
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion effect on the redox properties of copper ions in ionic liquids and deep eutectic solvents†
Evangelia
Daskalopoulou
,
Jennifer M.
Hartley
 *,
Rodolfo Marin
Rivera
,
Guillaume
Zante
and
Andrew P.
Abbott
*,
Rodolfo Marin
Rivera
,
Guillaume
Zante
and
Andrew P.
Abbott
School of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: jmh84@le.ac.uk
First published on 18th January 2023
Abstract
It has long been claimed that the anion of the DES or IL is critical for controlling the redox properties of metal ions. In this study we investigate the effect of different salt anions on the copper redox properties and speciation, and compare that with the effect of the different solvent anions, when a single copper salt is used in a range of solvents. It is shown that the effect of the solvent anion is much more significant than that of the salt anion on the redox properties. It is also found that copper species remain the same copper tetrachloride species despite the starting salt. An exception is seen for the copper(I) salt, which makes linear dichloride species, as well as the copper(II) acetate system, which displays concentration dependence. When the anion of the ionic liquid is changed, the copper species change correspondingly with the coordinating strength of the solvent anion, leading to a greater difference in redox response, which is due to the different species present. Thus, these speciation differences can be used to modify the redox potentials in the solution.
1. Introduction
Ionic liquids (ILs) and deep eutectic solvents (DESs) have often been described as designer solvents, as through judicious choice of cation and anion, they have the potential to be tailored to specific applications due to the wide variety of metal complexes that can be formed.1–7 The speciation, and hence reactivity, of these metal complexes is generally determined by the anionic component of the IL or DES,8 except when significantly stronger ligands such as cyanide are already present on the starting metal salt,9 or when the metal ion has an oxophilic character.10,11 In certain cases when the hydrogen bond donor (HBD) component of the DES is strongly coordinating, this can result in metal complexes being precipitated, such as nickel ions in the presence of oxalate, or lead ions in the presence of lactate.12 The use of ILs or DESs containing anions with greater coordination strength often results in metal complexes with higher stability constants, hence greater electrodeposition overpotentials are often required to electrowin metals from such solutions. This phenomenon is more commonly seen in aqueous plating baths, where strongly coordinating additives must be added to the solution to obtain the desired electrochemical properties. These additives include ammonia,13,14 ethylenediamine,14 chloride,15,16 citrate,17,18 glycine,19,20 and cyanide21,22 among others. One of the major benefits of using ILs or DESs instead of aqueous systems is that the desired ligands are naturally present in high concentrations as part of the solvent formulation, and do not have to compete with the 55.6 mol dm−3 water content in aqueous systems. Therefore, detrimental side reactions due to (oxy)hydroxide chemistry can be avoided.2There is inevitably a small amount of water present in ILs and DESs when exposed to atmospheric conditions because of the hygroscopic nature of these solvents. However, the presence of small amounts of water (<10 wt%) in traditional ILs and DESs have generally been found to not affect the speciation or the redox behaviour of the metal ions,23–26 instead mainly affecting the physical properties of the solvent, e.g. viscosity, conductivity, and density,27–30 along with electrochemical stability.31 Above ca. 30 to 40 wt% water content, the DES matrix breaks down to form an aqueous solution of DES components.32 Recently, systems formed from calcium chloride hexahydrate and ethylene glycol have been investigated, and remarkably, were found to have similar physicochemical properties to a DES formed from choline chloride and ethylene glycol.33,34 In each of the solvent compositions investigated, copper, cobalt, and iron chloride complexes were present, despite the significant proportion of water in these systems – ranging from 17.5 to 24 kg mol−1. This behaviour was proposed to be due to the water and ethylene glycol molecules being tightly solvated to the calcium cation.
Copper was selected as the model system to investigate as it has simple and well-defined electrochemical behaviour, and has been characterised in a wide range of IL and DES systems, with and without the presence of water.23,24,33,35–37 In this work, we aim to determine how the anion initially present on the copper salt affects copper redox behaviour and speciation in two eutectic systems made from EG:ChCl and CaCl2·6H2O:EG. Following this, the effect of solvent anion will be compared in six imidazolium-based ionic liquids, which were selected to have a range of anions of different coordination strength, and to also be suitable for use with ferricyanide as the internal standard.38 These anions were acetate (OAc−), tetrafluoroborate (BF4−), chloride (Cl−), dicyanamide (DCN−), hydrogen sulphate (HSO4−), and thiocyanate (SCN−).
2. Experimental
The solvents were prepared by the following methods. Choline chloride (ChCl) (Acros Organics, 99%) was mixed with the hydrogen bond donors (HBDs) in a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HBD:ChCl for ethylene glycol (EG) (Fisher, 98%), formic acid solution (Fisons, 90%), lactic acid solution (Fisher, 88%), and glycolic acid (Alfa Aesar, 98%), or in a molar ratio of 1:1 HBD:ChCl for glutaric acid (Aldrich, 99%), malonic acid (Alfa Aesar, 99%), and oxalic acid dihydrate (Alfa Aesar, 98%). This was followed by heating and stirring at 80 °C until a homogenous liquid was formed. The calcium chloride system was formed by mixing ethylene glycol and CaCl2·6H2O (Fluka, >97%) in a molar ratio of 1:1 CaCl2·6H2O:EG, followed by heating and stirring at 50 °C until a homogenous liquid was formed. The lower temperature was required to prevent solvent decomposition through water loss. The ionic liquids were all purchased from IoLiTec, with purities of > 98%, and used as received.
:1 HBD:ChCl for ethylene glycol (EG) (Fisher, 98%), formic acid solution (Fisons, 90%), lactic acid solution (Fisher, 88%), and glycolic acid (Alfa Aesar, 98%), or in a molar ratio of 1:1 HBD:ChCl for glutaric acid (Aldrich, 99%), malonic acid (Alfa Aesar, 99%), and oxalic acid dihydrate (Alfa Aesar, 98%). This was followed by heating and stirring at 80 °C until a homogenous liquid was formed. The calcium chloride system was formed by mixing ethylene glycol and CaCl2·6H2O (Fluka, >97%) in a molar ratio of 1:1 CaCl2·6H2O:EG, followed by heating and stirring at 50 °C until a homogenous liquid was formed. The lower temperature was required to prevent solvent decomposition through water loss. The ionic liquids were all purchased from IoLiTec, with purities of > 98%, and used as received.
Copper(II) acetate (Aldrich, 98%), copper(II) bromide (Aldrich, 99%), copper(II) chloride anhydrous (Fluorochem, 98%), copper(II) nitrate trihydrate (Sigma-Aldrich, 99%), copper(II) sulphate anhydrous (Scientific & Chemicals, 98%), and copper(I) thiocyanate (Hopkin & Williams, 98%) were dissolved in EG:ChCl or CaCl2·6H2O:EG to make solutions of 0.02 mol dm−3 concentration.
Cyclic voltammetry (CV) measurements of the copper salt-containing solutions were measured using an IVIUMnSTAT multichannel potentiostat/galvanostat, together with the corresponding Ivium software. A 3-electrode set-up was used, consisting of a 0.5 mm diameter Pt-disc working electrode, a Pt-flag counter electrode, and an aqueous 3.0 M KCl silver chloride reference electrode. The recorded data were then referenced to an internal standard of the [Fe(CN)6]3−/4− redox couple so that direct comparisons could be made between the different solvents.38 Data were measured at 20 °C, with a scan rate of 20 mV s−1. Formal electrode potentials were obtained from taking an average of oxidation and reduction onset potentials, and then applying the Nernst equation to correct for small changes in concentration and temperature.39
UV-vis spectroscopy was carried out using a Mettler Toledo UV5 Bio spectrometer using the same 0.02 mol dm−3 concentration solutions as for the voltammetry measurements. The cuvettes were 0.1 mm quartz glass slides.
3. Results and discussion
3.1 Effect of salt anion
Cyclic voltammetry of the six different copper salts dissolved in EG:ChCl (Fig. 1a) displayed very little change in redox behaviour for either the Cu2+/+ redox couple (+0.333 V ± 0.006 V vs. [Fe(CN)6]3−/4−) or for the Cu+/0 redox couple (−0.377 V ± 0.011 V vs. [Fe(CN)6]3−/4−). The electrochemical window of stability for the Cu+ species is ca. 0.7 V for all systems, indicating that the presence of these additional anions are not affecting the stability of Cu+ in solution, either by increasing or decreasing the complex stability, i.e. the Cu+ species are consistently [CuCl2]− across all systems. Small changes in the electrodeposition potential of copper are more likely related to how the salt anion impacts copper nucleation behaviour, rather than altering the complex stability of the copper species in solution. Slight changes were observed between the first scan and any subsequent scans, as would be expected after an alteration to the substrate (Fig. S2, ESI†).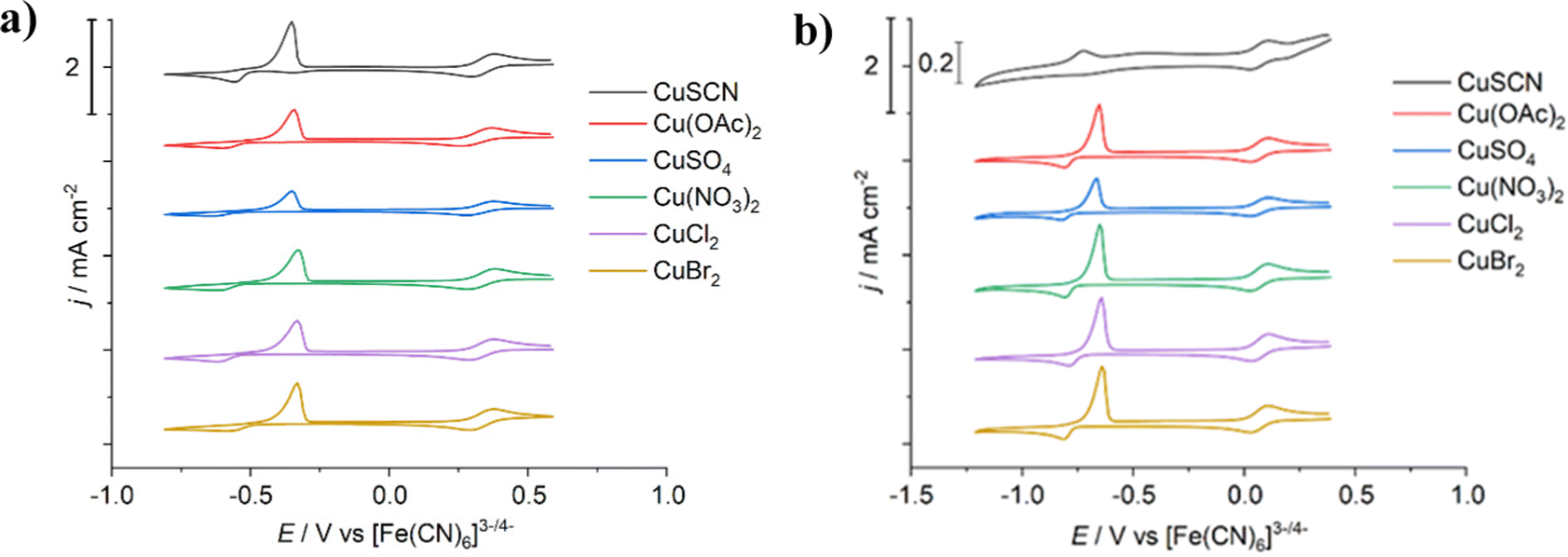 | ||
| Fig. 1 Cyclic voltammograms of the different copper salts (each 0.02 mol dm−3) in: (a) EG:ChCl, and (b) CaCl2·6H2O:EG at a 0.5 mm diameter Pt-disc, referenced to the [Fe(CN)6]3−/4− redox couple, with a scan rate of 20 mV s−1. Temperature = 20 °C. CVs offset for clarity. Presented data is the first scan. Note that CuSCN in CaCl2·6H2O:EG is on a different scale to the other systems. | ||
In the CaCl2·6H2O:EG system (Fig. 1b), the voltammetric behaviour of the copper salts is comparable to EG:ChCl. The main difference is a cathodic shift of ca. 0.3 V between the two solvents, which could either be due to different copper species being present in CaCl2·6H2O:EG due to the high water content of 21.3 mol kg−1, or more simply due to the presence of a substantial liquid junction potential. The redox potentials are Cu2+/+ = +0.069 V ± 0.001 V, and Cu+/0 = −0.630 V ± 0.025 V, both vs the [Fe(CN)6]3−/4− redox couple. Of all the copper salts, CuSCN had the greatest difference in the copper deposition potential at an apparent value of −0.571 V, which may in part be an effect of the poorer solubility of this salt (Fig. 2). This is in contrast to the redox behaviour after CuSCN was dissolved in EG:ChCl, where the redox couples have similar electrode potentials to the other salts investigated. Additionally, a significant change in current density was observed, most likely related to the poor solubility of CuSCN in CaCl2·6H2O:EG, as this solution was observed to be cloudy (Fig. S1b, ESI†). If the value obtained for CuSCN is excluded, the average Cu+/0 redox potential in CaCl2·6H2O:EG is −0.642 V ± 0.004 V.
 | ||
| Fig. 2 Redox potentials obtained from the different copper salts (each 0.02 mol dm−3) in: (a) EG:ChCl, and (b) CaCl2·6H2O:EG, presented as a function of salt anion. All potentials were corrected for small changes in concentration and temperature using the Nernst equation, and referenced to the [Fe(CN)6]3−/4− couple. | ||
UV-vis spectroscopy was carried out on 0.02 mol dm−3 solutions of the copper salts in the two solvents. Fig. 3 shows that for both EG:ChCl and CaCl2·6H2O:EG, the copper species present is [CuCl4]2−, except for the CuSCN system where [CuCl2]− is known to be present.8 This is despite the visual differences in colour between the concentrated solutions. For example, copper(II) acetate in EG:ChCl is a green colour (Fig. S1a, ESI†), which would usually indicate the presence of a mixed chloride–acetate complex, or the presence of a mixture of copper chloride and acetate species. Slight cloudiness was observed after dissolution of CuSO4 in CaCl2·6H2O:EG, and significant cloudiness was observed for the corresponding CuSCN system, either due to poor solubility of the copper salt, or due to complexation of the copper salt ligand with some component of the solvent, most likely the Ca2+ ion. As a major component of the CaCl2·6H2O:EG system is water, the solubility values of potential precipitating species in water are compared. For the CuSCN system, the most likely identity of the solid species is insoluble CuSCN, as the solubility of this species in water at room temperature is 4.1 × 10−6 mol kg−1.40 For the CuSO4 system, it is more likely that CaSO4 is present (solubility of <0.05 mol kg−1),41 rather than undissolved CuSO4 (solubility of >1 mol kg−1). While the molar extinction coefficient values for the spectra of these two systems are much smaller than for the other investigated systems, the spectra still correspond to the presence of copper chloride complexes. In the EG:ChCl system, the average absorbance maxima for the [CuCl4]2− species are 239.4(4), 291.5(2), and 406.1(4) nm, while absorbance maxima for the corresponding CaCl2·6H2O:EG systems are present as a shoulder at ca. 238 nm, along with peaks at 279(1) and 390.1(6) nm (Table S1, ESI†). This difference in absorbance maxima of 5 to 16 nm between the two solvents is indicative of a change in the solvation environment of the metal complex – in this case, the difference between a solvation environment of ethylene glycol and a solvation environment of a mixture of ethylene glycol and water. There is only minimal difference between the spectra of the different copper salts, indicating that the salt anion is not having an effect on copper speciation in solution. Therefore, any differences in redox potential must be related to anion-substrate effects rather than any changes in copper speciation. When considering the [CuCl2]− species, in EG:ChCl absorption maxima are present at 269.8 and 406.4 nm, whereas in CaCl2·6H2O:EG they are present at 263.8 and 391.4 nm. This is a solvatochromic shift of 6 to 15 nm, indicating that this species is also affected by the change in the solvation environment.
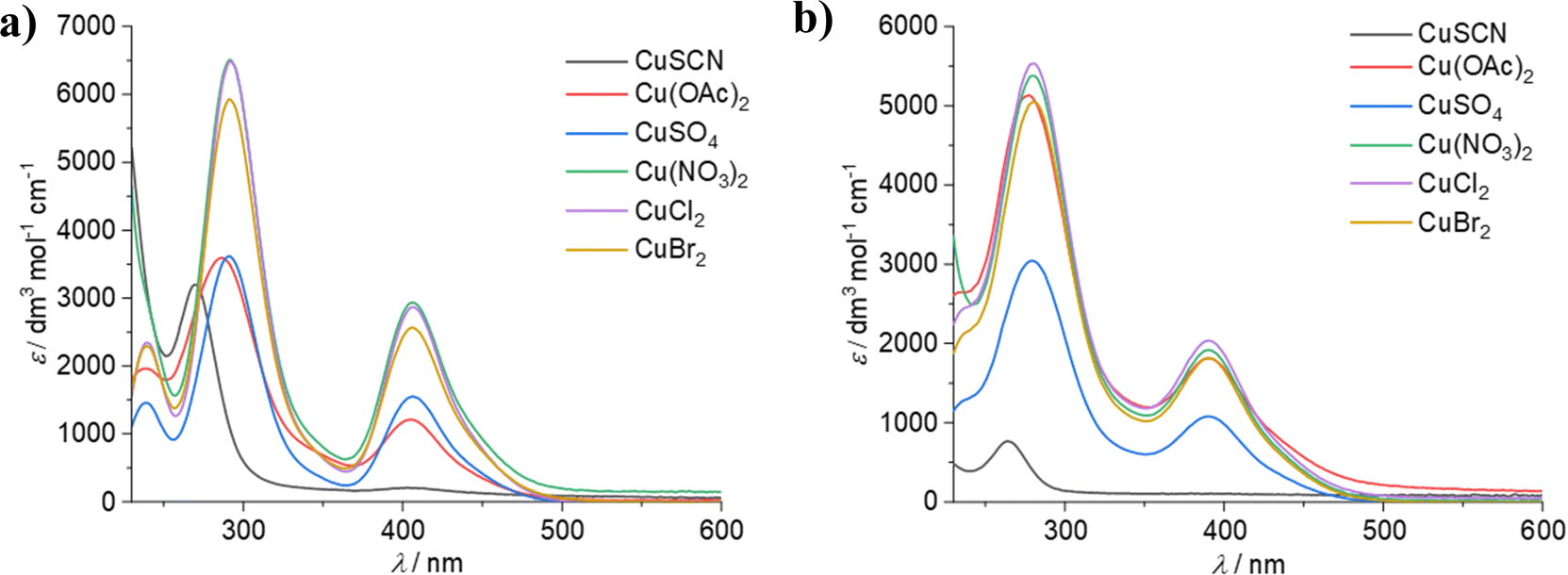 | ||
| Fig. 3 UV-vis spectra of the different copper salts in: (a) EG:ChCl, and (b) CaCl2·6H2O:EG. | ||
3.2 Effect of solvent anion
Having confirmed that salt anion generally does not affect speciation or electrochemistry of the Cu2+/+ or Cu+/0 redox couples in DES media based on EG:ChCl and CaCl2·6H2O:EG, solutions of copper(II) chloride were made up in a range of imidazolium-based ILs with anions of different coordination strength, including acetate (OAc−), tetrafluoroborate (BF4−), chloride (Cl−), dicyanamide (DCN−), hydrogen sulphate (HSO4−), and thiocyanate (SCN−). From the substantial difference in UV-vis spectra between these systems, it is clear that a wide range of species form (Fig. 4, Table S2, ESI†). Images of these solutions are presented in Fig. S4 (ESI†), where it is shown that for the case of [C2mim][BF4] there is a precipitation of copper, which indicates poor solubility of copper(II) chloride in this system. | ||
| Fig. 4 UV-vis spectra of copper(II) chloride dissolved in six different imidazolium-based ILs. The corresponding spectrum for EG:ChCl is included for comparison. | ||
In the liquids with weak anions such as tetrafluoroborate, mixed anion–chloride complexes are known to be present, whereas in the liquids with more strongly coordinating anions (acetate, chloride, dicyanamide, and thiocyanate) metal complexes with the solvent anion only form. For example, it is known that [Cu(NCS)2(SCN)]− forms in [C2mim][SCN], and that [CuCl3]− is present in [C6mim][Cl].8 In the [C4mim][DCN] system, a copper–dicyanamide complex is predicted to be present, as the UV-vis spectrum shows a characteristic peak at ca. 366 nm.36 If a mixed chloride–dicyanamide complex was present, a red-shift of this maximum would be expected, as observed for copper(II) species in tetrahexylammonium dicyanamide.42 The dicyanamide ligands are most likely coordinating through the terminal nitrogen moieties, rather than the central one,43–45 with the possibility to act as bridging ligands to form multinuclear complexes or a polymeric structure.
The spectra of CuCl2·2H2O and Cu(OAc)2·H2O dissolved in [C2mim][OAc] are almost identical, indicating that the species present are almost entirely coordinated to acetate (Fig. S3, ESI†). Absorbance maxima are present at 352 nm in both systems, but the longer wavelength absorbance relating to the d–d transition is at 667.5 nm for the chloride salt, compared to 659.5 nm for the acetate salt, meaning that there are slight solvation differences between the systems. In [C4mim][OAc] the blue copper acetate solution displays an absorbance maximum at 630 nm, whereas in a tetraalkyl phosphonium liquid, maxima are present at 370 nm and 690 nm.46 Metal acetate complexes are able to form dimers, as in the case of copper acetate dissolved in ethanol, with absorbance maxima at 250, 375 and 658 nm,47 suggesting the possibility of copper acetate dimers also being present in [C2mim][OAc], and potentially also in other ILs with the acetate anion.
Cyclic voltammograms were recorded in these imidazolium-based ILs (Fig. 5), confirming that the different copper species present have different electrochemical behaviour. In the case of [C4mim][DCN] and [C2mim][SCN], the Cu2+/+ and Cu+/0 redox couples are well-resolved, as seen in the case of EG:ChCl. There are, however, larger copper electrodeposition overpotentials in the ILs, indicating that thiocyanate and dicyanamide are more tightly complexed to the copper(I) ions than chloride. Interestingly, the relative redox potentials for the Cu2+/+ couple would tend to suggest that the copper(II) dicyanamide complex is more reactive than either the [CuCl4]2− or copper(II) thiocyanate complexes, which themselves have similar redox potentials. Supporting the claim that the anion controls speciation and electrochemical properties of the metal ion, the voltammetry of CuCl in [C2mim][DCN] on glassy carbon can be compared to [C4mim][DCN]. The Cu2+/+ and Cu+/0 redox couples are well-defined, with a copper(I) species stability window of ca. 1 V, similar to the present work.48
 | ||
| Fig. 5 Cyclic voltammograms of copper salts in EG:ChCl and six ILs at a Pt-disc working electrode, with a scan rate of 10 mV s−1. The current density was normalised to remove any current effects due to the different solvent viscosity values. CVs are referenced to the [Fe(CN)6]3−/4− redox couple. Presented data is the first scan. | ||
Physical properties of the ILs can also affect electrochemical responses of the systems, as seen for [C4mim][HSO4] and [C6mim][Cl], which are the most viscous liquids (464 and 18089 mPa s, respectively).49,50 A high solvent viscosity hinders mass transport to and from the electrode surface, resulting in the large electrodeposition overpotentials for copper relative to the other systems, along with a broadening of the anodic processes. Raising the temperature decreases solvent viscosity, and hence improves mass transport. At 102 °C, voltammetry of copper in [C6mim][Cl] shows well-resolved redox couples, again with a separation of ca. 1 V.51 Regarding the other ILs investigated in the present work, [C2mim][OAc] has a viscosity of 162 mPa s at 25 °C, while the remainder all have viscosity values in the range 24 to 37 mPa s,30,52,53 which are comparable to the EG:ChCl and CaCl2·6H2O:EG systems (37 and 40 mPa s, respectively).33,54
In the case of [C2mim][BF4] and [C2mim][OAc], complex electrochemistry is observed. The tetrafluoroborate anion is poorly coordinating, resulting in an insufficiency of suitable ligands to coordinate to the copper ions and maintain their stability and solubility in solution. This may cause copper salt precipitates at the electrode surface, which could be the cause of the complex anodic electrochemical behaviour. To counteract this issue, [C2mim][BF4] is commonly used with the addition of “free” chloride to help stabilise metal ions in solution.55,56 For [C2mim][OAc], two potential issues arise, as acetate is electroactive and there is also the possibility of copper acetate dimers being formed. This resulted in multiple reductive and oxidative processes that could not easily be assigned. In an acetic acid–methanol mixture, copper acetate dimers could be partially reduced to form purple mixed valence Cu(II)Cu(I) acetate complexes.57 Voltammetry of this system showed two reductive processes; an initial broad reduction, followed by electrodeposition of copper. On the reverse sweep, three oxidative processes were present, which were suggested to be oxidation of metallic copper to a Cu(I)Cu(I) complex, formation of the mixed valence complex, and formation of the fully oxidised Cu(II)Cu(II) acetate species. While the present investigation in [C2mim][OAc] shows a different arrangement of the redox processes, the underlying identities are likely to remain the same.
3.3 Effect of hydrogen bond donor
An alternative source of different electrochemical behaviour involves the type of HBD present in a DES system, as these can change the physical and chemical properties of the solvent, as well as being able to act as complexing agents.8,11,12,58 UV-vis spectra of copper chloride in a selection of DESs containing HBDs with different pKa values show that copper speciation is generally unaffected by the choice of HBD, except in the cases of the oxalic acid system where the precipitation of copper oxalates took place above 0.01 mol dm−3, and in the urea system where ammonia formation results in the formation of blue copper–ammonia complexes. Images of these solutions are shown in Fig. S4 (ESI†), with absorbance maxima values shown in Table S3 (ESI†). Even in a solution of fresh Urea:ChCl there is a small presence of non-chloride coordination, as seen by the difference in UV-vis spectra compared to all the other DESs (Fig. 6). The average absorbance maxima are 241(1) nm, 290(5) nm, and 405(1) nm. It should be noted that the shortest wavelength absorbance is not detectable in the most acidic DESs due to no signal below 250 nm. The greatest solvatochromic shifts were seen for the oxalic acid and urea systems, which are also the most coordinating towards both the copper ions and also to the chloride.59 Based on these spectroscopic data showing very little change in copper speciation, it would be reasonable to assume that there would also be very little change in the Cu2+/+ redox potentials in the different solvents. If speciation alone is affecting the redox potentials, the greatest variation would be expected for the urea and oxalic acid systems. This, however, was not the case.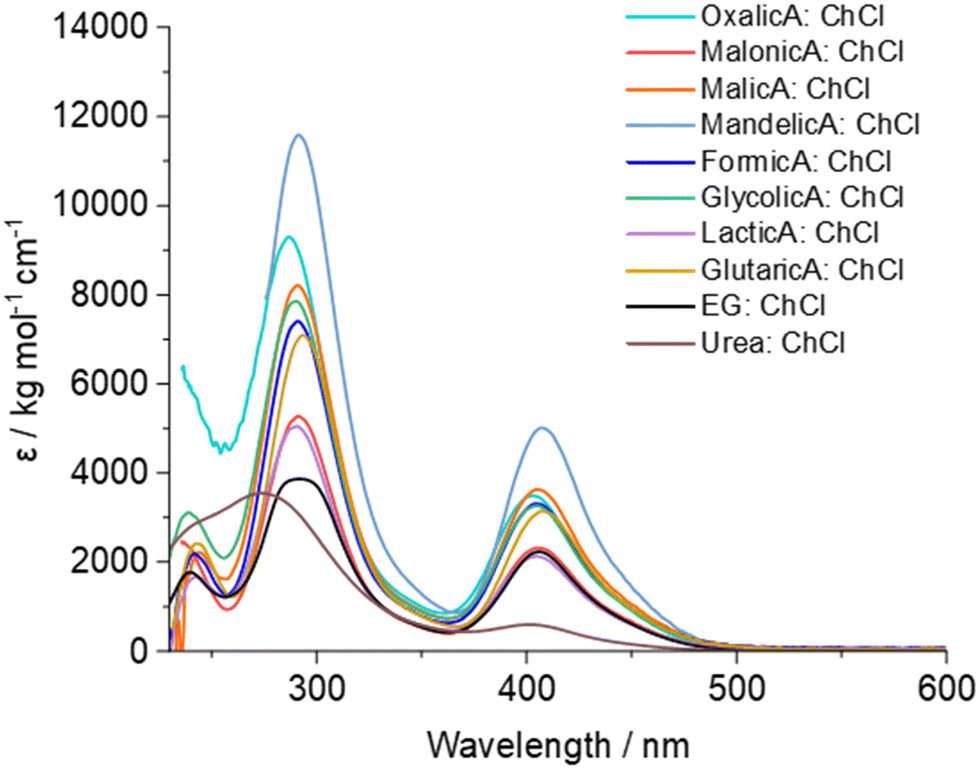 | ||
| Fig. 6 UV-vis spectra of copper(II) chloride in 10 different choline chloride-based DESs. | ||
Voltammetry of these solutions shows that both the Cu2+/+ and Cu+/0 redox couples are present and stable in all liquids (Fig. 7), although the Cu+/0 couple is poorly resolved in the systems with the most acidic HBDs due to solvent decomposition via hydrogen evolution at the Pt-disc working electrode. As the pKa of the HBD becomes smaller (more acidic), the potential window of stability of the DES contracts, as shown by the blank CVs in Fig. S5 (ESI†). Interestingly the type of HBD has a greater impact on the relative redox potentials of both redox couples than would be predicted from the effect of copper speciation alone. Fig. 8 shows the values of the Cu2+/+ and Cu+/0 redox couples for the different DES systems in relative acidity order of the HBDs. For the Cu2+/+ couple, there is a difference of up to 0.6 V between the different solvents, while for the Cu+/0 couple there is a difference of up to 0.4 V. This is approximately a 40 to 60 kJ mol−1 difference in complex stability between the neutral EG:ChCl and the most acidic OxA:ChCl systems. The copper redox potentials in the DESs formed of formic, glycolic, and lactic acids have similar values to each other. More specifically, the average redox potentials for the Cu+/0 couple is −0.620 V ± 0.040 V and for Cu2+/+ it is 0.064 V ± 0.042 V. Remarkably, the water content of the DES does not seem to play a major role in the redox properties of the copper redox couples, as the systems containing the greatest amount of water, OxA:ChCl (ca. 13.55 wt% water) and LacA:ChCl (11.4 wt% water), have very different redox potentials to each other.
 | ||
| Fig. 7 Cyclic voltammograms of copper(II) chloride (0.02 mol dm−3) in the 8 different DESs at a 0.5 mm diameter Pt-disc, referenced to the [Fe(CN)6]3−/4− redox couple, with a scan rate of 20 mV s−1. Temperature = 20 °C. CVs are offset for clarity, with EG:ChCl included as a comparison. Presented data is the first scan. | ||
 | ||
| Fig. 8 Redox potentials plotted as a function of HBD pKa. Values corrected for temperature and concentration using the Nernst equation. | ||
4. Conclusions
The effect of the anion on the redox properties of copper ions in six ILs with different anions and two eutectic systems was investigated. The changes in the redox potential of the Cu2+/+ and Cu+/0 redox couples of six different copper salts in the EG:ChCl and CaCl2·6H2O:EG systems were negligible. Minimal differences were observed between the UV-vis spectra of the copper(II) salts, with the wavelengths of the absorbance maxima varying by < 2 nm between the different salts. This leads to the conclusion that the anion of the salt is not affecting the speciation of copper(II) ions in solution. While there is a solvatochromic shift between the EG:ChCl and CaCl2·6H2O:EG systems of 10 to 20 nm, speciation remains quite similar despite the large differences in the water content between the two different DESs.Regarding the investigation of the copper(II) chloride in a range of imidazolium-based ILs with different anions, it was confirmed that there are some remarkable differences at the species formed, corresponding to different electrochemical responses. The electrochemical behaviour of these systems can also be affected by the physical properties of the ionic liquids, especially in the case of [C4mim][HSO4] and [C6mim][Cl], which are the most viscous liquids. Generally, electrochemistry in DESs is simpler than in ILs because of lower viscosity and fewer electroactive anions being present, such as acetate or hydrogen sulphate. Therefore, this study has shown that the effect of the anion on the starting salt is a lot less important for the redox potentials than the anion present in the solvent, as this solvent anion controls metal ion speciation. This offers the ability to use almost any starting salt in a DES (excepting particularly strong ligands such as cyanide) and retain the same electrochemical behaviour.
The effect of HBD type was also investigated, and it was found that while the copper speciation remained very similar across the range of DESs investigated, the relative redox potentials changed with the pKa value of the HBD. As the system became more acidic, the Cu+/0 redox potential shifted more anodic, and the Cu2+/+ redox potential shifted more cathodic. The potential difference between the neutral EG:ChCl system and the most acidic OxA:ChCl system is up to 0.6 V.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1) and the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement number 101026159, the Faraday Institution (award number FIRG027, project website: https://relib.org.uk/), and Johnson Matthey for funding this work.References
- P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- E. L. Smith, Trans. IMF, 2014, 91, 241–248 CrossRef.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- K. Binnemans and P. T. Jones, J. Sustainable Metall., 2017, 3, 570–600 CrossRef.
- P. J. Dyson, Transition Met. Chem., 2002, 27, 353–358 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- J. M. Hartley, C. M. Ip, G. C. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- A. D. Ballantyne, G. C. Forrest, G. Frisch, J. M. Hartley and K. S. Ryder, Phys. Chem. Chem. Phys., 2015, 17, 30540–30550 RSC.
- A. P. Abbott, A. A. Al-Barzinjy, P. D. Abbott, G. Frisch, R. C. Harris, J. Hartley and K. S. Ryder, Phys. Chem. Chem. Phys., 2014, 16, 9047–9055 RSC.
- J. T. M. Amphlett, Y. Lee, W. Yang, D. Kang, N. E. Sung, J. Park, E. C. Jung and S. Choi, ACS Omega, 2022, 7, 921–932 CrossRef CAS PubMed.
- I. M. Pateli, D. Thompson, S. S. M. Alabdullah, A. P. Abbott, G. R. T. Jenkin and J. M. Hartley, Green Chem., 2020, 22, 5476–5486 RSC.
- I. Rodriguez-Torres, G. Valentin and F. Lapicque, J. Appl. Electrochem., 1999, 29, 1035–1044 CrossRef CAS.
- I. C. Oh, H. Y. Kim, S. K. Hyun and Y. M. Byoun, Surf. Interface Anal., 2021, 53, 1035–1042 CrossRef CAS.
- W. Shao, G. Pattanaik and G. Zangari, J. Electrochem. Soc., 2007, 154, D201–D207 CrossRef CAS.
- Y. L. Kao, K. C. Li, G. C. Tu and C. A. Huang, J. Electrochem. Soc., 2005, 152, C605–C611 CrossRef CAS.
- A. C. Frank and P. T. A. Sumodjo, Electrochim. Acta, 2014, 132, 75–82 CrossRef CAS.
- A. Bigos, M. Wolowicz, M. Janusz-Skuza, Z. Starowicz, M. J. Szczerba, R. Bogucki and E. Beltowska-Lehman, J. Alloys Compd., 2021, 850, 156857 CrossRef CAS.
- B. Tadesse, M. Horne and J. Addai-Mensah, J. Appl. Electrochem., 2013, 43, 1185–1195 CrossRef CAS.
- M. C. Esteves, P. T. A. Sumodjo and E. J. Podlaha, Electrochim. Acta, 2011, 56, 9082–9087 CrossRef CAS.
- A. Dolati, M. Ghorbani and M. R. Ahmadi, J. Electroanal. Chem., 2005, 577, 1–8 CrossRef CAS.
- P. Wilkinson, Gold Bull., 1986, 19, 75–81 CrossRef CAS.
- A. Y. M. Al-Murshedi, J. M. Hartley, A. P. Abbott and K. S. Ryder, Trans. IMF, 2019, 97, 321–329 CrossRef CAS.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS PubMed.
- J. T. M. Amphlett and S. Choi, J. Mol. Liq., 2021, 332, 115845 CrossRef CAS.
- G. Li, D. M. Camaioni, J. E. Amonette, Z. C. Zhang, T. J. Johnson and J. L. Fulton, J. Phys. Chem. B, 2010, 114, 12614–12622 CrossRef CAS PubMed.
- A. Yadav, J. R. Kar, M. Verma, S. Naqvi and S. Pandey, Thermochim. Acta, 2015, 600, 95–101 CrossRef CAS.
- F. S. Mjalli and H. Mousa, Chin. J. Chem. Eng., 2017, 25, 1877–1883 CrossRef.
- E. P. Grishina, L. M. Ramenskaya, M. S. Gruzdev and O. V. Kraeva, J. Mol. Liq., 2013, 177, 267–272 CrossRef CAS.
- S. Fendt, S. Padmanabhan, H. W. Blanch and J. M. Prausnitz, J. Chem. Eng. Data, 2011, 56, 31–34 CrossRef CAS.
- A. M. O’Mahony, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Chem. Eng. Data, 2008, 53, 2884–2891 CrossRef.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- J. M. Hartley, J. Allen, J. Meierl, A. Schmidt, I. Krossing and A. P. Abbott, Electrochim. Acta, 2022, 402, 139560 CrossRef CAS.
- R. Marin Rivera, G. Zante, J. M. Hartley, K. S. Ryder and A. P. Abbott, Green Chem., 2022, 24, 3023–3034 RSC.
- I. B. Assaker and M. Dhahbi, J. Mol. Liq., 2011, 161, 13–18 CrossRef.
- S. Vanderaspoilden, J. Christophe, T. Doneux and C. Buess-Herman, Electrochim. Acta, 2015, 162, 156–162 CrossRef CAS.
- A. P. Abbott, K. El Ttaib, G. Frisch, K. J. McKenzie and K. S. Ryder, Phys. Chem. Chem. Phys., 2009, 11, 4269–4277 RSC.
- N. Frenzel, J. Hartley and G. Frisch, Phys. Chem. Chem. Phys., 2017, 19, 28841–28852 RSC.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- S. Ahrland, B. Tagesson, P. Salomaa, H. Svanholt, P. Hagenmuller and A. F. Andresen, Acta Chem. Scand., 1977, 31a, 615–624 CrossRef.
- R. Taherdangkoo, T. Meng, M. N. Amar, Y. Sun, A. Sadighi and C. Butscher, Rock Mech. Rock Eng., 2022, 55, 4391–4402 CrossRef.
- S. Boudesocque, A. Mohamadou, L. Dupont, A. Martinez and I. Déchamps, RSC Adv., 2016, 6, 107894–107904 RSC.
- O. Reckeweg, R. E. Dinnebier, A. Schulz, B. Blaschkowski, C. Schneck and T. Schleid, Z. Naturforsch. B, 2017, 72, 159–165 CrossRef CAS.
- M. Biswas, G. M. Rosair, G. Pilet and S. Mitra, Inorg. Chim. Acta, 2007, 360, 695–699 CrossRef CAS.
- O. Reckeweg, A. Schulz and F. J. DiSalvo, Z. Naturforsch. B, 2013, 68, 296–300 CrossRef CAS.
- M. Swadzba-Kwasny, L. Chancelier, S. Ng, H. G. Manyar, C. Hardacre and P. Nockemann, Dalton Trans., 2012, 41, 219–227 RSC.
- M. L. Tonnet, S. Yamada and I. G. Ross, Trans. Faraday Soc., 1964, 60, 840–849 RSC.
- M.-J. Deng, P.-Y. Chen, T.-I. Leong, I. W. Sun, J.-K. Chang and W.-T. Tsai, Electrochem. Commun., 2008, 10, 213–216 CrossRef CAS.
- A. Bhattacharjee, C. Varanda, M. G. Freire, S. Matted, L. M. N. B. F. Santos, I. M. Marrucho and J. A. P. Coutinho, J. Chem. Eng. Data, 2012, 57, 3473–3482 CrossRef CAS.
- E. Gómez, Begoña González, Á. Domínguez, E. Tojo and J. Tojo, J. Chem. Eng. Data, 2006, 51, 696–701 CrossRef.
- K. Shakeela, A. S. Dithya, C. J. Rao and G. R. Rao, J. Chem. Sci., 2015, 127, 133–140 CrossRef CAS.
- A. W. Taylor, P. Licence and A. P. Abbott, Phys. Chem. Chem. Phys., 2011, 13, 10147–10154 RSC.
- M. Larriba, P. Navarro, J. García and F. Rodríguez, J. Chem. Thermodyn., 2014, 79, 266–271 CrossRef CAS.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- P.-Y. Chen and I. W. Sun, Electrochim. Acta, 1999, 45, 441–450 CrossRef CAS.
- M. Morimitsu, Y. Nakahara, Y. Iwaki and M. Matsunaga, J. Min. Metall., Sect. B, 2003, 39, 59–67 CrossRef CAS.
- I. Toledo, M. Arancibia, C. Andrade and I. Crivelli, Polyhedron, 1998, 17, 173–178 CrossRef CAS.
- J. T. M. Amphlett, M. D. Ogden, W. Yang and S. Choi, J. Mol. Liq., 2020, 318, 114217 CrossRef CAS.
- A. P. Abbott, A. Y. M. Al-Murshedi, O. A. O. Alshammari, R. C. Harris, J. H. Kareem, I. B. Qader and K. Ryder, Fluid Phase Equilib., 2017, 448, 99–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04389k |
| This journal is © the Owner Societies 2023 |