
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction of SF5CF3via iridium catalysis: radical trifluoromethylation of aromatics†
Domenique
Herbstritt
and
Thomas
Braun
 *
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489, Berlin, Germany. E-mail: thomas.braun@chemie.hu-berlin.de
First published on 6th March 2023
Abstract
The greenhouse gas SF5CF3 acts as CF3 source for the photocatalytic trifluoromethylation of arenes on using [Ir(dtbbpy)(ppy)2]PF6 (4,4′-di-tert-butyl-2,2′-dipyridyl, ppy = 2-phenylpyridine) as catalyst. The trifluoromethylation of C6D6 in the presence of 1-octanol results in the concomitant generation of 1-fluorooctane, presumably by intermediate SF4.
The CF3 group plays an important role in medicinal chemistry in part due to their ability to increase the lipophilicity of compounds, and thereby enhance the rate of absorption and transport of drugs across the blood–brain barrier.1 In 1963 Bedard et al. reported on the thermal homolysis of the trifluoromethyl derivatives CF3I, CF3Br, CF3Cl to achieve a radical trifluoromethylation of halobenzenes.2,3 Silver trifluoroacetate and TiO2 as a photocatalyst were also used to form CF3 radicals in order to synthesize trifluoromethylated aromatics.4 Kamigata et al. demonstrated the trifluoromethylation of aromatics on using a ruthenium(II)phosphine complex as a catalyst and trifluormethanesulfonylchloride as CF3 source in a thermal reaction.5 In 2011 MacMillan et al. published a photocatalytic trifluoromethylation of aromatics with [Ru(phen)3]Cl2 (phen = phenanthroline) as catalyst again using F3CSO2Cl as a source for the trifluoromethyl group.6 The research group of Hu et al. described a photochemical trifluoromethylation reaction in which an electron donor–acceptor complex between an aromatic thiophenolate anion and trifluoromethylphenylsulfone initiates a single electron transfer, and therefore the generation of CF3 radicals.7 In other photocatalytic trifluoromethylations of aromatics CF3SO2Na8 or trifluoromethanesulfonic anhydride9 were used as trifluoromethyl radical source. Note also that in 2009 MacMillan et al. reported on an enantioselective trifluoromethylation of aldehydes via a photoredox process coupled with organocatalysis on using the Ir redox catalyst [Ir(dtbbpy)(ppy)2]PF6 (4,4′-di-tert-butyl-2,2′-dipyridyl, ppy = 2-phenylpyridine). In an reductive quenching cycle CF3I is being reduced to give a CF3 radical.10 Furthermore, [Ir(dtbbpy)(ppy)2]PF6 was used in a photocatalytic approach for the trifluoromethylation of alkynes.11 Note that apart from CF3 transfer other fluoroalkyl groups can be transferred as well via photocatalysis.12
In the last few years the activation of the greenhouse gas SF6 and its use as fluorinating agent has been studied extensively.13–18 Conversions are often initiated by an electron transfer to SF6 to yield SF6−. The latter can either transform into a SF5 radical and a fluoride or SF5− and a fluoro radical.16,17,19 Examples for the photochemical activation of SF6 include the application of the photocatalysts [Ru(phen)3]Cl2,6 4,4-dimethoxybenzophenone15 or [Ir(dtbbpy)(ppy)2]PF618 The SF6 derivative SF5CF3 has also a high global warming potential and a long atmospheric life-time.20 It is assumed that SF5CF3 is a breakdown product of SF6 in high-voltage equipment. The latter contains fluoropolymers which are sources for CF3 moieties that can react with SF5 radicals formed by high voltage discharges.20 However, electron transfer to SF5CF3 can result in the generation of SF5− and a CF3 radical. Thus, the formation of SF5− has been observed in low-energy attachment experiments by mass spectrometry.21,22 SF5CF3 activation processes are rare and in solution only one example is known in the literature, to the best of our knowledge.14
In this contribution we report on an unprecedented trifluoromethylation of aromatic compounds by photocatalytic generation of CF3 radicals from the greenhouse gas SF5CF3.
Irradiation of SF5CF3 at a wavelength of 353 nm in C6D6 in the presence of 4,4′-dimethoxybenzophenone as an organic photocatalyst (Scheme 1) led to the generation of small amounts of α,α,α-trifluorotoluene-d5. Similar, only traces of α,α,α-trifluorotoluene-d5 were observed when 10-phenylphenothiazine (Scheme 1) was used to activate SF5CF3. However, irradiation of SF5CF3 in the presence of the photoredox catalyst [Ir(dtbbpy)(ppy)2]PF6 (Scheme 1), triethylamine and cesium carbonate with a 456 nm LED lamp for 16 h in C6D6 yielded α,α,α-trifluorotoluene-d5 with a turnover number (TON) of 410 (TON are based on the concentration of the catalyst on using 1,4-difluorobenzene as internal standard) (Scheme 2). Note that without the presence of Et3N the conversion does not proceed. Attempts to use K2HPO4 as an alternative base failed. Triethylamine and cesium carbonate in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 led to the highest turnover numbers. 19F NMR experiments revealed the presence of DF when no Cs2CO3 was present. Using 4-methoxydiphenylamine and Cs2CO3 as bases in a ratio of 1:1 led to a decrease of the TON to 26. Note that with the photoredox catalyst [Ru(bpy)3](PF6)2 or [Ir(dFppy)3] lower TONs of 28 and 46, respectively, were observed in the presence of NEt3 and Cs2CO3 in a ratio of 1:1. Reactions with [Ir(dtbbpy)(ppy)2]PF6 as catalyst with only stoichiometric amounts of benzene in acetonitrile or dichloroethane as solvents gave α,α,α-trifluorotoluene-d5 only in very low yield. Mainly the generation of trifluoromethane and other minor unidentified products was observed (ratio CF3H to PhCF3: 71:1 in acetonitrile, 52:1 in dichloroethane). With dichloromethane and tetrahydrofurane as solvents in the presence of stoichiometric amounts of benzene did not lead to any product formation.
:1 led to the highest turnover numbers. 19F NMR experiments revealed the presence of DF when no Cs2CO3 was present. Using 4-methoxydiphenylamine and Cs2CO3 as bases in a ratio of 1:1 led to a decrease of the TON to 26. Note that with the photoredox catalyst [Ru(bpy)3](PF6)2 or [Ir(dFppy)3] lower TONs of 28 and 46, respectively, were observed in the presence of NEt3 and Cs2CO3 in a ratio of 1:1. Reactions with [Ir(dtbbpy)(ppy)2]PF6 as catalyst with only stoichiometric amounts of benzene in acetonitrile or dichloroethane as solvents gave α,α,α-trifluorotoluene-d5 only in very low yield. Mainly the generation of trifluoromethane and other minor unidentified products was observed (ratio CF3H to PhCF3: 71:1 in acetonitrile, 52:1 in dichloroethane). With dichloromethane and tetrahydrofurane as solvents in the presence of stoichiometric amounts of benzene did not lead to any product formation.
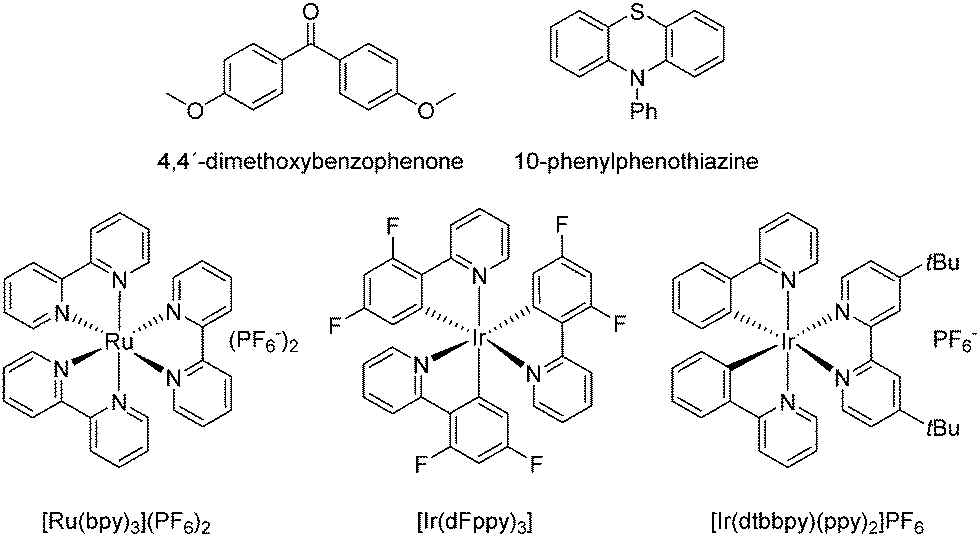 | ||
| Scheme 1 Catalysts studied for the reduction of SF5CF3. | ||
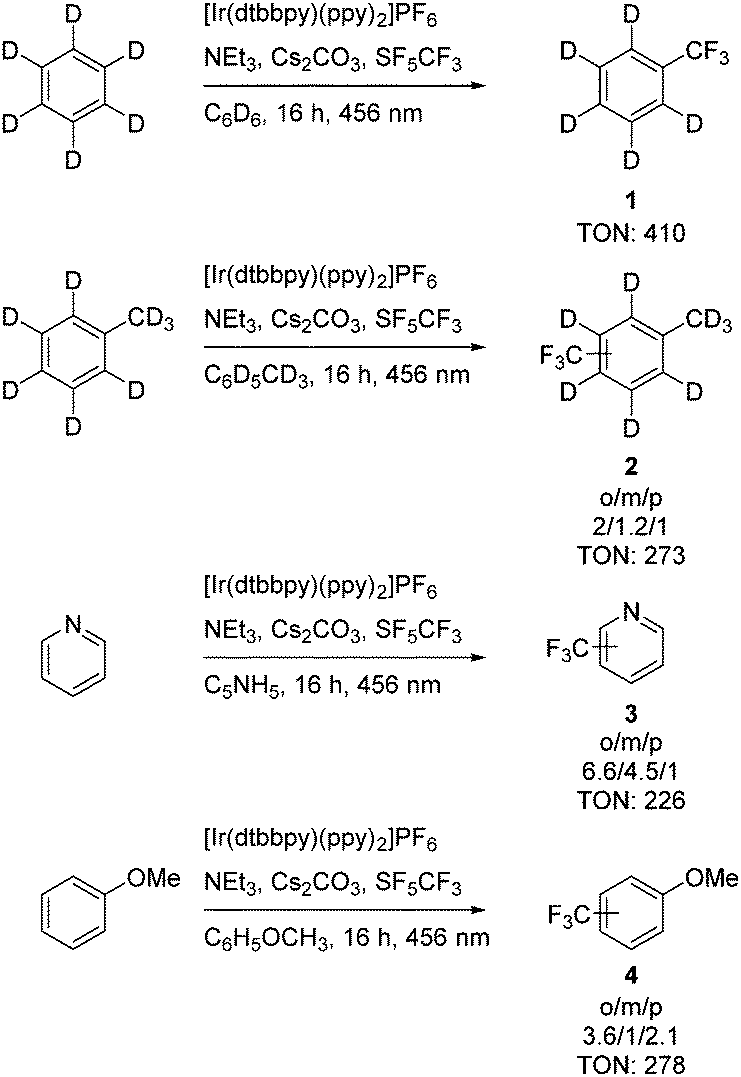 | ||
| Scheme 2 Trifluoromethylation of arenes on using [Ir(dtbbpy)(ppy)2]PF6 as photoredox catalyst. | ||
To expand the scope of the conversions, pyridine, anisole, 1,2,3,4-tetrafluorobenzene and deuterated toluene (Scheme 2) were studied towards a photochemical trifluoromethylation on using a stock solution of [Ir(dtbbpy)(ppy)2]PF as photocatalyst. All the conversions were run with the substrate as solvent. Selectivities can be explained when considering that the CF3 radical shows an electrophilic character.2,6 Transformations of toluene-d8 and anisole resulted mainly in the formation of isomers with the CF3 group in the ortho or para position. Pyridine gives mainly the ortho-product, but also considerable amounts of meta-trifluoromethylpyridine. Minor amounts of trifluoromethane were also detected. 1,2,3,4-Tetrafluorobenzene however did not show any reactivity under the standard conditions. The trifluoromethylated aromatics were characterized by their signals in the 19F NMR spectra for the CF3 groups in the para, meta or ortho positions as well as by GC-MS.
Mechanistically, it can be presumed that after irradiation at 456 nm a single electron transfer SET from the photocatalytic system to SF5CF3 leads to the SF5CF3− radical anion (Scheme 3). As mentioned above, the subsequent trifluoromethylation does not proceed without the presence of NEt3 as reductant. Thus, NEt3 can reduce the photoexcited species [Ir(dtbbpy)(ppy)2]+* generated after excitation of [Ir(dtbbpy)(ppy)2]+ in a reductive quenching cycle (Scheme 3). This step is followed by the SET from the reduced [Ir(dtbbpy)(ppy)2] (Epc = −1.51 V vs. SCE)23,24 to SF5CF3. Alternatively, [Ir(dtbbpy)(ppy)2]+* (Epc = −0.96 V vs. SCE)23,24 reduces SF5CF3 and the generated [Ir(dtbbpy)(ppy)2]2+ is then reduced by NEt3 in an oxidative quenching cycle. Note that photocatalytic conversions of [Ir(dtbbpy)(ppy)2]+ have a tendency to proceed via a reductive quenching cycle.10,25 However, the SF5CF3− radical anion then decomposes to give a CF3 radical and SF5−.22,26 The SF5− anion is not very stable and furnishes SF4 and fluoride.16,27 The CF3 radical reacts then with benzene forming a cyclohexadienyl radical. The generation of the latter in trifluoromethylation reactions has been proposed by Kamigata et al.5 and others.6,9,28 Oxidation of the cyclohexadienyl radical by the NEt3˙+ radical cation and subsequent deprotonation of the cyclohexadienyl cation by fluoride yields the trifluoromethylated aromatic product and NEt3·HF.2,6,9,29 Alternatively, hydrogen atom abstraction HAT from the cyclohexadienyl radical would give the intermediate Et3NH+, and in the presence of fluoride the same products.23 Cesium carbonate acts as an HF scavenger. There is no indication for the formation of HD or H2 along with an iminium ion from the NEt3 radical. However, the observed generation of trifluoromethane in acetonitrile or dichloromethane can involve HAT from the solvent or the NEt3˙+ radical. The latter is consistent with the fact that higher NEt3 concentrations deliver more CF3H.
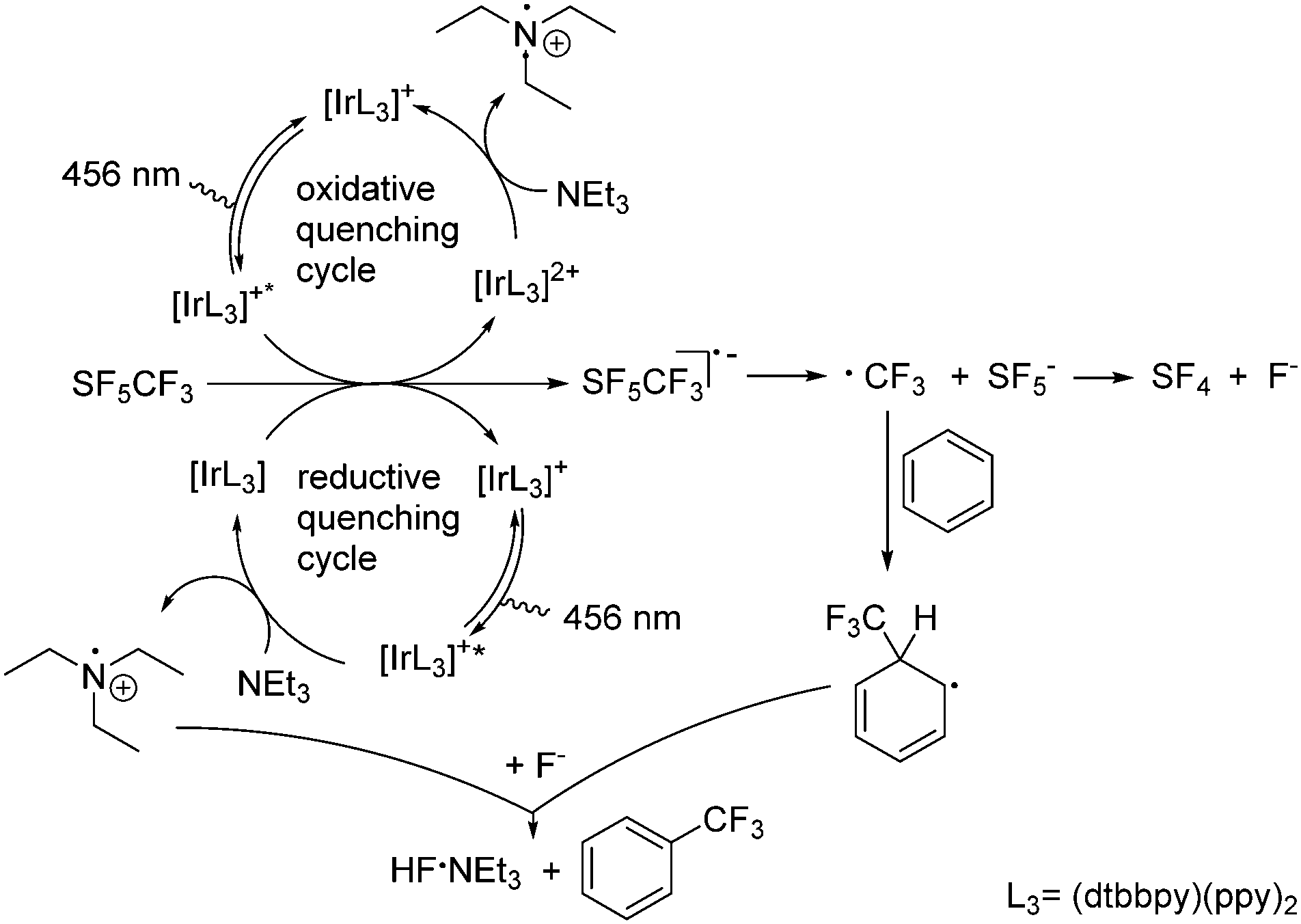 | ||
| Scheme 3 Proposed mechanism for the trifluoromethylation of arenes by SF5CF3. | ||
Furthermore, to confirm the intermediate formation of a CF3 radical, TEMPO (2,2,6,6-tetramethylpiperidinyloxyl) was added to the photocatalytic trifluoromethylation reaction of C6D6, and indeed TEMPO inhibited the conversion (Scheme 3). However, after 96 h TEMPO-CF3 as well as the α,α,α-trifluorotoluene-d5 were observed in a ratio of 1:2. The formation of SF4 was not observed by low temperature NMR experiments, presumably because of the presence of Cs2CO3. Nevertheless, when the trifluoromethylation of C6D6 was run in the presence of 1-octanol in the generation of 1-fluorooctane was observed, which was identified by GC-MS and 19F NMR spectroscopy (Scheme 4). The experiment suggests the deoxyfluorination of the alcohol by intermediate SF4.30 Hence, trifluoromethylation steps can be coupled with the fluorination of an alcohol. However, under the UV irradiation SF4 might also be further reduced yielding elemental sulfur, as it was also proposed by Nagorny et al. for the activation of SF6 in the presence of the organophotocatalyst 4,4-dimethoxybenzophenone.15 Indeed, when PPh3 was added to the reaction mixture after the photolytic formation of C6D5CF3, the generation of small amounts of SPPh3 (7) was observed after heating the reaction mixture for two hours at 60 °C, which might be due to the presence of sulfur (Scheme 4).
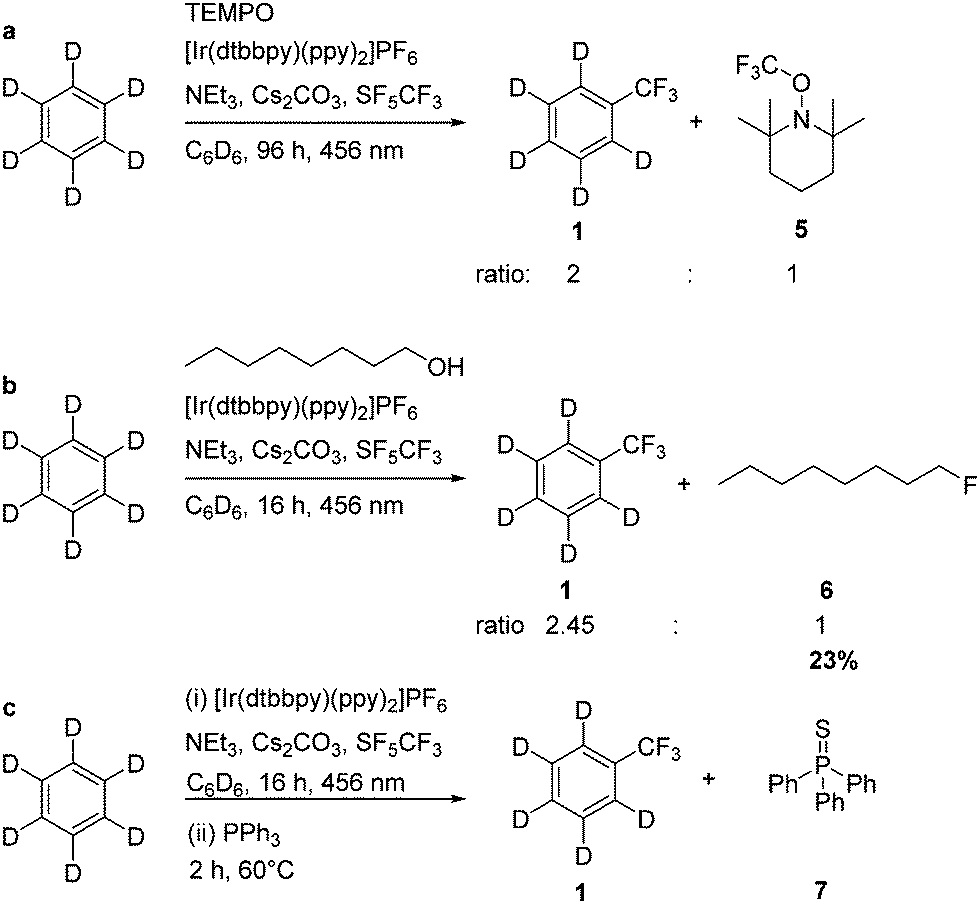 | ||
| Scheme 4 Quenching of the photocatalytic trifluoromethylation with TEMPO or 1-octanol (a and b); generation of Ph3P = S after addition of PPh3 to the reaction mixture (c). | ||
In conclusion, a catalytic photoredox process for the activation of the greenhouse gas SF5CF3 and a concomitant trifluoromethylation of aromatics were reported. SF5CF3 acts as source for CF3 radicals and at the same time SF4 and sulfur can be furnished. The former can be used for fluorination to couple the trifluoromehylation of aromatics with a fluorination of an alcohol. Note also that iridium photocatalyzed processes were applied for the defluorination of fluorinated organic substrates,31 which was not observed for the described transformations.
Experiments, data collection (NMR and GC-MS) and analysing the data was performed by D. Herbstritt. The manuscript was drafted by D. Herbstritt. T. Braun supervised the work, designed the project and contributed to manuscript writing.
We acknowledge financial support from the CRC 1349 funded by the Deutsche Forschungsgemeinschaft (German Research Foundation; Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – Projektnummer 387284271 – SFB 1349) The work was also supported by the DFG under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. G. Perrone, P. Vitale, A. Panella, A. Tolomeo and A. Scilimati, ed., Current and emerging applications of fluorine in medicinal chemistry, MedComm, 2015, vol. 11 Search PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- A. Studer, Angew. Chem., Int. Ed., 2012, 124, 9082–9090 CrossRef.
- E. S. Huyser and E. Bedard, J. Org. Chem., 1964, 29, 1588–1590 CrossRef CAS.
- C. Lai and T. E. Mallouk, J. Chem. Soc., Chem. Commun., 1993, 1359–1361 RSC.
- N. Kamigata, T. Ohtsuka, T. Fukushima, M. Yoshida and T. Shimizu, J. Chem. Soc., Perkin Trans. 1, 1994, 1339–1346 RSC.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- Z. Wei, Z. Lou, C. Ni, W. Zhang and J. Hu, Chem. Commun., 2022, 58, 10024–10027 RSC.
- C. Tian, Q. Wang, X. Wang, G. An and G. Li, J. Org. Chem., 2019, 84, 14241–14247 CrossRef CAS.
- Y. Ouyang, X.-H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2018, 57, 6926–6929 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS.
- T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937–1945 CrossRef CAS PubMed.
- (a) N. J. W. Straathof, S. E. Cramer, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2016, 128, 15778–15782 CrossRef; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed; (c) T. Liu, J. Liu, Y. Hong, H. Zhou, Y.-L. Liu and S. Tang, Synthesis, 2022, 1919–1938 Search PubMed; (d) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed; (e) S. Barata-Vallejo, S. M. Bonesi and A. Postigo, Org. Biomol. Chem., 2015, 13, 11153–11183 RSC.
- (a) L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 127, 10798–10802 CrossRef; (b) M. Wozniak, T. Braun, M. Ahrens, B. Braun-Cula, P. Wittwer, R. Herrmann and R. Laubenstein, Organometallics, 2018, 37, 821–828 CrossRef CAS; (c) M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571–2575 RSC; (d) F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 130, 5045–5049 CrossRef; (e) T. Eder, F. Buß, L. F. B. Wilm, M. Seidl, M. Podewitz and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202209067 CAS; (f) P. Tomar, T. Braun and E. Kemnitz, Chem. Commun., 2018, 54, 9753–9756 RSC; (g) P. Holze, B. Horn, C. Limberg, C. Matlachowski and S. Mebs, Angew. Chem., Int. Ed., 2014, 126, 2788–2791 CrossRef; (h) D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS; (i) B. S. N. Huchenski and A. W. H. Speed, Chem. Commun., 2021, 57, 7128–7131 RSC; (j) R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Chem. – Eur. J., 2021, 27, 6460–6464 CrossRef CAS PubMed; (k) A. Taponard, T. Jarrosson, L. Khrouz, M. Médebielle, J. Broggi and A. Tlili, Angew. Chem., Int. Ed., 2022, 61, e202204623 CrossRef CAS; (l) S. Kim and P. Nagorny, Org. Lett., 2022, 24, 2294–2298 CrossRef CAS PubMed; (m) C. Berg, T. Braun, M. Ahrens, P. Wittwer and R. Herrmann, Angew. Chem., Int. Ed., 2017, 56, 4300–4304 CrossRef CAS; (n) D. Dirican, M. Talavera and T. Braun, Chem. – Eur. J., 2021, 27, 17707–17712 CrossRef CAS PubMed; (o) D. Dirican, N. Pfister, M. Wozniak and T. Braun, Chem. – Eur. J., 2020, 26, 6945–6963 CrossRef CAS PubMed.
- L. Zámostná, T. Braun and B. Braun, Angew. Chem., Int. Ed., 2014, 53, 2745–2749 CrossRef PubMed.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Org. Lett., 2021, 23, 190–194 CrossRef CAS PubMed.
- G. Iakobson, M. Pošta and P. Beier, J. Fluorine Chem., 2018, 213, 51–55 CrossRef CAS.
- D. Rombach and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 132, 306–310 CrossRef.
- T. A. McTeague and T. F. Jamison, Angew. Chem., Int. Ed., 2016, 55, 15072–15075 CrossRef CAS PubMed.
- A. Akhgarnusch, R. F. Höckendorf and M. K. Beyer, J. Phys. Chem. A, 2015, 119, 9978–9985 CrossRef CAS PubMed.
- W. T. Sturges, T. J. Wallington, M. D. Hurley, K. P. Shine, K. Sihra, A. Engel, D. E. Oram, S. A. Penkett, R. Mulvaney and C. A. M. Brenninkmeijer, Science, 2000, 289, 611–613 CrossRef CAS PubMed.
- C. M. R. A. Kennedy, Int. J. Mass Spectrom., 2001, 206, vii–x CrossRef.
- W. Sailer, H. Drexel, A. Pelc, V. Grill, N. J. M. Eugen Illenberger, J. D. Skalny, T. Mikoviny, P. Scheier and T. D. Märk, Chem. Phys. Lett., 2001, 71–78 Search PubMed.
- L. Zhou, Molecules, 2021, 26, 7051 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl Jr., R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS.
- (a) R. Chim, R. Kennedy and R. Tuckett, Chem. Phys. Lett., 2003, 367, 697–703 CrossRef CAS; (b) S. Solovev, A. Palmentieri, N. D. Potekhina and T. E. Madey, J. Phys. Chem. C, 2007, 111, 18271–18278 CrossRef CAS.
- (a) J. T. Goettel, N. Kostiuk and M. Gerken, Angew. Chem., Int. Ed., 2013, 125, 8195–8198 CrossRef; (b) N. Kostiuk, J. T. Goettel and M. Gerken, Inorg. Chem., 2020, 59, 8620–8628 CrossRef CAS PubMed.
- C. F. Harris, C. S. Kuehner, J. Bacsa and J. D. Soper, Angew. Chem., Int. Ed., 2018, 57, 1311–1315 CrossRef CAS PubMed.
- J. Xie, X. Yuan, A. Abdukader, C. Zhu and J. Ma, Org. Lett., 2014, 16, 1768–1771 CrossRef CAS PubMed.
- G. A. Boswell Jr., W. C. Ripka, R. M. Scribner and C. W. Tullock, ed., Organic Reactions, Fluorination with Sulfur Tetrafluoride, 21st edn, 1974 Search PubMed.
- (a) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed; (b) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00495c |
| This journal is © The Royal Society of Chemistry 2023 |