
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the interaction sites in glucose and galactose using phenol as a probe†
Paúl
Pinillos‡
 ,
Ander
Camiruaga‡
,
Fernando
Torres-Hernández
,
Francisco J.
Basterrechea
,
Imanol
Usabiaga
* and
José A.
Fernández
*
,
Ander
Camiruaga‡
,
Fernando
Torres-Hernández
,
Francisco J.
Basterrechea
,
Imanol
Usabiaga
* and
José A.
Fernández
*
Department of Physical Chemistry, Fac. of Science and Technology, University of the Basque Country (UPV/EHU), Barrio Sarriena s/n, 48940, Spain. E-mail: josea.fernandez@ehu.es; i.usabiaga@outlook.es
First published on 7th February 2023
Abstract
Sugars, together with amino acids and nucleobases, are the fundamental building blocks of a cell. They are involved in many fundamental processes and they especially play relevant roles as part of the immune system. The latter is connected to their ability to establish a collection of intermolecular interactions, depending on the position of their hydroxyl groups. Here we explore how the position of the OH in C4, the anomeric conformation and the nature substituent affect the interaction with phenol, which serves as a probe of the preferred site for the interaction. Using mass-resolved excitation spectroscopy and density functional calculations, we unravel the structure of the dimers and compare their conformation with those found for similar systems. The main conclusion is that the hydroxymethyl group has a very strong influence, guiding the whole aggregation process and that the position of the substituent in C4 has a stronger influence on the final structure of the dimer than the anomeric conformation.
Introduction
Sugars, together with nucleobases, amino acids and lipids, are the building blocks of a cell. They play fundamental roles from energy storage to constituent molecules of some biopolymers, such as starch and cellulose.1,2 One of the most fascinating tasks they are involved in is the immune function. Combining several sugar units, cells create polysaccharides that present unique conformational and interactional landscapes, defined by the nature of their monosaccharide components.3 The corresponding receptors in the immune cells are able to sense, probe and recognize such configurations and determine if the cell is friend or foe. It is surprising how the immune system is able to distinguish even the smallest modification: the axial/equatorial position of a hydroxyl group in one of the monosaccharide constituents.4 In part, this ability relies on a kind of amplification effect produced by the hydrogen bond networks that extend along the whole glycan. Modification of the position of a given hydroxyl group from axial to equatorial or vice versa alters such a network. This facilitated the corresponding receptor to detect the structural differences.5,6To understand the whole recognition process, information regarding two aspects of the system is essential: a deep knowledge of the structure of mono- and poly-saccharides and of the interactions that they can establish with other molecules, such as for example, the lateral chains of amino acids. There are many research groups contributing to increasing the knowledge on the structure of saccharides. Since the pioneering works of Simons’ group using laser spectroscopy,7 the main advances have come from the high resolving power of microwave spectroscopy.8–15
On the other hand, the large size of the aggregates between monosaccharides and other molecules usually makes the use of other techniques to extract structural information necessary. Two techniques are commonly used: NMR and mass-resolved spectroscopy in jets. While the former gives important information regarding the behaviour of the saccharides in solution,16 the latter enables the characterization of the most stable structures in the absence of external perturbations.17 Yet, such studies are not easy: first, the saccharides usually do not contain a chromophore and therefore, it is necessary to modify the molecule to include an aromatic substituent. This is usually done in the anomeric carbon, to avoid spontaneous α/β isomerization. Certainly, monosaccharides present a linear form, especially in solution, which allow them to interconvert between anomers. Second, sugars cannot be transferred to the gas phase by simple warming. They usually require a desorption system, which introduces additional noise and limits the maximum signal intensity achievable. Signals become less stable and the spectra noisier, complicating their acquisition and interpretation. This is probably one of the reasons why not many groups have engaged in the study of the structure of sugar aggregates.
Our group has contributed to this field with the study of the aggregation preferences of glucose (Glc), galactose (Gal) and several derivatives with a collection of molecules: from sugar derivatives to drugs, using several mass-resolved laser spectroscopic techniques.5,6,18–20
Here, we extend those previous studies to the aggregation preferences of Gal derivatives with phenol (PhOH, Scheme 1) and compare the results obtained with those found for the aggregation of Glc derivatives with PhOH. As can be seen, Glc and Gal only differ in the position of the hydroxyl group attached to C4. Such a small structural difference produces a re-accommodation of the rest of OH groups in the molecule and a change in the orientation of the intramolecular hydrogen bond network, also modifying the way in which the sugar unit interacts with other molecules.
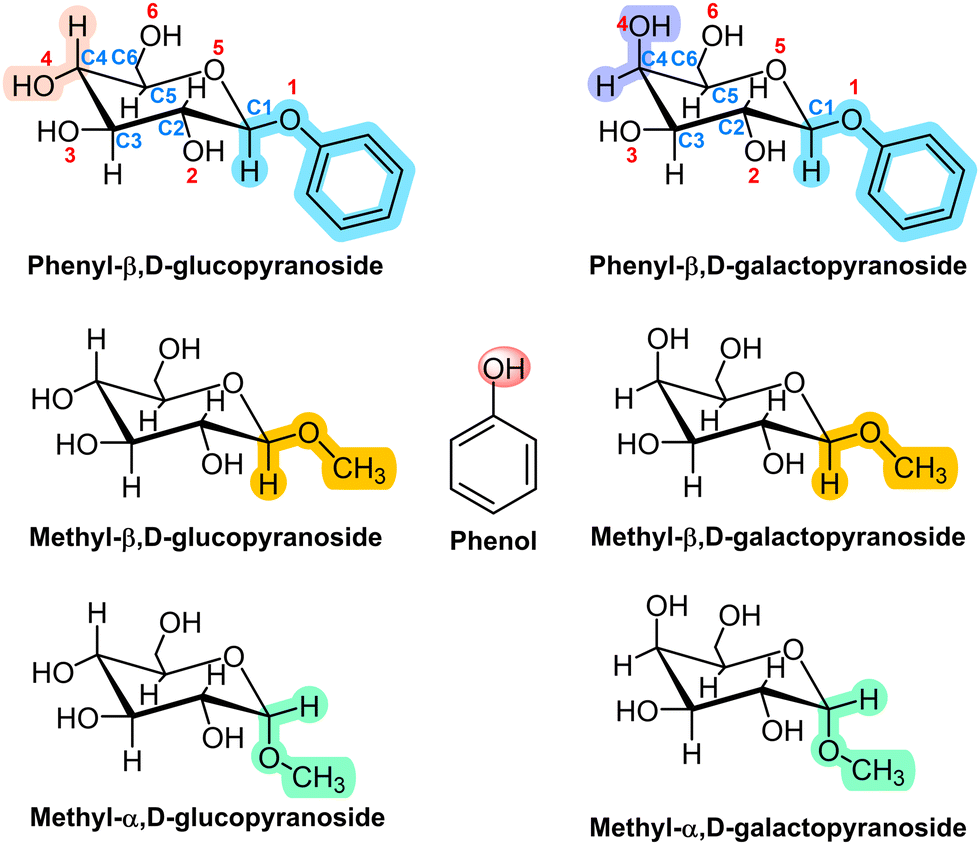 | ||
| Scheme 1 Structures of phenol, phenyl-β,D-glucopyranose, methyl-β,D-glucopyranose, methyl-α,D-glucopyranose and their galactose analogues. | ||
Two derivatives of each monosaccharide were included in this study: with an –OMe and with an –OPhe group in the anomeric carbon, either in the α or in the β configuration. The comparison between the structures of the aggregates with each of the two substituents allowed us to evaluate the tendency of the sugar towards stacking, a type of interaction necessary to understand the aggregation of nucleobases. On the other hand, past studies demonstrated that the subtle structural variation introduced by the α/β conformation of the anomeric substituent resulted in very different aggregation structures and interaction energy values.6 Therefore, we also analyse here the anomeric effect on the structure of the aggregates.
Methods
Experimental
The experimental set up has been extensively described in previous publications and therefore only the most relevant aspects will be offered here. The system is built around a linear time of flight mass spectrometer that acts as a mass-discriminant detector. The sample, consisting of a mixture of sugar derivatives, phenol and carbon nanotubes (MWCNT, Cheaptubes Inc.), was attached to a cylindrical sample holder as described in Usabiaga et al.,19 and placed on one side at the exit of a pulsed valve (General Valve Series 9). In this way, the sample desorbed by the IR photons from the ablation laser (Nd/YAG Quantel Brilliant B, 1064 nm 0.5–1 mJ per pulse focused on the sample) was picked by the expanding gas (Ar 12 bar, 99.999% purity, Praxair) at each valve opening, and cooled by the subsequent expansion. Under such conditions, aggregates formed and travelled as a dense molecular beam that was interrogated using a combination of pulsed ns lasers (UV: Quantel Q-Scan, Coumarin 540 A, 500 μJ per pulse and IR: LaserVision OPO/A, ∼7 mJ per pulse). One-color REMPI spectroscopy was used to obtain signal from each molecular aggregate, while IR/UV double resonance allowed us to extract conformer-selective structural information to compare with the computational predictions and offer a sound assignment for each species detected. Purity of the compounds studied: phenyl-β,D-glucopyranoside (β-PhGlc), 97%; phenyl-β,D-galactopyranoside (β-PhGal): 98%; methyl-β,D-glucopyranoside (β-MeGlc): ≥ 99%; methyl-β,D-galactopyranoside (β-MeGal): ≥ 98%; methyl-α,D-glucopyranoside (α-MeGlc): ≥ 99%; and methyl-α,D-galactopyranoside (α-MeGal): ≥ 99%. All of them were purchased from Sigma-Aldrich (Madrid, Spain).Computations
The computational procedure has been detailed in previous publications.21 Briefly, it was divided into three stages. First, several force fields (MMFFs,22 AMBER23 and OPLS3e24) were used to explore the conformational landscape of each dimer. The (usually) thousands of structures generated this way were grouped into families with a similar balance of interactions and which very likely present shallow barriers for isomerization. The lowest energy member of each family, together with some other selected structures were subjected to full optimization using Density Functional Theory (DFT) with M06-2X and B3LYP-D3 functionals and 6-311++G(d,p) basis functions: M06-2X/6-311++G(d,p) and B3LYP-D3/6-311++G(d,p).Once the final set of structures was obtained, their IR spectra were simulated using the normal modes generated in the corresponding calculation and an algorithm that takes into account the anharmonicity by introducing a parameter (0.953 for CHs and 0.9385 for OHs for the calculations at the M06-2X/6-311++G(d,p) level and 0.968 for CHs and 0.9535 for OHs for the calculations at the B3LYP-D3/6-311++G(d,p) level respectively). The broadening in the OH stretching transitions introduced by the formation of the hydrogen bonds was also included using a polynomial expression:
| Δν = 1 + (80 (3600 − XFreq)/(3600–3200)) |
Finally, the laser bandwidth was also introduced by convolving the spectrum (Lorentzian function for each frequency) with a Gaussian function of 5 cm−1 FWHM.
Results
Electronic excitation spectroscopy of the aggregates
Fig. 1 summarizes the 1-color REMPI spectra of the species studied in this work. The spectra of the monomers are well-resolved absorptions that contain contributions from several conformers. The spectrum of phenol, for example, has already been reported by several authors25–29 and is well-resolved, presenting some vibronic activity. The spectrum presented in Fig. 1 was recorded using the ablation system and therefore is not as cold as those recorded using other sources, but still, the 000 transition appears at 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 349 cm−1, in good agreement with published data.26–32
349 cm−1, in good agreement with published data.26–32
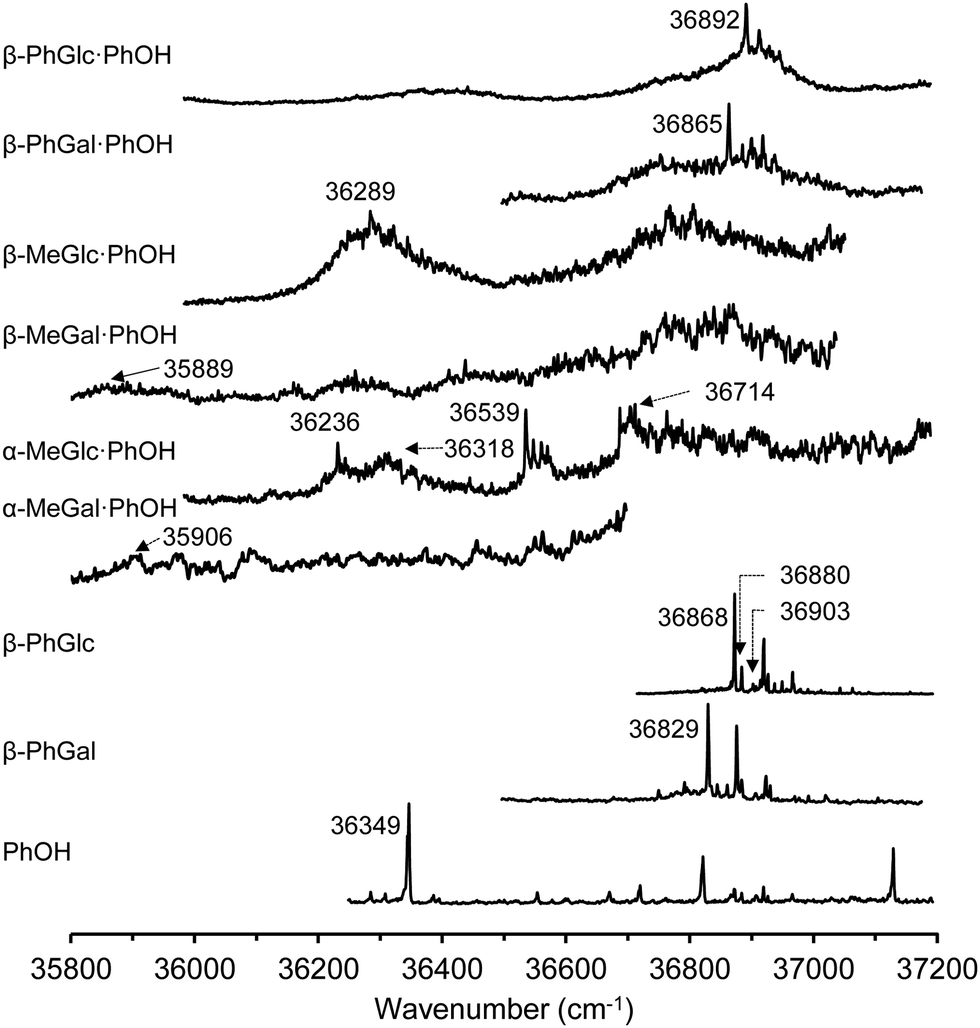 | ||
| Fig. 1 1-color REMPI spectra of phenol, β-PhGlc, β-PhGal, α-MeGal⋯PhOH, β-MeGlc⋯PhOH, β-MeGal⋯PhOH, β-MeGlc⋯PhOH, β-PhGal⋯PhOH and β-PhGlc⋯PhOH. 000 transitions and those wavenumbers used to record the IR/UV traces are indicated. | ||
Very close in energy but slightly to higher wavenumbers, the spectrum of PhGlc and PhGal appear. They are very similar, with well-defined transitions that hide the contribution from several conformational isomers.
The conformational variability of hexoses arises from the flexibility of the hydroxymethyl group and the orientation of the intramolecular hydrogen bond network: either clockwise or anticlockwise. Previous studies using mass-resolved excitation spectroscopy (MRES) already identified three isomers of PhGlc, differing in the relative orientation of the hydroxymethyl group,33 and another three isomers of PhGal, although in the latter, one of the isomers is based in a different orientation of the hydrogen bond (hbond) ribbon.34
In stark contrast to the spectra of the monomers, those of the dimers are unstructured absorptions, with a very limited number of discrete features. The broadening seems to be connected to the nature of the aggregation partner. Previous reports on the MRES of sugar units and on their aggregates demonstrated that even disaccharides tagged with a chromophore present discrete spectra.3 A background absorption starts appearing in the electronic spectra of the monohydrates35 and becomes more evident in the dihydrates36,37 to become unstructured absorptions in the dimers with larger molecules.38 However, the loss of vibronic structure does not hamper recording a clean and discrete mass-resolved IR spectrum, as demonstrated previously.38
Mass-resolved IR spectroscopy
Fig. 2 shows a comparison between the mass-resolved IR spectra of the dimers studied in this work and the predictions built using the normal mode analysis at the M06-2x/6-311++G(d,p) level. The rest of calculations may be found in the ESI.† The comparison in the figure shows an excellent agreement between simulations and experimental data. Such an agreement is magnified by the comparison with the predictions at the B3LYP-D3/6-311++G(d,p) level (ESI†). Although computations at the latter level also reproduce the general shape of the spectra, there is a larger discrepancy in the position of the bands, especially at the high wavenumber-end of the spectra, where the vibrations are less anharmonic and therefore, they are supposed to be better reproduced by the calculations.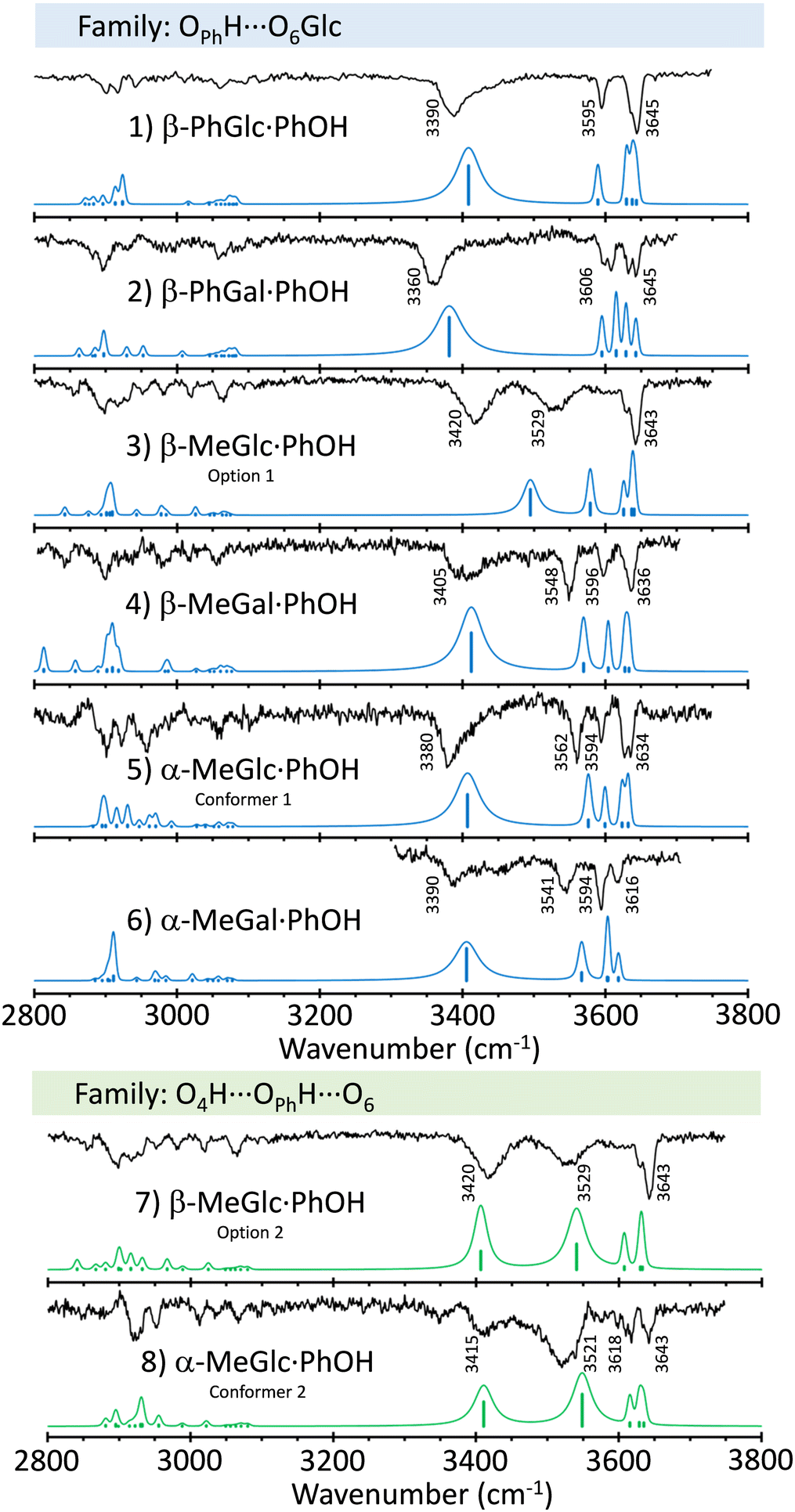 | ||
| Fig. 2 Mass-resolved IR spectra of PhOH aggregates with β-PhGlc, β-PhGal, β-MeGlc, β-MeGal, α-MeGlc and α-MeGal, together with the simulated spectra of the isomers to which they were assigned (in blue), built using the normal mode analysis computed at the M06-2x/6-311++G(d,p) level, and the procedure described in the Methods section. | ||
Slightly larger differences between experiment and predictions were found for β-MeGlc⋯PhOH, for which the calculations predict a smaller shift for the OH stretching of the phenolic hydroxyl group. Apparently, the experimental structure presents a stronger hbond than predicted. The simulation for the second most stable isomer (option 2 in the figure) better reproduces the position of the OPhH stretch (Fig. S5, ESI†), but at the expense of a worse agreement with the bands around 3600 cm−1. Those bands are due to the stretching of the OH groups involved in mild interactions and therefore, present the lowest anharmonicity. In consequence, they should be described more accurately by the computational methods used. In conclusion, we prefer to maintain the assignment to the most stable structure.
Interestingly, a single isomer was found for each aggregate, except for α-MeGlc⋯PhOH, for which the experimental trace recorded probing different wavenumbers point to the existence of at least two isomers. Determining the exact number of isomers for these systems is not an easy task, as the employment of double resonance techniques such as UV/UV hole burning is not possible, due to the unresolved nature of the electronic spectra. However, the IR spectra were recorded at several wavelengths, always obtaining the same results, except for the above-mentioned case of α-MeGlc⋯PhOH.
Assigned structures
A summary of the proposed assignments for the experimental spectra may be found in Fig. 3, while the complete set of computed structures is collected in the ESI.†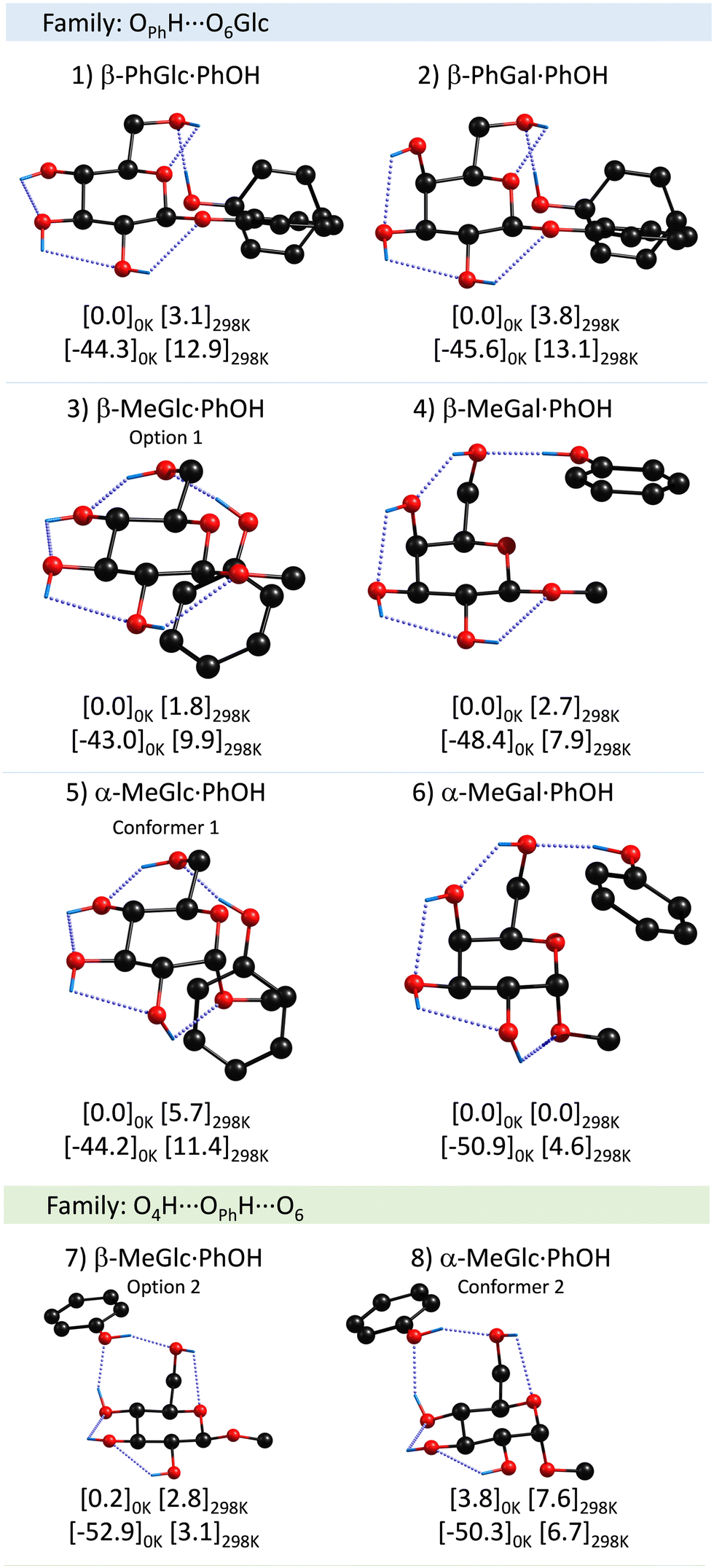 | ||
| Fig. 3 Assigned structures of PhOH aggregates with β-PhGlc, β-PhGal, β-MeGlc, β-MeGal, α-MeGlc and α-MeGal, computed at the M06-2x/6-311++G(d,p) level. Numbers in brackets are the relative energy and the binding energy in kJ mol−1 calculated at the specified temperatures. The rest of the computed structures may be found in the ESI.† | ||
There is a clear preference for phenol to act as a proton donor to the sugar unit, independently of the anomer and/or the anomeric substituent. Also interesting is the strong propensity for the interaction of PhOH with the hydroxymethyl group. Although the calculations demonstrate that several conformers with very similar stability are possible, it seems that the molecules always find a low-energy-barrier path to the global minimum, except for α-MeGlc⋯PhOH.
Interestingly, the axial/equatorial position of the hydroxyl moiety in C4 also modifies the attack angle of the phenol, through the alteration of the position of the hydroxymethyl group. Such modification is amplified and transmitted to the substituent, mainly because, in all the systems studied, there is a clear preference for maximizing the OPhH⋯O interaction while the CH⋯π contacts appear as secondary interactions. This is clearly seen comparing β-MeGlc and β-MeGal aggregates: in both cases PhOH “attacks” one of the lone pairs of the O6H. However, the latter is below the plane of the sugar ring in β-MeGlc to interact with O4H and above the plane in β-MeGal. This difference is mainly a rotation of the C4C5C6O6 dihedral angle, from −71 to 71 degrees, and results in a completely different orientation of the PhOH, which in the former interacts with the CringH atoms and with the CβH3 in the latter. A similar shift in the position of the PhOH was observed for α-MeGlc/Gal.
Discussion
In general, the assignment in all dimers studied here is very similar and corresponds to the same molecular family, in which phenol acts as a proton-donor to the O6 of sugar (the family in blue). A second family of structures, marked in green in Fig. 2 and 3, in which O4H from the sugar forms a secondary hydrogen bond with the phenolic oxygen, seems to be present at least for one of the dimers and it is in general very close in stability to the global minimum. This secondary family has the peculiarity that the binding energy is maximized, at the expense of a reduction on the stability of the sugar unit.Influence of the anomeric conformation and substituent on the aggregation process
Apart from the difference in the position of the substituent in C4, the monomers studied present two variations: the α/β anomeric conformation and the substituent at that position: either a MeO– or a PhO– group. In principle, one would expect the phenyl-substituted monosaccharides to give rise to stronger binding energies, as it gives the molecules the chance to establish a collection of additional interactions (ππ stacking or CH–π). One would expect those systems with the PhO– substituent to present a stronger binding energy, simply because the PhO– group is larger and therefore, more polarizable. Certainly, there is a substantial difference in the position of PhOH respect to the sugar unit, depending on the anomeric substituent. However, the calculations predict similar binding energy values for both sets of species, or even lower for the phenolic-substituted ones (Table 1).| Dimer | Binding energy 0 K (kJ mol−1) | σ(PhOH)/cm−1 | σ(OH)sugar/cm−1 |
|---|---|---|---|
| β-PhGlc⋯PhOH | −44.3 | 3390 | 3595, 3645 |
| β-PhGal⋯PhOH | −45.6 | 3360 | 3600, 3606, 3632, 3645 |
| β-MeGlc⋯PhOH | −43.0 | 3420 | 3529, 3642 |
| β-MeGal⋯PhOH | −48.4 | 3405 | 3548, 3596, 3636 |
| α-MeGlc⋯PhOH I | −44.2 | 3380 | 3562, 3594, 3634 |
| α-MeGlc⋯PhOH II | −50.3 | 3415 | 3521, 3618, 3643 |
| α-MeGal⋯PhOH | −50.9 | 3390 | 3541, 3594, 3616 |
The anomeric conformation does not seem to have a strong influence on the binding energy. The largest changes in these values seem to be related to the conformation of the substituent in C4, at least for the methyl-substituted species. It is clear from the table that the α-/β-MeGal species present ∼10% higher binding energy values than their homologous Glc species.
The shift in the position of the OH stretching band is usually a good indicator of the strength of a hydrogen bond. As can be seen in Fig. 4, two different correlations may be established between these two parameters: one for α-/β-MeGal plus β-MeGlc and a second linear correlation with a smaller slope for the rest of the species plus β-MeGlc. The second isomer assigned for α-MeGlc does not fit well in this general picture, although it is closer to the values for α-/β-MeGal. The steeper slope of the red fit, means that the increase in binding energy produced a smaller shift in the position of the stretching of the phenolic OH, indicating that the extra energy may come from other interactions instead of the reinforcement of the intermolecular hydrogen bond. Certainly, when the strength of the interactions is computed using the bond critical points, all the values fit to a single straight line (Fig. 5).
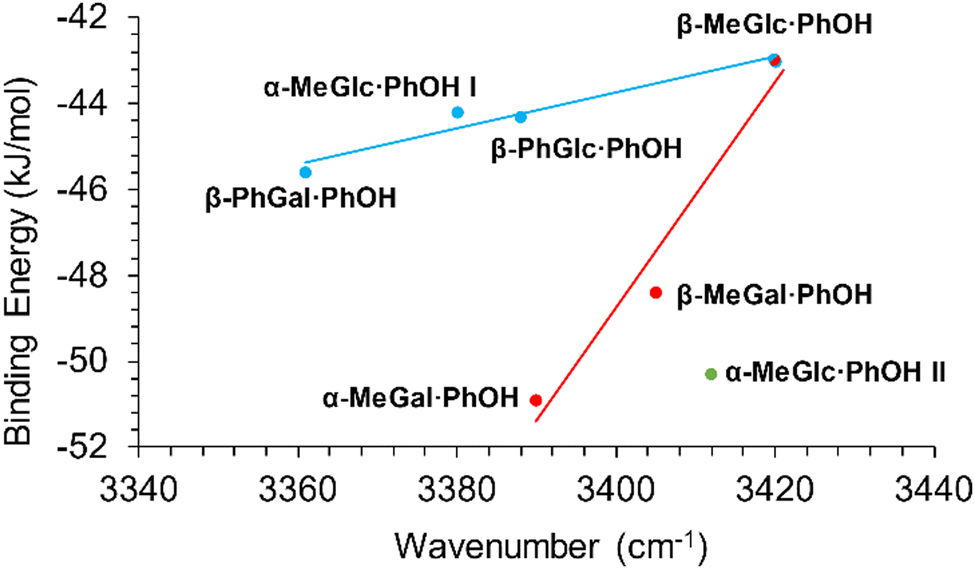 | ||
| Fig. 4 Binding energy of the dimers studied in this work vs. position of the stretching of the phenolic OH. | ||
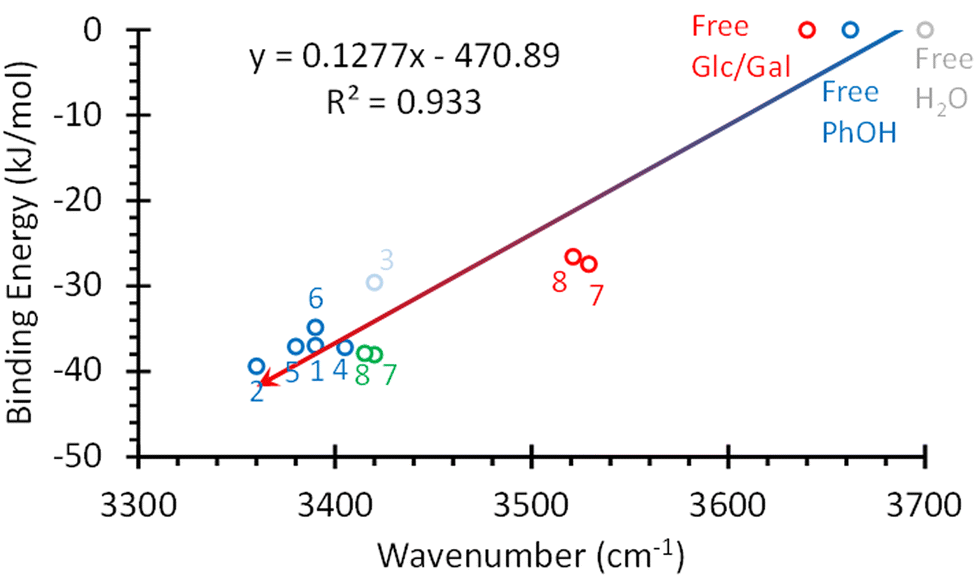 | ||
| Fig. 5 Strength of the hbonds in the systems studied in this work vs. shift in the position of the OH stretching. The hbond strength was estimated using the bond critical points. The numbers correspond to the structures in Fig. 3. The values calculated for the O4H⋯OPhH interaction in structures 7 and 8 were added in red. The red circle labelled as “Free Glc/Gal” corresponds to the values for the OH groups not directly bonded to PhOH in Glc and Gal. Values for PhOH and water OH stretching were also added for comparison. | ||
Comparison with similar systems: the influence of the stacking interaction
Most studies have centred on tackling the structure of sugar–water or sugar–aromatic aggregates. The reason behind the former studies is clear: water is the main constituent of the biological environment. On the other hand, sugar–aromatic interactions are of high relevance in biological environments: most of the sugar–protein interactions involve contacts with aromatic amino acids.39,40Comparison of the results obtained in the present work with those from the monohydrates may help us understand the influence of the interactions due to dispersive forces in the final structure of the aggregates. However, one of the problems encountered for such comparative study is that some of the published works are already close to 20 years old and ask for a re-investigation, especially due to the computational levels used to interpret the experimental results, which were not as accurate as the state-of-the-art functional. Certainly, the carbohydrate–aromatic interaction is considered a test for DFT calculations, because of the delicate balance between the interactions involving OH and CH groups.41
Nevertheless, there is a general agreement in that water inserts in the weakest intramolecular hbond of the sugar.42 In Glc and mannose, water tends to interact with O4H as a proton-acceptor and, at the same time, to donate to O6, forming an eight-membered ring.33,35,43,44 Additional studies on Glc-water found a secondary isomer with the water molecule trapped between the O6H and the ring's oxygen atom,34 which is precisely the preferred conformation of Gal monohydrates.36 Thus, in the case of sugar–water interaction, the preferred solvation site changes with the monosaccharide, while in the case of the dimers with phenol, the global minimum always has the phenol molecule acting as proton-donor to the suggar's O6H. This is true at least for the two monosaccharides studied in this work. This may be the result of several factors: phenol is a better proton-donor than water, and therefore, it may result more efficient for the system to place the phenol molecule at the tail of the hydrogen-bond network. It is also a poorer hydrogen bond acceptor, and therefore, it may not be able to insert between the O4H and O6, as water does in its interaction with Glc. Furthermore, adopting the position shown in Fig. 3, it may be able to maximize other secondary interactions.
To evaluate the relative weight of each of these factors it would be interesting to extend the studies on phenol–sugar dimers to other monosaccharides, in which water forms what has been termed as “insertion structures”: isomers in which water inserts between O3H and O4H or between O2H and O3H, breaking an intramolecular hydrogen bond.43,45–48
Regarding the sugar–aromatic interaction, several studies demonstrated that the optimal interaction takes place with the hydrogen atoms of C3, 4 and 5.41,49–54 These interactions are maximized in those monosaccharides in which all the OHs are on one side of the plane of the ring, such as in fucose.53 Interestingly, a computational study on fucose–phenol dimer using CCSD(T) and extrapolating to the basis set limit, predicted a structure for the global minimum substantially different from those in Fig. 3, with the phenol forming part of an O5H⋯OPhOHH⋯Oring hydrogen bond network and the aromatic ring pointing away from the sugar. Conversely, in all the systems studied here, phenol acts as a proton-donor to the hydroxymethyl group. Assuming that the authors did not overlook any isomer, an always present risk in this kind of complex system with a multitude of local minima, the reason for this difference with the dimers studied in this work may be the absence of a hydroxymethyl group in fucose. This functional group may completely modify the conformational landscape. Certainly, the global minimum of fucose–water presents an insertion structure, as mentioned above,46 differing from the structures found for Glc/Gal-water.33,37
Comparison between the structures in Fig. 3 and those found for monosaccharide–aromatic (aromatic = benzene, toluene or indole) aggregates40,41,49–54 shows that the hydroxyl group is a game changer. While in the former systems the aromatic always lies parallel to the saccharide ring, phenol–sugar interaction is guided by the optimization of the hydrogen bond. Only if the orientation of the O6H allows it, phenol can adjust the position of its aromatic ring to also maximize the interaction with the aliphatic hydrogens of the monosaccharide, such as in α-/β-Glc⋯PhOH. It is also worth mentioning that the interaction with phenol fixes the orientation of the hydroxymethyl group in a single position, collapsing the collection of rotamers observed for the monosaccharides into a single species. This is also in line with the observation of several authors, which connect interaction with the solvent with a simplification of the conformational landscape of the sugar units.36
Conclusions
The dimers of α-/β-MeGlc, α-/β-PhGlc, α-/β-MeGal and α-/β-PhGal with phenol have been characterized by a combination of laser spectroscopy in jets and DFT calculations. In all cases, the preferred interaction site is the hydroxymethyl group, with phenol acting as the proton donor. This is in stark contrast with the structure of monosaccharide-benzene/toluene dimers, which are mainly formed by CH⋯π interactions. The reason for such a different behaviour is the modulation introduced by the phenolic hydroxyl group: the formation of an intermolecular hydrogen bond seems to guide the whole aggregation process, imposing restrictions in the conformational landscape.The other determinant factor of the structure adopted in the aggregate is the position of the O4H, axial in galactose and equatorial in glucose. Such small difference propagates through the intermolecular hydrogen bond network and determines the orientation of the O6H group, to which phenol is anchored, strongly influencing its final position. This observation is in agreement with previous studies that attributed the intramolecular hydrogen bond network to a kind of amplification effect that facilitates the “reading” of the small structural differences between sugars by the receptor, improving the sensitivity and specificity of the sugar–receptor interaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants PGC2018-098561 and PID2021-127918NB-I00 funded by MCIN/AEI/10.13039/501100011033 and, by “ERDF A way of making Europe”. Grant IT1491-22 funded by the Basque Government. F. T. H. acknowledges financial support from Spanish Ministry of Science and Innovation under the FPI predoctoral program. A. C. would like to thank the Basque Government for a predoctoral fellowship. We also thank the SGIKER (UPV/EHU, MICIU-FEDER) for the computational and laser resources.Notes and references
- A. Varki, Glycobiology, 1993, 3, 97–130, DOI:10.1093/glycob/3.2.97.
- A. Lehninger, D. Nelson and M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, 2008 Search PubMed.
- P. Çarçabal, I. Hünig, D. P. Gamblin, B. Liu, R. A. Jockusch, R. T. Kroemer, L. C. Snoek, A. J. Fairbanks, B. G. Davis and J. P. Simons, J. Am. Chem. Soc., 2006, 128, 1976–1981, DOI:10.1021/ja055891v.
- D. Solís, N. V. Bovin, A. P. Davis, J. Jiménez-Barbero, A. Romero, R. Roy, K. Smetana Jr. and H. Gabius, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 186–235, DOI:10.1016/j.bbagen.2014.03.016.
- I. Usabiaga, J. González, I. León, P. F. Arnaiz, E. J. Cocinero and J. A. Fernández, J. Phys. Chem. Lett., 2017, 8, 1147–1151, DOI:10.1021/acs.jpclett.7b00151.
- I. Usabiaga, A. Camiruaga, A. Insausti, P. Çarçabal, E. J. Cocinero, I. León and J. A. Fernández, Front. Phys., 2018, 6 DOI:10.3389/fphy.2018.00003.
- J. P. Simons, Mol. Phys., 2009, 107, 2435–2458, DOI:10.1080/00268970903409812.
- E. R. Alonso, I. Peña, C. Cabezas and J. L. Alonso, J. Phys. Chem. Lett., 2016, 7, 845–850, DOI:10.1021/acs.jpclett.6b00028.
- I. Peña, L. Kolesnikova, C. Cabezas, C. Bermúdez, M. Berdakin, A. Simao and J. L. Alonso, Phys. Chem. Chem. Phys., 2014, 16, 23244–23250, 10.1039/c4cp03593c.
- C. Bermúdez, I. Peña, S. Mata and J. L. Alonso, Chem. – Eur. J., 2016, 22, 16829–16837, DOI:10.1002/chem.201603223.
- I. Peña, C. Cabezas and J. L. Alonso, Chem. Commun., 2015, 51, 10115–10118, 10.1039/C5CC01783A.
- J. L. Alonso, M. A. Lozoya, I. Pena, J. C. Lopez, C. Cabezas, S. Mata and S. Blanco, Chem. Sci., 2014, 5, 515–522, 10.1039/C3SC52559G.
- C. S. Barry, E. J. Cocinero, P. Çarçabal, D. P. Gamblin, E. Stanca-Kaposta, S. M. Remmert, M. C. Fernández-Alonso, S. Rudic, J. P. Simons and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 16895–16903, DOI:10.1021/ja4056678.
- E. J. Cocinero, A. Lesarri, P. Écija, A. Cimas, B. G. Davis, F. J. Basterretxea, J. A. Fernández and F. Castaño, J. Am. Chem. Soc., 2013, 135, 2845–2852, DOI:10.1021/ja312393m.
- E. J. Cocinero, A. Lesarri, P. Écija, F. J. Basterretxea, J. Grabow, J. A. Fernández and F. Castaño, Angew. Chem., Int. Ed., 2012, 51, 3119–3124, DOI:10.1002/anie.201107973.
- M. Fernández-Alonso del Carmen, D. Díaz, M. Á. Berbis, F. Marcelo, J. Cañada and J. Jiménez-Barbero, Curr. Protein Peptide Sci., 2012, 13, 816–830, DOI:10.2174/138920312804871175.
- A. M. Rijs and J. Oomens, Gas-Phase IR Spectroscopy and Structure of Biological Molecules, Springer International Publishing, Heidelberg, New York, Dordrecht, London, 2015 Search PubMed.
- A. Camiruaga, I. Usabiaga, A. Insausti, E. J. Cocinero, I. León and J. A. Fernández, Mol. BioSyst., 2017, 13, 1709–1712, 10.1039/C7MB00293A.
- I. Usabiaga, J. Gonzalez, P. F. Arnaiz, I. León, E. J. Cocinero and J. A. Fernández, Phys. Chem. Chem. Phys., 2016, 18, 12457–12465, 10.1039/C6CP00560H.
- A. Camiruaga, I. Usabiaga, A. Insausti, I. León and J. A. Fernández, Phys. Chem. Chem. Phys., 2017, 19, 12013–12021, 10.1039/C7CP00615B.
- I. Usabiaga, A. Camiruaga, C. Calabrese, A. Maris and J. A. Fernández, Chem. – Eur. J., 2019, 25, 14230–14236, DOI:10.1002/chem.201903478.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 730–748 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688, DOI:10.1002/jcc.20290.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2019, 15, 1863–1874, DOI:10.1021/acs.jctc.8b01026.
- H. Watanabe and S. Iwata, J. Chem. Phys., 1996, 105, 420–431 CrossRef CAS.
- A. Oikawa, H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1983, 87, 5083–5090, DOI:10.1021/j150643a009.
- K. Fuke and K. Kaya, Chem. Phys. Lett., 1983, 94, 97–101 CrossRef CAS.
- R. J. Stanley and A. W. Castleman, J. Chem. Phys., 1991, 94, 7744–7756 CrossRef CAS.
- D. Solgadi, C. Jouvet and A. Tramer, J. Phys. Chem., 1988, 92, 3313–3315, DOI:10.1021/j100323a001.
- H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1982, 86, 1768–1771, DOI:10.1021/j100207a007.
- N. Gonohe, H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1985, 89, 3642–3648, DOI:10.1021/j100263a015.
- G. Reiser, O. Dopfer, R. Lindner, G. Henri, K. Mullerdethlefs, E. W. Schlag and S. D. Colson, Chem. Phys. Lett., 1991, 181, 1–4 CrossRef CAS.
- P. Çarçabal, R. A. Jockusch, I. Hünig, L. C. Snoek, R. T. Kroemer, B. G. Davis, D. P. Gamblin, I. Compagnon, J. Oomens and J. P. Simons, J. Am. Chem. Soc., 2005, 127, 11414–11425, DOI:10.1021/ja0518575.
- J. P. Simons, R. A. Jockusch, P. ÇarÇabal, I. Hünig, R. T. Kroemer, N. A. Macleod and L. C. Snoek, Int. Rev. Phys. Chem., 2005, 24, 489–531, DOI:10.1080/01442350500415107.
- R. A. Jockusch, R. T. Kroemer, F. O. Talbot and J. P. Simons, J. Phys. Chem. A, 2003, 107, 10725–10732, DOI:10.1021/jp0351730.
- J. P. Simons, B. G. Davis, E. J. Cocinero, D. P. Gamblin and E. C. Stanca-Kaposta, Tetrahedron: Asymmetry, 2009, 20, 718–722 CrossRef CAS.
- E. J. Cocinero, E. C. Stanca-Kaposta, E. M. Scanlan, D. P. Gamblin, B. G. Davis and J. P. Simons, Chem. – Eur. J., 2008, 14, 8947–8955 CrossRef CAS PubMed.
- E. J. Cocinero, P. Çarçabal, T. D. Vaden, B. G. Davis and J. P. Simons, J. Am. Chem. Soc., 2011, 133, 4548–4557 CrossRef CAS PubMed.
- M. D. Díaz, M. Fernández-Alonso del Carmen, G. Cuevas, F. J. Cañada and J. Jiménez-Barbero, Pure Appl. Chem., 2008, 80(8), 1827–1835, DOI:10.1351/pac200880081827.
- M. Kumari, P. V. Balaji and R. B. Sunoj, Phys. Chem. Chem. Phys., 2011, 13, 6517–6530, 10.1039/C0CP02559C.
- R. K. Raju, A. Ramraj, I. H. Hillier, M. A. Vincent and N. A. Burton, Phys. Chem. Chem. Phys., 2009, 11, 3411–3416, 10.1039/B822877A.
- P. Çarçabal, E. J. Cocinero and J. P. Simons, Chem. Sci., 2013, 4, 1830–1836, 10.1039/C3SC50135C.
- N. Mayorkas, S. Rudić, E. J. Cocinero, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2011, 13, 18671–18678, 10.1039/C1CP22348H.
- N. A. Macleod, C. Johannessen, L. Hecht, L. D. Barron and J. P. Simons, Int. J. Mass Spectrom., 2006, 253, 193–200, DOI:10.1016/j.ijms.2006.01.031.
- I. Hünig, A. J. Painter, R. A. Jockusch, P. Çarçabal, E. M. Marzluff, L. C. Snoek, D. P. Gamblin, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2005, 7, 2474–2480, 10.1039/B504230E.
- L. Jin, J. P. Simons and R. B. Gerber, J. Phys. Chem. A, 2012, 116, 11088–11094, DOI:10.1021/jp303080k.
- P. Çarçabal, T. Patsias, I. Hünig, B. Liu, C. Kaposta, L. C. Snoek, D. P. Gamblin, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2006, 8, 129–136, 10.1039/B514301B.
- L. Jin, J. P. Simons and R. B. Gerber, Chem. Phys. Lett., 2011, 518, 49–54, DOI:10.1016/j.cplett.2011.11.008.
- Z. Su, E. J. Cocinero, E. C. Stanca-Kaposta, B. G. Davis and J. P. Simons, Chem. Phys. Lett., 2009, 471, 17–21 CrossRef CAS.
- E. Stanca-Kaposta, P. Çarçabal, E. J. Cocinero, P. Hurtado and J. P. Simons, J. Phys. Chem. B, 2013, 117, 8135–8142, DOI:10.1021/jp404527s.
- E. Cristina Stanca-Kaposta, D. P. Gamblin, J. Screen, B. Liu, L. C. Snoek, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2007, 9, 4444–4451, 10.1039/B704792D.
- R. K. Raju, A. Ramraj, M. A. Vincent, I. H. Hillier and N. A. Burton, Phys. Chem. Chem. Phys., 2008, 10, 6500–6508, 10.1039/B809164A.
- S. Tsuzuki, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2011, 115, 11256–11262, DOI:10.1021/jp2045756.
- S. Kozmon, R. Matuška, V. Spiwok and J. Koča, Chem. – Eur. J., 2011, 17, 5680–5690, DOI:10.1002/chem.201002876.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional computed structures and predicted IR spectra. See DOI: https://doi.org/10.1039/d2cp06036a |
| ‡ These two authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |