
Doubly ionized OCS bond rearrangement upon fragmentation – experiment and theory†
Mahmoud
Jarraya
abc,
Måns
Wallner
d,
Saida
Ben Yaghlane
b,
Emelie
Olsson
d,
Veronica
Ideböhn
d,
Richard J.
Squibb
d,
Jérôme
Palaudoux
 c,
Gunnar
Nyman
e,
Muneerah Mogren
Al-Mogren
f,
John H. D.
Eland
*g,
Raimund
Feifel
*d and
Majdi
Hochlaf
*a
c,
Gunnar
Nyman
e,
Muneerah Mogren
Al-Mogren
f,
John H. D.
Eland
*g,
Raimund
Feifel
*d and
Majdi
Hochlaf
*a
aUniversité Gustave Eiffel, COSYS/IMSE, 5 Bd Descartes, 77454, Champs Sur Marne, France. E-mail: majdi.hochlaf@univ-eiffel.fr
bLSAMA, Faculté des Sciences de Tunis, Université de Tunis El Manar, Tunis, 2092, Tunisia
cLCP-MR, Sorbonne Université – UMR 7614, 75231 Paris Cedex 05, France
dDepartment of Physics, University of Gothenburg, 412 58 Gothenburg, Sweden. E-mail: raimund.feifel@physics.gu.se
eDepartment of Chemistry and Molecular Biology, University of Gothenburg, 405 30 Gothenburg, Sweden
fDepartment of Chemistry, College of Sciences, King Saud University, PO Box 2455, Riyadh 11451, Saudi Arabia
gDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, Oxford University, Oxford, OX1 3QZ, UK. E-mail: john.eland@chem.ox.ac.uk
First published on 5th July 2023
Abstract
The dissociation of OCS2+ ions formed by photoionization of the neutral molecule at 40.81 eV is examined using threefold and fourfold electron–ion coincidence spectroscopy combined with high level quantum chemical calculations on isomeric structures and their potential energy surfaces. The dominant dissociation channel of [OCS]2+ is charge separation forming CO+ + S+ ion pairs, found here to be formed with low intensity at a lower-energy onset and with a correspondingly smaller kinetic energy release than in the more intense higher energy channel previously reported. We explain the formation of CO+ + S+ ion pairs at low as well as higher ionization energies by the existence of two predissociation channels, one involving a newly identified COS2+ metastable state. We conclude that the dominant CO+ + S+ channel with 5.2 eV kinetic energy release is reached upon OCS2+ → COS2+ isomerization, whereas the smaller kinetic energy release (of ∼4 eV) results from the direct fragmentation of OCS2+ (X3Σ−) ions. Dissociation of the COS2+ isomer also explains the existence of the minor C+ + SO+ ion pair channel. We suggest that isomerization prior to dissociation may be a widespread mechanism in dications and more generally in multiply charged ion dissociations.
1. Introduction
The photoionization and consequent ionic dissociations of carbonyl sulfide, OCS, have been studied extensively using a broad variety of methods. In single ionization, the spectrum of electronic states of OCS+ over a wide energy range1,2 is known, as are the initial state populations and the identities of the fragmentation as functions of ionizing photon energy.3 Coincidence measurements have shown what products, including fluorescence photons, arise from selected initially populated states,4 and how the initial state populations depend upon ionizing photon energy.5 Moreover, they give the kinetic energy released in each process and the probable states of the products.3Double and triple ionization of the OCS molecule have been popular hunting grounds for the investigation of three-body dissociation dynamics and selective bond breaking.6–9 For double ionization our knowledge of the fates of nascent OCS2+ ions formed by various means is extensive. Studies have included direct ionization by single photons,3,10–16 electron17–19 and ion impacts,20,21 indirect ionization by Auger processes,22–28 double-charge-transfer29,30 and recently strong-field ultrafast laser interaction-induced multiple ionization.31–39 The interpretation of the experimental work has been aided by extensive calculations of the state manifold40–45 and possible dication dissociation pathways or by performing real-time time-dependent density functional theory (rtTDDFT) and semi-classical surface-hopping dynamics.38 In particular, the multireference configuration interaction (MRCI) computations of OCS2+ electronic states by Brites et al.40 showed that relatively deep potential wells separated from dissociation by large potential barriers exist for the lowest states where long-lived dicationic rovibrational levels may be populated efficiently, so accounting for the observation of an intense OCS2+ ion signal in the photoionisation mass spectrum.40
Previous experimental and theoretical work has shown that OCS2+ ions, once formed by various ionization methods, undergo fragmentations to produce dominantly the CO+ + S+ ion pair and to a much smaller extent CS+ + O+ and SO+ + C+. For the dominant CO+ + S+ channel, the thermodynamical threshold for the formation of these fragments with respect to the OCS ground state was estimated at ∼27.5 eV.40 Nevertheless, they appear at higher energies, carrying out some kinetic energy release (KER). The most abundant KER peak and also the mean value are in the range of 5–6 eV so an appearance energy (AE) of 27.5 + 5.5 = 33 eV is inferred by assuming that the CO+ + S+ products are formed in their ground states at threshold. The KER distribution has been measured, it extends to below 4 eV and above 6 eV.38,46 In the work of Endo et al.38 a weak but distinct feature with KER of ∼4 eV was observed for this ion pair. For the same channel a discrepancy arose in the MRCI computations by Brites et al.,40 where the computed appearance energy (of 31.6 eV) was distinctly lower than the value then experimentally available. In contrast, a plausible mechanism was proposed by Brites et al.40 for the formation of the CS+ + O+ ion pair, which appears at high energies with respect to OCS2+(X3Σ−).44 Briefly, these authors ruled out the implication of the OCS2+ ground state in such a process. Instead, direct dissociation from the electronically excited OCS2+(13Π) ions or after predissociation via the OCS2+ quintet states allowed to explain the observation of these fragments with a KER of ∼6 eV as determined using photoelectron photoion photoion coincidence (PEPIPICO) technique.13,40
The minor channel producing the SO+ + C+ ion pair was reported by Wang and Vidal19 in 2003 using electron impact ionization, and was observed again in 2019 by Zhao et al.31 using strong field ionization. This ion pair can be formed only after bond rearrangement or by extreme bending of the molecules, as happens in CO2 yielding O2+ by Auger decay at the main long-lived π* pre-edge resonances47–49 or after intramolecular isomerization upon double ionization as evidenced for SO2 producing an O2+ + S+ pair.50 Interestingly, Zhao et al. observed that under strong field ionization conditions the angular distributions of CO+ + S+ and SO+ + C+ share the same behavior. In 2022, Endo et al.38 using ultrafast asymmetric laser fields identified both a major (high KER) and a minor (lower KER) CO+ + S+ channel, which may be the same two channels identified in the present work, despite the different excitation conditions. These channels exhibit a clear dependency of the asymmetry on the kinetic energy, and a phase shift of 270° between the two sub-channels of CO+ + S+ is deduced. These authors attributed this observation to a population transfer between the involved electronic states in the applied intense laser fields.
To fully picture the further dynamics of the OCS2+ dication formed upon double ionization of OCS, a direct measurement of the product ion yields as a function of the initial internal energy or state of the nascent dication is lacking. To achieve this, we here use threefold and fourfold electron–ion coincidence methods to examine the yield of the parent dication and of the ion-pair products, CO+ + S+ and O+ + CS+ from doubly ionized OCS as a function of ionization energy. Our measurements indicate a significantly lower first appearance energy (AE) of 31.7 ± 0.4 eV for the dominant CO+ + S+ ion pair, compared with those given by previous measurements, or inferred as a lower limit from measurements of the dominant KER by addition to the energy of the lowest asymptote. For this channel, our newly measured appearance energy agrees with the prediction of 31.6 eV from detailed MRCI calculations40 and with some other earlier measurements (cf.Table 1). This implies that the KER in dissociation from states near the threshold must be close to the calculated value of 4.2 eV, which is within the range of KER distributions where these have been measured. Moreover, our yield curves show that a process producing the same ion pair more abundantly starts at 34.1 ± 0.5 eV, which is consistent with the intense KER peak and the onsets observed in previous investigations. Overall, we find that the evidence for partial dissociation of OCS2+ at energies below 33 eV taken from the present measurements together with data from earlier photoelectron photoion coincidence (PEPICO) experiments3,46 and coincidence work using the Auger processes,28 is convincing, but we have no definite explanation for the discrepancy between this conclusion and the higher threshold in direct photoionization at variable wavelengths reported earlier by Millié et al.44 and by Masuoka and Doi.9 Participation by a less stable COS2+ was invoked more than once.31,38,40 For instance, a possible dissociation involving the COS2+ isomer may lead to the CO+ + S+ or the SO+ + C+ ion pairs. However, this isomer has not been characterized hitherto and its putative role on such unimolecular decomposition is not yet established.
| Ref. | CO+ + S+ | CS+ + O+ | SO+ + C+ | |||
|---|---|---|---|---|---|---|
| AE (eV) | KER (eV) | AE (eV) | KER (eV) | AE (eV) | KER (eV) | |
| a This work. KERs are inferred, not directly measured. b Time-of-flight photoelectron photoelectron coincidence (TOF-PEPECO). Ref. 40. c Femtosecond laser pulses. Ref. 39. d Photo-ion time-of-flight mass spectra synchrotron radiation. Ref. 25. e Photo-ion time-of-flight mass spectra synchrotron radiation. Ref. 26. f Ion momentum spectrometry and energy selected Auger electron–photoion–photoion coincidence techniques. Ref. 11. g Soft X-ray synchrotron radiation. Ref. 10. h Synchrotron radiation based photoion–photoion coincidence (PIPICO). Ref. 13. i Momentum imaging technique (electron collision). Ref. 17. j Ultrafast Asymmetric Intense Laser Fields. Ref. 38. k Photoion photoion coincidence (PIPICO). Ref. 44. l Auger electron–ion coincidences. Ref. 24. m Photoion photoion coincidence (PIPICO). Ref. 16. n Strong-field (laser) ionization. Ref. 31. | ||||||
| 31.7 ± 0.4 | ≤4.2 | 36.9 ± 0.5 | ca. 4 | |||
| 34.1 ± 0.5 | ≤6.5 | |||||
| 34 | 5.1 ± 0.5 | |||||
| 4.3 | 4.0 & 5.9 | |||||
| 5.7 | ||||||
| 8 | ||||||
| 6.3 & 6.9 & 7.6 | 7.6 & 8.3 | |||||
| 7 | 5 & 7 &10 | |||||
| 4.1 ± 0.5 | ∼6 | |||||
| 5 | ||||||
| 4.0 & 5.2 | ∼5.6 | |||||
| 33.5 ± 0.5 | 40.2 ± 1 | |||||
| 34/32 | ||||||
| 5.7 ± 1.5 | ||||||
| 5.5 | ||||||
In this work, we perform computations at a high level of theory to characterize stable structures of dicationic structures of [O,C,S] sum formula by screening the lowest A′ and A′′ singlet, triplet and quintet potential energy surfaces. These calculations confirm the existence of a (meta)stable COS2+ isomer. In addition to the potential energy surfaces (PESs) of OCS2+ available in ref. 40, we mapped the PESs of the lowest electronic states of COS2+ allowing us to re-examine the dissociation pathways. In particular, we examine bond rearrangement upon double ionization of OCS to model formation of the fragmentation products CO+ + S+ and SO+ + C+.
2. Methods
2.1. Experimental details
Multi-electron–ion coincidence measurements were carried out in Gothenburg at 40.81 eV photon energy using a time-of-flight (TOF) magnetic bottle instrument. The apparatus, which has been described before,51 allows for simultaneous detection of electrons with energy information and ions with mass/charge information. The sample molecule is let into the interaction region of the spectrometer as an effusive jet in the form of a hollow needle where it is intersected by wavelength-selected light in a crossed beam configuration. The helium discharge lamp producing the 40.81 eV photons was operated at a repetition rate of about 4 kHz with sufficiently low light flux, providing an ionization probability in accordance with classical coincidence conditions. All emitted electrons are guided by the divergent field of a strong ring magnet and a hollow conical pole piece, coupled to the homogenous field of a solenoid enveloping the length of a 2.2 m drift tube. A weak electric draw-out field ensures that all electrons arrive at the detector within 10 microseconds, and a linear gate is used to ensure that recorded signals arise only from real photoionization events, removing noise caused by triggering the electric field used to extract the ions. About 150 ns after ionization, an electric field is applied across the interaction region to extract cations through the ring magnet. The ions are accelerated further in a two-field configuration optimized for time-focusing conditions. At the time of the experiment the collection efficiency was about 55% for electrons and 29% for ions over the relevant mass range of 29 to 50 amu. The electron kinetic energy resolving power (E/ΔE) was about 25, and the mass resolution was about 50 for thermal ions. The OCS sample was commercially obtained, with impurities of about 0.3% of CS2 and a similar amount of CO2 as deduced from our spectra.2.2. Computational details
The ab initio computations were performed using the MOLPRO program suite.52 They correspond to the search for stable forms of [O,C,S]2+ sum formula and to mapping of the PESs of the newly identified isomers. We considered singlet, triplet and quintet spin multiplicities. For these calculations, the S atom is described using the aug-cc-pV(Q+d)Z basis set and the aug-cc-pVQZ basis set is adopted for oxygen and carbon atoms.53–55 In fact, the inclusion of diffuse and tight-d atomic functions is highly recommended for the correct description of sulfur containing molecular species.56–58For geometry optimizations, we use the partially spin restricted coupled cluster method including perturbative treatment of triple excitation (RCCSD(T)).59–61 This allows us to derive the structural parameters, harmonic frequencies, total energies and dissociation limits of all the species involved in this work (cf. ESI†).
For the PESs of the dicationic species, we use the complete active space self-consistent field (CASSCF) method62,63 followed by the internally contracted MRCI64–66 approach. At the CASSCF level, we consider all valence electrons and orbitals as active. All electronic states having the same spin-multiplicity are averaged together. In the MRCI calculations, we consider all configurations having a weight greater than 0.05 in the CI expansion. This results in >8 × 108 uncontracted configuration state functions (CSFs) while treating the singlet electronic states, >16 × 108 CSFs for the triplets and >1 × 109 CSFs for the quintets.
3. Results
3.1. Experimental results
To select double ionization events with known energy transfer in direct photoionization, it is necessary and sufficient to detect two electrons in coincidence and to measure the energy of each one. The yield of stable (or long-lived metastable) doubly charged parent ions is fully determined as a function of ionization energy by measurement of threefold electron–electron–ion (eei) coincidences. For cation pairs, although fourfold coincidence measurements of two electrons and two ions are generally necessary, the relatively more abundant threefold eei coincidences are sufficient for these too in simple cases like OCS, where no ambiguities arise. A threefold coincidence map for OCS double ionization is shown in Fig. 1. Despite some evident false coincidence interference at electron pair energies below the double ionization range of 29 to 40 eV, particularly for S+, formation of the parent dication and of the ion pairs CO+ + S+ and O+ + CS+ is clearly visible. The onsets of CO+ and S+ with TOF width characteristic of the KER in charge separation both occur at about 9 eV electron pair energy, equivalent to about 32 eV ionization energy, but increase strongly in intensity at about 6.5 eV electron pair energy or 34.5 eV ionization energy. There is no evidence in the present experiments for significant formation of the SO+ + C+ ion pair, despite the fact that the thermodynamic limit for its formation, at 30.396 eV (Table S2, ESI†) is well within reach, even allowing for about 5 eV kinetic energy release. The SO+ signal present, but not discernible, in the mass spectrum at 40.81 eV photon energy amounts to less than 0.05% of all electron–ion pairs.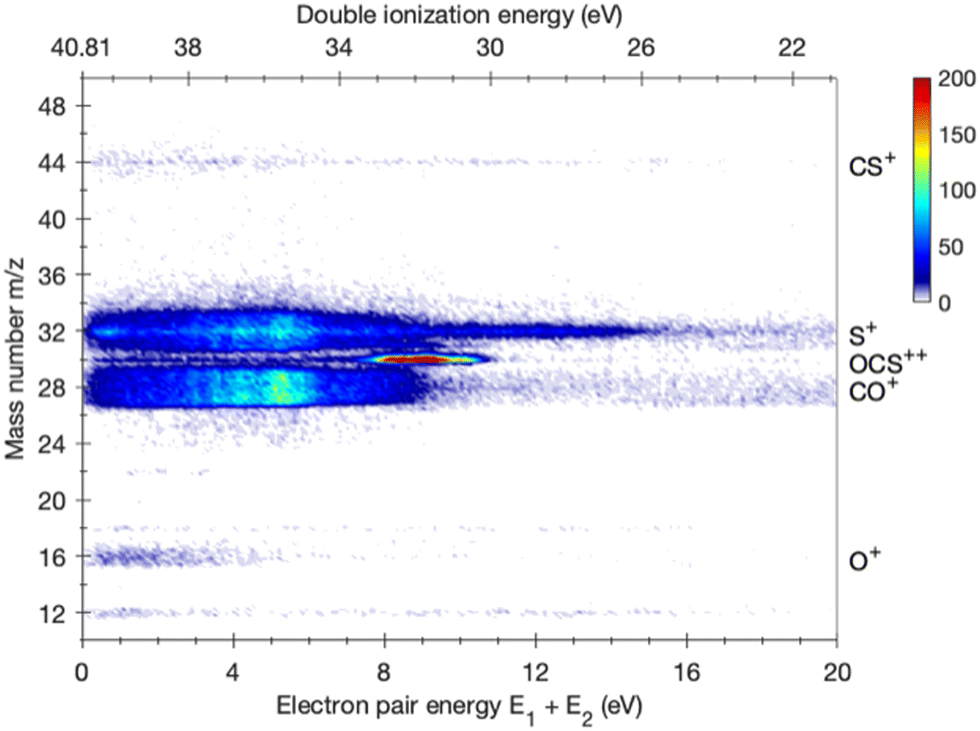 | ||
| Fig. 1 Electron–electron–ion coincidence map for OCS obtained at a photon energy of hν = 40.81 eV in the form of mass number against summed electron pair energy on the bottom axis and double ionization energy on the top axis. | ||
To convert the data to spectra of the yield in each channel as a function of ionization energy, we take selected projections from the same data, using cuts of each mass chosen to minimize overlaps. The results are shown on an ionization energy scale in Fig. 2 together with the spectral yield curve for CO+ + S+ and O+ + CS+ from fourfold eeii coincidences, where statistics are poor but there should be relatively fewer interfering false coincidences because of the stricter selectivity. An electron-only spectrum of OCS double photoionization obtained at the same photon energy with a more highly resolving electron-only spectrometer is included for comparison.
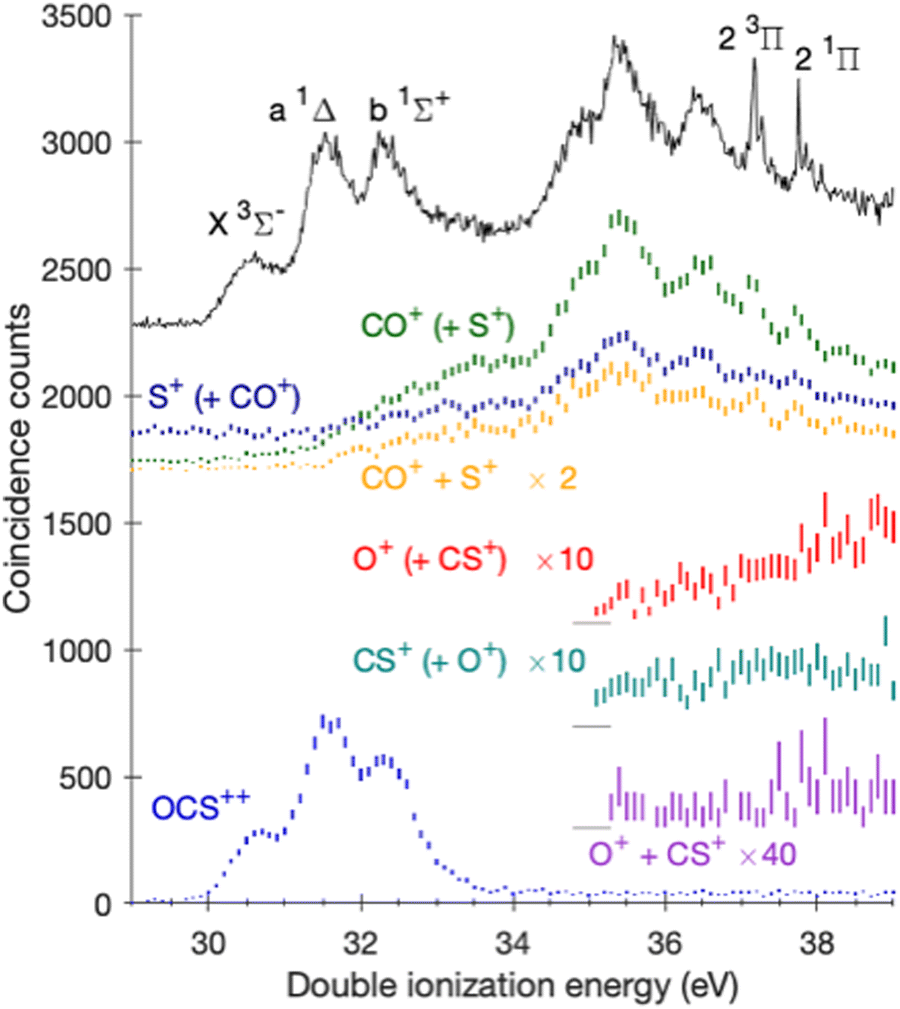 | ||
| Fig. 2 Double photoionization spectra coincident with the parent dication and with ion pairs projected from the data of Fig. 1, compared with a better resolved electron-only double photoionization spectrum (black curve) at the same photon energy.40 CO+ (green trace) and S+ (dark blue trace) data represents the CO+ + S+ pair and O+ (red trace) and CS+ (teal trace) data represents the O+ + CS+ pair. The aforementioned curves are threefold coincidences with two electrons and one ion, where the second ion is implied and shown in parenthesis. The CO+ + S+ pair (yellow trace) and the weak O+ + CS+ pair (purple trace) are from fourfold coincidences. The signals are magnified by the denoted factors and the curves display 2σ error bars. | ||
In Fig. 2 the parent OCS2+ dication yield shows a gradual rise from an apparent threshold of 29.5 eV before a stronger rise at 30.2 eV, near the established experimental double ionization energy of OCS of 30.0 ± 0.1.40 This tailing below the true onset is attributed to the modest electron energy resolution (ca. 0.4 eV at 10 eV electron energy). This spectrum shows three bands. As discussed in ref. 40, they correspond to the population of the X3Σ−, a1Δ and b1Σ+ states of OCS2+ after doubly ionizing neutral OCS. For energies >34 eV, the dication signal drops to zero because these dicationic states dissociate.
The CO+ + S+ ion pair has an apparent onset at 31.6 ± 0.2 eV. Taking the resolution into account, we estimate the real appearance energy of this main ion pair as 31.7 ± 0.4 eV. For O+ + CS+ there is a gradual onset starting at 37.0 ± 0.5 eV; this is not affected by the resolution, because of the low electron energies involved. The two dissociation thresholds estimated in this way are both considerably lower than the appearance energies of 33.5 ± 0.5 eV and 40.2 ± 1 eV measured using variable wavelength photoionization as reported earlier by Millié et al.44 and measured with low resolution as about 35 eV by Masuoka and Doi9 and by Masuoka et al.13 As stated above, the formation of the O+ + CS+ pair was already explained in ref. 40. The presently determined AE of about 37 eV and the known peak KER of about 4 eV confirm that mechanism.
The spectra of Fig. 2 show formation of the CO+ + S+ ion pairs at energy levels within the energy range of the a1Δ and b1Σ+ states of OCS2+ (Fig. 2).40 Hikosaka and Shigemasa,28 who prepared OCS2+ dications by Auger decay following S 2p core ionization also found that OCS2+ is partly dissociated in the same energy range. Morse et al.,3 using well-resolved single-electron–ion coincidences (ei) also found that all CO+ ions formed at ionization energies above 32 eV are in the form of CO+ + S+ ion pairs. Kaneyasu et al.24 using Auger electron–ion coincidences state in the text that the onset of the CO+ + S+ ion pair is at 34 eV, but their Fig. 3, from which this value is taken, clearly shows a weaker onset at 32 eV. The observation that partial dissociation occurs in the energy ranges of the a1Δ and b1Σ+ states is supported in our data and in the Auger-electron–ion coincidence data of Hikosaka and Shigemasa28 by the relative intensities of the three 3π2 states (i.e. X3Σ−, a1Δ and b1Σ+) in the parent dication channel compared with the full double ionization spectrum measured at the same photon energy. The b1Σ+ state in particular appears relatively less intense in the parent dication spectrum than in overall double photoionization, whether direct or by Auger decay.
 | ||
| Fig. 3 [O,C,S] sum formula doubly charged minimal structures as computed at the RCCSD(T)/aug-cc-pV(Q+d)Z level of theory. We locate the [CO + S]2+, [SO + C]2+ and [CS + O]2+ channels, where the fragments are in their ground states. These energies are given with respect to the neutral OCS (X1Σ+) ground state. The ZPE correction is also included. See Tables S1–S3 (ESI†) for more details. | ||
Further evidence can be sought in the KER in formation of the ion pair as a function of ionization energy, as the KER cannot exceed the available energy, i.e. the excess of ionization above the lowest asymptote of 27.43 eV for CO+(X2Σ+) + S+(4S) (Table S3, ESI†). If this ion pair is formed at our estimated threshold of 31.7 eV the maximum possible KER is 4.2 eV, a bit less than the most abundant KER of ca 5.5 ± 0.5 eV found in previous measurements at the same photon energy40 and also under different conditions of ionization (Table 1). The available energy in the center of the range where we see partial dissociation is 5 eV also well within the range of measured KER distributions.46 The mass resolution in our apparatus does not allow peak broadening by KER to be measured reliably in the individual ion peak widths for CO+ or S+, but ion-pair time difference peaks (t2 − t1 or photoion–photoion coincidence (PIPICO) spectra), have twice the broadening due to KER compared to single ions. Mass peak broadening due to timing jitter and to a spread in initial ionization position is also eliminated in time difference spectra, as both ions come from the same event. Because the mass numbers for CO+ and S+ are relatively close together, only the later part of the PIPICO peak (t(S+ backwards) – t(CO+ forwards)) is free from interfering signals near the t2 = t1 limit and the strong OCS2+ signal. In Fig. S1 (ESI†) we show two reconstituted PIPICO peaks for CO+ + S+ with ionization energies in the ranges 31–33 eV and 34–36 eV. To the extent that the poor statistics allow any conclusions, the peak shapes in Fig. 1 are consistent with the idea that ion pairs coincident with electron pairs of lower ionization energies carry smaller KER than those formed from higher energy ionization. The peak shapes are not well enough defined for quantitative deduction of the actual energy releases.
The sum of the parent OCS2+ peak and the yield of CO+ (representing the ion pair) evidently does not exactly match the full double ionization spectrum in shape, having lower intensity on the high ionization energy side. This difference is attributed to increasing losses of ion pairs as the KER increases following the increasing available energy. Other pertinent observations from Fig. 2 are that all the peaks in the well-resolved double ionization spectrum are also present in the coincidence data on the CO+ + S+ channel, and that the parent dication OCS2+ is not formed in detectable quantity at ionization energies above 34 eV. On this last point we disagree with Hikosaka and Shigemasa28 who reported the detection of metastable OCS2+ over a range of ionization energies including the major peaks between 36.5 and 37.5 eV. The lack of observed OCS2+ ions at the higher energies in our data agrees with the lack of any fluorescence emission from the excited OCS2+ ion in contrast to the behavior of its congeners CO22+ and CS22+.67
3.2. Theoretical results
Fig. 3 shows the stable structures located in the lowest singlet, triplet and quintet OCS2+ PESs. These isomers were found after screening the 3D-PESs for a wide range of nuclear configurations at the RCCSD(T)/aug-cc-pV(Q+d)Z level of theory. The corresponding structural, vibrational and total energies are listed in Table S1 (ESI†). In addition to the well-known linear OCS2+ (X3Σ−, a1Δ) dication, we characterize a cyclic form (denoted as cyc-COS2+ (11A′′)) and a linear isomer COS2+ (X3Σ−, a1Δ, a5Π). The existence of COS2+ was invoked, but so far not theoretically established. Indeed, it is identified here for the first time, as well as the cyclic form. Since the PESs of OCS2+ were mapped in ref. 40, we computed those of COS2+. Indeed, Fig. S4 and S5 (ESI†) display the one-dimensional cuts of the 3D-PESs of COS2+ along the SO, CO distances, respectively. Table S4 (ESI†) lists MRCI/aug-cc-pV(Q+d)Z double ionization energies of COS2+ with respect to the energy at the equilibrium geometry of OCS (X1Σ+).Fig. 3 reveals that the most stable form is OCS2+ (at 30.01 eV w.r.t. neutral OCS) followed in energy by COS2+ (at 32.03 eV) and the cyclic form (at 34.04 eV). This figure shows also that these isomers are all located above the lowest dissociation limit CO+(X2Σ+) + S+(4S) (at 27.42 eV). They are metastable species and thus they may undergo predissociation processes populating this charge separation channel or the upper ones either directly or after intramolecular isomerization processes involving the less stable [O,C,S]2+ isomeric forms. OCS2+ (a1Δ) and COS2+ (a1Δ, a5Π) correspond to electronically excited metastable dicationic species. For higher energies, other metastable electronic states may exist. From ref. 40, we do know indeed that OCS2+(b1Σ+) ions are also long-lived.
The metastable singlet species of Fig. 3 are lying within the lowest singlet PES, the triplets are on the lowest triplet PES and the quintet is located on the lowest quintet PES. Within the same PES, intramolecular isomerization processes may occur converting one form to another. We may expect the occurrence of spin–orbit intersystem conversions at the singlet–triplet and triplet–quintet crossings. To shed light on these conversions, we mapped the one-dimensional cuts of the 3D PESs of OCS2+, of cyc-COS2+ and of COS2+ along the bending coordinates as given in Fig. 4. This figure shows that OCS2+ isomerizes into cyc-COS2+ by bending the OCS2+ in-plane bending angle (Fig. 4(A)). cyc-COS2+ converts into COS2+ by bending the in-plane COS2+ angle (Fig. 4(C)). Indeed, cyc-COS2+(11A′′) corresponds to the 11A′′ component of the OCS2+ (11Δ)/COS2+ (11Δ) states split by the Renner–Teller effect for nonlinear configurations.
 | ||
| Fig. 4 MRCI/aug-cc-pV(Q+d)Z one dimensional cuts of the PESs of the singlet, triplet and quintet electronic states of the OCS2+ system along the in-plane θ angle (in A), of COS2+ along the in-plane φ (in B) and α angles (in C). The reference energy is that of OCS (X1Σ+) at equilibrium. The distances are kept fixed at their equilibrium values in OCS2+ (X3Σ−) in (A) and (C) and in COS2+ (X3Σ−) in (B). | ||
Fig. 4(B) shows the one-dimensional evolution of the electronic states of OCS2+ while varying the φ angle whereas the distance between the O and S atoms is kept fixed at 5.5 Bohr (= its equilibrium value in OCS(X1Σ+)). For φ = 0°, one can find the OCS2+ dication whereas for φ > 120° we have a loosely bound CO–S2+ dication since the O–S distance is too long, i.e. almost a CO+ + S+ ion pair. Therefore, this figure shows that OCS2+ leads to COS2+ while the S atoms roams around the CO moiety. However, a potential barrier needs to be overcome. Within the triplet PES, we compute a barrier of ∼2.25 eV for OCS2+ → COS2+. For OCS2+ (11Δ) → COS2+ (11Δ) and OCS2+ (11Σ+) → COS2+ (11Σ+) barriers of ∼2.76 and ∼2.47 eV are estimated, respectively. While these singlets are crossed by a quintet state for φ ∼ 40°–50°, these barriers are not lowered since these singlet–quintet conversions are forbidden by spin–orbit. Nevertheless, these barriers may be slightly lowered after relaxation of the internuclear distances.
With respect to neutral OCS (X1Σ+), the OCS2+ → COS2+ isomerization requires hν ∼ 32.26, 33.98 or 34.63 eV, depending on whether photo-double ionization populates the OCS2+ (X3Σ−) or the OCS2+ (11Δ) or the OCS2+ (11Σ+) states, respectively. For the formation of the CO+ + S+ ion pair, we have a priori two mechanisms: either direct dissociation of OCS2+ ions or dissociation of the newly identified COS2+ ion after OCS2+ → COS2+ intramolecular isomerization processes. As detailed in the introduction, the former was commonly proposed, but it does not fully account for the previously determined experimental observations and energetics (KERs, AEs), and the latter was not previously proposed for that purpose.
Fig. 5 (right part) presents the one-dimensional cuts of the 3D-PESs of the lowest singlet, triplet and quintet states of COS2+ by varying the SO distance. In the 28–35 eV energy range, we locate three potential wells corresponding to the COS2+ X3Σ−, 11Δ and 11Σ+ states at 32.17, 33.33 and 34.22 eV w.r.t. OCS(X1Σ+). The corresponding potential barriers are computed as 0.44, 0.72 and 0.96 eV, resulting in 32.62, 34.06 and 35.18 eV AEs w.r.t. the neutral ground state of OCS(X1Σ+). The triplet correlates adiabatically to the CO+(X2Σ+) + S+(4S) asymptote, whereas the 11Δ state correlates to the CO+ (X2Σ+) + S+(2D) asymptote. Although the 11Δ and 11Σ+ singlets are crossed by the 15Σ− state, the singlet–quintet spin–orbit conversion is forbidden. Thus, the COS2+(11Δ) ions should fragment to CO+ (X2Σ+) + S+(2D). The KER associated with the COS2+(X3Σ−) → CO+(X2Σ+) + S+(4S) reaction is computed to be 5.19 eV and that for COS2+(11Δ) → CO+(X2Σ+) + S+(2D) is 4.79 eV. While comparing to the OCS2+ isomer, Brites et al.40 computed the barrier to dissociation from the OCS2+ (X3Σ−) ground state to the lowest CO+ (X2Σ+) + S+ (4S) asymptote, correlating adiabatically to OCS2+ (X3Σ−) by C–S bond extension to be 1.6 eV (cf.Fig. 5), giving a predicted appearance energy of 31.6 eV and a kinetic energy release of 4.17 eV. The AE and KER for OCS2+(11Δ) → CO+(X2Σ+) + S+(2D) are 33.4 eV and 4.10 eV, respectively (cf.Table 2). For COS2+(11Δ) → CO+(X2Σ+) + S+ (2D), we calculate an AE = 34.06 eV and a KER of 4.79 eV. These sets of data are quite different. Nevertheless, the present experiments can probe these differences making it possible to characterize the unimolecular dissociation of OCS2+ and of COS2+ and its competition with the OCS2+–COS2+ isomerization. The COS2+(11Σ+) and OCS2+(11Σ+) states correlate to the CO+ (X2Σ+) + S+ (2Pu) upper limit. Once populated, these ions may form COS2+/OCS2+ (11Δ) ions and/or fragment to produce the CO+ (X2Σ+) + S+ (2D) ions after spin–orbit conversion by the 13Π or the 23Π states and/or fragment leading to CO+ (X2Σ+) + S+ (2P) ions.
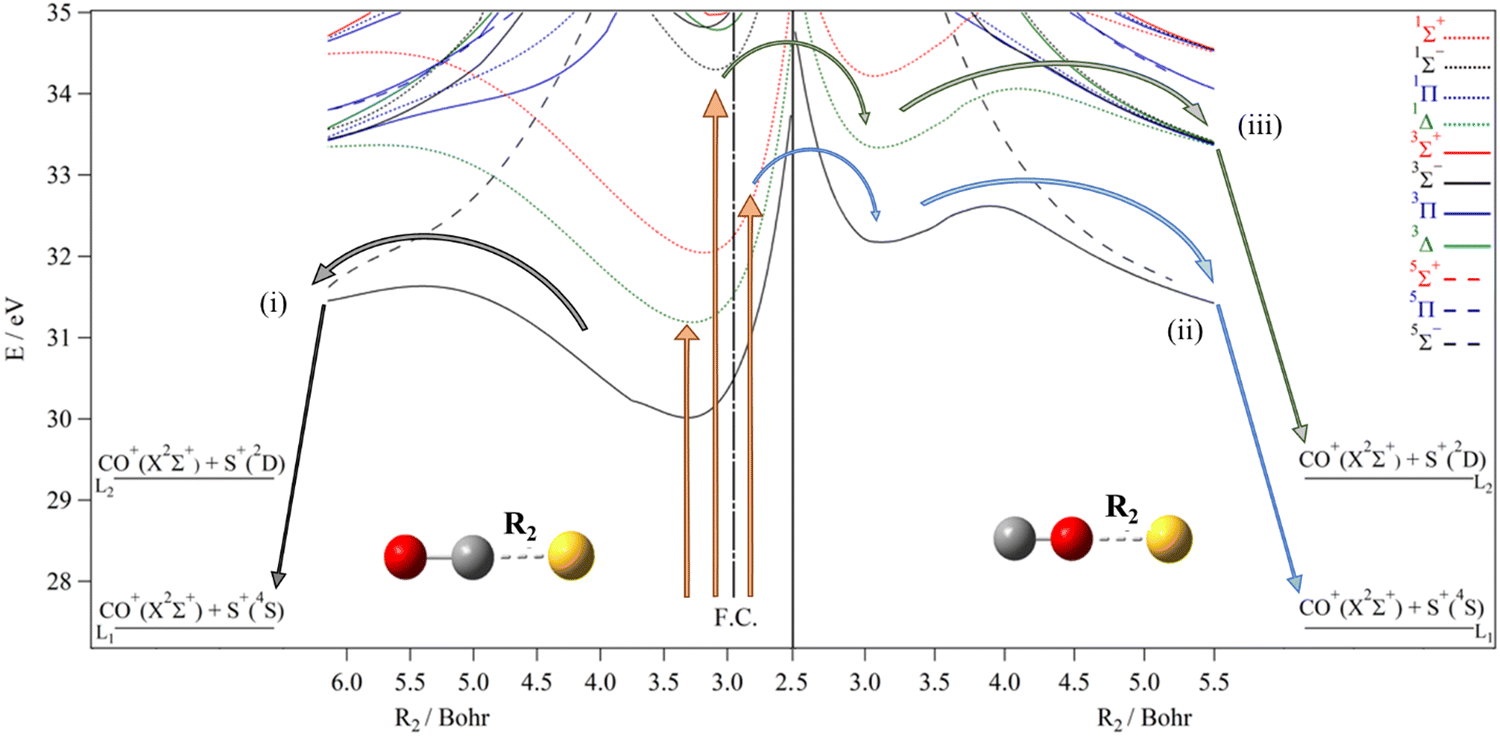 | ||
| Fig. 5 Right: MRCI/aug-cc-pV(Q+d)Z potential energy curves of the lowest electronic states of COS2+ for collinear configuration along the SO coordinates (R2), where the CO distance is kept at 2.288 Bohr (i.e. its equilibrium value in COS2+ (X3Σ−)). F. C. corresponds to middle of the Franck–Condon zone accessed from the ground state of OCS. Left: MRCI collinear one-dimensional cuts of the 3D-PESs of OCS2+ electronic states along the CS coordinate, where the CO distance is fixed at 2.185 Bohr (i.e. its equilibrium value in OCS (X1Σ+)).40 The reference energy is the energy at OCS (X1Σ+) minimum. The vertical arrows correspond to different photon energies used for discussing the mechanisms in the text. The clear and crossed circles correspond to allowed and forbidden spin–orbit conversions, respectively. | ||
| Reaction/channel | AE | KER | Ref. |
|---|---|---|---|
| a CASSCF/MRCI/spdfg cc-pV5Z. Ref. 40 b Multistate density-functional-theory method (MSDFT PBE0/aug-cc-pVTZ level of theory). Ref. 39 c This work. | |||
| CO + + S + | |||
| OCS2+ (X3Σ−) → CO+ (X2Σ+) + S+ (4S) | 31.6 | 4.17 | |
| 4.5 | |||
| OCS2+ (11Δ) → CO+ (X2Σ+) + S+ (2D) | 33.4 | 4.10 | |
| OCS2+ (11Σ+) → CO+ (X2Σ+) + S+ (2D) | 34.5 | 5.24 | |
| COS2+ (X3Σ−) → CO+ (X2Σ+) + S+ (4S) | 32.62 | 5.19 | |
| COS2+ (11Δ) → CO+ (X2Σ+) + S+ (2D) | 34.06 | 4.79 | |
| COS2+ (11Σ+) → CO+ (X2Σ+) + S+ (2D) | 35.18 | 5.91 | |
| CS + + O + | |||
| OCS2+ (X3Σ−) → CS+ (X2Σ+) + O+ (4S) | 35.37 | 4.07 | |
| 3.5 & 5.7 | |||
| OCS2+ (a1Δ) → CS+ (X2Σ+) + O+ (2D) | 38.52 | 3.89 | |
| OCS2+ (b1Σ+) → CS+ (X2Σ+) + O+ (2D) | 39.01 | 4.38 | |
| SO + + C + | |||
| COS2+ (X3Σ−) → SO+ (X2Π) + C+ (2P) | 35.49 | 5.10 | |
| COS2+ (11Δ) → SO+ (X2Π) + C+ (2P) | 35.69 | 5.29 | |
| COS2+ (11Σ+) → SO+ (X2Π) + C+ (2P) | 35.88 | 5.48 | |
Fig. 6 shows the one-dimensional cuts of the 3D PESs of the lowest singlet, triplet and quintet states of COS2+ while lengthening the CO distance for collinear configurations whereas the SO distance is kept fixed. These cuts are given with respect to OCS(X1Σ+) at equilibrium. These potentials correlate to the [SO + C]2+ dissociation limits. This figure shows that the lowest singlets and triplets correlate to the lowest asymptote SO+(X2Π) + C+(2P). Also, we found relatively deep potential wells for these states along the CO coordinate. Indeed, we compute potential barriers of 3.36, 2.34 and 1.64 eV for the X3Σ−, a1Δ and b1Σ+ states, resulting in AEs of 35.49, 35.69 and 35.88 eV and KERs of 5.1, 5.3 and 5.5 eV for the COS2+(X3Σ−, 11Δ and 11Σ+) → SO+(X2Π) + C+(2Pu) fragmentations, respectively. Moreover, the potentials exhibit a high density of electronic states located above 36 eV favoring their mutual interactions by vibronic couplings at their respective crossings or avoided crossings between states of the same spin multiplicity. Also the electronic states can interact by spin–orbit between singlets and triplets and between triplets and quintets at their crossings. These effects make internal conversions possible to populate the lower states.
 | ||
| Fig. 6 MRCI/aug-cc-pV(Q+d)Z potential energy curves of the lowest electronic states of COS2+ along the CO coordinates (R1) for collinear configurations. The SO distance is set to 3.172 Bohr, i.e. its equilibrium value in COS2+ (X3Σ−). The reference energy is that of OCS (X1Σ+) at equilibrium. Adapted from Fig. S4 (ESI†). | ||
4. Discussion
Table 1 summarizes the present and previous experimental determinations of the AEs and the associated KERs for the formation of the CO+ + S+, CS+ + O+ and SO+ + C+. As mentioned before, two appearance energies of 31.7 ± 0.4 eV and 34.1 ± 0.5 eV are measured for the formation of CO+ + S+, with KERs of ca 4 eV and 5.5 eV respectively. The former is a minor fragmentation channel upon formation of OCS2+ as are both the production of CS+ + O+ (AE ca. 37 eV) and SO+ + C+ (AE not known, KER reported as 5.5 eV31). We also give the corresponding data as deduced from the present MRCI computations in Table 2. At present, we focus on the dissociations from the lowest electronic states i.e. those lying below 36 eV.In 2008, Brites et al.40 computed one-dimensional potentials of OCS2+ along the internal coordinates, in particular along the CS distance leading to the CO+ + S+ fragments (cf.Fig. 5) and along the CO distance leading to CS+ + O+. Since then, the interpretation of the dynamics of the triatomic doubly charged ion formed upon single or multiple photon ionization of OCS has been based on these potentials. At that time only the mechanism leading to CO+ + S+ with an appearance energy of 34 eV was observed. It was suggested that the dissociation occurred from the OCS2+ a1Δ or b1Σ+ states to the lowest asymptote. Nevertheless, this mechanism was not convincing. Indeed, these two singlet states do not correlate adiabatically to the lowest asymptote, CO+(X2Σ+)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) + S+(4S), for which only the 13Σ− and 15Σ− states of OCS2+ correlate. However, Fig. 5 shows that there is no intersystem crossing or internal conversion between the a1Δ or b1Σ+ states and the ground triplet state. Also, the singlet–quintet conversion is forbidden although these singlets are crossed by the 15Σ− state (see Fig. 5). This means that the measured and computed KERs did not match for this channel. So, populating the lowest asymptote from singlet states is not possible, invalidating the fragmentation pathways proposed by Brites et al.40 for the observed appearance energy of 34 eV. Generally, previous works have not considered the possible involvement of all isomers of the [O, C, S]2+ molecular system when studying the fragmentation pathways.
+ S+(4S), for which only the 13Σ− and 15Σ− states of OCS2+ correlate. However, Fig. 5 shows that there is no intersystem crossing or internal conversion between the a1Δ or b1Σ+ states and the ground triplet state. Also, the singlet–quintet conversion is forbidden although these singlets are crossed by the 15Σ− state (see Fig. 5). This means that the measured and computed KERs did not match for this channel. So, populating the lowest asymptote from singlet states is not possible, invalidating the fragmentation pathways proposed by Brites et al.40 for the observed appearance energy of 34 eV. Generally, previous works have not considered the possible involvement of all isomers of the [O, C, S]2+ molecular system when studying the fragmentation pathways.
Our new experimental data and some previous ones show that the situation is more complicated for OCS2+ producing CO+ + S+ ions. To identify the OCS2+ electronic states decaying to CO+ + S+ we make use of the calculations of Brites et al.40 combined with our present theoretical data (cf.Fig. 5). For this channel, the angular distribution analysis by Zhao et al.31 and by Endo et al.38 revealed that the CO+ + S+ ion pair formed with AE of 34 eV and the SO+ + C+ ion pair have the same trend, whereas the CO+ + S+ ion pair formed with AE of 31.7 eV does not. This suggests that two triatomic dicationic species are at the origin of the formation of the CO+ + S+ ion pair depending on the photon energy. In particular, the dominant predissociation mechanism (AE ∼ 34 eV) may first follow a bond rearrangement mechanism via the formation of the COS2+ less stable isomer rather than the most stable form, OCS2+.
From Fig. 5 we identify four dissociation mechanisms leading to the CO+ + S+ limits with different AEs and KERs. First, the population of the OCS2+ ground state in the Franck–Condon zone leads directly to the lowest asymptote CO+(X2Σ+) + S+(4S) (pathway (i)). The computed AE of 31.6 eV and the KER of 4.17 eV of ref. 40 are close to the present and previous experimental measurements for this weak CO+ + S+ channel production. In particular, we reassign the weak feature associated with a KER of ∼4 eV detected in the recent study of the post-ionization dynamics of OCS in asymmetric laser fields by Endo et al.38 to the OCS2+ → CO+(X2Σ+) + S+(4S) reaction. Thus, the formation of CO+(X2Σ+) and S+(4S) fragments from OCS2+ is possible in contrast to previous statements. Nevertheless, this channel is not a major one as found previously and confirmed by the present and recent experiments.38 To explain the weak signal associated with this fragmentation, we suggest that only the presumably very weak Franck–Condon tail of the X state goes to CO+ + S+ since the initial population in this energy region is predominantly in the a1Δ and b1Σ+ states of OCS2+.
Pathway (ii) in Fig. 5 leads to CO+(X2Σ+) + S+(2D). This channel could be reached directly from OCS2+(a1Δ) with an AE of 33.4 eV and a KER of 4.10 eV. However, the involvement of the OCS2+(a1Δ) state can be ruled out since the corresponding AE differs with the measured value. We might also suggest the participation of OCS2+(b1Σ+), which is efficiently populated at these energies because of favorable Franck–Condon factors. Afterwards, OCS2+(b1Σ+) leads to CO+(X2Σ+) + S+(2D) upon spin–orbit conversion by the 13Π or 23Π states, which correlate adiabatically to this limit. The crossings of this singlet with these triplets occurs at ∼34 eV, which is thus the AE for this reaction. Such conversions are allowed since the respective CASSCF/cc-pVTZ spin–orbit integrals are evaluated to be ∼137 and ∼12 cm−1 at the 13Π and 23Π crossings with b1Σ+. A KER of ∼5 eV is evaluated (cf.Table 2), close to the presently measured value. This pathway however should be ruled out while forming OCS2+ by strong field ionization as in the experiments of Zhao et al.31 and Endo et al.38 Indeed, the formation of this ion pair is due to bond rearrangement in accordance with their anisotropy measurements. As stated in the introduction, the CO+ + S+ ions formed at 34 eV and those produced at 31.7 eV present indeed a phase shift of 270° between these two sub-channels. The involvement of the other isomeric forms of OCS2+ is thus required. For instance, we suggest the formation of COS2+. This dication can be formed after OCS2+ → COS2+ isomerization. Fig. 5 shows two plausible pathways, which are reported here for the first time. They are denoted as pathway (iii) and pathway (iv). Pathway (iii) corresponds to the population of the ground state of COS2+ and its subsequent dissociation. Table 2 shows that we compute an AE of 32.6 eV and a KER of 5.19 eV. Although the AE is slightly smaller than the measured one, we cannot rule out this mechanism. It involves the newly identified COS2+ isomer, which is reached after intramolecular isomerization on the ground triplet potential. Upon double ionization, OCS2+ ions are formed in the b1Σ+ state within the 32–33 eV energy range, which can be converted into the a1Δ state by internal conversion and then to the X3Σ− state by spin–orbit conversion. Afterwards, these ions isomerize into COS2+ species that then overcome the isomerization barrier between OCS2+(X3Σ−) and COS2+(X3Σ−) (cf.Fig. 4(B)). Once formed, COS2+(X3Σ−) ions possess enough energy to fragment since the potential barrier of COS2+(X3Σ−) is relatively low (0.44 eV). Alternatively, the OCS2+(11Δ) ions convert into COS2+(11Δ) ions. Fig. 4 shows that the potential barrier converting OCS2+ (a1Δ) into COS2+(11Δ) can be overcome at these energies. Then, the COS2+(11Δ) ions dissociate into CO+(X2Σ+) + S+(2D) for energies >34 eV and release a KER of ∼4.8 eV. Indeed, we compute an AE of 34.06 eV for this channel and a KER of 4.79 eV for COS2+(11Δ) → CO+ (X2Δ+) + S+ (2D). Both values are close to the experimental determinations of AE = 34 eV and KER 5.1–5.2 eV. Therefore, the S+ cation is produced into an electronically excited state and not in its ground state. This corresponds to pathway (iv) in Fig. 5.
For energies >35 eV, a high density of states is computed for OCS2+ and COS2+. This may favor the internal conversion processes and predissociations. For instance, the COS2+(11Σ+) ions may be produced from OCS2+. The COS2+(11Σ+) state is predissociated by the 13Π and 23Π states of COS2+ that correlate adiabatically to CO+(X2Σ+) + S+(2D). The 11Σ+–13Π and the 11Σ+–23Π spin–orbit integrals are evaluated 87 and 58 cm−1 at their crossings (i.e. at 34.9 and 35.1 eV), which allow such conversions. KERs ≥ 6 eV are expected as those listed in Table 2. Also, such large KERs can be obtained from the electronic states of OCS2+ located above 35 eV as suggested previously.38
For the weaker fragmentation channels, the formation of the CS+ + O+ ion pair is due to C–O bond breaking upon doubly ionizing OCS as is already explained using the potentials of Brites et al.,40 whereas, the formation of the SO+ + C+ products by Wang and Vidal and later by Zhao et al.31 is not. These authors showed that the further evolution of the OCS2+ may lead to SO+ + C+ ion pair. The measured branching ratio was very small (0.0253%) in comparison to the main dissociative double ionization channel (CO+ + S+) (of 26.8%).31 Strong bending or a bond rearrangement is required and here the newly identified COS2+ isomer may be invoked for explanation. Fig. 6 shows that the COS2+ (X3Σ−, 11Δ, 11Σ+) ions may dissociate for energies >35.5–35.9 eV, where the SO+ + C+ ions carry KERs of ∼5.1–5.5 eV. Our values are in good agreement with the experimental findings by Zhao et al.31 meaning that the bending mode leads to isomerization into one of the three lowest states of COS2+ and not into one of its repulsive states.
5. Conclusions
We investigated the OCS2+ dissociation mechanism by threefold and fourfold electron–ion coincidence spectroscopy and by advanced ab initio methods. The present experimental findings together with evidence from other coincidence works and supported by the theoretical findings can be summarized as follows. In the IE range 30.0–ca. 32 eV OCS2+ is formed in the X3Σ− and a1Δ states and remains undissociated on a multi-microsecond time scale. In the IE range of ca. 31–33 eV some metastable OCS2+ is formed mainly in the b1Σ+ state and while some remains as undissociated OCS2+, a part dissociates to CO+ + S+ with an average KER of ca. 4 eV. In the IE range above 33 eV and particularly strongly in the range 34–36 eV OCS2+ is initially formed in states derived from the π13π23, configuration (particularly 11Σ−) which are not individually resolved. The nascent OCS2+ ions dissociate to CO+ + S+ with a characteristic KER of about 5.5 eV via the less stable isomer COS2+, obtained after intramolecular isomerization. The O+ + CS+ ion pair is formed at IEs above 37 eV where multiple states of the parent dication are populated by double photoionization. Moreover, we identified a mechanism for the very minor SO+ + C+ dissociation channel, which operates by the same bond rearrangement mechanism as the main dissociative double ionization channel, i.e. via the COS2+ isomer. In sum, our work indicates that the OCS2+ ions may either dissociate directly or may undergo bond rearrangement before dissociation.The present work gives evidence for the formation of the less stable COS2+ isomer, which has been invoked previously but is characterized here for the first time. This isomer plays a key role in the dynamics of OCS upon double ionization. Its formation from OCS2+ requires intramolecular isomerization i.e. bond arrangement. The efficiency of pathway (iii) and pathway (iv) (cf.Fig. 5) suggests that this bond arrangement is rapid (at least faster than the fragmentation).
In a wider perspective, our work demonstrates the importance of low stability isomers and their coupling to close-lying electronic excited states. New reaction routes are identified in this way and rate coefficients may therefore depend on skeletal molecular structures and the relevant PESs as well as on internal energy (temperature). Applications of this fundamental finding are expected in organic chemistry, astrochemistry, cold chemistry, atmospheric chemistry, electrochemistry, catalysis, and exobiology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out while M. H. was Waernska Guest Professor at the University of Gothenburg. We thank the Swedish Research Council (grant number 2018-03731) and the Knut and Alice Wallenberg Foundation (grant number 2017.0104), Sweden, for financial support. Some computations were carried out using the Swedish National Infrastructure for Computing (SNIC) at Chalmers Centre for Computational Science and Engineering (C3SE) partially funded by the Swedish Research Council through grant no. 2018-05973. M. J. thanks a fellowship from the Tunisian ministry for higher education and research. The authors extend their appreciation to the Researchers supporting project number (RSPD2023R808) King Saud University, Riyadh, Saudi Arabia. This work was supported by the “ASTRONOMIE-ASTROPHYSIQUE program (CSAA)” of CNRS/INSU co-funded by CEA and CNES.References
- L. S. Wang, J. E. Reutt, Y. T. Lee and D. A. Shirley, J. Electron Spectrosc. Relat. Phenom., 1988, 47, 167 CrossRef CAS.
- D. M. P. Holland and M. A. MacDonald, Chem. Phys., 1990, 144, 279 CrossRef CAS.
- S. Morse, M. Takahashi, J. H. D. Eland and L. Karlsson, Int. J. Mass Spectrom. Ion Processes, 1999, 184, 67 CrossRef CAS.
- S. Taylor and J. H. D. Eland, Chem. Phys., 2005, 315, 8 CrossRef CAS.
- D. M. P. Holland and M. A. MacDonald, Chem. Phys., 1990, 144, 279 CrossRef CAS.
- T. Masuoka and I. Koyano, J. Chem. Phys., 1998, 95, 909 CrossRef.
- T. Masuoka, J. Chem. Phys., 1998, 98, 6989 CrossRef.
- J. H. D. Eland, M. Hochlaf, P. Linusson, E. Andersson, L. Hedin and R. Feifel, J. Chem. Phys., 2010, 132, 014311 CrossRef CAS PubMed.
- T. Masuoka and H. Doi, Phys. Rev. A: At., Mol., Opt. Phys., 1993, 47, 278 CrossRef CAS PubMed.
- A. Ramadhan, B. Wales, I. Gauthier, R. Karimi, M. MacDonald, L. Zuin and J. Sanderson, J. Phys. B: At., Mol. Opt. Phys., 2016, 49, 215602 CrossRef.
- K. Saha, S. B. Banerjee and B. Bapat, Chem. Phys. Lett., 2014, 607, 85 CrossRef CAS.
- R. I. Hall, L. Avaldi, G. Dawber, A. G. McConkey, M. A. MacDonald and G. C. King, Chem. Phys., 1994, 187, 125 CrossRef CAS.
- T. Masuoka, I. Koyano and N. Saito, J. Chem. Phys., 1998, 97, 2392 CrossRef.
- W. A. Bryan, W. R. Newell, J. H. Sanderson and A. J. Langley, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 74, 053409 CrossRef.
- J. Laksman, D. Ćolin, M. Gisselbrecht and S. L. Sorensen, J. Chem. Phys., 2010, 133, 144314 CrossRef CAS PubMed.
- D. M. Curtis and J. H. D. Eland, Int. J. Mass Spectrom. Ion Process., 1985, 63, 241 CrossRef CAS.
- Z. Shen, E. Wang, M. Gong, X. Shan and X. Chen, J. Chem. Phys., 2016, 145, 234303 CrossRef PubMed.
- R. G. Cooks, D. T. Terwilliger and J. H. Beynon, J. Chem. Phys., 2003, 61, 1208 CrossRef.
- P. Wang and C. R. Vidal, J. Chem. Phys., 2003, 118, 5383 CrossRef CAS.
- H. Kumar, P. Bhatt, C. P. Safvan and J. Rajput, J. Chem. Phys., 2018, 148, 064302 CrossRef PubMed.
- B. Wales, T. Motojima, J. Matsumoto, Z. Long, W.-K. Liu, H. Shiromaru and J. Sanderson, J. Phys. B: At., Mol. Opt. Phys., 2012, 45, 045205 CrossRef.
- Y. Hikosaka, P. Lablanquie, E. Shigemasa, T. Aoto and K. Ito, J. Phys. B: At., Mol. Opt. Phys., 2008, 41, 025103 CrossRef.
- L. Asplund, P. Kelfye, H. Siegbahn, O. Goscinski, H. Fellner-Feldegg, E. Hamrin, B. Blomster and K. Siegbahn, Chem. Phys. Lett., 1976, 40, 353 CrossRef CAS.
- T. Kaneyasu, M. Ito, K. Soejima, Y. Hikosaka and E. Shigemasa, J. Phys. B: At., Mol. Opt. Phys., 2015, 48, 125101 CrossRef.
- U. Ankerhold, B. Esser and F. von Busch, Chem. Phys., 1997, 220, 393 CrossRef CAS.
- K. Yoshiki Franzén, P. Erman, P. A. Hatherly, A. Karawajczyk, E. Rachlew and M. Stankiewicz, Chem. Phys. Lett., 1998, 285, 71 CrossRef.
- P. Bolognesi, P. O’Keeffe and L. Avaldi, J. Phys. Chem. A, 2009, 113, 15136 CrossRef CAS PubMed.
- Y. Hikosaka and E. Shigemasa, Int. J. Mass Spectrom., 2019, 439, 13 CrossRef CAS.
- J. Reid, Int. J. Mass Spectrom. Ion Processes, 1991, 110, 195 CrossRef.
- W. J. Griffiths and F. M. Harris, Int. J. Mass Spectrom. Ion Processes, 1989, 87, 349 CrossRef CAS.
- S. Zhao, B. Jochim, P. Feizollah, J. Rajput, F. Ziaee, K. P. Raju, B. Kaderiya, K. Borne, Y. Malakar, B. Berry, J. Harrington, D. Rolles, A. Rudenko, K. D. Carnes, E. Wells, I. Ben-Itzhak and T. Severt, Phys. Rev. Accel. Beams, 2019, 99, 053412 CAS.
- J. Rajput, et al. , Phys. Rev. Lett., 2018, 120, 103001 CrossRef CAS PubMed.
- B. Wales, et al. , J. Electron Spectrosc. Relat. Phenom., 2014, 195, 332 CrossRef CAS.
- C. Wu, et al. , Phys. Rev. Lett., 2013, 110, 103601 CrossRef PubMed.
- W. A. Bryan, W. R. Newell, J. H. Sanderson and A. J. Langley, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 74, 053409 CrossRef.
- J. H. Sanderson, T. R. J. Goodworth, A. El-Zein, W. A. Bryan, A. J. Langley and P. F. Taday, Phys. Rev. A: At., Mol., Opt. Phys., 2002, 65, 043403 CrossRef.
- S. L. Sorensen, M. Gisselbrecht, J. Laksman, E. P. Månsson, D. Céolin, A. Sankari and F. Afaneh, J. Phys.: Conf. Ser., 2014, 488, 012006 CrossRef CAS.
- T. Endo, K. M. Ziems, M. Richter, F. G. Fröbel, A. Hishikawa, S. Gräfe, F. Légaré and H. Ibrahim, Front. Chem., 2022, 10, 859750 CrossRef CAS PubMed.
- P. Ma, C. Wang, S. Luo, X. Li, W. Hu, J. Yu, X. Yu, X. Tian, Z. Qu and D. Ding, Phys. Rev. Accel. Beams, 2019, 99, 023423 CAS.
- V. Brites, J. H. D. Eland and M. Hochlaf, Chem. Phys., 2008, 346, 23 CrossRef CAS.
- V. Sekushin, R. Püttner, R. F. Fink, M. Martins, Y. H. Jiang, H. Aksela, S. Aksela and G. Kaindl, J. Chem. Phys., 2012, 137, 044310 CrossRef CAS PubMed.
- M. L. Langford, F. M. Harris, C. J. Reid, J. A. Ballantine and D. E. Parry, Chem. Phys., 1991, 149, 445 CrossRef CAS.
- D. Minelli, F. Tarantelli, A. Sgamellotti and L. S. Cederbaum, J. Chem. Phys., 1998, 107, 6070 CrossRef.
- P. Millié, I. Nenner, P. Archirel, P. Lablanquie, P. Foumier and J. H. D. Eland, J. Chem. Phys., 1998, 84, 1259 CrossRef.
- Y. J. Zhao, X. bin Shan, L. S. Sheng, Z. Y. Wang, J. Zhang and C. R. Yu, Chin. Phys. B, 2011, 20, 043201 CrossRef.
- S. Hsieh and J. H. D. Eland, J. Phys. B: At., Mol. Opt. Phys., 1997, 30, 415 CrossRef.
- G. Öhrwall, M. M. Sant’Anna, W. C. Stolte, I. Dominguez-Lopez, L. T. N. Dang, A. S. Schlachter and D. W. Lindle, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 4543 CrossRef.
- J. H. D. Eland, S. Zagorodskikh, R. J. Squibb, M. Mucke, S. L. Sorensen and R. Feifel, J. Chem. Phys., 2014, 140, 184305 CrossRef CAS PubMed.
- Y. Muramatsu, et al. , Phys. Rev. Lett., 2002, 88, 133002 CrossRef CAS PubMed.
- M. Wallner, M. Jarraya, E. Olsson, V. Ideböhn, R. J. Squibb, S. Ben Yaghlane, G. Nyman, J. H. D. Eland, R. Feifel and M. Hochlaf, Sci. Adv., 2022, 8, eabq5411 CrossRef CAS PubMed.
- J. H. D. Eland and R. Feifel, Chem. Phys., 2006, 327, 85 CrossRef CAS.
- H. Werner, P. Knowles, et al., MOLPRO version 2015 is a package of ab initio computations, cf., http://www.molpro.net Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1998, 96, 6796 CrossRef.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1998, 98, 1358 CrossRef.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244 CrossRef CAS.
- S. Ben Yaghlane, N.-E. Jaidane, C. E. Cotton, J. S. Francisco, M. Mogren Al Mogren, R. Linguerri and M. Hochlaf, J. Chem. Phys., 2014, 140, 244309 CrossRef CAS PubMed.
- S. Ben Yaghlane, C. E. Cotton, J. S. Francisco, R. Linguerri and M. Hochlaf, J. Chem. Phys., 2013, 139, 174313 CrossRef PubMed.
- T. Trabelsi, M. Mogren Al-Mogren, M. Hochlaf and J. S. Francisco, J. Chem. Phys., 2018, 149, 064304 CrossRef PubMed.
- C. Hampel, K. A. Peterson and H. J. Werner, Chem. Phys. Lett., 1992, 190, 1 CrossRef CAS.
- M. J. O. Deegan and P. J. Knowles, Chem. Phys. Lett., 1994, 227, 321 CrossRef CAS.
- P. J. Knowles, C. Hampel and H. J. Werner, J. Chem. Phys., 1998, 99, 5219 CrossRef.
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1985, 115, 259 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1998, 82, 5053 CrossRef.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1998, 89, 5803 CrossRef.
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1988, 145, 514 CrossRef CAS.
- K. R. Shamasundar, G. Knizia and H. J. Werner, J. Chem. Phys., 2011, 135, 054101 CrossRef CAS PubMed.
- S. Taylor and J. H. D. Eland, Chem. Phys., 2005, 315, 8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01688a |
| This journal is © the Owner Societies 2023 |