
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImaging rotational energy transfer: comparative stereodynamics in CO + N2 and CO + CO inelastic scattering
Zhong-Fa
Sun
 *a,
Roy J. A.
Scheidsbach
b,
Marc C.
van Hemert
c,
Ad
van der Avoird
d,
Arthur G.
Suits
e and
David H.
Parker
*ab
*a,
Roy J. A.
Scheidsbach
b,
Marc C.
van Hemert
c,
Ad
van der Avoird
d,
Arthur G.
Suits
e and
David H.
Parker
*ab
aAnhui Province Key Laboratory of Optoelectric Materials Science and Technology, Department of Physics, Anhui Normal University, Wuhu, Anhui 241000, China. E-mail: zfsun@ahnu.edu.cn
bDepartment of Molecular and Laser Physics, Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: parker@science.ru.nl
cLeiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
dTheoretical Chemistry, Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
eDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
First published on 6th June 2023
Abstract
State-to-state rotational energy transfer in collisions of ground ro-vibrational state 13CO molecules with N2 molecules has been studied using the crossed molecular beam method under kinematically equivalent conditions used for 13CO + CO rotationally inelastic scattering described in a previously published report (Sun et al., Science, 2020, 369, 307–309). The collisionally excited 13CO molecule products are detected by the same (1 + 1′ + 1′′) VUV (Vacuum Ultra-Violet) resonance enhanced multiphoton ionization scheme coupled with velocity map ion imaging. We present differential cross sections and scattering angle resolved rotational angular momentum alignment moments extracted from experimentally measured 13CO + N2 scattering images and compare them with theoretical predictions from quasi-classical trajectories (QCT) on a newly calculated 13CO–N2 potential energy surface (PES). Good agreement between experiment and theory is found, which confirms the accuracy of the 13CO–N2 potential energy surface for the 1460 cm−1 collision energy studied by experiment. Experimental results for 13CO + N2 are compared with those for 13CO + CO collisions. The angle-resolved product rotational angular momentum alignment moments for the two scattering systems are very similar, which indicates that the collision induced alignment dynamics observed for both systems are dominated by a hard-shell nature. However, compared to the 13CO + CO measurements, the primary rainbow maximum in the DCSs for 13CO + N2 is peaked consistently at more backward scattering angles and the secondary maximum becomes much less obvious, implying that the 13CO–N2 PES is less anisotropic. In addition, a forward scattering component with high rotational excitation seen for 13CO + CO does not appear for 13CO–N2 in the experiment and is not predicted by QCT theory. Some of these differences in collision dynamics behaviour can be predicted by a comparison between the properties of the PESs for the two systems. More specific behaviour is also predicted from analysis of the dependence on the relative collision geometry of 13CO + N2 trajectories compared to 13CO + CO trajectories, which shows the special ‘do-si-do’ pathway invoked for 13CO + CO is not effective for 13CO + N2 collisions.
1. Introduction
As an omnipresent gas phase molecular process, rotational energy transfer (RET) is of great interest and the subject of extensive investigations in various fields such as atmospheric, astrophysical, and combustion chemistry. Many collision systems involving small molecules have been studied in detail by experiment and theory, particularly for the NO and OH radicals, in both their ground and excited electronic states.1,2 The essence of understanding any collision is to determine how the reagents with specific initial conditions transform into specific products. By defining the initial pre-collision conditions such as the collision energy and the internal quantum states of the molecules, the scalar and vector properties of the products after collision can be measured experimentally. Theoretically, the collisional dynamics are predicted by performing quantum or quasi-classical calculations over potential energy surfaces (PESs) of the collision complex, which describe the underlying interaction between the two collision partners. Comparisons of experimental measurements with theoretical predictions can thus test the quality of scattering theory and of potential energy surfaces, and help create and improve empirical models and approximations.Compared to the more averaged scalar measurements such as collision rate coefficients and integral cross sections, stereodynamic measurements, especially state-to-state differential cross sections (DCSs) and/or scattering angle-resolved rotational angular momentum polarization, have proven particularly useful in providing rigorous tests of state-of-the-art theory.3 The last few decades have witnessed tremendous progress in the field of experimental study of stereodynamics in rotationally inelastic scattering; this was significantly driven by the development of the velocity map imaging (VMI)4 technique and its application to crossed molecular beam (CMB) experiments. The power of the CMB-VMI approach is derived from the ability to directly image the velocity vectors of scattered products independently of their position of formation onto a position-sensitive detector, thereby probing all product recoil angles simultaneously, i.e., pure angular distributions can be measured. In combination with laser-based state-selective spectroscopic probe methods (mainly resonance enhanced multiphoton ionization, (REMPI), the VMI technique yields an image that fully describes the scattering event, with the size of the image reflecting the recoil speed of scattered products into a specified channel as well as the angular distribution, yielding the state-resolved differential cross section (DCS). In addition, with appropriate choice of the REMPI spectroscopic transition and good control of the polarization of the probe laser (linearly or circularly polarized), measurement of state-to-state scattering angle-resolved rotational angular momentum alignment and orientation has become possible.5,6 The most extensive work using the CMB-VMI approach has been performed on inelastic scattering of NO with Rare gas (abbreviated Rg) atoms by the groups of Stolte, van de Meerakker, Chandler, Brouard, and Costen.7–17 Fully rotational state and fine-structure resolved state-to-state DCSs have been measured initially for NO(X) + Rg collision systems using hexapole state-selection for NO(X) preparation.12 Instead of using only a hexapole filter, the group of van de Meerakker successfully utilized Stark deceleration, which manipulates neutral polar molecules based on the Stark effect to generate a compact packet of NO(X) radicals with (almost) perfect quantum state purity and very well-controlled velocity before collision. The combination of Stark deceleration and VMI detection significantly enhances the resolution of scattering images and allowed direct observation of quantum diffraction oscillations in state-to-state DCSs for inelastic scattering between Stark decelerator-selected NO(X) and He, Ne, Ar atoms.16 With the advantage of the continuous tunability of the desired velocity offered by Stark deceleration, they also pushed the collision energy of NO(X) + He collision system firstly down to the ∼20 K regime and recently to the <1 K regime,18 and gained new insight into the PESs by probing scattering resonances in the state-to-state and parity-resolved DCSs.8,19 Imaging studies of collision-induced angular momentum polarization in NO(X) + Ar were pioneered by Chandler and Cline,5,9 followed by Brouard and co-workers both experimentally with improved resolution7 and theoretically by means of various calculations.11 Brouard et al. also extended detailed investigations on angular momentum polarization effects to inelastic scattering of NO(X) with other rare gas atoms such as Ne, Kr.6,10,14 Recently, imaging of the inelastic scattering of electronically excited NO with various collision partners has been studied by Costen and co-workers, including measurements of differential cross sections and collision-induced alignment.15,17,20
Most of the previous inelastic scattering studies have concentrated on molecule-atom inelastic scattering stereodynamics. This is because of experimental convenience and the availability of fully quantum theory. However, there are a limited number of environments where collisions between molecules and rare gas atoms are of importance in practice. By contrast, advanced studies of bimolecular inelastic scattering are much less common, and little is known about stereodynamics arising when two molecules approach each other. A molecular collision partner possesses internal degrees of freedom that allow the possibility to induce excitation of both partners (and also de-excitation of a reactant molecule that has initial rotational or vibrational population) in a scattering event. In the case of diatom-diatom inelastic scattering process, both partners may become rotationally excited after collision. Full characterization of this effect yields the so-called product-pair correlations, which provide great challenges for theory as well as experiment. Theoretically, the dimension of the PES required to describe the interaction of bimolecular collision system is increased, bringing about considerably more complexity for a numerically exact treatment, especially when the collision energy is larger than ∼700 cm−1. On the experimental side, it is very challenging to prepare pure initial state molecular beams for both molecular beam sources, and it is difficult to probe correlations between two products simultaneously at the detection stage. Study of identical molecule scattering, as in our recent paper21 on 12CO + 13CO, is even more demanding due to the need for isotope selective REMPI. In the case of velocity map ion imaging detection used in the current work, imaging the product(s) with a specific final state result in a projection of multiple concentric Newton rings arising from excitation to a range of rotational energy levels of the molecular collision partner(s), which may overlap each other partially or fully on the detector. Up until now, only limited experimental data is available on bimolecular inelastic scattering systems and most cases studied have been collisions with H2 or its isotopologue D2 as the molecular colliders, such as NO(X) + (ortho-)D2,22–25 NO(X) + (para-)H2,24 NO(A) + D2,26 O2 + H2,27,28 H2O + H2,29 ND3 + H2,30 CD3 + H2 and D2.31 The choice of H2 and D2 as the partner molecule is favourable in that they possess relatively large rotational energy spacing and in addition their ortho or para symmetry will be conserved during collision. Jointly, these conditions, in principle, allow restriction of the number of open pair-correlated product channels and thereby simplify the complexity of both experiment and theory. Rotational excitation of H2 or D2 is usually not observed at the lower collision energy in the experiments mentioned above. By contrast, rotational excitation of HD (which has no ortho–para restrictions) in collision with NO at 220 cm−1 collision energy has been observed,32 as discussed later in this section. Other than H2/D2, molecular nitrogen has been previously used as a collider in studies of inelastic scattering of diatomic and polyatomic molecules. Orr-Ewing and co-workers investigated the inelastic scattering of CD3 with N2 in comparison to Ar as collider; they observed evidence in the image for scattered CD3 products with coincident rotational excitation of the unobserved N2, but no further attempt was performed to analyse the product-pair correlation information in DCSs.33 The group of Costen studied the state-to-state rotational energy transfer for collisions of electronically excited NO(A) with N2 and showed that it is possible to extract stereodynamics information as a function of correlated internal energy in the products but only for coarse-grained amounts of energy transfer to N2 rotation.34 In the recent study mentioned above,32 correlated rotational excitation of NO–CO pairs at 220 cm−1 collision energy was also reported.
Carbon monoxide (CO) is another benchmark molecule, along with NO, for inelastic scattering stereodynamics study in the gas-phase. As the second most abundant molecule in the universe after molecular hydrogen, CO is of extremely high astrochemical and astrophysical relevance. It is commonly used as a tracer to extract astrophysical parameters of the interstellar medium (ISM) when local thermodynamic equilibrium (LTE) can be assumed; while in the environment where the LTE approximation breaks down, successful molecular modelling of the observed CO spectra is based on accurate collision rate coefficients of CO calculated from theory, the quality of which must be tested by experiment in laboratory. Alignment-free DCSs and/or polarization dependent (PD)DCS measurements for inelastic scattering processes involving CO are shown here to offer excellent tests for theoretical calculations. In our latest study of imaging rotational energy transfer in the 13CO + CO system,21 we have shown that under the right experimental conditions velocity map imaging (VMI) can be applied to study the state-to-state inelastic scattering of identical molecules using crossed-molecular-beam methods. Polarisation dependent images for scattered 13CO with CO were measured experimentally and the extracted pair-correlated DCSs were compared with predictions using quasi-classical scattering theory as well as close-coupling quantum mechanical theory; new effects regarding energy partitioning and propensity rules arising with the product pair correlation have been revealed for this bimolecular collision process. The good agreement between experiment and QCT theory along with close-coupled quantum mechanics (CC QM) for the 13CO + CO system confirmed the reliability of the experimental approach. The most striking result of ref. 21, revealed by QCT, was the concerted face-to-face/rotation/back-to-back or ‘do-si-do’ motion taken by colliding partners which end in the same high rotational final states without changing their initial direction, i.e., exhibiting strongly forward scattering. This only occurred when the initial orientation of each molecule was such that the carbon atoms collided first, where a significant 10–20% of the collisions met this criterion. As pointed out in ref. 21, forward scattering in the CO–CO system is due to an attractive force acting after a hard repulsive collision that rotates the pair of product molecules during their separation. This concept will be further evaluated for the kinematically equivalent 13CO–12CO and 13CO–N2 collisions in this paper where we now extend our CMB-VMI experimental approach to study the stereodynamics of state-to-state inelastic scattering of 13CO with N2 instead of CO as the collision partner. Collisions of 13CO with CO and N2 molecules appear particularly intriguing in that both collision partners have the same mass and similar chemical properties. Using the same settings for experimental conditions of molecular beams and of collision geometry, the two colliding systems will yield identical collision energies, thus identical kinematic conditions. CO and N2 are thus in this case rather similar collision partners for the primary 13CO molecule, and similar collision behaviour and cross sections may be expected at first glance. Deeper insight into the dynamics for these colliding systems could be gained by observing subtle differences in their collision behaviour and cross sections. In the current report, we present and discuss the complete measurements of DCSs and alignment moments for 13CO + N2 system in comparison to the full set of 13CO + CO data measured previously, and draw some general conclusions about the dynamics of rotational energy transfer in these kinematically equivalent colliding systems.
2. Methods
2.1 Experimental method
The CMB-VMI experimental apparatus has been described at length in ref. 21, so only the most relevant details are mentioned here. Two supersonically cooled molecular beams produced from two Nijmegen pulsed valves (NPVs)35 intersected at the centre of a differentially pumped scattering chamber. Molecular beam source conditions for the 13CO + N2 experiment were set the same as those of the 13CO + CO experiment in order to achieve nearly equivalent collision energies. A molecular beam of 5% 13CO seeded in Ar was formed from NPV1 with a stagnation pressure of 1.2 bar, acting as the primary beam. The isotopologue 13CO was used in 13CO + CO scattering in order to distinguish the scattered molecular products and for 13CO + N2 for a direct comparison of equal mass number 12C16O and 14N2. The secondary beam was generated by the same type of pulsed valve (NPV2) with a mixture of N2/He of the same concentration at the same backing pressure just as the secondary molecular beam containing CO/He used previously. Both valves operated at 10 Hz and the pulse duration at the scattering center was about 50 μs. After separately being skimmed through a 1.5 mm diameter skimmer placed 3 cm downstream from the nozzle of the valve, the two molecular beams crossed each other at an estimated crossing volume of 3 × 3 × 3 mm3 with a collision angle of 70°. Using product imaging, the distance (in pixels) from the mean position of the beamspot from the ionization center, defined as the zero lab-frame velocity in the plane of the detector was measured along with the mean speeds of the molecular beams: v(13CO/Ar) ≈ 670 m s−1, v(N2/He) ≈ 1,660 m s−1, jointly resulting in a mean collision energy of 1460 cm−1 for the 13CO + N2 colliding system, which is the same as that for the 13CO + CO system. Comparison between REMPI spectra measured for the primary beam and the corresponding simulation from Pgopher36 software indicated that the rotational temperature of the rotationally cooled 13CO/Ar molecular beam was less than 1.5 K, giving a predominant population of 13CO molecules in the lowest rotational state j = 0 (more than 90%). However, this situation is different for the nitrogen molecule. 14N atom has a nuclear spin of I = 1, and the nuclear spin statistics lead to a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the ortho14N2: para14N2 for the rotational states, where even j corresponds to ortho and odd j to para. After the rotational cooling during adiabatic expansion of the molecular beam, N2 molecules previously populated in even j states will be redistributed mainly on the lowest even j state (j = 0), and N2 in odd j states will fall into the lowest odd j state (j = 1), while the ratio of ortho:para will be retained. No independent measurements of the N2 rotational distribution were made, but considering the same experimental conditions for producing the N2 molecular beam in this experiment and the CO beam production previously, and considering the very similar rotational constants of N2 and CO, it is reasonable to assume that the secondary beam (N2/He) in the current experiment has a similar rotational temperature (Trot ≈ 3 K) as the CO/He beam had. Based on the simulation with the Pgopher program for an N2 spectrum at a rotational temperature of around 3 K, the rotational population of rotationally cooled N2 molecular beam is estimated to be j = 0 (∼60%), j = 1 (∼32%) and higher j states (∼8%). An encounter of a N2 molecule with a 13CO molecule can not only rotationally excite the 13CO molecule, but also induce rotation of the N2 molecule. However, because of the fact that the nuclear spin acts as a spectator to the collision process to a high level of approximation, there is a conservation of ortho/para character of N2 in the collision process and only Δj = even transitions are allowed.
:1 for the ortho14N2: para14N2 for the rotational states, where even j corresponds to ortho and odd j to para. After the rotational cooling during adiabatic expansion of the molecular beam, N2 molecules previously populated in even j states will be redistributed mainly on the lowest even j state (j = 0), and N2 in odd j states will fall into the lowest odd j state (j = 1), while the ratio of ortho:para will be retained. No independent measurements of the N2 rotational distribution were made, but considering the same experimental conditions for producing the N2 molecular beam in this experiment and the CO beam production previously, and considering the very similar rotational constants of N2 and CO, it is reasonable to assume that the secondary beam (N2/He) in the current experiment has a similar rotational temperature (Trot ≈ 3 K) as the CO/He beam had. Based on the simulation with the Pgopher program for an N2 spectrum at a rotational temperature of around 3 K, the rotational population of rotationally cooled N2 molecular beam is estimated to be j = 0 (∼60%), j = 1 (∼32%) and higher j states (∼8%). An encounter of a N2 molecule with a 13CO molecule can not only rotationally excite the 13CO molecule, but also induce rotation of the N2 molecule. However, because of the fact that the nuclear spin acts as a spectator to the collision process to a high level of approximation, there is a conservation of ortho/para character of N2 in the collision process and only Δj = even transitions are allowed.
In the experiment, single collision conditions are confirmed by monitoring the change in initial rotational population of molecules in the primary beam as a result of collision; depletion of the 13CO beam ground state by collision was measured to be less than 5%. The rotationally excited 13CO products after collision were ionized via a VUV REMPI scheme along the electronic A1Π − X1Σ+ transition using VUV radiation around 154 nm. The VUV REMPI scheme has been investigated in detail in ref. 37 and 38, therefore for experimental details we refer to the previous publications. The VUV radiation is one-photon resonant with the 13CO A1Π − X1Σ+ (0, 0) transition and this VUV REMPI scheme turns out to be very polarization-sensitive for measurements involving nascent CO.37,39,40
The ionized scattered products 13CO+ were extracted by the VMI ion optics, and the three-dimensional Newton sphere, after 85 cm free flight along the time-of-flight (TOF) tube, was projected onto a two-dimensional detector consisting of chevron MCPs and a phosphor screen. The VMI voltage setting was tuned to obtain the best focusing of the ion velocity on the image detector and TOF time gating was applied to select the required mass and suppress contributions from other ionized molecules (hydrocarbons or water) in the chamber to the ion images. The ion images were captured by a charge coupled device (CCD) camera and then transferred to a laboratory computer for acquisition and averaged with DaVis software (LaVision) using an event counting mode.
Overall differential cross sections and overall alignment moments were extracted from the experimental images following the extraction procedure described by Suits et al. in ref. 39. In this procedure, two types of angular distribution components (in-plane component; out-of-plane component) were extracted from H and V polarization images by integrating over an annulus covering the two-dimensional Newton ring and over a narrow stripe through the middle of the image along the relative velocity vector. In total, four sets of angular distribution components were obtained for each final rotationally excited state j′: HIP (horizontal, in-plane), HOOP (horizontal, out-of-plane), VIP (vertical, in-plane) and VOOP (vertical, out-of-plane).39 It is known that the overall detection probability function of an image depends not only on the alignment of product rotational angular momentum, but also the probe sensitivity factors which are determined by the actual experimental geometry of probe laser polarization. Solving the equations of the overall image detection probability linked to alignment moments and probe sensitivity factors result in algebraic combination of the four sets of components mentioned above, which yield the state-to-state alignment-free differential cross sections (DCSs) and the alignment moments A{k}q. In the current experiment, due to the large velocity difference between two incident molecular beams, the probe laser propagated at an angle of 130° with respect to the relative velocity vector of the colliding system; such experimental geometry has sensitivity to all alignment moments A{2}0, A{2}1+ and A{2}2+, and these were extracted and reported here. On the other hand, the pair-correlated differential cross sections, i.e., the rotational-state pair-correlated alignment-free moments were extracted from the radius-dependent angular distribution of the experimental images using an onion-peeling inversion method which is similar to that in ref. 41 and then summed to obtain the overall differential cross sections for comparison. Within the experimental error bar, the overall DCSs obtained from the summation of the rotational-state pair-correlated DCSs agree with the directly extracted overall DCSs, regarding the trend, shape and peak positions, etc. Since the rotational-state pair-correlated DCSs in the 13CO + CO inelastic scattering together with those in the 13CO + N2 collisions have been described and discussed in great detail in our previous publication,21 we do not concentrate on pair correlation in this article and instead we compare the stereodynamics of inelastic scattering in the 13CO + CO and the 13CO + N2 collisions based on the overall DCSs and alignment moments.
2.2 Theoretical method
For the 13CO–CO system the calculation of the potential energy surface and its parameterization such that it could be used in trajectory calculations followed the procedure described in detail in ref. 21. We follow the same approach for the calculation of the 13CO–N2 potential energy surface in this report. The general form of the PESs for both systems is in good agreement with those presented previously for 12CO–12CO42 and for 12CO–N2.43 In short, the coupled cluster singles and doubles with perturbative triplets [CCSD(T)] approach was used as implemented in the Molpro program (2012 version).44 The energies were corrected for basis set superposition errors (BSSE) by performing the calculations for the complex and the separate diatomic molecules in the full complex basis set. An augmented quadruple zeta (AVQZ) basis was used. For the CO–N2 potential energy surface 3465 unique geometries were considered. These numbers are related to 21 R values and an angular grid consisting of 7 θA, 7 θB and 6 Φ values, where R is the length of the R vector pointing from the center of mass of monomer A to that of B, θA and θA are angles between the vector R and the vectors rA and rB pointing along the bond axis of each monomer, and Φ is the dihedral angle between the two planes defined by the vectors R and rA, and R and rB. The angles were chosen in such a way that the angular dependence of the interaction energy was optimally described within the spherical expansion formalism. The 21 R values chosen were 2.49, 2.5, 2.625, 2.75, 2.875, 3.0, 3.125, 3.25, 3.5, 3.75, 4.0, 4.25, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 9.0, 15.0, 20.0 a0. The data from the first small R value was used to set the slope at small R of the cubic spline interpolation of the spherical expansion coefficients. The slope at the largest R value was set to zero for all coefficients except for the isotropic term and the low order multipole components. The slopes of the latter components were set in accord with the proper R dependence of the energy terms (1/R6 for the isotropic dispersion and 1/R(L+1) for the multipole-multipole interactions where L is sum of the multipole orders of the two diatomic molecules). For the calculation of the intermolecular interaction energy the bond lengths of the two diatomic molecules were kept fixed at their equilibrium values. Since the dynamics calculations run in 6 dimensions, the intermolecular potential was appended with two Morse oscillators with parameters chosen for optimal reproduction of the rotational levels of the dimers up to the collision energy of ∼1500 cm−1 (for CO re = 1.128 Å, β = 2.302 Å−1, De = 11.20 eV and for N2re = 1.098 Å, β = 3.444 Å−1, De = 9.90 eV).45,46 The dependence of the intermolecular energy on the bond lengths of the diatomic molecules was neglected. Indirect support for this approximation was provided by trajectory calculations of CO–Ar which showed little influence of this dependence on cross sections and alignment parameters. To aid comparison of the collision dynamics of CO–CO versus CO–N2, maps of the electrostatic moment expansions for both CO–N2 and CO–CO are presented later in this paper.The details of the quasi-classical trajectory calculations have been described in ref. 21 as well, hence only important points are summarized briefly here. The trajectory calculations were performed with the help of the Venus 2005 program.47 An interface was constructed to provide energies and forces from the spherical expansion representation of the potential energy surface. Forces were derived numerically with left- and right-hand finite differences of 10−4 Å in the 4 × 3 Cartesian coordinates that were converted to the Jacobi coordinates used in the spherical expansion. The Newton equations of motion were integrated with the Velocity–Verlet scheme with a time-step of 0.02 fs. At the start of a trajectory the molecules were set apart with 8 Å between their centers of mass. The orientations and bond lengths of the diatomic molecules were randomly sampled taking care that the vibrational motion for v = 0 corresponds to the quasi-classical description and initial rotation corresponds to j = 0 or j = 1. The impact parameter range was stepped from 0 to bmax = 7 Å with 0.1 Å steps. At each impact parameter 50000 trajectories, varying in vibrational coordinate and orientation of monomers described by v = 0, j = 0 and v = 0, j = 1 as mentioned above, were employed. A trajectory was considered finished when the R value exceeded 8 Å again. Then the coordinates and momenta of the molecules at the end of each trajectory were stored for later analysis. The collision energy was set at 1460 cm−1 in all cases. With the chosen settings the energy conservation was better than 2 cm−1.
The analysis consisted of the evaluation of state-to-state cross sections, pair-correlated state-to-state cross sections, state-to-state differential cross sections, pair-correlated differential cross sections and scattering angle dependent alignment moments.
(a) State-to-state cross sections (σj′): for final rotational state j′ values up to 30, σj′ was obtained by replacing the integral over impact parameter by the following sum with Δb the impact parameter step size (here equal to 0.1 Å)
 000 starting trajectories. The rotation level j′ was derived from the rotational energy Erot = j′(j′ + 1)/2μr2 with μ being the diatom reduced mass and r the instantaneous bond length. The real j′ value was rounded to the nearest integer value.
000 starting trajectories. The rotation level j′ was derived from the rotational energy Erot = j′(j′ + 1)/2μr2 with μ being the diatom reduced mass and r the instantaneous bond length. The real j′ value was rounded to the nearest integer value.
(b) Pair-correlated state-to-state cross sections (σj′,j′′): these are evaluated from 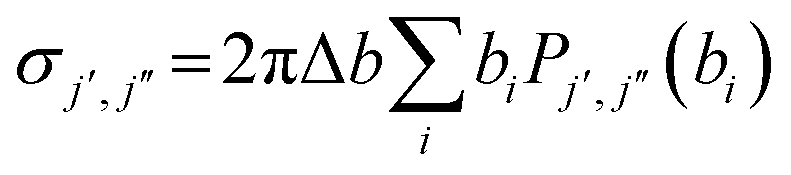 with
with  and Nj′,j′′(bi) equals the number of trajectories ending with j′ for the scattered CO product and j′′ for the scattered partner (i.e., N2) at the ith impact parameter.
and Nj′,j′′(bi) equals the number of trajectories ending with j′ for the scattered CO product and j′′ for the scattered partner (i.e., N2) at the ith impact parameter.
(c) State-to-state differential cross sections and pair-correlated differential cross sections. The state-to-state differential cross sections defined as σj′(θ) = dσj′/d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) cosθ were derived by binning the σj′ as function of the scattering angle θ in 40 equally spaced cosine intervals. The pair-correlated differential cross sections termed as σj′,j′′(θ) were derived by binning the σj′,j′′ of the product pair (j′, j′′) as function of the scattering angle θ.
cosθ were derived by binning the σj′ as function of the scattering angle θ in 40 equally spaced cosine intervals. The pair-correlated differential cross sections termed as σj′,j′′(θ) were derived by binning the σj′,j′′ of the product pair (j′, j′′) as function of the scattering angle θ.
(d) Scattering angle dependent alignment moments A{k}q: in accord with the experimental setup, the following alignment moments were calculated based on the formulas described in reference.48 In each cosine interval of the scattering angle, the alignment moments were determined by the averages:
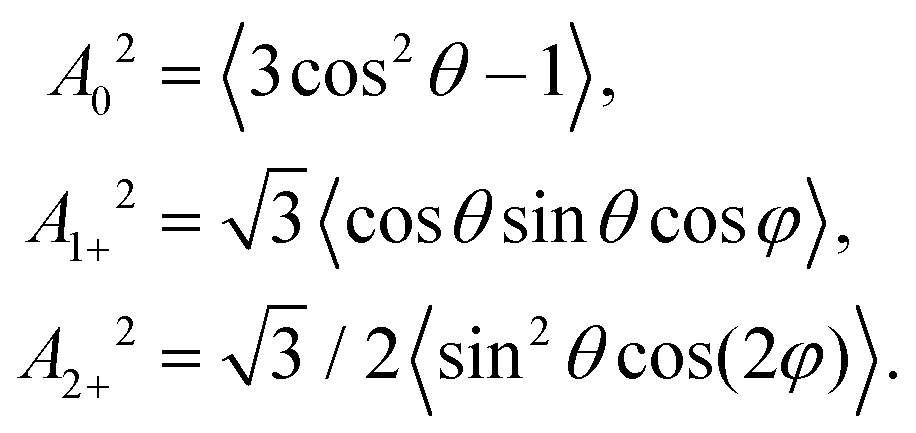
3. Results
Ref. 21 contains a presentation and discussion of pair-correlated scattering data for 13CO–CO collisions, along with a brief discussion of pair-correlated data for 13CO–N2 collisions. Pair correlation is a doubly-differential cross section measurement where angular information is recorded for separate radial regions in the scattering image, which of course relies on sufficient imaging resolution. A key experimental challenge is to account for the effects of collision induced alignment when nascent molecules are probed by linearly polarized detection lasers, in order to extract alignment-free pair correlated differential scattering cross sections. The alignment information needed for this separation could be reliably obtained from ‘normal’ rotational state dependent singly (radial-angle integrated) differential cross sections which are presented in this paper along with analysis of the collision induced alignment moments for 13CO–N2 and also 13CO–CO collisions.Scattered 13CO products from inelastic scattering with N2 were detected using a vertical and a horizontal polarized VUV probe laser via Q-branches in the 1 + 1′ + 1′′ REMPI spectra, and experimental 13CO+ velocity map images for the 13CO(j′) = 15, 17, 19, 22, 23 final states are presented in panel (a) of Fig. 1. These spectroscopic transitions used for detecting final states of scattered 13CO products are the same as those used in the 13CO + CO inelastic scattering experiment. Avoidance of overlap with the scattering signal due to collision with He carrier gas restricted study to states above j′ > 14, with the criteria of choosing strong Q-branch transitions free of overlap with other branches and perturbative states in the CO A-state spectrum. A Newton diagram that describes the experimental geometry is overlaid on the V-polarization image for the product final state j′ = 15, where the white arrows specify the velocity vectors of the colliders V13CO and VN2, the center of mass velocity VCM, while the red arrow indicates the initial relative velocity vector k. All images are presented in such a way that the initial relative velocity vector k is directed horizontally, with forward scattered angles positioned at the left side of the image and small segments around forward direction are masked because of the beamspot that coincides with the lab-frame velocity of the un-scattered 13CO molecular beam. The beamspot is well-separated from the scattered ion signal for most of the final j′ states and the space in-between grows as j′ increases.
 | ||
| Fig. 1 (a) Raw experimental velocity map images for the 13CO + N2 scattering process into the j′ = 15–23 final states of 13CO probed using (1 + 1′ + 1′′) VUV REMPI detection of 13CO. Images are presented such that the relative velocity vector is directed horizontally, with forward scattered angles positioned at the left side of the image. Small masked segments near the forward direction on the images are due to the beam spots of 13CO molecular beam. The polarization of the resonant VUV laser is indicated by ‘V Polarization’ for Elaser perpendicular to the collision plane and ‘H Polarization’ parallel to the collision plane. The sensitivity scale for the raw images is adjusted to make the main features visible. A Newton diagram for the scattering process is overlaid on the V polarization image for the 13CO j′ = 15 final state, where the velocity vectors of the two colliders (V13CO and VN2), the center-of-mass velocity (VCM) and the scattering angle (θ) are defined. The collision angle is 70° and the corresponding mean collision energy is 1460 cm−1. (b) The HIP, HOOP, VIP, and VOOP distribution components extracted from these orthogonal probe polarization images by integrating selected annulus and rectangle regions upon the images as described in ref. 39. | ||
A number of features in the raw experimental 13CO+ images can be seen directly. First of all, compared to CO + Rg inelastic scattering images we reported previously,38–40 the measured images of 13CO for inelastic scattering with N2 are much broader in the radial direction, which is attributed to the production of a range of final rotational levels of N2. Because of the small rotational constant of the N2 molecule, B0(N2) ∼ 2.0 cm−1, transitions involving rotational excitation in the collision partner N2 will cause the Newton rings for different N2 final states to (partially) blur with each other, resulting in the 13CO+ images with a broad distribution of Newton sphere radii. This image broadening effect has also been observed in the previous experiment of 13CO + CO inelastic scattering. In addition, we note that the overall size of the scattering image decreases as j′(13CO) increases because a larger amount of collision energy is required to transfer into rotational energy with excitation to a higher final rotational state of the products. When inspecting the experimental images of the same final j′(13CO) state obtained from V-polarization and H-polarization laser detection, clear differences can be seen, which indicates the strong collision induced alignment of the rotational angular momentum of the scattered products. Finally, as the detected j′(13CO) increases, the scattering images show forward scattering behaviour moving towards sideways and backward scattering eventually, for both V- or H-polarization detection.
For each final rotationally excited state j′, four sets of angular distribution components were obtained: HIP (horizontal, in-plane), HOOP (horizontal, out-of-plane), VIP (vertical, in-plane) and VOOP (vertical, out-of-plane), as shown in the panel b) of Fig. 1, and algebraic combination of them yield the state-to-state differential cross sections (DCSs) and alignment moments A{2}0, A{2}1+ and A{2}2+. The resulting state-to-state alignment-free DCSs for scattered 13CO into the j′ = 15–23 final states after collision with N2 at a mean collision energy of 1460 cm−1 are shown in the top row of Fig. 2, in comparison with theoretical predictions. We note here that the 13CO+ images and alignment-free DCSs from the current experiment on 13CO scattering with N2 look quite similar to those measured in the 13CO + CO inelastic scattering study with kinematically equivalent experimental settings: not only the image size and broadening effects, but also the angular distribution features of each image and their polarization dependence. Results for the image analysis for the state-to-state pair-correlated DCSs and alignment moments are thus expected to be similar to those for the 13CO + CO study. As shown in the 13CO + CO study described in ref. 21, analysis of the typical experimental images for the lowest detected final state of 13CO (j′ = 15) which have the largest image size and best resolution, yielded state-to-state pair-correlated DCSs and good agreement with theoretically predicted pair-correlation information had been observed. On the theoretical side, the weighted sum of these fully state-resolved pair-correlated DCSs over all the populated partner final CO rotational states, j′′(CO), results in the averaged DCS for a specific final 13CO rotational state, which show the overall preferential angular distribution of scattering as a function of j′(13CO) only. Similarly, the theoretical partner-state averaged alignment moments describe the overall alignment preference of rotational angular momentum of scattered 13CO products. In general, the overall DCSs and the overall alignment moments from the 13CO + CO experiment agree reasonably well with theoretical predictions. Ref. 21 contains a full discussion of impact parameter distributions and other dynamical aspects for CO–CO collisions which also apply to CO–N2 collisions. Therefore, only the overall DCSs and alignment moments from the 13CO + N2 experiment are shown here and compared to the corresponding overall data from the 13CO + CO system.
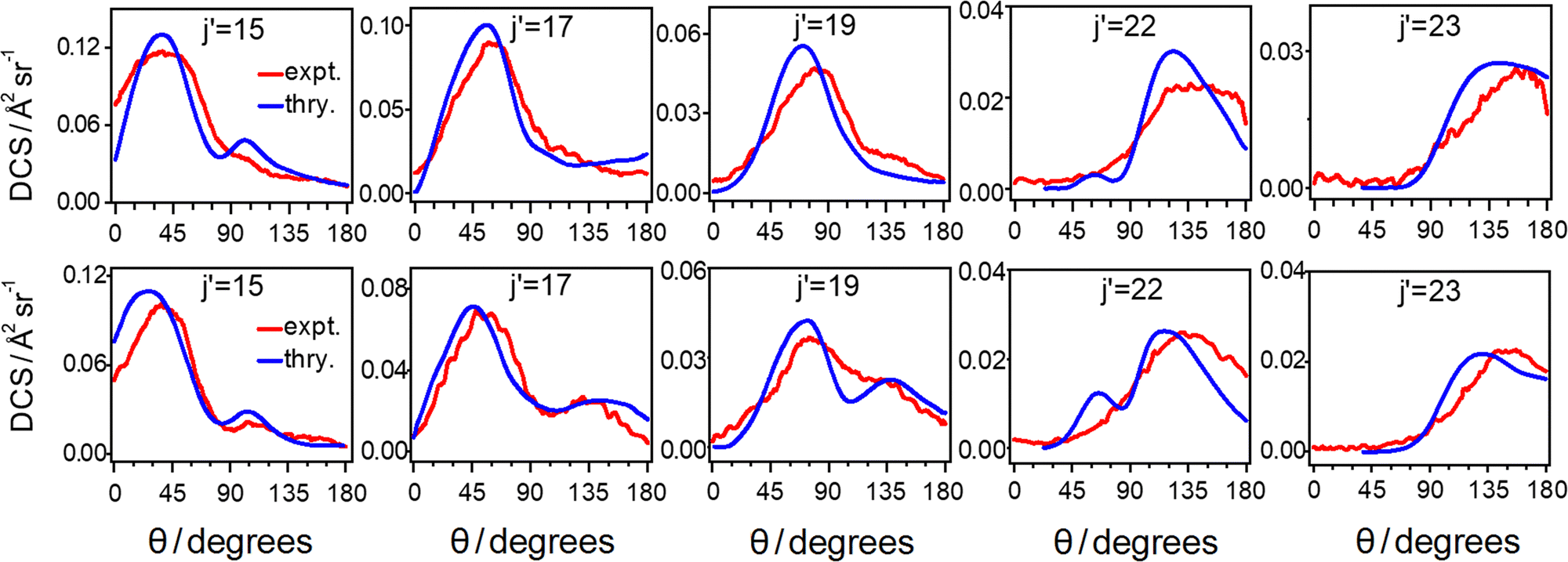 | ||
Fig. 2 Alignment-free differential cross sections for rotational energy transfer of 13CO to final states. Top row: experimental state-to-state alignment-free differential cross sections (red line) and theoretically averaged ones calculated by quasi-classical trajectory (QCT) calculations (blue line) for 13CO in collision with N2 molecules. Bottom row: experimental state-to-state alignment-free differential cross sections (red line) and theoretical averaged ones calculated by quasi-classical trajectory calculations (blue line) for 13CO in collision with CO molecules. In the comparison between experiment and theory, the experimental results are normalized to the QCT predicted data by setting the area integration of  from the experimental DCS equal to the QCT predicted DCS. from the experimental DCS equal to the QCT predicted DCS. | ||
As shown in the top row of Fig. 2, there is quite good general agreement between the experimental overall alignment-free DCSs and the theoretical averaged DCSs across all the final states of 13CO collided with N2. These DCSs display a gradual shift to increasingly larger scattering angles as j′(13CO) increases, which is consistent with the measured images in panel (a) of Fig. 1. Note that the DCSs predicted by QCT calculation go to zero in the classically forbidden region which is located at very forward scattering angles for the highest final states (j′ = 22, 23). The level of agreement between the experimentally extracted overall DCSs and the theoretically averaged DCSs for all final product states implies that the features of the 13CO–N2 PES which determine the DCS are quantitatively correct within the collision energy range sampled in this experiment.
The bottom row of Fig. 2 displays the state-to-state overall DCSs for 13CO products scattered into the same final states together with theoretical predicted DCSs, which are taken from the 13CO + CO inelastic scattering work. When comparing the results of the 13CO + N2 experiment and the 13CO + CO experiment in the context of state-to-state overall DCSs for scattered 13CO, it can be seen that the qualitative behaviour of the DCS is broadly similar in both overall structure aspect and absolute intensity aspect for the two collision partners. This indicates that the 13CO–N2 PES and the 13CO–CO PES are quite similar. However, inspection reveals that the primary maximum (maxima) of the DCSs for scattered 13CO with N2 collision partner appear(s) at consistently more backward scattering angles (≈15–20°) compared to the case of 13CO scattering with CO as collision partner. The maxima in the rotationally inelastic scattering angular distribution, to some extent, reflect the classical rotational rainbow positions, where collisions with different impact parameters result in scattering at the same final angle.
It is also apparent in Fig. 2 that for all the final states except the highest one, i.e., j′ = 23, the DCSs of scattered 13CO with CO collision partner clearly show a primary and secondary maximum in the DCSs, the latter of which become much less evident in the case of the 13CO + N2 scattering, especially for the final state range of j′ = 17–22. Previously, such double maxima were observed as well by Lorenz et al. and McBane et al. for the CO + Ne inelastic scattering,49,50 where the primary peak was attributed to scattering at the C end of the molecule and the secondary peak to either O-end scattering and/or supernumerary rainbows. This difference might be attributed to the different anisotropy of the collision partners with respect to the same primary collider 13CO. Because of the higher asymmetry of the CO molecule compared to the N2 molecule, from a simple classical view the difference between the C-end and O-end with respect to the primary collider 13CO in the CO–13CO potential is not present in the case of N2 with respect to 13CO in the N2–13CO potential. In addition, 13CO–CO shows enhanced forward scattering at θ = 0° compared to 13CO–N2, particularly for j′ = 15 products. Other details in the differences between the DCSs for 13CO–CO and 13CO–N2 are described in the Discussion section.
Turning to the alignment moments A{2}q, in order to compare the collision induced rotational angular momentum polarisation in the 13CO + N2 and 13CO + CO scattering processes clearly, experimental alignment moments for the 13CO + N2 system together with the results of QCT calculations are displayed in Fig. 3. The results for the 13CO + CO system from ref. 21 are shown in the same format style in Fig. 4. As shown there, the QCT-predicted classical PDDCSs also go to zero in the classically forbidden region, where the alignment-free DCSs are zero. Note that the signal-to-noise ratio of the V-polarization and H-polarization experimental images are also poor in the very forward scattering regions, thus the experimental alignment moment data in the range 0–30° are discarded. When concentrating on the alignment moments results for the 13CO + N2 system only, generally speaking, good agreement between experimental results and QCT calculated predictions can be seen for all the three alignment moments. For the A{2}0(θ) moments, even though the rapid oscillation behaviour in the very forward scattering region caused by quantum interferences cannot be predicted by QCT calculations, the amplitude and the overall shape of the QCT predicted A{2}0(θ) curve are well reproduced by experiment. And as the final state of scattered 13CO product j′ increases, both of the QCT calculated and experimental values are increasingly to the negative side. However, different from the CO + Ar, CO + He, and NO + Ar scattering studies, where the A{2}0(θ) moments were always found to be positive in the forward scattering region and to be at the negative limit (−1) in the backward region, the averaged A{2}0(θ) moments for 13CO scattered into such relatively high j′ states by collision with N2 molecules still reach the negative limit but with less positive values. This reveals strong ‘Frisbee’-like rotational alignment in the backward region and less ‘propeller’-like rotational alignment in the forward region. This difference of averaged A{2}0(θ) moments in the current case with respect to the molecule + Rg collision cases likely comes from the difference of alignment behaviour related to the range of rotational states of the N2 partner. The agreement between the experimental A{2}2+(θ) moments and the corresponding predictions from theory is also good, not only for the amplitude and the overall shape but also in the trends with increasing j′. Even though the agreement between the experimental and theoretical A{2}1+(θ) moments is not as good as those in the cases of the A{2}0(θ) and A{2}2+(θ) moments due to the lower precision of the experimental data from the analysis procedure, the experimental shape and trend of the A{2}1+(θ) moments reasonably agree with theory predictions. The overall good agreement of the theory calculations with the experimental alignment moments across all final states detected provides further evidence that the potential energy surface very accurately represents the interaction of the 13CO–N2 system.
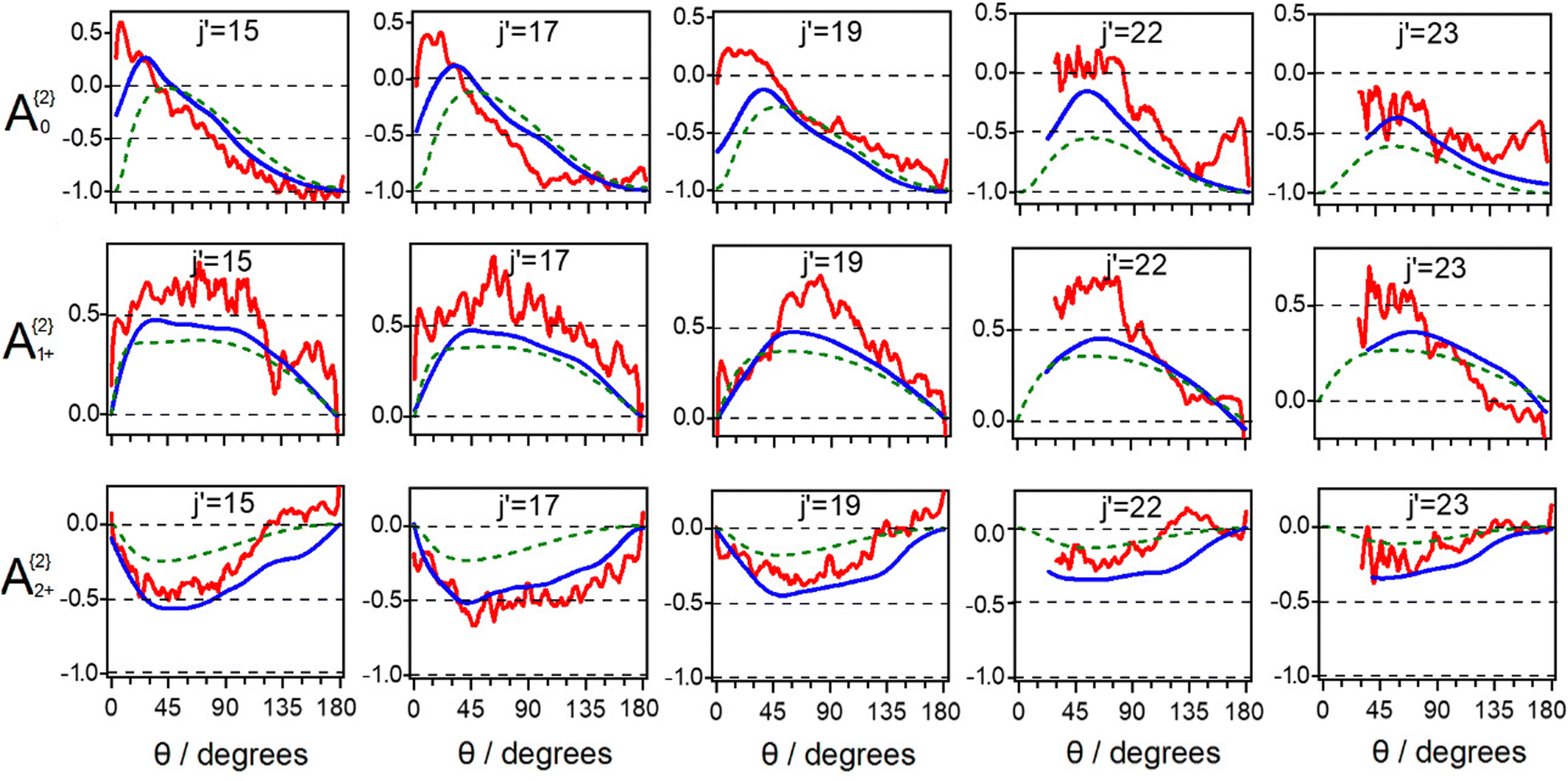 | ||
| Fig. 3 Angular momentum alignment moments A{2}q for rotational energy transfer of 13CO to final states 13CO j′ = 15–23 in collision with N2 molecules. Red line: overall alignment moments extracted from experimental polarization-dependent images; blue line: theoretically averaged alignment moments predicted by quasi-classical trajectory calculations. Predictions from the kinetic apse hard ellipsoid model are given by the green dashed lines. | ||
 | ||
| Fig. 4 Angular momentum alignment moments A{2}q for rotational energy transfer of 13CO to final states 13CO j′ = 15–23 in collision with CO molecules for the purpose of comparison with the 13CO + N2 system. Red line: overall alignment moments extracted from experimental polarization-dependent images; blue line: theoretically averaged alignment moments predicted by quasi-classical trajectory calculations. | ||
For both the 13CO + N2 and 13CO + CO systems the collision induced rotational alignment results suggest that the dominant-contribution originates from the repulsive part of the interaction potential of the colliding systems. However, only moderate and high final rotational states of the scattered products were experimentally probed here, where the hard-shell nature of the interaction is expected to mainly contribute to the collision induced alignment.
4. Discussion
The stereodynamic properties determined from differential cross sections for 13CO + N2versus13CO + CO, as well as the model and parameters describing collision induced alignment, are similar, which is consistent with the observed behavior in state-to-state DCSs for NO(X) and NO(A) in collision with D2 and He, where D2 and He are kinematically similar collision partners. For our systems, however, quite large differences in the pair-correlated data for 13CO + N2versus13CO + CO were described in ref. 21 for the forward scattering region. A significant component found there for 13CO + CO were products in symmetrically excited final states, i.e., j′(13CO) = j′′(CO) = 14, 15, 16, 17, which were not observed at all for the 13CO + N2 system. Note that these differences in the two systems appear strongly in the pair-correlated doubly differential cross section data, and are less obvious in the normal, singly DCS data, in Fig. 2, which illustrates the wider scope of dynamic information provided by pair correlation measurements. In speculations about the origin of these differences particularly in forward scattering for the two systems we first consider differences in their potential energy surfaces and then show that the (miss-)use of QCT provides additional hints.In our QCT calculations the PESs of both CO–CO and CO–N2 are represented by a set of expansion functions, following the convention given in ref. 51. A plot of the spherical expansion analysis of the two pair potentials is shown in Fig. 5. The isotropic terms (black, full line for CO–CO, dashed line for CO–N2) are rather similar, showing nearly the same well depth, while CO–N2 is slightly less repulsive. The dipole–dipole term is given in red, and is of course absent for CO–N2. The dipole quadrupole term is in blue, and the quadrupole–dipole in magenta (opposite in sign from the dipole–quadrupole term) is again absent for CO–N2. Finally, the quadrupole-quadrupole term (green) is somewhat smaller for CO–N2. These terms are not confined to electrostatic interactions but include also the short-range interactions with the same orientational dependence as the electrostatic multipole-multipole interactions. From Fig. 5 it is clear that considerations involving only the small dipole moment of CO and the lack thereof for N2 are misleading since quadrupole moments play an equally or more important role in the pair potentials. Note also that the collision energy of 1460 cm−1 is much higher than the equilibrium well depth of the two clusters. Short range (mainly repulsive) forces that depend strongly on the relative molecular orientations determine whether the attractive forces in the pair potential will be sampled during the collision. At 2.5 Å, for example, the least unfavourable orientation already has an energy more than twice the collision energy and other orientations are up to 35 times the collision energy. The forces calculated at the shortest distance possible should therefore also be correct.
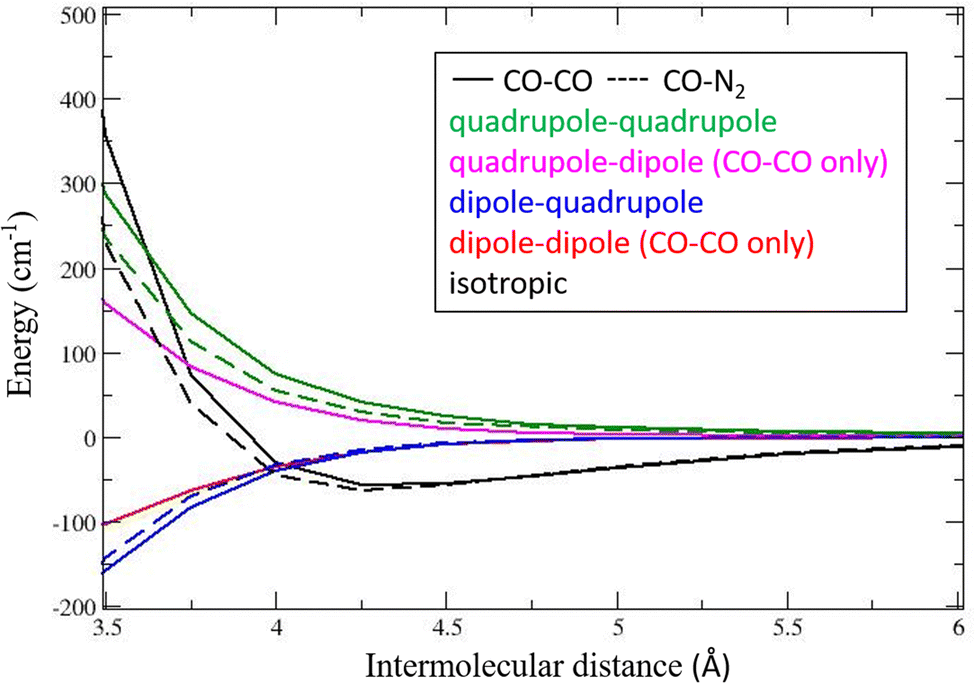 | ||
| Fig. 5 Spherical expansion of the pair potentials for CO–CO and CO–N2 in terms of electrostatic moment interactions. | ||
The balance and time development of repulsive and attractive forces is critical in explaining the exceptional forward scattering mechanism seen only in the 13CO + CO system. This is illustrated in Fig. 6, which tracks the interaction energy of a successful forward scattering with symmetric excitation (FSSE) collision at 1460 cm−1 between two CO molecules that start off with an initial orientation where the two C atom ends of the molecules strike each other. At the start of the collision, a repulsive interaction of ∼150 cm−1 takes place which is followed by an attractive interaction of ∼−100 cm−1, corresponding to an anti-geared rotation of the molecules around each other continuing to the point where the two O-atoms reach closest approach. Symmetric excitation may be favored for efficient anti-geared motion after the initial C-end collision. This attractive force after the large rotational energy transfer (j′ 13CO = j′ CO = 15, RET = 2 × 462 cm−1 = 924 cm−1) collision results in a torque of the collision pair during separation that effectively redirects the initially sideways recoil angle towards the forward direction, as illustrated schematically in the lower panel of Fig. 6 (from ref. 21). The concept of forward scattering due to attractive forces following large rotational energy transfer was recently52 applied with success to other molecule-molecule collision systems.
 | ||
| Fig. 6 Time development traces of the interaction energy for two similar trajectories for CO–CO (ref. 21) and CO–N2. | ||
A remaining question is why no FSSE channel was found in the CO + N2 system, despite the kinematic similarity with CO + CO. With QCT theory it is possible to force a CO + N2 collision to follow the motion of a trajectory leading to FSSE in a CO + CO collision. The trajectory data of the CO–CO collision described in Fig. 2 of ref. 21, which results in the potential energy plot with the black line in Fig. 6 was used to calculate the potential interaction for the case where CO collides with N2, given by the red line in Fig. 6. At each time step, the geometry is that of CO on CO but the potential is that for CO on N2. The trajectory is thus in the case of CO on N2 purely fictitious since the run is not on the own CO–N2 potential. Still, some insight is provided: at the start of the trajectory the CO–CO and CO–N2 potentials are rather similar. One might therefore assume that with the same initial geometry CO + N2 would follow a similar path as CO + CO. At 250 fs there is for both systems an increasing repulsive potential. In the case of CO + CO collision this repulsion is responsible for the typical (15, 15) forward scattering behavior. Since the repulsion is approximately 3 times smaller for CO + N2, the collisional kick is not large enough for excitation into e.g. (15, 15). In fact, only a miniscule number of CO + N2 trajectories on the actual CO–N2 potential energy surface (which sample all initial geometries) resulted in (15, 15) or similar high rotational excitation, and these contributed only to the primary rainbow scattering angle at ∼80 degrees.
5. Conclusion
State-to-state DCSs and scattering angle resolved alignment moments for rotational energy transfer for the 13CO + N2 inelastic scattering have been measured under kinematically equivalent conditions as 13CO + CO scattering and compared to the corresponding results of the 13CO + CO system. For each scattering system, there is good agreement between experimental measurements and theoretical predictions, which confirms that the calculated PESs represent the interactions in the colliding system very well. Comparison of the observed scattering stereodynamics for collision of 13CO with N2 and CO as collision partners provide extra information on the subtle differences between the two PESs. The broad similarities between the state-to-state DCSs and the qualitatively similar behaviours of the collision induced rotational alignment moments for the two collision partners indicate that the 13CO–N2 PES in general has a similar form as the 13CO–CO PES and that the dynamics follow predictions of a hard-shell collision model. Close inspection reveals that the primary rainbow maximum in DCSs for 13CO in collision with N2 appears at larger, more backward scattering angles compared to those from scattering with CO, implying that the 13CO–N2 PES is less anisotropic than the 13CO–CO PES. This cannot be simply attributed to the dipole–dipole interaction of the 13CO–CO system since dipole–quadrupole and quadrupole–quadrupole interactions are important for both 13CO + CO and 13CO + N2 and also short range repulsive forces (shape effects) play a role. The secondary maximum for scattering with N2 is also less obvious which may result from the configurational asymmetry of a CO molecule compared to N2. The doubly-differential product pair correlated DCS data for the two systems has previously revealed a new unexpected channel labelled forward scattering with symmetric excitation (FSSE) only for the 13CO + CO system. Trajectory analysis by QCT theory explained FSSE in 13CO + CO as a face-to-face/rotation/back-to-back or ‘do-si-do’ synchronous motion between the two colliding molecules. However, no FSSE is observed for the kinematically similar 13CO + N2 system. By forcing 13CO + N2 to follow the initial 13CO + CO trajectory of an FSSE collision using QCT, it appears that it may be difficult for 13CO + N2 trajectories to reach a repulsive wall high enough to cause the deeply inelastic energy transfer.Conflicts of interest
The authors declare no competing interests.Acknowledgements
ZFS is supported by the National Natural Science Foundation of China (no. 22103003), Anhui Provincial Natural Science Foundation (no. 2208085Y02) and the Natural Science Research Project of Education Department of Anhui Province (no. KJ2021ZD0008). This project has moreover received funding from the University Annual Scientific Research Plan of Anhui Province (no. 2022AH010013) and the open foundation of the state key laboratory of molecular reaction dynamics in DICP, CAS (no. SKLMRD-K202204). DHP, RJAS and MCvH acknowledge the NWO-Dutch Astro-chemistry Network II project number 648.000.024, and the NWO-TOP project number 715.013.002 for support. AGS acknowledges the US National Science Foundation under award CHE-1955239. All the authors thank Prof. Gerrit C. Groenenboom and Prof. Sebastiaan Y. T. van de Meerakker (RU Nijmegen) for helpful discussions on this paper. We thank Prof. Mark Brouard (University of Oxford) for offering a program that allows the calculations of the alignment moments based on the kinematic apse model that was coded by B. Hornung.References
- G. Paterson, M. L. Costen and K. G. McKendrick, Int. Rev. Phys. Chem., 2012, 31, 69–109 Search PubMed.
- H. Chadwick, M. Brouard, T. Perkins and F. J. Aoiz, Int. Rev. Phys. Chem., 2014, 33, 79–123 Search PubMed.
- M. L. Costen, S. Marinakis and K. G. McKendrick, Chem. Soc. Rev., 2008, 37, 732–743 RSC.
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- K. T. Lorenz, D. W. Chandler, J. W. Barr, W. W. Chen, G. L. Barnes and J. I. Cline, Science, 2001, 293, 2063–2066 CrossRef CAS PubMed.
- H. Chadwick, B. Nichols, S. D. S. Gordon, B. Hornung, E. Squires, M. Brouard, J. Kłos, M. H. Alexander, F. J. Aoiz and S. Stolte, J. Phys. Chem. Lett., 2014, 5, 3296–3301 CrossRef CAS PubMed.
- M. Brouard, H. Chadwick, C. J. Eyles, B. Hornung, B. Nichols, F. J. Aoiz, P. G. Jambrina and S. Stolte, J. Chem. Phys., 2013, 138, 104310 CrossRef CAS PubMed.
- S. N. Vogels, J. Onvlee, S. Chefdeville, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, Science, 2015, 350, 787–790 CrossRef CAS PubMed.
- J. I. Cline, K. T. Lorenz, E. A. Wade, J. W. Barr and D. W. Chandler, J. Chem. Phys., 2001, 115, 6277–6280 CrossRef CAS.
- M. Brouard, H. Chadwick, S. D. S. Gordon, B. Hornung, B. Nichols, J. Kłos, F. J. Aoiz and S. Stolte, J. Chem. Phys., 2014, 141, 164305 CrossRef PubMed.
- M. Brouard, H. Chadwick, C. J. Eyles, B. Hornung, B. Nichols, F. J. Aoiz, P. G. Jambrina, S. Stolte and M. P. de Miranda, J. Chem. Phys., 2013, 138, 104309 CrossRef CAS PubMed.
- A. Gijsbertsen, H. Linnartz, G. Rus, A. E. Wiskerke, S. Stolte, D. W. Chandler and J. Kłos, J. Chem. Phys., 2005, 123, 224305 CrossRef CAS PubMed.
- M. S. Elioff and D. W. Chandler, J. Chem. Phys., 2002, 117, 6455–6462 CrossRef CAS.
- M. Brouard, H. Chadwick, C. J. Eyles, B. Hornung, B. Nichols, J. M. Scott, F. J. Aoiz, J. Kłos, S. Stolte and X. Zhang, Mol. Phys., 2013, 111, 1759–1771 CrossRef CAS.
- T. R. Sharples, T. F. M. Luxford, D. Townsend, K. G. McKendrick and M. L. Costen, J. Chem. Phys., 2015, 143, 204301 CrossRef PubMed.
- A. von Zastrow, J. Onvlee, S. N. Vogels, G. C. Groenenboom, A. van der Avoird and S. Y. T. van de Meerakker, Nat. Chem., 2014, 6, 216–221 CrossRef CAS PubMed.
- J. D. Steill, J. J. Kay, G. Paterson, T. R. Sharples, J. Kłos, M. L. Costen, K. E. Strecker, K. G. McKendrick, M. H. Alexander and D. W. Chandler, J. Phys. Chem. A, 2013, 117, 8163–8174 CrossRef CAS PubMed.
- T. de Jongh, Q. Shuai, G. L. Abma, S. Kuijpers, M. Besemer, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, Nat. Chem., 2022, 14, 538–544 CrossRef CAS PubMed.
- T. de Jongh, M. Besemer, Q. Shuai, T. Karman, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, Science, 2020, 368, 626–630 CrossRef CAS PubMed.
- T. F. M. Luxford, T. R. Sharples, K. G. McKendrick and M. L. Costen, J. Chem. Phys., 2016, 145, 174304 CrossRef PubMed.
- Z. F. Sun, M. C. van Hemert, J. Loreau, A. van der Avoird, A. G. Suits and D. H. Parker, Science, 2020, 369, 307–309 CrossRef CAS PubMed.
- M. S. Westley, K. T. Lorenz, D. W. Chandler and P. L. Houston, J. Chem. Phys., 2001, 114, 2669–2680 CrossRef CAS.
- A. Gijsbertsen, H. Linnartz and S. Stolte, J. Chem. Phys., 2006, 125, 133112 CrossRef PubMed.
- Z. Gao, S. N. Vogels, M. Besemer, T. Karman, G. C. Groenenboom, A. van der Avoird and S. Y. T. van de Meerakker, J. Phys. Chem. A, 2017, 121, 7446–7454 CrossRef CAS PubMed.
- T. de Jongh, T. Karman, S. N. Vogels, M. Besemer, J. Onvlee, A. G. Suits, J. O. F. Thompson, G. C. Groenenboom, A. van der Avoird and S. Y. T. van de Meerakker, J. Chem. Phys., 2017, 147, 013918 CrossRef PubMed.
- T. F. M. Luxford, T. R. Sharples, D. Townsend, K. G. McKendrick and M. L. Costen, J. Chem. Phys., 2016, 145, 084312 CrossRef PubMed.
- S. Chefdeville, Y. Kalugina, S. Y. T. van de Meerakker, C. Naulin, F. Lique and M. Costes, Science, 2013, 341, 1094–1096 CrossRef CAS PubMed.
- C. K. Bishwakarma, G. van Oevelen, R. Scheidsbach, D. H. Parker, Y. Kalugina and F. Lique, J. Chem. Phys., 2018, 149, 121101 CrossRef PubMed.
- C. H. Yang, G. Sarma, D. H. Parker, J. J. ter Meulen and L. Wiesenfeld, J. Chem. Phys., 2011, 134, 204308 CrossRef PubMed.
- O. Tkáč, A. K. Saha, J. Loreau, Q. L. Ma, P. J. Dagdigian, D. H. Parker, A. van der Avoird and A. J. Orr-Ewing, Mol. Phys., 2015, 113, 3925–3933 CrossRef.
- O. Tkáč, Q. L. Ma, C. A. Rusher, S. J. Greaves, A. J. Orr-Ewing and P. J. Dagdigian, J. Chem. Phys., 2014, 140, 204318 CrossRef PubMed.
- G. Tang, M. Besemer, J. Onvlee, T. Karman, A. van der Avoird, G. C. Groenenboom and S. Y. T. van de Meerakker, J. Chem. Phys., 2022, 156, 214304 CrossRef CAS PubMed.
- O. Tkáč, Q. L. Ma, M. Stei, A. J. Orr-Ewing and P. J. Dagdigian, J. Chem. Phys., 2015, 142, 014306 CrossRef PubMed.
- T. F. M. Luxford, T. R. Sharples, K. G. McKendrick and M. L. Costen, J. Chem. Phys., 2017, 147, 013912 CrossRef PubMed.
- B. Yan, P. F. H. Claus, B. G. M. van Oorschot, L. Gerritsen, A. T. J. B. Eppink, S. Y. T. van de Meerakker and D. H. Parker, Rev. Sci. Instrum., 2013, 84, 023102 CrossRef CAS PubMed.
- C. M. Western, A Program for Simulating Rotational, Vibrational and Electronic Structure, version 8.0, University of Bristol, 2014, https://pgopher.chm.bris.ac.uk Search PubMed.
- Z. F. Sun, A. D. von Zastrow and D. H. Parker, J. Chem. Phys., 2017, 147, 013909 CrossRef CAS PubMed.
- L. Song, G. C. Groenenboom, A. van der Avoird, C. K. Bishwakarma, G. Sarma, D. H. Parker and A. G. Suits, J. Phys. Chem. A, 2015, 119, 12526–12537 CrossRef CAS PubMed.
- A. G. Suits, C. K. Bishwakarma, L. Song, G. C. Groenenboom, A. van der Avoird and D. H. Parker, J. Phys. Chem. A, 2015, 119, 5925–5931 CrossRef CAS PubMed.
- Z. F. Sun, C. K. Bishwakarma, L. Song, A. van der Avoird, M. C. van Hemert, A. G. Suits, G. C. McBane and D. H. Parker, Phys. Chem. Chem. Phys., 2019, 21, 9200–9211 RSC.
- M. Brouard, S. D. S. Gordon, A. H. Boyle, C. G. Heid, B. Nichols, V. Walpole, F. J. Aoiz and S. Stolte, J. Chem. Phys., 2017, 146, 204304 CrossRef CAS PubMed.
- L. A. Surin, D. N. Fourzikov, T. F. Giesen, S. Schlemmer, G. Winnewisser, V. A. Panfilov, B. S. Dumesh, G. W. M. Vissers and A. van der Avoird, J. Phys. Chem. A, 2007, 111, 12238–12247 CrossRef CAS PubMed.
- L. A. Surin, I. V. Tarabukin, S. Schlemmer, Y. N. Kalugina and A. van der Avoird, J. Chem. Phys., 2018, 148, 044313 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- J. A. Coxon and P. Hajigeorgiou, J. Chem. Phys., 2004, 121, 2992–3008 CrossRef CAS PubMed.
- K. P. Huber and G. H. Herzberg, Constants of Diatomic Molecules, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, 2020.
- X. Hu, W. L. Hase and T. Pirraglia, J. Comput. Chem., 1991, 12, 1014–1024 CrossRef CAS.
- M. P. de Miranda, F. J. Aoiz, L. Banares and V. S. Rabános, J. Chem. Phys., 1999, 111, 5368–5383 CrossRef CAS.
- G. C. McBane and S. M. Cybulski, J. Chem. Phys., 1999, 110, 11734–11741 CrossRef CAS.
- K. T. Lorenz, D. W. Chandler and G. C. McBane, J. Phys. Chem. A, 2002, 106, 1144–1151 CrossRef CAS.
- M. C. van Hemert, J. Chem. Phys., 1983, 78, 2345–2354 CrossRef CAS.
- M. Besemer, G. Tang, Z. Gao, A. van der Avoird, G. C. Groenenboom, S. Y. T. van de Meerakker and T. Karman, Nat. Chem., 2022, 2022, 1–6 Search PubMed.
| This journal is © the Owner Societies 2023 |