
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe interplay between size, shape, and surface segregation in high-entropy nanoalloys†
Florent
Calvo

Université Grenoble Alpes, CNRS, LiPhy, 38000 Grenoble, France. E-mail: florent.calvo@univ-grenoble-alpes.fr; Tel: +33-476514592
First published on 14th June 2023
Abstract
The respective influences of particle shape and size on the energetic stability of five-component multimetallic nanoparticles have been computationally investigated for AlCuFeCrNi and AuCuPdNiCo mixtures at equiconcentration. Using available embedded-atom model potentials, exchange Monte Carlo simulations possibly assisted with systematic quenching, we explore tools to approach ideal phase equilibrium in such high-entropy nanoalloys. In particular, we show how deviations to ideal solid solution behaviors can be characterized using percolation analyses, and how the contribution of alloying fluctuations at finite temperature can be inferred to evaluate the entropy of mixing in such nonideal cases. An approximation to the entropy of mixing based on pair correlations only is also found to capture the behavior of the thermodynamical mixing entropy quite well, and can be used as an order parameter of mixing. While the AlCuFeCrNi mixture appears to mix reasonably well in all cases considered, cobalt and nickel segregate significantly in AuCuPdNiCo nanoparticles, deviating strongly from ideal random mixtures. A simple Gaussian regression model applied to a coarse distribution of concentrations is found to correctly predict conditions for optimising the mixing thermodynamical properties of the miscible AlCuFeCrNi nanoparticle.
1 Introduction
Over the last couple of decades, the synergistic physical and chemical effects resulting from the combination of two or more metals together at the nanoscale have been evidenced by many groups, and nanoalloys have emerged as a community of their own, with interest ranging from purely basic to mostly applied considerations.1–6 Recent progress in this field includes more robust but also more diverse synthesis methods with convincing efforts toward mass production, access to more colorful palettes of characterization tools, but also more efficient plasmonic nanostructures or versatile nanocatalysts. In parallel, computational advances have enabled the modeling of larger and more complex nanoalloys, paving the way towards increasingly realistic simulations over longer time scales and on a more statistical ground.7One way of increasing the complexity, and thus potentially also the tunability, is to consider a larger number of elements to be combined in the nanoparticles. In the bulk limit, such so-called high-entropy alloys (HEAs) composed of 5 or more metals8–17 have been found to exhibit enhanced mechanical properties18–25 and a greater resistance against corrosion26 than single metal materials, making them useful notably for the nuclear industry.27–30 Low-dimensional HEA thin films have further been shown to provide ideal coating materials31–35 owing to their good thermal stability and slow diffusion,36,37 and some HEA mixtures have also been suggested to exhibit enhanced magnetic38–40 and superconducting41 properties.
Another field where HEA compounds have found potential applications is that of energy, notably for hydrogen storage,42,43 as well as catalysis44–56 where it is hoped that catalytic efficiency will be improved owing to the superior stability of electrodes made from HEA under the harsh conditions of reactors. Additionally, it is hoped with HEA materials that a significant fraction of expensive or rare elements used in conventional catalysts can be eliminated in favor of more common metals.
Such promises of HEA materials have unsurprisingly led several groups to explore them at the nanoscale, either as nanowires57 or as nanoparticles.58–61 In recent years the synthesis of various high-entropy alloy nanoparticles, or high-entropy nanoalloys (HENAs) made from AuCuPdNiCo,62 CoNiPtAuCu,63 AlCoCrCuFeNi,64 the so-called Cantor alloy FeCrMnNiCo,65–67 and even up to 8 different elements68 were successfully reported.
HENAs present various exciting challenges from a fundamental point of view. While in bulk HEA systems, kinetic control often prevails over thermodynamic factors; at the nanoscale the numerous surface atoms can diffuse much faster and favor the latter, and there is experimental evidence that some metals segregate at the free surfaces of HEA materials, at least for the Cantor alloy.69 In HEA nanoparticles, the sought presence of chemical disorder, known to often favor face-centered cubic crystals in the bulk limit, thus interferes here with the presence of free surfaces of various types, as well as edges and vertices whose relative contribution increases as the size decreases. In classical thermodynamical models such as the CALPHAD approach,70,71 HEAs are typically treated by incorporating appropriate terms to describe the propensity for mixing between unlike elements in the Gibbs free energy, either as polynomials in the fractions xi or as xi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logxi in the mixing enthalpy and mixing entropy, respectively.72,73 Finite-size corrections can also be incorporated into such models, usually through appropriate surface energy terms that scale linearly with the inverse particle size. However, surface energies themselves depend on a number of factors, mainly the facet orientation (Miller indices) but also the surface composition. Besides surface composition, surface energies depend on size through a curvature correction known as the Tolman length, further dependent itself on composition.74
logxi in the mixing enthalpy and mixing entropy, respectively.72,73 Finite-size corrections can also be incorporated into such models, usually through appropriate surface energy terms that scale linearly with the inverse particle size. However, surface energies themselves depend on a number of factors, mainly the facet orientation (Miller indices) but also the surface composition. Besides surface composition, surface energies depend on size through a curvature correction known as the Tolman length, further dependent itself on composition.74
In the present contribution, we follow an atomistic point of view and explore the very basic properties of HENAs from an equilibrium perspective through finite temperature Monte Carlo (MC) simulations. In particular, we aim at identifying stable structural motifs as a function of size and characterize the extent of chemical disorder using thermodynamical and also topological indicators. Our approach is complementary to earlier efforts in which molecular dynamics (MD) simulations were performed to shed light onto the synthesis and relaxation mechanisms in low-dimensional HEA systems,75–80 and also to various works on HEAs under periodic boundary conditions.81–84 The level of modeling used here, which relies on known many-body force fields, allows reasonably large statistics to be accumulated over broad size ranges and even composition to be varied.
As our main findings, a roadmap is suggested to model HENAs at finite temperature and address their expected equilibrium thermodynamics, including the propensity for mixing of the various elements and ways to determine the entropy of mixing without assuming empirical interaction rules or correcting deviations to ideal solid solutions, either from thermodynamic integration or more approximately from nearest-neighbor pair correlation functions.85 Two types of mixtures were chosen to illustrate contrasted behaviors arising from combining different metals together. The AuCuPdNiCo set of metals has already been explored at the nanoscale by the Yacaman group62 who highlighted the importance of grain boundaries. The AlCuFeCrNi mixture has previously been studied experimentally86 and found in MD simulations to mix rather homogeneously.79,80 The Monte Carlo approach we use here circumvents the time scale issue of molecular dynamics to a significant extent, and allows us to approach HENAs closer to their true equilibrium state.
In the next section, we briefly describe the set of computational tools employed to model the HENAs of the two metal mixtures, the methodology chosen to address specifically the influences of shape and size on their relative stability, and especially the thermodynamical and structural probes used to characterize chemical disorder at finite temperature. Section 3 presents our results at fixed equiconcentrations in the five elements, discussing successively the energetic properties, surface structure, connectivity analyses and the entropy of mixing. Section 4 attempts to correlate two specific descriptors of the propensity for mixing, this time for fixed nanoparticle size and shape, but varying the composition. Section 5 finally provides some concluding discussion and ends the paper.
2 Methods
Our approach to the modeling of high-entropy nanoalloys relies on a fully atomistic, off-lattice description based on well-established many-body interatomic potentials of the embedded-atom model (EAM) type.87 Traditional approaches to the computational determination of nanoalloy structure88 based on such potentials are likely to be poorly efficient in multi-element systems with excessively rugged energy landscapes such as HENAs. Here we thus refrain from attempting any systematic global optimization, but instead turn to a more coarse-grained perspective already used since the 1990s to identify stable structural motifs of increasingly large nanoparticles.89–93HENAs based on well-defined shapes and in the approximate size range 100–10000 atoms were constructed, assigning elements randomly in this initial lattice according to a prescribed composition. Classical Monte Carlo simulations at finite temperature T were then performed with translational moves allowing the lattice to deform, and swap moves allowing the chemical ordering to relax, the moves being accepted through a conventional Metropolis–Hasting probability. In some cases discussed below, multiple trajectories at different temperatures were also carried out, with occasional swap moves between configurations from random pairs of adjacent replicas.94 Such parallel tempering Monte Carlo simulations allow a much faster equilibration and convergence to thermal equilibrium, and also suggest a means to evaluate the entropy of mixing (vide infra).
Our simulations at finite temperatures imply observables of a statistical nature. We primarily considered the internal energy U = 〈E〉, where E(R) is the potential energy of configuration R, as the main indicators of relative stability among structural motifs. Structural probes were implemented as well, using a simple connectivity criterion rij < rc to identify if atoms i and j separated by the distance rij lie below the cut-off distance rc = 3.1 Å. For each atom i, a coordination number Nc allows us to identify whether this atom belongs to the surface of the nanoparticle (Nc < 10) or else to the core (for which Nc = 12 for the three chosen structural motifs). The average propensity for each surface atom to be of any elemental type X can also be determined, from which the corresponding surface fractions psurfX are obtained after cumulating the probabilities over the entire set of surface atoms.
The Monte Carlo framework further allows us to look more specifically into the influence of having a particular element at the surface on the overall energetic stability of the nanoparticles. Here we introduce two types of bias in the simulations, to explore energy landscape hypersurfaces in which either (i) all the atoms of a given type X are constrained to be at the surface or (ii) no atom of a given type X is allowed to reside at the surface. In both cases, the Monte Carlo moves (translations and atom swaps) are restricted in such a way as to preserve the desired constraint: in case (i) atoms of type X can still move but they must stay at the surface; in case (ii) atoms of type X cannot migrate to the surface from the core. The two types of constraints (i) and (ii) are extreme cases of a more general hypersurface sampling in which the probability of having X atoms at the surface of the nanoparticle is kept fixed. While they are straightforward to implement in the Monte Carlo procedure, it must be kept in mind that the bias they introduce modifies the thermal properties that could be determined at equilibrium. In the present work, we use these biased simulations to explore the underlying inherent structures of the energy landscape, and help decipher the complex interplay between mixing and surface segregation.
To quantify the propensity of the various elements towards segregation or aggregation within the nanoparticles, a statistical approach was considered in which rigorous comparison can be made to the behavior expected for an ideal (noninteracting) solid solution on the same nanoparticle. Here a percolation perspective, as already used in the physics of the Potts model95,96 is followed by considering connected clusters of a given elemental type. For any configuration R, the connectivities among all atoms of type X are first determined. Clusters of this type are then identified,97 two atoms belonging to the same cluster if they can be linked by successive chains of nearest-neighbors, all being of type X. The analysis is repeated for all atom types and the distributions of fragments are obtained. We focus notably on two complementary properties, namely the largest fragment of type X within the nanoparticle, and the number of individual fragments irrespective of their size.
In practice, only nanoparticles with complete shells with icosahedral (ICO), Marks’ decahedral (DEC), and truncated octahedral (TO) shapes were considered. For each HENA particle, the Monte Carlo simulations consisted of 106 Monte Carlo sweeps following 106 equilibration sweeps, the connected cluster analysis being performed as post-processing from 104 evenly spaced configurations only. Individual translational moves were attempted with 90% probability, biparticle swaps between random pairs of unlike elements being attempted with 10% probability. Additional MC simulations were performed under periodic boundary conditions, at the same temperature but zero pressure (variable cell size) to evaluate the specific effects of dimensionality but also to provide limiting values for the surface fractions of atoms in the limit of infinite slabs.
For the two mixtures considered, AlCuFeCrNi and AuCuPdNiCo, the same embedded-atom model proposed by Zhou and coworkers87 and developed for multimetallic compounds was used. This potential was specifically extended to chromium and its alloys in a subsequent publication.98 This model has been used by many authors in the recent years to simulate high-entropy alloys.99–104
3 Shape and size effects
In this section we only consider high-entropy nanoalloys at equiconcentration in the five AlCuFeCrNi or AuCuPdNiCo elements. We present and discuss successively the results obtained on the two types of HENAs, identifying the most stable structural motif, then focusing on the structural analysis through the distribution of surface atoms and elemental connectivity. We also describe how simulations conducted at two different temperatures provide a way to evaluate the contribution of fluctuations in chemical ordering to the mixing entropy in the system.3.1 AlCuFeCrNi mixture
Fig. 1 shows the variations of U/N, the internal energy per atom at 300 K obtained for the N-atom AlCuFeCrNi mixtures at equiconcentration with icosahedral (N = 147–10179), decahedral (N = 75–9062), or truncated octahedral (N = 201–9201) shapes. For any structural type, these variations follow a generic polynomial behavior in powers of N−1/3 well known in single-component nanoparticle systems89–93| U/N = U0 − aN−1/3 − bN−2/3 + O(1/N) | (1) |
 | ||
| Fig. 1 Relative energies of AlCuFeCrNi nanoalloys at equiconcentration and at 300 K, as a function of N−1/3 and for truncated octahedral (TO), Marks’ decahedral (DEC) and Mackay icosahedral (ICO) shapes. Energies are reported relative to the large-size limiting behavior E(N → ∞) ≃ −3.283 − 1.169N−1/3 eV per atom inferred from the results obtained from the largest TO nanoparticle and the bulk fcc sample. | ||
Having identified truncated octahedral HENA particles to be the most stable among the three considered motifs, we focus on HENAs of this specific shape in the following. Fig. 2 shows the proportions of surface atoms of each elemental type, again as a function of inverse particle radius N−1/3. Additional simulations of the bidimensional face-centered cubic slab with (111) surfaces exposed under periodic boundary conditions were performed to evaluate the same quantities in the N → ∞ limit. This figure clearly shows that the five elements behave differently regarding their propensity for surface segregation, and that the trend between finite-size TO nanoparticles and the extended slab seems smooth and essentially regular. While aluminum avoids being exposed at the surface even in the very small (201-atom) NPs, copper becomes increasingly prone to surface segregation as size increases, at the expense of the three remaining metals. In all nanoparticles simulated here, chromium and iron are the next contributors of surface atoms, but as will be shown below they occupy rather different sites as copper does, explaining the trends towards larger sizes.
 | ||
| Fig. 2 Proportion of surface atoms in AlCuFeCrNi truncated octahedral nanoalloys, at equiconcentration and at 300 K, as a function of N−1/3. The values at infinite size were obtained from periodic simulations on a 4000-atom fcc slab with (111) surfaces. | ||
The statistical analysis in terms of connected fragments also shows major differences among elements in AlCuFeCrNi HENAs. In Fig. 3(a) we have represented the distributions of the largest connected fragments, while panel (b) shows the distributions of the number of connected fragments, irrespective of their size. In both panels, and in addition to the Monte Carlo results, we have superimposed the predictions of lattice sampling on the 2406-atom truncated octahedron in which atomic interactions are neglected altogether. The corresponding distributions should be understood as the results for an ideal, noninteracting solid solution. Since a fixed number of atoms has to be distributed into the same lattice, the largest connected fragment and the number of connected fragments are somewhat anticorrelated to one another: large numbers of fragments necessarily imply that they must be small in order to accommodate the amount of available material. Comparison with the distributions predicted for the ideal solid solution allows the mixing propensity of each individual element to be quantified within the nanoalloy. For the present HENA particle, and from the point of view of fragment distributions, chromium thus appears as the element that behaves the most similarly to the solid solution (many small fragments), while both nickel and aluminum depart the most from ideality (few but large fragments). Iron and copper appear as intermediate cases, with particularly broad distributions of their largest fragments that suggest significant fluctuations caused in particular by finite temperature effects.
 | ||
| Fig. 3 Connected fragments analysis of 2406-atom AlCuFeCrNi nanoalloys with truncated octahedral shape, at 300 K. (a) Distribution of the largest fragment size; (b) distribution of the number of connected fragments. In addition to element-resolved distributions, the results obtained for the ideal solid solution nanoparticle are superimposed as black curves. | ||
From the Monte Carlo simulations, systematic local minimizations were performed to identify particularly stable HENA structures. For the present 2406-atom truncated octahedral NP, the lowest energy minimum is depicted in Fig. 4 along with the five elemental representations associated with hiding all other elements from the structure. For this nanoparticle, chromium atoms are clearly located at vertices and edges, this lower coordination being consistent with their higher propensity for desorbing. Copper occupies the (111) facets in priority, surface impurities being mostly of the iron type. As the truncated octahedral NP grows larger, the relative fraction of surface atoms belonging to the (111) facets increases, explaining the monotonic trend found for these two elements in Fig. 2. Aluminum and nickel both strongly favor interior locations, and in this low-energy structure only a few nickel atoms are occasionally found at the surface, but strictly no aluminum atom.
 | ||
| Fig. 4 Visual depiction of elemental distributions in the most stable structure found for the 2406-atom truncated octahedral AlCuFeCrNi nanoalloy. | ||
From a more statistical perspective, Fig. 5 shows the distributions of energies obtained by quenching configurations sampled by the Monte Carlo simulations, and compared with the corresponding distributions produced from the biased Monte Carlo trajectories in which elements are constrained to (a) all lie at the surface; or (b) all lie in the core. Imposing an element to lie at the surface acts as a major energetic constraint, and Fig. 5 confirms the structural trends identified in the previous figures. For example, copper and aluminum show the most extreme opposite behaviors, leading to more favorable or unfavorable minima, respectively, with respect to unbiased sampling. The results obtained for copper, in particular, could be interesting for a more realistic attempt at identifying configurations lying deeper in the energy landscape. The similar distributions found for chromium and iron are also consistent with their partial occupation of surface sites, the case of nickel lying again in between those of iron and aluminum.
 | ||
| Fig. 5 Distributions of inherent structure energies obtained from biased Monte Carlo simulations of 2406-atom truncated octahedral AlCuFeCrNi nanoalloys, at 300 K, in which (a) all atoms of a given elemental type are prescribed to lie at the surface; (b) no atom of a given elemental type is allowed to lie at the surface. The results of unbiased simulations are superimposed as black curves. | ||
When no atom of a given element type is allowed to be at the surface, the resulting distributions are less contrasted, apart from copper which definitely leads to the least stable minima. More surprisingly, aluminum is not found to produce the lowest-energy distribution in this case, while nickel does with iron behaving similarly. The finding that, apart from copper, the distributions obtained in the two panels (a) and (b) of Fig. 5 are not mirror images of each other as the elements are varied was not anticipated. In particular, it suggests synergistic effects between alloying and strain release that must operate at the surface, involving especially the aluminum atoms.
One natural issue of interest with multi-element alloys is the contribution of mixing to the thermodynamical properties. In ideal solid solutions with a fully random distribution of elements, contributions to the entropy of the form xlnx, with x the composition of a given element, are added to the Gibbs free energy, polynomial corrections being further added to account for interactions among elements that cause deviations from ideality.72,73 In HEA nanoparticles, the free surfaces also contribute to thermodynamical functions, typically through 1/R ∝ N−1/3 contributions. In liquid alloys, the prefactor of such contributions varies essentially linearly with the alloy composition,74 however in the present case of solid nanoparticles with well-defined surfaces, the surface energies are expected to depend non trivially on composition at least through the preference of the various elements to alloy with one another and occupy different surface or core sites.
To obtain further insight into the complex interplay between alloying effects and surface energies, we next show that it is possible to evaluate the contribution of mixing fluctuations to the entropy for the present HENA particles, which we will refer to simply as the mixing entropy in what follows. In bulk HEAs, Gao and Widom have shown that the mixing entropy can be evaluated from the pair correlation functions at equilibrium.85 This approach cannot be straightforwardly extended to low-dimensional systems although useful approximations can be implemented, as discussed below. Here we focus first on a purely thermodynamical approach.
As our starting point, we show in Fig. 6(a) the variations with temperature of the internal energy in the 100–500 K range, as obtained from parallel tempering Monte Carlo simulations on the 2406-atom truncated octahedral alloy, performed with or without random particle swaps, configuration exchanges being still allowed between random pairs of adjacent trajectories. In both simulations, the initial configuration allocated for all replicas is the lowest-energy structure identified in the previous systematic local optimization step, as discussed above.
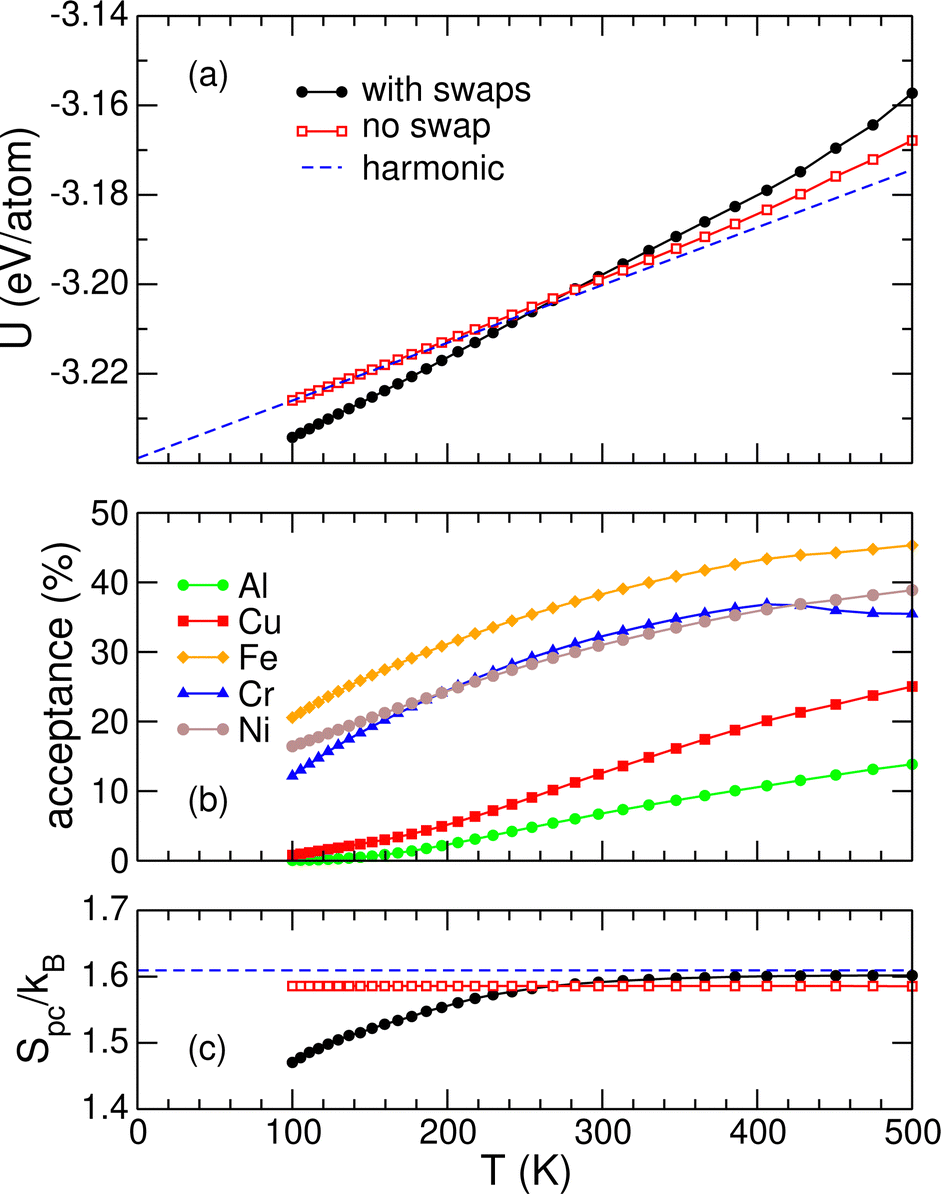 | ||
| Fig. 6 (a) Internal energy of the 2406-atom truncated octahedral AlCuFeCrNi nanoalloy as a function of increasing temperature, as predicted by parallel tempering Monte Carlo simulations with (black symbols) or without (red squares) particle swap moves. The dashed blue line highlights the harmonic prediction; (b) element-resolved Metropolis acceptance ratios of the swapping moves; (c) pair correlation entropy Spc. The dashed blue line is the ideal solid solution value of kBln5. | ||
The internal energies are markedly different depending on whether particle swaps are allowed, the slope that quantifies the heat capacity being much higher when fluctuations in alloying are accounted for. In Fig. 6(b) the probabilities of accepting such particle swaps in the parallel tempering simulations are shown as a function of temperature, for all elements involved. We note that the absolute values of these probabilities depend on the details of the Monte Carlo process of selecting atoms for exchange, and here no attempt was made to use smarter selecting rules, as achieved e.g. in our previous work106 employing preferential swapping.
The values obtained for the exchange probability of swapping unlike elements are generally high (above 10%) for chromium, iron, and nickel, lower for aluminum and copper but still significant, indicating a clear tendency for alloying fluctuations at room temperature and above, even for those elements. For chromium, a minor drop in the acceptance probability is found above 400 K as the manifestation of the increasing desorption of this element.
The contribution of alloying fluctuations to the thermodynamical properties, clearly seen in the internal energy of Fig. 6(a), can be quantified also in the canonical entropy. In this purpose, we first define the mixing contribution Cmixv to the heat capacity as the difference between the heat capacities Cv and C(0)v obtained for the fluctuating system and those for the non-mixed system, as predicted by the parallel tempering Monte Carlo simulations with and without particle swaps, respectively:
| Cmixv(T) = Cv(T) − C(0)v(T). | (2) |
 | (3) |
| U(T) ≃ U(0)(T) + γ + αT + O(T2), | (4) |
 | (5) |
| U(0)(T) = ≃κkBT/2, | (6) |
 | (7) |
Evaluating the contribution of alloying fluctuations to the mixing entropy thus requires two simulations to be performed at different temperatures, both allowing particle swaps, and this was achieved for the present systems by repeating the Monte Carlo simulations at 100 K, for the same series of truncated octahedral, decahedral, and icosahedral nanoparticles. The relative entropies of mixing obtained from these additional simulations are represented in Fig. 7 as a function of inverse particle size N−1/3. Here again one extra simulation was also performed for the periodic system, at 100 K, to determine the corresponding value in the bulk limit. The variations reported in Fig. 7 are generally increasing with size, at least above 500–1000 atoms. In all cases the values obtained lie significantly below the theoretical bulk limit of 5kB × (1/5)ln5 ≃ 1.61kB per atom expected in the absence of interactions, but are much closer to the corresponding value of about 0.75kB per atom found under periodic boundary conditions. Low-entropy minima found for the decahedral and icosahedral particles could suggest that these structural motifs, at equiconcentration, can lock chemical orderings more efficiently than their respective neighboring sizes due to optimal allocation of surface and core sites.
 | ||
| Fig. 7 Relative entropies of mixing of AlCuFeCrNi nanoalloys at equiconcentration and at 300 K, as a function of N−1/3 and for truncated octahedral (TO), Marks’ decahedral (DEC) and Mackay icosahedral (ICO) shapes. The dashed line highlights the results obtained from the largest TO nanoparticle and the bulk sample. | ||
While the mixing entropy can be evaluated using only limited amounts of computational resources, a simpler alternative can be designed based on the structural information contained in the pair correlation functions.85 We thus introduce a pair correlation entropy Spc as an order parameter defined from the probabilities pij that elements i and j are nearest neighbors:
 | (8) |
Fig. 6(c) shows the variations of Spc of the 2406-atom truncated octahedral AlCuFeCrNi mixture with increasing temperature, as obtained from the parallel tempering Monte Carlo simulations with and without atom swap. In the absence of atom swap, the pair correlation entropy remains constant, equal to its value in the starting minimum but lower than the ideal solution value of kBln5. However, when atom swaps are allowed, much lower values are obtained at low temperatures, that are indicative of a lesser degree of mixing.
One virtue of Spc is that this quantity can also be defined for any static configuration R, and can thus play the role of a mixing order parameter, phase separated systems being associated with lower values relative to randomly mixed systems. However, it should be kept in mind that the pair correlation entropy is strongly nonlinear in the probabilities pij, hence the value of Spc from the probabilities accumulated over a sample is not the average of individual values associated with each member of the sample.
3.2 AuCuPdNiCo mixture
The very same tools used in the previous section for the AlCuFeCrNi mixture were used for the high-entropy nanoalloys made of the AuCuPdNiCo metals, still at equiconcentration, and we follow here the same line of presentation and discussion of the results. Fig. 8 thus shows the variations with inverse particle radius of the internal energy at 300 K for the three different structural motifs considered. The baseline connecting the values obtained for the largest truncated octahedral particle and for the periodic fcc sample was again chosen to shift the internal energies, however for the present metals TO particles are found to be significantly higher in energy than both decahedral and, especially, icosahedral particles. | ||
| Fig. 8 Relative energies of AuCuPdNiCo nanoalloys at equiconcentration and at 300 K, as a function of N−1/3 and for truncated octahedral (TO), Marks’ decahedral (DEC) and Mackay icosahedral (ICO) shapes. Energies are reported relative to the large-size limiting behavior E(N → ∞) ≃ −3.283 − 1.169N−1/3 eV per atom inferred from the results obtained from the largest TO nanoparticle and the bulk fcc sample. | ||
The variations of the surface proportions with inverse particle radius, obtained for all icosahedral particles, are represented in Fig. 9. The values obtained for the (111) slab, superimposed in the N → ∞ limit, are also shown and connected to the corresponding data predicted for the largest, 9201-atom truncated octahedral particle. The discontinuities found between the values obtained for the largest icosahedral and truncated octahedral particles generally emphasize the role of the different surfaces, and in particular the presence of surfaces other than (111) in the TO particle, on the propensity of the various elements to occupy such surface sites. For this mixture, the surface ratios obtained for the slab are also quite at variance with those obtained for the largest nanoparticles, especially for copper which displays a strong segregation towards the (111) surface, occupying about 95% of it. In nanoparticles, the surface is much more mixed in copper, gold and palladium, cobalt and nickel exhibiting a stronger preference towards the subsurface positions.
 | ||
| Fig. 9 Proportion of surface atoms in AuCuPdNiCo icosahedral nanoalloys, at equiconcentration and at 300 K, as a function of N−1/3. The values at infinite size obtained from simulations on a 4000-atom slab are connected by dashed lines to the data found for the largest truncated octahedral nanoparticle with 9201 atoms. | ||
Focusing now on the 2057-atom icosahedral HENA of the AuCuPdNiCo mixture, we show in Fig. 10 the distribution of connected fragments obtained at 300 K obtained from the Monte Carlo simulation, once again in comparison with the reference distributions found for an ideal solid solution on this icosahedral lattice. As was the case for the AlCuFeCrNi mixture, the percolation analysis reveals that the various metals can behave quite differently regarding their mixing or segregation propensities. Here nickel and especially cobalt tend to form few but large fragments, while gold makes numerous but very small fragments with 10 atoms or less. Copper and palladium provide intermediate cases, both with a stronger segregation tendency than the ideal solid solution, i.e. with fewer but larger fragments too.
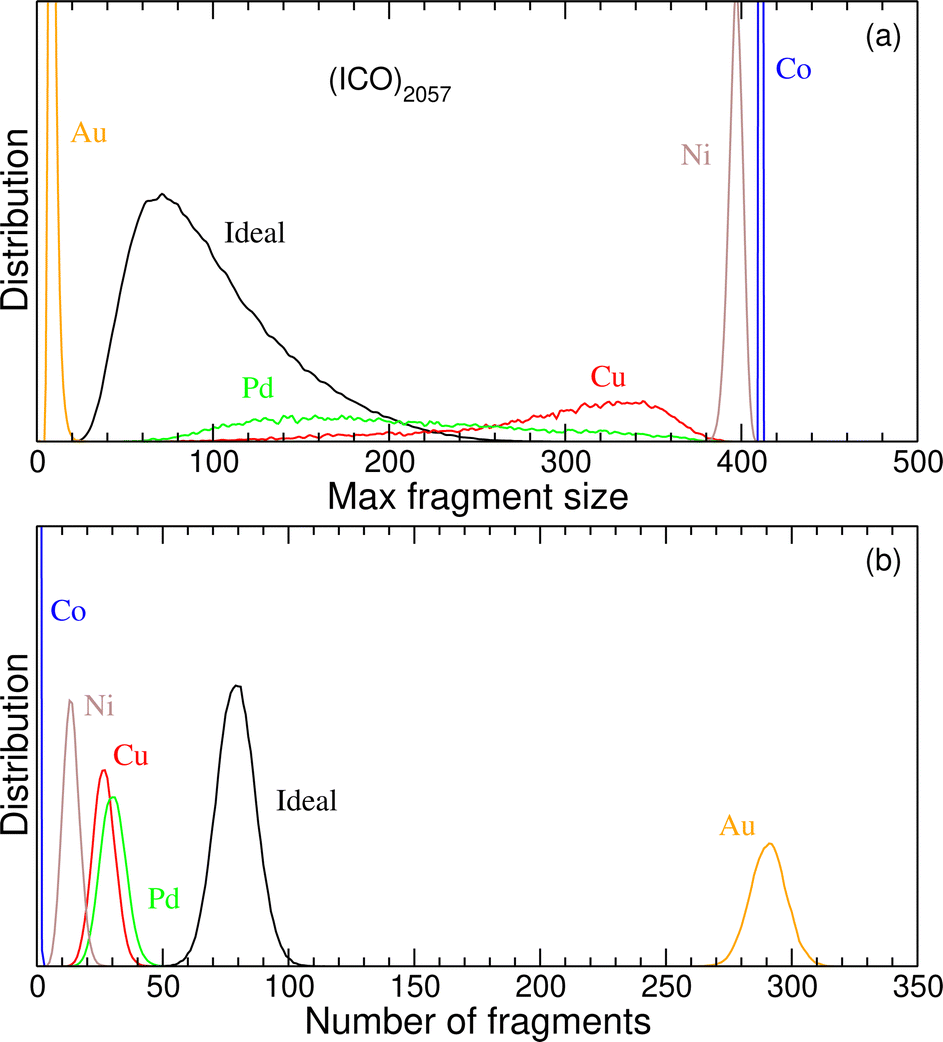 | ||
| Fig. 10 Connected fragments analysis of 2057-atom AuCuPdNiCo nanoalloys with Mackay icosahedral shape, at 300 K. (a) Distribution of the largest fragment size; (b) distribution of the number of connected fragments. In addition to element-resolved distributions, the results obtained for the ideal solid solution are superimposed as black curves. | ||
Systematic local optimization of the Monte Carlo configurations was then conducted, the lowest-energy structure being depicted in Fig. 11 with the allocation of each individual element being highlighted using their own specific color. These plots show that, at this size, copper occupies all edges and vertices sites, palladium the centers of the (111) facets, nickel forms a tightly connected core, cobalt prefers subsurface positions but also as a highly connected cluster, and gold distributes itself much more homogeneously than all other atoms as many diluted small fragments, also lying at the surface.
 | ||
| Fig. 11 Visual depiction of elemental distributions in the most stable structure found for the 2057-atom Mackay icosahedral AuCuPdNiCo nanoalloy. | ||
Repeating the Monte Carlo simulations with additional biases on the surface allocations of atoms leads to contrasted results relative to the previous alloy. Fig. 12 shows the corresponding distributions of local energy minima obtained without any bias, (a) imposing that a given element always occupies surface sites or (b) that this element never lies at the surface. For this alloy, either constraint appears to deteriorate the overall energetic stability of the nanoparticle, and such global surface biases are thus not expected to ease the global optimization problem. All distributions in Fig. 12(a) are rather sharp and are ordered consistently with the surface preferences seen in Fig. 9, with palladium (high) and cobalt (low) providing the two extremes on the scale expressing the preference towards the surface. Preventing an element from occupying the surface sites comparatively has little effect, except for gold and copper which are the most disfavored elements under this constraint. Interestingly, and as was the case for the AlCuFeCrNi mixture, the distributions obtained after applying the two opposite constraints on the surface atoms do not mirror one another in their positions relative to the unbiased reference distribution.
 | ||
| Fig. 12 Distributions of inherent structure energies obtained from biased Monte Carlo simulations of 2057-atom Mackay icosahedral AuCuPdNiCo nanoalloys, at 300 K, in which (a) all atoms of a given elemental type are prescribed to lie at the surface; (b) no atom of a given elemental type is allowed to lie at the surface. The results of unbiased simulations are superimposed as black curves. | ||
To further validate our approach for evaluating the contribution of alloying fluctuations to the mixing entropy, we have represented in Fig. 13(a) the variations of the internal energy of the 2057-atom icosahedral HENA with increasing temperature, as predicted by parallel tempering Monte Carlo simulations in the 100–500 K range, again allowing or disabling swap moves between unlike atoms. Fig. 13(b) shows the corresponding probabilities of accepting such swap moves and involving each of the five elements individually. Finally, Fig. 13(c) displays the pair correlation entropy Spc obtained from the nearest-neighbor probabilities, atom swap moves being again either allowed or disabled. The generic behavior of the internal energy is similar to that found in the AlCuFeCrNi HENA, namely that alloying fluctuations increase the slope and are thus associated with a higher heat capacity. However, the pair correlation entropy is generally much lower than in the AlCuFeCrNi mixture, consistently with the lesser extent of segregation of the AuCuPdNiCo nanoparticles. We use the same procedure as above to evaluate the contribution of alloying fluctuations to the mixing entropy using eqn (5) with Cmixv estimated from eqn (7), noticing however that the variations in U(T) seen in Fig. 13(a) are more prone to anharmonicities, with not just a higher slope but also some second-order contribution.
 | ||
| Fig. 13 (a) Internal energy of the 2057-atom Mackay icosahedral AuCuPdNiCo nanoalloy as a function of increasing temperature, as predicted by parallel tempering Monte Carlo simulations with (black symbols) or without (red squares) particle swap moves. The dashed blue line highlights the simple harmonic behavior; (b) element-resolved Metropolis acceptance ratios of the swapping moves; (c) pair correlation entropy Spc. The dashed blue line is the ideal solid solution value of kBln5. | ||
With respect to the AlCuFeCrNi alloy, the success of swapping moves is comparatively much less likely, probabilities falling below 10% even at the highest temperature considered of 500 K. This finding is consistent with the greater disparity amongst elements seen, e.g. in the percolation analysis of Fig. 10, thus underlying that the present alloy departs even more from the ideal solid solution.
The contribution of alloying fluctuations to the mixing entropy was determined for all AuCuPdNiCo HENAs following the same procedure as in the previous section, repeating the Monte Carlo simulations but at 100 K, using the most stable low-energy structure identified from the periodic quenches at 300 K to speed up convergence. The results are represented in Fig. 14 as a function of inverse particle radius N−1/3 for nanoparticles of the three structural motifs. The values obtained for the mixing entropy are markedly lower than those reported in Fig. 7, which is another manifestation of the lower tendency of the AuCuPdNiCo mixture towards random alloying, even in the bulk limit, and consistently with the lower values exhibited by the pair correlation entropy. While the variations found for the TO and ICO nanoparticles are again mostly monotonic with only shallow minima, a more complex behavior is found for the decahedral particles, with a maximum at N = 389 and a deep minimum for N = 3274–4371 atoms. Intriguingly, and unlike the results for icosahedra shown in Fig. 9, the surface ratios for the decahedral motif do not display any particular non-monotonicity in their variations with size, for any of the five elements (results not shown). The mixing entropy found here for the decahedral motif thus appears to be extremely sensitive to the structural details of the nanoparticle, and the present results are confirmed by the pair correlation entropy, as shown in Fig. S2 in the ESI.† Here it would probably be useful to identify appropriate descriptors to interpret to which extent is the feature found near 4000 atoms caused by specific chemical orderings.
 | ||
| Fig. 14 Relative entropies of mixing of AuCuPdNiCo nanoalloys at equiconcentration and at 300 K, as a function of N−1/3 and for truncated octahedral (TO), Marks’ decahedral (DEC) and Mackay icosahedral (ICO) shapes. The dashed line highlights the results obtained from the largest TO nanoparticle and the bulk sample. | ||
4 Mixing energy versus mixing entropy
High-entropy alloys are usually designed near equiconcentration in the various elements in order to maximize the likelihood of synergistic effects. In this section we explore the effects of alloy composition on the relative stability of HENA particles, fixing the size and the shape to be those suggested at equiconcentration. It would be clearly cumbersome to vary the compositions of the five elements continuously, and we follow instead a coarse-grained perspective by considering only variations in individual concentrations by steps of 20%. For five elements, this leads to 126 possible combinations, including monometallic cases, for which individual Monte Carlo simulations were also performed.Nanoparticles in the size range N = 2000–3000 with complete shells were chosen to address compositional effects for the AlCuFeCrNi and AuCuPdNiCo sets of metals, namely truncated octahedra with 2406 atoms and Mackay icosahedra with 2057 atoms, respectively. To quantify the effects of alloying on the thermodynamical properties of those HENAs, we use the entropy of mixing that measures the contribution of alloying fluctuations, as defined above in eqn (5). As explained in the previous section, evaluating the mixing entropy ΔSmix requires performing two Monte Carlo simulations with particle swaps allowed at two temperatures. In practice the simulation at 300 K is carried out first, followed by systematic local optimization, and the simulation at 100 K is then performed, initiated at the lowest-energy configuration determined from quenches of the 300 K trajectory.
An internal energy of mixing, Umix is also introduced by subtracting from the internal energy of the alloyed nanoparticle the weighted contributions from the pure systems,107
 | (9) |
![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0078_20d1.gif) = {x1,…,x5} the set of compositions (with x1+⋯+x5 = 1), U() being the internal energy at composition and Ui the internal energy of the pure nanoparticle with element of type i. Negative values for the energy of mixing indicate that alloying stabilizes the nanoparticle.
= {x1,…,x5} the set of compositions (with x1+⋯+x5 = 1), U() being the internal energy at composition and Ui the internal energy of the pure nanoparticle with element of type i. Negative values for the energy of mixing indicate that alloying stabilizes the nanoparticle.
The correlations between the energy of mixing and the relative entropy of mixing are shown in Fig. 15(a) and (b) for the AlCuFeCrNi and AuCuPdNiCo HENAs, respectively. From these scatter plots, we have tried to make sense of specific patterns by highlighting the monometallic and bimetallic systems. By definition, monometallic particles have a zero energy of mixing, and should also be characterized with a zero mixing entropy since atom swaps do not operate for them. The slightly positive values obtained for ΔSmix originate from anharmonicity effects and provide an estimate for the residual error obtained by applying eqn (7). From Fig. 15 we can thus estimate that the mixing entropies due to alloying fluctuations are accurate only up to about 0.1kB per atom due to anharmonicities.
 | ||
| Fig. 15 Correlation between the mixing energy and the relative entropy of mixing obtained at 300 K for nanoalloys with compositions varying by steps of 20%. (a) 2406-atom truncated octahedra of AlCuFeCrNi mixtures; (b) 2057-atom Mackay icosahedra of AuCuPdNiCo mixtures. In both cases, the binary alloys are depicted with colored symbols, while the results for the pure nanoparticles are circled. In (a) the predictions of a simple Gaussian regression model maximizing the entropy of mixing or minimizing the energy of mixing are also highlighted. | ||
A striking difference between the results obtained for the two sets of alloys is the much lower energies of mixing (in magnitude) of the AlCuFeCrNi compounds, but this is obviously related to the stronger binding energies of the heavier elements having more electrons.
In their vast majority, binary alloys display lower mixing entropies than arbitrary alloys with 3 or more elements, with the notable exception of some Cu–Cr systems lying on the energetically defavorable side Umix > 0. In general, we find that these low mixing entropies are associated with strong phase segregation of the core–shell type, with little or no thermal fluctuation in chemical ordering at the interface. Examples of such behavior are the Al–Cu and Au–Co alloys in Fig. 15(a) and (b), respectively. We note that the energies of mixing are not always correlated to the mixing entropies, due to the size mismatch between the different metals that can produce core–shell structures that are energetically strongly stabilized (Al–Cu and Au–Co) or only marginally (Fe–Ni).
The more frequent occurrence of core–shell structures in the AuCuPdNiCo compounds, at least for the binary cases, is consistent with the lowest degree of mixing found in the previous section at equiconcentration. We have attempted to rationalize compositional effects using ideas from unsupervised machine learning, predicting the energies and entropies of mixing of similar HENA particles but with arbitrary composition, through a simple Gaussian regression on the numerical data at the coarse-grained scale. More precisely, we denote by {i} the 126 composition vectors that map the entire composition space by steps of 20% and by xi,α the proportion of element α in composition vector i, such that  . The property A() at arbitrary composition is taken as a linear combination over the values known at the coarse compositions i, using composition itself as the sole descriptor:
. The property A() at arbitrary composition is taken as a linear combination over the values known at the coarse compositions i, using composition itself as the sole descriptor:
 | (10) |
,i) is the kernel measuring the degree of similarity between the two compositions, the parameter δ entering the Gaussian function being taken as 0.15.
Applying this simple regression rule to the dataset of the AlCuFeCrNi HENAs, the energy of mixing is found to be minimized for e = (xAl = 0.58, xCu = 0.21, xFe = 0, xCr = 0.21, and xNi = 0), while the mixing entropy is maximized for s = (xAl = 0.18, xCu = 0.36, xFe = 0, xCr = 0.23, and xNi = 0.23). Additional Monte Carlo simulations were performed for these specific ternary and quaternary compositions, the results of which are superimposed in Fig. 15(a). The outcome of these simulations agrees with the expectations in both cases, indicating that these specific HENA particles have optimal energies or entropies of mixing with respect to the coarse dataset. The results obtained by considering the pair correlation entropy instead of the thermodynamical mixing entropy, shown in Fig. S3 of the ESI,† are essentially similar. Spc appears here particularly sensitive to the number of chemical elements present in the mixture, the correlation plots showing clear progressions in Spc as the number of elements increases. Here again, the Gaussian regression model performs well, and predicts that the pair correlation entropy is maximized exactly at equiconcentration.
The regression model was also applied to the AuCuPdNiCo alloy, but at the compositions predicted to minimize Umix or to maximize ΔSmix, the results of the additional simulations do not agree that well, finding energies of mixing that lie off the range of Fig. 15(b), on either side. However, when the pair correlation entropy is used, the model works satisfactorily (see Fig. S4 in the ESI†), and predicts that Spc is maximized slightly off equiconcentration, namely for s = (xAu = 0.22, xCu = 0.20, xPd = 0.20, xNi = 0.19, and xCo = 0.19), the mixing energy being minimized for the ternary mixture with e = (xAu = 0.26, xCu = 0.16, xPd = 0.0, xNi = 0.0, and xCo = 0.58). The latest result is close, but not exactly identical, to the direct simulations. The poor ability of the regression model for this alloy provides another interesting clue about the different behaviors of the two alloys, and especially the much stronger compositional effects at play in the AuCuPdNiCo mixture with its greater tendency towards phase separation.
5 Concluding remarks
The inherent chemical complexity associated with bringing together numerous metals, combined with the presence of free surfaces with different orientations, constitutes an unprecedented challenge for nanoalloys. In practical syntheses and applications of such nanoparticles, it is obvious that kinetics plays a key role in stabilizing certain structures under experimental conditions. However, the thermal equilibrium point of view adopted in the present work should be seen as a necessary step on the (likely bumpy) road towards understanding HENA particles. Ignoring kinetics is not only a simplifying assumption, it is actually rather common practice in metallurgy with the classical approaches to alloy phase diagrams such as the CALPHAD community, in which phases are identified by minimizing free energy, irrespective of the material preparation.The computational approach followed here rests on atomistic simulations and follows previous efforts combining semi-empirical models with well-established Monte Carlo frameworks, including the occasional use of the parallel tempering strategy.106 In the size range of interest, covering 100–10000 atoms, solving the global optimization problem is probably hopeless and we restricted our investigation to specific structural motifs. To obtain insight into the interplay between alloying, surface segregation, and the essential effects of thermal fluctuations, a number of analysis tools were specifically introduced. The distributions of monometallic fragments for a fixed nanoparticle lattice can be compared to the reference distributions expected for ideal (noninteracting) solid solutions, which in turn allows the various elements to be classified according to their propensity towards alloying or segregation. Such a percolation analysis could be extended to identify more specifically the locally ordered arrangements, as a complement to more conventional tools such as common neighbor analyses, by specifically looking for pairs between specified elements, possibly assisted by robust geometrical descriptors such as the smooth overlap of atomic positions.108
Besides percolation analyses, the interplay between alloying and surface segregation could be addressed by introducing biases into the Monte Carlo process, enabling the investigation of potential energy hypersurfaces in which atoms of a given element are constrained to reside at the surface or in the core of the nanoparticle. Systematic local minimizations performed to remove lattice fluctuations generally confirm the strong influence of surface allocations on the relative stability of the nanoparticle, especially for the least miscible AuCuPdNiCo alloy. For the more miscible AlCuFeCrNi system, such hypersurface constraints were found as a possible way to facilitate the global optimization problem. Here also the bias introduced in the Monte Carlo simulations could be generalized, e.g. to examine separately the various contributions of vertices, edges, subsurface atoms, or facets with different Miller indices.
The extent of alloying and, even more importantly, of the associated thermal fluctuations on the relative stability of the nanoparticles, could also be discussed by extracting the specific contribution of alloying fluctuations to the mixing entropy, through a simple computational procedure involving Monte Carlo simulations at two temperatures. The mixing entropy was generally found to be lower than the limiting value expected for noninteracting solid solutions, and to exhibit rather smooth size effects for the AlCuFeCrNi mixture, and more puzzling variations in the case of the AuCuPdNiCo compound under the decahedral arrangement. An approximation to the true mixing entropy based on the probabilities of nearest-neighbor elements was found to capture the thermodynamical data rather well, and could find some additional use as a mixing order parameter for static configurations.
Finally, we considered the effects of alloy composition, for a fixed size and shape of the HENA particles, by mapping the coarse-grained composition space. The two thermodynamical indicators of energy and entropy of mixing generally appear poorly correlated to one another, although simpler compositions such as binary alloys are usually associated with lower mixing entropies. A simple regression model was found to perform satisfactorily in predicting compositions that optimize either the energy or the entropy of mixing, but only for the AlCuFeCrNi miscible case.
The importance of mixing was addressed here for individual nanoparticles, but could of course be also evaluated in other nanostructures such as nanowires or thin films. In addition, it could also be of interest in the interaction between nanostructures, which are responsible, e.g. for their self-assembly. One natural extension of the present work could thus consist in assessing the role of the mixing entropy on the effective potential of mean force between two HENAs, using Monte Carlo methods already established for conventional nanoparticles109 and naturally extended here to account for mixing fluctuations.
The main limitation of the present exploratory work lies in the underlying embedded-atom model employed for the present HENAs, which despite a long and successful history in the field of bulk alloys may not be accurate enough for compounds with a high surface/volume ratio. However, here also the challenge for modeling is quite significant, as surface energies of alloys, for which measurements are scarce, are likely to depend on the composition itself. The EAM potential used here as the main engine of our simulations covers many metals but relies on a functional form with dedicated combination rules that allow any alloy of the fitted element to be represented, without introducing dedicated parameters between unlike elements. A greater transferability, but also at a much heavier cost, could be achieved by treating pair combinations separately. Alternatively, the increasingly popular machine learned potentials110 could offer a valuable alternative, but only provided that surfaces are also treated appropriately, and even possibly on a comparable footing as bulk properties. This effort would naturally fit along the same lines as the recent, growing interest in using ideas from data-driven science towards improving the design and understanding of HEAs,39,111–114 including at the nanoscale.115–117 Irrespective of the functional form, the training set has to be significantly large to account for the various, low-symmetry configurations of nanoscale systems. The major computational effort associated with building this training set might be better addressed in a distributed form or at the scale of the community. In any case, it would probably be useful to first evaluate the performance of first-principles methods for such systems.118
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Some of the results presented in this work were obtained on the computing center Pôle Scientifique de la Modélisation Numérique (PSMN) in Lyon, which we gratefully acknowledge.Notes and references
- R. L. Johnston, Faraday Discuss., 2008, 138, 1–433 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- D. Alloyeau, C. Mottet and C. Ricolleau, Nanoalloys: synthesis, structure and properties, Engineering Materials, Springer, London, 2012 Search PubMed.
- R. L. Johnston and J. P. Wilcoxon, Metal nanoparticles and nanoalloys, Frontiers of Nanoscience, Elsevier, Oxford, 2012 Search PubMed.
- M. M. Mariscal, O. Oviedo and E. P. M. Lieva, Metal clusters and nanoalloys, Springer, New York, 2013 Search PubMed.
- F. Calvo, Nanoalloys: From fundamentals to emergent applications, Elsevier, Oxford, 2nd edn, 2020 Search PubMed.
- E. Janssens, Faraday Discuss., 2022, 242, 1–550 Search PubMed.
- B. Cantor, I. T. H. Chang, P. Knight and A. J. B. Vincent, Mater. Sci. Eng., 2004, 375–377, 213–218 CrossRef.
- J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsay and S.-Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- M.-H. Tsai and J.-W. Yeh, Mater. Res. Lett., 2014, 2, 107–123 CrossRef.
- Y. Zhang, T. T. Zuo, Z. Tang, M. C. Gao, K. A. Dahmen, P. K. Liaw and Z. P. Lu, Prog. Mater. Sci., 2014, 61, 1–93 CrossRef CAS.
- W. Ji, W. Wang, H. Wang, J. Zhang, Y. Wang, F. Zhang and Z. Fu, Intermetallics, 2015, 56, 24–27 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Mater. Today, 2016, 19, 349–362 CrossRef CAS.
- E. J. Pickering and N. G. Jones, Int. Mater. Rev., 2016, 61, 183–202 CrossRef CAS.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- M. C. Gao, High-entropy alloys: fundamentals and applications, Springer, Berlin, 2018 Search PubMed.
- X. H. Yan and Y. Zhang, Scr. Mater., 2020, 187, 188–193 CrossRef CAS.
- M. A. Hemphill, T. Yuan, G. Y. Wang, J. W. Yeh, C. W. Tsai, A. Chuang and P. K. Liaw, Acta Mater., 2012, 60, 5723–5734 CrossRef CAS.
- A. Gali and E. P. George, Intermetallics, 2013, 39, 74–78 CrossRef CAS.
- B. Gludovatz, A. Hohenwarter, D. Catoor, E. H. Chang, E. P. George and R. O. Ritchie, Science, 2014, 345, 1153–1158 CrossRef CAS PubMed.
- K. M. Youssef, A. J. Zaddach, C. Niu, D. L. Irving and C. L. Koch, Mater. Res. Lett., 2014, 3, 95–99 CrossRef.
- Y. Zou, J. Ma and R. Spolenak, Nat. Commun., 2015, 6, 7748 CrossRef PubMed.
- G. Laplanche, A. Kostka, O. M. Horst, G. Eggeler and E. P. George, Acta Mater., 2016, 118, 152–163 CrossRef CAS.
- X. Xian, Z.-H. Zhong, L.-J. Lin, Z.-X. Zhy, C. Chen and Y.-C. Wu, Rare Met., 2018, 41, 1015–1021 CrossRef.
- Z. Li, S. Zhao, R. O. Ritchie and M. A. Meyers, Prog. Mater. Sci., 2019, 102, 296–345 CrossRef CAS.
- D. Raabe, C. Cemal, H. Springer and M. Bausch, Steel Res. Int., 2015, 86, 1127–1138 CrossRef CAS.
- S. C. Middleburgh, D. M. King and G. R. Lumpkin, R. Soc. Open Sci., 2015, 2, 140292 CrossRef CAS PubMed.
- F. Granberg, K. Nordlund, M. W. Ullah, K. Jun, C. Lu, H. Bei, L. Wang, F. Djurabekova, W. J. Weber and Y. Zhang, Phys. Rev. Lett., 2016, 116, 135504 CrossRef CAS PubMed.
- G. L. Zhang, K. S. Ming, J. L. Kang, Q. Huang, Z. J. Zhang, X. R. Zheng and X. F. Bi, Electrochim. Acta, 2018, 279, 19–23 CrossRef CAS.
- S. Sonal and J. Lee, Metals, 2021, 11, 1980 CrossRef CAS.
- B. R. Braeckman, F. Boydens, H. Hidalgo, P. Dutheil, M. Jullien, A.-L. Thomann and D. Depla, Thin Solid Films, 2015, 580, 71–76 CrossRef CAS.
- W. Li, P. Liu and P. K. Liaw, Mater. Res. Lett., 2018, 6, 199–229 CrossRef CAS.
- L. R. Shaginyan, V. F. Britun, N. A. Krapivka, S. A. Firstov, A. V. Kotko and V. F. Gorban, Powder Metall. Met. Ceram., 2018, 57, 293–300 CrossRef CAS.
- Y.-C. Hsu, C.-L. Li and C.-H. Hsueh, Entropy, 2019, 22, 2 CrossRef PubMed.
- N. I. M. Nadzri, D. S. C. Halin, M. M. A. B. Abdullah, S. Joseph, M. A. A. M. Salleh, P. Vizureanu, D.-P. Burduhos-Nergis and A. V. Sandu, Coatings, 2022, 12, 1842 CrossRef.
- K.-Y. Tsai, M.-H. Tsai and J.-W. Yeh, Acta Mater., 2013, 61, 4887–4897 CrossRef CAS.
- J. Dabrowa, M. Zajusz, W. Kucza, G. Cieslak, K. Berent, T. Czeppe, T. Kulik and M. Danielewski, J. Alloys Compd., 2019, 783, 193–207 CrossRef CAS.
- C.-Z. Yao, P. Zhang, M. Liu, G.-R. Li, J.-Q. Ye, P. Liu and Y.-X. Tong, Electrochim. Acta, 2008, 53, 8359–8365 CrossRef CAS.
- E. W. Huang, G. Y. Hung, S. Y. Lee, J. Jain, K. P. Chang, J. J. Chou, W. C. Yang and P. K. Liaw, Crystals, 2020, 10, 18 Search PubMed.
- C. L. P. Pavithra, R. K. S. K. Janardhana, K. M. Reddy, C. Murapaka, J. Joardar, B. V. Sarada, R. R. Tamboli, Y. Hu, Y. Zhang, X. Wang and S. R. Dey, Sci. Rep., 2021, 11, 8836 CrossRef CAS PubMed.
- S. Vrtnik, P. Koželj, A. Meden, S. Maiti, W. Steurer, M. Feuerbacher and J. Dolinšek, J. Alloys Compd., 2017, 695, 3530–3540 CrossRef CAS.
- M. Sahlberg, D. Karlsson, C. Zlotea and U. Jansson, Sci. Rep., 2016, 6, 36770 CrossRef CAS PubMed.
- P. Edalati, R. Floriano, A. Mohammadi, Y. Li, G. Zepon, H. W. Li and K. Edalati, Scr. Mater., 2020, 178, 387–390 CrossRef CAS.
- W. Zhang, R. Tang, Z. B. Yang, C. H. Liu, H. Chang, J. J. Yang, J. L. Liao, Y. Y. Yang and N. Liu, J. Nucl. Mater., 2018, 512, 15–24 CrossRef CAS.
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed.
- Z. J. Dai, T. Lu and Y. Pan, J. Power Sources, 2019, 430, 104–111 CrossRef.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castellio, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3.3, 834–845 CrossRef.
- T. Loffler, A. Savan, A. Garzon-Manjon, M. Meischein, C. Scheu, A. Ludwig and W. Schihmann, ACS Energy Lett., 2019, 4, 1206–1214 CrossRef.
- T. Loffler, A. Savan, H. Meyer, M. Meischein, V. Strotkotter, A. Ludwig and W. Schihmann, Angew. Chem., Int. Ed., 2020, 59, 5844–5850 CrossRef PubMed.
- J. K. Pedersen, T. A. A. Batchelor, A. Bagger and J. Rossmeisl, ACS Catal., 2020, 10, 2169–2176 CrossRef CAS.
- H. Zheng, G. Luo, A. Zhang, X. Lu and L. He, ChemCatChem, 2020, 13, 806–817 CrossRef.
- G. Feng, F. Ning, J. Song, H. Shang, K. Zhang, Z. Ding, P. Gao, W. Chu and D. Xia, J. Am. Chem. Soc., 2021, 143, 17117–17127 CrossRef CAS PubMed.
- L. Yu, K. Zheng, C. Li, X. Lin, H. Liu, W. Shi, J.-J. Qiu, Y. Yuan and Y. Yao, Carbon Energy, 2022, 4, 731–761 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- Y. Hinuma and K. Mori, Sci. Technol. Adv. Mater.: Methods, 2023, 3 DOI:10.1080/27660400.2022.2161807.
- H. Xu, Z. Jin, Y. Zhang, X. Lin, G. Xie, X. Liu and H.-J. Qiu, Chem. Sci., 2023, 13, 771–790 RSC.
- Y. Sun, W. Zhang, Q. Zhang, Y. Li, L. Gu and S. Guo, Matter, 2023, 6, 193–205 CrossRef CAS.
- W. A. Z. R. A. K. Putri, M. R. Abukhadra and Y. G. Ko, Nano Energy, 2023, 110, 108362 CrossRef.
- D. Modupeola and P. Popoola, Front. Energy Res., 2023, 11 DOI:10.3389/fenrg.2023.1149446.
- Y. Zhou, X. Shen, T. Qian, C. Yan and J. Lu, Nano Res., 2023, 13 DOI:10.1007/s12274-023-5419-2.
- C. L. P. Pavithra and S. R. Dey, Nano Select, 2023, 4, 48–78 CrossRef CAS.
- A. Lehr, J. J. Velázquez-Salazar, J. M. Montejano-Carrizales, S. Mejia-Rosales, R. Mendoza-Cruz, L. Bazan-Diaz and M. J. Yacaman, Faraday Discuss., 2023, 242, 10–22 RSC.
- A. Barbero, C. M. D. Silva, N. O. Pena, N. Kefane, A. Jaafar, M. Thorey, H. Bouaia, J. Nelayah, G. Wang, H. Amara, C. Ricolleau, V. Huc and D. Alloyeau, Faraday Discuss., 2023, 242, 129–143 RSC.
- S. Singh, N. Wanderka, B. S. Murty, U. Glatzel and J. Banhart, Acta Mater., 2011, 59, 182–190 CrossRef CAS.
- N. Kumar, C. S. Tiwary and K. Biswas, J. Mater. Sci., 2018, 54, 13411–13423 CrossRef.
- F. Waag, Y. Li, A. R. Ziefuss, E. Bertin, M. Kamp, V. Duppel, G. Marzun, L. Kienle, S. Barcikowski and B. Gökce, RSC Adv., 2019, 2, 18547 RSC.
- T. Löffler, F. Waag, B. Kökce, A. Ludwig, S. Barcikowski and W. Schuhmann, ACS Catal., 2021, 11, 1014–1023 CrossRef.
- N. L. N. Broge, A. D. Bertelsen, F. Sø ndergaard-Pedersen and B. B. Iversen, Chem. Mater., 2023, 35, 144–153 CrossRef CAS.
- J. Ledieu, M. Feuerbacher, C. Thomas, M.-C. de Weerd and S. Sturm, Acta Mater., 2021, 209, 116790 CrossRef CAS.
- L. Kaufman and H. Bernstein, Computer Calculation of Phase Diagrams, Academic Press, New York, 1970 Search PubMed.
- C. Zhang, F. Zhang, S. Chen and W. Cao, JOM, 2012, 64, 839–845 CrossRef CAS.
- O. Redlich and A. T. Kister, Ind. Eng. Chem., 1948, 40, 345–348 CrossRef.
- R. O. Williams, Calphad, 1991, 15.1, 1–10 CrossRef.
- F. Calvo, J. Chem. Phys., 2012, 136, 154701 CrossRef CAS PubMed.
- L. Xie, P. Brault, A. L. Thomann and J. M. Bauchire, Appl. Surf. Sci., 2013, 285, 810–816 CrossRef CAS.
- L. Xie, P. Brault, A. L. Thomann, X. Yang, Y. Zhang and G. Y. Shang, Intermetallics, 2016, 68, 78–86 CrossRef CAS.
- M. Widom, J. Mater. Res., 2018, 33, 2881–2898 CrossRef CAS.
- O. I. Kushnerov, J. Phys. Electron., 2019, 27, 41–46 CrossRef PubMed.
- C. Tang, P. Ren and X. Chen, Phys. Lett. A, 2019, 383, 2290–2295 CrossRef CAS.
- Z. M. Zeng, J. F. Zhao, X. F. Zhou, J. H. Li and B. D. Liang, Chem. Phys., 2019, 517, 126–130 CrossRef CAS.
- J. Li, Q. Fang, B. Liu, Y. Liu and Y. Liu, RSC Adv., 2016, 80, 76409–76419 RSC.
- I. A. Alhafez, C. J. Ruestes, E. M. Bringa and H. M. Urbassek, J. Alloys Compd., 2019, 803, 618–624 CrossRef.
- G. Lin, J. Guo and P. Ji, Phys. Chem. Chem. Phys., 2021, 23, 19482–19493 RSC.
- I. Toda-Caraballo, J. S. Vróbel, D. Nguyen-Manh, P. Pérez and P. E. J. Rivera-Díaz-del-Castillo, JOM, 2017, 69, 2137 CrossRef.
- M. C. Gao and M. Widom, J. Phys. Chem. B, 2018, 122, 3550 CrossRef CAS PubMed.
- M. Akkas and F. R. A. A. Sudani, Sci. Sintering, 2021, 53, 19–35 CrossRef CAS.
- X. W. Zhou, R. A. Johnson and H. N. G. Wadley, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144113 CrossRef.
- G. Rossi and R. Ferrando, J. Phys.: Condens. Matter, 2009, 21, 084208 CrossRef CAS PubMed.
- J. Xie, J. A. Northby, D. L. Freeman and J. D. Doll, J. Chem. Phys., 1989, 91, 612 CrossRef CAS.
- B. W. van de Waal, J. Chem. Phys., 1989, 90, 3407 CrossRef CAS.
- C. L. Cleveland and U. Landman, J. Chem. Phys., 1991, 94, 7376 CrossRef CAS.
- H. S. Lim, C. K. Ong and F. Ercolessi, Surf. Sci., 1992, 269/270, 1109 CrossRef.
- J. P. K. Doye and F. Calvo, Phys. Rev. Lett., 2001, 86, 3570 CrossRef CAS PubMed.
- R. H. Swendsen and J.-S. Wang, Phys. Rev. Lett., 1986, 57, 2607 CrossRef PubMed.
- M. Aydn, Y. Gündüç and T. Çelik, Phys. A, 1996, 230, 651–657 CrossRef.
- M. Aydn, T. Çelik and Y. Gündüç, Int. J. Mod. Phys. C, 1997, 8, 1081–1084 CrossRef.
- J. F. H. Stillinger, J. Chem. Phys., 1963, 38, 1486–1494 CrossRef.
- Z. Lin, R. A. Johnson and L. V. Zhigilei, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 214108 CrossRef.
- S. I. Rao, C. Woodward, T. P. Parthasarathy and O. Senkov, Acta Mater., 2017, 134, 188–194 CrossRef CAS.
- S. Chen, Z. H. Aitken, Z. Wu, Z. Yu, R. Banerjee and Y.-W. Zhang, Mater. Sci. Eng., A, 2020, 773, 138873 CrossRef CAS.
- L. Wang, W. Liu, B. Zhu, W. Chen, F. Zhang, B. Liu, J. Liu, J. Zhou and Y. Zhao, J. Mater. Res. Technol., 2021, 14, 2071–2084 CrossRef CAS.
- Y. H. Niu, D. Zhao, B. Zhu, S. B. Wang, Z. X. Wang and H. W. Zhao, Comput. Mater. Sci., 2022, 215, 111787 CrossRef CAS.
- D. Utt, S. Lee, Y. Xing, H. Jeong, A. Stokowski, S. H. Oh, G. Dehm and K. Albe, Nat. Commun., 2022, 13, 4777 CrossRef CAS PubMed.
- T. Cheng, G. Wei, S. Jiang, J. Zhang, Y. Wang, P. Liu, M. Hong, E. Guo, F. Zhong, G. Cai, C. Jiang and F. Ren, Acta Mater., 2023, 248, 118765 CrossRef CAS.
- T. Gheno, F. Jomard, C. Desgranges and L. Martinelli, Materiala, 2018, 3, 145–152 CrossRef CAS.
- F. Calvo, E. Cottancin and M. Broyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 121406(R) CrossRef.
- F. Otto, Y. Yang, H. Bei and E. P. George, Acta Mater., 2013, 61, 2628–2638 CrossRef CAS.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184115 CrossRef.
- F. Calvo and F. Spiegelmann, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 10949 CrossRef CAS PubMed.
- J. Behler, J. Chem. Phys., 2016, 145, 170901 CrossRef PubMed.
- B. Ruiz-Yo, ACS Comb. Sci., 2016, 18, 596–603 CrossRef PubMed.
- Y. Lederer, C. Toher, K. S. Vecchio and S. Curtarolo, Acta Mater., 2018, 159, 364–383 CrossRef CAS.
- Y. Yao, Z. Liu, P. Xie, Z. Huang, T. Li, D. Morris, Z. Finfrock, J. Zhou, M. Jiao, J. Gao and Y. Mai, Sci. Adv., 2020, 6, eeaz0510 CrossRef PubMed.
- N. Linton and D. S. Aidhy, APL Mach. Learn., 2023, 1, 016109 CrossRef.
- Y. Yao, Z. Huang, T. Li, H. Wang, Y. Liu, H. S. Stein, Y. Mao, J. Gao, M. Jiao, Q. Dong and J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6316–6322 CrossRef CAS PubMed.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, J. Zhiyong, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, 6589 CrossRef PubMed.
- D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumara, O. Seo, I. Gueye, J. Kim, L. S. R. Kumara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 3365–3369 CrossRef CAS PubMed.
- F. Tian, L. Delczeg, N. Chen, L. K. Varga, J. Shen and L. Vitos, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085128 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Corresponding data obtained using the pair correlation entropy instead of the thermodynamical entropy of mixing, and atomic coordinates of all lowest-energy structures. See DOI: https://doi.org/10.1039/d3cp01869e |
| This journal is © the Owner Societies 2023 |