
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium catalysed oligomerisation in cement-forming silicate systems†
Mohammed S.
Salha
 ac,
Rickey Y.
Yada
b,
David H.
Farrar
c,
Gregory A.
Chass
*bcd,
Kun V.
Tian
*bce and
Enrico
Bodo
a
ac,
Rickey Y.
Yada
b,
David H.
Farrar
c,
Gregory A.
Chass
*bcd,
Kun V.
Tian
*bce and
Enrico
Bodo
a
aDepartment of Chemistry, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185, Roma, Italy
bFaculty of Land and Food Systems, The University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada
cDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, Ontario L8S 4M1, Canada
dSchool of Physical and Chemical Sciences, Queen Mary University of London, London, E1 4NS, UK. E-mail: g.chass@qmul.ac.uk
eDepartment of Chemical Science and Pharmaceutical Technologies, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185, Roma, Italy. E-mail: kun.tian@uniroma1.it
First published on 8th December 2022
Abstract
Alumino-silicates form the backbone of structural materials including cements and the concrete they form. However, the nanoscale aspects of the oligomerisation mechanisms elongating the (alumino-)silicate chains is not fully clarified; the role of aluminium in particular. Herein, we explore and contrast the growth of silicate and alumino-silicate oligomers by both neutral and anionic mechanisms, with focus on the influence of Al on oligomer structure and stability. Further, the spontaneity of chain lengthening in the absence and presence of Al of differing coordination (Al-IV, V, VI) was characterised. Result trends showed Al-IV facilitating oligomerisation in neutral conditions, with respect to Si only systems, effectively promoting longer chain formation and stabilisation. The anionic pathway similarly showed Al reducing the overall energetic barriers to oligomerisation. In both conditions, Al's coordinative and structural flexibility, at O–Al–O hinge points in particular, was responsible for the lowering of the energetic expense for oligomerisation. The results and implications resolved herein are informative for chain formation and stability for bulk material properties of alumino-silicate materials such as cements, where the aluminosilicate systems are dominated by short chains of 2–5 units in length.
1. Introduction
The initial stages of nucleation and growth in aluminosilicate crystals and glasses play a vital role in predicting and controlling their final structures and properties; fracture toughness in particular.1 The epitome of manufactured material durability is evidenced in Roman structures such as the Pantheon and Colosseum, with their ∼2000 years of endurance testament to this. It has been documented that durability was the first consideration for material selection in lieu of the modern inclination for easy flow, tailored slump and rapid setting.2 Low water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :solid ratios and invested workmanship also contributed to the enduring success. On the atomic scale, these practices effectively optimised the nucleation and growth of the crystalline and amorphous phases in the concrete, achieving high toughness, resistance to water and chemical attacks and weathering effects.2 Contrastingly, ordinary portland cement (OPC) typically lasts ∼50 years (with steel reinforcement),3 partly due to inadequate compaction, as well as relatively high water:solids ratios to facilitate flow into the forms employed,4 reducing the need for manual compaction.
:solid ratios and invested workmanship also contributed to the enduring success. On the atomic scale, these practices effectively optimised the nucleation and growth of the crystalline and amorphous phases in the concrete, achieving high toughness, resistance to water and chemical attacks and weathering effects.2 Contrastingly, ordinary portland cement (OPC) typically lasts ∼50 years (with steel reinforcement),3 partly due to inadequate compaction, as well as relatively high water:solids ratios to facilitate flow into the forms employed,4 reducing the need for manual compaction.
Atomistically, a key to the longevity of Roman concrete was the use of ground volcanic ash, which contains high amounts of aluminium. Modern literature points to high Al cements having increased durability, due to Al substitution into the main binding phases.5
Cement hydration involves a range of complex physical and chemical processes, with the main hydration product being calcium silicate hydrate (C–S–H), the nanoscopic structure of which is not yet comprehensively characterised. Studies employing non-destructive techniques such as small angle neutron scattering (SANS) have helped to establish some of the more quantitative properties, such as the Ca/Si ratio being ∼1.7 and the density ∼2.6 g cm−3.6 C–S–H's short-chain dominated structuring has also been characterised with 29Si and 27Al nuclear magnetic resonance (NMR), resolving the fractions of Si with differing connectivity: Q0 ≈ 10%, Q1 ≈ 66.7%, Q2 ≈ 23.3% and Q3–4 ≈ 0–1%,7 where Qn denotes the n-number of bridging oxygen atoms (i.e. non-terminal OH) the central Si or Al atom is bound to. The number of bound bridging oxygens influences the local electronic environments and thus the shielding and NMR chemical shift. For example, Si(OH)4 would be labelled as a Q0 unit, a bridged dimer [(HO)3Si–O–Si(OH)3] would be said to contain two Q1 silicates, whilst a trimer [(HO)3Si–O–Si(OH)2–OSi(OH)3] would equate to two Q1 silicates and one Q2 (Q1–Q2–Q1).
Essentially, these silicate oligomers make up the foundation of the C–S–H structure, with calcium setting in and around a myriad of “layered” silicate chains.8 The Ca2+ cation interacts with the electron-density concentrated at the O-atoms in the OH-groups (HOδ−). The H2O present in C–S–H exists in two forms: electrostatically bound to the HOδ− groups like Ca2+ and physically bound water which remains in the C–S–H and the concrete; essentially trapped in the structure.9 This inhomogeneous mix of differing structural elements, themselves of differing chemical identities, polarizability, physical sizes and shapes, densities, distributions & concentrations, result in a disordered system with a multitude of diverse surfaces and interfaces.
Atomic-level fracture in concrete and mortar occurs along stark interfaces between different phases.10 Chemical attack can trigger or exacerbate fracture by causing reactions in the cement that lead to mineral formation. Among these minerals is ettringite, whose growth inside concrete induces a series of micro stresses11 due to its mechanical properties. The rigidity of the silicate chains, due to the geometrically invariable Si-tetrahedra, gives the cement most of its strength and thus ability to support load without deformation.12 Yet, this same rigidity is responsible for the inherent brittleness, lacking toughness and thus vulnerability to fracture. Toughness being the ability of a material to undergo limited deformation through local dissipation of stresses.4
Contrastingly, Al in cements can exist in different geometries from 3-coordinated trigonal planar (albeit very rare) through 4-coordinate tetrahedral, 5-coordinate trigonal bipyramidal or square-based pyramidal, to 6-coordinate octahedral.13 The additional degrees of freedom and flexibility of the bonds and angles of the O–Al–O/Al–O–Si units contributes to the improved mechanical properties of Al-rich cement14; similar to other metal-containing functional material systems.15 Al is shown to substitute into Q2 bridging positions in silicate chains and the pronounced broadening and convolution of peaks in Al-NMR with respect to Si-NMR evidences increased geometric and configurational flexibility, hence Al helping to create hinge-points at the centre of a chain helping to dissipate external stresses.16 Al has also been shown to promote merging processes that increase the mean chain length (MCL) in cement.17 The silicate chain length in C–S–H tends to follow a ‘3n − 1’ rule (n = 1, 2, 3…) with a dreierketten arrangement, where the chain repeats every three units.18 Chain lengths of n = 2, 5, 8 are shown to be the most stable, including those with Al substitution.19 This spurred our interests to characterise the structures and stabilities of pentamers formed from silicate dimers and monomers, in the presence and absence of Al substitution (Scheme 1).
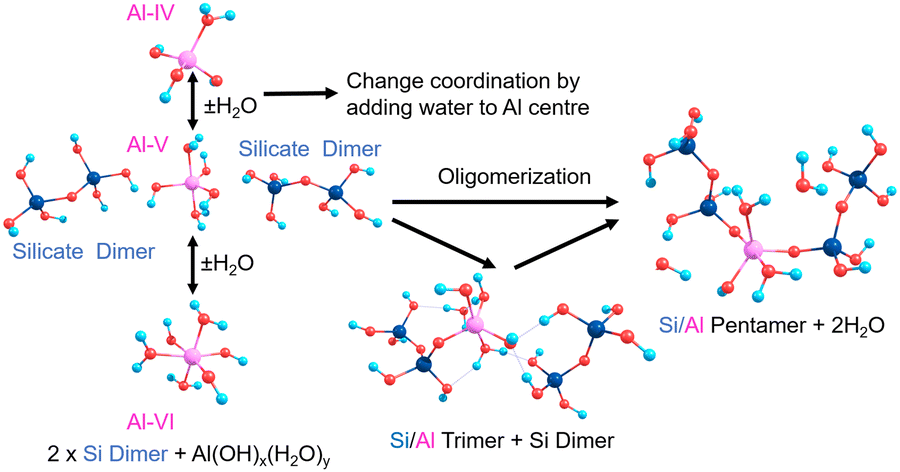 | ||
| Scheme 1 Silicate pentamerisation initiating from two silicate dimers with Al positioned as a bridging atom, with manifold coordinations (Al-IV, V, VI). | ||
2. Mechanisms of silicate oligomerisation
Silicate oligomerisation mechanisms were characterised by Pereira et al. in 1998 employing ab initio modelling for silicate dimerisations20 and supporting a mechanism akin to 2nd order nucleophilic substitution (SN2). This two-step process involved nucleophilic attack forming a charged complex followed by elimination of a differing ligand from the complex. Specifically, despite showing the SN2 mechanism to exhibit low energy barriers for the condensation of Si(OH)4 and Si(OH)4H+, Pereira et al. stated that other mechanisms “should still be possible and may occur simultaneously in the solution”.20The current work focused on neutral and anionic mechanisms for silicate oligomerisation building on established results,17–20 with models of silicate monomers and their Al-IV, V, VI counterparts, oligomerising to Si/Al pentamers with Q2 Al.5 Details are shown in Scheme 2 (top/green), wherein an exemplar for neutral oligomerisation is shown using a 5-coordinate Al transition state complex (Al-V or Al-5). Therein, oligomerisation initiates via hydrogen bonds (H-bonds) forming between the reactants (RCT), drawing them closer together and aligning them for reaction. The transition state (TS) for the neutral mechanism involves the simultaneous formation of the (Al)–O–Si bond while breaking the Al–O(–H) bond as part of a 4-membered transition state. This proton transfer leads to the formation of water (condensation) and the Si–O–Al trimer (PRD1). This is then repeated with another Si-dimer to form the Si/Al pentamer (PRD2). A relatively high energy barrier is predicted for the transition state since the mechanism is predicated on molecular rearrangement, as opposed to an explicit nucleophilic attack. Work on silicate species by Zhang et al. showed neutral pH to favour linear oligomerisation whereas high pH favours ring closure.18
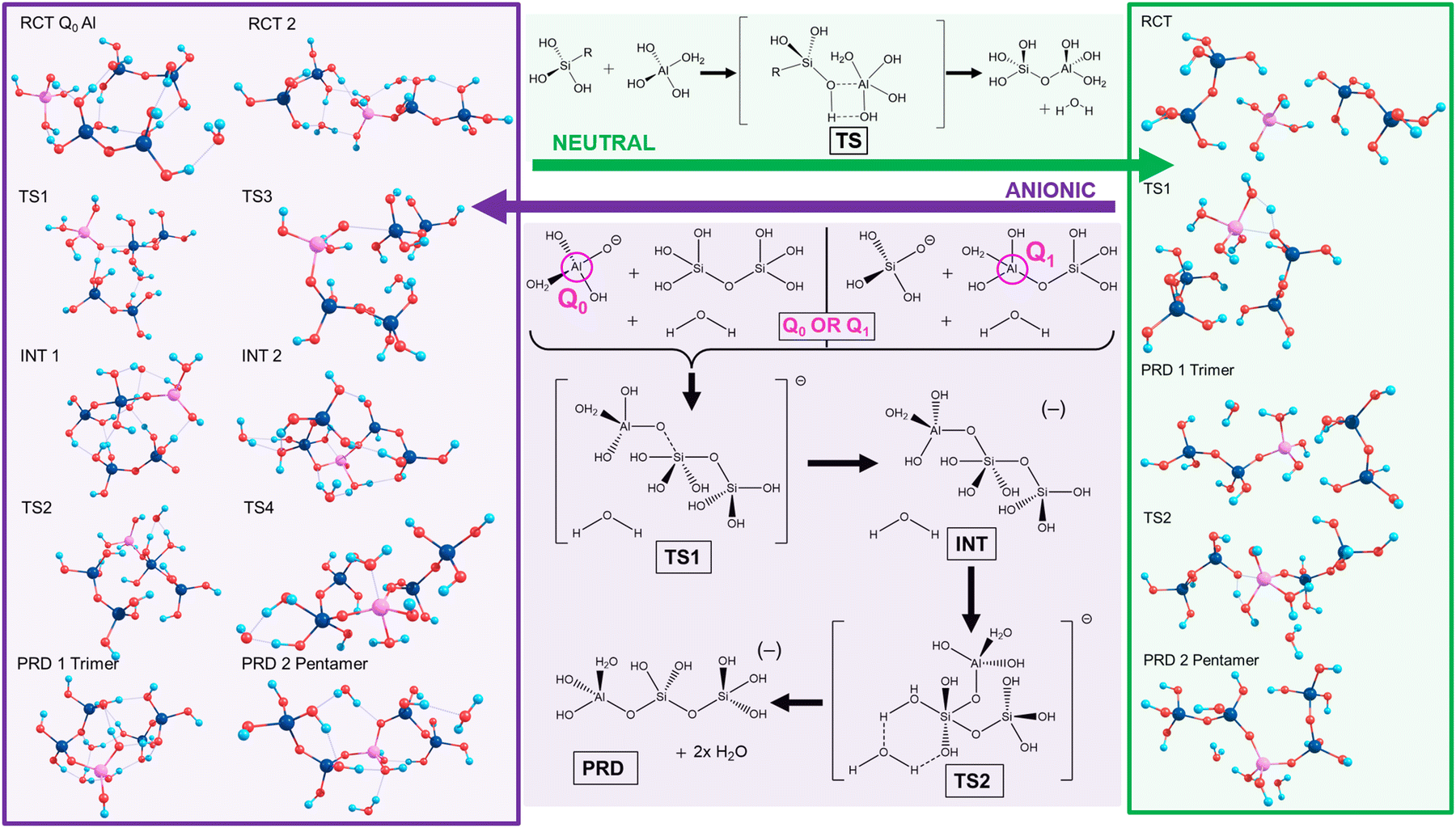 | ||
| Scheme 2 Neutral mechanism (top/green outline) for Si-dimer trimerization (dimer + monomer → trimer) with Al-IV monomer is shown. Further reaction with an additional Si-dimer generates the Si–Al-pentamer (trimer + dimer → penatmer). The anionic “lateral attack” mechanism (bottom/purple outline) is shown for the Al-IV case, wherein an explicit water molecule helps stabilise a 6-membered transition state. The scheme displays initial oligomerization between an Si–Al dimer (Q1 Al) and an Si monomer; the alternate case with a Si-dimer attacked by an anionic Al monomer (Q0, Al) was also modelled. | ||
The anionic mechanism (Scheme 2, bottom/purple) may also be operative in cements due to their high pH environments (pH ∼ 11–14).21 Tracking the anionic example in Scheme 2 (purple), deprotonation of Si/Al OH groups generates Si(OH)3O(−). The Si anion initiates attack from the deprotonated silicate oxygen forming the first transition state (TS1), leading to a penta-coordinate Al-V intermediate (INT1). This is followed by the appearance of a second transition state (TS2) with that leads to the first product (PRD 1). This second TS is catalysed by a separate, explicit water molecule.22 The resultant charged trimer product repeats the process reacting with another dimer to form a pentamer (PRD2).
Scheme 2 highlights the asymmetry of Al substitution, since the anionic mechanism could potentially proceed inversely, using a Q0 Al (monomer) to attack an Si dimer, leading to a TS centred on the Si atom. This work models both these potential reaction pathways in the case of the anionic mechanism: (1) Q1 Al: An Al–Si dimer being attacked by an Si monomer; (2) Q0 Al: starting from Q0 Al attacking an Si dimer.
Towards exploring structure and energetics of both the neutral and anionic mechanisms, we employed a bare molecular cluster (BMC) approach23 to generate the geometries based on each step of the reactions (Scheme 2) and geometry-optimised these to the relevant critical point along their reaction profiles (structures located at minima or 1st-order TS on their hypersurfaces). Such non-periodic models allow for configurational and conformational freedom for all constituent and reacting components during chemical transformations and in set structures. Work by Trinh et al.24 revealed that the explicit inclusion of H2O changed the kinetics with respect to the gas-phase, thus further justifying their inclusion as opposed to a solely implicit solvent method. The free energy for each step of the reaction was determined from which relative values were determined (ΔGrel); this with respect to the energy of the starting reactants, and set to the ‘zero’ (ΔG = 0). For the neutral mechanism, the optimised structures are shown in Scheme 2 (right-hand side, green-shading) in the case of a tetra-coordinated Al-IV. The associated negative frequency motions in the neutral TS structures are illustrated in Fig. 1, involving the proton transfer.
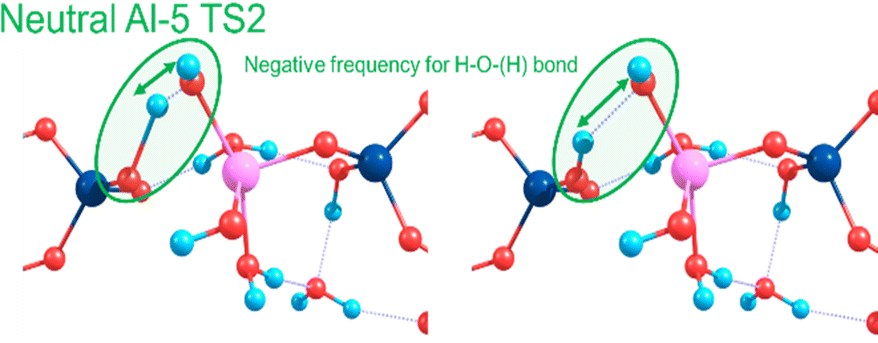 | ||
| Fig. 1 Atomic motions along the negative frequency mode during the proton transfer from Si–OH to form Al OH2 as part of the second transition state for the neutral mechanism using Al-5. | ||
The same approach was used to model the anionic mechanism (ten structures each profile), for all four cases, including the following: (1) Si-only: to compare relative free-energies in the absence of Al (i.e. Si vs. Al) as well as for differing Al-coordinations, (2) Al-IV; (3) Al-V; (4) Al-VI. In the case where the reactive Si monomer anion [Si(OH)3O(−)] attacks an Si–Al dimer, forcing the Al atom on the acceptor dimer raising coordination from Al-IV → Al-V (Q1, Al pathway). As Al can shift its coordination more easily than Si we compared the opposite case, wherein a nucleophilic anionic Al-monomer [Al(OH)3O(−)] attacks a Si-dimer (Q0, Al-pathway). Optimised geometries for the tetra-coordinated Al-IV pathway are shown in Scheme 2 (left-hand side, purple-shading). Fig. 2 shows the 6 membered ring with proton transfer occurring from the first Si–OH group through the catalytic water molecule and onto the second Si–OH group forming Si–OH2.
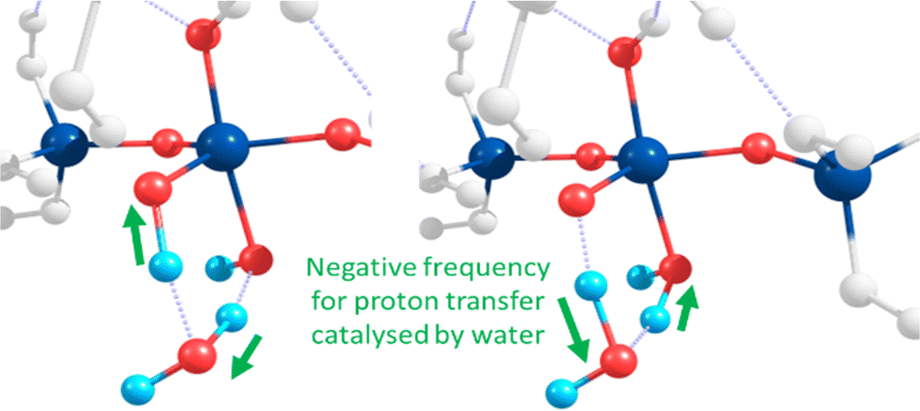 | ||
| Fig. 2 Atomic motions along the negative frequency mode in the 6 membered transition state involved in TS2 and TS4 of the anionic mechanism. Here the TS is localised on the pentacoordinate Si intermediate. | ||
2.3 Computational details
The Gaussview 5.025 graphical tool was used to construct the initial models, ensuring oxygens were bridging or terminal –OH groups.The Gaussian 09 (G09) program package26 was used for all computations in this work. The models were optimised using the density functional theory DFT B3LYP method with D3 dispersion correction and a with a 6-31G(d,p) basis set (B3LYP-D3/6-31G(d,p)), employing 6 × Cartesian d-orbitals (vs. 5 × spherical ones) and bondi radii; the latter shown to more accurately determine aqueous solvation free energies of ions and anions.27–30 Together with the explicit water molecules involved in the reactions, the reactive clusters were modelled in implicit water solvent employing the Polarizable Continuum Model (PCM) method.31 Analytical frequencies were computed on the geometry-optimised structures to confirm the identity of each structure as residing at minima or 1st order saddle points on their respective potential energy hypersurfaces (PEHSs). Thermochemical parameters and entropy contributions (at 300 K) were determined and used to determine free-energies. For reproducibility of thermodynamic and kinetic trends, Anionic Si and Anionic Q1 Al models were also determined with the CAM-B3LYP/6-311G(d,p) level.
3. Results and discussion
3.1 Neutral energetics
The calculated energies of the compounds involved in the reaction of Scheme 2 are depicted in Fig. 3 and in the ESI.† The magenta, green and rust-red lines mark the reaction pathways for starting coordinations for Al-IV, Al-V and Al-VI, respectively. For comparison, the blue line reports the Si-only pathway (no Al substitution). In the case of the neutral mechanism, substitution of the nucleophilic anionic Si monomer [Si(OH)3O(−)] for its Al complement [Al(OH)3O(−)] lowers the free-energy (ΔGrel) barriers by 72.1(124.9–52.8) and 111.1(146.5–35.4) kJ mol−1 for TS1 and TS2, respectively. This indicates that Al acts as a more efficient chain linker than Si and its substitution more spontaneously increases mean chain length (MCL).14 The high energy barriers along the Si pathway arise from its fixed tetrahedral coordination resulting in higher energies required to accommodate changes in geometry along reaction pathways (i.e. 4 → 5 coordinate). As Si in aluminosilicates is present as tetrahedral centres, ∼125 kJ mol−1 is required to form the pentacoordinate Si in the transition states (TSs). In addition, forcing Si to attain a penta-coordinate structure results in the appearance of parasitic bond breaking processes either producing H2O or dissociating a Si–O bond.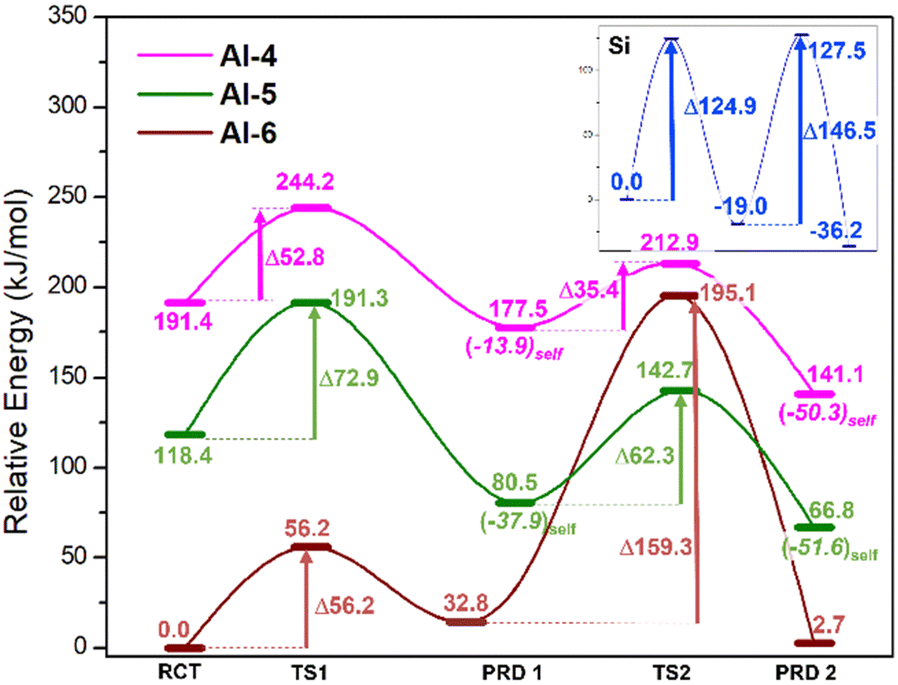 | ||
| Fig. 3 Neutral mechanism: relative free energy (ΔGrel) for differing Al-coordinations with respect to reactants: Al-IV (magenta) Al-V (green) and Al-VI (rust-red). Inset: Si-only mechanism (blue) devoid of an Al-centre. Isodesmic comparison of free energies is possible between differing Al-coordinations by adding or subtracting the energy of one water molecule, generating the relative-relative free-energies (ΔΔGrel). | ||
With respect to Al systems, the coordination temporarily increases at the TSs; Al-IV → Al-V and Al-V → Al-VI TSs. The barrier of TS1 for the Al-IV path is lower than that of Al-V by ∼20.1 kJ mol−1 (52.8 vs. 72.9 kJ mol−1, Fig. 3), implying that Al-IV monomers are more likely to form Si/Al trimers. Pathways that initiate with an Al-VI monomer cannot supersaturate Al to adopt a heptacoordinated coordination (Al-VII) and instead lose a water ligand in favour of Al–O–Si bond formation. The molecular rearrangement forms this Al-VI TS, leaving a free H2O molecule. In a bulk cementitious environment water molecules generated locally as-such could be adsorbed in the capillary pores, else interact with Ca2+ cations or possibly react to form Ca(OH)2 (portlandite).
In these cementitious clusters, water molecules form H-bond networks with terminal Si–OH groups on the chains. These “extra” H-bonds result in the Al-VI reaction pathway having the lowest energy barriers even though the rearrangement process (where Al-VI spontaneously swaps ligands) is unlikely to take place as part of oligomerisation. The barrier associated with Al-IV → Al-V transition (magenta TS2 = +35.4) is 26.9 kJ mol−1 lower than the Al-V → Al-VI barrier (green TS2 = +62.3, Fig. 3). This leaves Al-IV as the optimal starting point for neutral oligomerisation.
3.2 Anionic mechanism
Results are disseminated in Fig. 4 and the ESI.† Therein, TS structures for the anionic mechanism were attempted using Al-IV, Al-V and Al-VI, only the tetracoordinate ion yielded stable TS structures. Trinh et al.'s work21 confirmed that the switch from gas phase to solvent phase significantly lowers the energy barriers for the TS2 and TS4 steps, as proton transfer was no longer reliant on the aforementioned intramolecular rearrangement. This energetic lowering of TS/TS4 renders SiO–Si bond formation at TS1 and TS3 more energetically demanding. This is due to these ‘water removal’ TS barriers being more difficult to surmount due to the hydrogen bonding stabilising the preceding steps; something that cannot be described solely with implicit solvent methods.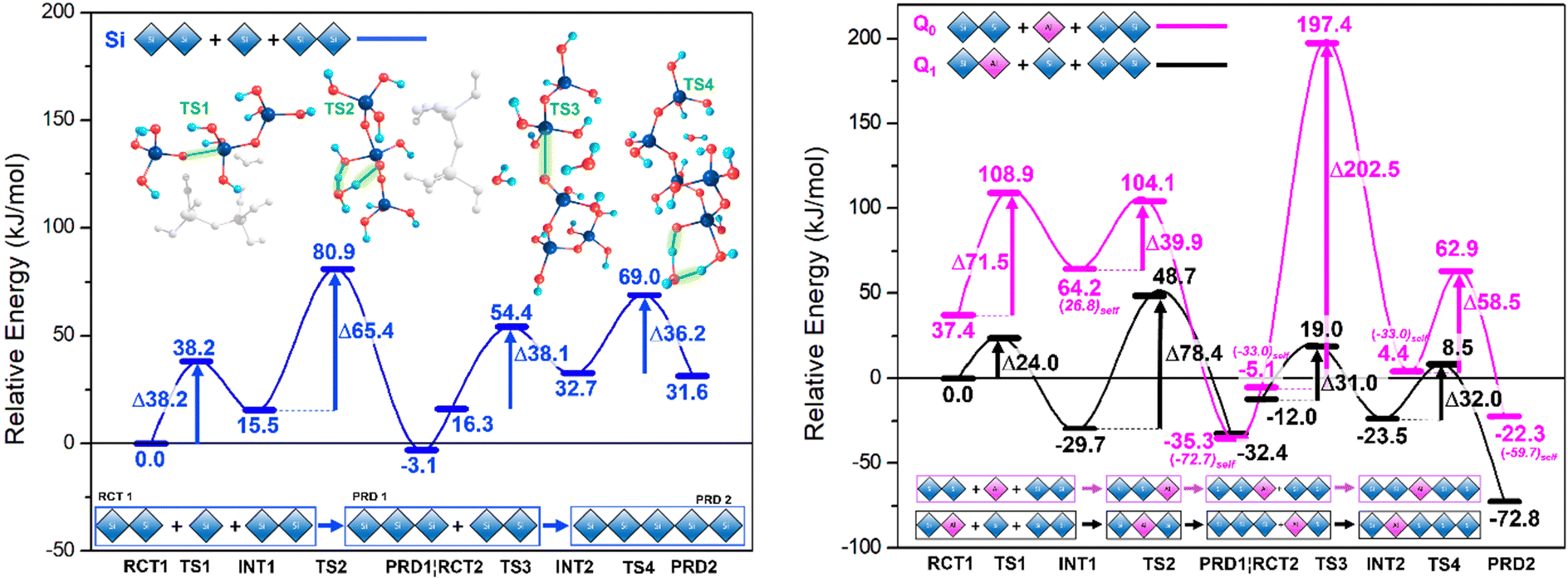 | ||
| Fig. 4 Energetics for anionic mechanism. Coloured lines represent the reaction pathway from dimer to trimer (PRD1) to pentamer (PRD2). Energy barriers for each TS are labelled with an accompanying illustration highlighting key atoms involved. All energies for Si are plotted relative to the Si RCT1 while Q0 and dimer are plotted relative to Q1 Al RCT 1. Calculated energy values determined with the B3LYP-D3/6-31G(d,p) method, which includes gd3 empirical dispersion correction, and employing 6 cartesian d-orbitals, together with the PCM solvation method and Bondi radii. | ||
Rather, there is need for inclusion of H2O particles and all reactants involved in the reaction profile at each step of the reaction (explicit H2O molecules + additional Si dimer), rather than energetic summation of individual components.
Such ‘all in’ method conserves stoichiometry across the profile. The rate determining step (RDS) must be considered for each system individually, for example TS2 for Q0 Al and Q1 Al are centred on different atoms (Si vs. Al respectively). In contrast to the work by Trinh et al., Fig. 4 shows the Si TS1 for trimerisation with a free energy barrier that sits lower than the subsequent water removal step (Si TS2).24 This may be explained by a hydrogen bond network formed between the Si monomer, dimer, the explicit water and the ‘spare’ (extra) Si-dimer, itself directly stabilising the anionic monomer [Si(OH)3O(−)] and lowering the free energy (Fig. 4, left-side). In Si TS3, this ‘spare’ Si-dimer makes part of the linear chain, thus leaving only water molecules available for H-bonding, in agreement with the literature where SiO Si bond formation is the RDS,24,33 albeit only slightly (TS3 ∼1.9 kJ mol−1 higher than TS4).
Both Q1 and Q0 Al pathways echo this trend with energy barriers for TS1 lower than that of TS2 (Fig. 4). Once the ‘spare’ dimer is incorporated into the chain, the barrier for the subsequent water removal step (TS4) drops to around half of the value of the preceding water-removal step (TS2). Stabilisation of the TS for the Si/AlO–Si bond formation step hinges on the relative stability of the Si/Al–O− anion and the ability of the Si/Al accepting centre to shift from 4- to 5-fold coordination. Al substitution employing [Al(OH)2(H2O)O(−)] anions as opposed to [Si(OH)3O(−)], to conserve charge.
Shifting the position of Al substitution from the attacking Q0 monomer (Fig. 4, magenta) to the accepting Q1 Si/Al dimer (Fig. 4, black), lowers the TS1 barrier by ∼14.2 kJ mol−1 relative to the Si-only pathway; previously 33.3 kJ mol−1 higher. This implies that initial short chain oligomerisation may be possible with Al if nucleophilic attack from [Si(OH)3O(−)] is feasible at the Al centre (Q1 pathway). As in the neutral mechanism, Al-substitution facilitates chain merging (trimer + dimer → pentamer) due to ease of coordination change at the Al-centre, the anionic mechanism is more spontaneous (lower free-energy) with reactive Al-centres.
While initial chain oligomerisation (TS2, dimer + monomer) remains the RDS along the Q1 Al path, the ensuing steps are all facilitated with an average barrier of ∼31.5 kJ mol−1. This implies that the effect Al has on chain merging begins after short chains (n = 2–3) have formed and that the position of Al in said chains is paramount to efficient oligomerisation. Kinetically speaking the trimerisation phase is roughly as viable with Si as it is for Q1 Al, with the discrepancies between TS1 and TS2 cancelling each other out However, in moving to pentamerisation Q1 Al exhibits TS3 and TS4 energy barriers that are 7.1 and 4.2 kJ mol−1, lower, respectively, than those of the Si-only pathway. Hence, the Q1 Al pathway is kinetically favoured for chain oligomerisation. Analysing these pathways from a thermodynamic perspective reveals a trough in the PES of each system at PRD 1 (the trimer). The trimers are the lowest energy products in all but the Q1 Al pathway, which has its PRD 2 (pentamer) 40.4 kJ mol−1 lower than PRD 1. This not only highlights the Q1 Al pathway as optimal for oligomerizing to longer chains over time but reveals the most stable bridging position for Al substitution into this pentamer (non-central Q2). Further, that oligormerisation of the Si-only systems would equilibrate at PRD1 with trimers dominating the system and sluggish movement beyond this stage; similarly for the Q0 Al pathway. Another noteworthy property of the Al substituted paths is that their 5-fold intermediates are more stable than the reactant steps that precede them (e.g. INT 1 < RCT 1). Al-V is stable and thus the Q1 Al pathway is both kinetically and thermodynamically favourable.
3.3 Neutral vs. anionic mechanisms
Whilst the Si neutral trimerisation barrier is higher in energy (∼21.3 kJ mol−1) than its anionic analogue, the pentamerisation step is 72.2(146.5–38.1) kJ mol−1 higher. This supports the idea that oligomerisation predominantly follows the anionic mechanism. The rationale that the abnormally low energy of anionic Si TS1 is a result of conformation and increased H-bonding (see above) and is in agreement with the literature.22 The barriers to trimerisation (RCT → PRD1) and pentamerisation (PRD1 → PRD2) for Al-IV are 52.8 and 35.4 kJ mol−1 in the neutral mechanism whereas the anionic Q1 Al set has corresponding barriers of 102.4 (Q1 TS1 +TS2) and 63.0 (Q1 TS3 and TS4). The Q0 Al set has even higher barriers at 111.4 (Q0 TS1 and TS2) and 261.0 kJ mol−1 (Q0 TS3 + TS4). The results herein confirm a preference for neutral conditions in the case of Al. Other works on cements indicate that Al increases mean chain length and considering that setting cements are very alkaline environments with high pHs, we expect Al to prefer the anionic path.20This is perhaps due to the contributions from Ca2+ ions that would be present in cement, as the introduction of free cations to these specific transition states have been shown to further inhibit chain merging; raising the TS1/TS3 energy barriers, in agreement with previous works.32 Our results indicate a kinetic preference for longer chains under anionic conditions with Q1 Al substitution and that this preference is further amplified in neutral conditions. Al helps to maintain chain length once it is established; the Si/Al pentamer ∼72.8 kJ mol−1 more stable than its starting configuration, whilst the Si-only pentamer is ∼31.6 kJ mol−1 less stable that its starting components (Fig. 4, ΔGrel of RCT vs. PRD2). Simulation results at higher level of theory (ESI†) confirm the thermodynamic and kinetic trends uncovered and presented herein (Table 1).
| Reaction path | RCT → PRD 1 (kJ mol−1) | PRD1 → PRD 2 (kJ mol−1) | Overall |
|---|---|---|---|
| Neutral Al-4 | 52.8 | 35.4 | −50.3 |
| Anionic Al-Q1 | 24.0 (TS1) | 31.0 (TS3) | −72.8 |
| 78.4 (TS2) | 32.0 (TS4) | ||
| Anionic Al-Q0 | 71.5 (TS1) | 202.5 (TS3) | −22.3 |
| 39.9 (TS2) | 58.5 (TS4) | ||
| Anionic Si | 38.2 (TS1) | 38.1 (TS3) | +31.6 |
| 65.4 (TS2) | 36.2 (TS4) |
4. Conclusions
Herein, we have demonstrated that Al substitution promotes alumino-silicate chain growth in neutral conditions. Substitution of Si with Al-IV, V and VI monomers lowering the energy barriers for generating trimeris and pentamers. Our findings are in agreement with the literature, that Si oligomerisation is more likely to proceed via an anionic mechanism.22 This work shows how Al substitution can further increase the kinetic and thermodynamic preference for oligomerisation to longer chains under anionic conditions, whilst also facilitating extension of chains in neutral conditions. The initial Si/AlO–Si formation step is generally the most difficult (RDS), with its free energy greatly influenced by the degree of hydrogen bonding stabilising the reactive anion. The water removal step is eased by the presence of Al due to its ability to shift coordination (Al-IV ↔ Al-V ↔ Al-VI). This highlights the dynamical role of Al-centres as coordinatively flexible ‘fulcrums’ helping distribute force and mitigate structural failure in such cementitious type materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MS and EB acknowledge PRACE and the Julich Supercomputing Centre for helping support computational requirements needed to complete this work. The authors acknowledge EU-Horizon 2020 and BEIS, UK for supporting the FUNMIN project (ACT, No. 299668) and providing the means to extending ongoing work to cementitious materials. The STFC and RAL-ISIS are thanked for funding neutron beam work (RB1710285, RB1710444, RB1710447, RB2010426, RB2010696), results informing on trends reflected in this work. KVT thanks Prof. Bruno Botta (Sapienza University of Rome) for mentoring, in addition to the Department of Chemistry and Chemical Biology (McMaster University), the Faculty of Land and Food Systems (University of British Columbia) and the Natural Sciences and Engineering Research Council, Canada (RGPIN 04598, RYY) for support. GAC thanks the departments of Chemistry at McMaster University and the University of Hong Kong for supporting his Adjunct and Honorary Professorships, respectively, as well as the LFS at UBC for his Affiliate Professorship.References
- T. Komatsu and T. Honma, Nucleation and crystal growth in laser-patterned lines in glasses, Front. Mater., 2016, 3(7), 1–8 Search PubMed.
- N. J. Delatte, Lessons from Roman Cement and Concrete, J. Prof. Issues Eng. Educ. Pract., 2001, 127(3), 109–115 CrossRef.
- R. A. De Medeiros-junior, M. G. De Lima and M. H. F. De Medeiros, Service life of concrete structures considering the effects of temperature and relative humidity on chloride transport, Environment, Development and Sustainability, Dordrecht, 2014, 17, 5, 1–17 Search PubMed.
- R. O. Ritchie, The conflicts between strength and toughness, Nat. Mater., 2011, 10(11), 817–822 CrossRef CAS.
- M. D. Jackson, et al., Unlocking the secrets of Al-tobermorite in Roman seawater concrete, Am. Mineral., 2012, 98(10), 1669–1687 CrossRef.
- A. J. Allen, J. J. Thomas, A. J. Allen, J. J. Thomas and H. M. Jennings, Composition and Density of Nanoscale Calcium – Silicate – Hydrate in Cement Composition and density of nanoscale calcium – silicate – hydrate in cement, no. May 2014, 2007 Search PubMed.
- R. J. M. Pellenq, et al., A realistic molecular model of cement hydrates, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(38), 16102–16107 CrossRef CAS PubMed.
- M. Bauchy, M. J. A. Qomi, F. J. Ulm and R. J. M. Pellenq, Order and disorder in calcium-silicate-hydrate, J. Chem. Phys., 2014, 140(21), 214503 CrossRef CAS.
- H. N. Bordello, L. P. Aldridge and A. Desmedt, Water dynamics in hardened ordinary portland cement paste or concrete: From quasielastic neutron scattering. J. Phys. Chem. B 110, 17966–, J. Phys. Chem. B, 2006, 110(36), 17966–17976 CrossRef.
- M. Torsæter, J. Todorovic and A. Lavrov, Structure and debonding at cement-steel and cement-rock interfaces: Effect of geometry and materials, Constr. Build. Mater., 2015, 96, 164–171 CrossRef.
- A. Quennoz and K. L. Scrivener, Interactions between alite and C3A-gypsum hydrations in model cements, Cem. Concr. Res., 2013, 44, 46–54 CrossRef CAS.
- E. Gartner, I. Maruyama and J. Chen, A new model for the C-S-H phase formed during the hydration of Portland cements, Cem. Concr. Res., 2017, 97, 95–106 CrossRef CAS.
- K. V. Tian, G. A. Chass and D. Di Tommaso, Simulations reveal the role of composition into the atomic-level flexibility of bioactive glass cements, Phys. Chem. Chem. Phys., 2016, 18(2), 837–845 RSC.
- Q. Zheng, J. Jiang, J. Yu, X. Li and S. Li, Aluminum-Induced Interfacial Strengthening in Calcium Silicate Hydrates: Structure, Bonding, and Mechanical Properties, 2020 Search PubMed.
- X. X. Zhang, P. Alvarez-Lloret, G. Chass and D. Di Tommaso, Interatomic potentials of Mg ions in aqueous solutions: structure and dehydration kinetics, Eur. J. Mineral., 2019, 31(2), 275–287 CrossRef CAS.
- I. G. Richardson, Nature of C-S-H in hardened cements, Cem. Concr. Res., 1999, 29(8), 1131–1147 CrossRef CAS.
- E. L’Hôpital, B. Lothenbach, G. Le Saout, D. Kulik and K. Scrivener, Incorporation of aluminium in calcium-silicate-hydrates, Cem. Concr. Res., 2015, 75, 91–103 CrossRef.
- A. Ayuela, J. S. Dolado, I. Campillo, Y. R. De Miguel, E. Erklzia and D. Sánchez-Portal, et al., Silicate chain formation in the nanostructure of cement-based materials, J. Chem. Phys., 2007, 127(16), 164710 CrossRef CAS PubMed.
- H. Manzano, J. S. Dolado and A. Ayuela, Aluminum incorporation to dreierketten silicate chains, J. Phys. Chem. B, 2009, 113(9), 2832–2839 CrossRef CAS.
- J. C. G. Pereira, C. R. A. Catlow and G. D. Price, Silica condensation reaction: An ab initio study, Chem. Commun., 1998, 1387–1388 RSC.
- X. Q. Zhang, T. T. Trinh, R. A. Van Santen and A. P. J. Jansen, Mechanism of the initial stage of silicate oligomerization, J. Am. Chem. Soc., 2011, 133(17), 6613–6625 CrossRef CAS PubMed.
- T. T. Trinh, A. P. J. Jansen and R. A. Van Santen, Mechanism of oligomerization reactions of silica, J. Phys. Chem. B, 2006, 110(46), 23099–23106 CrossRef CAS.
- K. V. Tian, et al., Periodic vs. molecular cluster approaches to resolving glass structure and properties: Anorthite a case study, J. Non-Cryst. Solids, 2016, 451, 138–145 CrossRef CAS.
- T. T. Trinh, A. P. J. Jansen, R. A. Van Santen and E. Jan Meijer, The role of water in silicate oligomerization reaction, Phys. Chem. Chem. Phys., 2009, 11(25), 5092–5099 RSC.
- A. B. Nielsen and A. J. Holder, Gauss View 5.0, User's Reference, GAUSSIAN Inc.: Pittsburgh, 2009 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. H. Mu, et al., Competing mechanisms, substituent effects, and regioselectivities of nickel-catalyzed [2+ 2+ 2] cycloaddition between carboryne and alkynes: A DFT Study, J. Org. Chem., 2015, 80(18), 9108–9117 CrossRef CAS PubMed.
- W. H. Mu, G. A. Chasse and D.-C. Fang, High level ab initio exploration on the conversion of carbon dioxide into oxazolidinones: The mechanism and regioselectivity, J. Phys. Chem. A, 2008, 112(29), 6708–6714 CAS.
- M. Zoltán, G. A. Chass and I. G. Csizmadia, Systemic energy management by strategically located functional components within molecular frameworks, determined by systems chemistry, J. Phys. Chem. B, 2009, 113(30), 10308–10314 Search PubMed.
- M. T. Pedersen, et al., Phase separation in an ionomer glass: Insight from calorimetry and phase transitions, J. Non-Cryst. Solids, 2015, 415, 24–29 CrossRef CAS.
- Y.-M. Chen, G. A. Chass and D.-C. Fang, Between a reactant rock and a solvent hard place–molecular corrals guide aromatic substitutions, Phys. Chem. Chem. Phys., 2014, 16(3), 1078–1083 RSC.
- A. Pavlova, T. T. Trinh, R. A. Van Santen and E. J. Meijer, Clarifying the role of sodium in the silica oligomerization reaction, Phys. Chem. Chem. Phys., 2013, 15(4), 1123–1129 RSC.
- T. T. Trinh, A. P. J. Jansen, R. A. Van Santen and E. Jan Meijer, The role of water in silicate oligomerization reaction, J. Phys. Chem. C, 2009, 113(7), 2647–2652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03918d |
| This journal is © the Owner Societies 2023 |