
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstitution (CH3, Cl, or Br) effects of the imidazolate linker on benzene adsorption kinetics for the zeolitic imidazolate framework (ZIF)-8†
Ryohei
Yagi
and
Takahiro
Ueda
 *
*
Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan. E-mail: ueda@chem.sci.osaka-u.ac.jp
First published on 12th July 2023
Abstract
Herein, the time dependence of benzene adsorption uptake was examined for ZIF-8, Cl-ZIF-8, and Br-ZIF-8 and analysed using an intra-crystalline (Fick's) diffusion model, yielding the diffusion coefficient and saturated adsorption amount of benzene. The saturated adsorption amount of benzene decreased in the order of ZIF-8, Cl-ZIF-8, and Br-ZIF-8. Notably, ZIF-8, with an intermediate pore volume among the three specimens, accommodated the greatest number of molecules (5.5 molecules per micropore). The activation energy, Ea, and the pre-exponential factor, D0, for benzene diffusion increased in the order of ZIF-8, Cl-ZIF-8, and Br-ZIF-8. These findings suggest that the 2-methylimidazolate moiety forms an effective attraction interaction with benzene molecules. The D0 values also yielded the activation entropy, ΔS‡, in the transition state when a benzene molecule passed through a six-membered ring aperture. The ΔS‡ values at 303 K were negative, and their absolute values increased in the order of Br-ZIF-8, Cl-ZIF-8, and ZIF-8. Considering the degree of freedom of translation and rotation of the benzene molecule and the vibration and disorder of the linker, we found that the differences in ΔS‡ were caused by the dynamic local structure of the six-membered ring aperture among the ZIF-8 analogues. Furthermore, infrared spectroscopy revealed a low-wavenumber shift of the C–H stretching band in both the imidazolate moiety and adsorbed benzene molecules. A solid-state 13C-nuclear magnetic resonance spectrum presented a downfield shift of 13C resonance peaks in the imidazolate moiety, suggesting that CH/π interactions reasonably explain the intermolecular interaction between the imidazolate moiety (including the methyl group) and π-electrons of benzene.
Introduction
Metal–organic frameworks, including imidazole derivatives as organic ligands, are referred to as zeolitic imidazolate frameworks (ZIFs). These ZIFs have zeolite-like topologies (such as SOD, A-type zeolite (LTA), and RHO zeolitic structures), in which the Zn(II) or Co(II) ions are tetrahedrally coordinated by imidazole derivatives via nitrogen atoms.1,2 ZIFs are attracting attention as zeolite alternatives owing to their versatile skeletal topology, hydrophobic pores, skeletal flexibility, and chemical and thermal stability.3,4 Among them, ZIF-8, which is composed of Zn2+ ions and 2-methylimidazolate anions, is represented by the formula [Zn(C4H5N2)2]n and has the same sodalite structure as LTA. The crystal has a cubic lattice belonging to the I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group, with a lattice constant of a = 1.701 nm (298 K), Z = 12 and a unit lattice volume of 4921 nm.3,5 The crystal also contains micropores with a diameter of 1.16 nm, which are three-dimensionally connected by eight six-membered ring apertures with a diameter of 0.34 nm. These micropores can accommodate various molecules and are expected to be applied in various fields, such as gas storage and separation, removal of harmful substances, catalysis and drug delivery systems.6–10
3m space group, with a lattice constant of a = 1.701 nm (298 K), Z = 12 and a unit lattice volume of 4921 nm.3,5 The crystal also contains micropores with a diameter of 1.16 nm, which are three-dimensionally connected by eight six-membered ring apertures with a diameter of 0.34 nm. These micropores can accommodate various molecules and are expected to be applied in various fields, such as gas storage and separation, removal of harmful substances, catalysis and drug delivery systems.6–10
ZIF-8 is thermally stable up to 550 °C. By contrast, it is chemically stable in NaOH aqueous solutions under alkaline conditions; neutral electrolyte solutions such as NaCl and LiCl; and various solvents such as H2O, MeOH, and benzene. However, acidic solutions decompose and dissolve ZIF-8. For mechanical stability, non-uniform pressures such as during ball milling induce amorphisation.11 In addition, negative thermal expansion is observed in ZIF-8.12,13 All of these changes are characterised by the flexibility of the framework caused by the Zn–Im–Zn bond character. This feature also significantly affects the gas adsorption behaviour of ZIF-8. The adsorption amount increases rapidly at a certain pressure for the adsorption of N2 and Ar.14,15 The change in orientation of the 2-methylimidazolate linker expands the six-membered ring apertures (swing effect), resulting in pore diameter expansion from 0.34 nm to 0.40 nm.5,15,16 This phenomenon, called ‘gate adsorption’, is an adsorption mechanism caused by the flexibility of the framework in ZIF-8.17–19
Meanwhile, diffusion coefficients have also been reported for some molecules larger than the expanded six-membered ring aperture.20 Furthermore, from the viewpoint of the activation energy of the diffusion of gas molecules, the molecular sieving effect on a typical LTA induces a rapid increase in activation energy as the kinetic diameter of the gas molecule approaches the aperture size. By contrast, the activation energy in ZIF-8 increases linearly in proportion to the kinetic diameter.21 This fact suggests that ZIF-8 regulates the degree of expansion of the six-membered ring aperture by recognising the size of a gas molecule, which is a unique characteristic of adsorption in ZIF-8 that is not observed in activated carbon and typical zeolites. Such characteristic and unique molecular diffusion behaviour also originates from the flexibility of the framework based on the twisting motion of the 2-methylimidazolate linkers around the Zn–Im–Zn axis, which is referred to as the swing effect. Recently, far-infrared spectroscopy and neutron inelastic scattering experiments have suggested that a type of phonon mode (cooperative large-amplitude vibration of an organic linker) excited by the six-membered ring apertures causes the transient expansion of the aperture.22 In addition, a low-frequency oscillation mode accompanying rotational motion of the linker has been observed by terahertz spectroscopy,23 suggesting that the swing effect may trigger gate adsorption. Solid-state deuterium nuclear magnetic resonance (NMR) spectrum24 and the second moment analysis of the wide-line 1H-NMR spectrum25 also reveal large-amplitude flipping motion of the 2-methylimidazolate linker with a flipping angle of 55°; that is, the swing effect of the 2-methylimidazolate linkers causes continuous and transient fluctuations in the six-membered ring apertures through the thermal motion of the linkers. This effect plays an important role in molecular diffusion,26 guest dynamics,27 and elastic properties28 of the ZIF-8 crystal lattice; however, the swing effect has not been clearly observed as a structural change in the crystal structure.
In our most recent studies, through temperature-dependent 1H-NMR spin-lattice relaxation time (1H T1) measurements, we found that the adsorption of bulky molecules resulted in a slowdown in both the rotation of the methyl group and the large-amplitude flipping of the linkers.25 For example, in the absence of adsorbed bulky molecules, 1H T1 could not distinguish the rotation of the methyl group and flipping of the 2-methylimidazolate moiety from each other and yielded a frequency of 0.58 THz as each motional mode at room temperature. When a saturated amount of CCl4 adsorbed onto ZIF-8, 1H T1 distinguished the rotation of the methyl group and flipping of the linker, and the frequency of each motional mode decreased to 0.24 and 0.06 THz, respectively, at room temperature. Based on our results, we proposed the transient gate mechanism of ZIF-8 for the adsorption of bulky molecules. In this mechanism, the adsorption of bulky molecules triggers the reduction in the frequency of the thermal motion of the linkers, resulting in prolongation of the transient expansion time of the apertures and promotion of molecular adsorption.25 Thus, by considering the swing effect from the viewpoint of the molecular motion of the linkers, the adsorption behaviour and diffusion mechanism at the molecular level can be elucidated and understood.
However, to control the adsorption process and diffusion behaviour of ZIF-8 based on the swing effect, a systematic and quantitative discussion regarding (1) the local structure of the six-membered ring aperture and (2) the intermolecular interaction between the organic linkers and the adsorbate molecule is essential. In our previous study, the diffusion coefficients and activation energies of molecular diffusion were evaluated for the adsorption process of various bulky molecules onto ZIF-8.29 The intermolecular interactions between the 2-methylimidazolate linkers and adsorbates were quantitatively examined, revealing that the large-amplitude flip motion of the linkers was necessary to explain those values. Our aim now is to quantitatively evaluate the intermolecular interaction between the adsorbate molecule and organic linker and to elucidate the local structure of the six-membered ring apertures when a bulky molecule is adsorbed in the micropores of ZIF-8 and its analogues. For this purpose, we examined the substituent effect of the functional group on the imidazolate moiety on the adsorption behaviour, as well as molecular diffusion. Thus, we focused on two analogues of ZIF-8, [Zn(C3H2N2Cl)2]n (Cl-ZIF-8) and [Zn(C3H2N2Br)2]n (Br-ZIF-8), which are composed of a 2-halogeno-substituted imidazolate ligand (C3H2N2X; X = Cl, Br). These crystals exhibit isomorphism with ZIF-8. Both Cl-ZIF-8 and Br-ZIF-8 were reported first by Li et al., who investigated the properties of kinetic separation between propane and propene.30 Thereafter, the behaviour of CO2 capture/separation has been studied by density functional theory calculations and grand canonical Monte Carlo simulations.31,32 In addition, molecular sieving properties based on the hydrophobic/hydrophobic nature of the materials have been evaluated by combining molecular simulations and a quantitative structure–property correlation approach.33 Furthermore, Chaplais et al. recently reported the detailed crystal structures of these materials,34 both of which have an isomorphic crystal structure with ZIF-8 and two types of disorders in the orientation of the 2-halogeno-substituted imidazolate linker. The substituent effects on the lattice parameters and porosity have been observed as follows: the lattice cell volume increases in the order of functional groups CH3 < Cl < Br. The space in the unit cell accessible by N2 increases in the order Cl < CH3 < Br, whereas the pore volume (cm3 g−1) decreases in the order Br < Cl < CH3.
In this study, the adsorption kinetics of Cl-ZIF-8 and Br-ZIF-8 were analysed using benzene as the bulk adsorbate. Through the time dependence of benzene uptake, we examined the effect of the 2-halogeno-substitutent on the adsorption behaviour. Based on the saturated amount of benzene, the intermolecular structure of benzene in the micropores was discussed to provide insights into the intermolecular interactions between the adsorbate molecules and organic linkers. Next, we examined the local structure of the six-membered ring apertures when a benzene molecule passed through the aperture via the activation parameters (activation energies and exponential pre-factors) obtained from the temperature dependence of benzene diffusivity. Furthermore, we obtained information on the intermolecular interaction between organic linkers and benzene by infrared absorption spectrum and solid-state 13C-NMR spectrum measurements. To the best of our knowledge, this study is the first to investigate the role of a substituent group in the adsorption kinetics for ZIF-8 analogues. We believe that this study will provide new and additional insights for understanding the adsorption mechanism of bulky molecules into ZIF-8 analogues.
Experimental
Chemicals
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99.0%), 2-methylimidazole (C4H6N2, 98%), 2-chloroimidazole (C3H2N2Cl, 96%), ammonium hydroxide, NH4OH (28 wt%), and ethanol (C2H5OH, 99.5%) were purchased from FUJIFILM Wako Pure Chemical Co. 2-Bromoimidazole (C3H2N2Br, 97%) was purchased from Fluoro Chem Ltd. All chemicals were used without further purification.Synthesis
Characterisations
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. The SEM images and size distribution of the crystallites are shown in Fig. S4 and S5 (see ESI†).
000×. The SEM images and size distribution of the crystallites are shown in Fig. S4 and S5 (see ESI†).
Adsorption rate measurements
The samples (ZIF-8, Cl-ZIF-8, and Br-ZIF-8) were packed into an aluminium pan for TG-DTA measurement after pretreatment (evacuation at 200 °C and 2.7 Pa for 12 h). The specimens used were ca.6 mg ZIF-8, ca.8 mg Cl-ZIF-8, and ca.10 mg Br-ZIF-8. The Al pan was placed in a closed container together with 3 mL of benzene (99.5%). Air coexisting in the container affects the adsorption of benzene vapour less because N2 and O2 at ambient temperature are supercritical gases. This container was fixed to a pedestal constructed from heat-insulating Styrofoam and then stored in a thermostatic chamber at 30 °C, 40 °C, and 50 °C, controlled within ±0.5 °C. The sample weight was measured at appropriate time intervals and recorded until a constant value was obtained. This procedure was repeated thrice, and the results were averaged to evaluate the experimental error.Results and discussion
Time dependence of benzene uptake
Fig. 1(a), (b), and (c) show the time dependence of the adsorption uptake of benzene measured at 303, 313, and 323 K, respectively. The sample weight of each specimen increased rapidly at the initial stage of adsorption and asymptotically approached the saturated value. In the order of ZIF-8, Cl-ZIF-8, and Br-ZIF-8, the saturated adsorption amount of benzene decreased, whereas the increasing rate of benzene uptake to the saturated adsorption amount increased. This trend is consistent with the order of Winstein–Holness A-value37 (AMe = 1.7, ACl = 0.43, ABr = 0.38), which is a useful indicator of the substituent effect on reaction selectivity. Therefore, the difference between the adsorption rate and the saturated adsorption amount of benzene among ZIF-8, Cl-ZIF-8, and Br-ZIF-8 is considered to result from the effect of the substituent group on the linker. For a more quantitative discussion on the substituent effect, the diffusion coefficient and saturated adsorption amount were obtained using the appropriate diffusion model for the adsorption process.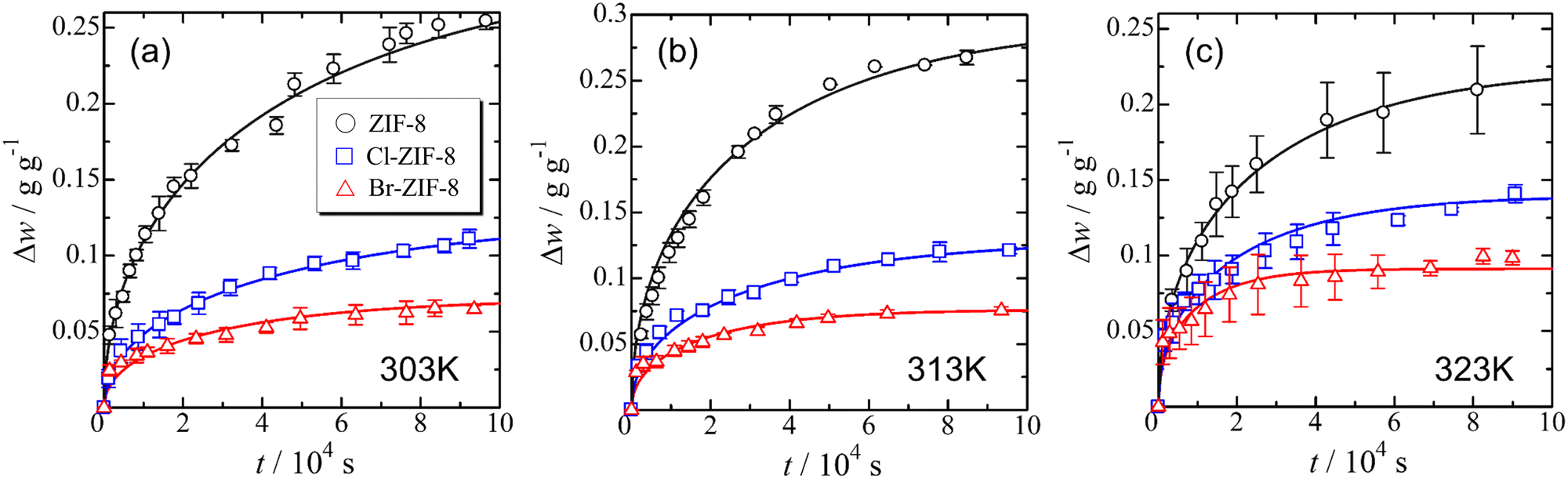 | ||
| Fig. 1 Time dependence of the adsorption uptake of benzene at 303 (a), 313 (b) and 323 K (c). The solid lines represent the result of the least-square fitting of eqn (2) based on the intra-crystalline (Fick) diffusion model. | ||
The time dependence of the adsorption uptake is generally denoted by a macroscopic diffusion phenomenon of benzene molecules from the vapour phase to crystallites. Two typical diffusion models exist, namely the surface barrier and intra-crystalline (Fick's) diffusion model.29,38–41 In the surface barrier model, the diffusion rate is controlled by the penetration resistance on the surface of the crystallites. The time dependence of the uptake, Δw(t), obeys the first-order reaction law and is given by the following equation:
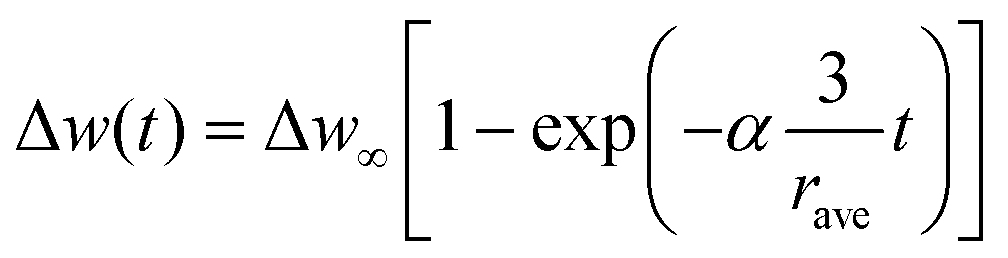 | (1) |
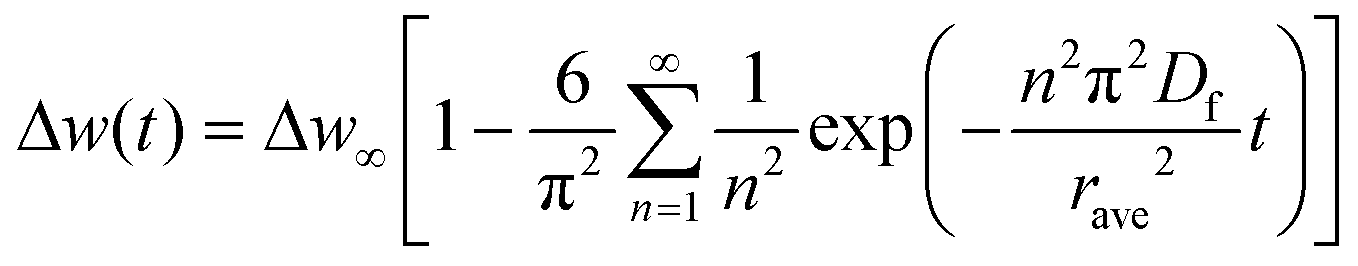 | (2) |
| T/K | k ads./10−5 s−1 | r ave/nm | D f/10−19 m2 s−1 | Δw∞/g g−1 | N ads/cage | |
|---|---|---|---|---|---|---|
| The errors estimated in kads, rave, Df, and Δw∞ are presented in parentheses. The errors in Df are mainly caused by the errors in the average particle sizes. | ||||||
| ZIF-8 | 303 | 1.5 (±0.1) | 235 (±50) | 0.84 (±0.4) | 0.29 (±0.01) | 5.5 |
| 313 | 2.4 (±0.1) | 1.3 (±0.5) | 0.29 (±0.01) | 5.5 | ||
| 323 | 2.9 (±0.3) | 1.6 (±0.7) | 0.22 (±0.02) | 4.3 | ||
| Cl-ZIF-8 | 303 | 1.6 (±0.1) | 585 (±100) | 5.6 (±2) | 0.13 (±0.01) | 2.8 |
| 313 | 2.5 (±0.1) | 8.7 (±3) | 0.13 (±0.01) | 2.8 | ||
| 323 | 3.8 (±0.1) | 13.2 (±5) | 0.14 (±0.01) | 3.1 | ||
| Br-ZIF-8 | 303 | 2.6 (±0.1) | 320 (±50) | 2.7 (±0.9) | 0.07 (±0.01) | 2.1 |
| 313 | 4.2 (±0.1) | 4.4 (±1.5) | 0.08 (±0.01) | 2.3 | ||
| 323 | 8.1 (±0.2) | 8.4 (±2.5) | 0.09 (±0.02) | 2.7 | ||
As shown in Table 1, the diffusion coefficient of Cl-ZIF-8 is larger than that of Br-ZIF-8, although the adsorption rate exhibits the opposite trend. This finding possibly reflects the difference in particle size between the specimens (rave = 585 nm for Cl-ZIF-8 and rave = 320 nm for Br-ZIF-8) because the average particle size is critically affected by the evaluation of the diffusion coefficient from the adsorption rate. In addition, the diffusion coefficient in the appropriate temperature region is given by the balance between the exponential pre-factor D0 and the activation energy Ea. Depending on both D0 and Ea values as described below, Cl-ZIF-8 may also have a larger diffusion coefficient than Br-ZIF-8 in the observed temperature range (303 K ≤ T ≤ 323 K). In the following sections, based on the saturated adsorption amount of benzene and the diffusion coefficients, we discuss the intermolecular structure of benzene, adsorbent–adsorbate interactions, and the diffusion behaviour of benzene in each specimen.
Saturated adsorption amount of benzene
Fig. 2 shows a plot of the average number (Nads) of benzene and nitrogen molecules occupying a micropore versus micropore volume. The Nads values of benzene used the saturated adsorption amounts at 303 K. The Nads value of the N2 molecule increases linearly against the pore volume, whereas for the benzene molecule, ZIF-8 with an intermediate pore volume among the three specimens can accommodate the greatest number of molecules in the micropore (5.5 molecules per micropore). The slope of this plot yields the effective density of the adsorbate in the micropores. For nitrogen, the slope is 17.4 molecules nm−3, which agrees well with the bulk density at the triple-point of N2. This result is as expected because the pore volume is determined from the N2 adsorption isotherms at 77 K. By contrast, the slope of the line passing through the data point between Br-ZIF-8 and Cl-ZIF-8 is 2.3 molecules nm−3, which corresponds to approximately 36% of the bulk density for liquid benzene. This indicates that the effective volume accessible to benzene molecules is approximately 36% of the pore volume determined from the N2 adsorption isotherms. Approximating that the micropore shape is a sphere, the reduction in pore volume corresponds to shortening the radius of the sphere by 70.8%. This apparent shrinkage of the micropores is caused by the difference in effective size between the nitrogen and benzene molecules. One of the Lenard–Jones parameters, σ, that is, the distance where the potential energy becomes zero, is 3.70 Å for N2 and 5.26 Å for liquid benzene.42 The ratio of these values is 0.703, which is consistent with the reduction in the radius of the empty sphere; that is, the slope of the line passing through the data point between Br-ZIF-8 and Cl-ZIF-8 is valid as the effective density of the adsorbed benzene. Thus, the large uptake of benzene by ZIF-8 is an anomaly in comparison with those of Cl-ZIF-8 and Br-ZIF-8, implying a specific attractive interaction between the methyl group and benzene.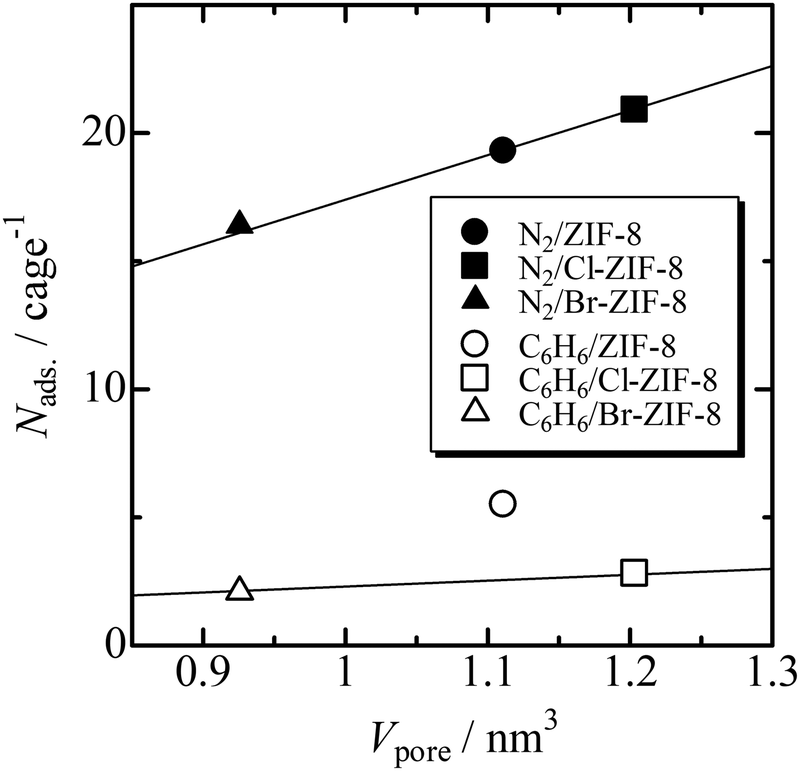 | ||
| Fig. 2 Average number (Nads) of benzene and nitrogen molecules occupying a micropore plotted against the volume of a micropore (Vpore) determined from the N2 adsorption isotherm. The Nads values of benzene are determined from the saturated adsorption amounts at 303 K. The effective density of adsorbate molecules in the micropore is derived from the slope of each dataset. | ||
In our previous study, we found that for bulky molecules, the stable adsorption sites were located beside the six-membered ring aperture.25,29 Therefore, we estimated the orientation of a benzene molecule adsorbed on the stable site, as well as the distance between the stable site and the surface of the six-membered ring aperture, using the MM method. MM calculations were performed using the Avogadro software,36 and the universal force field was used as the molecular force field.43 Some orientations and distances with similar stable energies within several kilojoules per mole were obtained depending on the initial orientation of benzene.
In addition, Cl-ZIF-8 and Br-ZIF-8 have disordered structures in the linker orientations.34 In the crystal, each specimen has two different linker orientations with approximately equal populations. Some typical examples of the stable configuration are shown in Fig. S8 in the ESI.† The angle between the aperture surface and the molecular plane of benzene, as well as the distance between the aperture surface and the centre of gravity of the benzene molecule, was evaluated by averaging several orientations and distances for all possible structures of the six-membered ring apertures, as listed in Table 2.
| Orientation of linker | R Benzene-ring /nm | θ Benzene-ring/° | |
|---|---|---|---|
| a R Benzene-ring is the distance measured between the centre of molecular plane and the aperture plane. | |||
| ZIF-8 | 0.2893 | 40.66 | |
| Cl-ZIF-8 | Orientation 1 | 0.3133 | 7.546 |
| Orientation 2 | 0.2705 | 5.581 | |
| Average | 0.2919 | 6.563 | |
| Br-ZIF-8 | Orientation 1 | 0.3111 | 12.57 |
| Orientation 2 | 0.2867 | 4.512 | |
| Average | 0.2989 | 8.512 | |
The stable sites occupied by benzene molecules become distant from the six-membered ring aperture surface in the following order: ZIF-8, Cl-ZIF-8, and Br-ZIF-8. In addition, the angle formed by the molecular plane of benzene with respect to the aperture surface of the six-membered ring was less than 10° for Cl-ZIF-8 and Br-ZIF-8, suggesting that the benzene molecule is adsorbed at an orientation in which the molecular plane lies along the aperture surface. By contrast, in ZIF-8, the angle between the molecular plane of benzene and the aperture surface is approximately 40°, indicating that the benzene molecule is adsorbed at an orientation slightly erect with respect to the six-membered ring aperture; that is, the pore walls of ZIF-8 can interact with more benzene molecules than Cl-ZIF-8 and Br-ZIF-8. Furthermore, as denoted later, the CH/π interaction between the methyl protons and π electrons of benzene is considered to contribute to the attractive interaction between the benzene molecule and the ZIF-8 pore wall. These interactions should effectively increase the saturated adsorption amount of benzene in ZIF-8. The interaction between the imidazolate linker and the benzene molecule was also investigated by infrared spectroscopy and solid-state 13C-NMR spectroscopy, as described below.
Temperature dependence of the diffusion coefficient
The diffusion coefficient depends on the temperature in all samples. Therefore, the Arrhenius plot of the diffusion coefficient can be used to determine the activation parameters. The Arrhenius equation represents the diffusion coefficient as follows: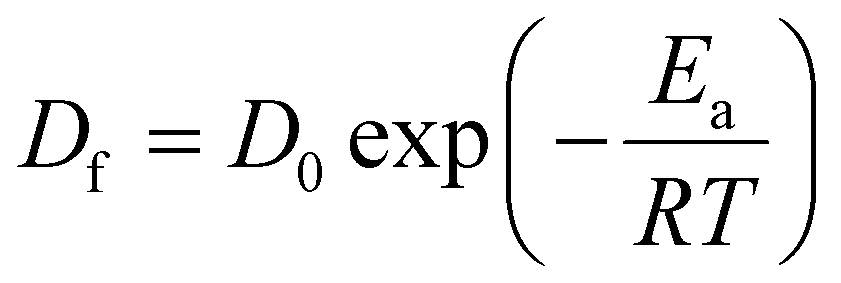 | (3) |
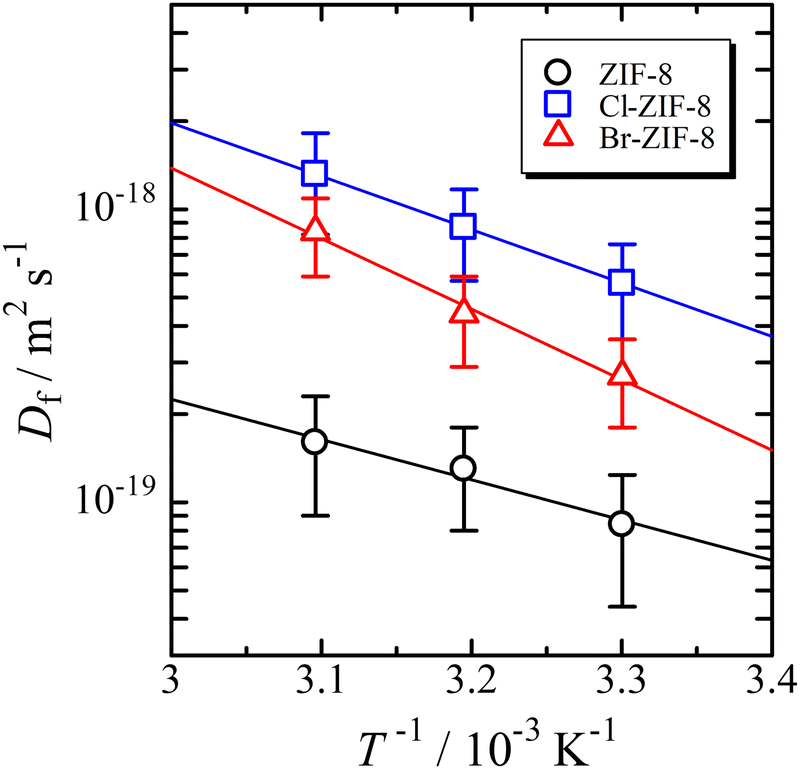 | ||
| Fig. 3 Arrhenius plot of the diffusion coefficient. The solid lines are the results of the optimisation of eqn (3) to the data using the least-squares method. | ||
| E a/kJ mol−1 | D 0/m2 s−1 | ΔS‡/J K−1 mol−1 | |
|---|---|---|---|
| The errors estimated in Ea, D0, and ΔS‡ are presented in parentheses. The Ea and D0 values are evaluated from the Arrhenius plot of kads. The errors in Df are mainly caused by the errors in the average particle sizes. | |||
| ZIF-8 | 27 (± 2) | (4 ± 2) × 10−15 | −190 (± 7) |
| Cl-ZIF-8 | 35 (± 1) | (6.5 ± 2.4) × 10−13 | −149 (± 3) |
| Br-ZIF-8 | 46 (± 2) | (2.4 ± 0.8) × 10−11 | −138 (± 3) |
Both the activation energy and pre-exponential factor increase in the following order: ZIF-8, Cl-ZIF-8, and Br-ZIF-8. The change in activation energy reflects the interaction between benzene and the six-membered ring aperture when a benzene molecule passes through the aperture. The activation energy between Cl-ZIF-8 and Br-ZIF-8 reflects the difference in the van der Waals radius of halogen (rCl = 175 pm and rBr = 183 pm);44 that is, the bromine atom induces a larger repulsive interaction with benzene than the chlorine atom, leading to a larger Ea value for Br-ZIF-8. Meanwhile, the methyl group has a van der Waals radius rCH3= 207 pm, which is estimated using a CH bond distance of 97 pm. This radius is larger than those of chlorine and bromine atoms. Nevertheless, the activation energy of ZIF-8 is smaller than that of Cl-ZIF-8. This finding suggests the occurrence of an attractive interaction that stabilises the transition state of benzene passing through the six-membered ring aperture, which exerts a more effective influence than the repulsive interaction with the methyl group. One of the possibilities for such interactions is the CH/π interaction. This consideration is consistent with the discussion of the saturated adsorption amount mentioned above.
The pre-exponential factor can be discussed based on the transition state theory introduced by Eyring.45 According to transition state theory, the diffusion coefficient is represented as follows:
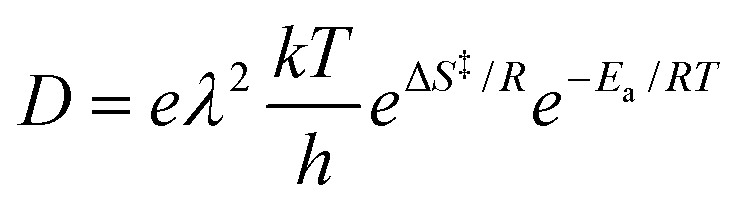 | (4) |
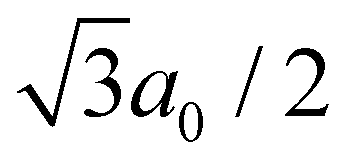 as a lattice constant of a0. By comparing eqn (4) to the Arrhenius equation, the following formula is obtained as a pre-exponential factor, D0:
as a lattice constant of a0. By comparing eqn (4) to the Arrhenius equation, the following formula is obtained as a pre-exponential factor, D0: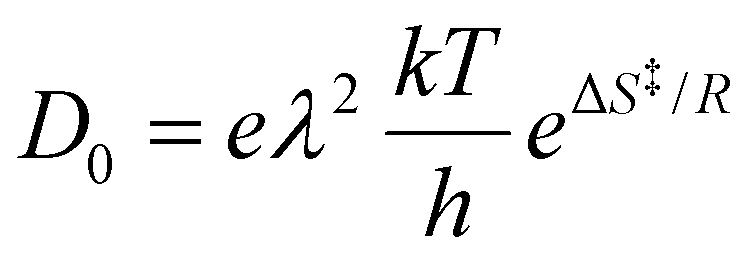 | (5) |
During adsorption, benzene molecules lose the degree of freedom of translation, which is accompanied by a significant reduction in entropy. This entropy change is similar to that of condensation from the vapour to liquid phase. The enthalpy of vapourisation at the standard boiling point of benzene (Tb = 353.3 K) is 33.9 kJ mol−1,46 given that the entropy of evaporation of benzene is 96 J K−1 mol−1. Thus, the entropy changes of approximately −100 J K−1 mol−1 in ΔS‡ may be associated with the translation of benzene. Considering the partition function of translation and rotation, we can estimate the entropy change of benzene between the gas phase and transition state at which a benzene molecule is trapped on the six-membered ring aperture. Logically, a benzene molecule loses three degrees of freedom in translation (−160 J K−1 mol−1) and in rotation modes (−82.7 J K−1 mol−1) by trapping at an aperture. These contributions are expected to result in an entropy reduction of −242.7 J K−1 mol−1 at 303 K (details of the evaluation are given in ESI†). By contrast, the frequency of the torsional vibration of the linker (flipping motion) is reduced to 1/10 by the interaction of bulky molecules, such as CCl4 and benzene, with the six-membered ring aperture.24,25 This feature increases the entropy on the six-membered ring aperture by 20 J K−1 mol−1 for a linker because the lower energy induces an increase in the number of vibrational states occupying the same temperature. Therefore, this contribution increases the entropy to 120 J K−1 mol−1 in the transition state. These three contributions yield a ΔS‡ of −122.7 J K−1 mol−1, corresponding to the experimental value of −138 J K−1 mol−1 for Br-ZIF-8.
Chaplais et al. reported that the imidazolate linkers in ZIF-8 and Cl-ZIF-8 have two orientations corresponding to gate adsorption when N2 adsorbs into the micropores, whereas the orientation of the linkers in Br-ZIF-8 does not change.34 Therefore, in both ZIF-8 and Cl-ZIF-8, a linker that is dynamically disordered between two orientations fixes its orientation, thereby reducing entropy. Assuming that two orientations for a linker fix to one orientation, this contribution is expected to result in an entropy reduction of −34.6 J K−1 mol−1 for a six-membered ring aperture directly interacting with a benzene molecule. For ZIF-8 and Cl-ZIF-8, this contribution is added to the above three contributions, resulting in a ΔS‡ of −157.3 J K−1 mol−1. This calculation can explain the difference in the ΔS‡ value between Br-ZIF-8 and Cl-ZIF-8.
Furthermore, in ZIF-8, the rotation of the methyl group also contributes to entropy change. The adsorption of benzene increases the activation energy of methyl rotation (or reorientation) from 1.3 kJ mol−1 to 5.1 kJ mol−1,24 implying a pseudo-low-temperature effect; that is, the interaction between the methyl groups and benzene molecules cools the methyl group from the free rotor to the restricted reorientating rotor. Consequently, the entropy of a methyl group for rotation is reduced from 27.2 J K−1 mol−1 to 9.13 J K−1 mol−1 (= R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln3). Therefore, this effect results in an entropy reduction of −108.6 J K−1 mol−1 for 6Me groups per aperture. This contribution can explain the difference in ΔS‡ between ZIF-8 and Cl-ZIF-8.
ln3). Therefore, this effect results in an entropy reduction of −108.6 J K−1 mol−1 for 6Me groups per aperture. This contribution can explain the difference in ΔS‡ between ZIF-8 and Cl-ZIF-8.
Thus, the differences in ΔS‡ among ZIF-8, Br-ZIF-8, and Cl-ZIF-8 are caused by the dynamic local structure of the six-membered ring aperture. The magnitude of ΔS‡ significantly affects the diffusion coefficients in the temperature range from 303 K to 323 K, suggesting a significant interaction between benzene and the six-membered ring aperture.
Insights from spectroscopy
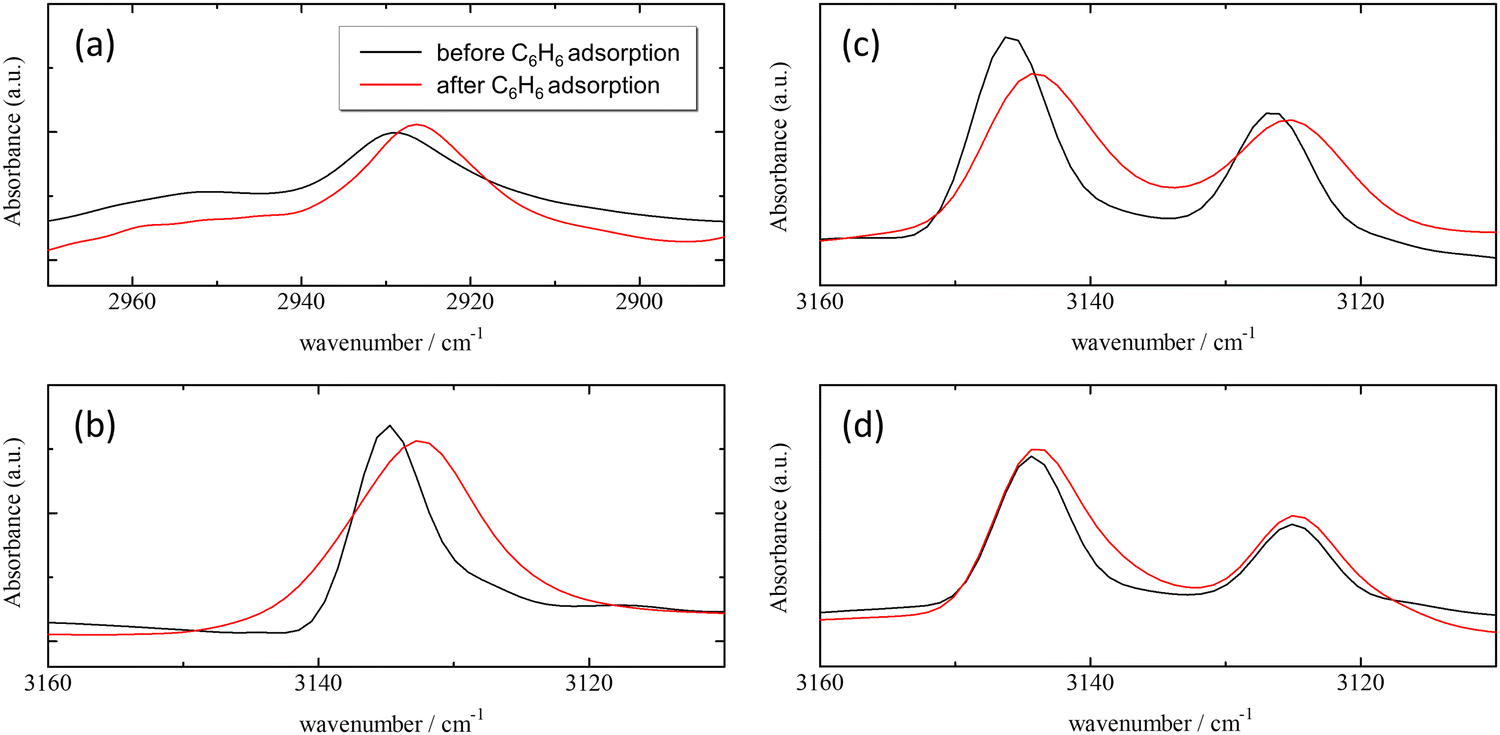 | ||
| Fig. 4 Infrared spectra in the CH stretching band regions of ZIFs; methyl group (a) and ring proton (b) of ZIF-8, Cl-ZIF-8 (c), and Br-ZIF-8 (d). The double lines of the CH stretching band in Cl-ZIF-8 and Br-ZIF-8 originate from the two different orientations of the linker molecules, which result from structural disorder in their crystal structure. | ||
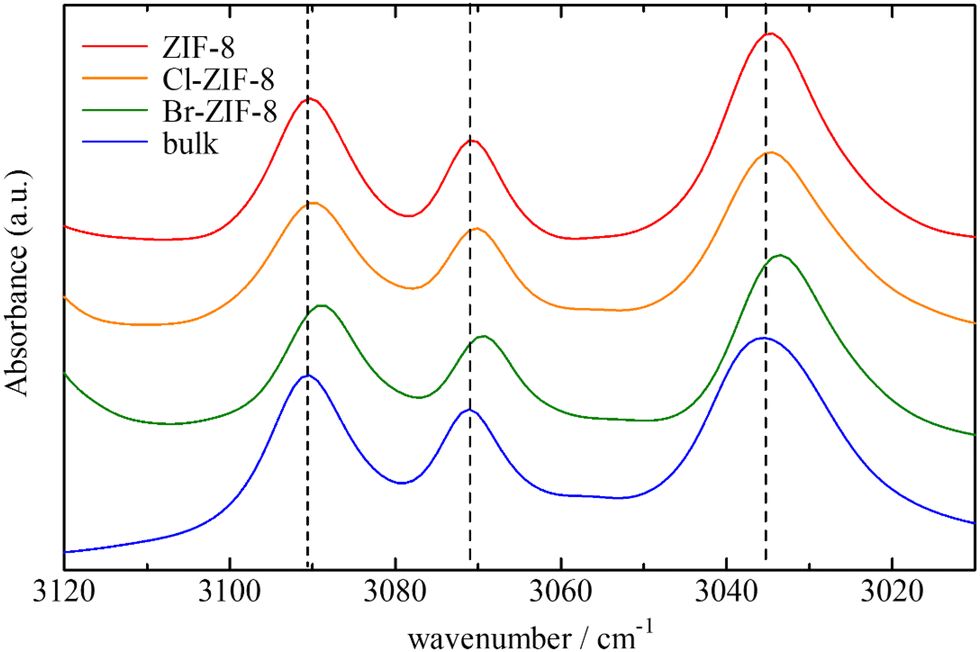 | ||
| Fig. 5 IR spectra in the CH stretching band regions of benzene in the ZIFs and bulk phase. | ||
The CH stretching bands of both ZIFs and benzene underwent a remarkable red-shift in the wavenumber range of 1–3 cm−1 by benzene adsorption. These shifts demonstrate attractive interactions with protons in the imidazole ring and in benzene. One of the plausible candidates for the attractive interactions acting on the aromatic and aliphatic protons is the C–H/π interaction, which has been observed in a number of organic compounds, metal complexes, and molecular assemblies.52–58
In both ZIF-8 and Cl-ZIF-8, the CH stretching band at carbon positions 4 and 5 in the imidazole ring notably shifts to the low-wavenumber side under benzene adsorption conditions. In addition, in ZIF-8, the CH stretching band of the methyl group also shifts to the low-wavenumber side. By contrast, no shift is observed in the corresponding bands for Br-ZIF-8. These results imply that the protons of the imidazole ring in ZIF-8 and Cl-ZIF-8 effectively interact with the π electrons of benzene molecules through CH/π interactions, whereas in Br-ZIF-8, the proton of the imidazole ring does not interact with benzene molecules. Furthermore, the methyl protons also interact with the π electrons of benzene, providing a more effective potential for benzene adsorption in ZIF-8.
Meanwhile, from the viewpoint of benzene molecules, the adsorption of benzene to ZIFs is dominated by the adsorption potential, which is derived by summing up the electrostatic and van der Waals interactions between all atoms in ZIFs and benzene molecules. However, the CH/π interactions between protons in benzene and π electrons of the imidazole ring will affect the molecular orientation of benzene adsorbed into the micropore of each ZIF specimen because intermolecular CH/π interactions are found in aromatic group pairs within 3 Å and in CH3 and an aromatic ring pair within 2.9 Å.52–58 Consequently, benzene molecules facilitate orientations that allow the imidazole ring to stand, dominating the local structure of benzene beside the six-membered ring aperture.
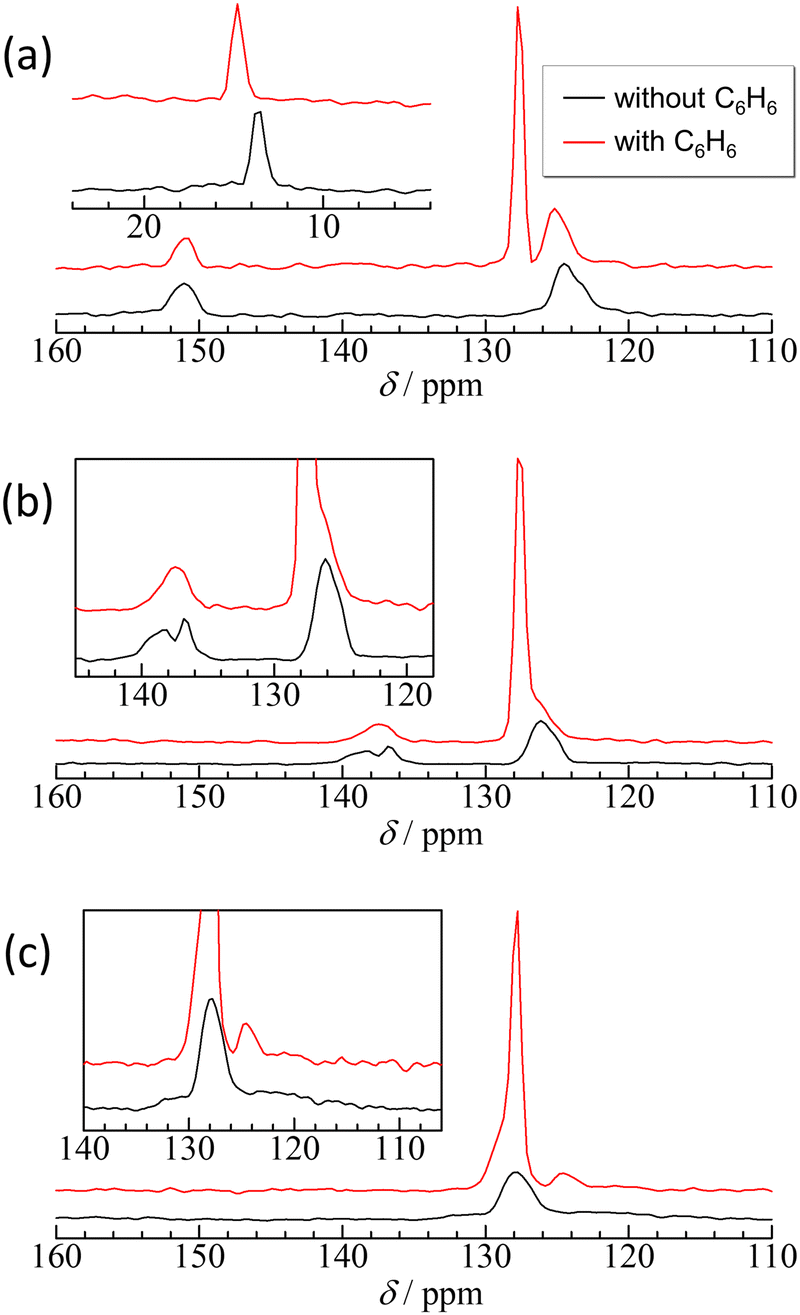 | ||
| Fig. 6 High-resolution solid-state 13C NMR spectra recorded under both magic-angle sample spinning and high-power 1H decoupling conditions. The insets depict the extension view around the chemical shift of methyl carbon in ZIF-8 and of imidazole carbons in Cl-ZIF-8 and Br-ZIF-8. | ||
In ZIF-8, the resonance peak of the imidazole ring is asymmetric, as expected, to determine whether the 13C–14N residual dipolar splitting59 or the existence of different environments for aromatic C–H carbons and the quaternary carbon. However, the 13C–14N residual dipolar splitting in ZIF-8 is estimated to be <0.2 ppm (see ESI†), which is too small to explain the full width at half maximum of the observed peaks. Therefore, the asymmetric peak implies different environments on the aromatic C–H carbons, probably caused by the different orientations of the linkers. After adsorption of benzene, the resonance peak of benzene shows an upfield shift of 1 ppm in comparison with bulk benzene, whereas the resonance peaks of the frameworks show a downfield shift of approximately 1 ppm, except for the quaternary carbon. These shifts are an evidence of the intermolecular interactions between benzene and the 2-methylimidazole moiety being able to consider the following two types of interactions: The upfield shift (decrease in the chemical shift value) of the aromatic carbons of benzene suggests π–π interactions between benzene and the 2-methylimidazole ring.60 By contrast, the downfield shift of methyl and aromatic C–H carbons implies CH/π interactions of these protons with the π electrons of benzene.61,62
In Cl-ZIF-8 and Br-ZIF-8, the resonance peaks of the aromatic CH carbons are also asymmetric similar to those in ZIF-8 but with smaller splitting, implying the existence of different orientations of the linker. Notably, the resonance of the quaternary carbon of Cl-ZIF-8 was split into two peaks, suggesting that the 2-chloroimidazole linker was disordered into two orientations. After the adsorption of benzene, the resonance peak of the quaternary carbon in Cl-ZIF-8 merges to one peak, indicating that benzene adsorption eliminates the disorder of the linker orientation. By contrast, the resonance of the quaternary carbon of Br-ZIF-8 broadened from 120 ppm to 140 ppm. This broadening may be caused by the dynamic disorder of the linker orientation34 and/or 13C–79/81Br residual dipolar splitting.63–65 The 13C–79/81Br residual dipolar splitting in Br-ZIF-8 is estimated to be 15 ppm for 81Br and 19 ppm for 79Br (see ESI†), which is consistent with the observed linewidth for the quaternary carbon of Br-ZIF-8. However, the adsorption of benzene notably made the broadened resonance peak in Br-ZIF-8 narrow and clear, suggesting that the adsorption of benzene slows down the motion of the dynamically disordered linkers. Both cases make it possible to reduce the entropy of the linker orientation, supporting the discussion regarding the activation entropy. The adsorption of benzene affected the local structure of the 2-chloroimidazole and 2-bromoimidazole linkers. After the adsorption of benzene onto Cl-ZIF-8, as well as onto Br-ZIF-8, the resonance peak of benzene also made an upfield shift of 1 ppm in comparison with bulk benzene, suggesting π–π interactions60 between benzene and 2-chlorolimidazole and/or 2-bromoimidazole rings.
Conclusions
The time dependence of the benzene adsorption uptake of ZIF-8, Cl-ZIF-8, and Br-ZIF-8 was analysed at 303, 313, and 323 K, and the adsorption was well reproduced by an intra-crystalline (Fick's) diffusion model. The diffusion coefficient and saturated adsorption amount of benzene were determined by optimising the model to experimental data.The saturated adsorption amount of benzene decreased in the order of ZIF-8, Cl-ZIF-8, and Br-ZIF-8. Notably, ZIF-8, which possesses an intermediate pore volume among the three specimens, can accommodate the greatest number of molecules in the micropore (5.5 molecules per micropore). This saturated amount of benzene was much greater than that predicted from the effective density of the adsorbed benzene. MM calculations revealed the closest distance between benzene and the six-membered ring aperture surface and the large tilting angle of the benzene molecule with respect to the aperture surface in ZIF-8. These findings suggest that the 2-methylimidazolate moiety forms an effective attraction interaction with benzene molecules. In this context, the CH/π interaction between the methyl proton and the π electrons of benzene is considered to contribute to the attractive interaction between the benzene molecule and ZIF-8 pore wall.
Both the activation energy and pre-exponential factor increased in the order ZIF-8, Cl-ZIF-8, and Br-ZIF-8. ZIF-8 exhibited the smallest Ea value, despite the expected effective steric hindrance caused by having the largest methyl group among the three specimens. This result suggests the occurrence of an attractive interaction that stabilises the transition state of benzene passing through the six-membered ring aperture, which is consistent with the discussion of the saturated adsorption amount. Furthermore, the pre-exponential factor, D0, gives the activation entropy, ΔS‡, in the transition state when a benzene molecule passes through a six-membered ring aperture. The ΔS‡ values at 303 K were negative, and their absolute values increased in the order of Br-ZIF-8, Cl-ZIF-8, and ZIF-8. Considering the degree of freedom of translation and rotation of the benzene molecule, as well as the vibration and disorder of the linker, we found that the differences in ΔS‡ were caused by the dynamic local structure of the six-membered ring aperture among the ZIF-8 analogues. The magnitude of ΔS‡ significantly affects the diffusion coefficients in the temperature range from 303 K to 323 K, suggesting a significant interaction between benzene and the six-membered ring aperture.
Infrared spectroscopy revealed that the aromatic C–H in ZIF-8 and Cl-ZIF-8 interacted with the π-electrons in benzene through CH/π interactions, whereas in Br-ZIF-8, the interaction acting on the aromatic C–H might be considerably small. This feature of Br-ZIF-8 is considered to originate from the large steric hindrance around the aromatic C–H because of the larger atomic radius of the bromine atom compared with other functional groups. By contrast, the low-wavenumber shift of the C–H stretching band in benzene suggests that the benzene molecules interact attractively with the π-electrons in the linkers through CH/π interactions. The CH/π interactions between methyl protons and the π-electrons of benzene and between aromatic C–H protons in the frameworks and the π-electrons of benzene were also confirmed by the downfield shift of the 13C resonance peaks for the corresponding carbons. Meanwhile, the π–π interactions between benzene and the 2-methylimidazole ring and between benzene molecules were confirmed by the upfield shift (decrease in the chemical shift value) of the 13C resonance peaks of benzene carbons. Thus, these spectroscopic features revealed that the intermolecular interactions between the linkers and benzene increased in the order of Br-ZIF-8 ≪ Cl-ZIF-8 < ZIF-8. This difference in intermolecular interactions is expected to dominate the saturated amount of benzene adsorption.
In summary, this study reveals that the CH/π interaction between the 2-substituted imidazole ligand of X-ZIF-8 and adsorbate molecule provides an energetic advantage when the benzene molecule passes through the six-membered ring aperture and accelerates adsorption. In particular, methyl groups were more effective than halogen groups in the CH/π interaction and also played an important role in the energetic stabilisation of benzene molecules in the pore. This finding suggests that the adsorption of molecules with π-electron systems on ZIF-8 is more favourable than that with non-π-electron compounds. In the future, to further clarify this point, we intend to examine the dependence of the number of π-electrons in the adsorbate on the adsorption behaviour into ZIF-8 by studying the adsorption rate of six-membered ring alicyclic hydrocarbons similar to benzene molecules.
Author contributions
Ryohei Yagi: conceptualisation, methodology, validation, formal analysis, investigation, data curation, writing – original draft, and visualisation. Takahiro Ueda: conceptualisation, methodology, validation, resources, software, formal analysis, visualisation, writing – review & editing, supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (Grant Number JP21K04979). This work was the result of using research equipment shared in the MEXT Project for promoting the public utilisation of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0441200021. The authors thank Dr Naoya Inazumi and Dr Yasuto Todokoro of the Analytical Instrument Facility, Graduate School of Science, Osaka University, for their helpful and useful advice, guidance and instructions concerning solid-state NMR measurements.Notes and references
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. Y. Yaghi, Acc. Chem. Res., 2010, 43(1), 58 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS PubMed.
- L. B. D. Bourg, A. U. Ortiz, A. Boutin and F.-X. Coudert, APL Mater., 2014, 2, 124110 CrossRef.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- W. Morris, C. J. Stevens, R. E. Taylor, C. Dybowski, O. M. Yaghi and M. A. Garcia-Garibay, J. Phys. Chem. C, 2012, 116, 13307 CrossRef CAS.
- P. Cheng and Y. H. Hu, J. Phys. Chem. C, 2014, 118, 21866 CrossRef CAS.
- K. Eum, K. C. Jayachandrababu, F. Rashidi, K. Zhang, J. Leisen, S. Graham, R. P. Lively, R. R. Chance, D. S. Sholl, C. W. Jones and S. Nair, J. Am. Chem. Soc., 2015, 137, 4191 CrossRef CAS PubMed.
- S. El-Hankari, J. Aguilera-Sigalat and D. Bradshaw, J. Mater. Chem. A, 2016, 4, 13509 RSC.
- D. M. Polyukhov, A. S. Poryvaev, A. S. Sukhikh, S. A. Gromilov and M. V. Fedin, ACS Appl. Mater. Interfaces, 2021, 13, 40830 CrossRef CAS PubMed.
- H. Zhao, H. Ye, J. Zhou, G. Tang, Z. Hou and H. Bai, ACS Appl. Mater. Interfaces, 2020, 12, 49431 CrossRef CAS PubMed.
- L. Zhang and Y. H. Hu, J. Phys. Chem. C, 2011, 115, 7967 CrossRef CAS.
- V. Eroshenko, Y. Grosu, N. Tsyrin, V. Stoudenets, J.-M. Nedelec and J.-P. E. Grolier, J. Phys. Chem. C, 2015, 119, 10266 CrossRef CAS.
- A. F. Sapnik, H. S. Geddes, E. M. Reynolds, H. H.-M. Yeung and A. L. Goodwin, Chem. Commun., 2018, 54, 9651 RSC.
- L. Zhang, Z. Hu and J. Jiang, J. Am. Chem. Soc., 2013, 135, 3722 CrossRef CAS PubMed.
- H. Tanaka, S. Ohsaki, S. Hiraide, D. Yamamoto, S. Watanabe and M. T. Miyahara, J. Phys. Chem. C, 2014, 118, 8445 CrossRef CAS.
- C. O. Ania, E. García-Perez, M. Haro, J. J. Gutierrez-Sevillano, T. Valdes-Solis, J. B. Parra and S. Calero, J. Phys. Chem. Lett., 2012, 3, 1159 CrossRef CAS PubMed.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900 CrossRef CAS PubMed.
- T. Tian, M. T. Wharmby, J. B. Parra, C. O. Ania and D. Fairen-Jimenez, Dalton Trans., 2016, 45, 6893 RSC.
- F.-X. Coudert, ChemPhysChem, 2017, 18, 2732 CrossRef CAS PubMed.
- C. Zhang, R. P. Lively, K. Zhang, J. R. Johnson, O. Karvan and W. J. Koros, J. Phys. Chem. Lett., 2012, 3, 2130 CrossRef CAS PubMed.
- K. Zhang, R. Lively, C. Zhang, R. Chance, W. Koros, D. Sholl and S. Nair, J. Phys. Chem. Lett., 2013, 4, 3618 CrossRef CAS.
- M. R. Ryder, B. Civalleri, T. D. Bennett, S. Henke, S. Rudić, G. Cinque, F. Fernandez-Alonso and J.-C. Tan, Phys. Rev. Lett., 2014, 113, 215502 CrossRef PubMed.
- N. Y. Tan, M. T. Ruggiero, C. Orellana-Tavra, T. Tian, A. D. Bond, T. M. Korter, D. Fairen-Jimenez and J. A. Zeitler, Chem. Commun., 2015, 51, 16037 RSC.
- D. I. Kolokolov, A. G. Stepanov and H. Jobic, J. Phys. Chem. C, 2015, 119, 27512 CrossRef CAS.
- T. Ueda, M. Nakai and T. Yamatani, Adsorption, 2017, 23, 887 CrossRef CAS.
- A. Awadallah-F, F. Hillman, S. A. Al-Muhtaseb and H.-K. Jeong, J. Mater. Sci., 2019, 54, 5513 CrossRef CAS.
- A. E. Khudozhitkov, H. Zhao, A. Ghoufi, S. S. Arzumanov, D. I. Kolokolov, G. Maurin and A. G. Stepanov, ACS Appl. Mater. Interfaces, 2021, 13, 33685 CrossRef CAS PubMed.
- D. Radhakrishnan and C. Narayana, J. Chem. Phys., 2015, 143, 234703 CrossRef PubMed.
- T. Ueda, T. Yamatani and M. Okumura, J. Phys. Chem. C, 2019, 123, 27542 CrossRef CAS.
- K. Li, D. H. Olson, J. Seidel, T. J. Emge, H. Gong, H. Zeng and J. Li, J. Am. Chem. Soc., 2009, 131, 10368 CrossRef CAS PubMed.
- H. Amrouche, S. Aguado, J. Perez-Pellitero, C. Chizallet, F. Siperstein, D. Farrusseng, N. Bats and C. Nieto-Draghi, J. Phys. Chem. C, 2011, 115, 16425 CrossRef CAS.
- J. Hu, Y. Liu, J. Liu and C. Gu, Fuel, 2017, 200, 244 CrossRef CAS.
- H. Amrouche, B. Creton, F. Siperstein and C. Nieto-Draghi, RSC Adv., 2012, 2, 6028 RSC.
- G. Chaplais, G. Fraux, J.-L. Paillaud, C. Marichal, H. Nouali, A. H. Fuchs, F.-X. Coudert and J. Patarin, J. Phys. Chem. C, 2018, 122, 26945 CrossRef CAS.
- B. Chen, F. Bai, Y. Zhu and Y. Xia, Microporous Mesoporous Mater., 2014, 193, 7 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminformatics, 2012, 4, 17 CrossRef CAS PubMed.
- B. S. Winsten and N. J. Holness, J. Am. Chem. Soc., 1955, 77, 5562 CrossRef.
- S. Tanaka, K. Fujita, Y. Miyake, M. Miyamoto, Y. Hasegawa, T. Makino, S. Van der Perre, J. C. Saint Remi, T. Van Assche, G. V. M. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2015, 119, 28430 CrossRef CAS.
- J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in nanoporous materials, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- L. Zhang, C. Chmelik, A. N. C. van Laak, J. Kärger, P. E. de Jongh and K. P. de Jong, Chem. Commun., 2009, 6424 RSC.
- S. Sircar and J. R. Hufton, Adsorption, 2000, 6, 137 CrossRef CAS.
- E. Wilhelm and R. Battino, J. Chem. Phys., 1971, 55, 4012 CrossRef CAS.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- S. Glasstone, K. J. Laidler and H. Eyring, The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena, McGraw-Hill, New York, USA, 1941 Search PubMed.
- M. V. Roux, M. Temprado, J. S. Chickos and Y. Nagano, J. Phys. Chem. Ref. Data, 2008, 37(4), 1855 CrossRef CAS.
- Y. Hu, H. Kazemian, S. Rohani, Y. Huang and Y. Song, Chem. Commun., 2011, 47, 12694 RSC.
- B. Xu, D. Xie, Y. Mei, Z. Kang, R. Wang and D. Sun, Phys. Chem. Chem. Phys., 2017, 19, 27178 RSC.
- C. Wu, D. Xie, Y. Mei, Z. Xiu, K. M. Poduska, D. Li, B. Xu and D. Sun, Phys. Chem. Chem. Phys., 2019, 21, 17571 RSC.
- S. Wang, Sci. Rep., 2020, 10, 17875 CrossRef CAS PubMed.
- M. A. Palafox, Int. J. Quantum Chem., 2000, 77, 661 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- S. Tsuzuki, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2012, 108, 69 RSC.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 3746 CrossRef CAS.
- R. M. Kumar, M. Elango, R. Parthasarathi, D. Vijay and V. Subramanian, J. Chem. Sci., 2012, 124, 193 CrossRef CAS.
- M. Li, G. Qing, Y. Xiong, Y. Lai and T. Sun, Sci. Rep., 2015, 5, 15742 CrossRef CAS PubMed.
- G. Platzer, M. Mayer, A. Beier, S. Brüschweiler, J. E. Fuchs, H. Engelhardt, L. Geist, G. Bader, J. Schörghuber, R. Lichtenecker, B. Wolkerstorfer, D. Kessler, D. B. McConnell and R. Konrat, Angew. Chem., Int. Ed., 2020, 59, 14861 CrossRef PubMed.
- S. Morita, A. Fujii, N. Mikami and S. Tsuzuki, J. Phys. Chem. A, 2006, 110, 10583 CrossRef CAS PubMed.
- K. Eichele, M. D. Lumsden and R. E. Wasylishen, J. Phys. Chem., 1993, 97, 8909 CrossRef CAS.
- Y. Zhang, B. Ji, A. Tian and W. Wang, J. Chem. Phys., 2012, 136, 141101 CrossRef PubMed.
- S. Scheiner, Chem. Phys. Lett., 2019, 714, 61 CrossRef CAS.
- G. Li, L. Huang, X. Yi, Y.-K. Peng, S. C. E. Tsang and A. Zheng, Chem. Commun., 2018, 54, 13435 RSC.
- R. K. Harris and A. C. Olivieri, Prog. Nucl. Magn. Reson. Spectrosc., 1992, 24, 435 CrossRef CAS.
- A. C. Olivieri, L. Frydman and L. E. Diaz, J. Magn. Reson., 1987, 75, 50 CAS.
- A. C. Olivieri, J. Magn. Reson., 1989, 81, 201 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of TG-DTA diagrams, nitrogen adsorption isotherms, PXRD patterns, SEM images, size distribution of crystallites, comparison of the data optimisation of a surface barrier model and an intra-crystalline (Fick's) diffusion model, typical examples of the model structure and the stable configuration of a benzene molecule adsorbed on a 6-membered ring aperture, Arrhenius plot of the adsorption rate constant, infrared spectra and results of the deconvolution of the 13C-NMR resonance peaks. Tables of BET surface area and pore volume, average particle size, desorption amount of benzene, results of the non-linear least-squared data fitting for Arrhenius plot, Band assignment of IR spectra and 13C chemical shift data. Additional explanation for evaluation of activation entropy of benzene trapped by six-membered ring aperture and 13C–14N and 13C–79/81Br residual dipolar splitting. See DOI: https://doi.org/10.1039/d3cp01662e |
| This journal is © the Owner Societies 2023 |