
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origin of the enantioselectivity of alcohol dehydrogenase†
Jiahui
Zhou‡
a,
Tao
Han‡
b,
Shahbaz
Ahmad
 acd,
Derek
Quinn
cd,
Thomas S.
Moody
cd,
Qi
Wu
*b and
Meilan
Huang
*a
acd,
Derek
Quinn
cd,
Thomas S.
Moody
cd,
Qi
Wu
*b and
Meilan
Huang
*a
aSchool of Chemistry and Chemical Engineering, Queen's University, David Keir Building, Stranmillis Road, Belfast BT9 5AG, Northern Ireland, UK. E-mail: m.huang@qub.ac.uk
bDepartment of Chemistry, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: wuqi1000@163.com
cAlmac Sciences, Department of Biocatalysis and Isotope Chemistry, Almac House, 20 Seagoe Industrial Estate, Craigavon BT63 5QD, Northern Ireland, UK
dArran Chemical Company Limited, Unit 1 Monksland Industrial Estate, Athlone, Co. Roscommon, Ireland
First published on 6th November 2023
Abstract
Alcohol dehydrogenases (ADH) are a family of enzymes that catalyse the interconversion between ketones/aldehydes and alcohols in the presence of NADPH cofactor. It is challenging to desymmetrise the substituted cyclopentane-1,3-dione by engineering an ADH, while the reaction mechanism of the metal independent ADH remains elusive. Here we measured the conversion of a model substrate 2-benzyl-2-methylcyclopentane-1,3-dione by LbADH and found it predominately gave the (2R,3R) product. Binding mode analysis of the substrate in LbADH from molecular dynamics simulations disclosed the origin of the enantioselectivity of the enzyme; the opening and closing of the loop 191–205 above the substrate are responsible for shaping the binding pocket to orientate the substrate, so as to give different stereoisomer products. Using QM/MM calculations, we elucidated the reaction mechanism of LbADH. Furthermore, we demonstrated the reaction profile corresponding to the production of different stereoisomers, which is in accordance with our experimental observations. This research here will shed a light on the rational engineering of ADH to achieve stereodivergent stereoisomer products.
Introduction
Alcohol dehydrogenases (ADH) can catalyse the reversible reduction of carbonyl compounds to yield alcohols in the presence of the cofactor NADPH. The desymmetrisation of substituted cyclopentane-1,3-dione was previously studied by chemical1–3 and enzymatic catalysts.4,5 Although high enantioselectivity was achieved by carbonyl reductase,4,5 it is not known how the substrate would adapt itself in the catalytic pocket to give a certain stereoisomer product. Therefore, it is crucial to understand the preferred binding of the substrate in the enzyme in order to rationally engineer the enzyme to achieve stereodivergent products.There are two types of ADHs, including Zinc containing and non-metal ADHs. The mechanism of zinc-ADH catalysed reduction mechanism was reported previously,6,7 however, to the best of our knowledge, the mechanism of the reduction of carbonyl compounds with metal-independent ADHs remains elusive. In this research, we expressed an ADH enzyme from Lactobacillus brevis (LbADH) and measured its conversion of the substrate. 2-Benzyl-2-methylcyclopentane-1,3-dione was chosen as a model substrate because the reduction would give four different enantiodivergent products and hence enable the desymmetrisation of the substrate. Our kinetic experiments showed that the (2R,3R) and (2S,3S) products were observed with a ratio of 9 to 1, while the other two chiral products (2S,3R) or (2R,3S) were not observed.
MD simulations disclosed that the loop 191–205 above the substrate could exhibit either open or closed conformation, which is responsible for the production of different stereoisomer products. Further hybrid quantum mechanics/molecular mechanics (QM/MM) calculations disclosed the mechanism of the reduction catalysed by LbADH. We elucidated that the reaction starts with the rate-limiting step, i.e., the concerted hydride transfer from NADPH to the substrate carbonyl carbon and proton transfer from Tyr155 to the substrate carbonyl oxygen. The reaction then proceeds with a concerted proton relay; deprotonated Tyr155 phenolate abstracts a proton from NADP+ ribose, which in turn abstracts a proton from the nearby catalytic acid Lys159. The second step is barrierless for the major product (2R,3R), but it needs to overcome a notable barrier for the minor product (2S,3S) and an even higher barrier for the inaccessible product (2S,3R) or (2R,3S), implicating the origin of the enantioselectivity of the enzyme. Additionally, we found the reaction coordinate in the reactant (i.e., the distance between the NADPH hydride to the carbonyl carbon) is correlated to the barrier for the rate-limiting step.
Experimental methods
Protein expression
100 μL stored bacteria containing LbADH gene in pET-28a was first inoculated in 5 mL LB medium (containing 50 μg mL−1 kanamycin) and was shaken at 200 rpm overnight as preculture. The preculture was used to inoculate large culture (250 mL LB + 50 μg mL−1 kanamycin in 1 L shake flasks) at 37 °C for about 4 hours until OD600 at 0.6–0.8. After cooling at 4 °C for 0.5 h, 1 mM isopropyl β-thiogalactopyranoside (IPTG) was added to induce LbADH expression. The culture was allowed to express at 18 °C for 18 h with shaking at 200 rpm. Then, cells were harvested by centrifugation at 9000 rpm and 4 °C for 10 min, and the supernatants were discarded. The cells were resuspended in 50 mM Tris–HCl buffer (pH 7.5) containing 100 mM NaCl and 2 mM MgCl2 (1 g wet cell in 5 mL 50 mM Tris–HCl buffer) and stored in −80 °C. The cells were repeated freezing and thawing for 3 times, and then released the target proteins by sonication. The cell debris was removed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min at 4 °C, and the crude enzyme solutions were stored at −80 °C.
000 rpm for 20 min at 4 °C, and the crude enzyme solutions were stored at −80 °C.
Protein purification
The crude enzyme solution was filtered and loaded on a GE Healthcare HisTrap FF Crude column (5 mL) preequilibrated with 50 mM Tris–HCl buffer (pH 7.5) containing 100 mM NaCl, 2 mM MgCl2 and 25 mM imidazole. The enzyme was eluted by 50 mM Tris–HCl buffer containing 100 mM NaCl, 2 mM MgCl2 and 250 mM imidazole. The proteins were dialyzed by 50 mM Tris–HCl buffer (pH 7.5) containing 100 mM NaCl and 2 mM MgCl2 for 12 h at 4 °C.Determination of kinetic parameters
Kinetic parameters of LbADH were measured by monitoring the decrease of NADPH following the absorbance at 340 nm using the spectrophotometry method (SHIMADZU-UV-2550). The activity assay was performed in a mixture (3 mL) containing 50 mM Tris–HCl buffer (pH 7.5), an appropriate amount of the enzyme and varying concentration of 2-benzyl-2-methylcyclopentane-1,3-dione (0–8 mM) with 5% (final) dimethyl sulfoxide (DMSO) as a cosolvent. NADPH was added to a final concentration of 100 μM as the last component to start the reaction.Computational methods
Molecular docking
In the docking studies, the crystal structure of wild-type Lactobacillus brevis alcohol dehydrogenase (LbADH) (PDB: 1ZK4)8 was employed, which contains the NADPH co-factor and the acetophenone substrate. The substrate acetophenone was removed while the NADPH cofactor was retained to construct the apo-protein. A 100 ns MD simulation was conducted for the apo-protein using AMBER 20,9 including energy minimization and equilibration steps. Representative structures were selected by cluster analysis of MD trajectories using CPPTRAJ10 module of the Amber 20 for the docking study. The model substrate in this study 2-benzyl-2-methylcyclopentane-1,3-dione was constructed using GaussView 611 and optimized at the B3LYP/6-31G(d) level. The optimized substrate was docked in the simulated apo protein using Autodock12 and then the top docked poses with favourable binding energies and appropriate orientation (that would lead to the respective stereoisomeric ketols) were selected for the subsequent MD simulations.Molecular dynamics (MD) simulations
The parameters for the substrate and the NADPH co-factor were calculated based on the optimized geometry at the B3LYP/6-31G(d) level of theory using Gaussian 1613 with the PCM solvation model. Electrostatic potential (ESP) was derived at the same level according to the Merz–Singh–Kollman scheme and was fit using RESP in the antechamber module of AMBER 209 coupled with the general AMBER force-field GAFF2.14 The H++ server15 was used to predict the protonation states of titratable amino acids and the FF19SB16 force field in AMBER 20 was used to obtain the parameters for the enzyme.The enzyme-substrate complexes were solvated in a pre-equilibrated cuboid box of TIP3P17 water molecules, and any protein atom was at least 10 Å to from the edge of the cuboid box. The system was then neutralised by adding 8 Na+ counterions using the tLEaP program implemented in AMBER 20. A harmonic restraint force constant of 100 kcal−1 Å−2 was applied to the solute molecules and ions to minimise the solvent molecules, followed by 1000-step steepest descend and 1000-step conjugate gradient unrestrained minimisation. A cut-off of 10 Å was applied for non-bonded Lennard-Jones potential and electrostatic interactions. Hydrogen bonds were constrained using the SHAKE algorithm during all MD simulations. Under constant volume and periodic boundary conditions, a progressive heating was performed from 0 K to 300 K in 100 ps (5000 steps with a step size of 0.02 ps), followed by 1 ns equilibration using NPT ensemble at 300 K. A harmonic restraint of 5 kcal mol−1 was applied to the solute at both equilibration stages. After the equilibration stages, a 200 ns production MD simulation was run using an NPT ensemble at 300 K and 1 bar. Three replicas of MD simulations were run for each system.
Hybrid quantum mechanics/molecular mechanics (QM/MM) calculations
The snapshots used for the QM/MM calculations were obtained from the CPPTRAJ cluster analysis10 of the MD trajectory, and then three cluster structures of each system were optimized using 2500-step steepest descend and 2500-step conjugate gradient algorithms, with a harmonic restraint force constant of 100 kcal−1 Å−2 applied on the heavy atoms.The substrate, truncated NADPH, Tyr155, Lys159, and Ser142 are included in the QM region. During the geometry optimization, residues within 6 Å of the QM region were allowed to move freely, while the remaining residues were kept frozen (Fig. S1, ESI†). The hydrogen link atoms were used to saturate the dangling bond at the QM/MM boundary. The QM/MM calculations were performed using the electronic embedding model within the Gaussian13 ONIOM. Geometries of all intermediates and transition states were optimized at the B3LYP/6-31G(d) level. Frequency calculations within the harmonic approximation were used to verify the nature of all intermediates and transition states. Free energy and enthalpic corrections were carried out by computing harmonic frequencies analytically at 298.15 K. For each step on the reaction profile, thermochemical correction terms δEG were carried out as a difference of the reaction energy (ΔEB3LYP/6-31G(d)) and the corresponding free energy (ΔGB3LYP/6-31G(d)):
| δEG = ΔGB3LYP/6-31G(d) − ΔEB3LYP/6-31G(d) |
Potential energy surface scans were conducted to locate the transition states at the same level, i.e., B3LYP-D3(BJ)/6-31G(d). Transition states were confirmed through visual inspection of the imaginary frequency modes as well as intrinsic reaction coordinate (IRC) calculations.
The corrected free energies (ΔG) were calculated as follows:
| ΔG = ΔESP + ΔEG |
The single point energy of the optimized intermediates and transition states were also compared using the large basis set, i.e., 6-311+G(2d,2p) and def-2TZVPP.
Results and discussion
Enantioselectivity of alcohol dehydrogenase
2-Benzyl-2-methylcyclopentane-1,3-dione was chosen as a model substrate for LbADH, because the reduction would introduce desymmetrisation of the substrate. The wild-type LbADH was expressed and purified and then kinetic parameters of the enzyme were measured by monitoring the consumption of NADPH.The stereoisomeric distribution of the products for WT-LbADH was measured as following. Sodium phosphate buffer (1 ml, 100 mM, pH 7.0) containing 10% DMSO (v/v), 10 mM substrate, 30 mM glucose, 0.5 mM NADP+, 0.5–2 mg of LbADH and 4 U GDH were shaken at 30 °C for overnight. Then the reaction was stopped by the addition of an equal amount of ethyl acetate. The organic phase was separated and the solvent was removed; the resulting sample was analysed by chiral HPLC to determine the enantiomeric excess value of the alcohol products.
The reduction of the substrate yielded 90% and 10% stereoisomeric (2R,3R) and (2S,3S) products, respectively, whereas no (2S,3R) nor (2R,3S) product was obtained (Fig. 1 and Scheme 1).
 | ||
| Fig. 1 The stereoisomeric distribution of the products from WT-LbADH catalyzed reaction. The enantiomeric excess values of the four different stereoisomer alcohol products (2R,3R), (2S,3S), (2S,3R), (2R,3S) were determined by chiral HPLC. | ||
 | ||
| Scheme 1 LbADH catalyzed reduction of the model substrate 2-benzyl-2-methylcyclopentane-1,3-dione. | ||
The opening/closing of loop 191–205 regulates the enantioselectivity
A catalytic tetrad comprised of Asn113, Ser142, Tyr155 and Lys159 was proposed in the previous crystal structure study of LbADH.8 However, the relative distances between Ser142, Lys159 and Tyr155 are not ideal for the reaction to take place, indicating the crystal structure may not be catalytically active conformation. The binding mode of the substrate 2-benzyl-2-methylcyclopentane-1,3-dione in the catalytic site of the enzyme was decided by combining molecular docking and molecular dynamics simulations. Molecular docking was performed and the substrate poses that would lead to different stereoisomer products were retained, including the most dominant (2R,3R) and the less dominant (2S,3S) product, as well as the (2S,3R) which was not obtained by the WT enzyme. These enzyme-substrate complexes were then subjected to MD simulations. RMSD analysis showed the simulations reached equilibrium (Fig. S2, ESI†).In the proximity of catalytic sites of all the structures, Ser142 stabilizes the substrate carbonyl group by a hydrogen bond (Fig. 2 and Fig. S3, ESI†); Tyr155 is positioned toward the substrate carbonyl to serve as an H-bond donor and also forms H-bond with the NADPH ribose hydroxyl group, which is, in turn, H-bonded to Lys159.
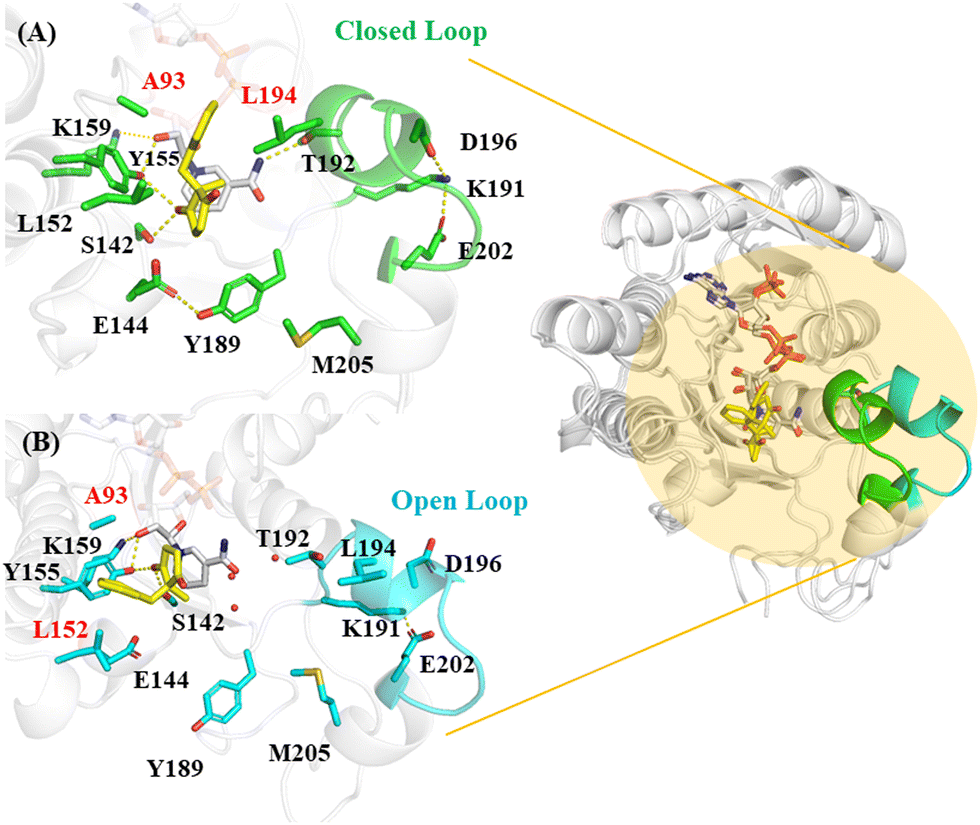 | ||
| Fig. 2 MD simulated LbADH in complex with the substrate (A) Substrate in closed loop 191–205 (in green) leading to the (2R,3R) product; (B) substrate in open loop (in cyan) leading to the (2S,3S) product. The hydrophobic residues Ala93 and Leu194/Leu152 are involved in the hydrophobic interactions with the substrate benzene. | ||
The binding of the substrate in the enzyme is mediated by Glu144, which results in different substrate orientations. In the substrate pose that leads to the main (2R,3R) product, Glu144 forms a hydrogen bond with Tyr189 (Fig. 2A and Fig. S3, ESI†), which is stabilized by the methyl group of Met205. The benzene ring of the substrate is nested between the surrounding Ala93 and Leu194, forming hydrophobic interactions with them. Thr192 forms H-bond with the amide NH2 of the NADPH nicotinamide. The loop 191–205 adjacent to Tyr189 displays a closed conformation, with Asp196 interacting with Lys191 by ionic interactions, which in turn tethered by Glu202 (Fig. 2).
In the substrate pose that leads to the minor (2S,3S) product (Fig. 2B), the sidechain of Glu144 is turned away from the catalytic pocket. The loss of the H-bond between Glu144 and Tyr189 caused the loop K191-M205 to display a significant conformational change to adopt an open conformation. The opening of the loop makes Leu194 become far away from the substrate, such that the substrate was able to move freely in the binding pocket and eventually stabilized by the favourable hydrophobic interactions with the surrounding Leu152 and Ala193 with its benzene ring. The opening of the loop also caused the loss of the H-bond between Thr192 and NADPH. Meanwhile, Asp196 on the loop becomes exposed and its interaction with Lys191 is lost, leaving the latter interacting only with Glu202. A water flux moved in the catalytic site with the opening of the loop (Fig. 2B).
Thus we demonstrated that the loop191–205 region is largely stabilized in the substrate pose leading to the dominant (2R,3R) product (Fig. S2B, ESI†), compared to other substrate poses that lead to minor product (2S,3S) or do not give the corresponding chiral product such as (2S,3R).
Reaction profile of enantioselective reduction by ADH
So far, the reaction mechanism of NADPH-dependant ADH remains elusive. To elucidate the reaction mechanism of the LbADH and the reaction profiles of ADH for desymmetrisation of the substituted cyclopentane-1,3-dione to produce different stereoisomers, the representative structures from MD simulations of the enzyme-substrate complexes were minimized and the minimized structures are similar to the initial representative structures from cluster analysis based on their RMSD values (Table S1, ESI†). These structures were selected as the starting structures for the subsequent mechanism study by QM/MM calculations.LbADH is homologous to RasADH3 and there is 5 AA difference between the two homologous enzymes. Previously, Chen et al. studied RasADH and suggested that Ser137, Tyr150, and Lys154 (corresponding to Ser142, Tyr155, Lys159 in LbADH) may participate in the catalytic process, along with the nicotinamide ring of NADPH.3 However, the exact catalytic process of ADH is not known.
From our MD simulations, Tyr155 and Lys159 are located in the proximity of the substrate and NADPH; therefore, these two residues were included in the QM region, along with the substrate, the NADPH cofactor. Tyr155 and Lys159 were truncated to keep the sidechain and NADPH was truncated to keep the nicotinamide riboside part in the QM/MM calculations.
Potential energy surface scans were conducted for the enzyme-substrate complex structures that would lead to different stereoisomers, i.e. the most dominant (2R,3R) (90% distribution among all products), the less dominant (2S,3S) (10% distribution), and also a chiral product (2S,3R) that was not observed from the kinetic experiments.
Benchmarking calculations were conducted for the DFT methods, namely B3LYP (6-31G*) and M06-2X (6-31G*) (Fig. S4, ESI†). For the substrate pose that lead to the dominant (2R,3R) stereoisomer, after the carbonyl group of the 1,3-dione substrate is reduced and Tyr155 is deprotonated, the deprotonated Tyr155 in I1 abstracts a proton from the nicotinamide riboside hydroxyl, which spontaneously abstract a proton from Lys159 (barrierless). Interestingly, for the substrate pose that leads to the minor product (2S,3S) or the unachievable product (2S,3R), the deprotonated Tyr155 in I1 needs to overcome a notable barrier to be reprotonated (Fig. S4b and S5, ESI†).
The dispersion in the DFT method may affect the energy barrier.21–23 For example, QM/MM calculations for cytochrome P450 catalysed reactions showed that dispersion correction may reduce the barrier of hydrogen abstraction significantly by around 5 kcal mol−1.21 To examine if dispersion is necessary to consider the polarisation effect for ADH, we conducted bench marking calculations with dispersion effect using B3YLP-D3(BJ) and M06-2X-D3 with the 6/31G* basis set.
Since M06-2X implicitly includes dispersion energy, as expected, including Grimme's correction did not cause significant change, whereas notable differences were observed with the B3LYP functional (Fig. 3 and Fig. S4, ESI†), highlighting the importance of including dispersion for B3LYP.
 | ||
| Fig. 3 Free energy profile for the enzyme-substrate complexes with the substrate poses that would lead to the (A) (2R,3R) product or (B) (2S,3S) product. Geometry optimizations and frequency calculations were conducted using B3YLP-D3(BJ), M06-2X-D3 and ωB97X-D with the 6/31G* basis set. | ||
Energy barrier values calculated by M06-2X-D3 and ωB97X-D are similar, whereas lower barrier values were obtained by using B3LYP-D3(BJ) (Fig. 3). It is worth noting that for (2R,3R), the TS2 couldn’t be located using B3LYP or M06-2X/M06-2X-D3 (Fig. S4a, ESI† and Fig. 3a), thus combining B3LYP with dispersion is recommended for geometry optimizations. The two transition states TS1 and TS2 were subsequently verified by frequency calculations and IRC analysis (Fig. S6, ESI†). Further, energy corrections were conducted with two larger basis sets for the optimized geometries, namely, Def-2TZVPP and 6-311+G(2d,2p), and consistent energies were obtained (Fig. 4). For the following discussion, the results obtained by B3LYP-D3(BJ)/6-31G* with ZPE correction at the same level and single point energy corrections at Def-2TZVPP basis set (i.e. B3LYP-D3(BJ)/Def-2TZVPP//B3LYP-D3(BJ)/6-31G*) were employed.
 | ||
| Fig. 4 Free energy profile for the enzyme-substrate complexes with the substrate poses leading to (A) (2R,3R) product or (B) (2S,3S) product. Geometry optimizations and frequency calculations were calculated based on the optimized geometries using B3LYP-D3(BJ) with the 6-31G* basis set, and the free energies were calculated with single point correction with Def-2TZVPP and 6-311+G(2d,2p) basis sets. | ||
The kinetic study of LbADH showed a KM value of 3.18 (mM) and a kcat value of 0.0114 s−1, which corresponds to a reaction barrier of 20.10 kcal mol−1 through the calculation of the Eyring equation. Our calculations show that the rate determining step of the ADH-catalysed reduction is the concerted hydride transfer from 4CH2 of NADPH nicotinamide to the substrate and proton transfer from Tyr155 to the substrate carbonyl. The formation of the major product (2R,3R) proceeds through a transition state TS1 with a barrier of 13.5 kcal mol−1 (Fig. 4). Then the reaction proceeds with a barrierless and concerted proton relay composed of the proton abstraction from NADPH ribose to the phenolate Tyr155 and proton transfer from Lys159 to the sugar. The bond-breaking distance between the attacking hydrogen and C4 increases to 1.36 Å in the transition state TS1, whereas the bond-formation distance between the C4H and the substrate carbon decreases to 1.44 Å in TS1 (Fig. 5).
 | ||
| Fig. 5 Optimized geometries in the reaction pathways conducted using B3LYP-D3(BJ) with the 6/31G* basis set. (a) Substrate pose leading to the dominant (2R,3R) product and (b) substrate pose that leads to the minor (2S,3S) product. (The number in parentheses in the reactant complex conformation is the bonding angle between the attacking hydrogen on C4 of NADPH and the carbonyl carbon of the substrate). | ||
The formation of the minor product (2S,3S) also goes through two transition states with similar barriers of 8.7 kcal mol−1 and 8.8 kcal mol−1, respectively (Fig. 4). The difference in the product distribution may be attributed to the different reaction pathways; the formation of the (2S,3S) requires surpassing a second barrier involving the reprotonation of Tyr155 facilitated by NADP+ and Lys159; hence the process is less energetically favourable compared to the formation of the major (2R,3R) product. We also ran a potential energy surface scan for the substrate pose that would lead to the (2S,3R) product, which was not obtained from the kinetic experiment. Our calculations show the production of the (2S,3R) product needs to go through a much high energy barrier of 24.2 kcal mol−1 for the rate limiting step and a second barrier of 2.6 kcal mol−1 (Fig. S7, ESI†), indicating the formation of such product is energetically prohibited and thus not liable, which is in agreement with our experimental observations.
Interestingly, during the formation of the main product (2R,3R), we observed that I1 transformed into a slightly lower-energy intermediate I1′. Notably, the catalytic acid Lys159 in I1′ became closer to the nicotinamide riboside hydroxyl of NADP+ than I1 (1.53Å vs. 2.12 Å), and also the bond angle is more favourable for proton transfer (165° vs. 110°), making the deprotonation from Lys159 to the NADPH ribose and the reprotonation of the Tyr155 easier to occur (Fig. 5a). The bond angle is 144° (the distance is 1.83Å) in the minor product (2S,3S) (Fig. 5b) and 126° (the distance is 2.43 Å) in the inaccessible product (2S,3R) (Fig. S8, ESI†). This indicates that the bond distance and bond angle involved in the H-bond between Lys159 and NADPH ribose is positively related to the reaction barrier of the second reaction step the proton reply.
Reaction mechanism of ADH catalyzed reduction
Our QM/MM calculations disclosed that the ADH catalyzed reduction reaction proceeds with a hydride transfer from the C4 methylene of NADPH nicotinamide to the carbonyl carbon atom of the substrate that is accompanied by the proton transfer from Tyr155 to the substrate carbonyl oxygen, to give the reduced product and deprotonated Tyr155 (I1) (Scheme 2). The reaction proceeds with an abstraction of a proton from the nicotinamide riboside hydroxyl by Tyr155 to resume its protonated state while the deprotonated sugar abstracts a proton from the nearby Lys159, which functions as a catalytic acid (Scheme 2). Notably, the previous hypothesis for RasADH4 proposed that the reprotonation of Tyr150 is fulfilled by the proton transfer from Lys154 (Lys159 in LbADH) to the phenolate oxygen of Tyr150 (Tyr155 in LbADH). Our calculations disclosed that the reprotonation of Tyr155 is facilitated by the nicotinamide riboside OH with the aid of the nearby lysine residue Lys159, thus presenting a complete reaction profile of the ADH-catalyzed reduction.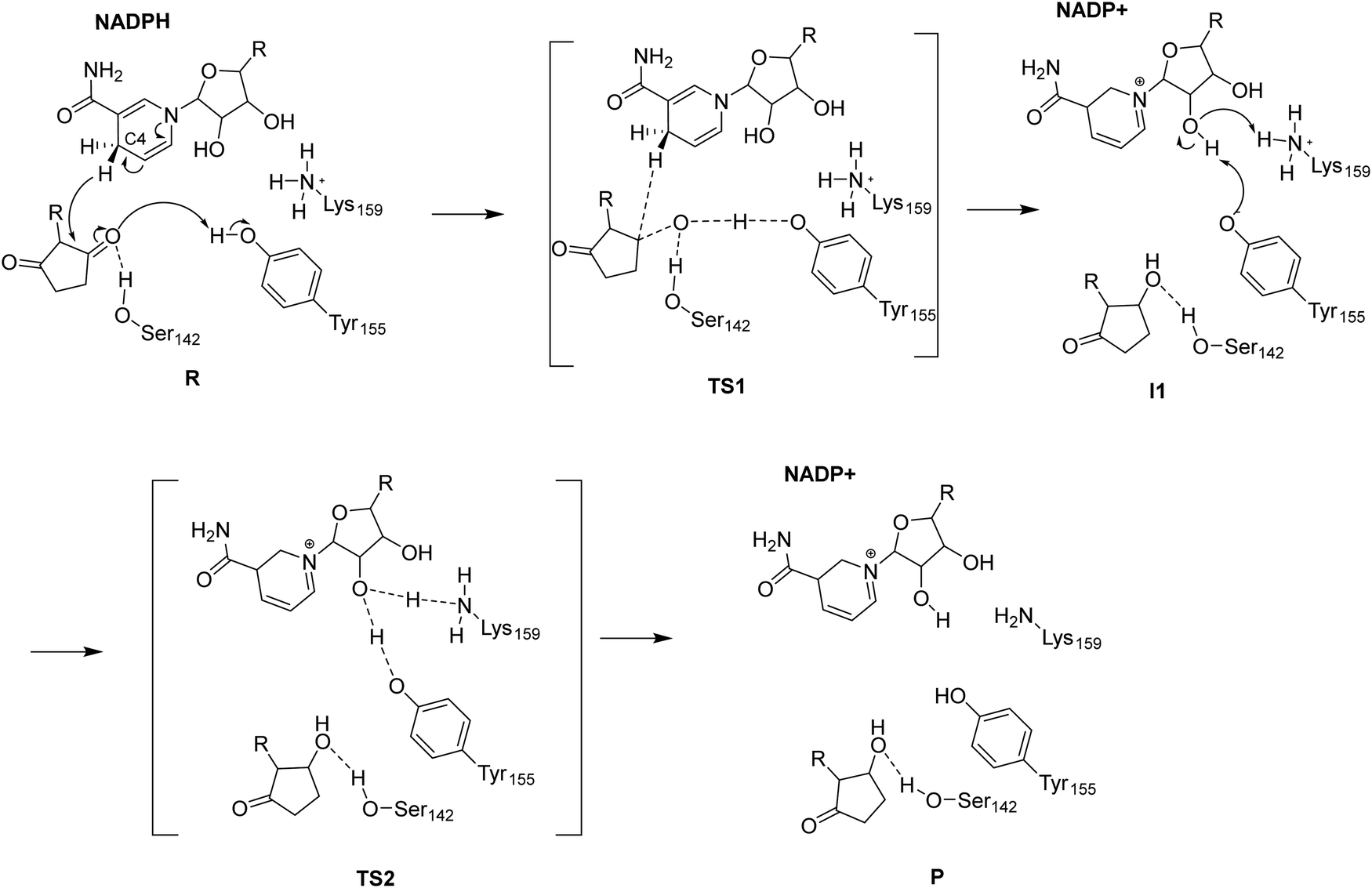 | ||
| Scheme 2 Reaction mechanism of ADH catalyzed reduction. | ||
Energy barrier of the reaction rate-determining step is linearly related to the associated reaction coordinate
The rate-limiting step in the reduction of the substrate is the hydride transfer from C4 of the nicotinamide moiety of NADPH to the substrate carbonyl carbon. The QM/MM optimized structures showed the reaction coordinate associated with this step (the bond-formation distance) is smallest for the complex that yields the (2S,3S) product, as compared to those producing (2R,3R), and (2S,3R) products (2.09 Å vs. 2.50 Å vs. 3.27 Å) (Fig. 5 and Fig. S8, ESI†). Since the reaction barriers for rate-determining step increase for the enzyme-substrate complexes that would lead to the (2R,3R), (2S,3S) to (2S,3R) products, we propose there is a relationship between the reaction coordinate and the energy barrier of the rate-limiting step and observed a linear correlation (Fig. 6). This correlation was consistently observed across all the optimization methods employed including B3LYP, B3LYP-D3(BJ), M06-2X, M06-2X-D3, and ωB97X-D. Thus, the reaction coordinate for the rate-determining step in the substrate can be directly used to estimate the reaction energy barrier and predict the reaction kinetics without the need to conduct a complete PES scan using computing-demanding QM/MM calculations. | ||
| Fig. 6 Energy barrier for the rate-limiting step is linear correlated with the reaction coordinate (the distance between the C4 methylene hydrogen of NADPH and the substrate carbonyl carbon atom). | ||
Conclusion
Reduction of a carbonyl compound by an ADH enzyme can give different stereoisomer products. However, the catalytic pocket preference for the enantioselectivity of ADH is not fully understood, nor is the exact mechanism of the ADH-catalysed reduction. Here we measured the conversion of a model substrate 2-benzyl-2-methylcyclopentane-1,3-dione by LbADH and found the enzyme can selectively transformed the substrate and gave four different stereoisomer products with different ratio, with (2R,3R) as the major product and (2S,3S) as the minor produce, while the other two stereoisomer products (2S,3R) and (2R,3S) were not obtained.We studied the binding of a model substrate 2-benzyl-2-methylcyclopentane-1,3-dione in an ADH enzyme represented by LbADH by molecular docking and MD simulations and demonstrated the orientation of the substrate is regulated by the opening/closing of the loop 191–205, which would lead to the different stereoisomer products. The calculations are in agreement with our kinetic studies.
We further elucidated the reaction mechanism of LbADH using by QM/MM calculations. In the first reaction step, the hydrogen of NADPH C4 methylene attacks the carbonyl carbon of the substrate, which is accompanied by a concerted proton transfer from the hydroxyl group of Tyr155. The second reaction step is also a concerted process, where the proton of the ribose hydroxyl of NADP+ is relayed between Lys159 and the phenolate Tyr155, giving rise to neutral Tyr155 and neutral Lys159. A correlation was observed between the reaction coordinate and the reaction barrier of the rate-determining step, indicating the reaction coordinate can be used as an important but easily achievable feature for large-scan screening of the enantioselective ADH variants in enzyme engineering. The understanding of preferred substrate poses in the ADH enzyme leading to different stereoisomers would provide structural basis for further rationally engineering of the enzyme to acquire enantiodivergent products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z. acknowledges the financial support of Royal Society International Exchange (IEC\NSFC\191454), Queen's University Belfast (QUB) and Chinese Scholarship Council. The authors acknowledge the financial support from INVEST NI Research and Development Programme, partly financed by the European Regional Development Fund under the Investment for Growth and Jobs Programme 2021–2027. The authors are grateful for the computing resources from QUB high performance computing Tier2 computing resource funded by EPSRC (EP/T022175). We also acknowledge Chenggong Hui for the helpful discussion on IRC analysis.References
- Q. Gong, J. Wen and X. Zhang, Chem. Sci., 2019, 10, 6350–6353 RSC.
- X.-L. Qin, A. Li and F.-S. Han, J. Am. Chem. Soc., 2021, 143, 2994–3002 CrossRef CAS PubMed.
- V. B. Patil, S. B. Jadhav, J. B. Nanubolu and R. Chegondi, Org. Lett., 2022, 24, 8233–8238 CrossRef CAS PubMed.
- X. Chen, H. Zhang, M. A. Maria-Solano, W. Liu, J. Li, J. Feng, X. Liu, S. Osuna, R.-T. Guo, Q. Wu, D. Zhu and Y. Ma, Nat. Catal., 2019, 2, 931–941 CrossRef CAS.
- M. L. Contente, F. Dall’Oglio, F. Annunziata, F. Molinari, M. Rabuffetti, D. Romano, L. Tamborini, D. Rother and A. Pinto, Catal. Lett., 2020, 150, 1176–1185 CrossRef CAS.
- G. V. Dhoke, M. D. Davari, U. Schwaneberg and M. Bocola, ACS Catal., 2015, 5, 3207–3215 CrossRef CAS.
- H. Zhang, L. Zhu, J. Feng, X. Liu, X. Chen, Q. Wu and D. Zhu, Catal. Sci. Technol., 2022, 12, 5841–5849 RSC.
- N. H. Schlieben, K. Niefind, J. Müller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2005, 349, 801–813 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao and A. P. A. Kollman, AMBER 2020, University of California, San Francisco, 2020 Search PubMed.
- D. R. Roe and T. E. Cheatham, III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- T. A. K. Roy Dennington and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- X. He, V. H. Man, W. Yang, T.-S. Lee and J. Wang, J. Chem. Phys., 2020, 153, 114502 CrossRef CAS PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- R. P. P. Neves, P. A. Fernandes and M. J. Ramos, ACS Catal., 2016, 6, 357–368 CrossRef CAS.
- F. E. Medina, R. P. P. Neves, M. J. Ramos and P. A. Fernandes, Phys. Chem. Chem. Phys., 2017, 19, 347–355 RSC.
- H. Mitsuizumi and S. Mori, J. Phys. Chem. B, 2021, 125, 4998–5008 CrossRef CAS PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Chem. Theory Comput., 2012, 8, 4637–4645 CrossRef CAS PubMed.
- R. J. O’Reilly, B. Chan, M. S. Taylor, S. Ivanic, G. B. Bacskay, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2011, 133, 16553–16559 CrossRef PubMed.
- M. Egli and T. P. Lybrand, J. Am. Chem. Soc., 2019, 141, 4445–4452 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04019d |
| ‡ Equal contributions. |
| This journal is © the Owner Societies 2023 |