
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of presolvated and solvated electrons with CO2 in water up to 118 bar at 298 and 308 K†
Denis S.
Dobrovolskii
 ,
Mehran
Mostafavi
and
Sergey A.
Denisov
*
,
Mehran
Mostafavi
and
Sergey A.
Denisov
*
Insitute de Chimie Physique UMR 8000, CNRS/Université Paris-Saclay, Bâtiment 349, Orsay, 91405, France. E-mail: sergey.denisov@universite-paris-saclay.fr; Fax: +33169156188; Tel: +33169156171
First published on 25th May 2023
Abstract
The reactivity of electrons in the CO2-water system was evaluated through picosecond electron pulse radiolysis at different gas pressures (ranging from 1 to 118 bar) and temperatures (25 and 35 °C) coupled with UV-vis transient spectroscopy. A custom-made spectroscopic cell was utilized for these experiments, which allowed for regulation of temperature and pressure. The scavenging of electrons was measured directly at gas pressures even in the supercritical state, and the results showed a non-monotonic dependence of electron reactivity with CO2 concentration, in agreement with the changing molar concentration of CO2 in water under varying pressure.
1 Introduction
The capture and subsequent chemical transformation of CO2 is a global issue. There are several ways to reduce it: electrochemical, photochemical, e.g., the use of a hydrated electron (ehyd−), which is the most reducing species in solution (standard reduction potential of −2.9 V NHE), is one of the effective approaches for CO2/CO2˙− (−2.14 V NHE) reduction without using any catalyst at room tempertature. Although electrochemical1 and photoelectrochemical2 approaches have been developed to reduce CO2 using ehyd− chemistry, their faradaic efficiency remains low. A promising alternative method involves the use of high-energy ionizing radiation to generate reducing radicals directly in water at room temperature. This method eliminates the need for a catalyst but requires appropriate chemical conditions to maximize CO2 conversion yield.3 A better understanding of CO2 reduction processes may lead to the development of methods for reducing carbon dioxide emissions by converting it into useful compounds or energy.This study extends previous research4 by investigating the reactivity of electrons dissolved in a CO2–water solution at increased pressure (1 to 118 bar) and temperature (25, 35 °C) using picosecond electron pulse radiolysis. Unlike the previous work, which only covered pressures up to 52 bar, this study expands the analysis to include measurements of reaction rate in the supercritical state of CO2 (P = 72.9 bar, T = 30.85 °C) in water. It is important to clarify that the measurements were conducted in water with dissolved CO2 gas. Throughout all experimental conditions, the water was not in a supercritical state; rather, the CO2 existed as a separate phase on top of the water as a gas, liquid, and supercritical liquid, respectively, of the pressure.
2 Experimental
2.1 Pulse radiolysis setup
The ELYSE picosecond pulse radiolysis facility at the Institute de Chimie Physique, Université Paris-Saclay was used to track the formation yields and decay kinetics of solvated electrons, es−. We employed a pulse-probe setup with a broad supercontinuum probe and charge-coupled device (CCD) detection for transient absorption measurements. The absorption measurements in the near-UV to visible region were conducted using a configuration previously detailed in ref. 4 The setup included a supercontinuum generated by focusing a laser source (@790 nm, 1 μJ into a CaF2 disk. The electron pulses were delivered at a repetition frequency of 5 Hz, with an electron energy of 6–8 MeV, a charge of 6 nC, and an average dose per pulse of 100 Gy4–6 This setup enabled the recording of the entire transient spectra between 350–780 nm, independent of shot-to-shot fluctuations and long-term drifts of the electron source.4–6The experimental setup is illustrated in Fig. 1S (ESI†), using a schematic illustration. The setup comprises a high-pressure cell filled with distilled water (1.5 ml) to approximately 3/4 of its capacity. CO2 gas is introduced into the cell at high pressure through a single channel, regulated by a NEM-B207-01D syringe pump, CETONI GmbH. The system has a pressure capacity of up to 200 bar. However, the highest reached value in our was 120 bar for safety reason. The measurements taken in a static cell were compared to those taken in a circulating solution under the same thermodynamic conditions and the results were found to be similar. As a result, all subsequent studies were conducted in a static solution. The high-pressure cell was maintained at a regulated temperature using a MINISTAT CC Cryothermostat, which was monitored by an RTD-100 sensor and an Agilent 34411A Digital Multimeter. Temperature was regulated using a closed water loop. The system was thermodynamically equilibrated for 30 minutes after the gas pressure was applied before the measurement.
The production of a hydrated electron, ehyd−, following a 7 ps electron pulse is associated with the scavenging of the pre-solvated electron, epre−, by the solute. The degree of scavenging of epre− during the pulse duration increases with decreasing ehyd− production. The reactivity of epre− with the solute can be determined by adjusting the solute concentration, and is commonly expressed as the C37 value, which represents the solute concentration at which 37% of the initial ehyd− remains.7,8 The C37 value is an useful comparative parameter for describing the ultrafast long-range reactions such as epre− with solutes. The radiolytic yield of ehyd− in the studied water-CO2 solution was calculated using eqn (1)–(2) provided in a previous study.4 The equations are explained in the supplementary information, ESI.†
The dose deposited per pulse was determined from the measurements of the absorbance of ehyd− (Aeaq− (λ, t)) in neat water and verified before each series of experiments, considering the initial yield of the solvated electrons, measured at 10 ps, to be G10ps = 4.4 × 10−7 mol J−1.5,7
Table S1 and S2 (ESI†) present the calculated dose factor F for 25 °C and 35 °C respectively. The energy absorption of ionizing radiation is influenced by the quantity of electrons in the substance. At low concentrations of dissolved material, the solvent absorbs most of the energy. However, in highly concentrated solutions, the dissolved substance start to absorb significant amount of the energy, resulting in an increase in excess electron production with increasing CO2 concentrations. In this way, the yield of the excess electron is altered in the presence of high CO2 concentration by less than 2%.
3 Results and discussion
The absorption of the ehyd− in solutions of CO2 was measured by pump–probe spectroscopy in subnano- and nanosecond timescale, in order to determine the reactivity of epre− and ehyd− with CO2 at two different temperatures, 25 °C and 35 °C (Fig. 1).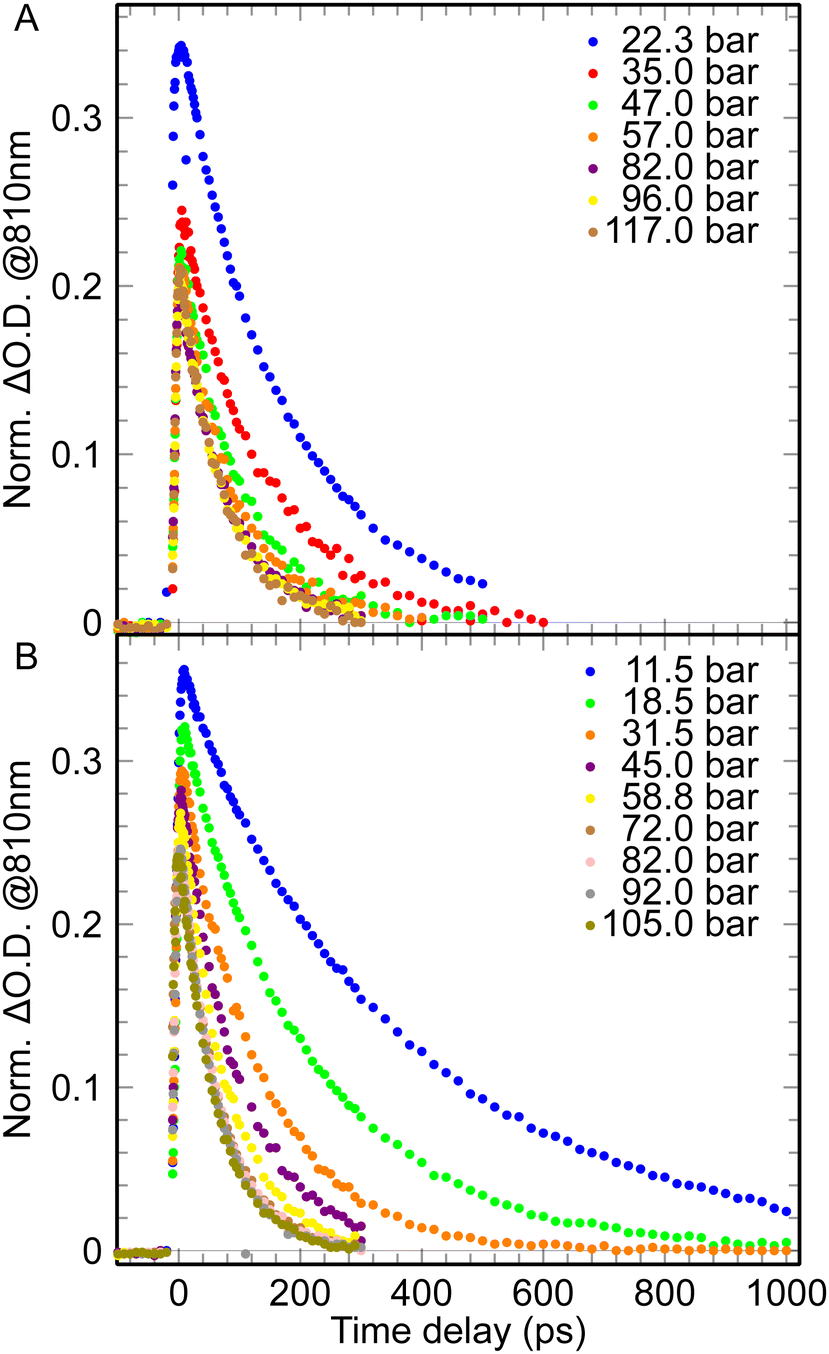 | ||
| Fig. 1 The pulse radiolysis data of a water– CO2 system at different pressures for nanosecond timescale: A – for 25 °C; B – for 35 °C. | ||
The dependence of the pulse radiolysis data of the water–CO2 system demonstrates similar behavior for normal and supercritical state (for P > 63 bar and T = 35 °C). With pressure increase the lifetime of ehyd− drops down (the observed rate of its disappearance increases, Fig. 1, 2) and the initial formation yield of ehyd− decreases (Fig. 3).
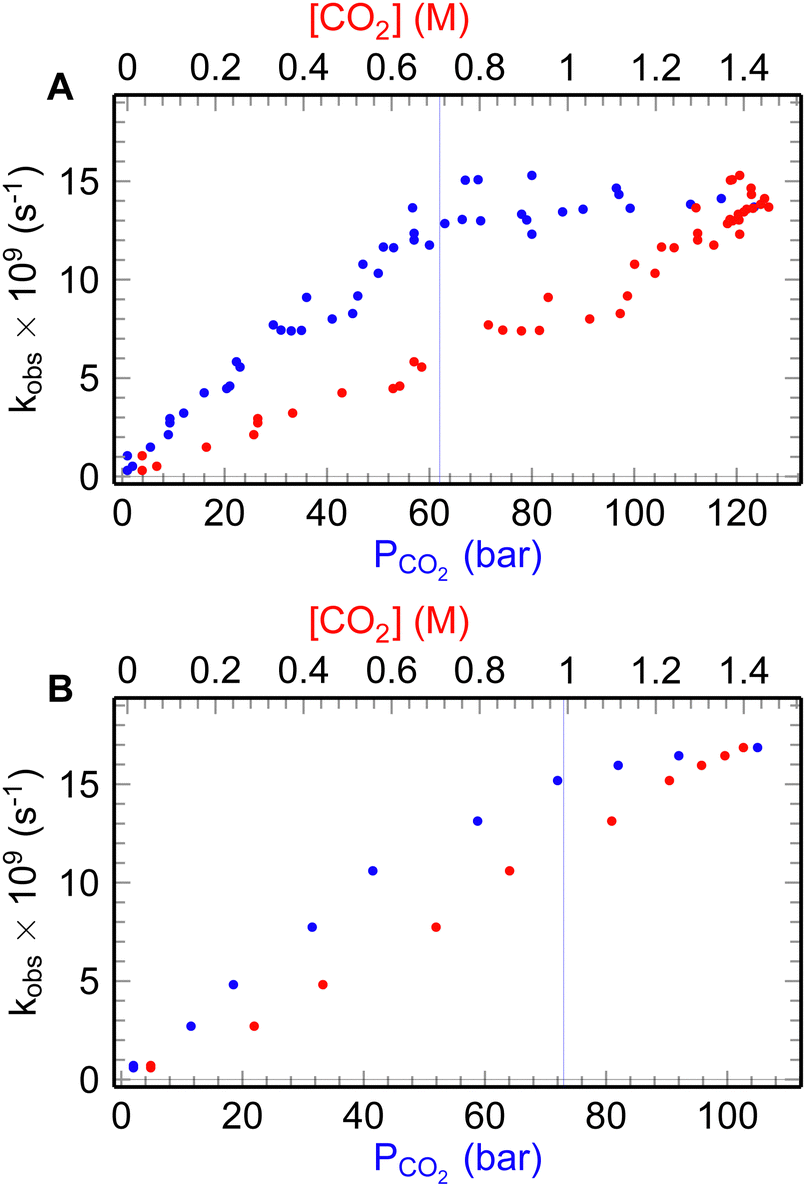 | ||
| Fig. 2 The ehyd− decay rate (blue dots) at different pressures and concentrations of CO2 (red dots): A – for 25 °C; B – for 35 °C. | ||
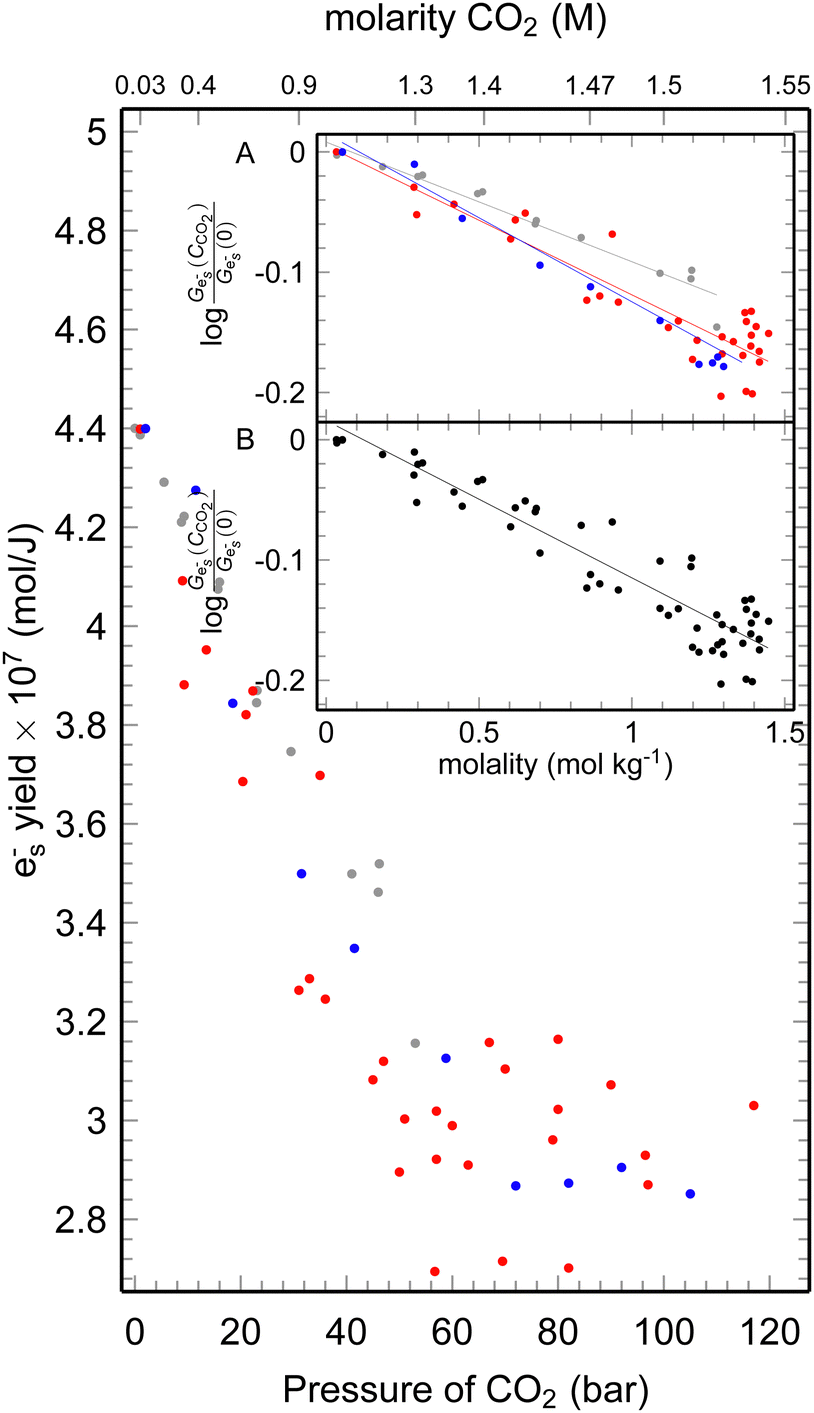 | ||
| Fig. 3 The yield of ehyd− at 10 ps after the 7 ps electron pulse at different CO2 pressures, the gray dots data up to 51 bars;4 red dots – data at 25 °C, blue dots – data at 35 °C. Inset: Plot of the ehyd− yield fraction remaining vs. the concentration of the CO2 A – 25 °C, B – 35 °C. | ||
As the pressure of CO2 increases, its concentration in water also increases. The solubility of CO2 is known to exhibit a non-linear dependence,9–11 and the actual Henry's law is no longer applicable above ca. 20 bar.‡ Moreover, beyond the liquidus pressure, the concentration of CO2 increases even more slowly with an increase in pressure (Fig. S2, ESI†).
As it can be seen from the Fig. 2 and 3 the dependencies of ehyd− lifetime and ehyd− initial yields at 10 ps after the pulse demonstrate the same behavior, reaching the plateau after the liquidus pressure, both for normal and supercritical liquid state, namely 25 °C and 35 °C, 53 and 63 bars, respectively.
The rate of decay of ehyd− below 60 bar increases from 0.1 to 13 × 10−9 s−1 as pressure increases, remaining almost constant afterwards. For the supercritical state, it slightly increases from 15 to 17 × 10−9 s−1. However, there is a tendency for the value to plateau as in the normal liquid state. The decay rate constants of ehyd− were determined up to the point of liquidus for experimental temperatures as: K25°C = 8.1 × 109 M−1 s−1 and K35°C = 10.2 × 109 M−1 s−1, respectively. These values are consistent with literature data.4,12
The leveling off of decay rates can be attributed to the saturation of CO2aq concentration, as depicted in Fig. S2 (ESI†). The concentration of CO2aq reach values up to 1.4 M above 60 bars (Fig. S2, ESI†). It should be noted that under the experimental conditions, CO2aq is present almost at its non-protonated form, as evidenced by the solutions pH going low as 3. For additional information refer to the ESI† of our previous study.4
As shown in Fig. 3, the formation yield of ehyd− 10 ps after the pulse initially drops from 4.4 to 3 × 10−7 mol J−1 upon reaching 60 bars, and subsequently slow further decrease continues with pressure rise. It is noteworthy that the dependence of electron yield remains unchanged in the supercritical state. The formation yield of ehyd− is deacresing with the pressure increase due to the interaction between epre− and CO2aq, forming CO2˙−.
The scavenging efficiency of CO2 towards epre− is inversely proportional to its C37 value. The initial yield of ehyd− decreases in the presence of a high concentration of scavengers and the fraction ehyd− could be described by the expression f = exp(−[S]/C37). The C37 value is determined for each temperature and are C2537 = 3.3 mol kg−1; C3537 = 3.6 mol kg−1, for 25 °C, 35 °C, respectively. In the previous4 measurement the value of 4.2 was determined for room temperature, that is erroneous (Fig. 3), that could be explained by the smaller range of used pressures. However, the C37 concentrations are not achievable since even at ultrahigh pressures (>500 bar) the CO2aq can only reach ca. 1.8 M.
The experimental data indicates that after reaching pressures higher than 60 bar, the solvated electrons’ rate of disappearance is reduced; the same is observed for the initial yield change with pressure for all temperatures, that correlates with the molar concentration of carbon dioxide (Fig. S2, ESI†).11,13
Our new experimental data is in a good agreement with previously published one4 describing reactivity of CO2 with ehyd− and epre− in water under atmosphere and elevated pressures (below 53 bar) at room temperatures.
Under supercritical conditions, CO2 molecules have been observed to form neutral clusters, which exhibit a diverse size distribution that is influenced by changes in pressure and temperature. As a result, a broad range of cluster anions could be present in supercritical CO2. Considering the fast reaction kinetics observed in electron transfer and the slow diffusivity of larger clusters, it is probable that in low-pressure scenarios, the primary electron donor is a small cluster, such as a (CO2)2− dimer.14 It could be inferred that the behavior of CO2 reactivity with excess electrons might be influenced in the water phase, when CO2 is in its supercritical state above the water phase. Specifically, clusters of CO2 molecules could potentially emerge in water, leading to an impact on the reactivity involving es− and epre− with CO2. However, our experimental results do not indicate that.
In addition, we would like to state that there is no detectable spectral shift observed for either the normal or supercritical state with an increase in pressure (Fig. S3, ESI†).
The direct implication of this work could be related to corrosion in steel caused by water dissolved in supercritical CO2 that is a widely acknowledged issue.15–17 The corrosion rate is largely determined by the ability of CO2 to form anions. As international programs for CO2 storage and transportation move towards liquid forms, the problem of pipe and storage facility corrosion will need to be addressed. The radiolysis technique provides a powerful research tool to investigate the fundamental properties of radicals in solutions and mixtures, and may bring the engineers to an effective solution of mentioned issue. Given that the dimerization of the CO2 anion radical results in the creation of oxalate,3 it is reasonable to anticipate not only corrosion of steel pipes transporting liquid CO2, but also the deposition of oxalate on walls of the tubes in the presence of natural ionizing radiation.
4 Conclusions
The reactivity of epre− in CO2-water system was studied in the pressure range of 1–118 bar at two temperatures 25, 35 °C. Additionally, studies were carried out for the supercritical state (for P > 63 bar, 35 °C). The C37 values were determined to be 3.3 and 3.6 mol kg−1, respectively, for 25 °C and 35 °C. Such concentrations for these systems are not achievable for practical study, so they could be used as reference concentrations demonstrating the scavenge rate of epre− in water solution for CO2. In additional, the rates of the reaction between ehyd− and CO2 were determined to be 8.1 × 109 M−1 s−1 and 10.2 × 109 M−1 s−1, respectively, for 25 °C and 35 °C. These results are agreement with previously published data.4Author contributions
D. Dobrovolskii contributed to the project by data curation, formal analysis, visualization, resources, investigations and writing the original draft. S. Denisov's contributions included conceptualization, data curation, methodology, project administration, investigations, funding acquisition, formal analysis, resources, software, supervision, validation, visualization, conceptualization and writing for review and editing. M. Mostafavi's contributions comprised conceptualization, funding acquisition, methodology and writing for review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support provided by the Labex PALM DISCOVER (2018, University of Paris-Saclay) is gratefully acknowledged by the authors. The authors also extend their gratitude to Pierre JEUNESSE and the mechanical workshop of ISMO UMR8214 for their contribution to the realization of the high-pressure cell. Additionally, the authors would like to thank Jean-Philippe LARBRE, Audrey GAYRAL, and Pierre JEUNESSE for their technical support of the ELYSE platform.References
- M.-Y. Lee, K. T. Park, W. Lee, H. Lim, Y. Kwon and S. Kang, Crit. Rev. Environ. Sci. Technol., 2020, 50, 769–815 CrossRef CAS.
- A. U. Pawar, C. W. Kim, M.-T. Nguyen-Le and Y. S. Kang, ACS Sustainable Chem. Eng., 2019, 7, 7431–7455 CrossRef CAS.
- C. Hu, S. A. Gharib, Y. Wang, P. Gan, Q. Li, S. A. Denisov, S. L. Caer, J. Belloni, J. Ma and M. Mostafavi, Chem. Phys. Chem., 2021, 22, 1900–1906 CrossRef CAS PubMed.
- S. A. Denisov and M. Mostafavi, Phys. Chem. Chem. Phys., 2021, 23, 5804–5808 RSC.
- J. L. Marignier, V. Waele, H. Monard and F. Gobert, Radiat. Phys. Chem., 2006, 75, 1024–1033 CrossRef CAS.
- J. Belloni, et al. , Nucl. Instrum. Methods Phys. Res., Sect. A, 2005, 539, 527–539 CrossRef CAS.
- F. Wang, U. Schmidhammer, J. P. Larbre, Z. Zong, J. L. Marignier and M. Mostafavi, Phys. Chem. Chem. Phys., 2018, 20, 15671–15679 RSC.
- Y. Muroya, et al. , Radiat. Phys. Chem., 2005, 72, 169–172 CrossRef CAS.
- R. F. Weiss, Mar. Chem., 1974, 2, 203–215 CrossRef CAS.
- N. Spycher, K. Pruess and J. Ennis-King, Geochim. Cosmochim. Acta, 2003, 67, 3015–3031 CrossRef CAS.
- Z. Duan, R. Sun, C. Zhu and I. M. Chou, Mar. Chem., 2006, 98, 131–139 CrossRef CAS.
- A. Lisovskaya and D. M. Bartels, Radiat. Phys. Chem., 2016, 158, 61–63 CrossRef.
- A. Hebach, A. Oberhof and N. Dahmen, J. Chem. Eng. Data, 2004, 49, 950–953 CrossRef CAS.
- N. M. Dimitrijevic, D. M. Bartels, C. D. Jonah and K. Takahashi, Chem. Phys. Lett., 1999, 309, 61–68 CrossRef CAS.
- Z. Cui, S. Wu, S. Zhu and X. Yang, Appl. Surf. Sci., 2006, 252, 2368–2374 CrossRef CAS.
- B. McGrail, H. Schaef, V. Glezakou, L. Dang and A. Owen, Energy Procedia, 2009, 1, 3415–3419 CrossRef CAS.
- Y.-S. Choi and S. Nešić, Int. J. Greenhouse Gas Control, 2011, 5, 788–797 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01535a |
| ‡ The CO2 concentration were determined according to the literature data.9, 10, 11 |
| This journal is © the Owner Societies 2023 |