
Epoxide activation by a silver phosphonate for heterogeneous catalysis of CO2 cycloaddition†
Chao-Ying
Gao‡
 *ab,
Chen
Mao‡
ab,
Yang
Yang
a,
Ning
Xu
a,
Jinglin
Liu
a,
Xiaohong
Chen
a,
Jinghai
Liu
*ab and
Limei
Duan
*ab
*ab,
Chen
Mao‡
ab,
Yang
Yang
a,
Ning
Xu
a,
Jinglin
Liu
a,
Xiaohong
Chen
a,
Jinghai
Liu
*ab and
Limei
Duan
*ab
aCollege of Chemistry and Materials Science, Inner Mongolia Minzu University (IMMN), Tongliao 028043, Inner Mongolia, People's Republic of China. E-mail: chaoyinggao@163.com; jhliu2008@sinano.ac.cn; duanlmxie@126.com
bInner Mongolia Key Lab of Carbon Nanomaterials, Nano Innovation Institute (NII), Inner Mongolia Minzu University (IMMN), Tongliao 028000, People's Republic of China
First published on 10th November 2022
Abstract
Hydrothermal reaction of silver nitrate and a tetraphosphonic acid (H8L) affords a novel coordination polymer, with the general formula, [Ag10(H3L)2(H2O)2(4,4′-bipy)], denoted as compound 1. It is built from extended ⋯Ag–O–P–O⋯ chains and organic linkers to form a 3D network. In its structure, three of four phosphonyl groups of the organic ligand are protonated, endowing compound 1 with rich Brønsted acid sites. With Brønsted and Lewis acidic sites, the catalytic activity of 1 was investigated through cycloaddition reaction of CO2 and epoxides, and the results displayed an excellent performance. Besides, the heterogeneous nature of compound 1 was proved by five cycle experiments with no significant compromise on the activity, indicating the silver phosphonate as an excellent catalyst for CO2 chemical fixation.
Metal phosphonate phases, including coordination polymers (CPs) and metal organic frameworks (MOFs), have been attracting continuous interest in solid chemistry ever since the first report on zirconium phenylphosphonate.1 Phosphonates, mainly used as building blocks in materials science, can exist in different states (i.e. fully deprotonated R–PO32− and monoprotonated R–PO3H−), and coordinate to various metals ions from a range of monovalent cations like Li+ and hexavalent ones like W6+. The diversified coordination modes and the strong affinity of the phosphonate ligands lead to abundant metal phosphonates with versatile structural motifs2 and interesting properties, such as proton-conduction,3,4 magnetism,5 fluorescence sensing,6etc. Furthermore, metal phosphonates also have shown high thermal and chemical stability,2 which greatly improves their commercial or industrial applications. As far as we know, the monoprotonated R–PO3H− functionalities are usually maintained in produced structural units,2–4 and the acidity of P–OH groups is inimitable. From the above considerations, metal phosphonates are considered to be good candidates for Brønsted acid based heterogeneous catalysts. However, metal phosphonates are not prone to crystalline and considerably rarer in reports than their carboxylate-based counterparts.2 Synthesizing metal phosphonates and investigating their potential applications are worthy of exploration and of scientific significance.
Silver catalysts are widely used in both organic reactions and the chemical industry as Lewis acids.7–9 Due to their d10 specific electronic configuration, silver catalysts have shown a characteristic σ- and/or π-activation in discrete reactions.10–12 Recently, heterogeneous silver-based catalysts by immobilization of silver nanoparticles or silver atoms into porous MOFs have been reported.13–15 Ag@MOF composites are usually prepared by multi-step postsynthesis under strongly acidic or strongly oxidizing conditions (such as concentrated sulfuric acid and concentrated nitric acid). Such harsh conditions can only be applied to very few ultra-stable MOFs (such as Cr-MIL-101, UiO-66, etc.).13,14 Meanwhile, it is difficult to explore the relevant catalytic mechanism for composite catalysts due to the lack of intuitive structural characterization (such as single crystal X-ray diffraction analysis). The above shortcomings are considered to be avoided by building up Ag(I) node based MOF catalysts: (1) the geometric coordination configuration of the Ag(I) sites can be intuitively determined; (2) the synthesis conditions are mild and the operation is simple. However, the stability of such materials have always puzzled chemists: Ag(I) metal centers usually undergo hydrolysis and lead to the degeneration of catalysts.16 Therefore, the application of MOFs with Ag(I) nodes in the field of heterogeneous catalysis is still relatively rare.10,11
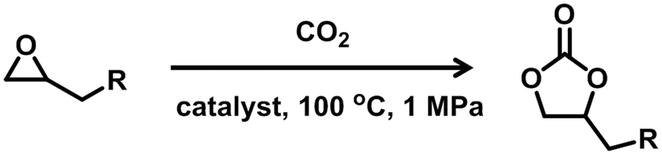
Considering the advantages of metal phosphonates on structural stability and diversity and the excellent specific activation ability of Ag(I) compounds, it is very meaningful to develop Ag(I) phosphonates and apply them in heterogeneous catalysis. Herein, H8L ((ethene-1,1,2,2-tetrayltetrakis(benzene-4,1-diyl))tetraphosphonic acid) (Scheme 1) was employed as the ligand and a Ag(I) phosphonate was successfully obtained under hydrothermal conditions. The cycloaddition of epoxides with CO2 to produce cyclic carbonates is one of the most promising ways for CO2 fixation,17–20 owing to it being an economical and green reaction. It is interesting that the compound exhibits high catalytic activity in the coupling of CO2 with epoxides. To the best of our knowledge, this is the first example of a Ag(I) phosphonate coordination polymer applied in CO2 chemical fixation.
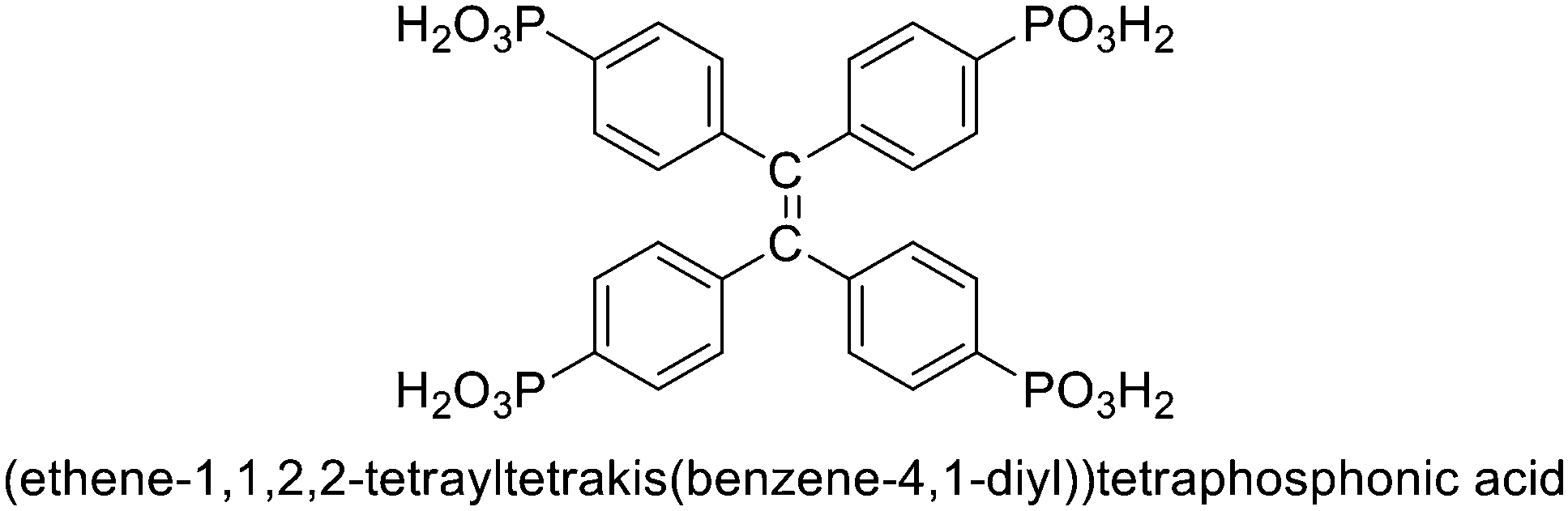 | ||
| Scheme 1 The structure of H8L. | ||
Experimental section
Synthetic procedure for [Ag10(H3L)2(H2O)2(4,4′-bipy)] 1
The ligand H8L (0.25 mmol, 0.162 g), silver nitrate (AgNO3) (0.5 mmol, 0.085 g), and 4,4′-bipy (0.5 mmol, 0.079 g) were suspended in 20 mL deionized water solution and stirred for 30 min. The mixture was transferred and sealed in a 25 mL Teflon-lined reactor. Then, the reactor was sealed in a stainless steel vessel and heated in a programmable oven at 180 °C for 3 days, followed by slow cooling to room temperature. Light yellow block-shaped crystals were obtained and collected via filtration, washed with ethanol and then with water and air-dried to give 1 in 58% yield, based on AgNO3. The formula of compound 1 was mainly determined by single-crystal X-ray diffraction, combining charge equilibrium consideration, thermogravimetric analysis (TGA) and elemental analysis. Elemental analysis: calculated for Ag10P8C62N2O26H50 (MW 2565.53): C, 29.03; N, 1.09; H, 1.96. Found: C, 29.15; N, 1.06; H, 1.98.IR: (KBr pellet, cm−1): 3422 (m), 3092 (w), 3044 (w), 1605 (s), 1535 (w), 1491 (m), 1418 (m), 1358 (s), 1327 (s), 1225 (m), 1130 (s), 1036 (S), 959 (m), 899 (m), 810 (s), 725 (w), 677 (s), 640 (w), 582 (w), 536 (m).
CO2 cycloaddition experiments
In a representative process, CO2 cycloaddition with an epoxide was performed in a 20 mL autoclave reactor with a magnetic stirrer. The vessel was placed in an oil bath and was purged with CO2 up to 1 MPa under a constant pressure. The general reaction in this work: 20 mmol of epoxide and 0.01 mmol of catalyst under solvent-free conditions with tetrabutylammonium bromide (n-Bu4NBr, 0.3 mmol) as a co-catalyst under 1 MPa pressure at 373 K.The loading of the catalyst is a 0.05% ratio based on the epoxide. After a given reaction time, a small amount of supernatant was taken out and used for Gas Chromatography (GC) analysis for calculating the conversion of the epoxide. At the end of the reaction, an aliquot of the reaction mixture was dissolved in CDCl3 and then filtered through a syringe filter (PTFE) for 1H NMR analyses. The product cyclic carbonate was confirmed by 1H NMR spectroscopy, and the yield was determined by GC and 1H NMR.
In a recycling experiment, the catalyst was isolated by centrifugation after the reaction, washed with dichloromethane three times and dried in air. The recovered catalyst was reused in the following reactions under the same conditions as those in the first run.
Results and discussion
Structure characterization
X-ray single-crystal diffraction analysis revealed that the compound crystallizes in the triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). The asymmetric unit consists of five crystallographically independent Ag+ ions, one deprotonated H3L5−, half a 4,4′-bipy molecule and one water molecule (Fig. S1†). Ag1 and Ag5 are in a tetrahedral coordination environment and surrounded by four oxygen atoms originating from four different L ligands, respectively. Ag2 is coordinated by three oxygen atoms from three L ligands. Ag3 is linked by three oxygen atoms from three L ligands and one nitrogen atom from 4,4-bipy. Ag4 is connected by four oxygen atoms from three L ligands and one coordinating water molecule (Fig. 1b). In turn, for the L ligand, three PO3C groups are protonated with two of them as μ3-η1,η2, η0 bridges and one μ4-η1,η3, η0, and the fourth PO3C group is fully deprotonated with the μ7-η2,η2,η3 mode (Fig. S1†). The interconnection of neighbouring Ag+ ions by the phosphonate oxygen atoms and nitrogen atoms results in infinite one-dimensional chains (Fig. 1), which are further connected by the organic linkers L and 4,4′-bipy to form a three-dimensional (3D) extended dense network (Fig. 1a).
(Table S1†). The asymmetric unit consists of five crystallographically independent Ag+ ions, one deprotonated H3L5−, half a 4,4′-bipy molecule and one water molecule (Fig. S1†). Ag1 and Ag5 are in a tetrahedral coordination environment and surrounded by four oxygen atoms originating from four different L ligands, respectively. Ag2 is coordinated by three oxygen atoms from three L ligands. Ag3 is linked by three oxygen atoms from three L ligands and one nitrogen atom from 4,4-bipy. Ag4 is connected by four oxygen atoms from three L ligands and one coordinating water molecule (Fig. 1b). In turn, for the L ligand, three PO3C groups are protonated with two of them as μ3-η1,η2, η0 bridges and one μ4-η1,η3, η0, and the fourth PO3C group is fully deprotonated with the μ7-η2,η2,η3 mode (Fig. S1†). The interconnection of neighbouring Ag+ ions by the phosphonate oxygen atoms and nitrogen atoms results in infinite one-dimensional chains (Fig. 1), which are further connected by the organic linkers L and 4,4′-bipy to form a three-dimensional (3D) extended dense network (Fig. 1a).
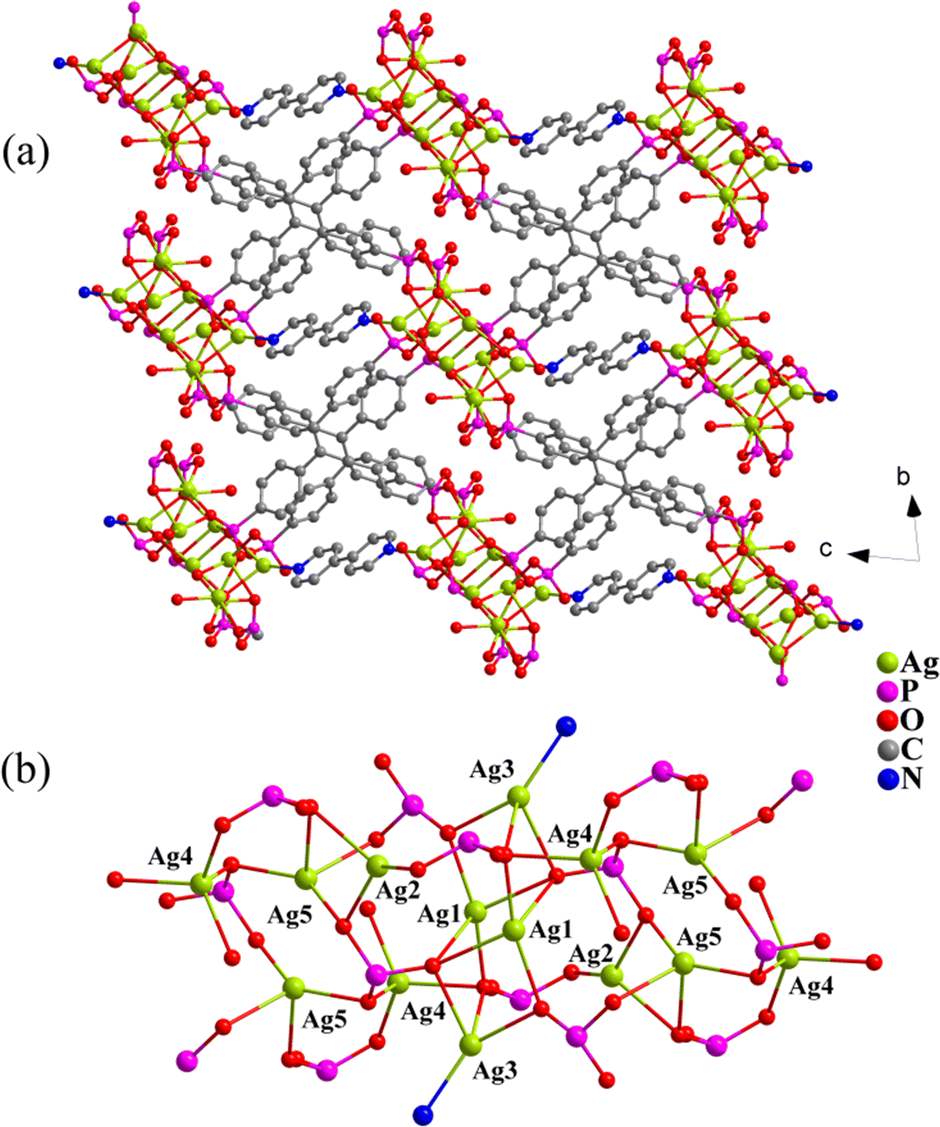 | ||
| Fig. 1 A view of the 3D architecture of compound 1 in a ball–stick model (a) and the representation of the ⋯Ag–O–Ag⋯ chain (b). | ||
Powder X-ray diffraction analysis
To check the purity and bulk phase synthesis of this crystal compound, the block shaped yellow single crystals were ground in a mortar with a pestle, and then the PXRD data of the powder sample were collected. Little difference was observed in the peak intensities of the two patterns, which could be ascribed to the different orientations of crystallites in the bulk material.21–23 The powder diffraction plot of silver phosphonate showed that the experimental peak positions substantially match those in the simulated pattern (Fig. 2), indicating the purity of the crystal.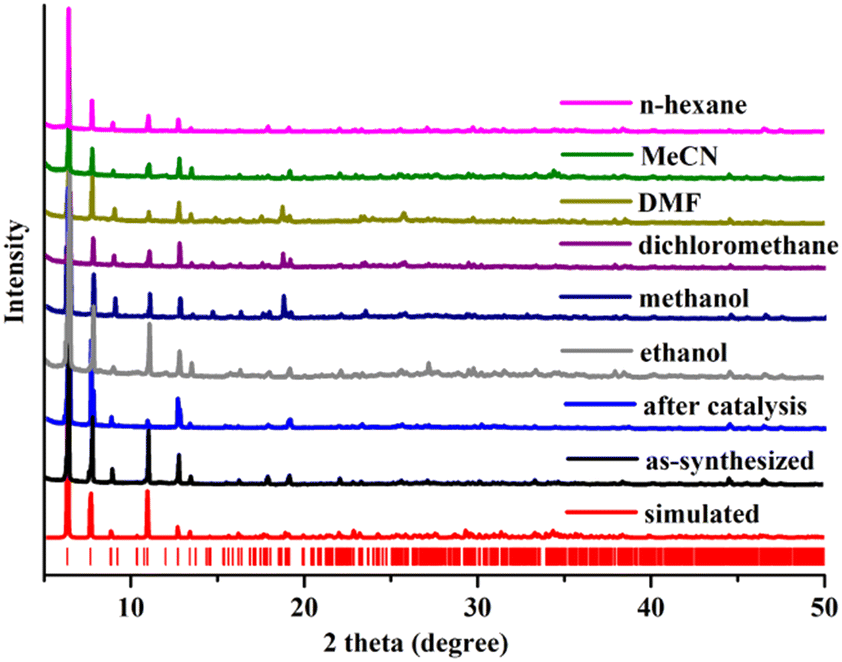 | ||
| Fig. 2 PXRD patterns of the compound upon different treatments. | ||
The stability test of the as-synthesized silver phosphonate was performed in different organic polar and nonpolar solvents for 24 h. PXRD patterns of the crystals after soaking in methanol, ethanol, dichloromethane, DMF (N,N′-dimethylformamide), acetonitrile and n-hexane solvents are in good agreement with the as-synthesized PXRD pattern (Fig. 2), suggesting the structural integrity of the silver phosphonate in solvent treatment.
Cycloaddition of epoxides with CO2
Microporous MOFs are a kind of attractive catalyst in the cycloaddition of CO2 and epoxides to produce cyclic carbonates, in which the reactive roles are commonly attributed to metal centres (as Lewis acid sites) and the porosity (providing the reaction space and surface area).24–30 However, hydrogen bonding moieties (as a Brønsted acid) at the solid/liquid interface have also shown a promoting role in effecting cycloaddition.31–34 Exploration of the coordination modes of the MOF ligand is a promising way to achieve reactive functional groups, and metal phosphonates are ideal candidates due to the inimitable P–OH groups maintained during typical framework assembly.2–4 With rich P–OH groups embedded in the silver phosphonate, compound 1 was investigated as a heterogeneous catalyst in the cycloaddition of CO2 with epoxides.The initial research employed epichlorohydrin as a model substrate to probe the optimized reaction conditions in the chemical conversion of CO2 into carbonates. The corresponding results and reaction conditions are listed in Table 1. The control experiments were carried out in the presence of n-Bu4NBr as a co-catalyst, referring to previous reports,24–30 with different reaction times. The representative amount of reactants is 20 mmol of epoxides and 0.3 mmol of co-catalyst n-Bu4NBr with a loading of 0.5 mol‰ ratio of silver phosphonate. When the reaction was performed under ambient conditions (entries 1–3 in Table 1), the yields were so low with 1.7%, 4.6% and 7.5% for 3 h, 6 h, and 12 h, respectively. Then, the CO2 pressure was increased to 1 MPa (entries 4–6 in Table 1); only a 17.8% yield was obtained even after 12 hours. When the temperature was gradually raised to 60 °C (entries 7–9 in Table 1) and 80 °C (entries 10–12 in Table 1), the yields were improved largely, with 65.8% and 94.3% for 12 hours, respectively. Afterwards, the temperature was increased to 100 °C, and the substrates were almost completely converted to cyclic carbonates after 6 h of reaction (entry 6), suggesting that temperature has a greater effect than pressure in such a reaction. Accordingly, the subsequent reactions were carried out at 100 °C under a 1 MPa CO2 reaction atmosphere, with n-Bu4NBr as the co-catalyst. Besides, AgNO3 was also employed as a catalyst for comparison (entry 15), and the yield was about 89.0%, which is a little lower than that obtained from compound 1 under similar conditions. The results manifest that the Lewis acid indeed can promote the coupling of CO2 with epoxides, as well as the superiority of silver phosphate. It is worth noting that AgNO3 as a homogeneous catalyst lacks recyclability.
|
|
||||
|---|---|---|---|---|
| Entry | T (°C) | P (MPa) | t (h) | Yied (%) |
| Reaction conditions: epichlorohydrin (20 mmol), catalyst (0.01 mmol) and n-Bu4NBr (0.3 mmol) under various conditions. Yields except for the final ones were calculated by GC using n-dodecane as an internal standard substance, and the final products and corresponding conversions were determined by 1H NMR spectroscopy. | ||||
| 1 | 25 | 0.1 | 3 | 1.7 |
| 2 | 25 | 0.1 | 6 | 4.6 |
| 3 | 25 | 0.1 | 12 | 7.5 |
| 4 | 25 | 1 | 3 | 5.9 |
| 5 | 25 | 1 | 6 | 9.8 |
| 6 | 25 | 1 | 12 | 17.8 |
| 7 | 60 | 1 | 3 | 20.9 |
| 8 | 60 | 1 | 6 | 50.3 |
| 9 | 60 | 1 | 12 | 65.8 |
| 10 | 80 | 1 | 3 | 68.9 |
| 11 | 80 | 1 | 6 | 90.6 |
| 12 | 80 | 1 | 12 | 94.3 |
| 13 | 100 | 1 | 3 | 93.6 |
| 14 | 100 | 1 | 6 | >99 |
| 15 | 100 | 1 | 3 | 89.3 |
To check the catalytic generality of the silver phosphonate, epoxide substrates with different sizes and functionalities were examined in the chemical fixation of CO2 into cyclic carbonates under the optimized conditions. The corresponding catalytic results are shown in Table 2. When the chlorine atom was substituted for a bromine atom, a slight increase was observed in the CO2 conversion with 95.6% within 3 hours, and also a complete conversion was observed within six hours. However, epoxybutane was converted to the corresponding cyclic carbonate with a lower conversion, 57.1%, 83.4%, 92.9%, and >99% for 3, 6, 9, and 12 hours, respectively. The above phenomena may be associated with the different electron-withdrawing or electron-donating substituents. Electron-withdrawing groups commonly facilitate nucleophilic attack during the ring opening of epoxides.28 The electronegativity order is –Br > –Cl > –CH3; as a consequence, the conversion of CO2 in the same amount of time is reduced in turn. When a larger sized epoxide, styrene oxide (SO), was employed, a lower decrease was observed in the product yields with 30.3%, 42.3%, 53.7% and 68.5% for 3, 6, 9, and 12 hours, respectively. The phenomenon in which the CO2 conversion decreases with the molecule size increase of epoxide substrates is consistent with most reports.28 When the phenyl group was substituted for a phenoxymethyl group, the reaction conversion was largely improved, with 83.0%, 90.2%, 96.4%, and >99% for 3, 6, 9, and 12 hours. Compared with SO, the higher yield for the allyl glycidyl is also attributed to the fact that the phenoxymethyl group is an electron-withdrawing substituent. The turnover number (TON) and turnover frequency (TOF) values according to the five kinds of epoxide substrates are also listed in Table 2. To better understand the activity of the silver phosphonate, comparisons with some MOF-based catalysts are listed in Tables S3 and S4,† in which epichlorohydrin and styrene oxide were chosen as the representative of small and large sized substrates, respectively. It was found that the TON and TOF values of the silver phosphonate for all the employed epoxides are even higher than those of some conventional porous MOF-based catalysts (Tables S3 and S4†). At the same time, in order to estimate the maximized conversion using the silver phosphonate catalyst, cycloaddition reactions were carried out using a larger amount of epichlorohydrin (40, 60, 100 mmol) and catalyst (0.01 mmol) under a constant pressure of 1 MPa (Table 2, entries 1a–1c) and at 100 °C. When the amount of epichlorohydrin is 100 mmol, the TON and TOF values are up to 8700 per mole of catalyst and 1450 per mole of catalyst per hour, proving that the silver phosphonate is a promising catalyst for the synthesis of cyclic carbonates from epoxides and CO2 for the chemical industry.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | t (h) | Yield (%) | TON | TOF |
| Reaction conditions: epoxide (40, 60, 100 mmol for 1a–1c, respectively; 20 mmol for entries 2–5), catalyst (0.01 mmol) and n-Bu4NBr (0.3 mmol) under CO2 (1 MPa), 100 °C. Yields except for the final ones were calculated by GC using n-dodecane as an internal standard substance, and the final products and corresponding conversions were determined by 1H NMR spectroscopy. | |||||
| 1a |
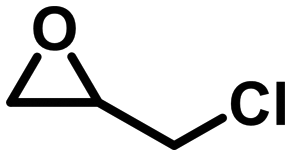
|
6 | >99 | 4000 | 666.7 |
| 1b | 6 | 97.2 | 5832 | 972.0 | |
| 1c | 6 | 87.0 | 8700 | 1450.0 | |
| 2 |
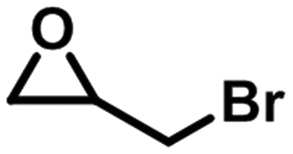
|
3 | 95.6 | 1912 | 637.3 |
| 6 | >99 | 2000 | 333.3 | ||
| 3 |
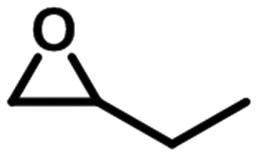
|
3 | 57.1 | 1142 | 380.7 |
| 6 | 83.4 | 1668 | 278 | ||
| 9 | 92.9 | 1858 | 206.4 | ||
| 12 | >99 | 2000 | 166.7 | ||
| 4 |
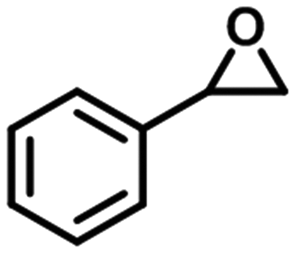
|
3 | 30.3 | 606 | 202.0 |
| 6 | 42.3 | 846 | 141.0 | ||
| 9 | 53.7 | 1074 | 119.3 | ||
| 12 | 68.5 | 1370 | 114.2 | ||
| 5 |
|
3 | 83.0 | 1660 | 553.3 |
| 6 | 90.2 | 1804 | 300.7 | ||
| 9 | 96.4 | 1928 | 214.2 | ||
| 12 | >99 | 2000 | 166.7 | ||
One crucial quality for heterogeneous catalysts is recyclability to evaluate their practical applications in industry. The reusability of the silver phosphonate was determined using epichlorohydrin as the reaction substrate. The reaction was performed under the optimized reaction conditions using 60 mmol epoxide and 0.01 mmol of catalyst for 6 h. After the reaction, the catalyst was centrifuged, washed three times with dichloromethane, and air-dried. The recovered catalyst was reused for subsequent reactions under the same conditions as those in the first run. After five cycles, there is no significant drop in the conversion, indicating that the catalytic activity of the silver phosphonate was still maintained. The powder X-ray diffraction (PXRD) pattern of the catalyst after five rounds was still in good agreement with that of the as-prepared one, showing structural integrity (Fig. 2). At the end of the reaction, the mixture filtrate was examined by inductively coupled plasma (ICP) analysis. No Ag leaching indicates the heterogeneous nature of the catalytic reaction. Besides, scanning electron microscopy (SEM) coupled with energy dispersive X-ray spectroscopy (EDS) was used to determine the morphologies and elemental composition of the samples before and after catalysis (Fig. 3), the results of which further proved the heterogeneous nature of the catalyst.
 | ||
| Fig. 3 SEM morphologies and element analysis of the samples before (a) and after catalysis (b). | ||
Based on previous reports,24–30 a plausible mechanism was proposed for the CO2 cycloaddition. As illustrated in Scheme S1,† firstly, the oxygen atom of the epoxide is activated by the metal centres and hydrogen bonding to initiate the epoxy ring. Then, Br− from n-Bu4NBr attacks the carbon atom of the activated epoxide, resulting in the epoxy ring opening. Subsequently, an epoxide intermediate is generated through the reaction between CO2 and the oxygen anion of the open ring. Finally, the ring closing leads to the final cyclic carbonate. At the same time, the catalyst is regenerated.
Conclusions
In summary, a new silver phosphonate has been developed, and its catalytic activity was well investigated through the coupling reaction of CO2 with epoxides. The high product yields of cyclic carbonates indicate its efficient activity. Besides, the heterogeneous nature of the silver phosphonate is also well verified through reusability experiments and filtration tests. After five runs, its catalytic activity is still maintained, indicating that the silver phosphonate is an excellent candidate for CO2 chemical fixation.Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22061033, 21961025, 21961024), the Chinese Scholarship Council (CSC, No. 202008155035), the Natural Science Foundation of Inner Mongolia (2019MS02003, 2019MS02004), the Fundamental Research Funds for the Provincial Universities of Inner Mongolia Autonomous Region (2022), and the Doctoral Scientific Research Foundation of Inner Mongolia Minzu University (BS462).Notes and references
- G. Alberti, U. Costantino, S. Allulli and N. Tomassini, J. Inorg. Nucl. Chem., 1978, 40, 1113–1117 CrossRef CAS.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS PubMed.
- S. Kim, K. W. Dawson, B. S. Gelfand, J. M. Taylor and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 963–966 CrossRef CAS PubMed.
- S. S. Bao, G. K. H. Shimizu and L. M. Zheng, Coord. Chem. Rev., 2019, 378, 577–594 CrossRef CAS.
- D. Zeng, M. Ren, S. S. Bao and T. Zheng, J. Mol. Struct., 2022, 1248, 32767 CrossRef.
- Z. Hu, M. Tsai, H. Sung and J. Wu, J. Solid State Chem., 2021, 299, 122178 CrossRef CAS.
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC.
- Z. Chang, X. Jing, C. He, X. Liu and C. Duan, ACS Catal., 2018, 8, 1384–1391 CrossRef CAS.
- X. Wang, Z. Chang, X. Jing, C. He and C. Y. Duan, ACS Omega, 2019, 4, 10828–10833 CrossRef CAS PubMed.
- Z. Zhou, C. He, L. Yang, Y. Wang, T. Liu and C. Y. Duan, ACS Catal., 2017, 7, 2248–2256 CrossRef CAS.
- L. Yang, Y. Dou, Z. Zhou, D. P. Zhang and S. Wang, NANO, 2019, 9, 1566 CAS.
- X. H. Liu, J. G. Ma, Z. Niu, G. M. Yang and P. Cheng, Angew. Chem., Int. Ed., 2015, 54, 988–991 CrossRef CAS.
- N. N. Zhu, X. H. Liu, T. Li, J. G. Ma, P. Cheng and G. M. Yang, Inorg. Chem., 2017, 56, 3414–3420 CrossRef CAS PubMed.
- G. Dutta, A. K. Jana, D. K. Singh, M. Eswaramoorthy and S. Natarajan, Chem. – Asian J., 2018, 13, 2677–2684 CrossRef CAS.
- C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- G. Cai, M. Ding, Q. Wu and H. L. Jiang, Natl. Sci. Rev., 2020, 7, 37–45 CrossRef CAS.
- M. Ding and H. L. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- M. Ding, S. Chen, X. Q. Liu, L. B. Sun, J. Lu and H. L. Jiang, ChemSusChem, 2017, 10, 1898–1903 CrossRef CAS.
- M. Ding and H. L. Jiang, Chem. Commun., 2016, 52, 12294–12297 RSC.
- J. M. Chin, E. Y. Chen, A. G. Menon, H. Y. Tan, A. T. S. Hor, M. K. Schreyer and J. Xu, CrystEngComm, 2013, 15, 654–657 RSC.
- J. Hafizovic, M. Bjorgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- D. Chakraborty, A. Ghorai, A. Chowdhury, S. Banerjee and A. Bhaumik, Chem. – Asian J., 2021, 16, 1562–1569 CrossRef CAS PubMed.
- Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS.
- Y. Gu, B. A. Anjali, S. Yoon, Y. Choe, Y. G. Chung and D. W. Park, J. Mater. Chem. A, 2022, 10, 10051–10061 RSC.
- H. Lv, L. Fan, H. Chen, X. Zhang and Y. Gao, Dalton Trans., 2022, 51, 3546–3556 RSC.
- K. Liu, S. Jiao, H. Zhao, F. Cao and D. Ma, Green Chem., 2021, 23, 1766–1771 RSC.
- J. Dong, P.-F. Shi, P. Cheng and B. Zhao, J. Am. Chem. Soc., 2015, 137, 15988–15991 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, X. Wang, R. J. Drout, M. R. Mian, R. Cao, K. Ma, Q. Xia, Z. Li and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 4302–4431 CrossRef CAS PubMed.
- M. Y. Li, F. Wang and J. Zhang, Cryst. Growth Des., 2020, 20, 2866–2870 CrossRef CAS.
- A. C. Kathalikkattil, D. W. Kim, J. Tharun, H. G. Soek, R. Roshan and D. W. Park, Green Chem., 2014, 16, 1607–1616 RSC.
- Y. Yang, C. Y. Gao, H. R. Tian, J. Ai, M. Xue and Z. M. Sun, Chem. Commun., 2018, 54, 1758–1761 RSC.
- C. Y. Gao, Y. Yang, N. Xu, J. Liu, L. Duan, X. Chen and M. Bao, Cryst. Growth Des., 2021, 21, 1413–1417 CrossRef CAS.
- J. Sun, J. Y. Ren, S. J. Zhang and W. G. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic refinement details, additional tables and structural figures, PXRD and TG curves, and 1H NMR spectra. CCDC 2203855. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01240e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |