
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A SIFT-MS study of positive and negative ion chemistry of the ortho-, meta- and para-isomers of cymene, cresol, and ethylphenol†
Stefan J
Swift
 *a,
Nikola
Sixtová
a,
Maroua
Omezzine Gnioua
ab and
Patrik
Španěl
a
*a,
Nikola
Sixtová
a,
Maroua
Omezzine Gnioua
ab and
Patrik
Španěl
a
aJ. Heyrovsky Institute of Physical Chemistry of CAS, v.v.i, Dolejškova 2155/3, 182 23 Prague, Czechia. E-mail: stefan.swift@jh-inst.cas.cz
bFaculty of Mathematics and Physics, Charles University, Ke Karlovu 3, 120 00 Prague, Czechia
First published on 20th June 2023
Abstract
Selected Ion Flow Tube Mass Spectrometry (SIFT-MS) is a soft ionisation technique based on gas phase ion–molecule reaction kinetics for the quantification of trace amounts of volatile organic compound vapours. One of its previous limitations is difficulty in resolving isomers, although this can now be overcome using different reactivities of several available reagent cations and anions (H3O+, NO+, O2+˙, O−˙, OH−, O2−˙, NO2−, NO3−). Thus, the ion–molecule reactions of these eight ions with all isomers of the aromatic compounds cymene, cresol and ethylphenol were studied to explore the possibility of their immediate identification and quantification without chromatographic separation. Rate coefficients and product ion branching ratios determined experimentally for the 72 reactions are reported. DFT calculations of their energetics confirmed the feasibility of the suggested reaction pathways. All positive ion reactions proceeded fast but largely did not discriminate between the isomers. The reactivity of the anions was much more varied. In all cases, OH− reacts by proton transfer forming (M–H); NO2− and NO3− were unreactive. The differences observed for product ion branching ratios can be used to identify isomers approximately.
1. Introduction
SIFT-MS is a powerful soft mass spectrometric technique that allows for the quantification of volatile organic compounds (VOCs) in the gas-phase, usually air. Whilst analyses of mixtures of multiple analytes are possible, it is currently not easy to analyse isomeric or isobaric species present simultaneously within a matrix. However, as the range of reagent ions available to SIFT-MS has been recently expanded to include anions (H3O+, NO+, O2+˙, O−˙, OH−, O2−˙, NO2−, NO3−), there is potential to use differing ionic products, produced from the range of reactions to facilitate simultaneous quantification, of isobaric species using SIFT-MS.Aromatic compounds are an essential chemical class which are analysed in gas-phase complex matrices including human breath;1–3 headspace of samples such as cosmetics4 and food stuffs;5 ambient air for air quality analysis for environmental monitoring purposes;6–9 as well as investigating ion–molecule reactions, at the centre of the SIFT-MS technique. In particular, aromatic compounds are often of great interest in the field of atmospheric chemistry and air pollution research as many of these species are known to be highly toxic and are released from anthropogenic emissions into the atmosphere, diminishing air quality. Some aromatic compounds may also however be found in human breath2 and are known to be potential breath biomarkers for certain diseases, such as various cancers,10 COPD,11 and polycystic ovarian syndrome.12
Previous studies of ion molecule reactions of H3O+, NO+ and O2+˙ (in an He carrier gas) have been carried out for several aromatic compounds with ortho-, meta- and para-positioning of chemical groups. This included the three isomers of xylene13 and the ortho- and para-isomers of F, Cl, Br and I containing halotoluenes14 which showed very minor differences in the branching ratios in the O2+˙ reaction. Also, work has been conducted15 which investigated the H3O+, NO+ and O2+˙ reactions with ortho-, meta-, and para-cresol, using He carrier gas. Although the separate quantification of the ortho-, meta-, and para-cresol isomers was not possible on the basis of this work when using just the positive reagent ions, recent developments in the SIFT-MS technique has now allowed for the inclusion of negative reagent ions (OH−, O2−˙, O−˙, NO2−, NO3−).16,17 The inclusion of the extra five negative reagent ions means that the classical way of determining the relative reaction rate coefficients (in which all ions are injected at once into the flow tube) is not possible. The inclusion of these negative reagent ion species to SIFT-MS thus warrants further investigation into whether isomers can be identified or even quantified separately within a mixture, based on their differing chemistry with the available reagent ion species.
To survey the potential of the negative ions to differentiate ortho-, meta- and para-isomers, we chose cymene, cresol, and ethylphenol as they are of particular interest in both atmospheric chemistry and disease diagnosis. Cresol is known to be an oxidation product of toluene,18–20 anthropogenically emitted from vehicle exhaust emissions, for example;21 the alkylbenzene, cymene, is known to be emitted from municipal solid waste treatment plants22 and landfill,22 as well as formed secondarily in the atmosphere;23 and 4-ethylphenol is known to be found in food stuffs, such as red wine.24 These compounds have therefore been the basis for our investigation into the ion–molecule reactions occurring when the reagent ions produced by the SIFT-MS instrument interact with these analytes, and how the chemistry changes when functional groups are found in varying positions around the aromatic ring.
The objective of this work was therefore to understand the differences in ion chemistry between the ortho-, meta-, and para-isomers of compounds for the ion–molecule reactions which occur within the flow-tube. Furthermore, to develop on from the previous work we have updated the kinetics library of the ion–molecule reactions, now with negative ions included (as well as within the N2 carrier gas).
2. Experimental
The principles of SIFT-MS are based on ion–molecule reactions which occur in a flow-tube between reagent ion species and neutral analyte compounds, as to produce varying product ions (even if the species are isobaric or isomeric). The reagent ions are produced in an ion source when a mixture of air and water is presented with microwave radiation from a magnetron to produce a plasma ion soup. The ions in the plasma are attracted towards the first quadrupole and enter the quadrupole mass filter, as to selectively filter one reagent ion to undergo reactions with the neutral analyte species in the flow tube (with either a He or N2 carrier gas). The product ions are directed through a second quadrupole which is used to analyse the sample matrix. The product ions then hit a detector which sends an electrical signal through to a computer, for which a software package allows for the conversion of this signal to gas-phase concentrations, when the branching ratios and kinetic parameters for a reaction are known.2.1 Instrument and standards
One of the latest models of the Syft Voice200 series of instruments, the Voice200infinity (Syft TechnologiesTM, New Zealand), was used to analyse 9 aromatic compounds for their branching ratios and rate coefficients when presented with 8 different reagent ion species. In the previous work by Wang et al.,15 a SIFT-MS instrument was used with the three positive reagent ions (H3O+, NO+ and O2+˙) to investigate ion–molecule reactions. In recent years however (since 2015),16 Syft TechnologiesTM have developed the Voice200infinity instrument which increases the number of possible reagent ions used by this analytical technique, to an extensive 8 reagent ions (with the addition of OH−, O2−˙, O−˙, NO2− and NO3−).16 With a larger number of reagent ions to choose from, a much greater scope of potential ion–molecule reactions is possible and the prospect of measuring ortho-, meta- and para-isomers individually, increases.To investigate ion–molecule reactions using a SIFT-MS instrument it is preferable to use a container into which analyte standards are injected that doesn't equalise the flow of air and in which the volume of the vessel is able to change easily, without changing the internal pressure. A nalophan sample bag was therefore used in which the bags were filled with dry pure air (generated by zero air generator Parker Hannifin Manufacturing Ltd, Model 636273000) and were spiked separately with the 9 different aromatic compounds, used for SIFT-MS analysis. Nalophan is known to release ethylene glycol,25 although it is not believed that this would interfere with the aromatic species investigated in this study. Nine different aromatic compounds were analysed, for which a large concentration of standards headspace (from a saturated atmosphere of having injected droplets of standard liquid in to a blank nalophan bag) of each species was injected using a syringe into the nalophan bags, followed by subsequent dilutions. Note that CO2 produced in the zero air generator was not removed and was thus present in all experiments at a small concentration.
The compounds analysed were ortho-cymene, meta-cymene and para-cymene, ortho-cresol, meta-cresol, para-cresol, ortho-ethylphenol, meta-ethylphenol and para-ethylphenol (all purchased from Sigma-Aldrich as analytical standards). The structures of these compounds are shown in Fig. 1.
 | ||
| Fig. 1 The structures of the nine compounds investigated in this work. | ||
A passivated needle (Syft TechnologiesTM) attached to the heated sample injection inlet (cone at 323 K) was pierced into the nalophan bag to sample the gaseous standard compound. Some of the standards were solid at room temperature (para-cresol and para-ethylphenol) and therefore to keep the species in the gaseous phase, the sampling capillary was heated to 323 K. The analyte molecules then entered a flow of N2 within the instrument flow tube which was at a temperature of 393 K.
The Voice200infinity is able to produce the required reagent ions from the ion source under three separate conditions required for the positive ion mode (H3O+, NO+ and O2+˙), the first negative mode (OH− and O2−˙) as well as the second negative mode (O−˙, NO2− and NO3−). After being selected by the quadrupole mass filter, the reagent ions also enter the flow tube and react with the analyte ions. N2 was the carrier gas used inside the flow tube at a pressure of 410 mTorr. Note that when injecting H3O+ into the N2 carrier gas the relative signals of the reagent ions and their hydrates were 70–80% H3O+, 15–30% of H3O+H2O and less than 2% H3O+(H2O)2. Despite the large mass difference between N2 and He, the branching ratios and rate coefficients for a range of compounds was found not to change between these two carrier gases,26 although the use of N2 made it necessary to need to dry the instrument out at the start of the day due to the internal condensation of water vapour while the instrument was in standby mode, overnight.
2.2 Determination of product ion branching ratios
The branching ratios were determined by first obtaining full scan spectra in the m/z range 15 to 250, for each reagent ion, for each of the 9 compounds analysed. The major product ions in each of the spectra were identified and an ion list was produced for each compound for the application of the Selected Ion Monitoring (SIM) mode on the Voice200infinity instrument.The SIM scans were obtained for a nalophan bag filled with zero air (blank), followed by a sequence of different concentrations of neutral analyte species, after injection of the compound headspace into the nalophan bag. The SIM scan mode is able to switch between the three different ion source conditions efficiently. At a constant concentration, 50 measurement data points were obtained for each product ion species across all source conditions (and therefore all reagent ions). This was repeated for several dilutions to obtain data across a range of concentrations.
Only the last 20 data points of each set of product ions were used for data analyses, as the source conditions required time to stabilise the reagent ion concentrations when switching between modes (N.B. this is graphically shown in Fig. S1 within the ESI†). The average across these data points was taken and the standard error (SE) was calculated. For each reagent ion, the branching ratios were calculated by summing the total product ion signals and dividing the individual signals by the total product ion count.
In most cases, only one product ion was found, although more complex chemistry was observed with some of the negative reagent ion species. For species which observed more than one reagent ion product, the calculated branching ratios at each concentration were plotted against the count rates of the known major product ions. The intercept of the trend was then taken as the true branching ratio of the reaction.27
2.3 Determining reaction rate coefficients
The ion source in the Voice200infinity instrument operates in three separate conditions. These are positive mode; negative mode dry; and negative mode wet. Therefore, it is not possible to inject all the ions simultaneously into the flow tube. As a result, the relative decay curves cannot be compared to the decay and regeneration of H3O+ (known to react via proton transfer to the analyte, at the collisional rate coefficient, kc). To obtain the reaction rate coefficients, the ratio of the total products [P] was divided by the total reagents species (including their hydrates) [R] and the [P]/[R] for each reagent ion, at different stable concentrations (using nalophan bags), for each reaction, were then compared to the reaction of the analyte with H3O+ (which occurs at the collisional rate).The relative slopes of the [P]/[R] were compared to H3O+, to give the relative rates of reaction between the reagent ions and the analyte molecule. The relative rates were then multiplied by the kc of H3O+ to calculate the rate coefficient of reaction. The collisional rates were calculated by using the method described by Su and Chesnavich.28
2.4 DFT calculations
All quantum chemistry calculations were performed using the ORCA 5.0.1 software. Molecular geometry forms of all neutral ortho-, meta- and para-isomers of aromatic molecules, their protonated forms as well as the product ions resulting from their reaction with the positive and negative reagent ions were first drawn using the AVOGADRO software. These were then further optimised using the ORCA programme with the B3LYP DFT and 6-311++G(d,p) basis set, with a D4 correction. This level of theory was also used to calculate the normal mode vibrational frequencies and thermodynamic quantities of the structures of the neutral molecules and reagent ions. The total enthalpies of all neutral molecules and ions were thus calculated for standard temperature and pressure (298.15 K, 1 atm). The calculations were performed for several feasible structures of each of the ions and the lowest energy structure was then chosen. Enthalpy and entropy changes were then calculated for the neutral and ionic reactants, as well as the products of all reactions. Proton affinities, polarizabilities and dipole moments were also obtained using this DFT method.3. Results and discussion
The branching ratio and reaction rate kinetics results which are required for the accurate quantification of gas-phase concentrations were carried out under similar conditions in which the Voice200infinity instrument is routinely used by the majority of its users.It's known that most of these compounds give only one product ion when using the H3O+ and NO+ ions, although a range of different products were produced when reacting these analyte species in the negative ion mode. The branching ratios calculated for the 9 compounds on their reaction with the available reagent ions in the Voice200infinity are shown in Table 1. As can be seen in Table 1, many reactions resulted in a single product which makes individual calculation simpler, although causes the accurate separate quantification of the three isomeric species for each compound to be more challenging.
| Compound | H3O+ | NO+ | O2+˙ | O−˙ | OH− | O2−˙ | NO2− | NO3− | |
|---|---|---|---|---|---|---|---|---|---|
| a — Indicates no reaction. | |||||||||
| Cymene | ortho- | 135 C10H15+ (94) | 134 C10H14+˙ (100) | 134 C10H14+˙ (10) | —a | 133 C10H13− (88) | — | — | — |
| 93 C7H9+ (6) | 119 C9H11+ (90) | 119 C8H7O− (12) | |||||||
| meta- | 135 C10H15+ (98) | 134 C10H14+˙ (100) | 134 C10H14+˙ (23) | — | 133 C10H13− (99) | — | — | — | |
| 93 C7H9+ (2) | 119 C9H11+ (77) | 119 C8H7O− (1) | |||||||
| para- | 135 C10H15+ (96) | 134 C10H14+˙ (100) | 134 C10H14+˙ (10) | — | 133 C10H13− (97) | — | — | — | |
| 93 C7H9+ (4) | 119 C9H11+ (90) | 119 C8H7O− (3) | |||||||
| Cresol | ortho- | 109 C7H8OH+ (100) | 108 C7H8O+˙ (100) | 108 C7H8O+˙ (98) | 109 C6H5O2− (2) | 107 C7H7O− (100) | 140 C7H8O·O2−˙ (16) | 154 C7H8O·NO2− (100) | 170 C7H8O·NO3− (100) |
| 107 C7H7O+ (2) | 107 C7H7O− (83) | 107 C7H7O− (84) | |||||||
| 106 C7H6O−˙ (15) | |||||||||
| meta- | 109 C7H8OH+ (100) | 108 C7H8O+˙ (100) | 108 C7H8O+˙ (99) | 109 C6H5O2− (2) | 107 C7H7O− (95) | 140 C7H8O·O2−˙ (29) | 154 C7H8O·NO2− (100) | 170 C7H8O·NO3− (100) | |
| 107 C7H7O+ (1) | 107 C7H7O− (92) | 93 C6H5O− (5) | 107 C7H7O− (71) | ||||||
| 106 C7H6O−˙ (6) | |||||||||
| para- | 109 C7H8OH+ (100) | 108 C7H8O+˙ (100) | 108 C7H8O+˙ (98) 107 C7H7O+ (2) | 109 C6H5O2− (1) | 107 C7H7O− (97) | 140 C7H8O·O2−˙ (39) | 154 C7H8O·NO2− (100) | 170 C7H8O·NO3− (100) | |
| 107 C7H7O− (83) | 93 C6H5O− (3) | 107 C7H7O− (61) | |||||||
| 106 C7H6O−˙ (16) | |||||||||
| Ethylphenol | ortho- | 123 C8H10OH+ (100) | 122 C8H10O+˙ (100) | 122 C8H10O+˙ (33) | 121 C8H9O− (95) | 121 C8H9O− (100) | 154 C8H10O·O2−˙ (25) | 168 C8H10O·NO2− (100) | — |
| 107 C7H7O+ (67) | 119 C8H7O− (5) | 121 C8H9O− (75) | |||||||
| meta- | 123 C8H10OH+ (100) | 122 C8H10O+˙ (100) | 122 C8H10O+˙ (76) | 121 C8H9O− (97) | 121 C8H9O− (100) | 154 C8H10O·O2−˙ (48) | 168 C8H10O·NO2− (100) | — | |
| 107 C7H7O+ (24) | 119 C8H7O− (3) | 121 C8H9O− (52) | |||||||
| para- | 123 C8H10OH+ (100) | 122 C8H10O+˙ (100) | 122 C8H10O+˙ (35) | 121 C8H9O− (89) | 121 C8H9O− (100) | 154 C8H10O·O2−˙ (55) | 168 C8H10O·NO2− (100) | — | |
| 107 C7H7O+ (65) | 119 C8H7O− (11) | 121 C8H9O− (45) | |||||||
The ion products for each reaction were determined by indicating the highest m/z ion signals and correlating these with the signal at the m/z value of the known major ion product (based on the reagent ion used) at several different concentrations of the aromatic standard gas. Using the identified product ions for each process, the branching ratios and kinetics were calculated for each species in a N2 carrier gas, over eight different positive and negative reagent ions. The results for these reaction rate coefficients are shown in Table 2.
| Compound | PA (kJ mol−1) | I.E. (eV) | Polarizability α, Å3 (×10−24 cm3) | Dipole moment D, Debye | H3O+ [kc] | NO+k [kc] | O2+˙ k [kc] | O−˙ k [kc] | OH−k [kc] | O2−˙ k [kc] | NO2−k [kc] | NO3−k [kc] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All the PA, α and D values have been calculated from theoretical DFT calculations using the B3LYP DFT and the basis set 6-311++G(d,p) with the D4 correction.a Taken from the NIST database.29b — Indicates no reaction. | |||||||||||||
| Cymene | ortho- | 717 | 17.4 | 0.68 | [2.6] | 2.2 [2.1] | 2.0 [2.1] | —b | 0.3 [2.7] | — | — | — | |
| meta- | 710 | 17.6 | 0.34 | [2.5] | 2.2 [2.0] | 2.0 [2.0] | — | 0.1 [2.6] | — | — | — | ||
| para- | 731 | 8.29a | 17.7 | 0.06 | [2.4] | 2.2 [2.0] | 2.0 [2.0] | — | 0.1 [2.6] | — | — | — | |
| Cresol | ortho- | 752 | 8.46a | 12.6 | 1.06 | [2.4], [2.8] | 2.2 [2.0], 2.2 [2.3] | 1.8 [2.0], 2.2 [2.3] | 0.3 [2.6] | 1.1 [2.5] | 0.9 [2.0] | 0.1 [1.7] | 0.0 [1.5] |
| meta- | 754 | 8.29a | 12.7 | 1.07 | [2.4], [2.8] | 2.1 [2.0], 2.0 [2.3] | 1.8 [2.0], 2.1 [2.3] | 0.2 [2.6] | 0.9 [2.5] | 0.8 [2.0] | 0.1 [1.7] | 0.0 [1.6] | |
| para- | 759 | 8.34a | 12.7 | 1.46 | [2.6], [2.8] | 2.3 [2.2], 2.2 [2.3] | 2.0 [2.1], 2.2 [2.3] | 0.3 [2.8] | 1.0 [2.8] | 0.8 [2.1] | 0.1 [1.9] | 0.0 [1.7] | |
| Ethylphenol | ortho- | 756 | 14.5 | 1.11 | [2.6] | 2.1 [2.1] | 1.9 [2.1] | 0.3 [2.8] | 1.0 [2.7] | 0.9 [2.1] | 0.2 [1.8] | — | |
| meta- | 756 | 14.5 | 1.07 | [2.5] | 2.3 [2.1] | 2.0 [2.0] | 0.3 [2.7] | 0.9 [2.7] | 0.9 [2.0] | 0.2 [1.8] | — | ||
| para- | 758 | 7.84a | 14.6 | 1.44 | [2.7], [2.8] | 2.6 [2.3], 2.4 [2.3] | 2.5 [2.2], 2.4 [2.2] | 0.3 [2.9] | 1.2 [2.9] | 1.1 [2.2] | 0.2 [1.9] | — | |
3.1 Positive ions
The three available positive ions, H3O+, NO+ and O2+˙ are the basis of most of the ion–molecule reactions using SIFT-MS known to date, up until 2015.16 Four of the aromatic compounds presented in this study (ortho-, meta- and para-cresol as well as para-ethylphenol) have earlier been investigated using He carrier gas at 300 K in previous work15 when negative ions were not available for SIFT-MS studies. We therefore extend the knowledge of ion–molecule reactions with positive reagent ions, by elaborating the library for the other aromatic species presented in this study.In comparison to the previous work conducted in He for the cresol isomers as well as 4-ethylphenol (Table 2), the collisional rate coefficients (kc) are slightly lower for the work in N2 with a flow tube temperature of 393 K. This is predominantly due to the increase in carrier gas temperature. The present experimental rate coefficients are also similar to the previous work in He (296–300 K).15 This agrees with the findings of our previous comparison study, which showed that rate coefficients conducted in He and N2 carrier gases (using a Profile 3 instrument) do not significantly change with carrier gas.26 The previous k values are generally slightly higher (see Table 2) which is most likely down to the increased flow tube temperature of the Voice200infinity.
Note that for cymene, a minor fragment product ion is formed at m/z 93, alongside the major proton transfer product.
| H3O+ + C10H14 → C10H15+ + H2O | (1a) |
| → C7H9+ + C3H7OH | (1b) |
The production of a small amount of C7H9+ from cymene is caused by the loss of the C3H6 group from the main benzene π-system. Alkyl groups are known to be electron donating and are more so, the larger the carbon chain. The electron donating group enriches the π-system and makes the molecule more susceptible to electrophilic attack by the H3O+ ion, resulting in formation of a fragment at m/z 93 (C7H9+). Table 1 also shows that the branching ratios are different between the different isomers of cymene, for m/z 135 (C10H15+) and m/z 93 (C7H9+). Although m/z 93 is a minor product ion, the difference in branching ratios indicates some possibility for identification of these species from the H3O+ SIFT-MS data. Table 1 shows that the ortho- (6%) isomer produces the greatest fraction of C7H9+, followed by the para- (4%) and meta- (2%) species. The difference in branching ratios may be somewhat influenced by the different exothermicities of reaction (1b) for the different isomers due to the relative positioning of the methyl group with respect to the isopropyl group.
Electron donating groups are known to cause the next additional substituent to add to the para- or ortho-positions of benzene rings. This is because there is a greater stabilisation of the cation, when the positive charge is located on the carbon (in the benzene ring) which is attached directly to the isopropyl group (as the isopropyl group is electron donating). When H+ adds to the ortho- or para-position of the cymene (with respect to the isopropyl group), this causes a resonance structure where the positive charge is able to spend some time on the carbon which is directly attached to the isopropyl group. This therefore makes the H+ addition to either the ortho- or para-position of the aromatic (with respect to the isopropyl group) more favourable (Fig. 2).
 | ||
| Fig. 2 Resonance stabilisation structures of the cation intermediates of the ortho- (top) para- (middle) and meta- (bottom) cymene species. | ||
When the H+ adds to the aromatic species, having the CH3 group in the meta-position with respect to the isopropyl group means that the delocalised positive charge does not dwell on the aromatic carbon directly attached to the isopropyl group. There is therefore a higher transition energy associated with this mechanism resulting in the lowest branching ratio (m/z 93, 2%) being associated with the meta-cymene species.
The ortho-cymene isomer produces the highest fraction of the C7H9+ fragment, which is most likely due to the isopropyl and methyl groups being adjacent to one another. This would cause a greater electron density to persist between carbons 1 and 2 on the benzene ring. The greater fraction of electron density would allow for better stability for the cation intermediate when the positive charge is located on carbon 1 and therefore the loss of the isopropyl cation is more susceptible in the ortho-cymene species.
However, no fragmentation was seen for any of the cresol or ethylphenol isomers.
| H3O+ + C7H8O → C7H8OH+ + H2O | (2) |
| H3O+ + C8H10O → C8H10OH+ + H2O | (3) |
| NO+ + C10H14 → C10H14+˙ + NO˙ | (4) |
| NO+ + C7H8O → C7H8O+˙ + NO˙ | (5) |
| NO+ + C8H10O → C8H10O+˙ + NO˙ | (6) |
3.1.3.1 Cymene. The two product ions formed from the interaction of O2+˙ with cymene are the molecular radical cation C10H14+˙ (m/z 134) and a fragment ion formed after the loss of a CH3˙ (C9H11+, m/z 119), formed by the reactions:
| O2+˙ + C10H14 → C10H14+˙ + O2 | (7a) |
| → C9H11+ + CH3˙ + O2 | (7b) |
The relative branching ratios for the separate isomers may however potentially be explained by resonance stabilisation of the transition intermediate. In the ortho- and para-isomers, the positive charge is able to become stabilised by the +I effect of the isopropyl group (assuming the methyl group is lost) which gives this transition state a lower relative enthalpy of formation, compared to that of the meta-isomer. This therefore explains the lower branching ratio of the C9H11+ (m/z 109) fragment ion product for the meta-isomer compared to the ortho- or para-isomers (Table 1).
3.1.3.2 Cresol. The O2+˙ reactions with all three cresol isomers predominantly produce a single charge transfer product (agreeing with the previous work in He);15
| O2+˙ + C7H8O → C7H8O+˙ + O2 | (8a) |
| → C7H7O+ + H˙ + O2 | (8b) |
3.1.3.3 Ethylphenol. The main product ions were the radical cation C8H10O+˙ and the fragment ion, C7H7O+ (m/z 107):
| O2+˙ + C8H10O → C8H10O+˙ + O2 | (9a) |
| → C7H7O+ + CH3˙ + O2 | (9b) |
This is however confirmed by the previous work in He.15 Analogues to cymene, the percentage of the unfragmented charge transfer product is seen to be largest in the meta-isomer (a trend similar to the EI spectra).
This may be explained by a +M mesomeric effect which is induced by the OH group in ethylphenol. The OH group causes the shown resonance structures within the three isomers of ethylphenol (Fig. 3).
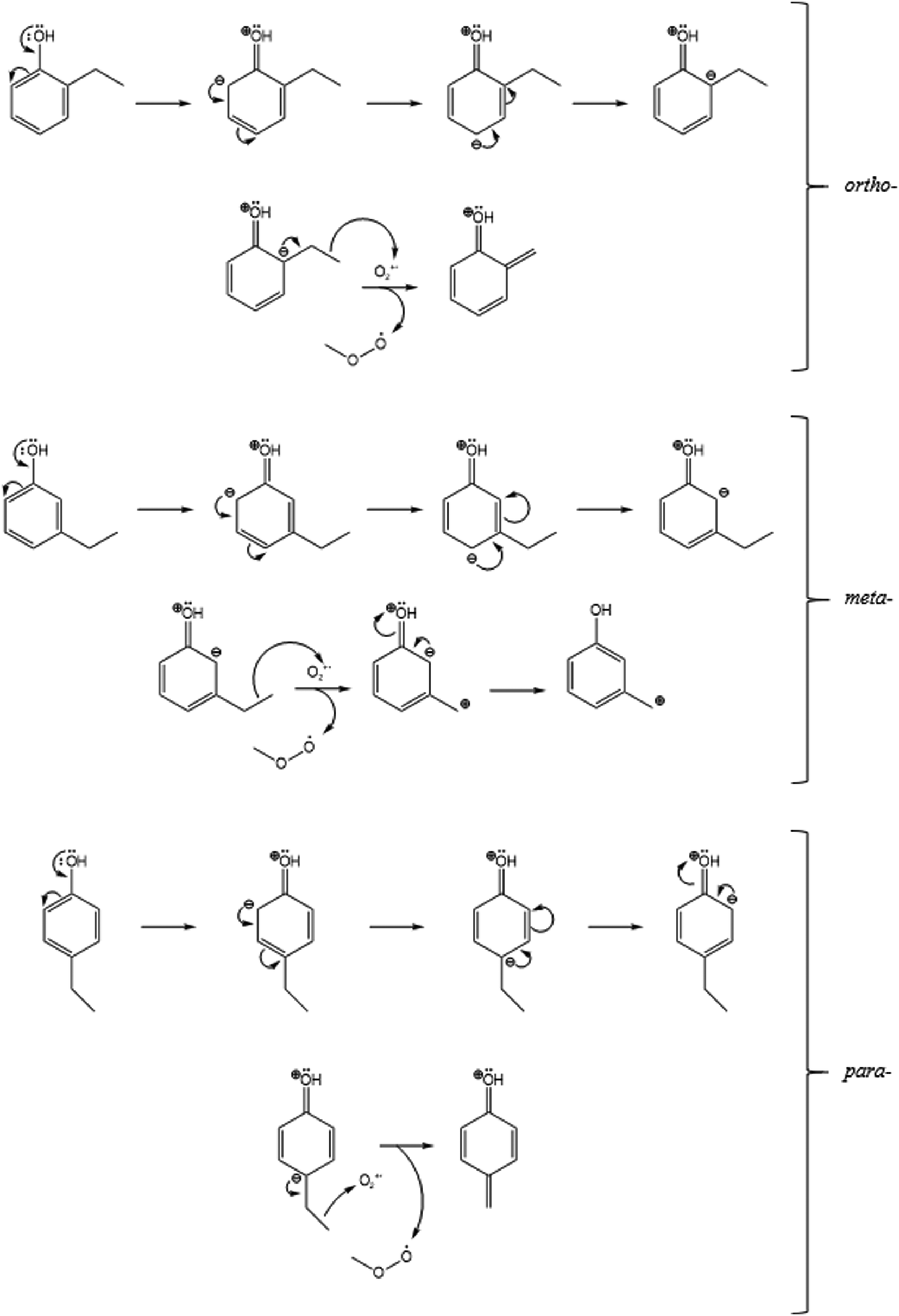 | ||
| Fig. 3 The mesomeric effect and the resonance structures of the ortho- (top) and meta- (middle) and para- (bottom) isomers of ethylphenol (for reaction (9b)), followed by reaction with the O2+˙ reagent ion. | ||
These resonance forms show that the negative charge may dwell on both the ortho- and para-carbons, relative to the OH group (Fig. 3). As a result, the negative charge may be more stable around the ethyl groups in the ortho- and para-positions, making these intermediates more prone to attack by the positive O2+˙ ion. This may be the reason for the higher C7H7O+ (m/z 107) in the ortho- and para-isomers.
Furthermore (and very similar to the NO+ reactions) little difference is seen between the relative rates of the isomeric species of each compound, although for both cresol and ethylphenol, the fastest reaction occurs when the methyl group is located in the para-position, which is most likely due to a reduction in sterical hindrance here.
3.2 Negative ions
The negative ion reactions with the aromatic compounds included in this study have not been studied (to the best of our knowledge) to date. In order to extend the number of possible ways to measure these aromatic isomers, it is necessary to extend the number of possible ion–molecule reaction pathways and therefore the differing ion products, rates and branching ratios which label a process. In this work, we therefore report the reaction rate coefficients and branching ratios and describe the chemistry occurring within the flow tube for the nine compounds with the negative reagent ions available in the Voice200infinity SIFT-MS instrument. DFT calculations of the enthalpy changes were carried out to support the proposed reaction pathways.Overall, it is seen that the negative ions have a significantly slower reaction compared to the positive ions (see Table 2). This is most likely due to the over-crowding of the negative π-system which hinders the negative ion chemistry. In addition, it is seen that these negative reagent ion–molecule reaction processes only produce either one or two ion products (including adducts).
3.2.1.1 Cymene. The rate coefficients O−˙ reactions with all three isomers was immeasurably slow (<10−11 cm3 s−1) and therefore they are not given in Table 2. Nevertheless, very low count rates of the C10H13− product ion were detected:
| O−˙ + C10H14 → C10H13− + OH˙ | (10) |
3.2.1.2 Cresol. The reactions of O−˙ with C7H8O show the production of three product ion species. The product ions were identified as:
| O−˙ + C7H8O → C6H5O2− + CH3˙ | (11a) |
| → C7H7O− + OH˙ | (11b) |
| → C7H6O−˙ + H2O | (11c) |
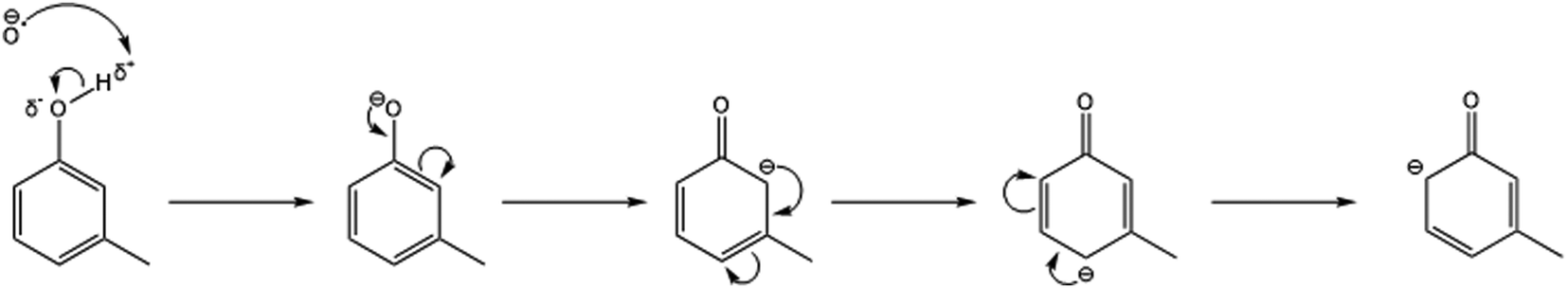 | ||
| Fig. 4 Resonance stabilisation structures of the reaction anion intermediate when the O−˙ nucleophile attacks the δ+ carbon within the meta-cresol isomer. | ||
In doing so, this would produce a resonance structure in which the negative charge does not dwell on the meta-carbon at any point. As a result, the +I effect from the methyl group does not repel against the negative charge (whereas for the ortho- and para-isomers this does happen). Correspondingly, this causes the meta-cresol transition state to be the most energetically favourable and therefore explains why the meta-cresol isomer produces the largest fraction of the proton transfer product (C7H7O−).
The second most abundant product is C7H6O−˙ (reaction (11c)), in which H2O is produced as a neutral product. The ΔH values for this reaction were −297 kJ mol−1 (ortho-), −299 kJ mol−1 (meta-) and −302 kJ mol−1 (para-). This channel is thus more exothermic compared to the production of C7H7O− because of the energetically favourable production of an H2O molecule (reaction (11c)) compared to OH˙ radical (reaction (11b)). Despite this, it has a considerably lower branching ratio. This may be explained by a likely increased transition energy required to produce an intermediate transition state to allow for the simultaneous removal of two H-atoms.
The other reaction pathway produced C6H5O2− (reaction (11a)). Table 1 shows that this product is minor and is present at ≤2% across the isomers. This reaction exhibited ΔH values of −218 kJ mol−1 (ortho-), −220 kJ mol−1 (meta-) and −222 kJ mol−1 (para-). The low branching ratio of (11a) could be due to a barrier in the process. Note that the charge transfer reaction forming C7H8O−˙ would be endothermic for all three isomers (according to our DFT results) by 159 kJ mol−1 (ortho- and meta-) and 160 kJ mol−1 (para-).
3.2.1.3 Ethylphenol. The O−˙ reaction with ethylphenol formed the same two product ion species for each isomer, C8H9O− is the major (proton transfer); and C8H7O− (triple H transfer) is the minor product.
| O−˙ + C8H10O → C8H9O− + OH˙ | (12a) |
| → C8H7O− + (H2O + H˙) | (12b) |
Analogously to cresol, the meta-isomer produces the highest proportion of the proton transfer product (C8H9O−), due to a similar charge delocalisation structure. As a result, the meta-ethylphenol anion transition state exhibits the lowest energy compared to the other two isomers, as the inductive effect induced by the ethyl group isn’t repelled by the negative charge. This therefore produces the lowest energy transition state out of the three isomers and explains why the C8H9O− product has the highest branching ratio for the meta-ethylphenol isomer.
The rate coefficient for all three reactions is about ten times slower than kc (Table 2), which may be due to production of neutral radicals, in both pathways. When compared to cresol, the ethylphenol reactions are slower by ca. 2–3 times, which may be explained by the methyl group inducing a lower +I effect compared to the ethyl group, which inherently enriches the π-system with negative charge resulting in ethylphenol being more prone at repelling the negatively charged reagent.
3.2.2.1 Cymene. The reactions of OH− with the cymene isomers are observed with C10H13− as the major product in all three reactions by proton transfer (most likely from the isopropyl group, although it may also be taken from the methyl constituent).
| OH− + C10H14 → C10H13− + H2O | (13a) |
| → C8H7O− + C2H6 + H2 | (13b) |
Table 1 shows that a 12% fraction of the minor product, C8H7O− is produced in the ortho-isomer, although its production is negligible in the meta- and para-isomers. This is reflected in the DFT results which show that the ΔH value for ortho-cymene is −44 kJ mol−1 which is more negative compared to the meta-isomer (−38 kJ mol−1) or para-isomer (−38 kJ mol−1). The reason for this may be down to sterical hinderance and the causation of a higher energy intermediate when the isopropyl and methyl groups are adjacent to each other.
3.2.2.2 Cresol. The reaction of OH− with cresol proceeds again mostly by proton transfer from the cresol OH group and a minor product, C6H5O− is observed for the meta- and para-isomers:
| OH− + C7H8O → C7H7O− + H2O | (14a) |
| → C6H5O− + CH3OH | (14b) |
Meta- and para-cresol reactions have a minor product, C6H5O−. This is most likely from the addition of the OH− reagent ion to the δ+ carbon (attached to the phenolic OH group) to produce a transition state (Fig. 5). This would produce an anion intermediate which is of significantly higher energy compared to the stable major anion product, C7H7O−. Although possible to be formed, this minor product is only seen at branching ratio percentages of 5% for meta-cresol and 3% for para-cresol. Note that in comparison with (14a) the ΔH values are less negative for (14b) (ortho-, meta- and para-isomers were −113 kJ mol−1, −115.07 kJ mol−1 and −118 kJ mol−1, respectively).
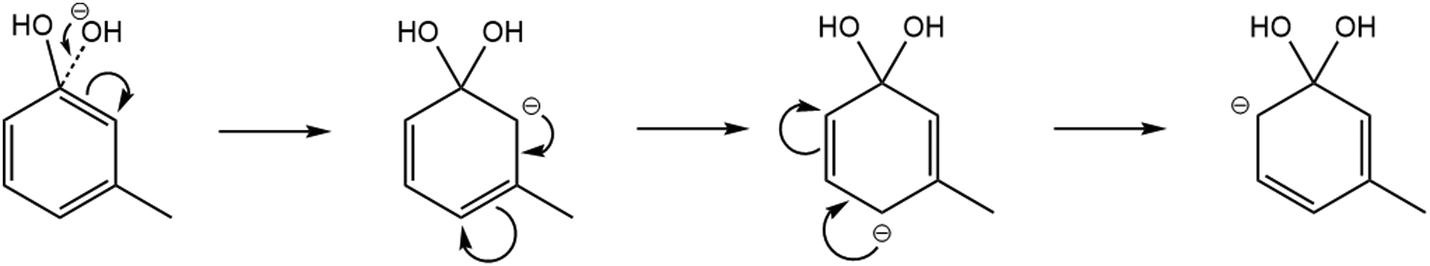 | ||
| Fig. 5 Resonance stabilisation structures of the reaction anion intermediate when the OH− nucleophile attacks the δ+ carbon within the meta-cresol isomer. | ||
The mechanism shown in Fig. 5 shows the resonance structure of the possible transition states, in which the negative charge may be located in either an ortho- or para-location. Although the finishing mechanistic steps are not clear for this process, this would explain why a slightly higher branching ratio is seen for the meta-cresol constituent, as there is no repulsion between the +I inductive effect (by the methyl group) and the delocalised negative charge. There is however a 0% branching ratio of this minor product for ortho-cresol, which is most likely down to sterical hinderance causing too high energy for the transition state.
3.2.2.3 Ethylphenol. OH− reacts with each isomer of ethylphenol by proton transfer.
| OH− + C8H10O → C8H9O− + H2O | (15) |
3.2.3.1 Cymene. No reaction was observed between O2−˙ and cymene. This is not surprising as cymene is a hydrocarbon, without any electronegative atoms.
3.2.3.2 Cresol. The product ions produced on the reaction of O2−˙ with cresol indicated association31 in parallel with proton transfer:
| O2−˙ + C7H8O + M → C7H8O·O2−˙ + M | (16a) |
| → C7H7O− + HO2˙ + M | (16b) |
The slower rate coefficients (Table 2) may therefore be explained by the much more dominant pathway (reaction (16b)) possessing very low changes in enthalpy of reaction.
3.2.3.3 Ethylphenol. The O2−˙ reaction is similar to that of cresol, parallel proton transfer and three body association.
| O2−˙ + C8H10O + M → C8H10O·O2−˙ + M | (17a) |
| → C8H9O− + HO2˙ + M | (17b) |
| NO2− + C7H8O + M → C7H8O·NO2− + M | (18) |
| NO2− + C8H10O + M → C8H10O·NO2− + M | (19) |
| NO3− + C7H8O + M → C7H8O·NO3− + M | (20) |
4. Conclusion
In this work, we investigated positive and negative ion chemistry that could facilitate SIFT-MS analyses of some isomeric aromatic species. We have experimentally determined the reaction rate coefficients and ion product branching ratios for 72 reactions. The observed trends were substantiated by DFT calculations of the reaction thermochemistry and, to some degree, rationalised by suggested mesomeric and inductive effects and steric hindrance.The results of this study revealed the consistency of chemistry which occurs between the SIFT-MS positive and negative reagent ions and selected aromatic species. It was found that, generally, OH− reactions lead to the (M–H)− product; O2−˙ reactions lead to the (M–H)− product and adduct (although they are very slow); O−˙ produces complicated chemistry which needs to be further investigated; and NO2− and NO3− are unreactive with these species. The differences observed for product ion branching ratios can be used to identify isomers approximately. These results, therefore, highlight the need to investigate negative ion chemistry across different chemical classes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Czech Science Foundation (Grantová Agentura České Republiky, GACR; Project No. 21-25486S) and from the Praemium Academiae funding by the Czech Academy of Sciences. We would also like to thank Nicholas Demarais at SyftTM for his useful contribution to the discussion around this manuscript.References
- M. Phillips, K. Gleeson, J. M. B. Hughes, J. Greenberg, R. N. Cataneo, L. Baker and W. P. McVay, Volatile organic compounds in breath as markers of lung cancer: a cross-sectional study, Lancet, 1999, 353, 1930–1933 CrossRef CAS PubMed.
- T. Issitt, L. Wiggins, M. Veysey, S. T. Sweeney, W. J. Brackenbury and K. Redeker, Volatile compounds in human breath: critical review and meta-analysis, J. Breath Res., 2022, 16, 024001 CrossRef PubMed.
- P. Španěl and D. Smith, Progress in SIFT-MS: breath analysis and other applications, Mass Spectrom. Rev., 2011, 30, 236–267 CrossRef PubMed.
- J. B. Sharmeen, F. M. Mahomoodally, G. Zengin and F. Maggi, Essential Oils as Natural Sources of Fragrance Compounds for Cosmetics and Cosmeceuticals, Molecules, 2021, 26, 666 CrossRef CAS PubMed.
- K. Dryahina, S. Som, D. Smith and P. Spanel, Characterization of spoilage-related volatile organic compounds in packaged leaf salads, Flavour Fragrance J., 2020, 35, 24–33 CrossRef CAS.
- V. S. Langford, SIFT-MS: Quantifying the Volatiles You Smell…and the Toxics You Don't, Chemosensors, 2023, 11, 111 CrossRef CAS.
- M. Ghislain, N. Costarramone, T. Pigot, M. Reyrolle, S. Lacombe and M. Le, Bechec, High frequency air monitoring by selected ion flow tube-mass spectrometry (SIFT-MS): influence of the matrix for simultaneous analysis of VOCs, CO2, ozone and water, Microchem. J., 2020, 153, 104435 CrossRef CAS.
- A. M. Yeoman, M. Shaw, N. Carslaw, T. Murrells, N. Passant and A. C. Lewis, Simplified speciation and atmospheric volatile organic compound emission rates from non-aerosol personal care products, Indoor Air, 2020, 30, 459–472 CrossRef CAS PubMed.
- M. Ghislain, M. Reyrolle, J.-M. Sotiropoulos, T. Pigot, H. Plaisance and M. Le, Bechec, Study of the Chemical Ionization of Organophosphate Esters in Air Using Selected Ion Flow Tube-Mass Spectrometry for Direct Analysis, J. Am. Soc. Mass Spectrom., 2022, 33, 865–874 CrossRef CAS PubMed.
- D. Ge, J. Zhou, Y. Chu, Y. Lu, X. Zou, L. Xia, Y. Liu, C. Huang, C. Shen, L. Zhang, H. Wang and Y. Chu, Distinguish oral-source VOCs and control their potential impact on breath biomarkers, Anal. Bioanal. Chem., 2022, 414, 2275–2284 CrossRef CAS PubMed.
- A. Gaida, O. Holz, C. Nell, S. Schuchardt, B. Lavae-Mokhtari, L. Kruse, U. Boas, J. Langejuergen, M. Allers, S. Zimmermann, C. Vogelmeier, A. R. Koczulla and J. M. Hohlfeld, A dual center study to compare breath volatile organic compounds from smokers and non-smokers with and without COPD, J. Breath Res., 2016, 10, 026006 CrossRef CAS PubMed.
- M. S. Ataabadi, S. Bahmanpour, S. Yousefinejad and S. Alaee, Blood volatile organic compounds as potential biomarkers for poly cystic ovarian syndrome (PCOS): an animal study in the PCOS rat model, J. Steroid Biochem. Mol. Biol., 2023, 226, 106215 CrossRef CAS PubMed.
- P. Španěl and D. Smith, Selected ion flow tube studies of the reactions of H3O+ NO+, and O2+ with several aromatic and aliphatic hydrocarbons, Int. J. Mass Spectrom., 1998, 181, 1–10 CrossRef.
- P. Španěl and D. Smith, Selected ion flow tube studies of the reactions of H3O+, NO+, and O2+ with several aromatic and aliphatic monosubstituted halocarbons, Int. J. Mass Spectrom., 1999, 189, 213–223 CrossRef.
- T. S. Wang, P. Španěl and D. Smith, A selected ion flow tube study of the reactions of H3O+, NO+ and O2+ with some phenols, phenyl alcohols and cyclic carbonyl compounds in support of SIFT-MS and PTR-MS, Int. J. Mass Spectrom., 2004, 239, 139–146 CrossRef CAS.
- D. Hera, V. S. Langford, M. J. McEwan, T. I. McKellar and D. B. Milligan, Negative Reagent Ions for Real Time Detection Using SIFT-MS, Environments, 2017, 4, 16 CrossRef.
- M. Ghislain, N. Costarramone, J. M. Sotiropoulos, T. Pigot, R. Van Den Berg, S. Lacombe and M. Le, Bechec, Direct analysis of aldehydes and carboxylic acids in the gas phase by negative ionisation selected ion flow tube mass spectrometry: quantification and modelling of ion–molecule reactions, Rapid Commun. Mass Spectrom., 2019, 33, 1623–1634 CrossRef CAS PubMed.
- T. J. Frankcombe, OH-Initiated Oxidation of Toluene. 3. Low-Energy Routes to Cresol and Oxoheptadienal, J. Phys. Chem. A, 2008, 112, 1572–1575 CrossRef CAS PubMed.
- J. Zhang, M. Choi, Y. Ji, R. Zhang, R. Zhang and Q. Ying, Assessing the Uncertainties in Ozone and SOA Predictions due to Different Branching Ratios of the Cresol Pathway in the Toluene-OH Oxidation Mechanism, ACS Earth Space Chem., 2021, 5, 1958–1970 CrossRef CAS.
- A. M. Persico and V. Napolioni, Urinary p-cresol in autism spectrum disorder, Neurotoxicol. Teratol., 2013, 36, 82–90 CrossRef CAS PubMed.
- J. Faber, K. Brodzik, A. Gołda-Kopek and D. Łomankiewicz, Benzene, toluene and xylenes levels in new and used vehicles of the same model, J. Environ. Sci., 2013, 25, 2324–2330 CrossRef CAS PubMed.
- A. I. Moreno, N. Arnáiz, R. Font and A. Carratalá, Chemical characterisation of emissions from a municipal solid waste treatment plant, Waste Manage., 2014, 34, 2393–2399 CrossRef CAS PubMed.
- A. Gratien, S. N. Johnson, M. J. Ezell, M. L. Dawson, R. Bennett and B. J. Finlayson-Pitts, Surprising Formation of p-Cymene in the Oxidation of α-Pinene in Air by the Atmospheric Oxidants OH, O3, and NO3, Environ. Sci. Technol., 2011, 45, 2755–2760 CrossRef CAS PubMed.
- A. P. Pollnitz, K. H. Pardon and M. A. Sefton, Quantitative analysis of 4-ethylphenol and 4-ethylguaiacol in red wine, J. Chromatogr. A, 2000, 874, 101–109 CrossRef CAS PubMed.
- R. B. Michalcikova, K. Dryahina, D. Smith and P. Španěl, Volatile compounds released by Nalophan; implications for selected ion flow tube mass spectrometry and other chemical ionisation mass spectrometry analytical methods, Rapid Commun. Mass Spectrom., 2020, 34, e8602 Search PubMed.
- P. Španěl, S. J. Swift, K. Dryahina and D. Smith, Relative influence of helium and nitrogen carrier gases on analyte ion branching ratios in SIFT-MS, Int. J. Mass Spectrom., 2022, 476, 116835 CrossRef.
- K. Sovová, K. Dryahina and P. Španěl, Selected ion flow tube (SIFT) studies of the reactions of H3O+, NO+ and O2˙+ with six volatile phytogenic esters, Int. J. Mass Spectrom., 2011, 300, 31–38 CrossRef.
- T. Su and W. J. Chesnavich, Parametrization of the ion–polar molecule collision rate constant by trajectory calculations, J. Chem. Phys., 1982, 76, 5183–5185 CrossRef CAS.
- The National Institute of Standards and Technology (NIST) Chemistry WebBook, SRD 69, (http://webbook.nist.gov/).
- S. J. Swift, D. Smith, K. Dryahina, M. O. Gnioua and P. Španěl, Kinetics of reactions of NH4+ with some biogenic organic molecules and monoterpenes in helium and nitrogen carrier gases: a potential reagent ion for selected ion flow tube mass spectrometry, Rapid Commun. Mass Spectrom., 2022, 36, e9328 CrossRef CAS PubMed.
- R. B. Michalčíková and P. Španěl, A selected ion flow tube study of the ion molecule association reactions of protonated (MH+), nitrosonated (MNO+) and dehydroxidated (M–OH)(+)) carboxylic acids (M) with H2O, Int. J. Mass Spectrom., 2014, 368, 15–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02123h |
| This journal is © the Owner Societies 2023 |