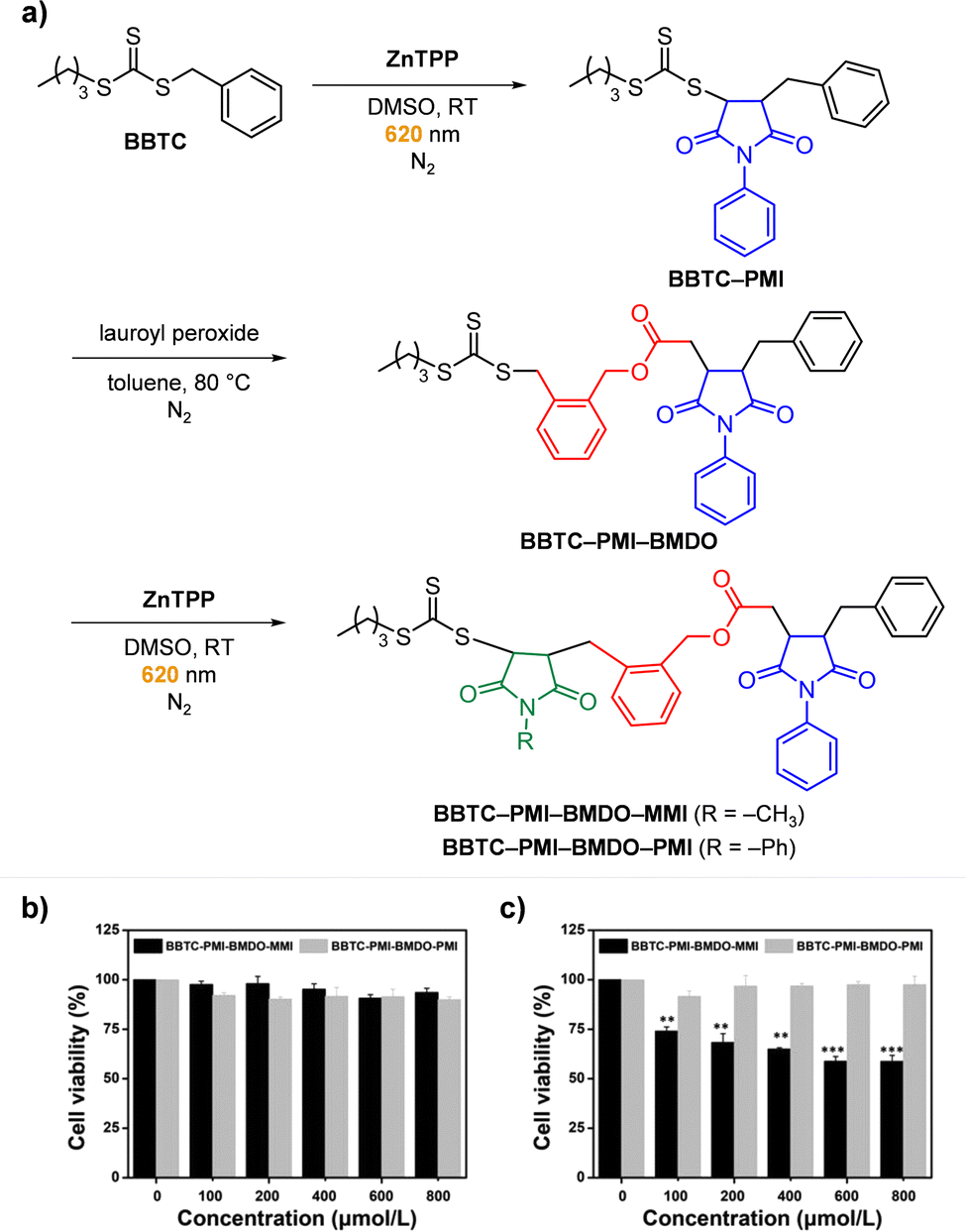 | ||
| Scheme 22 (a) Synthetic scheme for the trimers BBTC-PMI-BMDO-MMI and BBTC-PMI-BMDO-PMI. Cell viability of the two trimers against (b) human normal cell line (HUVEC) and (c) human cancer cell line (MCF-7) after 24 hours of incubation. Adapted with permission from (b) and (c) ref. 274 (Copyright 2021 American Chemical Society). | ||
4.3.2.1. Design of the CTA. Designing an appropriate initiator is one of the simplest ways to obtain polymers with complex architectures (Scheme 23). For example, star or multi-arm polymers have been synthesized from multi-arm CTAs.265,276–279 Multi-arm TTCs were prepared from thiols, carbon disulfide, and scaffold molecules with multiple bromines which underwent substitution reactions, whereas multi-arm xanthates were prepared using xanthogenate instead of thiol and carbon disulfide. Multi-arm polymers have been examined for various applications. Chapman, Boyer, and coworkers prepared a series of star polymers from two, three, or four-arm CTAs to investigate the relationship between the structures and binding affinities of polymers to protein.265 Gormley, Chapman, and coworkers employed simple and oxygen-tolerant HTP polymerization to build a library of multivalent polymers possessing a strained alkyne at each arm end, which may be useful for conjugating biomolecules via strain-promoted azide-alkyne cycloaddition (SPAAC).276 Boyer, Corrigan, Zhang, and coworkers controlled the mechanical properties of 3D printed objects in a broad range by optimizing the compositions (i.e., the ratio and concentrations of multi-arm CTAs possessing different number of arms) of 3D printable resins.279 Finally, Chapman and coworkers analyzed the effects of the type of monomers (hydrophobic, hydrophilic, and charged) and their order of arrangements on the sizes and assemblies of multiblock star polymers in solution.277
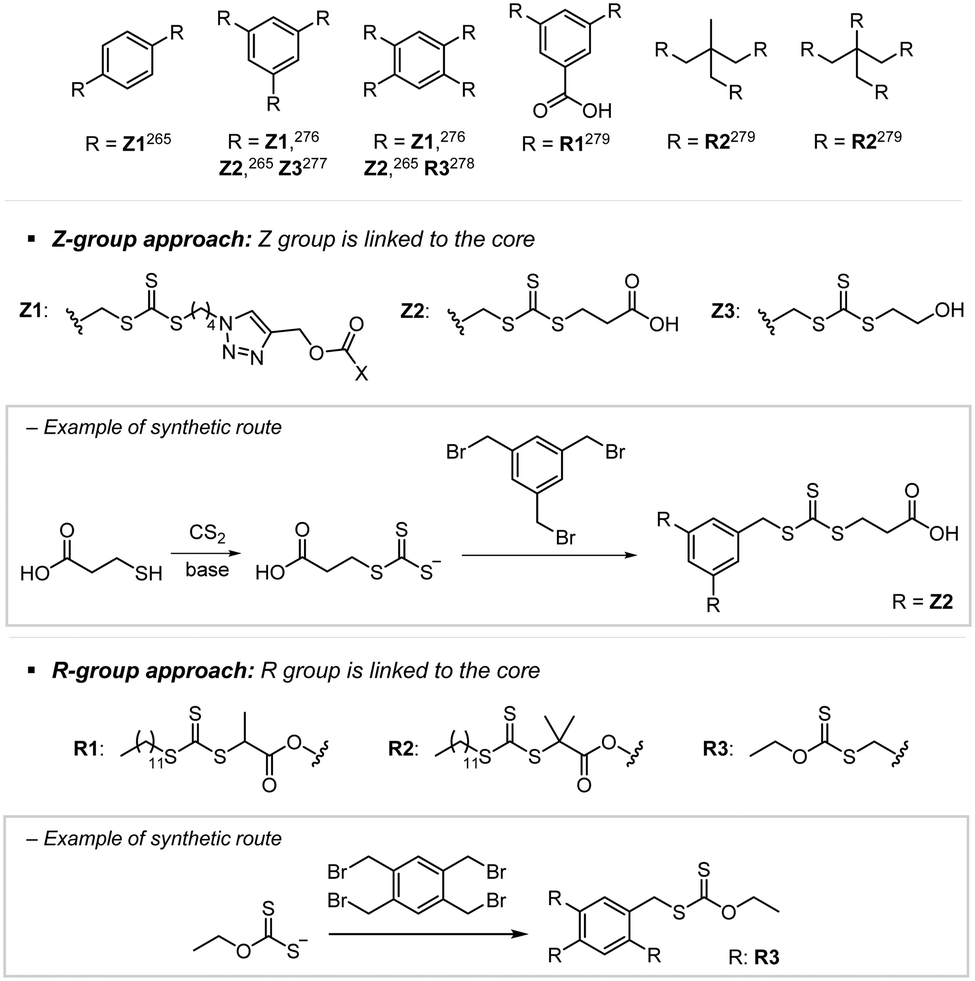 | ||
| Scheme 23 Examples of multi-arm CTAs and the corresponding synthetic routes. | ||
It is noted that the core of star polymers can be linked to either the R or Z group of the CTA (Scheme 23). In the Z-group approach, the polymers grow from the fragmented R group liberated from the core, whereas in the R-group approach, the polymers grow from the core. For controlled polymerization, the Z-group approach is suitable to attach short arms at low grafting density because steric hindrance limits reattachment and chain transfer of propagating chains to the core Z group. The R-group approach suffers from biomolecular radical termination between either two star polymers (star–star coupling) or a star polymer and an exogenous macroradical rather than from the steric hindrance. Star–star coupling results in higher molecular weight byproducts, and reattachment of the exogenous macroradical instead of the Z group leads to the loss of CTA functionality and livingness, preventing further polymerization from dead chains to broaden the dispersity. Therefore, the synthesis of well-defined star polymers with (ultra)high molecular weights or multiple blocks with preserved end-group fidelity has particularly been challenging. Photocontrolled RAFT polymerization in the absence of exogenous initiating radicals thus emerged as a successful solution to the abovementioned problem as observed for photoiniferter polymerization by Qiao and coworkers278 and PET-RAFT polymerization by Chapman and coworkers (Fig. 13).277
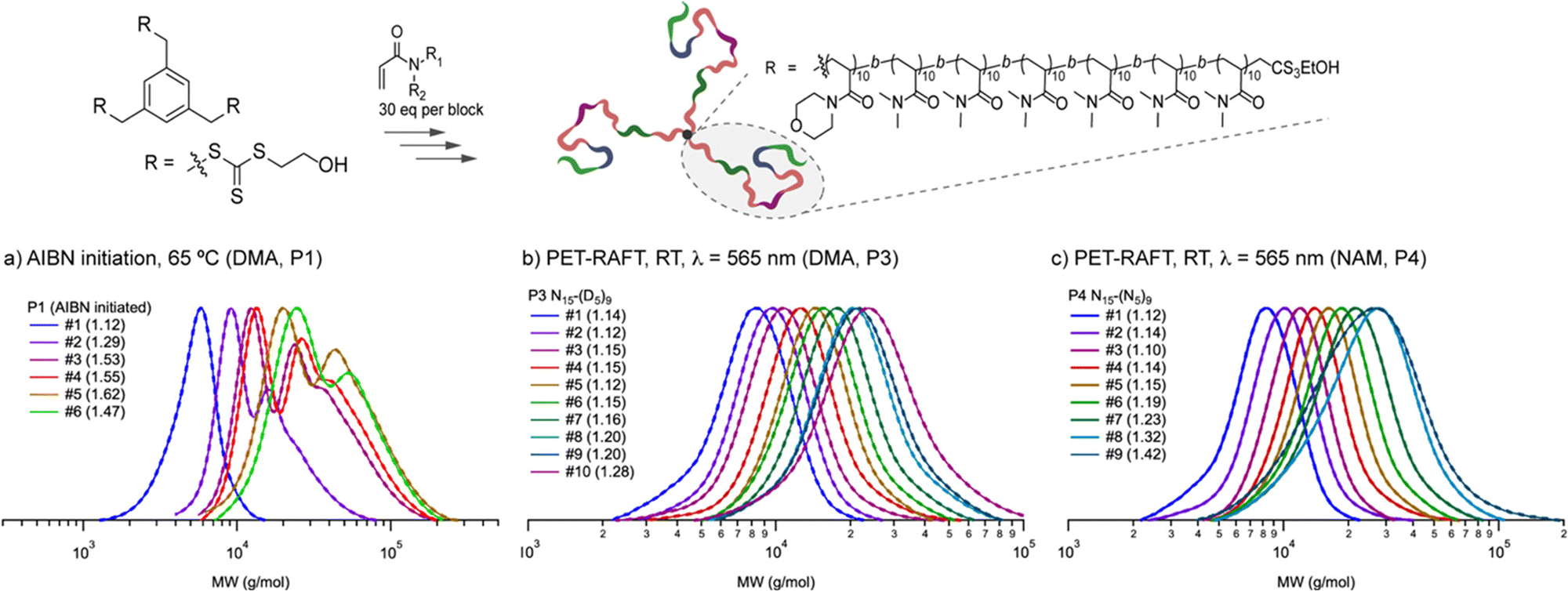 | ||
| Fig. 13 Synthesis of multiblock star polymers by thermal and ZnhTPP-catalyzed PET-RAFT polymerization. MWDs after each chain extension are shown below. Adapted with permission from ref. 277 (Copyright 2022 American Chemical Society). | ||
4.3.2.2. Design of the monomer. Other unique advantages of photocontrolled RAFT polymerization include spatiotemporal control, selectivity (which needs to be satisfied for the productive activation of the RAFT process), and orthogonality (to other polymerization methods). Carefully designed reaction conditions or systems along with properly functionalized monomers have enabled the preparation of complex polymeric architectures such as graft, comb-like, bottlebrush, and (hyper)branched polymers. For example, so-called “inimer” contains initiating moieties for two polymerizations, which can be activated selectively and in series (Scheme 24). Initially, as a mono“mer” for RAFT polymerization, the vinyl moiety comprises a polymer backbone while the other initiating moiety remains unreacted in the side chain. By changing the reaction conditions (e.g., irradiation wavelength) or adding different types of monomers, the remaining “ini”tiator is then activated for another polymerization to transform the linear polymer into polymers with grafting chains or other high complexities.
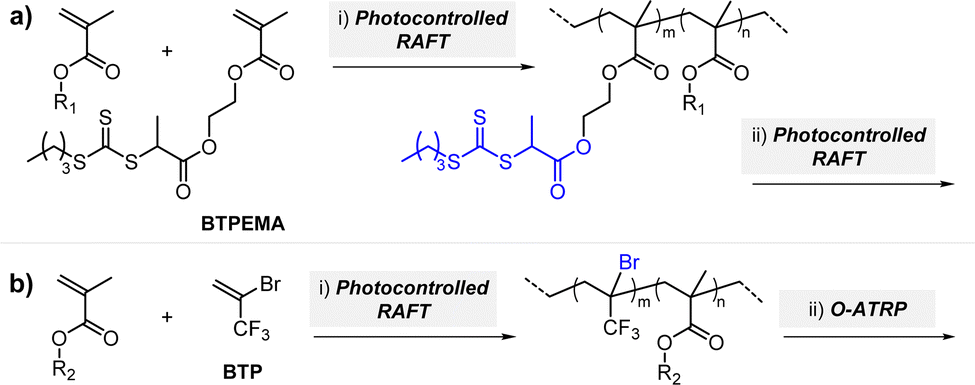 | ||
| Scheme 24 Example of functionalized monomers and their stepwise activation to control polymer architectures.106,211 | ||
Boyer, Xu, and coworkers synthesized graft copolymers via two selectively activated PET-RAFT polymerizations (Scheme 8 and 24a).106 The reaction mixture contained two pairs of PC and CTA. The methacrylic polymer backbone was synthesized by the PheoA-catalyzed PET-RAFT polymerization of MMA and another methacrylate embedded with a TTC (BTPEMA) from CPADB under red light irradiation (690 nm, 2 mW cm−2). Then, MA was added to the reaction mixture for the ZnTPP-catalyzed PET-RAFT polymerization from the TTCs under green light irradiation (530 nm, 0.6 mW cm−2) to graft acrylic chains along the backbone. Herein, selective activation was possible as (i) the irradiation wavelengths were carefully set to avoid overlap in the absorption profiles of the two PCs PheoA and ZnTPP, and (ii) the pair of PheoA–CPADB and ZnTPP–TTC exhibited specific PC–CTA interaction. Selective excitation of one PC led to concomitant selective activation of one CTA within the reaction mixture. Switching the irradiation wavelength enabled sequential two-step polymerization in one-pot. However, it is noted that the second monomer MA should be added after the polymerization of MMA from CPADB because otherwise the insertion of MA into CPADB would block further propagation due to the change in C–S bond dissociation energy. Boyer, Hawker, Moad, and coworkers coupled PET-RAFT polymerization with photoiniferter polymerization (Fig. 14a).280 At first, the methacrylic backbone of MMA and BTPEMA was prepared from CDTPA via green light-mediated photoiniferter polymerization (520 nm, 4.4 W m−2) at 60 °C. Model polymerization of MMA confirmed that after the addition of MMA into CDTPA or BTPA, fragmentation of the RAFT intermediate was only favored for CDTPA owing to higher C–S bond dissociation energy. Subsequently, the unreacted BTPA moieties in the side chains were polymerized with DMAm. In this step, despite the increase in reaction temperature and/or irradiation at shorter wavelengths (i.e., with higher energy), the rate of polymerization was not sufficiently high. Thus, PET-RAFT polymerization using ZnTPP was conducted instead. This one-pot approach could be operated even in one-pass using a flow reactor. In a one-pass reaction system, the solution of DMAm and ZnTPP for the second reaction was easily added before the reaction mixture flows to the second batch, and irradiation of each batch was separately turned on and off. Moreover, by simply varying the injection volume, concentration, flow rate, and/or irradiation time, the synthesis of diverse graft and branched copolymers possessing backbones and/or side chains with different lengths and backbones with different numbers of grafted chains was achieved. Matyjaszewski and coworkers utilized two sequential photoiniferter polymerizations for the synthesis of comb-like and bottlebrush polymers.110 The polymerization of the methacrylic backbone from CDTPA under green light irradiation (520 nm, 4.25 mW cm−2) was followed by the polymerization of MA or DMAm from pendant BTPA of polymerized BTPEMA under blue light irradiation (465 nm, 6.5 mW cm−2). Depending on the MMA/BTPEMA ratio in the first polymerization, the grafting density of acrylic side chains in the resulting polymer was facilely controlled so that the homopolymer of “inimer” was transformed into a bottlebrush polymer.
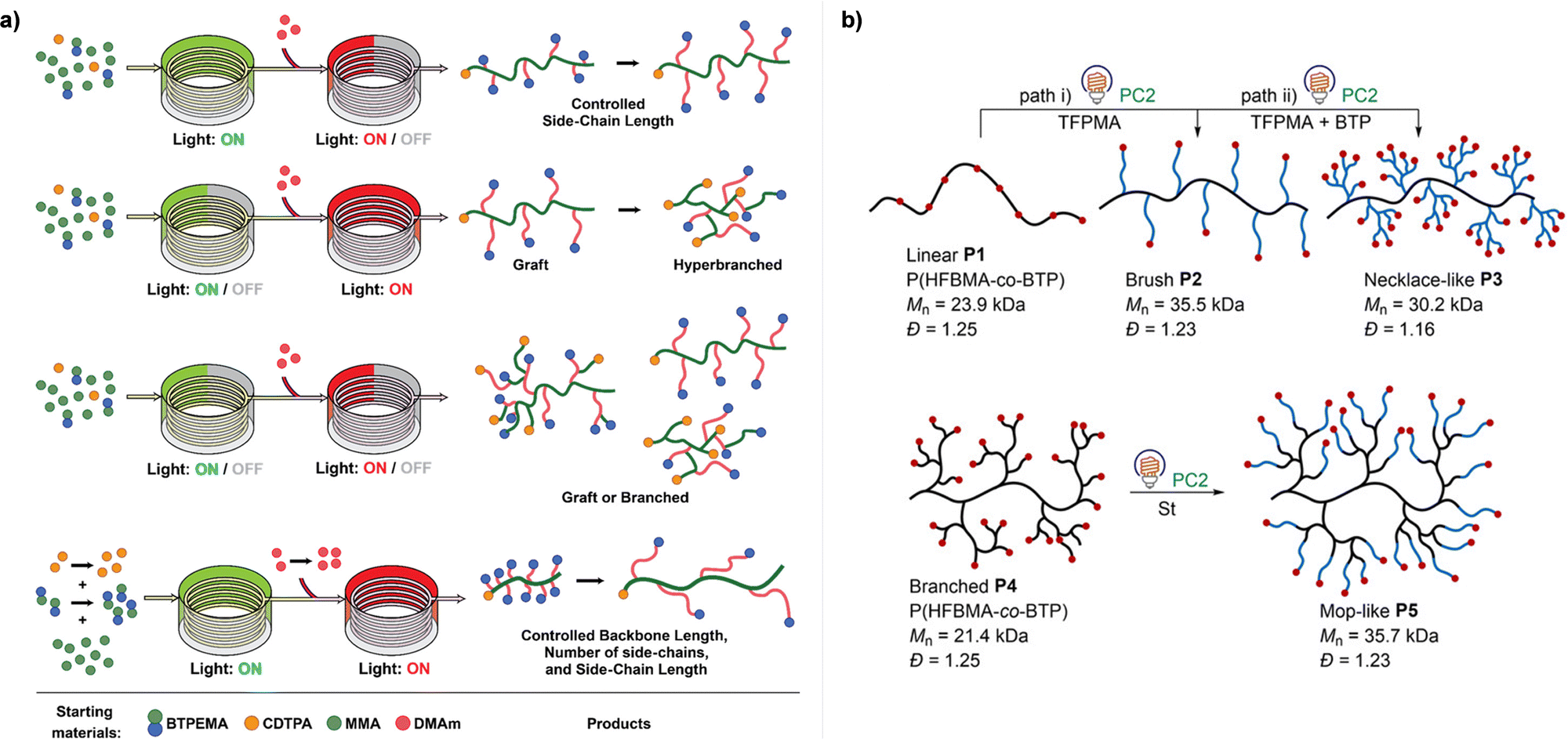 | ||
| Fig. 14 Diverse polymer architectures obtained by (a) switching each reactor on and off or manipulating the parameters (e.g., flow rate and injection volume) in flow chemistry and (b) combination of different photomediated RDRP processes. Adapted with permission from (a) ref. 280 (Copyright 2021 American Chemical Society) and (b) ref. 211 (Copyright 2020 John Wiley & Sons, Inc.). | ||
The second polymerization from the initiating moiety of the inimer after the first polymerization of vinyl moieties need not be limited to visible-light-driven RAFT polymerization. The orthogonality of visible-light-driven RAFT polymerization to other polymerization methods and the accompanying ability to interconvert two orthogonal processes for the facile synthesis of multiblock copolymers possessing various (non)vinyl monomers has been discussed in Section 4.3.1.2. Chen and coworkers designed an inimer containing both vinyl and alkyl bromide functionalities for photocontrolled divergent synthesis of polymers with a wide variety of topologies (Scheme 24b).211 The reaction system comprised two PCs (EY and a phenothiazine derivative), fluorinated-TTC, methacrylate monomers bearing different numbers of fluorine atoms, and 2-bromo-trifluoropropene (BTP) as an inimer (Fig. 14b). Owing to the different excited-state redox potentials, reduction of bromine for O-ATRP after the polymerization of BTP could only be achieved using the phenothiazine-derived PC, whereas TTC could be reduced by both PCs for PET-RAFT polymerization. Therefore, EY-catalyzed PET-RAFT polymerization of the linear copolymer of BTP and other monomers followed by phenothiazine derivative-catalyzed O-ATRP afforded branched polymers in which bromine at any location served as a branching point. Variation in the ratio of BTP and other monomers in copolymerization, target DP, and/or location of bromine led to various polymer topologies such as linear, brush, necklace-like, branched, and mop-like.
 | ||
| Scheme 25 Coordination of Y(OTf)3 to the amide functionality to increase the isotacticity of poly(DMAm).282 | ||
Boyer and coworkers introduced flow chemistry in combination with the use of ZnTPP as a PC.288–290 In PET-RAFT polymerization of acrylamides in a continuous flow reactor, polymers of different molecular weights were generated by simply adjusting the flow rates, concentration of the CTA, and intensity or wavelength of the light source (blue, green, or red).288 Blending of the prepared polymers resulted in the target MWD as per the calculated theoretical curves (Fig. 15a)288 or the mathematically modelled fitting which is the so called computer-guided approach.290 The polymer retained high end-group fidelity so that polymerization from the blended poly(DMAm) led to successful copolymerization with NAM or benzyl acrylate (BzA) and yielded a block copolymer with tailored compositional gradients (Fig. 15b).289 The second monomer was easily added from a separated line (pump) in the flow setup. However, to fully utilize the aforementioned simplicity and robustness of the flow chemistry technique for dispersity control, fluid dynamics or profiles and parameters such as tube diameter, volume, flow rate, and reactant volume have to be carefully investigated and optimized.290
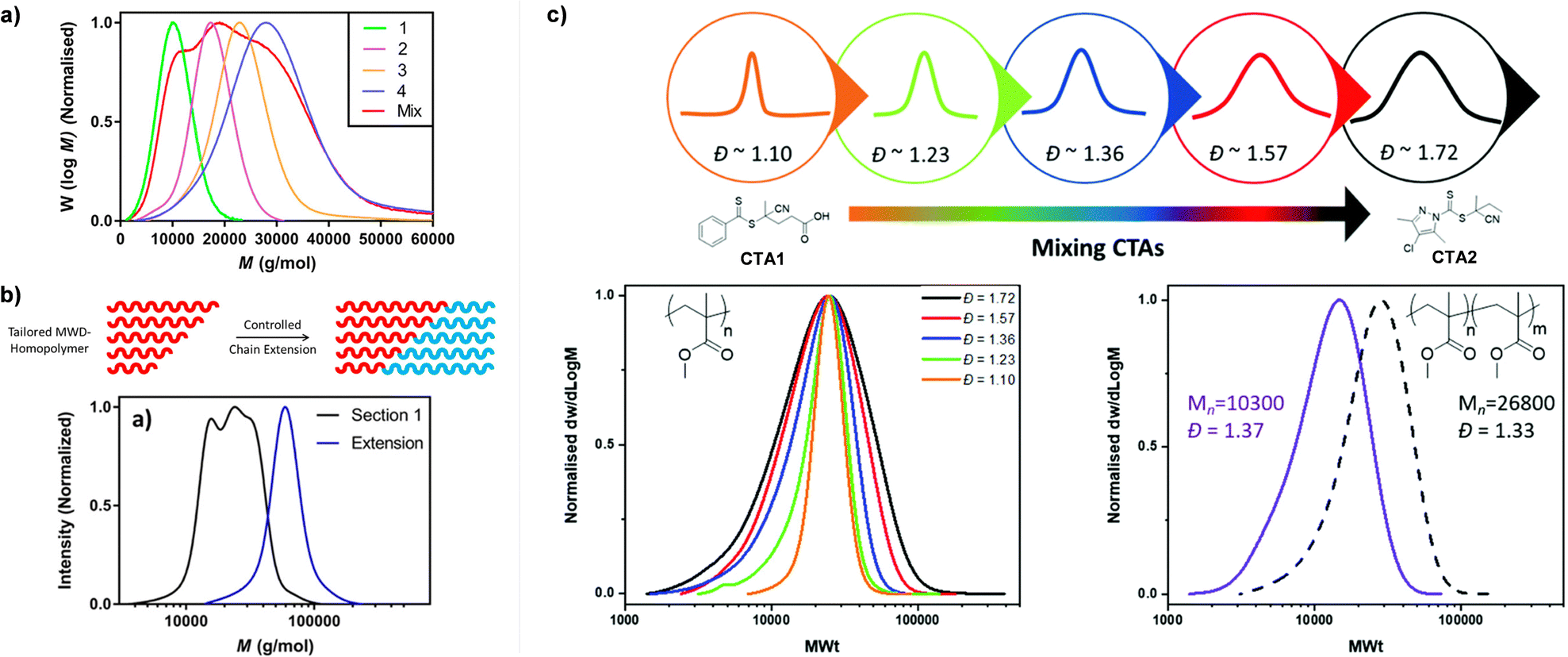 | ||
| Fig. 15 (a) MWDs for monomodal poly(DMAm)s (1–4) and their blended mixture. (b) Narrowing the initially broad MWD through chain extensions. (c) The variation in dispersity as two CTAs were mixed in different ratios (aligned by the peak molecular weight value). Poly(MMA) prepared from 10 mol% CTA1 and 90 mol% CTA2 was successfully extended with MMA. Adapted with permission from (a) ref. 288 (Copyright 2017 American Chemical Society), (b) ref. 289 (Copyright 2018 American Chemical Society), and (c) ref. 292 (Copyright 2020 The Royal Society of Chemistry). | ||
In the case of PET-RAFT polymerization in batch, Anastasaki and coworkers mixed two CTAs at varying ratio,291 as previously applied for AIBN-initiated conventional RAFT polymerization by the same research group.292 In EY-catalyzed PET-RAFT polymerization in DMSO under blue light irradiation (465 nm, 12 W), two CTAs possessing different transfer constants or reactivities (dictated by R and mostly Z group) were mixed at varying ratios to obtain polymers with any intermediate targeted value of dispersity (Fig. 15c). One CTA with the higher transfer constant afforded polymers with low dispersity (as the lower limit), whereas another CTA with the lower transfer constant afforded polymers with high dispersity (as the higher limit). Apart from the discrepancy in reactivities, two CTAs should separately provide monomodal MWD, high end-group fidelity, and agreement between theoretical and experimental molecular weights for polymerization of a given monomer. The method was effective for MMA, MA, and DMAm in which dispersity was tunable across a wide range (1.08–1.82). Konkolewicz and coworkers expanded the abovementioned method to poly(vinyl ketone)s.293 Herein, after the generation of initiating radicals through Norrish chemistry of vinyl ketone monomers under blue light irradiation (450 nm, 7.9 mW cm−2), either homopolymers of phenyl or methyl vinyl ketone or copolymers of ethyl acrylate were synthesized from the mixture of TTC (higher activity) and xanthate (lower activity). Dispersity was increased with the ratio of xanthate and was able to be modulated further owing to the photodegradable nature of vinyl ketone under UV light irradiation.
Anastasaki and coworkers also utilized their strategy of mixing CTAs to prepare two homopolymers each possessing a wide range of dispersities and blended them to provide polymers with unrivalled precise control of both dispersity and MWD shape.294 Moreover, the strategy was still valid for a single CTA with switchable reactivity, rather than the mixture of two CTAs.295 Herein, the reactivity of the CTA (methyl 2-[methyl(4-pyridinyl)carbamothioylthio]propionate) was in situ switched back and forth by the addition of acid and base to the reaction mixture. It was still only reported for VA-044 initiated thermal RAFT polymerization; nevertheless, it is notable that on-demand control over dispersity of the sequence-defined multiblock copolymer of acrylamides was accomplished by simply adjusting acidic and basic conditions along with the subsequent monomer addition.
Supramolecular perylene diimide/cucurbit[7]uril complex/TEOA under green light irradiation (535 nm, 5 W) induced aqueous polymerization of DMAm from TTC with molecular weight up to 1.04 × 106 g mol−1 and a dispersity of 1.33 after 8 hours.297 This photoinitiating system was so efficient that only 1 ppm was required, but the role of TEOA was claimed to be rather complex, including initiation of RAFT polymerization after its single-electron oxidation by excited-state perylene diimide. Photoenzymatic RAFT polymerization enabled the synthesis of UHMW polymers of unconjugated NVP, N-vinylcaprolactam with rather narrow dispersity (Đ = ∼1.39)164 and of monomers with low kp (e.g., OEGMA)165 under violet light irradiation (405 nm, 11.5 W). These exceptional reactivities were attributed to the generation of stable radicals in low concentration by GOx/glucose as the PC, and the high reducing power of excited-state FADH− to efficiently conduct the polymerization process.
On the other hand, Chen and coworkers reported a mechanically distinct strategy for the synthesis of UHMW fluoropolymers from TTCs (up to 3.05 × 106 g mol−1 and Đ ≤ 1.12 after 2–8 hours) using Ir(ppy)3 in DMSO under white light irradiation (33 mW cm−2) (Fig. 16a).190 Fluoropolymers from a polymer chemistry perspective have been appreciated due to their functionally diverse properties,298 but here strong and selective fluorine–fluorine interaction also played a key role in excellent regulation over propagation (Fig. 16b). As depicted, at first, the CTA with a lipophilic DMAm block as the R group (denoted as R2) and an alkyl chain as the Z group (denoted as R1) stabilized the resulting emulsion with a fluorophilic interior where fluoromonomers were located. As some portions of the CTA (propagating group, PG) participated in polymerization, fluorine–fluorine interaction between the grown polymers and the monomers resulted in phase separation. The remaining intact CTA (supporting group, SG) residing in the outer solvent phase had no access to the monomer but to RAFT equilibrium, providing the polymerization of fluoromonomers from PG with the additional control and livingness. Replacing Ir(ppy)3 with a PC possessing a fluoro-substituent significantly reduced monomer conversion, evidencing that single-electron transfer between the excited-state PC and CTA and subsequent polymerization occurred in the outer solvent phase. This distinctive spatial confinement strategy for the preparation of well-defined UHMW fluoropolymers was compatible with fluorinated (meth)acrylates and methacrylamides as well.
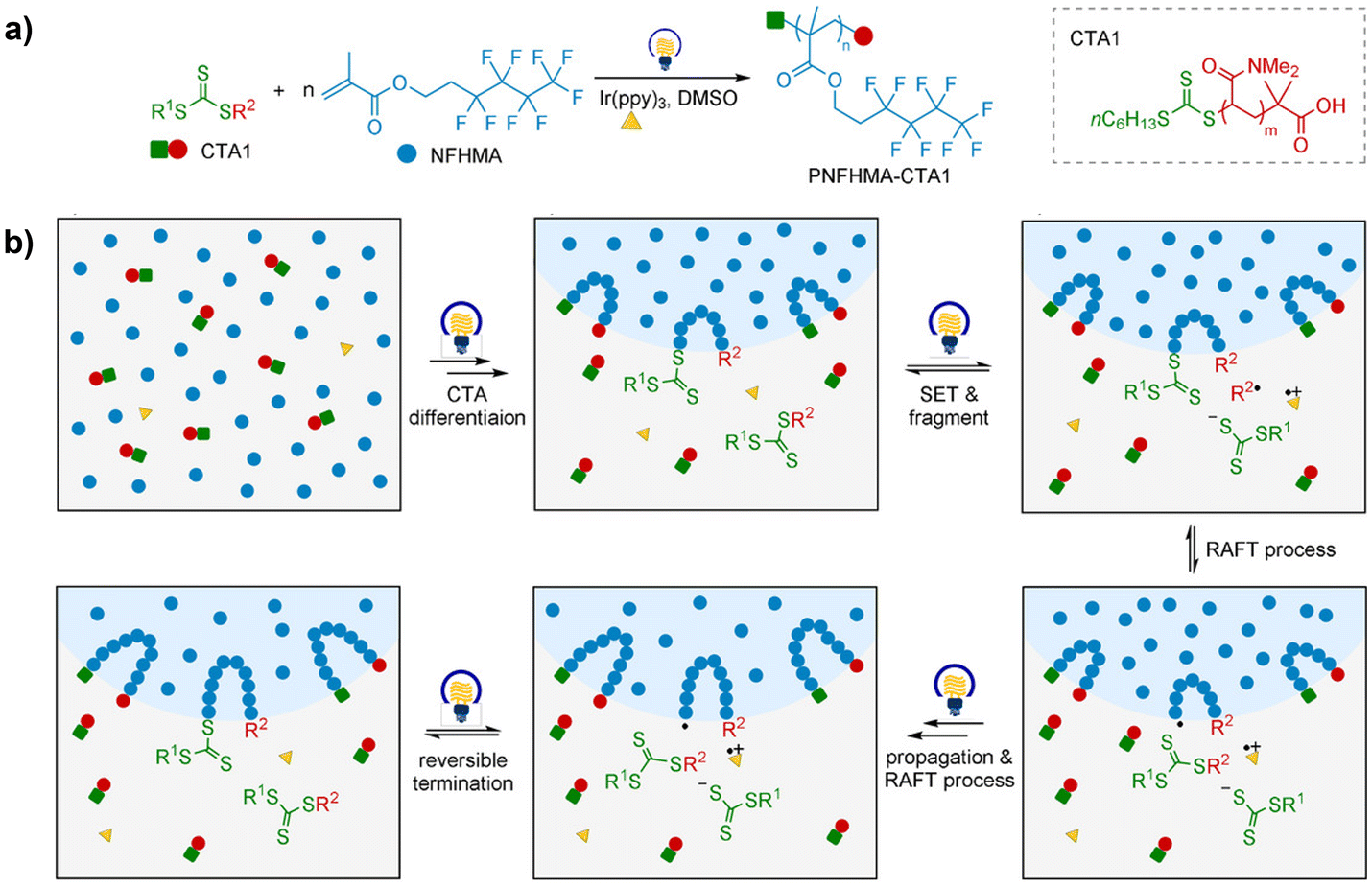 | ||
| Fig. 16 (a) Synthetic scheme of UHMW fluoropolymers and (b) mechanistic concept for CTA differentiation for regulation over propagation. Adapted with permission from ref. 190 (Copyright 2020 John Wiley & Sons, Inc.). | ||
Boyer and coworkers reported the first photo-PISA that was mediated by Ru(bpy)3Cl2 under blue light irradiation (460 nm, 0.7 mW cm−2).192 Up to 100 ppm of Ru(bpy)3Cl2 was loaded for the dispersion polymerization of solvophobic benzyl methacrylate (BzMA) from the solvophilic poly(OEGMA)-based macro-CTA. The dispersity of the polymer was maintained at lower than 1.3 despite the presence of unreacted or dead chains with lower molecular weight than the theoretical values and increase in the viscosity of the solution. ZnTPP was the very first PC for oxygen-tolerant photo-PISA and under red or yellow light irradiation at longer wavelength (635 and 560 nm, respectively).301 Oxygen tolerance was achieved by the combination of ZnTPP as a singlet oxygen generator and ascorbic acid301 or 9,10-dimethylanthracene146 as a singlet oxygen quencher, so as to conduct the reaction in an open vessel at ultralow volumes (40 μL scale) without prior deoxygenation. However, only in the presence of 9,10-dimethylanthracene, perfect temporal control of polymerization was observed. Furthermore, ascorbic acid in larger amount seemed to affect self-assembly behavior. Very recently, Boyer and coworkers for the first time reported oxygen-tolerant photo-PISA under NIR irradiation (730 nm, 12 mW cm−2) using ZnPCS4− in combination with TEOA.173
Meanwhile, ZnTPP could still generate singlet oxygen after its encapsulation in the hydrophobic core of the vesicle via dialysis of the reaction mixture into water, implying the potential use of the resulting vesicle as a photodynamic therapeutic agent.301 It was insolubility of ZnTPP in water that enabled the encapsulation. Similarly, doxorubicin, a hydrophobic antitumor drug, was encapsulated in spherical polymeric NPs.302 Herein, doxorubicin was added into the reaction mixture from the beginning of the polymerization. While the polymerization process did not significantly affect the bioactivity of doxorubicin, the polymerization kinetics were improved, implying the potential photocatalytic capability of doxorubicin for photomediated polymerization.
ZnTMPyP, as a water-soluble analogue of ZnTPP, allowed aqueous dispersion polymerization,147 and simplified photo-PISA by eliminating the preparation of solvophilic macro-CTAs which undergo chain extension with solvophobic monomers.200,201 Instead, hydrophilic OEGMA and hydrophobic core-forming diacetone acrylamide (DAAm) were put together for gradient copolymerization based on preferential incorporation of OEGMA over DAAm owing to different reactivities of methacrylates and acrylamides for polymerization.201 If both solvophilic and solvophobic blocks are prepared from the same monomer families, slow injection of solvophobic monomers into the reaction mixture gradually changed the ratio of the solvophobic- to solvophilic block length within a polymer chain, which led to PISA.200 Lastly, Chen and coworkers controlled the size of raspberry-like NPs with a poly(pentafluorostyrene) core by light intensity.303 At around 330 ppm of Ir(ppy)3 and the same target DP, strong irradiation (33 W cm−2) as compared to weak light irradiation (0.56 W cm−2) activated more CTAs to decrease the polymer molecular weight at the same monomer conversion, and then to reduce the size of NPs. The effect of intensity and wavelength of light irradiated on polymerization control (e.g., molecular weight and end group fidelity) and thus the morphologies of synthesized NPs has been reported for several cases.301,303
Organic PCs such as PTH,304 EY,135,170,305 Rose Bengal,306 and Rhodamine 6G307 have also been employed but mostly in combination of tertiary amines to achieve oxygen-tolerant reaction systems and enhance the reaction rate. Oxygen tolerance in photo-PISA particularly allows one to conduct the reaction in a continuous flow reactor with simple optimization of reaction parameters.135,305,308
Soh, Hawker and coworkers reported in situ grafting-to conjugation of poly(acrylamide)s onto several biomolecules (L-phenylalanine, 5-[(2-aminoethyl)amino]-naphthalene-1-sulfonic acid sodium salt (EDANS), and bovine serum albumin (BSA)) via Ru(bpy)3Cl2/sodium ascorbate-catalyzed PET-RAFT polymerization under blue light irradiation (470 nm, 80 mW cm−2).150 The copolymerization of NAM and N-acryoylsulfosuccinimide (NASS) with the target DP of 40 reached 95% conversion after 15 minutes even without prior deoxygenation. This short reaction time minimized the hydrolysis of succinimides and allowed in situ preparation of bioconjugates via the addition of biomolecules to the polymerization mixture.
To functionalize the surface of live cells, more rapid kinetics under milder reaction conditions are required to reduce the potential harm to the cells. Soh, Hawker and coworkers employed EY/TEA-catalyzed PET-RAFT polymerization under blue light irradiation (465 nm, 5.0 mW cm−2) for grafting-from functionalization of live yeast and mammalian cells.67 After 10 minutes of Ar purging and 5 minutes of irradiation, the cell surface was successfully functionalized with polyacrylamides bearing poly(ethylene glycol) (PEG) side chains while maintaining both high control over polymerization and high cell viability. Copolymerization with azido- or biotin-incorporated monomers allowed post-polymerization functionalization of the cells via copper-free SPAAC or streptavidin conjugation chemistry, respectively, which is promising to manipulate the cell surface and regulate cell–cell interactions. Although this early demonstration required Ar purging and the use of blue light, the recently reported examples with novel modified PCs have improved the reaction system toward a wide range of irradiation wavelengths. Qiao, Tang, Fu, and coworkers synthesized SA-TCPP for TEOA-cocatalyzed PET-RAFT polymerization of OEGMA (Mn = 480 g mol−1) in the presence of mouse fibroblast cells (NIH 3T3) in a 96-well plate.223 After 45 minutes of red light irradiation, the monomer conversion and cell viability were 11 and 46%, respectively. Subsequently, a ZnTPP-derived 2D nanosheet reported by Cai, Li, and coworkers was employed for the ascorbic acid-cocatalyzed PET-RAFT polymerization of OEGA (Mn = 480 g mol−1) in the presence of human embryonic kidney 293 cells.149 The monomer conversion was 60.8–73.2% resulting in polymers with a rather broad dispersity (1.21–1.32) without negatively impacting the cell viability after two hours of irradiation under red light (635 nm, 3.6 mW cm−2). Although the reaction conditions have to be further optimized, these preliminary studies suggested that PET-RAFT polymerization is suitable for cytocompatible in vivo polymerization in cellular applications.
Barner-Kowollik, Ng, Weil, and coworkers synthesized DNA–polymer conjugates from CTA-terminated single-stranded DNA (ssDNA) (Fig. 17a).311 EY with the aid of ascorbic acid as a singlet oxygen quencher under blue light irradiation (470 nm, 4.0 mW cm−2) was employed to avoid problematic deoxygenation at ultralow reaction volumes (20 μL). However, good control over dispersity could only be achieved after the freeze–pump–thaw process despite the use of ascorbic acid. The synthesized ssDNA–polymer conjugates could be hybridized with complementary ssDNA, and the size of the hybridized structure was increased with the size of the conjugated polymer. Lipid–polymer conjugates, prepared from CTA-terminated lipid using EY under blue light irradiation (465 nm, 5.0 mW cm−2), could self-assemble and were used to mediate assemblies of gold NPs.312
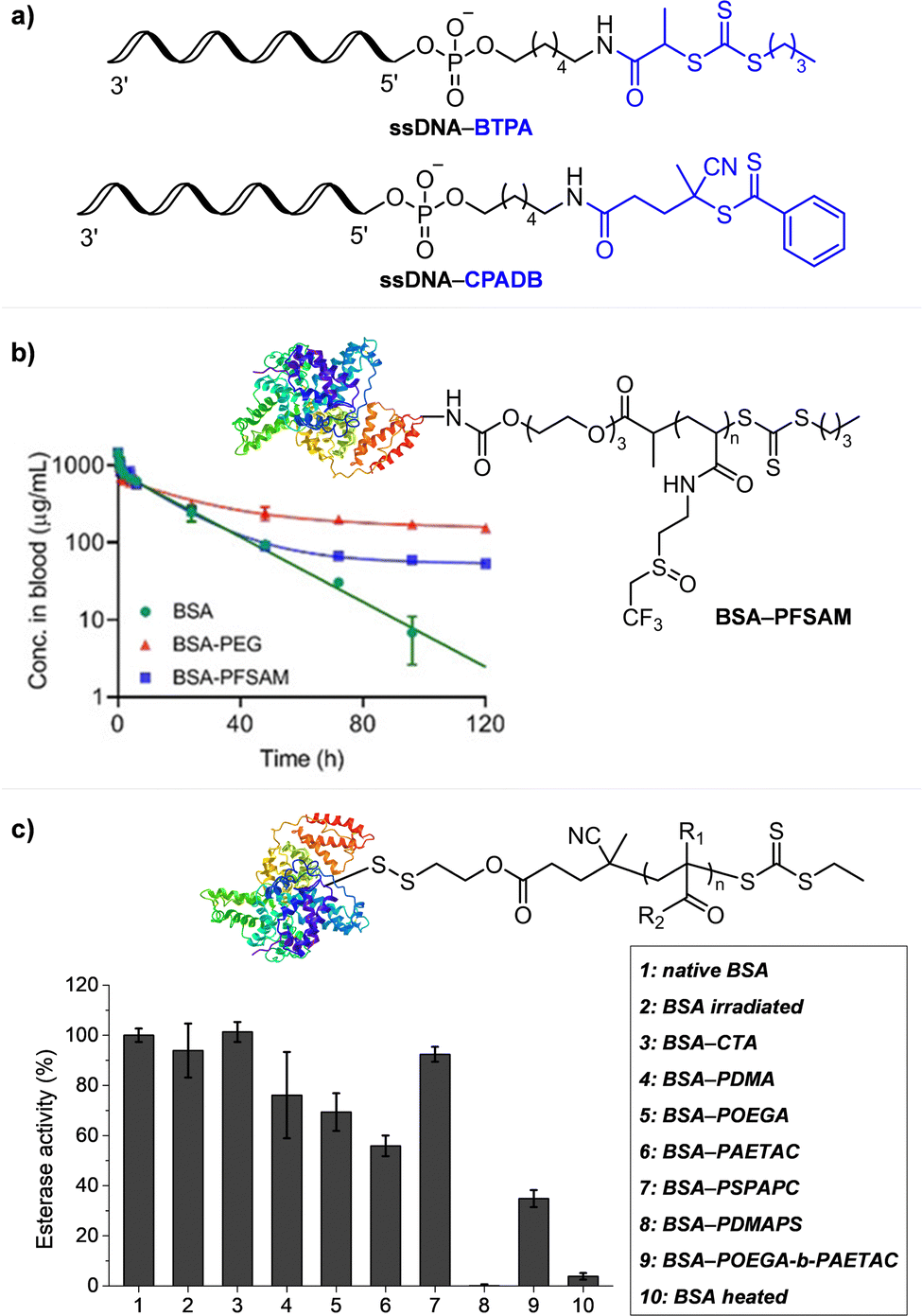 | ||
| Fig. 17 (a) Schematic representation of ssDNA–CTA conjugates for synthesis of ssDNA–polymer conjugates.311 (b) Pharmacokinetics and (c) enzymatic activities of BSA and various BSA–polymer conjugates. Adapted with permission from (b) ref. 70 (Copyright 2020 John Wiley & Sons, Inc.) and (c) ref. 176 (Copyright 2022 John Wiley & Sons, Inc.). | ||
Lastly, proteins are also of great interest. Once a polymer is conjugated to a protein, the properties of the protein would be influenced as in the case of a protein attached to PEG, being widely used in the clinic worldwide.313,314 PEG enhances the pharmacokinetic/dynamic properties while reducing the immunogenicity of therapeutic proteins.315 Therefore, owing to the large diversity of polymers and the increasing desire to utilize proteins for the degradation of plastics,316 for capture of heavy metal ions,317,318 and as catalysts in organic chemical synthesis,319 researchers are attracted to develop novel protein–polymer conjugates (PPCs). To reduce the potential harm to the structural integrity of proteins and preserve their enzymatic activities after polymerization, PET-RAFT polymerization that operates under mild reaction conditions and offers oxygen tolerance has been particularly recognized as a promising method.
Averick and coworkers studied the effects of the hydrophobicity and molecular weight of grafted acrylamide polymers on the enzymatic activities of two lipases (Candida antarctica lipase B and Thermomyces lanuginosus lipase).141 EY/TEMED-catalyzed PET-RAFT polymerization from the grafted TTC in DMF was performed. Hydrophobic polyacrylamides led to a molecular weight-dependent increase in the activities and stability of lipases, so that the resulting hydrophobic hybrids could be immobilized on a glass slide while retaining the enzymatic activities. Whittaker and coworkers utilized EY with a tertiary amine to attach hydrophilic and low-fouling fluoropolymers to BSA as a model protein for in vivo quantitative tracking of polymer-conjugated therapeutics by 19F magnetic resonance imaging (MRI) and extending the pharmacokinetics (e.g., blood circulation time) (Fig. 17b).70 Another hydrophilic sulfoxide-containing polymer improved the pharmacokinetics of lysozymes by reducing macrophage cellular uptake.320 Although the listed applications of PPCs still have been mainly limited to therapeutics, many researchers focused on how to use PET-RAFT polymerization as a more efficient platform for preparation of PPCs, which would allow extension of the diversity of protein-based functional materials in the near future. For example, macromolecular and heterogeneous PCs for their facile purification after the reaction,247,321,322 or PCs operating under irradiation at longer wavelength (i.e., with lower energy) were developed.
Ever since Boyer and coworkers employed Ru(bpy)3Cl2 under blue light irradiation for PET-RAFT polymerization from BSA macroinitiator which was chemically modified to possess TTC moieties,193 Ru(bpy)3Cl2,323 EY and its derivatives,68,70,141,247,249,320,321 ZnTPP derivative,322 and a novel purely organic PC176 have been discovered and utilized. Although EY is biocompatible and has facilitated the use of green LEDs as an irradiation source, tertiary amines68,70,141,247,320 and/or ascorbic acid247,321 need to be added to achieve oxygen tolerance. Photodegradation of EY is another issue.141,247,324 Averick and coworkers observed photodegradation of EY in the presence of TEMED as an amine cocatalyst in DMF after up to 60 min of irradiation at 460 nm.141 Sumerlin and coworkers observed photobleaching of EY under irradiation at 458 nm (0.6 mW cm−2) and 515 nm (0.5 mW cm−2), which was attributed to one of the possible side reactions caused by ascorbic acid at high concentration.247 Due to the lack of singlet oxygen quenchers under aqueous conditions, the ZnTPP derivative where zinc porphyrin was integrated into a MOF (MOF-525-Zn) required purging with Ar, in contrast to the excellent oxygen tolerance of ZnTPP observed in the presence DMSO which acted as both a solvent and a singlet oxygen quencher.322 Although these water-insoluble and heterogeneous PCs can be easily purified after the reaction and even reused, a water-soluble PC that enables oxygen-tolerant polymerization without additives is also beneficial; nevertheless, it has been limited to Ru(bpy)3Cl2. In this regard, Kwon and coworkers in collaboration with Gierschner and Koo developed a highly efficient water-soluble purely organic PC 3DP-MSDP-IPN.176 Efficient generation of triplet excited states, substantially negative  (−1.56 V vs. Ag/AgCl), and highly stable radical cation of the PC synergetically afforded oxygen tolerance (or even acceleration of polymerization kinetics by oxygen present at limited concentration; for a detailed explanation, please see Section 3.4). Thus, polymerization was conducted using the BSA macroinitiator in ambient and aqueous environments without additives under green light irradiation (515 nm, 10 mW cm−2). A variety of acrylamide/acrylate monomers, including hydrophilic and ionic (cationic, anionic, and zwitterionic) monomers, were polymerized from the protein macroinitiator to affect the enzymatic activities to varying degrees as compared to native BSA (Fig. 17c). However, the authors observed that the molecular weights of the polymers cleaved from the protein were smaller than the corresponding theoretical values and presumed that via reactions between the excited-state PC and disulfide bonds of BSA, thiyl radicals might be generated and became additional reaction sites for the propagating radical intermediates. Although the formation of thiyl radicals has not yet been detected, the selectivity issue for the PC, by PET, to selectively activate the CTA over other reactive groups within proteins (e.g., TTCs over disulfide bonds) needs to be further investigated to design better photocatalytic systems for future bioapplications. Development of novel PCs and expansion of the available water-soluble and/or biocompatible PCs, rather than Ru(bpy)3Cl2 and EY, would stimulate related research and allow the synthesis of polymers comprising various monomers rather than acrylamide monomers which are extensively preferred because of their more rapid polymerization kinetics than those of other monomer families.
(−1.56 V vs. Ag/AgCl), and highly stable radical cation of the PC synergetically afforded oxygen tolerance (or even acceleration of polymerization kinetics by oxygen present at limited concentration; for a detailed explanation, please see Section 3.4). Thus, polymerization was conducted using the BSA macroinitiator in ambient and aqueous environments without additives under green light irradiation (515 nm, 10 mW cm−2). A variety of acrylamide/acrylate monomers, including hydrophilic and ionic (cationic, anionic, and zwitterionic) monomers, were polymerized from the protein macroinitiator to affect the enzymatic activities to varying degrees as compared to native BSA (Fig. 17c). However, the authors observed that the molecular weights of the polymers cleaved from the protein were smaller than the corresponding theoretical values and presumed that via reactions between the excited-state PC and disulfide bonds of BSA, thiyl radicals might be generated and became additional reaction sites for the propagating radical intermediates. Although the formation of thiyl radicals has not yet been detected, the selectivity issue for the PC, by PET, to selectively activate the CTA over other reactive groups within proteins (e.g., TTCs over disulfide bonds) needs to be further investigated to design better photocatalytic systems for future bioapplications. Development of novel PCs and expansion of the available water-soluble and/or biocompatible PCs, rather than Ru(bpy)3Cl2 and EY, would stimulate related research and allow the synthesis of polymers comprising various monomers rather than acrylamide monomers which are extensively preferred because of their more rapid polymerization kinetics than those of other monomer families.
Meanwhile, without grafting and conjugation, polymers can also affect biomolecules. In polymer–protein hybrids, certain polymers could be hybridized with certain proteins, thereby helping to maintain the protein activities in non-native environments such as organic solvents316,325,326 and thermal denaturing conditions.266 Synthetic antimicrobial polymers that mimic the peptides with antimicrobial activities were discovered by investigating the structure–activity relationships of a large library of polymers composed of different categories of monomers (e.g., hydrophobic, hydrophilic, and cationic) with varying compositions263,327 and compositional drifts along the polymer backbone,264 or by incorporating additional functional monomers into the polymers.328 Wong, Boyer and coworkers synthesized a novel monomer (acrylated ZnTPP) that not only self-catalyzed the oxygen-tolerant PET-RAFT polymerization of monomers, but also enhanced the production of reactive oxygen species to promote antimicrobial activities.328 Gianneschi and coworkers synthesized enzyme-responsive and pro-apoptotic peptide-modified vinyl monomers to prepare cytotoxic peptide brush polymers, which disrupted the mitochondrial membrane and triggered the death of cancer cells.69 Later, copolymerization of these peptide-functionalized monomers with hydrophobic acrylamide monomers, and subsequent PISA resulted in synthesis of peptide brush polymer NPs, proposing their applications for peptide delivery systems.329 Regardless of the applications, the discovery of high-performing synthetic polymers has been boosted by PET-RAFT polymerization (mostly catalyzed by ZnTPP in DMSO due to its excellent oxygen tolerance), specifically in combination with HTP synthesis (please see Section 4.4.4) and very recently with machine learning approaches and automated polymer chemistry,266,330–332 which has enabled the facile preparation of a large number of polymers at a time.
4.4. Advanced techniques
Surface-initiated PET-RAFT (SI-PET-RAFT) polymerization was firstly demonstrated by Pester, Boyer and coworkers (Fig. 18a).334 Given the well-known oxygen tolerance of ZnTPP-catalyzed PET-RAFT polymerization in DMSO, the polymerization from a CTA-functionalized silicon oxide surface was successfully performed without prior deoxygenation. The polymerization mixture deposited on the surface was covered with a glass coverslip and irradiated at 405 or 590 nm (1.1 μm cm−2). As expected, PET-RAFT polymerization provided polymer brushes with uniform thickness and high end-group fidelity, allowing the synthesis of diblock copolymers by chain extension of the prepared polymer brushes. Fabrication of a patterned surface via spatially controlled polymerization using a photomask was also demonstrated.
 | ||
| Fig. 18 Schematic representation of (a) spatially controlled SI-PET-RAFT polymerization using a photomask, (b) stepwise polymerization for fabrication of a multilayer architecture, and (c) spatially controlled SI-PET-RAFT polymerization of fluorescent monomers using a digital micromirror device (DMD). Adapted with permission from (a) ref. 334 (Copyright 2019 American Chemical Society), and (b) and (c) ref. 335 (Copyright 2021 John Wiley & Sons, Inc.). | ||
Since then, ZnTPP in combination with DMSO as a solvent has been widely utilized for oxygen-tolerant SI-PET-RAFT polymerization under violet,335 green,336–338 or yellow light irradiation.338 A water-soluble ZnTPPS4− under green light irradiation339 or EY in the presence of TEOA under blue140,340 or green light irradiation138,341 expanded the polymerization media to aqueous systems. The lack of oxygen tolerance in the absence of TEOA required prior degassing342 or enzymatic degassing strategy using GOx and glucose.163 Using these various PCs, SI-PET-RAFT polymerization has been employed for fabrication of antifouling surfaces for biomedical and industrial applications,138,140,336–338,340 polycarbonate surface with tuned hydrophilicity,339 organic–inorganic hybrid materials,235,341–344 and functional polymeric materials.335
Antifouling surfaces were prepared from hydrophilic, zwitterionic (e.g., carboxybetaine methacrylamide (CBMAm)140,340), and low-fouling monomers (e.g., sulfoxide-336 or fluorine monomers337) to inhibit adsorption of biomolecules such as bacteria, proteins, and cells. Besides its role as a PC, ZnTPP that remained after the polymerization was effective as an antifouling agent through photodynamic inactivation of bacteria owing to its ability to generate singlet oxygen under irradiation.337 The presence of residual ZnTPP was ascribed to covalent incorporation of ZnTPP into the polymer chain by radical-mediated polymerization of the reduced porphyrin macrocycle. This photodynamic activity of the residual ZnTPP, on the other hand, likely caused a significant cytotoxicity to mouse macrophage-like (RAW 264.7) cells, implying the need for the use of organic PCs in biomedical applications.336
Organic–inorganic hybrid materials were provided via grafting-from synthesis of polymers from mesoporous silica NPs,342 mesoporous silica films,343 gold-coated silicon wafers,140 indium tin oxide (ITO)/gold-coated electrodes,341 NaYF4:Tm/Yb UCNPs,344 and CdSe QDs.235 On the ITO/gold-coated electrode surface, the CTA possessing an electrochemically active carbazole moiety was immobilized by an electrodeposition method.341 NaYF4:Tm/Yb UCNPs344 and CdSe QDs235 acted as both substrates and PCs for SI-PET-RAFT polymerization, where the synthesized polymers were tethered to the surfaces of nanocomposites via ligand exchange between the initial ligands of nanocomposites and TCT groups of the polymers. Encapsulation of gold NPs by the poly(dimethylaminoethyl acrylate (DMAEA))-tethered surface341 and subsequent immobilization of antibodies onto the prepared polymer brushes140 implied that SI-PET-RAFT polymerization would become a promising strategy for the development of organic–inorganic hybrid biosensors.
Very recently, Hudson, Pester, and coworkers employed ZnTPP-catalyzed SI-PET-RAFT polymerization under violet irradiation (405 or 415 nm) for fabrication of multilayer thin films for organic electrodes.335 Using various acrylic monomers comprised of thermally activated delayed fluorescence (TADF) emitters, stepwise polymerization from CTA-functionalized ITO provided multilayer architectures of hole-transport layer (HTL), emissive layer (EML), and electron-transport layer (ETL), mimicking the structure of multilayer organic light-emitting diodes (OLEDs) (Fig. 18b). Moreover, combined with the recently emerged digital projection lithography technique using arrayed digital micromirror devices (DMDs), a spatial control over complex polymer architectures at high resolution was imparted (Fig. 18c)
A bare surface without prior functionalization with a CTA was also utilized for the preparation of a polyvinyl alcohol (PVA)-based hydrogel. The PVA-based hydrogel was immersed in the reaction mixture of PC (i.e., Ru(bpy)3Cl2,345,346 fluorescein,347 EY,348 and aluminum phthalocyanine chloride349), CTA, monomers, and TEA. It was proposed that alkoxyl radicals generated from PVA by a one-electron oxidized PC or by irradiation participated in PET-RAFT polymerization. Zwitterionic monomers such as CBMA were polymerized to provide antifouling properties to PVA-based materials for biomedical applications.345–349 Thermoresponsive poly(NIPAm) brushes provided surface properties which were adjustable by temperature.347
 | ||
| Fig. 19 (a) Synthesis of a 4D printed object capable of swelling- and desolvation-induced actuation owing to difference in mechanical properties of two layers by the spatially resolved light doses. (b) Original stl file image and 3D printed theater complex. Adapted with permission from (a) ref. 142 (Copyright 2019 John Wiley & Sons, Inc.) and (b) ref. 362 (Copyright 2021 American Chemical Society). | ||
ZnTPP then extended the operation wavelength to red (635 nm, 0.5 mW cm−2) although 3D printing was performed under a N2 atmosphere to achieve a reasonable maximum build speed of 0.1 cm h−1 due to the long inhibition period in the presence of air.365 Nevertheless, there is still a need to develop new PCs capable of achieving faster polymerization, which would enable 3D printing with a reasonable build speed. To overcome the slow printing speed achieved by PET-RAFT polymerization, Norrish type I photoinitiator diphenyl (2,4,6-trimethylbenzoyl) phosphine oxide (TPO) replaced the PC, which resulted in the fabrication of 3D printing objects at a faster printing rate (up to 9 cm h−1).366 The incorporation of the CTA conferred additional properties to the 3D printed objects, including capability for surface modification366 and self-healing property.367 More recently, the incorporation of macro-CTAs in the 3D printing resins enabled the preparation of 3D printed materials containing defined nanostructures via polymerization-induced microphase separation.368–370 This technique would open opportunities for on-demand fabrication of nanostructured polymer materials with tunable mechanical properties and ion conductivity for energy storage applications.371,372
Among visible-light-driven RAFT polymerizations, PET-RAFT polymerization was the first to be performed in a continuous flow reactor, owing to its superior oxygen tolerance.145 In contrast, there were a few reports employing flow chemistry techniques to (UV light-induced) photoiniferter379–381 and photomediated cationic RAFT polymerizations, owing to their sensitivity to oxygen.382 Oxygen tolerance is not a prerequisite for flow chemistry techniques; nevertheless, the high surface-area-to-volume of tubing reactors may facilitate the diffusion of oxygen into the reaction mixtures, causing a negative impact on oxygen-sensitive reactions.264 In this regard, the more rapid polymerization kinetics of PET-RAFT polymerization compared to other polymerizations is also beneficial for operationally reasonable reaction conditions.380
Flow chemistry has been applied to rapid syntheses of homopolymers209 or multiblock copolymers,136,204 photo-PISA135,305 and gram-scale synthesis of discrete pentamers via sequential SUMI.273 Theoretically, the synthesis of polymers within minutes could enable their large-scale production (e.g., multi-gram within a day). In addition to enhanced polymerization kinetics, the molecular weights and monomer compositions can be controlled by simply adjusting the compositions of the reaction mixtures, flow rates (i.e., residence time), and irradiation times. In the case of block copolymer synthesis, different monomers can be sequentially added in order at defined times through separate channels (Fig. 20). Furthermore, in a one-pass reaction system, each batch can be irradiated at a different wavelength and separately turned on and off, such that multiple polymerization reactions are sequentially and selectively activated during the reaction in one pass. This approach was then employed to the synthesis of diverse polymers with complex architectures.280 At last, dispersity control was achieved via temporally regulated initiation and/or adjustment of the reaction parameters (e.g., flow rate and wavelength and intensity of irradiation).288–290
 | ||
| Fig. 20 Schematic representation and photographs of a continuous flow reactor for the block copolymer synthesis. The reactor system is composed of two separated syringe pumps for two tubing reactors, a micromixer tee, and a back-pressure regulator (BPR). Adapted with permission from ref. 136 (Copyright 2019 American Chemical Society). | ||
Nevertheless, to fully realize the potential of flow chemistry for expanding the current lab-scale polymerization system into the industrial scale, the influences of fluid dynamics and reactor parameters (such as tubing diameter and operation method) on polymerization control must be thoroughly investigated.204,290 For example, in the synthesis of high-molecular-weight polymers, the polymer properties were inconsistent because the polymers caused reactor fouling and affected the fluidic profiles of the reaction mixture due to low Reynolds number.204 Homogeneity of the reaction mixture within flow could be improved by applying a plug packed of low-cost and easily accessible silicon oxide beads.383
 | ||
| Fig. 21 Combinatorial approach for the discovery of antimicrobial polymers prepared by high-throughput PET-RAFT polymerization. Adapted with permission from ref. 386 (Copyright 2018 John Wiley & Sons, Inc.). | ||
Among PCs, ZnTPP dissolved in DMSO demonstrating extreme oxygen tolerance without additives was extensively employed. ZnTPP-catalyzed HTP polymerization has enabled the simple and efficient construction of a library of polymers without prior deoxygenation.262–266,276,387–390Via preparation and investigation of a large combinatorial library of polymers, researchers efficiently optimized polymerization conditions262,389 and screened structure–activity relationships of polymers synthesized for antimicrobial applications,155,263,264 polymer–protein binding interactions,265,266 conjugation of biomolecules,276 preparation of a glycopolymer library,390 and design of single-chain polymer NPs.388 Other than ZnTPP, EY was also employed for photo-PISA in a 96-well plate, but ascorbic acid was necessary to achieve oxygen tolerance.170
Apart from its outstanding oxygen tolerance in DMSO, ZnTPP also showed promise in characterization of polymerization at low volumes.387 Boyer and coworkers noticed that fluorescence emission bands of ZnTPP changed owing to incorporation of ZnTPP into the polymer chain during polymerization. The linear relationship between fluorescence emission and monomer conversion allowed online monitoring of polymerization within a few minutes using plate readers, which was consistent with the results provided by NMR analysis. Herein, polymerization and direct characterization were conducted at below 40 μL scale in a 382-well plate. This ultralow volume is typically difficult to sample for NMR analysis that is carried out to determine monomer conversion in polymerization in batch. Therefore, the merge of HTP synthesis in well-plates and simple online monitoring of polymerization, combined with machine learning approaches and automated polymer chemistry,266,330,331,391 is expected to accelerate the discovery of novel functional polymers.392,393
5. Photoiniferter polymerization
In the absence of additives and/or PCs, simplicity and efficiency of the reaction system would be further increased. Moreover, the negative impacts of remaining additives on polymer properties will no longer become a concern. Nevertheless, since the first application of DTCs by Otsu and coworkers,40 the operating wavelength has been limited to the UV region. Because UV light can lead to undesired decomposition of the CTA and loss of controllability of polymerization, CTAs that can be cleaved by milder visible light have been extensively explored, and the relevant applications have recently emerged. Accordingly, the very recent review article reported by Hartlieb covered the concept, history, and various applications of photoiniferter polymerization irrespective of the irradiation wavelength.56 In this section, particular emphasis is placed on the examples harnessing visible light rather than UV light.5.1. CTAs for polymerization
Theoretically, visible-light-induced photocleavage of the C–S bond is possible for xanthates or TTCs and DTBs owing to their n → π* electronic transitions. The n → π* electronic transition of TTCs and DTBs occurs under blue and green light irradiation, respectively (Fig. 22).394 The red-shift effect for DTBs is due to a resonance in the Z group. In contrast, both n → π* and π → π* electronic transitions of xanthates395 and DTCs occur under UV light irradiation because of electron-rich oxygen and nitrogen in the Z group, respectively. Although n → π* electronic transitions at longer wavelengths are beneficial due to lower energies, their spin-forbidden nature and consequently small molar extinction coefficient make it challenging to achieve visible-light-driven photoiniferter polymerization at a reasonable polymerization rate. Therefore, the recently reported photoiniferter polymerization from TTCs still mostly harnesses UV light to target π → π* electronic transition-derived homolysis of the C–S bond regardless of the type of monomers (methacrylates,396 acrylates,33,379 acrylamides,33,58,397 and other monomers380,397). The use of a continuous flow reactor and/or high reaction temperature helped to improve the practicality and polymerization kinetics. Johnson and coworkers adapted a flow reactor for UV light irradiation (352 nm)-induced polymerization of acrylates and acrylamides.379 Junker, Abetz, and coworkers increased the reaction temperature to 120 °C for polymerization of isoprene and styrene in a flow reactor.380 On the other hand, in this section, we focus on the visible-light-driven photoiniferter polymerization using TTCs, DTBs, xanthates, and others. Scheme 26 depicts the monomers employed thus far.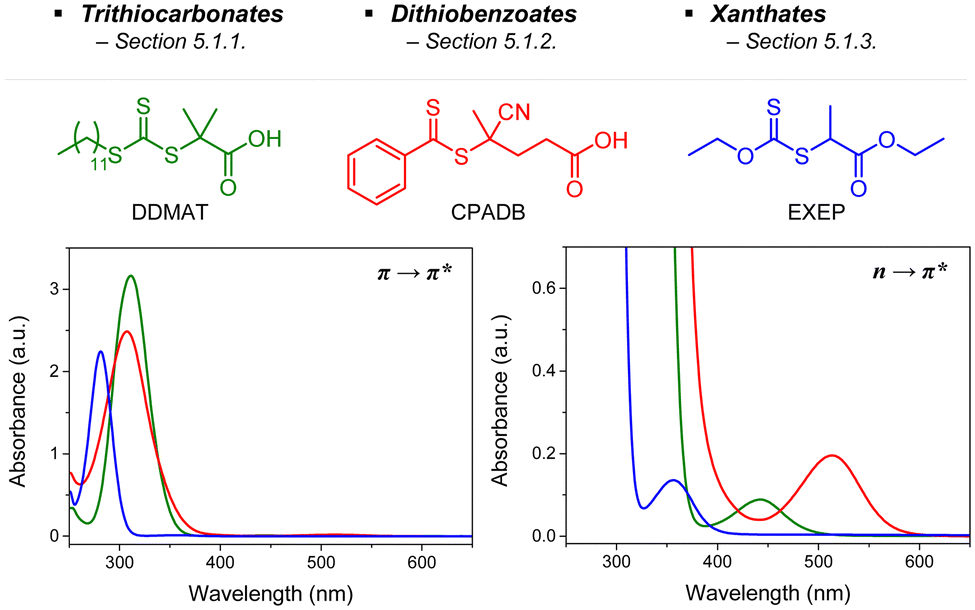 | ||
| Fig. 22 UV/Vis absorption spectra of DDMAT (green), CPADB (red), and EXEP (blue) in DMSO at 0.2 and 2.0 mM concentration to present π → π* and n → π* electronic transitions, respectively. | ||
 | ||
| Scheme 26 Chemical structures and abbreviations of monomers employed for visible-light-driven photoiniferter polymerization. | ||
The monomer scope was expanded to methacrylates,110,157,280,381,399,400 acrylates,33,88,110,134,157,401–408 acrylamides,33,110,157,404,407,409 AN,410 2-vinylpyridine (2-VP),411 styrene,404,406 and fluoro(meth)acrylates412 (Table 4). Compared to UV light, the use of milder visible light (460 nm, 4.8 W) prohibited the self-initiation of MA although the induction period was increased by four times.33 The authors ascribed the increase in the induction period to the prolonged time of addition of the first monomer to the CTA. Then, the same authors reported that the induction period was rather related to the R-group stability or inactivation of residual oxygen or trace impurities within the reaction mixture before the polymerization.398 TTCs with better fragmenting R groups demonstrated significantly reduced induction periods for initiation, whereas during propagation the R-group effect became less important owing to the similar acrylate-inserted R groups of all TTCs. Junker and coworkers also discovered that in the polymerization of MMA under blue light irradiation (450 nm, 14.4 W), the adduct of CTA with one, two, or three MMAs resulted in polymers with lower dispersity.381 However, to eliminate the need for the preparation of SUMI adducts, the authors suggested to render the initiation process effective by increasing photon flux in a continuous flow reactor. To achieve fast reaction kinetics, the reaction temperature was also increased to 90 °C, which was sufficiently low not to cause the autoinitiation of MMA or degradation of TTC. As reported by Cameron, Saito, and coworkers, temperature control is particularly important at high irradiation powers.409 In the aqueous polymerization of acrylamides such as DMAm under irradiation (402 or 451 nm, and 6, 26, 104, or 208 W), high irradiation powers increased not only photon flux and photolysis, but also the reaction temperature, which considerably reduced the polymerization time from 12 hours to 11 minutes, while broadening the dispersity of polymers. Dispersity was narrowed by controlling the temperature to prevent hydrolytic degradation of the CTA and concomitant formation of low molecular weight dead chains.
| CTA (common name) | Irradiation wavelength | Monomer | |||
|---|---|---|---|---|---|
| Methacrylates | Acrylates | Acrylamides | Others | ||
| a Performed in a continuous flow reactor, at 90 °C. | |||||

|
Violet (405 nm) | PEGDA | |||
| Blue (455–472 nm) | BzMA | n-Bu, t-Bu, BzA, CHA, DA, EA, EHA, HEA, MA, AHFiP | St | ||
| Green (520, 530 nm) | n-BMA, BzMA, BTPEMA, DMAEMA, GMA, HEMA, MMA | ||||
|
Blue (450 nm)a | t-BMA, DEGMA, EMA, HEMA, MMA | |||
|
Blue (460 nm) | GMA, HEMA, OEGMA | |||
| Green (530 nm) | |||||
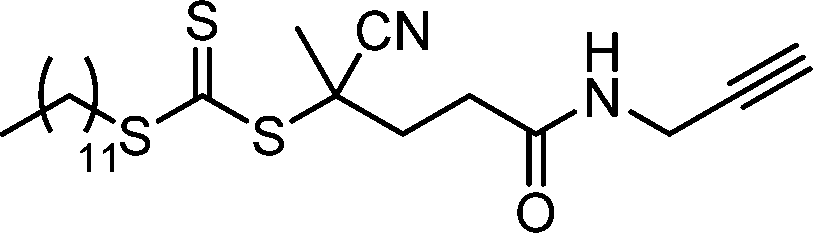
|
Blue (460 nm) | OEGA | |||
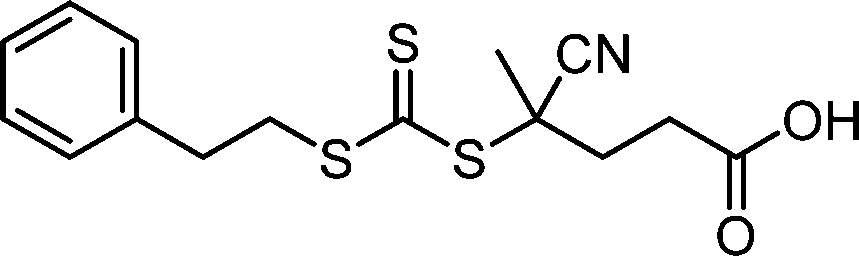
|
White | FATRIFE, FAHFiP, MAF-TBE | |||
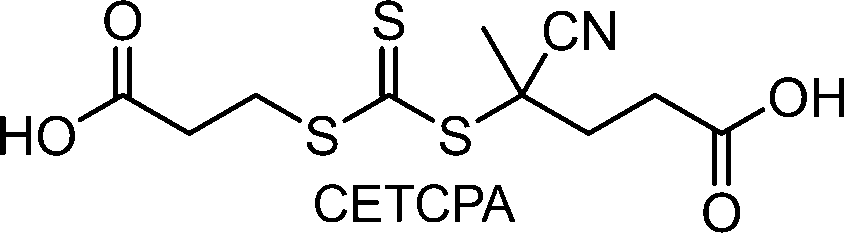
|
Blue (455 nm) | DMAPS, HEA, OEGA | |||
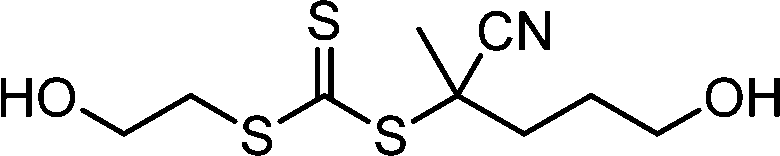
|
Blue (455 nm) | n-BA, MA | |||
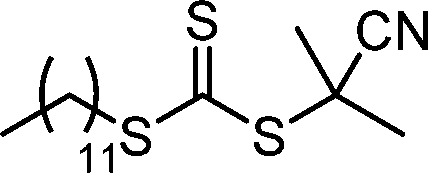
|
Blue (460, 470 nm) | MA | AN | ||
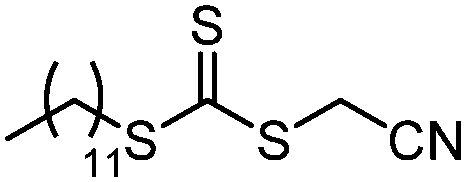
|
Blue (472 nm) | n-BA | |||
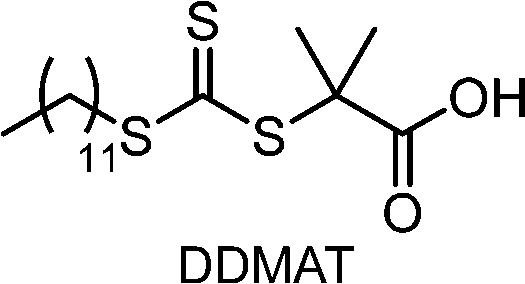
|
Violet (420 nm) | 2-VP | |||
| Blue (450–460 nm) | n-BA, MA | NAM | St | ||
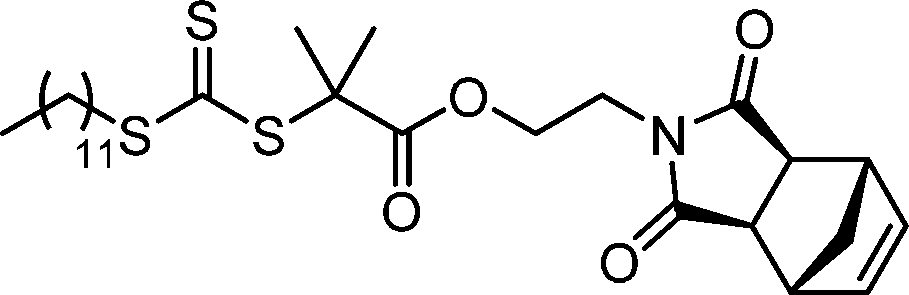
|
Blue (450 nm) | n-BA | |||
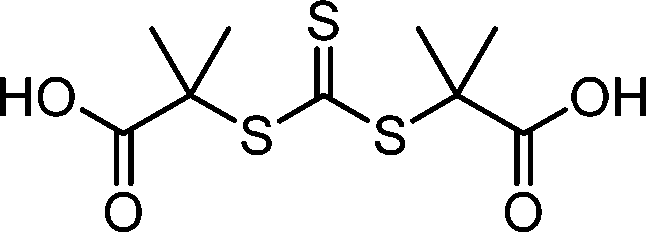
|
Violet (402 nm) | Am, DMAm | |||
| Blue (450, 451 nm) | MA | Am | |||
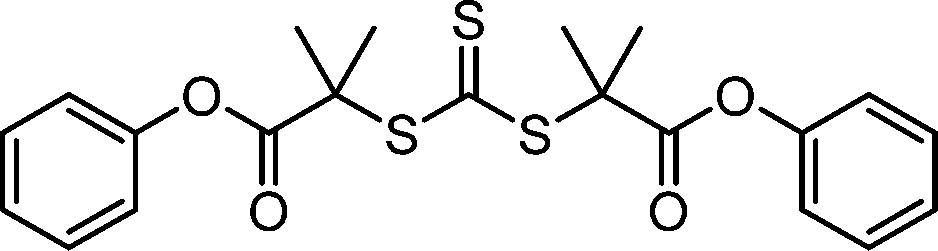
|
Blue (450 nm) | MMA | n-BA, t-BA, DEGA, MA | Am, DMAm, NIPAm | |
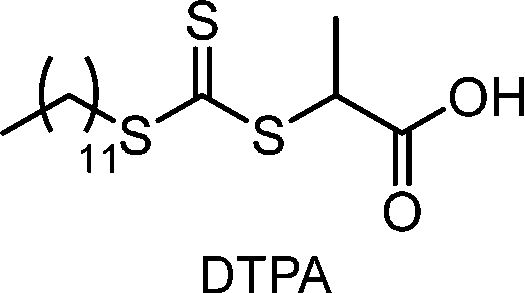
|
Blue (460 nm) | MA | |||
|
Blue (460, 480 nm) | MA | |||
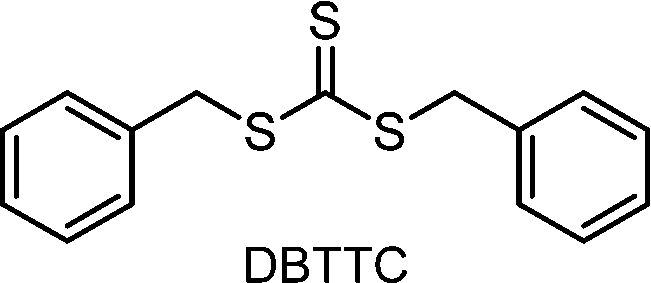
|
White | FATRIFE | |||
| Violet (405 nm) | OEGA, TEGDA | ||||
| Blue (460 nm) | n-BA, OEGA, TEGDA | ||||
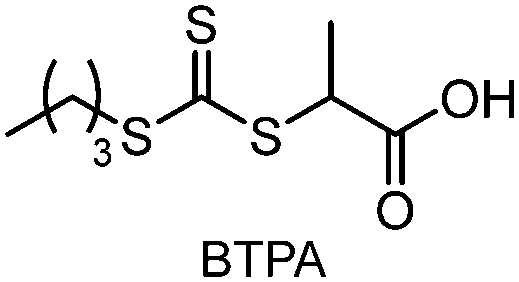
|
Blue (460 nm) | MA, t-BA, EGDA | NIPAm | ||
|
Green (520 nm) | MA | DMAm | ||
|
Blue (453 nm) | DMAm | |||
Very recently, Sumerlin and coworkers reported that irradiation with visible light to target n → π* electronic transitions led to more efficient and well-controlled polymerization than irradiation with UV light to target π → π* electronic transitions, if the light intensity was identical (0.6 mW cm−2).407 Due to the spin-forbidden nature, n → π* electronic transitions show a much smaller molar extinction coefficient than π → π* electronic transitions. Nevertheless, the polymerization results and the radical-trapping experiment using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) confirmed that n → π* electronic transitions led to the higher quantum yield of photolysis of the C–S bond, higher radical concentration, and more rapid polymerization. This result was consistent with xanthates.
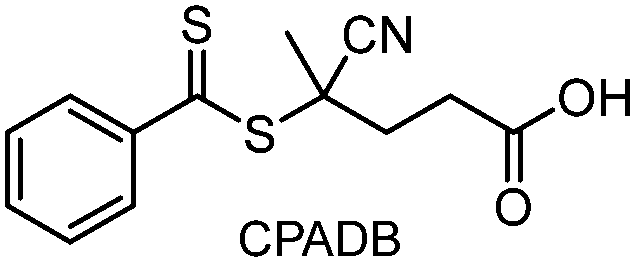
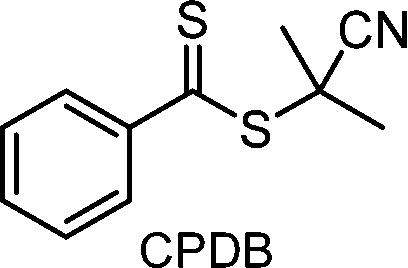
Meanwhile, Sumerlin and coworkers prepared a UHMW polymer via alternative copolymerization of styrene and benzyl maleimide or maleic anhydride under 450 nm irradiation.413 2-(dimethylamino)ethyl methacrylate (DMAEMA) also underwent polymerization under irradiation with a household lamp (14 W, 2.4 mW cm−2), but here the acceleration effect of the amine incorporated in DMAEMA on the polymerization mechanism needs to be considered. Electron transfer from the amine to excited-state CPADB generated the CPADB anion and facilitated the polymerization as in the case of amine-catalyzed polymerization using TTCs71,151,152 and DTB.72 By the addition of TEA, polymerization of MMA from CPDB even under green (520 nm, 5.9 mW cm−2) or sunlight irradiation was realized.72 Recently, Konkolewicz and coworkers reported that an electron-donating substituent at the para position of the benzyl Z group of CPDB dramatically accelerated the polymerization of MMA under blue light irradiation (440 nm, 11.6 mW cm−2) in the absence of an amine additive.87 The monomer conversion (85%) was comparable to the conversion in polymerization using 1 ppm of Ir(ppy)3 (i.e., PET-RAFT polymerization). The polymerization rate (kappp = 0.2 h−1) was much faster than that using CPDB (kappp = 0.017 h−1) despite the slight deviation in molecular weight control (Đ = 1.28) and end-group fidelity. The investigation of five CPDB derivatives confirmed the similar molar extinction coefficient at irradiation wavelength, but their Hammett Sigma parameters (σp) and E0reds were decreased by the presence of electron-donating substituents. The linear relationship between logarithm of kappp and σp or E0red and σp proposed that homolysis of the C–S bond (i) could be enhanced by electronics, (ii) might involve the formation of partial positive charge on the DTB group in the transition state, and thus (iii) the polymerization was accelerated by the electron-donating para methoxy substituent.
| CTA (common name) | Irradiation wavelengths | Monomer | |||
|---|---|---|---|---|---|
| Methacrylates | Acrylates | Acrylamides | Others | ||
|
Violet (401 nm) | DMAm | |||
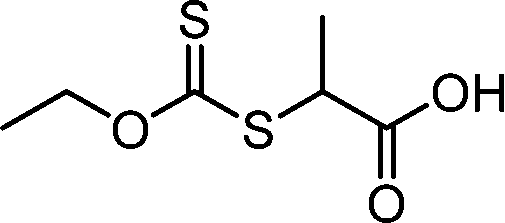
|
Blue (460–470 nm) | VAc | |||
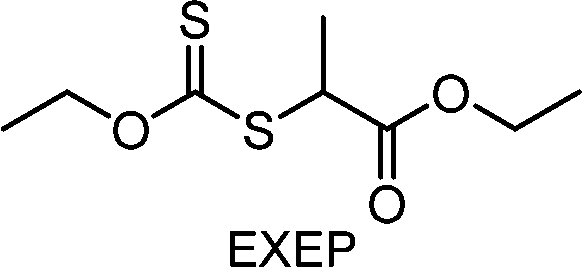
|
Blue (460–470 nm) | VAc | |||
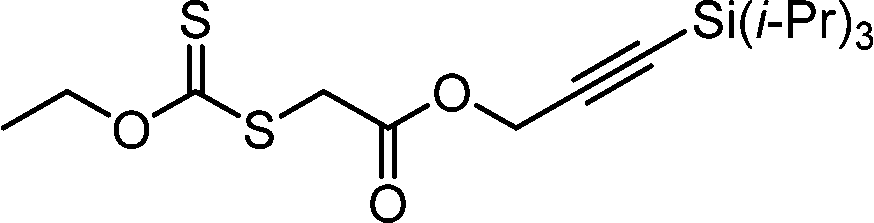
|
Blue (460–470 nm) | VAc | |||
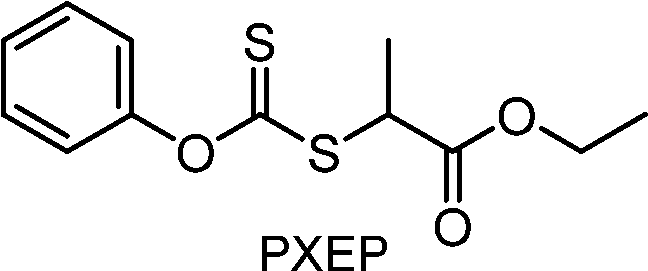
|
Violet (390 nm) | n-BA, t-BA | NIPAm | ||
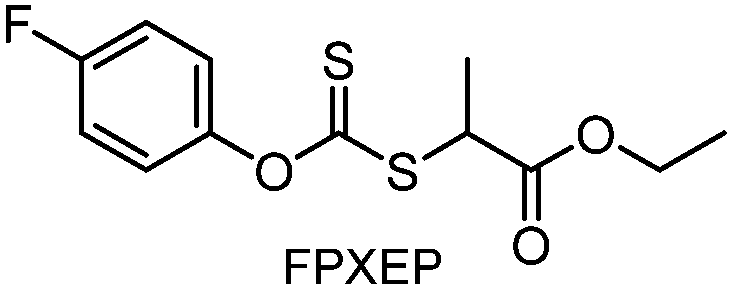
|
Violet (390 nm) | n-BA, MA | NIPAm | VAc | |
|
Violet (391 nm) | n-BA | |||
| Blue (465 nm) | |||||
| Violet (391 nm) | MA | St | |||
|
Violet (391 nm) | VAc | |||
| Blue (465 nm) | NVP | ||||
|
Violet (391 nm) | NVP, VAc | |||
| CTA (common name) | Irradiation wavelengths | Monomer | |||
|---|---|---|---|---|---|
| Methacrylates | Acrylates | Acrylamides | Others | ||
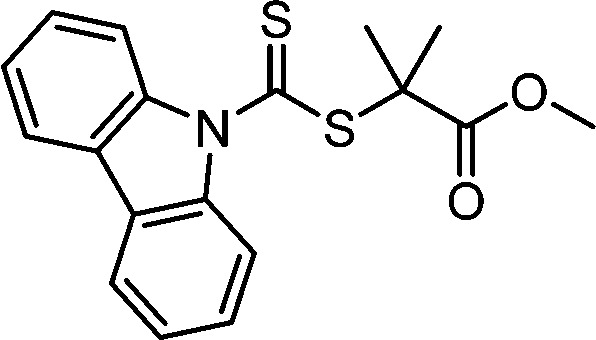
|
Blue (472 nm) | n-BA | DMAm | ||
| Green (525 nm) | |||||
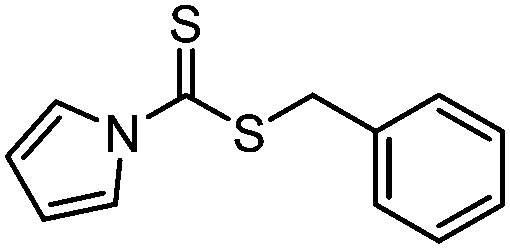
|
Blue (460 nm) | MA | |||
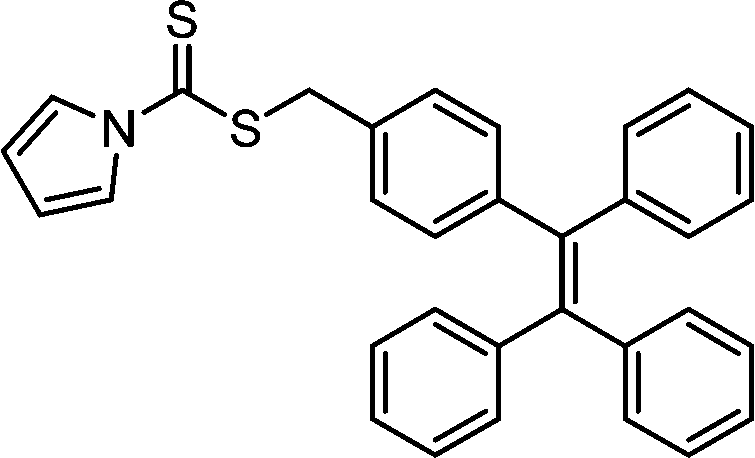
|
Blue (460 nm) | MMA | n-BA, MA, OEGA, isooctyl acrylate | St | |
5.2. Recent developments and applications
Visible-light-driven photoiniferter polymerization has recently been employed in numerous applications; nevertheless, the range of applications is still narrower than those reported for PET-RAFT polymerization owing to the lack of oxygen tolerance of the reaction system and the decreased polymerization rate caused by the absence of a PC. For example, photomediated expansion of the polymer network through incorporation of additional monomers was significantly efficient in the presence of PTH in terms of reaction rate and controllability of polymerization.208 Moreover, direct photolysis of TTC under visible-light irradiation, which was less efficient than EY/TEA-catalyzed PET-RAFT polymerization, was impractical for 3D printing application where a sufficiently fast photocuring (i.e., a high build speed) is required.134 In this section, the reported examples of applications that were realized solely by visible-light-driven photoiniferter polymerization are described. The other examples which were aided by PET-RAFT polymerization are mentioned in the relevant subsections throughout Sections 4.3 and 4.4. Herein, the applications driven by UV light are basically excluded, whereas several noteworthy examples which show implications for their expansion to visible-light-driven reaction systems are mentioned. For the applications under UV light irradiation, please refer to the review article by Hartlieb.56If different types of monomers have to be involved in each block, the monomer that would become a better leaving group needs to be polymerized first to favor fragmentation and chain extension with the second monomer during addition–fragmentation in the RAFT process. If the order is inverted, the second monomer with a better leaving group, instead of the first block, is likely to favor fragmentation and propagation, and will be homo-polymerized in the presence of an exogenous radical derived from external initiators. As a result, a mixture of homopolymers rather than the desired uniform block copolymers comprising two types of monomers is provided. Sumerlin and coworkers surmised that this restriction regarding the order of blocks in a copolymer would be resolved via photoiniferter polymerization.418 Photoiniferter polymerization that is mediated by (i) direct photolysis of the C–S bond, which does not require an exogenous radical initiator, and (ii) reversible termination rather than degenerative chain transfer is thus expected not to restrict the sequence of blocks in copolymers based on the difference in leaving group abilities. Initially, DMAm was polymerized as the first block from TTC by conventional RAFT polymerization or from xanthate by photoiniferter polymerization. As expected, subsequent photoiniferter polymerization of poly(DMAm)-xanthate under UV light irradiation (365 nm, 5.0 mW cm−2) successfully extended poly(DMAm)-xanthate with MMA, whereas thermally initiated polymerization provided a mixture of homopolymers of DMAm and MMA. Poly(DMAm)-TTC was also extended with MMA under blue light irradiation, but the rate of photolysis was slower. Using xanthate that was efficiently cleaved under UV light irradiation as a CTA, not only DMAm but also MA and N-vinylcarbazole were used as the first blocks and were extended by MMA.
Diblock copolymers of poly(acrylamide)s and polyethers were synthesized by combined photoiniferter polymerization under UV light irradiation (365 nm, 3.5 mW cm−2) and t-Bu-P2/triethylborane-catalyzed ROP at room temperature.122 As TTCs remained intact under the reaction conditions of ROP, either sequentially or simultaneously performed photoiniferter polymerization and ROP from the mono-hydroxyl-functionalized TTC provided the diblock copolymers. α,ω-Di-hydroxyl-functionalized TTC provided the α,ω-di-hydroxyl-functionalized polyacrylates that could serve as building blocks for polyurethane (PU).88 After the preparation of poly(n-BA) (Mn,GPC = 3100 g mol−1 and Đ = 1.03) under blue light irradiation (455 nm, 5 mW cm−2), thermoplastic PU elastomer was synthesized from the mixture of 5 mol% poly(n-BA) and 95 mol% poly(tetramethylene ether glycol) as a soft segment, isophorone diisocyanate as a hard segment, and ethylene glycol as a chain extender. The prepared elastomer surprisingly overcame the strength-elongation and robustness-self-healing ability trade-off relationships typically observed in elastomers. As various monomers can be incorporated as a part of soft segments to influence phase separation behavior and chain mobility within the polymer network, the development of thermoplastic PU elastomers with unprecedented properties was expected. In addition, via copper-catalyzed azide–alkyne reaction, polymers prepared from α-alkyne-functionalized TTC under blue light irradiation (460 nm, 4 mW cm−2) could be linked to bioactive peptides possessing the N-terminal azide functionality.408 Thus, high end-group fidelity under mild reaction conditions of visible-light-driven photoiniferter polymerization was highlighted.
In the absence of a PC, sequence-defined oligomers could be prepared via an iterative method rather than sequential SUMI (Fig. 23).419 Boyer and coworkers in collaboration with Moad and Hawker confirmed the high end-group fidelity of the monoadduct of MA and CDTPA and functionalized the TTC moiety. Aminolysis of TTC into thiol was followed by a thiol–ene reaction with a second monomer containing a hydroxy group (2-hydroxyethyl acrylate (2-HEA)). Thereafter, the hydroxy group underwent esterification with CDTPA to introduce a new CDTPA for another SUMI using DMAm as a third monomer. Finally, aminolysis, thiol–ene reaction with a fourth monomer (2-hydroxy butyl acrylate), and esterification with CDTPA were repeated to insert BzA as the last monomer to provide a sequence-defined pentamer. Although the monomer scope was expanded to acrylates and acrylamides, pre-functionalization of the carboxylic acid group of the starting CDTPA with a 3-trimethylsiyl protecting group to block its esterification with the lately introduced monomers with hydroxy groups and isolation of each intermediate can be cumbersome in this strategy.
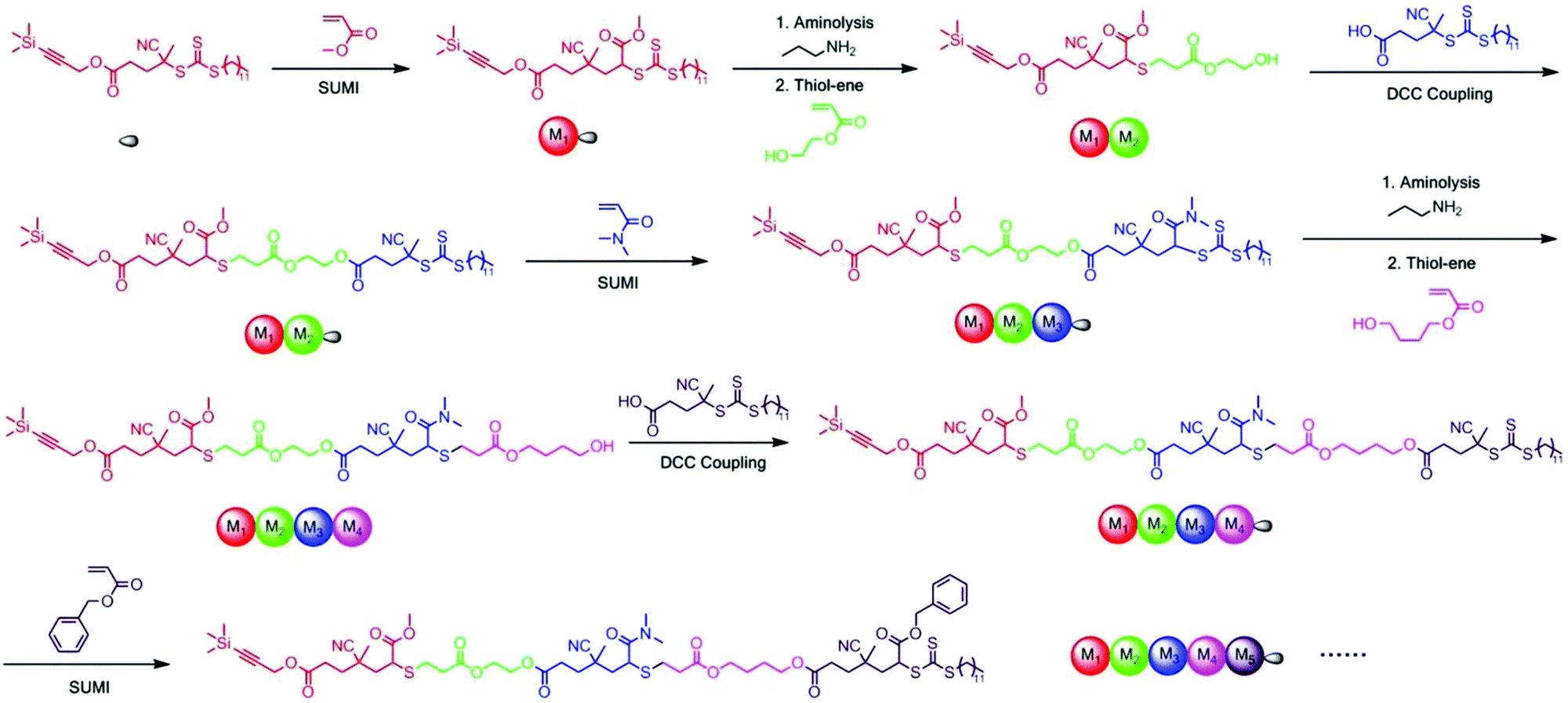 | ||
| Fig. 23 Schematic representation of the synthesis of a sequence-defined pentamer via iterative SUMI, aminolysis, thiol–ene, and esterification reactions. Adapted with permission from ref. 419 (Copyright 2017 The Royal Society of Chemistry). | ||
Sumerlin and coworkers expanded the monomer scope to monomers with relatively low kp such as (meth)acrylates and styrene in DMSO.413 In DMSO, monomer concentrations should be higher than those in water to achieve sufficiently high polymerization kinetics. In particular, synthesis of UHMW poly(MMA) from TTC under UV light irradiation (365 nm, 7.0 mW cm−2) required the addition of tertiary amine PMDETA. It was presumed that in the absence of PMDETA, the TTC sulfanyl radical abstracted the hydrogen atom from the propagating methacryloyl chain end, thereby affording a dead chain via disproportionation. The lower kp of styrene needed to be compensated by alternatively copolymerizing styrene with maleic anhydride or maleimide via charge-transfer complex formation. However, because of absorption and background initiation from the charge-transfer complex in the UV region, TTC was replaced with DTB to shift the irradiation wavelength from 365 nm to 450 nm.
 | ||
| Fig. 24 (a) 3D printed objects. (b) Welding two 3D printed balls by adding fresh TMPTA on the interface and subsequent irradiation. (c) SEM images of the 3D printed bridge before and after post-modification with fluorescein-conjugated TMPTA. The fluorescein-embedded layer was visualized by fluorescence lifetime imaging microscopy (FLIM). Adapted with permission from (a) ref. 403 (Copyright 2020 The Royal Society of Chemistry), (b) ref. 161 (Copyright 2022 American Chemical Society), and (c) ref. 424 (Copyright 2021 John Wiley & Sons, Inc.). | ||
Open-to-air 3D printing by the DLP technique under violet light irradiation (405 nm, 2.0 mW cm−2) was performed using EXEP.161 DFT calculations and ESR spin-trapping experiments confirmed that photolysis of EXEP under irradiation at 325–480 nm was more efficient than that of structurally similar TTC due to the lower bond dissociation energy of EXEP (38.06 kcal mol−1vs. 41.5 kcal mol−1). Although oxidation-induced loss of end-group fidelity of polymers was previously observed for photoiniferter polymerization using EXEP under blue light irradiation (460–470 nm),162 the end-group fidelity of polymers within 3D printed objects seemed to be preserved allowing post-functionalization with the freshly added PEGDA and trimethyloylpropane triacrylate (TMPTA) or welding, without an additional introduction of the photoinitiator or PC (Fig. 24b). When this photoiniferter polymerization was combined with cationic RAFT polymerization using diphenyliodonium hexafluorophosphate onium salt, welding of 3D printed objects was realized by either photoiniferter polymerization under violet light irradiation (405 nm, 60 mW cm−2) or ZnCl2-mediated conventional cationic RAFT polymerization without irradiation.423 Herein, polymerization (i.e., 3D printing) was initiated by photolysis of two types of CTAs which were expected to orthogonally proceed radical and cationic RAFT polymerization, which would allow tailoring of the mechanical properties of polymeric materials by adjusting the ratio of radically and cationically polymerizable monomers.
DTC bearing conjugated carbazole in the Z group was also applied to prepare 3D microstructures by the direct laser writing technique in the presence of air.424 Photoiniferter polymerization of n-BA under blue light irradiation (480 nm) followed by subsequent photoiniferter polymerization with TMPTA under green light irradiation (532 nm) provided mechanically stable 3D microstructures. Owing to the preserved DTC units, fabrication of multilayered microstructures comprised of multiple polymeric materials via repeated polymerization was successfully realized. Post-modification by polymerization with thermoresponsive NIPAm provided the printed object capable of heating-induced shrinkage and cooling-induced swelling, whereas polymerization with fluorescein-conjugated TMPTA allowed the visualization of the modified layer by fluorescence lifetime imaging microscopy (FLIM) (Fig. 24c).
6. Photomediated cationic RAFT polymerization
6.1. PCs for polymerization
A strongly oxidizing pyrylium salt was first employed for the photomediated cationic RAFT polymerization of vinyl ethers under blue light irradiation (450 nm) (Scheme 27).43 The excited-state pyrylium salt oxidized the vinyl ether-derived CTA to generate a carbocation intermediate and reversible generation of the carbocations enabled the regulation of cationic RAFT polymerization by light. iBVE and other vinyl ethers (i.e., ethyl, 2-chloroethyl, n-propyl, and n-butyl-vinyl ethers) were polymerized from DTC and TTC, respectively. In contrast to PET-RAFT polymerization, photomediated cationic RAFT polymerization required CTAs possessing vinyl ether as the R group. However, imperfect temporal control and monomer conversion in the absence of irradiation were observed in further studies, which was ascribed to the decomposition of the PC. Moreover, the experimental molecular weights of polymers were significantly lower than the theoretical value when the authors targeted to synthesize polymers with molecular weight above 20 kg mol−1.Other oxidizing PCs including the Ru complex and acridinium salt, however, were unsuccessful for the polymerization of iBVE from DTCs.90 Thereafter, Kamigaito, Ohkubo, and coworkers discovered that acridinium salts at doubled catalyst loadings to that of the previous study90 could catalyze the polymerization from TTC at −40 °C despite imperfect temporal control,91 where the polymerization rate was somehow affected by the type of counter-anions of acridinium salts which possessed the same structure and similar redox potentials. Although the polymerization of the chloroethyl vinyl monomer from DTC was uncontrolled because the high 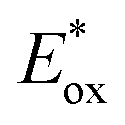 (2.06–2.25 V vs. SCE) oxidized the chloroethyl substituent of the monomer or the amino substituent of the CTA, acridinium salts extended the polymerization wavelength from blue to white and green.
(2.06–2.25 V vs. SCE) oxidized the chloroethyl substituent of the monomer or the amino substituent of the CTA, acridinium salts extended the polymerization wavelength from blue to white and green.
The imperfect temporal control (i.e., monomer conversion in the dark), observed particularly at high monomer conversion, that was noticed for both pyrylium and acridinium salts was ascribed to low stability of the PC or high 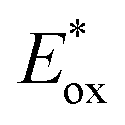 . Instead, Ir complexes with enhanced stability demonstrated no monomer conversion in the dark.425 Moreover, appropriate redox potentials of Ir complexes enabled the synthesis of high-molecular-weight polymers owing to the well-balanced rates of activation and deactivation steps, and the preferential oxidation of the CTA over that of vinyl ether monomers. Owing to more reducing E0red, Ir complex in contrast to pyrylium salt efficiently generated the thiolate anion that recaps the propagating cation intermediates and completely prevented the polymerization in the dark.426 Herein, a thioacetal instead of a dithioester was used as a CTA, but the mechanism was technically identical except that the sulfonium intermediate participated in degenerative chain transfer. The importance of an appropriate redox potential was also verified by Liao and coworkers in the case of bisphosphonium salt (
. Instead, Ir complexes with enhanced stability demonstrated no monomer conversion in the dark.425 Moreover, appropriate redox potentials of Ir complexes enabled the synthesis of high-molecular-weight polymers owing to the well-balanced rates of activation and deactivation steps, and the preferential oxidation of the CTA over that of vinyl ether monomers. Owing to more reducing E0red, Ir complex in contrast to pyrylium salt efficiently generated the thiolate anion that recaps the propagating cation intermediates and completely prevented the polymerization in the dark.426 Herein, a thioacetal instead of a dithioester was used as a CTA, but the mechanism was technically identical except that the sulfonium intermediate participated in degenerative chain transfer. The importance of an appropriate redox potential was also verified by Liao and coworkers in the case of bisphosphonium salt (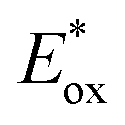 = 2.01–2.06 V vs. SCE).427 The bisphosphonium salt allowed the polymerization of a variety of vinyl ethers from TTC with excellent temporal control at low catalyst loadings (in the ppm range) under sunlight irradiation. Similarly, the monophosphonium salt synthesized by the same research group exhibited excellent temporal control.428 The relationship between the structure of the PC and polymerization activity was explored based on high modularity of the structure of the PC and the concomitantly widened redox window. The controllability of polymerization was dependent on the successful oxidation of the CTA by the excited-state PC, which was influenced by the redox potential, lifetime, and ΦFL of the excited-state PC.
= 2.01–2.06 V vs. SCE).427 The bisphosphonium salt allowed the polymerization of a variety of vinyl ethers from TTC with excellent temporal control at low catalyst loadings (in the ppm range) under sunlight irradiation. Similarly, the monophosphonium salt synthesized by the same research group exhibited excellent temporal control.428 The relationship between the structure of the PC and polymerization activity was explored based on high modularity of the structure of the PC and the concomitantly widened redox window. The controllability of polymerization was dependent on the successful oxidation of the CTA by the excited-state PC, which was influenced by the redox potential, lifetime, and ΦFL of the excited-state PC.
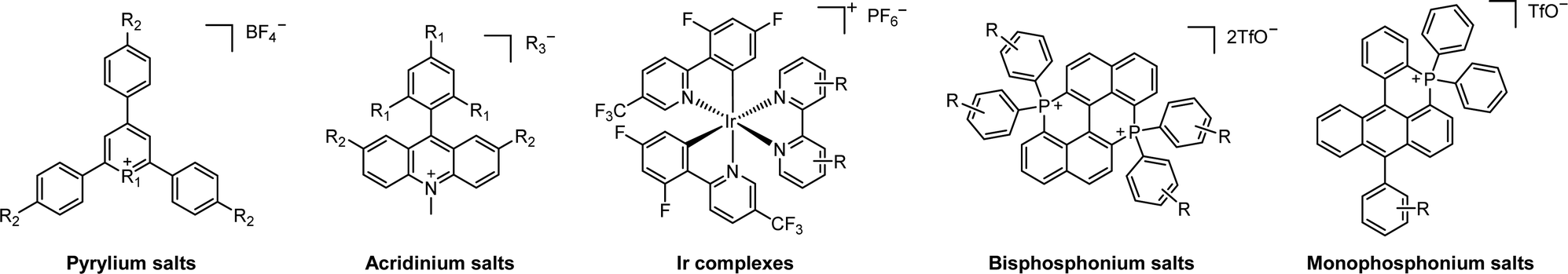 | ||
| Scheme 27 Chemical structures of PCs employed for photomediated cationic RAFT polymerization. | ||
On the other hand, cationic RAFT polymerization under NIR light irradiation (788 nm, 14 mW cm−2) was induced by Fe2(Cp)2(CO)4 in combination with alkyl bromide as an initiating system,429 where in situ-generated FeCp(CO2)Br upon irradiation oxidizes a carbon-centered radical to provide an initiating cation via a mechanic process similar to photoinduced radical oxidation/addition/deactivation (PROAD) that was previously reported by Yagci and coworkers.430 Zhu, Perrier, and coworkers employed Mn(CO)10/alkyl bromide431 or Mn(CO)5Br alone under blue light irradiation (440 nm, 10 mWcm−2).432 Mn(CO)5Br, a bench-stable PC with reduced sensitivity to oxygen compared to the Mn(CO)10/alkyl bromide system, for the first time facilitated the polymerization of iBVE without prior deoxygenation either in a batch or continuous flow reactor.
6.2. Applications
Selective activation of Ir(ppy)3-catalyzed PET-RAFT and pyrylium salt-catalyzed photomediated cationic RAFT polymerization of MA and iBVE, respectively, by simply changing the irradiation wavelength was applied in one-pot synthesis of a multiblock copolymer (Table 2, entry 1).124 According to the absorption profile of PCs, absorption of Ir(ppy)3 and subsequent PET-RAFT polymerization of MA under blue light (450 nm), and absorption of the pyrylium salt and subsequent photomediated cationic RAFT polymerization of iBVE under green light (520 nm) were expected. However, partially overlapping absorption profiles of two PCs also resulted in photomediated cationic RAFT polymerization at 450 nm and the problematic synthesis of tapered block copolymers. Fors and coworkers took advantage of the simultaneous PET-RAFT and photomediated cationic RAFT polymerizations to modulate the mechanical properties of thermoset polymers.126 Cross-linking of 1,4-butanediol divinyl ethers under green light irradiation (525 nm, 142 mW cm−2) generated the polymer network, whereas by switching to blue light (456 nm, 108 mW cm−2), butanediol diacrylates were also incorporated into the network and increased the cross-linking density and Young's modulus. Thus, the mechanical properties of the polymer network became easily adjustable by the amount of diacrylates, irradiation time and light intensity, and via spatially controlled PET-RAFT polymerization by applying a photomask. Nevertheless, photomediated cationic RAFT polymerization is still at an early stage for numerous applications owing to the narrow scope of monomers, PCs, and solvents and lack of oxygen tolerance in the reaction system. The recent development of PCs92 or use of continuous flow reactors382 combined with further studies would expand the utility of the polymerization.7. Summary and outlook
Among photocontrolled RAFT polymerization reactions, visible-light-driven RAFT polymerization which harnesses visible light or even sunlight has drawn attention in various fields including polymer chemistry, materials science, and bioengineering to name a few. As visible light is an environmentally benign and easily accessible energy source and visible-light photocatalysis provides oxygen tolerance that simplifies the reaction design by eliminating laborious deoxygenation, visible-light-driven RAFT polymerization is expected to be soon utilized at an industrial scale and face the new era in the near future. In particular, PET-RAFT polymerization with highly efficient oxygen tolerance has been successfully adapted for bioapplications, surface functionalization, 3D/4D printing, and HTP synthesis in readily accessible reactor system designs without prior degassing.In addition, polymers synthesized by RAFT polymerization have recently been subjected to depolymerization, drawing attention to a sustainable future of polymer chemistry. Anastasaki and coworkers reported radical-mediated, catalyst-free, and near-quantitative depolymerization of various methacrylic polymers.433 Thermally induced homolytic cleavage of the C–S bond at 120 °C generated chain-end radicals and released monomers from the end of the polymer. Depolymerization of the polymer became favored over re-insertion of the released monomers under highly dilute conditions (5 mM relative to the monomer; 200 times lower than that of typical polymerization conditions). Again, via visible-light-driven photocatalysis both in the presence434 and absence of a PC,435 faster rates and higher conversions of depolymerization were achieved despite the temperature being lowered to 100 °C. The improved reaction efficiencies were attributed to more efficient generation of chain-end radicals via the combined thermally and photo-induced homolytic cleavage of the C–S bond. In all cases, high end-group fidelity of polymers provided by RAFT polymerization was essential for efficient depolymerization. These initial demonstrations of depolymerization are expected to be applied for a wider scope of polymers (i.e., prepared from non-methacrylic monomers and various CTAs) under milder reaction conditions. Combined with the synthesis of degradable vinyl polymers via copolymerization with macrocyclic allylic sulfones,100,185 radical-mediated depolymerization would provide further opportunities for polymer chemistry.
However, there is still room for improvement. First, although the interest in comprehensively elucidating mechanistic backgrounds using computational chemistry and kinetic modeling has increased, it is currently limited to only a few types of PCs and/or CTAs. Only when the discrepancy in each combination of PC and/or CTA is considered, a fully optimized reaction system for the intended applications may be designed.436 Second, compared to PET-RAFT polymerization, the applications of photoiniferter and photomediated cationic RAFT polymerizations are still limited by the lack of oxygen tolerance and a rather narrow range of operational irradiation wavelengths and solvents for the polymerization reactions. Regardless of the polymerization method, a fundamental understanding of the mechanisms of the photomediated process (e.g., the involvement of singlet or triplet excited states, the transfer of electron or energy, and the fate of the reaction intermediates) would improve the reaction system. Third, although the introduction of flow chemistry has enhanced the scalability of visible-light-driven RAFT polymerizations, the design of a reactor that could provide an accurate control over the light intensity (i.e., photon flux) and reaction temperature is necessary for consistency of polymer properties. This influence of the reactor geometry and volume on PET-RAFT polymerization was also reported for a cylindrical batch system.437
In summary, we have provided herein a brief history, progress, and future challenges of photocontrolled RAFT polymerization. Recent studies on degradation and thermally or photo-induced depolymerization of polymers synthesized via RAFT polymerization, which potentially provide closed-loop recycling of the polymers synthesized by RAFT polymerization, have also been mentioned. Therefore, photocontrolled RAFT polymerization holds significant promise in polymer chemistry ranging from an environmentally friendly synthesis to a green future of polymers such as commercial vinyl polymers including plastics.438 We anticipate that this review will provide guidance on how and to what extent the photocontrolled RAFT polymerization could be utilized, and attract researchers from various fields to achieve the full potential of the method, which pose a greater impact on our society in line with the ever-growing need for environmental compatibility.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work at SNU was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (2022R1A2C2011627 and 2021R1A5A1030054) and the Challengeable Future Defense Technology Research and Development Program (No. 912909601) of Agency for Defense Development in 2022. Professor Cyrille Boyer (or C.B.) is the recipient of an Australian Research Council Australian Laureate Fellowship (FL220100016) funded by the Australian government.References
- L. J. Malone and T. Dolter, Basic Concepts of Chemistry, John Wiley & Sons, Inc., 2008 Search PubMed.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- B. König, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- T. Rigotti and J. Alemán, Chem. Commun., 2020, 56, 11169–11190 RSC.
- V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- J. Chiefari, Y. K. (Bill) Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- S. Dadashi-Silab, M. Atilla Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878–2888 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- R. Ran, Y. Yu and T. Wan, J. Appl. Polym. Sci., 2007, 105, 398–404 CrossRef CAS.
- H. Wang, Q. Li, J. Dai, F. Du, H. Zheng and R. Bai, Macromolecules, 2013, 46, 2576–2582 CrossRef CAS.
- Y. Kwak and K. Matyjaszewski, Macromolecules, 2010, 43, 5180–5183 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, Controlled Radical Polymerization: Mechanisms, American Chemical Society, Washington, DC, 2015, vol. 1187, pp. 247–267 Search PubMed.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- C. Aydogan, G. Yilmaz, A. Shegiwal, D. M. Haddleton and Y. Yagci, Angew. Chem., Int. Ed., 2022, 61, e202117377 CrossRef CAS PubMed.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- T. Otsu and M. Yoshida, Macromol. Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- T. Otsu, M. Yoshida and T. Tazaki, Macromol. Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS PubMed.
- S. Sugihara, N. Konegawa and Y. Maeda, Macromolecules, 2015, 48, 5120–5131 CrossRef CAS.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 15535–15538 CrossRef CAS PubMed.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS PubMed.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 1830–1844 RSC.
- V. Bellotti and R. Simonutti, Polymers, 2021, 13, 1119 CrossRef CAS PubMed.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef PubMed.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed.
- C. A. Figg, J. D. Hickman, G. M. Scheutz, S. Shanmugam, R. N. Carmean, B. S. Tucker, C. Boyer and B. S. Sumerlin, Macromolecules, 2018, 51, 1370–1376 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- Q. Wang, F.-Y. Bai, Y. Wang, F. Niu, Y. Zhang, Q. Mi, K. Hu and X. Pan, J. Am. Chem. Soc., 2022, 144, 19942–19952 CrossRef CAS PubMed.
- E. E. Stache, V. Kottisch and B. P. Fors, J. Am. Chem. Soc., 2020, 142, 4581–4585 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- L. T. Strover, A. Cantalice, J. Y. L. Lam, A. Postma, O. E. Hutt, M. D. Horne and G. Moad, ACS Macro Lett., 2019, 8, 1316–1322 CrossRef CAS PubMed.
- F. Izadi, E. Arthur-Baidoo, L. T. Strover, L.-J. Yu, M. L. Coote, G. Moad and S. Denifl, Angew. Chem., Int. Ed., 2021, 60, 19128–19132 CrossRef CAS PubMed.
- Z. Wu, K. Jung, C. Wu, G. Ng, L. Wang, J. Liu and C. Boyer, J. Am. Chem. Soc., 2022, 144, 995–1005 CrossRef CAS PubMed.
- C. Yu, J. Song, T. I. Kim, Y. Lee, Y. Kwon, J. Kim, J. Park, J. Choi, J. Doh, S. K. Min, S. Cho and M. S. Kwon, ACS Catal., 2023, 13, 665–680 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O’Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537–545 CrossRef CAS PubMed.
- B. S. Tucker, M. L. Coughlin, C. A. Figg and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 452–457 CrossRef CAS PubMed.
- H. Sun, W. Choi, N. Zang, C. Battistella, M. P. Thompson, W. Cao, X. Zhou, C. Forman and N. C. Gianneschi, Angew. Chem., Int. Ed., 2019, 58, 17359–17364 CrossRef CAS PubMed.
- C. Fu, B. Demir, S. Alcantara, V. Kumar, F. Han, H. G. Kelly, X. Tan, Y. Yu, W. Xu, J. Zhao, C. Zhang, H. Peng, C. Boyer, T. M. Woodruff, S. J. Kent, D. J. Searles and A. K. Whittaker, Angew. Chem., Int. Ed., 2020, 59, 4729–4735 CrossRef CAS PubMed.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC.
- M. L. Allegrezza, Z. M. DeMartini, A. J. Kloster, Z. A. Digby and D. Konkolewicz, Polym. Chem., 2016, 7, 6626–6636 RSC.
- B. Nomeir, O. Fabre and K. Ferji, Macromolecules, 2019, 52, 6898–6903 CrossRef CAS.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- J.-H. Back, Y. Kwon, H. Cho, H. Lee, D. Ahn, H.-J. Kim, Y. Yu, Y. Kim, W. Lee and M. S. Kwon, Adv. Mater., 2022, 2204776 CrossRef PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- S. S. Skourtis, C. Liu, P. Antoniou, A. M. Virshup and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8115–8120 CrossRef CAS PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- P. Seal, J. Xu, S. Luca, C. Boyer, S. C. Smith, S. De Luca, C. Boyer and S. C. Smith, Adv. Theory Simulations, 2019, 2, 1900038 CrossRef.
- M. D. Thum, S. Wolf and D. E. Falvey, J. Phys. Chem. A, 2020, 124, 4211–4222 CrossRef CAS PubMed.
- J. Christmann, A. Ibrahim, V. Charlot, C. Croutxé-Barghorn, C. Ley and X. Allonas, ChemPhysChem., 2016, 17, 2309–2314 CrossRef CAS PubMed.
- N. Corrigan, J. Xu, C. Boyer and X. Allonas, ChemPhotoChem, 2019, 3, 1193–1199 CrossRef CAS.
- L. R. Kuhn, M. L. Allegrezza, N. J. Dougher and D. Konkolewicz, J. Polym. Sci., 2020, 58, 139–144 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- M. L. Allegrezza, N. De Alwis Watuthanthrige, Y. Wang, G. A. Garcia, H. Ren and D. Konkolewicz, Polym. Chem., 2020, 11, 6129–6133 RSC.
- C. Yu, J.-K. Ha, J. Ahn, J. Lee, J. Choi, T. Chang, S. K. Min and M. S. Kwon, Chemrxiv, 2023 DOI:10.26434/chemrxiv-2023-dl842.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- Q. Michaudel, T. Chauviré, V. Kottisch, M. J. Supej, K. J. Stawiasz, L. Shen, W. R. Zipfel, H. D. Abruña, J. H. Freed and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 15530–15538 CrossRef CAS PubMed.
- M. Matsuda, M. Uchiyama, Y. Itabashi, K. Ohkubo and M. Kamigaito, Polym. Chem., 2022, 13, 1031–1039 RSC.
- R. J. Sifri, Y. Ma and B. P. Fors, Acc. Chem. Res., 2022, 55, 1960–1971 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2017, 50, 1832–1846 CrossRef CAS.
- M. Chen, S. Deng, Y. Gu, J. Lin, M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2017, 139, 2257–2266 CrossRef CAS PubMed.
- Y. Huang, X. R. Zhang, S. Ye, J. Le Li, X. Li and T. Cai, Nanoscale, 2019, 11, 13502–13510 RSC.
- C. Wu, H. Chen, N. Corrigan, K. Jung, X. Kan, Z. Li, W. Liu, J. Xu and C. Boyer, J. Am. Chem. Soc., 2019, 141, 8207–8220 CrossRef CAS PubMed.
- X. Li, Y. C. Zhang, Y. Zhao, H. P. Zhao, B. Zhang and T. Cai, Macromolecules, 2020, 53, 1550–1556 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Polym. Chem., 2016, 7, 6437–6449 RSC.
- L. Zhang, C. Wu, K. Jung, Y. H. Ng and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 16811–16814 CrossRef CAS PubMed.
- W. Wang, Z. Zhou, D. Sathe, X. Tang, S. Moran, J. Jin, F. Haeffner, J. Wang and J. Niu, Angew. Chem., Int. Ed., 2022, 61, e202113302 CAS.
- X. Li, S. Ye, Y. C. Zhang, H. P. Zhao, Y. Huang, B. Zhang and T. Cai, Nanoscale, 2020, 12, 7595–7603 RSC.
- X. Li, J. Le Li, W. G. Huang, X.-Z. Zhang, B. Zhang and T. Cai, Nanoscale, 2018, 10, 19254–19261 RSC.
- L. Hu, Q. Wang, X. Zhang, H. Zhao, Z. Cui, P. Fu, M. Liu, N. Liu, S. He, X. Pang and X. Qiao, RSC Adv., 2020, 10, 6850–6857 RSC.
- C. A. Cox, A. N. Ogorek, J. P. Habumugisha and J. D. Martell, J. Am. Chem. Soc., 2023, 145, 1818–1825 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- J. Xu, S. Shanmugam, C. Fu, K. F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef CAS PubMed.
- S. Houshyar, D. J. Keddie, G. Moad, R. J. Mulder, S. Saubern and J. Tsanaktsidis, Polym. Chem., 2012, 3, 1879–1889 RSC.
- J. Vandenbergh, G. Reekmans, P. Adriaensens and T. Junkers, Chem. Commun., 2013, 49, 10358–10360 RSC.
- J. Xu, Macromolecules, 2019, 52, 9068–9093 CrossRef CAS.
- S. Shanmugam, J. Cuthbert, T. Kowalewski, C. Boyer and K. Matyjaszewski, Macromolecules, 2018, 51, 7776–7784 CrossRef CAS.
- C. Wu, K. Jung, Y. Ma, W. Liu and C. Boyer, Nat. Commun., 2021, 12, 478 CrossRef CAS PubMed.
- N. Corrigan, M. Ciftci, K. Jung and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 1748–1781 CrossRef CAS PubMed.
- P. Lu, D. Ahn, R. Yunis, L. Delafresnaye, N. Corrigan, C. Boyer, C. Barner-Kowollik and Z. A. Page, Matter, 2021, 4, 2172–2229 CrossRef CAS.
- N. Corrigan and C. Boyer, ACS Macro Lett., 2019, 8, 812–818 CrossRef CAS PubMed.
- Z. Zhang, T.-Y. Zeng, L. Xia, C.-Y. Hong, D.-C. Wu and Y.-Z. You, Nat. Commun., 2018, 9, 2577 CrossRef PubMed.
- M. J. Supej, B. M. Peterson and B. P. Fors, Chem, 2020, 6, 1794–1803 CAS.
- A. Nikolaev, Z. Lu, A. Chakraborty, L. Sepunaru and J. Read de Alaniz, J. Am. Chem. Soc., 2021, 143, 12278–12285 CrossRef CAS PubMed.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228–1234 CrossRef CAS PubMed.
- L. Xia, Z. Zhang and Y.-Z. You, Polym. J., 2020, 52, 1323–1331 CrossRef CAS.
- K. Satoh, Z. Sun, M. Uchiyama, M. Kamigaito, J. Xu and C. Boyer, Polym. J., 2020, 52, 65–73 CrossRef CAS.
- Y. Xia, G. M. Scheutz, C. P. Easterling, J. Zhao and B. S. Sumerlin, Angew. Chem., Int. Ed., 2021, 60, 18537–18541 CrossRef CAS PubMed.
- C. Fu, J. Xu, M. Kokotovic and C. Boyer, ACS Macro Lett., 2016, 5, 444–449 CrossRef CAS PubMed.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 10665–10668 CrossRef CAS PubMed.
- J. C. Theriot, G. M. Miyake and C. A. Boyer, ACS Macro Lett., 2018, 7, 662–666 CrossRef CAS PubMed.
- Y. Ma, V. Kottisch, E. A. McLoughlin, Z. W. Rouse, M. J. Supej, S. P. Baker and B. P. Fors, J. Am. Chem. Soc., 2021, 143, 21200–21205 CrossRef CAS PubMed.
- M. Fromel, E. M. Benetti and C. W. Pester, ACS Macro Lett., 2022, 415–421 CrossRef CAS PubMed.
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS.
- I.-H. Lee, E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Polym. Chem., 2017, 8, 3351–3356 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2016, 49, 9345–9357 CrossRef CAS.
- H. Lu, Y. Huang, E. Zhang, Y. Liu, F. Lv, L. Liu, Y. Ma and S. Wang, ACS Macro Lett., 2021, 10, 996–1001 CrossRef CAS PubMed.
- M. Fromel, D. M. Sweeder, S. Jang, T. A. Williams, S. H. Kim and C. W. Pester, ACS Appl. Polym. Mater., 2021, 3, 5291–5301 CrossRef CAS.
- A. Bagheri, C. W. A. Bainbridge, K. E. Engel, G. G. Qiao, J. Xu, C. Boyer and J. Jin, ACS Appl. Polym. Mater., 2020, 2, 782–790 CrossRef CAS.
- N. Zaquen, W. A. A. W. Azizi, J. Yeow, R. P. Kuchel, T. Junkers, P. B. Zetterlund and C. Boyer, Polym. Chem., 2019, 10, 2406–2414 RSC.
- N. Zaquen, A. M. N. B. P. H. A. Kadir, A. Iasa, N. Corrigan, T. Junkers, P. B. Zetterlund and C. Boyer, Macromolecules, 2019, 52, 1609–1619 CrossRef CAS.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- C. W. A. Bainbridge, K. E. Engel and J. Jin, Polym. Chem., 2020, 11, 4084–4093 RSC.
- A. R. Kuzmyn, L. W. Teunissen, P. Fritz, B. van Lagen, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 9, 2101784 CrossRef CAS.
- M. Kovaliov, M. L. Allegrezza, B. Richter, D. Konkolewicz and S. Averick, Polymer, 2018, 137, 338–345 CrossRef CAS.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- Q. Fu, Q. Ruan, T. G. McKenzie, A. Reyhani, J. Tang and G. G. Qiao, Macromolecules, 2017, 50, 7509–7516 CrossRef CAS.
- E. Paola Fonseca Parra, B. Chouchene, J.-L. Six, R. Schneider and K. Ferji, ACS Appl. Polym. Mater., 2021, 3, 3649–3658 CrossRef.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS PubMed.
- S. Xu, G. Ng, J. Xu, R. P. Kuchel, J. Yeow and C. Boyer, ACS Macro Lett., 2017, 6, 1237–1244 CrossRef CAS PubMed.
- Y. Huang, W. L. Guo, J. C. He, X. Li and T. Cai, Macromol. Rapid Commun., 2022, 43, 2200020 CrossRef CAS PubMed.
- J. Niu, Z. A. Page, N. D. Dolinski, A. Anastasaki, A. T. Hsueh, H. T. Soh and C. J. Hawker, ACS Macro Lett., 2017, 6, 1109–1113 CrossRef CAS PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- C. Stubbs, T. Congdon, J. Davis, D. Lester, S.-J. Richards and M. I. Gibson, Macromolecules, 2019, 52, 7603–7612 CrossRef CAS PubMed.
- Y. Huang, Y. Sun, Y. Weng and W. Zhang, ChemistrySelect, 2022, 7, e202201583 CAS.
- J. Wang, M. Rivero, A. Muñoz Bonilla, J. Sanchez-Marcos, W. Xue, G. Chen, W. Zhang and X. Zhu, ACS Macro Lett., 2016, 5, 1278–1282 CrossRef CAS PubMed.
- Y. Zheng, Y. Luo, K. Feng, W. Zhang and G. Chen, ACS Macro Lett., 2019, 8, 326–330 CrossRef CAS PubMed.
- Y. Zhou, C. Gu, L. Zheng, F. Shan and G. Chen, Polym. Chem., 2022, 13, 989–996 RSC.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- C.-Y. Li and S.-S. Yu, Macromolecules, 2021, 54, 9825–9836 CrossRef CAS.
- J. Li, C. Ding, Z. Zhang, X. Pan, N. Li, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2017, 38, 1600482 CrossRef PubMed.
- K. M. Burridge, N. De Alwis Watuthanthrige, C. Payne, R. C. Page and D. Konkolewicz, J. Polym. Sci., 2021, 59, 2530–2536 CrossRef CAS.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620–1628 CrossRef CAS.
- C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2015, 36, 2181–2185 CrossRef CAS PubMed.
- S. E. Seo, E. H. Discekici, Y. Zhang, C. M. Bates and C. J. Hawker, J. Polym. Sci., 2020, 58, 70–76 CrossRef CAS.
- R. Li and Z. An, Angew. Chem., Int. Ed., 2020, 59, 22258–22264 CrossRef CAS PubMed.
- F. Zhou, R. Li, X. Wang, S. Du and Z. An, Angew. Chem., Int. Ed., 2019, 58, 9479–9484 CrossRef CAS PubMed.
- J. Lalevée, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2003–2010 CrossRef.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- D. Ahn, L. M. Stevens, K. Zhou and Z. A. Page, ACS Cent. Sci., 2020, 6, 1555–1563 CrossRef CAS PubMed.
- D. Ahn, L. M. Stevens, K. Zhou and Z. A. Page, Adv. Mater., 2021, 33, 2104906 CrossRef CAS PubMed.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- L. Yu, Y. Wei, Y. Tu, S. Lin, Z. Huang, J. Hu, Y. Chen, H. Qiao and W. Zou, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2437–2444 CrossRef CAS.
- H. Yang, Z. Lu, X. Fu, Q. Li, Y. Zhao, L. Xiao and L. Hou, Polym. Chem., 2022, 13, 4776–4781 RSC.
- Z. Wu, W. Fang, C. Wu, N. Corrigan, T. Zhang, S. Xu and C. Boyer, Chem. Sci., 2022, 13, 11519–11532 RSC.
- J. Sun, S. Ren, H. Zhao, S. Zhang, X. Xu, L. Zhang and Z. Cheng, ACS Macro Lett., 2023, 12, 165–171 CrossRef CAS PubMed.
- H. Yang, Z. Lu, X. Fu, Q. Li, L. Xiao, R. Zhao, Y. Zhao and L. Hou, Polym. Chem., 2021, 12, 6998–7004 RSC.
- Y. Lee, Y. Kwon, Y. Kim, C. Yu, S. Feng, J. Park, J. Doh, R. Wannemacher, B. Koo, J. Gierschner and M. S. Kwon, Adv. Mater., 2022, 34, 2108446 CrossRef CAS PubMed.
- C. Lv, C. He and X. Pan, Angew. Chem., Int. Ed., 2018, 57, 9430–9433 CrossRef CAS PubMed.
- O. R. Wilson and A. J. D. Magenau, ACS Macro Lett., 2018, 7, 370–375 CrossRef CAS PubMed.
- Y. Peng, S. Liu, L. Wang, Y. Xu, Z. Wu and H. Chen, Macromol. Rapid Commun., 2022, 43, 2100920 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, Science, 2016, 352, 1053–1054 CrossRef CAS PubMed.
- J. T. Trotta and B. P. Fors, Synlett, 2016, 702–713 CAS.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 CrossRef CAS PubMed.
- Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 6028–6043 CrossRef CAS.
- W. Wang, B. Rondon, Z. Wang, J. Wang and J. Niu, Macromolecules, 2023, 56, 2052–2061 CrossRef CAS.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS.
- J. A. Reeves, N. De Alwis Watuthanthrige, C. Boyer and D. Konkolewicz, ChemPhotoChem, 2019, 3, 1171–1179 CrossRef CAS.
- P. Yang, P. Pageni, M. P. Kabir, T. Zhu and C. Tang, ACS Macro Lett., 2016, 5, 1293–1300 CrossRef CAS PubMed.
- G. Li, W. Feng, N. Corrigan, C. Boyer, X. Wang and J. Xu, Polym. Chem., 2018, 9, 2733–2745 RSC.
- H. Gong, Y. Gu, Y. Zhao, Q. Quan, S. Han and M. Chen, Angew. Chem., Int. Ed., 2020, 59, 919–927 CrossRef CAS PubMed.
- Q. Yang, V. Ladmiral and B. Améduri, ChemPhotoChem, 2019, 3, 1095–1099 CrossRef CAS.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS PubMed.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- C. Wu, S. Shanmugam, J. Xu, J. Zhu and C. Boyer, Chem. Commun., 2017, 53, 12560–12563 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- Z. Yajun, Z. Shuaishuai, L. Xiaojing, Z. Xiaoyu, X. Jing, X. Bijin, W. Yong, Z. Xingping and X. Xiaolin, CCS Chem., 2021, 4, 122–131 Search PubMed.
- J. Xu, S. Shanmugam and C. Boyer, ACS Macro Lett., 2015, 4, 926–932 CrossRef CAS PubMed.
- Y. Zhou, Z. Zhang, C. M. Reese, D. L. Patton, J. Xu, C. Boyer, A. Postma and G. Moad, Macromol. Rapid Commun., 2020, 41, 1900478 CrossRef CAS PubMed.
- S. Xu, N. Corrigan and C. Boyer, Polym. Chem., 2021, 12, 57–68 RSC.
- S. Xu, T. Zhang, R. P. Kuchel, J. Yeow and C. Boyer, Macromol. Rapid Commun., 2020, 41, 1900493 CrossRef CAS PubMed.
- L. Shen, Q. Lu, A. Zhu, X. Lv and Z. An, ACS Macro Lett., 2017, 6, 625–631 CrossRef CAS PubMed.
- W. Wang, S. Zhong, G. Wang, H. Cao, Y. Gao and W. Zhang, Polym. Chem., 2020, 11, 3188–3194 RSC.
- N. Corrigan, L. Zhernakov, M. H. Hashim, J. Xu and C. Boyer, React. Chem. Eng., 2019, 4, 1216–1228 RSC.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read De Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS PubMed.
- M. W. Lampley and E. Harth, ACS Macro Lett., 2018, 7, 745–750 CrossRef CAS PubMed.
- H. Gong, Y. Zhao, X. Shen, J. Lin and M. Chen, Angew. Chem., Int. Ed., 2018, 57, 333–337 CrossRef CAS PubMed.
- Q. Quan, H. Wen, S. Han, Z. Wang, Z. Shao and M. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 24319–24327 CrossRef CAS PubMed.
- Y. Zhao, M. Ma, X. Lin and M. Chen, Angew. Chem., Int. Ed., 2020, 59, 21470–21474 CrossRef CAS PubMed.
- K. Jiang, S. Han, M. Ma, L. Zhang, Y. Zhao and M. Chen, J. Am. Chem. Soc., 2020, 142, 7108–7115 CrossRef CAS PubMed.
- Q. Quan, Y. Zhao, K. Chen, H. Zhou, C. Zhou and M. Chen, ACS Catal., 2022, 12, 7269–7277 CrossRef CAS.
- C. Wu, N. Corrigan, C.-H. Lim, K. Jung, J. Zhu, G. Miyake, J. Xu and C. Boyer, Macromolecules, 2019, 52, 236–248 CrossRef CAS PubMed.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner, L. Lüer and M. S. Kwon, Macromolecules, 2019, 52, 5538–5545 CrossRef CAS.
- B.-H. He, M. Lu, C.-X. Yang, Y. Liu, E. Liang and G.-X. Wang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2018, 55, 583–587 CrossRef CAS.
- W. Zhang, X. Zhang, Y. Ma, D. Chen and W. Yang, Macromol. Chem. Phys., 2019, 220, 1900022 CrossRef.
- Q. Yang, X. Zhang, Y. Ma, D. Chen and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2072–2079 CrossRef CAS.
- Q. Yang, X. Zhang, W. Ma, Y. Ma, D. Chen, L. Wang, C. Zhao and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 229–236 CrossRef CAS.
- H. Tao, L. Xia, G. Chen, T. Zeng, X. Nie, Z. Zhang and Y. You, Polymers, 2019, 11, 892 CrossRef CAS PubMed.
- H. Cao, G. Wang, Y. Xue, G. Yang, J. Tian, F. Liu and W. Zhang, ACS Macro Lett., 2019, 8, 616–622 CrossRef CAS PubMed.
- Q. Ma, W. Wang, L. Zhang and H. Cao, Macromol. Rapid Commun., 2022, 43, 2200122 CrossRef CAS PubMed.
- S. Allison-Logan, Q. Fu, Y. Sun, M. Liu, J. Xie, J. Tang and G. G. Qiao, Angew. Chem., Int. Ed., 2020, 59, 21392–21396 CrossRef CAS PubMed.
- L. Zhang, G. Ye, X. Huo, S. Xu, J. Chen and K. Matyjaszewski, ACS Omega, 2019, 4, 16247–16255 CrossRef CAS PubMed.
- S. Han, T. Qiu, C. Xiong, X. Li and L. Guo, Macromolecules, 2022, 55, 5314–5325 CrossRef CAS.
- L. Xia, B.-F. Cheng, T.-Y. Zeng, X. Nie, G. Chen, Z. Zhang, W.-J. Zhang, C.-Y. Hong and Y.-Z. You, Adv. Sci., 2020, 7, 1902451 CrossRef CAS PubMed.
- Z. Lu, H. Yang, X. Fu, Y. Zhao, L. Xiao, Z. Zhang and L. Hou, Macromol. Rapid Commun., 2021, 42, 2100384 CrossRef CAS PubMed.
- J. Jiang, G. Ye, Z. Wang, Y. Lu, J. Chen and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 12037–12042 CrossRef CAS PubMed.
- T. Zhang, J. Yeow and C. Boyer, Polym. Chem., 2019, 10, 4643–4654 RSC.
- J. Jiang, G. Ye, F. Lorandi, Z. Liu, Y. Liu, T. Hu, J. Chen, Y. Lu and K. Matyjaszewski, Angew. Chem., Int. Ed., 2019, 58, 12096–12101 CrossRef CAS PubMed.
- E. Liang, M. Liu, B. He and G.-X. Wang, Adv. Polym. Technol., 2018, 37, 2879–2884 CrossRef CAS.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Angew. Chem., Int. Ed., 2019, 58, 1828–1832 CrossRef CAS PubMed.
- K. P. McClelland, T. D. Clemons, S. I. Stupp and E. A. Weiss, ACS Macro Lett., 2020, 9, 7–13 CrossRef CAS PubMed.
- Y. Liang, H. Ma, W. Zhang, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, Polym. Chem., 2020, 11, 4961–4967 RSC.
- Y. Zhu and E. Egap, Polym. Chem., 2020, 11, 1018–1024 RSC.
- Q. Wang, L. Hu, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, ACS Appl. Mater. Interfaces, 2020, 12, 42161–42168 CrossRef CAS PubMed.
- Y. Zhu, Y. Liu, K. A. Miller, H. Zhu and E. Egap, ACS Macro Lett., 2020, 9, 725–730 CrossRef CAS PubMed.
- C. Ding, J. Wang, W. Zhang, X. Pan, Z. Zhang, W. Zhang, J. Zhu and X. Zhu, Polym. Chem., 2016, 7, 7370–7374 RSC.
- Y. Zhu and E. Egap, ACS Polym. Au, 2021, 1, 76–99 CrossRef CAS PubMed.
- Y. Chu, N. Corrigan, C. Wu, C. Boyer and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15245–15253 CrossRef CAS.
- Y. Chu, Z. Huang, K. Liang, J. Guo, C. Boyer and J. Xu, Polym. Chem., 2018, 9, 1666–1673 RSC.
- Y. Huang, X. Li, J. Le Li, B. Zhang and T. Cai, Macromolecules, 2018, 51, 7974–7982 CrossRef CAS.
- Y. Chu, Z. Huang, R. Liu, C. Boyer and J. Xu, ChemPhotoChem, 2020, 4, 5201–5208 CrossRef CAS.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779–790 CrossRef CAS.
- K. Bell, S. Freeburne, A. Wolford and C. W. Pester, Polym. Chem., 2022, 13, 6120–6126 RSC.
- K. Bell, S. Freeburne, M. Fromel, H. J. Oh and C. W. Pester, J. Polym. Sci., 2021, 59, 2844–2853 CrossRef CAS.
- R. A. Olson, J. S. Levi, G. M. Scheutz, J. J. Lessard, C. A. Figg, M. N. Kamat, K. B. Basso and B. S. Sumerlin, Macromolecules, 2021, 54, 4880–4888 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, A. B. Korpusik, R. A. Olson, C. A. Figg and B. S. Sumerlin, Polym. Chem., 2021, 12, 2205–2209 RSC.
- X. Li, Y. C. Zhang, S. Ye, X. R. Zhang and T. Cai, J. Mater. Chem. A, 2020, 8, 25363–25370 RSC.
- Y. Zhao, S. Shao, J. Xia, Y. Huang, Y. C. Zhang, X. Li and T. Cai, J. Mater. Chem. A, 2020, 8, 9825–9831 RSC.
- C. Chen, G. A. Zhou, H. R. Zhang, X. Tang, J. N. Cheng, Y. H. Zhao, X. Li and T. Cai, Chem. Eng. J., 2021, 424, 130395 CrossRef CAS.
- M. Zhao, S. Zhu, X. Yang, Y. Wang, X. Zhou and X. Xie, Macromol. Rapid Commun., 2022, 43, 2200173 CrossRef CAS PubMed.
- F. Li, Y. Yu, H. Lv, G. Cai and Y. Zhang, Polym. Chem., 2021, 12, 2258–2270 RSC.
- F. Li, Y. Yu, H. Lv, Y. Wan, X. Gao, Y. Li and Y. Zhang, Eur. Polym. J., 2022, 178, 111519 CrossRef CAS.
- Y. Zhu, D. Zhu, Y. Chen, Q. Yan, C.-Y. Liu, K. Ling, Y. Liu, D. Lee, X. Wu, T. P. Senftle and R. Verduzco, Chem. Sci., 2021, 12, 16092–16099 RSC.
- H. Yang, Z. Lu, X. Fu, Q. Li, L. Xiao, Y. Zhao and L. Hou, Eur. Polym. J., 2022, 173, 111306 CrossRef CAS.
- L. Zhang, X. Shi, Z. Zhang, R. P. Kuchel, R. Namivandi-Zangeneh, N. Corrigan, K. Jung, K. Liang and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 5489–5496 CrossRef CAS PubMed.
- L. Zhang, G. Ng, N. Kapoor-Kaushik, X. Shi, N. Corrigan, R. Webster, K. Jung and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 22664–22671 CrossRef CAS PubMed.
- X. Li, Y. Huang, W. F. Wei, W. L. Guo, Z. Luo, J. Xu and T. Cai, Chem. Eng. J., 2022, 434, 134692 CrossRef CAS.
- V. P. Beyer, J. Kim and C. R. Becer, Polym. Chem., 2020, 11, 1271–1291 RSC.
- C. Fu, J. Xu, L. Tao and C. Boyer, ACS Macro Lett., 2014, 3, 633–638 CrossRef CAS PubMed.
- G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White and C. Boyer, Macromolecules, 2018, 51, 7600–7607 CrossRef CAS.
- P. R. Judzewitsch, L. Zhao, E. H. H. Wong and C. Boyer, Macromolecules, 2019, 52, 3975–3986 CrossRef CAS.
- P. R. Judzewitsch, N. Corrigan, F. Trujillo, J. Xu, G. Moad, C. J. Hawker, E. H. H. Wong and C. Boyer, Macromolecules, 2020, 53, 631–639 CrossRef CAS.
- A. J. Gormley, J. Yeow, G. Ng, Ó. Conway, C. Boyer and R. Chapman, Angew. Chem., Int. Ed., 2018, 57, 1557–1562 CrossRef CAS PubMed.
- M. J. Tamasi, R. A. Patel, C. H. Borca, S. Kosuri, H. Mugnier, R. Upadhya, N. S. Murthy, M. A. Webb and A. J. Gormley, Adv. Mater., 2022, 34, 2201809 CrossRef CAS PubMed.
- Q. Ma, X. Zhang, Y. Jiang, J. Lin, B. Graff, S. Hu, J. Lalevée and S. Liao, Polym. Chem., 2022, 13, 209–219 RSC.
- C. Boyer, M. Kamigaito, K. Satoh and G. Moad, Prog. Polym. Sci., 2023, 138, 101648 CrossRef CAS.
- Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 CrossRef CAS PubMed.
- S. Lin, L. Zhang, Z. Huang, P. V. Kumar and J. Xu, Macromolecules, 2019, 52, 7157–7166 CrossRef CAS.
- L. Zhang, S. Lin and J. Xu, Macromolecules, 2022, 55, 2463–2474 CrossRef CAS.
- A. Aerts, R. W. Lewis, Y. Zhou, N. Malic, G. Moad and A. Postma, Macromol. Rapid Commun., 2018, 39, 1800240 CrossRef PubMed.
- Z. Huang, N. Corrigan, S. Lin, C. Boyer and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1947–1955 CrossRef CAS.
- Y. Yang, K. Yu, S. Liu, J. Yan, H. Lai, F. Xing and P. Xiao, Macromolecules, 2021, 54, 10923–10930 CrossRef CAS.
- L. Zhang, R. Liu, S. Lin and J. Xu, Chem. Commun., 2021, 57, 10759–10762 RSC.
- Z. Li, S. Kosuri, H. Foster, J. Cohen, C. Jumeaux, M. M. Stevens, R. Chapman and A. J. Gormley, J. Am. Chem. Soc., 2019, 141, 19823–19830 CrossRef CAS PubMed.
- H. Foster, M. H. Stenzel and R. Chapman, Macromolecules, 2022, 55, 5938–5945 CrossRef CAS.
- S. Allison-logan, F. Karimi, Y. Sun, T. G. McKenzie, M. D. Nothling, G. Bryant and G. G. Qiao, ACS Macro Lett., 2019, 8, 1291–1295 CrossRef CAS PubMed.
- X. Shi, J. Zhang, N. Corrigan and C. Boyer, Mater. Chem. Front., 2021, 5, 2271–2282 RSC.
- N. Corrigan, F. J. Trujillo, J. Xu, G. Moad, C. J. Hawker and C. Boyer, Macromolecules, 2021, 54, 3430–3446 CrossRef CAS.
- A. J. Teator, T. P. Varner, P. C. Knutson, C. C. Sorensen and F. A. Leibfarth, ACS Macro Lett., 2020, 9, 1638–1654 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed.
- N. Li, D. Ding, X. Pan, Z. Zhang, J. Zhu, C. Boyer and X. Zhu, Polym. Chem., 2017, 8, 6024–6027 RSC.
- S.-H. Shim, M. Ham, J. Huh, Y.-K. Kwon and Y.-J. Kwark, Polym. Chem., 2013, 4, 5449–5455 RSC.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- S. Xu, F. J. Trujillo, J. Xu, C. Boyer and N. Corrigan, Macromol. Rapid Commun., 2021, 42, 2100212 CrossRef CAS PubMed.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- N. Corrigan, A. Almasri, W. Taillades, J. Xu and C. Boyer, Macromolecules, 2017, 50, 8438–8448 CrossRef CAS.
- N. Corrigan, R. Manahan, Z. T. Lew, J. Yeow, J. Xu and C. Boyer, Macromolecules, 2018, 51, 4553–4563 CrossRef CAS.
- K. Liu, N. Corrigan, A. Postma, G. Moad and C. Boyer, Macromolecules, 2020, 53, 8867–8882 CrossRef CAS.
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968–4972 RSC.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- T. Nwoko, N. De Alwis Watuthanthrige, B. Parnitzke, K. Yehl and D. Konkolewicz, Polym. Chem., 2021, 12, 6761–6770 RSC.
- R. Whitfield, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2021, 60, 19383–19388 CrossRef CAS PubMed.
- M.-N. Antonopoulou, R. Whitfield, N. P. Truong, D. Wyers, S. Harrisson, T. Junkers and A. Anastasaki, Nat. Chem., 2022, 14, 304–312 CrossRef CAS PubMed.
- Z. An, ACS Macro Lett., 2020, 9, 350–357 CrossRef CAS PubMed.
- Y. Yang and Z. An, Polym. Chem., 2019, 10, 2801–2811 RSC.
- B. Ameduri, Chem. – Eur. J., 2018, 24, 18830–18841 CrossRef CAS PubMed.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O’Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 RSC.
- J. Yeow, S. Shanmugam, N. Corrigan, R. P. Kuchel, J. Xu and C. Boyer, Macromolecules, 2016, 49, 7277–7285 CrossRef CAS.
- L. Zhang, L. Xie, S. Xu, R. P. Kuchel, Y. Dai, K. Jung and C. Boyer, Biomacromolecules, 2020, 21, 3887–3897 CrossRef CAS PubMed.
- S. Han, Y. Gu, M. Ma and M. Chen, Chem. Sci., 2020, 11, 10431–10436 RSC.
- J. Zhou, C. Hong and C. Pan, Mater. Chem. Front., 2017, 1, 1200–1206 RSC.
- N. Zaquen, H. Zu, A. M. N. B. P. H. A. Kadir, T. Junkers, P. B. Zetterlund and C. Boyer, ACS Appl. Polym. Mater., 2019, 1, 1251–1256 CrossRef CAS.
- A. B. Korpusik, Y. Tan, J. B. Garrison, W. Tan and B. S. Sumerlin, Macromolecules, 2021, 54, 7354–7363 CrossRef CAS.
- C. Lin, S. K. Katla and J. Perez-Mercader, J. Photochem. Photobiol., A, 2021, 406, 112992 CrossRef CAS.
- J. Wang, X. Hu, N. Zhu and K. Guo, Chem. Eng. J., 2021, 420, 127663 CrossRef CAS.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142–5156 RSC.
- M. S. Messina, K. M. M. Messina, A. Bhattacharya, H. R. Montgomery and H. D. Maynard, Prog. Polym. Sci., 2020, 100, 101186 CrossRef CAS PubMed.
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Biomacromolecules, 2019, 20, 212–221 CrossRef CAS PubMed.
- A. Watanabe, J. Niu, D. J. Lunn, J. Lawrence, A. S. Knight, M. Zhang and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1259–1268 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- E. M. Pelegri-O’Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- A. Abuchowski, T. van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- W.-H. Leung, P.-K. So, W.-T. Wong, W.-H. Lo and P.-H. Chan, RSC Adv., 2016, 6, 106837–106846 RSC.
- M. Liu, L. Jia, Z. Zhao, Y. Han, Y. Li, Q. Peng and Q. Zhang, Chem. Eng. J., 2020, 390, 124667 CrossRef CAS.
- A. J. Russell, S. L. Baker, C. M. Colina, C. A. Figg, J. L. Kaar, K. Matyjaszewski, A. Simakova and B. S. Sumerlin, AIChE J., 2018, 64, 3230–3245 CrossRef CAS.
- Y. Yu, W. Xu, X. Huang, X. Xu, R. Qiao, Y. Li, F. Han, H. Peng, T. P. Davis, C. Fu and A. K. Whittaker, ACS Macro Lett., 2020, 9, 799–805 CrossRef CAS PubMed.
- W. L. Guo, Y. Zhou, B. Duan, W. F. Wei, C. Chen, X. Li and T. Cai, Chem. Eng. J., 2022, 429, 132120 CrossRef CAS.
- Y. Huang, X. Li, Y. C. Zhang, Z. Shi, L. Zeng, J. Xie, Y. Du, D. Lu, Z. Hu, T. Cai and Z. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 44488–44496 CrossRef CAS PubMed.
- C. Ma, X. Liu, G. Wu, P. Zhou, Y. Zhou, L. Wang and X. Huang, ACS Macro Lett., 2017, 6, 689–694 CrossRef CAS PubMed.
- G. Szczepaniak, J. Jeong, K. Kapil, S. Dadashi-Silab, S. S. Yerneni, P. Ratajczyk, S. Lathwal, D. J. Schild, S. R. Das and K. Matyjaszewski, Chem. Sci., 2022, 13, 11540–11550 RSC.
- B. Panganiban, B. Qiao, T. Jiang, C. DelRe, M. M. Obadia, T. D. Nguyen, A. A. Smith, A. Hall, I. Sit, M. G. Crosby and P. B. Dennis, Science, 2018, 359, 1239–1243 CrossRef CAS PubMed.
- C. DelRe, B. Chang, I. Jayapurna, A. Hall, A. Wang, K. Zolkin and T. Xu, Adv. Mater., 2021, 33, 2105707 CrossRef CAS PubMed.
- P. R. Judzewitsch, T.-K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559–4564 CrossRef CAS PubMed.
- P. R. Judzewitsch, N. Corrigan, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 24248–24256 CrossRef CAS PubMed.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Angew. Chem., Int. Ed., 2020, 59, 19136–19142 CrossRef CAS PubMed.
- A. J. Gormley and M. A. Webb, Nat. Rev. Mater., 2021, 6, 642–644 CrossRef CAS PubMed.
- M. Tamasi, S. Kosuri, J. DiStefano, R. Chapman and A. J. Gormley, Adv. Intell. Syst., 2020, 2, 1900126 CrossRef PubMed.
- R. Upadhya, A. Punia, M. J. Kanagala, L. Liu, M. Lamm, T. A. Rhodes and A. J. Gormley, ACS Appl. Polym. Mater., 2021, 3, 1525–1536 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- M. Li, M. Fromel, D. Ranaweera, S. Rocha, C. Boyer and C. W. Pester, ACS Macro Lett., 2019, 8, 374–380 CrossRef CAS PubMed.
- J. Poisson, A. M. Polgar, M. Fromel, C. W. Pester and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 19988–19996 CrossRef CAS PubMed.
- X. Xu, X. Huang, Y. Chang, Y. Yu, J. Zhao, N. Isahak, J. Teng, R. Qiao, H. Peng, C.-X. Zhao, T. P. Davis, C. Fu and A. K. Whittaker, Biomacromolecules, 2021, 22, 330–339 CrossRef CAS PubMed.
- G. Ng, P. Judzewitsch, M. Li, C. W. Pester, K. Jung and C. Boyer, Macromol. Rapid Commun., 2021, 42, 2100106 CrossRef CAS PubMed.
- G. Ng, M. Li, J. Yeow, K. Jung, C. W. Pester and C. Boyer, ACS Appl. Mater. Interfaces, 2020, 12, 55243–55254 CrossRef CAS PubMed.
- M. Fromel and C. W. Pester, Macromolecules, 2022, 55, 4907–4915 CrossRef CAS.
- A. R. Kuzmyn, L. W. Teunissen, M. V. Kroese, J. Kant, S. Venema and H. Zuilhof, ACS Omega, 2022, 7, 38371–38379 CrossRef CAS PubMed.
- L.-H. Rong, X. Cheng, J. Ge, O. K. Krebs, J. R. Capadona, E. B. Caldona and R. C. Advincula, ACS Appl. Polym. Mater., 2022, 4, 6449–6457 CrossRef CAS.
- T. Joshi and L. Nebhani, Surf. Interfaces, 2022, 29, 101764 CrossRef CAS.
- C. Förster, L. Veith and A. Andrieu-Brunsen, RSC Adv., 2022, 12, 27109–27113 RSC.
- L. Hu, Q. Hao, L. Wang, Z. Cui, P. Fu, M. Liu, X. Qiao and X. Pang, Polym. Chem., 2021, 12, 545–553 RSC.
- J. Zhou, L. Ye, Y. Lin, L. Wang, L. Zhou, H. Hu, Q. Zhang, H. Yang and Z. Luo, J. Appl. Polym. Sci., 2019, 136, 47653 CrossRef.
- J. Zhou, Y. Lin, L. Ye, L. Wang, L. Zhou, H. Hu, Q. Zhang, H. Yang and Z. Luo, Macromol. Res., 2019, 27, 1144–1154 CrossRef CAS.
- J. Zhou, Y. Sun, Z. Huang, Z. Luo and H. Hu, J. Appl. Polym. Sci., 2021, 138, 51395 CrossRef CAS.
- Y. Lin, L. Wang, J. Zhou, L. Ye, H. Hu, Z. Luo and L. Zhou, Polymer, 2019, 162, 80–90 CrossRef CAS.
- J. Zhou, Y. Lin, L. Wang, L. Zhou, B. Yu, X. Zou, Z. Luo and H. Hu, Colloids Surf., A, 2021, 617, 126369 CrossRef CAS.
- H. Kodama, Rev. Sci. Instrum., 1981, 52, 1770–1773 CrossRef.
- C. W. Hull, US Pat., 4575330, 1986 Search PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- T. T. Wohlers, W. A. (Firm), R. I. Campbell, O. Diegel, J. Kowen, R. Huff, N. Mostow, I. Fidan, D. L. Bourell, J. Van Rensburg and others, Wohlers Report 2022: 3D Printing and Additive Manufacturing Global State of the Industry, Wohlers Associates, 2022 Search PubMed.
- H. N. Chia and B. M. Wu, J. Biol. Eng., 2015, 9, 4 CrossRef PubMed.
- T. J. Wallin, J. Pikul and R. F. Shepherd, Nat. Rev. Mater., 2018, 3, 84–100 CrossRef.
- A. Paolini, S. Kollmannsberger and E. Rank, Addit. Manuf., 2019, 30, 100894 Search PubMed.
- C. Sun, N. Fang, D. M. Wu and X. Zhang, Sens. Actuators, A, 2005, 121, 113–120 CrossRef CAS.
- X. Zheng, J. Deotte, M. P. Alonso, G. R. Farquar, T. H. Weisgraber, S. Gemberling, H. Lee, N. Fang and C. M. Spadaccini, Rev. Sci. Instrum., 2012, 83, 125001 CrossRef PubMed.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32, 1903850 CrossRef CAS PubMed.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593–611 CrossRef CAS.
- V. A. Bobrin, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater. Technol., 2022, 2201054 Search PubMed.
- Z. Zhang, N. Corrigan and C. Boyer, Macromolecules, 2021, 54, 1170–1182 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- A. Bagheri, H. Ling, C. William Anderson Bainbridge and J. Jin, ACS Appl. Polym. Mater., 2021, 3, 2921–2930 CrossRef CAS.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 8839–8850 CrossRef CAS PubMed.
- Z. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202114111 CAS.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2107643 CrossRef CAS PubMed.
- X. Shi, V. A. Bobrin, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202206272 CAS.
- V. A. Bobrin, Y. Yao, X. Shi, Y. Xiu, J. Zhang, N. Corrigan and C. Boyer, Nat. Commun., 2022, 13, 3577 CrossRef CAS PubMed.
- D. Melodia, A. Bhadra, K. Lee, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, Small, 2023, 2206639 CrossRef PubMed.
- K. Lee, Y. Shang, V. A. Bobrin, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2204816 CrossRef CAS PubMed.
- R. M. Myers, D. E. Fitzpatrick, R. M. Turner and S. V. Ley, Chem. – A Eur. J., 2014, 20, 12348–12366 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 RSC.
- N. Zhu, X. Hu, Z. Fang and K. Guo, ChemPhotoChem, 2018, 2, 831–838 CrossRef CAS.
- B. L. Buss and G. M. Miyake, Chem. Mater., 2018, 30, 3931–3942 CrossRef CAS PubMed.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- C. Sambiagio, M. Ferrari, K. van Beurden, N. della Ca’, J. Van Schijndel and T. Noël, Org. Lett., 2021, 23, 2042–2047 CrossRef CAS PubMed.
- F. Zhong, Y. Zhou and M. Chen, Polym. Chem., 2019, 10, 4879–4886 RSC.
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Macromolecules, 2019, 52, 3–23 CrossRef CAS.
- C. Stubbs, K. A. Murray, T. Ishibe, R. T. Mathers and M. I. Gibson, ACS Macro Lett., 2020, 9, 290–294 CrossRef CAS PubMed.
- S.-J. Richards, A. Jones, R. M. F. Tomás and M. I. Gibson, Chem. – A Eur. J., 2018, 24, 13758–13761 CrossRef CAS PubMed.
- J. Yeow, S. Joshi, R. Chapman and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 10102–10106 CrossRef CAS PubMed.
- R. Upadhya, N. S. Murthy, C. L. Hoop, S. Kosuri, V. Nanda, J. Kohn, J. Baum and A. J. Gormley, Macromolecules, 2019, 52, 8295–8304 CrossRef CAS PubMed.
- G. Ng, K. Jung, J. Li, C. Wu, L. Zhang and C. Boyer, Polym. Chem., 2021, 12, 6548–6560 RSC.
- M. Nagao, Y. Kimoto, Y. Hoshino and Y. Miura, ACS Omega, 2022, 7, 13254–13259 CrossRef CAS PubMed.
- S. Kosuri, C. H. Borca, H. Mugnier, M. Tamasi, R. A. Patel, I. Perez, S. Kumar, Z. Finkel, R. Schloss, L. Cai, M. L. Yarmush, M. A. Webb and A. J. Gormley, Adv. Healthcare Mater., 2022, 11, 2102101 CrossRef CAS PubMed.
- R. Upadhya, S. Kosuri, M. Tamasi, T. A. Meyer, S. Atta, M. A. Webb and A. J. Gormley, Adv. Drug Delivery Rev., 2021, 171, 1–28 CrossRef CAS PubMed.
- F. Soheilmoghaddam, M. Rumble and J. Cooper-White, Chem. Rev., 2021, 121, 10792–10864 CrossRef CAS PubMed.
- J. Li, M. Zhang, J. Zhu and X. Zhu, Polymers, 2019, 11, 1722 CrossRef CAS PubMed.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 RSC.
- P. Lertturongchai, M. I. A. Ibrahim, A. Durand, P. Sunintaboon and K. Ferji, Macromol. Rapid Commun., 2020, 41, 2000058 CrossRef CAS PubMed.
- T. G. McKenzie, L. P. da, M. Costa, Q. Fu, D. E. Dunstan and G. G. Qiao, Polym. Chem., 2016, 7, 4246–4253 RSC.
- K. Jung, C. Boyer and P. B. Zetterlund, Polym. Chem., 2017, 8, 3965–3970 RSC.
- A. Bagheri, Z. Sadrearhami, N. N. M. Adnan, C. Boyer and M. Lim, Polymer, 2018, 151, 6–14 CrossRef CAS.
- B. Cabannes-Boué, Q. Yang, J. Lalevée, F. Morlet-Savary and J. Poly, Polym. Chem., 2017, 8, 1760–1770 RSC.
- A. Bagheri, C. Bainbridge and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 1896–1904 CrossRef CAS.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641–647 RSC.
- K. J. Arrington and J. B. Matson, Polym. Chem., 2017, 8, 7452–7456 RSC.
- T. G. McKenzie, E. H. H. Wong, Q. Fu, A. Sulistio, D. E. Dunstan and G. G. Qiao, ACS Macro Lett., 2015, 4, 1012–1016 CrossRef CAS PubMed.
- V. Tkachenko, C. Matei Ghimbeu, C. Vaulot, L. Vidal, J. Poly and A. Chemtob, Polym. Chem., 2019, 10, 2316–2326 RSC.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 14–19 CrossRef CAS PubMed.
- A. Rasines Mazo, T. N. Tran, W. Zhang, Y. Meng, A. Reyhani, S. Pascual, L. Fontaine, G. G. Qiao and S. Piogé, Polym. Chem., 2020, 11, 5238–5248 RSC.
- R. W. Lewis, R. A. Evans, N. Malic, K. Saito and N. R. Cameron, Polym. Chem., 2018, 9, 60–68 RSC.
- J. Li, C. Ding, Z. Zhang, J. Zhu and X. Zhu, React. Funct. Polym., 2017, 113, 1–5 CrossRef CAS.
- J. Luo, M. Li, M. Xin, W. Sun and W. Xiao, Macromol. Chem. Phys., 2016, 217, 1777–1784 CrossRef CAS.
- Q. Yang, M. Guerre, V. Ladmiral and B. Ameduri, Polym. Chem., 2018, 9, 3388–3397 RSC.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS PubMed.
- J. Li, X. Pan, N. Li, J. Zhu and X. Zhu, Polym. Chem., 2018, 9, 2897–2904 RSC.
- Y. Wang, M. Wang, L. Bai, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2020, 11, 2080–2088 RSC.
- S. Liu, Y. Cheng, H. Zhang, Z. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 6274–6278 CrossRef CAS PubMed.
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537–1546 RSC.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS PubMed.
- C. Fu, Z. Huang, C. J. Hawker, G. Moad, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 4637–4643 RSC.
- S. Shanmugam, G. Ross, C. Y. Mbuncha and A. Santra, Polym. Chem., 2021, 12, 6705–6713 RSC.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS PubMed.
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165–5172 CrossRef CAS.
- B. Zhao, J. Li, Z. Li, X. Lin, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 7181–7192 CrossRef CAS.
- X. Wu, B. Gross, B. Leuschel, K. Mougin, S. Dominici, S. Gree, M. Belqat, V. Tkachenko, B. Cabannes-Boué, A. Chemtob, J. Poly and A. Spangenberg, Adv. Funct. Mater., 2022, 32, 2109446 CrossRef CAS.
- V. Kottisch, M. J. Supej and B. P. Fors, Angew. Chem., Int. Ed., 2018, 57, 8260–8264 CrossRef CAS PubMed.
- R. J. Sifri, A. J. Kennedy and B. P. Fors, Polym. Chem., 2020, 11, 6499–6504 RSC.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed.
- Z. Yang, J. Chen and S. Liao, ACS Macro Lett., 2022, 11, 1073–1078 CrossRef CAS PubMed.
- J. Li, M. Chen, X. Lin, Q. Li, W. Zhang, G. Jin, X. Pan, J. Zhu and X. Zhu, ACS Macro Lett., 2020, 9, 1799–1805 CrossRef CAS PubMed.
- M. Ciftci, Y. Yoshikawa and Y. Yagci, Angew. Chem., Int. Ed., 2017, 56, 519–523 CrossRef CAS PubMed.
- J. Li, A. Kerr, S. Häkkinen, T. Floyd, M. Zhang, X. Pan, X. Zhu, S. Perrier and J. Zhu, Polym. Chem., 2020, 11, 2724–2731 RSC.
- J. Li, A. Kerr, Q. Song, J. Yang, S. Häkkinen, X. Pan, Z. Zhang, J. Zhu and S. Perrier, ACS Macro Lett., 2021, 10, 570–575 CrossRef CAS PubMed.
- H. Suk Wang, N. P. Truong, Z. Pei, M. L. Coote, A. Anastasaki, H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 1390–1395 CrossRef CAS PubMed.
- S. V. Wanasinghe, M. Sun, K. Yehl, J. Cuthbert, K. Matyjaszewski and D. Konkolewicz, ACS Macro Lett., 2022, 11, 1156–1161 CrossRef CAS PubMed.
- P. N. Kurek, A. J. Kloster, K. A. Weaver, R. Manahan, M. L. Allegrezza, N. De Alwis Watuthanthrige, C. Boyer, J. A. Reeves and D. Konkolewicz, Ind. Eng. Chem. Res., 2018, 57, 4203–4213 CrossRef CAS.
- Y. Lee, C. Boyer and M. S. Kwon, Nat. Rev. Mater., 2022, 7, 74–75 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |