
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative dihydrogen activation at a Na(I)2/Mg(I)2 ensemble†
Han-Ying
Liu
,
Samuel E.
Neale
 ,
Michael S.
Hill
*,
Mary F.
Mahon
,
Claire L.
McMullin
* and
Benjamin L.
Morrison
,
Michael S.
Hill
*,
Mary F.
Mahon
,
Claire L.
McMullin
* and
Benjamin L.
Morrison
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bth.ac.uk
First published on 1st March 2023
Abstract
[{SiNDipp}MgNa]2 ({SiNDipp} = {CH2SiMe2N(Dipp)}2; Dipp = 2,6-i-Pr2C6H3) reacts directly with H2 to provide a heterobimetallic hydride. Although the transformation is complicated by the simultaneous disproportionation of magnesium, computational density functional theory (DFT) studies suggest that this reactivity is initiated by orbitally-constrained 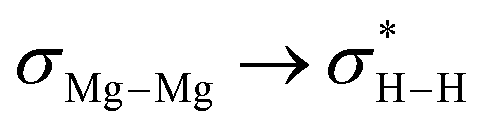 and
and 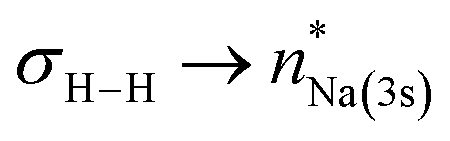 interactions between the frontier MOs of both H2 and the tetrametallic core of [{SiNDipp}MgNa]2.
interactions between the frontier MOs of both H2 and the tetrametallic core of [{SiNDipp}MgNa]2.
The binding and cleavage of the strong (436 kJ mol−1) H–H σ-bond of dihydrogen at mid and late transition metal centres provided a fulcrum for both a fundamental understanding of d-block complex reactivity and the development of numerous catalytic and stoichiometric processes.1,2 This chemistry is enabled by the narrowly spread manifold of nd valence orbital energies involved in the synergic activation of the H2 molecule (Fig. 1a).
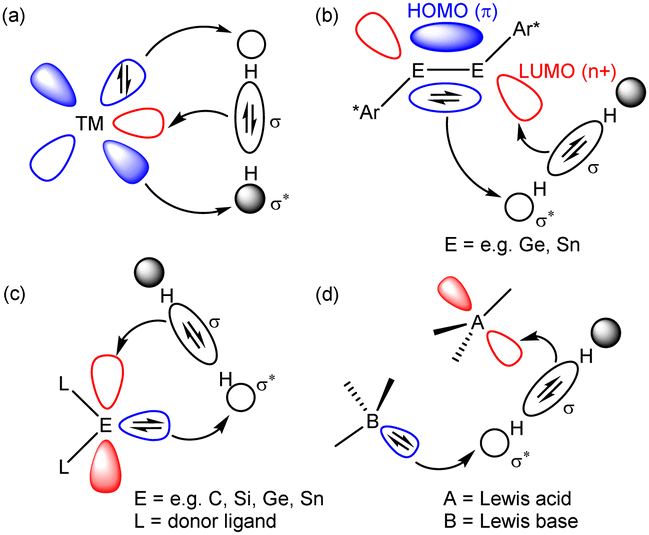 | ||
| Fig. 1 Modes of synergic H2 activation by (a) a typical late transition metal, (b) heavier group 14 element alkyne analogues, (c) tetrelenes and (d) an archetypal FLP. | ||
As articulated in Power's influential review of 2010,3 typical s- and p-block compounds do not generally display the modest separation in valence orbital energies (≤4 eV) required to effect similar interactions. From 2005 onwards, however, it was shown that a selection of multiply bonded and carbenoid compounds derived from, primarily, the heavier elements of group 13–15 in low formal oxidation states can provide frontier orbitals with an energetic and spatial disposition appropriate for H–H oxidative addition (Fig. 1b and c).4–7 Although semantics might identify frustrated Lewis pairs (FLPs) as a separate sub-category of main group chemistry,8,9 a commonly considered mode of H2 activation (Fig. 1d) attributes a significant degree of complementarity.10 From this viewpoint, therefore, the reactivity of an FLP toward dihydrogen may be rationalised in an analogous manner, albeit one in which the frontier orbitals are introduced as spatially separated basic (HOMO) and acidic (LUMO) components.
There is now significant precedent for orbitally unconstrained (i.e. highly polarised) H–H σ-bond metathesis at the Ae–X (Ae = alkaline earth, Mg, Ca, Sr, Ba) bonds of group 2 derivatives in their conventional 2+ oxidation state.11 The thermodynamic viability of H2 addition to the Mg–Mg bonds of Jones and co-workers’ β-diketiminato (e.g.1–3, Fig. 2) and guanidinato Mg(I) complexes (ΔH ≈ 24 kcal mol−1) was also assessed soon after their initial report in 2007.12 Despite the synthesis of more than 20 further comparable species, however, and an intense exploration of Mg(I) compounds as potent reducing reagents,13 reductive H2 activation at an isolable Mg–Mg bonded molecule, either thermal or photoactivated, remains to be described.14 In contrast, Harder and co-workers have recently reported that treatment of both the unique Mg(0) species, [(BDI*)MgNa]2 (4; BDI* = HC{C(t-Bu)N(DiPeP)}2; DiPeP = 2,6-(3-pentyl)-phenyl) and the mixed oxidation state [Mg(I)/Mg(0)] compound, [(BDI*)MgMgMg(BDI*)] (5), derived therefrom, with H2 (1.5 bar) generates the magnesium hydride, [(BDI*)MgH] (3) in ca. 30% isolated yield (Fig. 2).15 Although no further products were identified from the reaction of 4, the transformation of 5 was noted to result in the extrusion of metallic magnesium. In neither case, however, was the potential mode of H2 activation assessed.
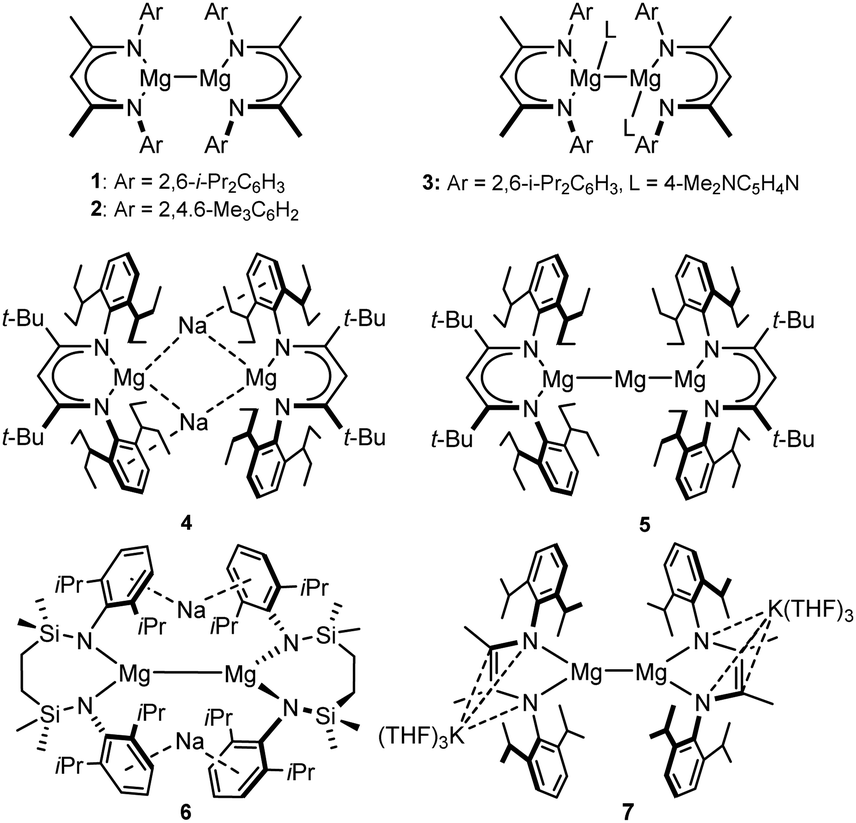 | ||
| Fig. 2 Selected examples of Mg–Mg bonded magnesium(I) derivatives. | ||
Inspired in part by the synthesis of 4, we have described the topologically-related, [{SiNDipp}MgNa]2 (6; [{SiNDipp} = {CH2SiMe2N(Dipp)}2; Dipp = 2,6-i-Pr2C6H3)] (Fig. 2).16 Although 6 bears a resemblance to the previously reported species of Yang and co-workers (e.g.7),17 the redox-innocence of the {SiNDipp} ligands and the N-aryl encapsulation of the sodium cations provides significant electronic discrimination. This is reflected in the structure of 6 through an elongation of the Mg–Mg bond [3.2124(11) Å (6) versus 2.9370(18) Å (7)]. Computational (NBO, QTAIM) analysis also identified a degree of electronic cooperativity between the magnesium and sodium centres. This manifests as a pronounced yellow colour arising from an absorption at 409 nm (3.0 eV) attributed to a transition between the Mg–Mg σ-bond (HOMO), arising from overlap of the magnesium 3s wavefunctions, and a LUMO largely represented by an out-of-phase combination of the sodium 3s atomic orbitals (Fig. 3a).18
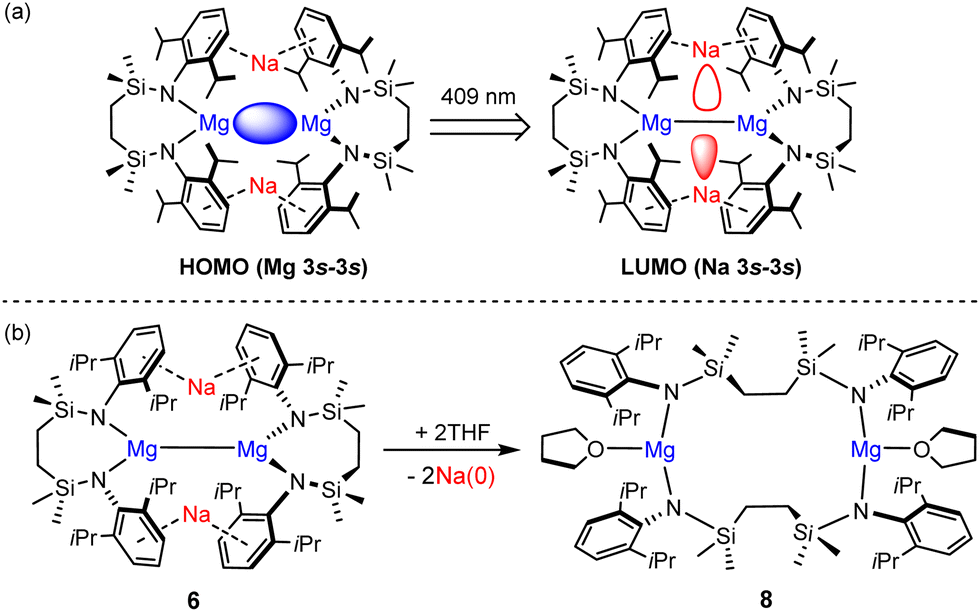 | ||
| Fig. 3 (a) Representations of the calculated frontier molecular orbitals of 6; (b) reactivity of compound 6 toward THF to form 8 and elemental sodium. | ||
The behaviour of 6 supports an interpretation of its bonding as a tetrametallic ensemble. Most strikingly, treatment with non-reducible bases such as THF (Fig. 3b) results in the selective extrusion of metallic sodium and oxidation of the Mg(I) centres to the more conventional Mg(II) state.18 Although these processes are also characterised by a structural reorganisation of the chelated diamide spectator ligand to form the macrocyclic species 8, computational studies indicated that intramolecular electron transfer is expedited via the Mg(I)-derived HOMO and Na(I)-derived LUMO. The narrow separation in energy between these frontier orbitals (ca. 3 eV) is more reminiscent of the low oxidation state p-block species (typically 2–3 eV) depicted in Fig. 1b and c than previously reported Mg–Mg bonded derivatives (≥4 eV).12 With these observations in mind, therefore, it was speculated that the alkali metal-centred frontier orbitals of 6 may also facilitate a cooperative reaction with H2.
Although treatment of a degassed benzene solution of 6 with H2 (2 bar) provided no evidence of an observable reaction over 12 hours at room (ca. 25 °C) temperature, monitoring by 1H NMR spectroscopy indicated conversion to a predominant new compound (9) was induced after heating at 40 °C for 3 days. This process occurred with the formation of a black metallic precipitate. Compound 9 was isolated as colourless single crystals from hexane at room temperature. The resultant X-ray diffraction analysis (Fig. 4) revealed that compound 9 is a centrosymmetric heterobimetallic hydride species. The hydride ligands of 9 were located and refined with a riding Uiso value to be trigonally encapsulated by a still chelated diamidomagnesium centre and two sodium cations. The sodium atoms are bound in a η6-fashion by each of the Dipp substituents of the chelated {SiNDipp} ligands, but are differentiated by their interactions with an additional diamide dianion that now adopts a {Na2-μ-κ1-N,μ-κ1-N′-Na2′} bridging mode reminiscent of those to Mg in the macrocyclic species 8.18 While Na1/Na11 engage via further polyhapto interactions with C31–C36 (and C311–C361) comprising the Dipp substituents of the bridging dianion, the coordination spheres of Na2/Na21 are completed by N3/N31. The Mg1–N1 [1.9795(13) Å] and Mg1–N2 [1.9711(14) Å] bonds of 9 are significantly shorter than the Mg–N distances observed in 6 (avg. 2.08 Å),16 consistent with the oxidation of Mg(I) to Mg(II). While several amido-derived Na/Mg hydrides have been reported to result from either β-C–H elimination or metal amide/Si–H metathesis,19 compound 9 is the first such species in which the hydride ligands arise from the direct activation of dihydrogen. Although its presence could not be identified by 1H NMR spectroscopy in d6-benzene solution, dihydrogen was confirmed as the source of the hydride ligands of 9 by performance of a further reaction of 6 with D2. This latter process provided similar observations and resulted in the isolation of 9-d2, which was characterised by a singlet signal at δ 4.16 ppm as the sole observable resonance in its 2H NMR spectrum in benzene.
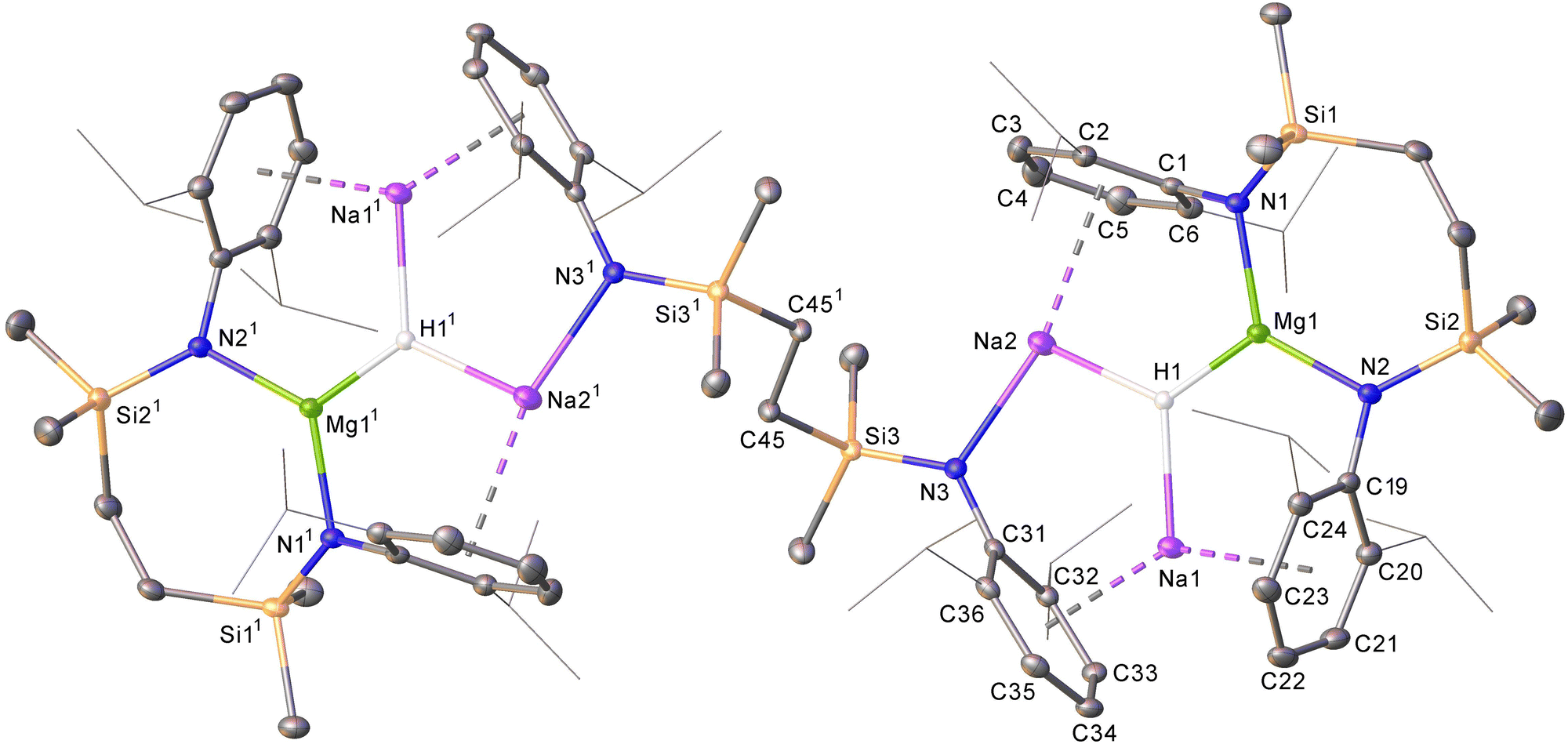 | ||
| Fig. 4 Displacement ellipsoid (30% probability) plot of compound 9. H atoms, apart from H1 and H11, and occluded hexane solvent have been omitted and iso-propyl groups are shown as wireframe for clarity. Selected bond lengths (Å) and angles (°): Mg1–N1 1.9795(13), Mg1–N2 1.9711(14), Na1–C22 2.7776(18), Na1–C33 2.7840(16), Na1–C34 2.7344(16), Na2–N3 2.2895(14), Na2–C1 3.1096(16), Na2–C2 3.0148(17), Na2–C3 2.9391(19), Na2–C4 2.948(2), Na2–C5 3.023(2), Na2–C6 3.1209(18), N1–Mg1–N2 126.44(6), N1–Mg1–N2 126.44(6). Operations to generate symmetry equivalent atoms: 1 − x, 2 − y, 1 − z. | ||
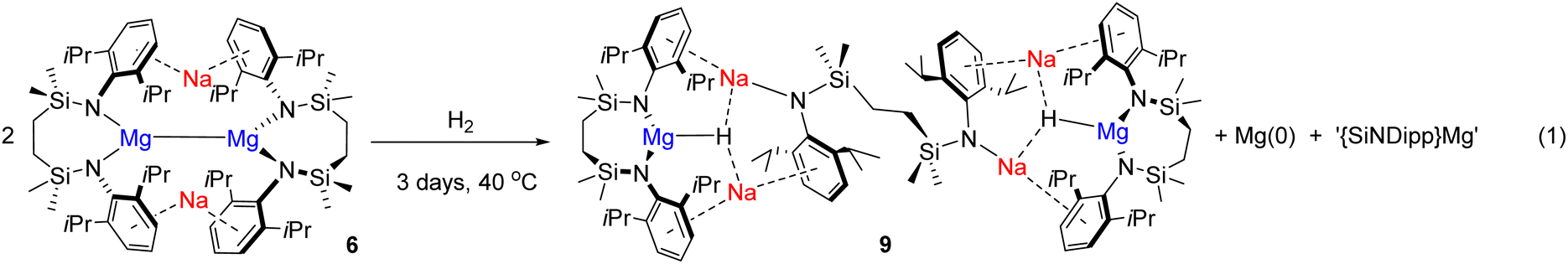
Acid digestion and quantitative analysis by ICP-OES revealed that the solid residue deposited during the reaction of 6 with H2 comprised magnesium as the sole constituent s-block metal with levels of sodium below the detection limit of the technique (see ESI†). Furthermore, the quantity of magnesium was consistent with the reaction stoichiometry shown in eqn (1), which we surmise results from the formal disproportionation of the Mg(I) centres of compound 6.
 | (1) |
DFT calculations (BP86-D3BJ/BS2(benzene)//BP86/BS1 level of theory, see the ESI† for full details) were performed to assess the kinetics of H2 addition to 6 (denoted as I in the computational study) and the structure of the resulting H2 adduct. Initial H2 addition was identified to take place viaTS(I–II) and a barrier of +18.5 kcal mol−1 to form II (+14.1 kcal mol−1). Subsequent H2 reorientation via a low-lying saddle point, TS(II–III) (+13.6 kcal mol−1), affords a more stable adduct, III (+11.0 kcal mol−1), in which a terminus of the H2 molecule is directed towards the Mg–Mg σ bond (Fig. 5a).20
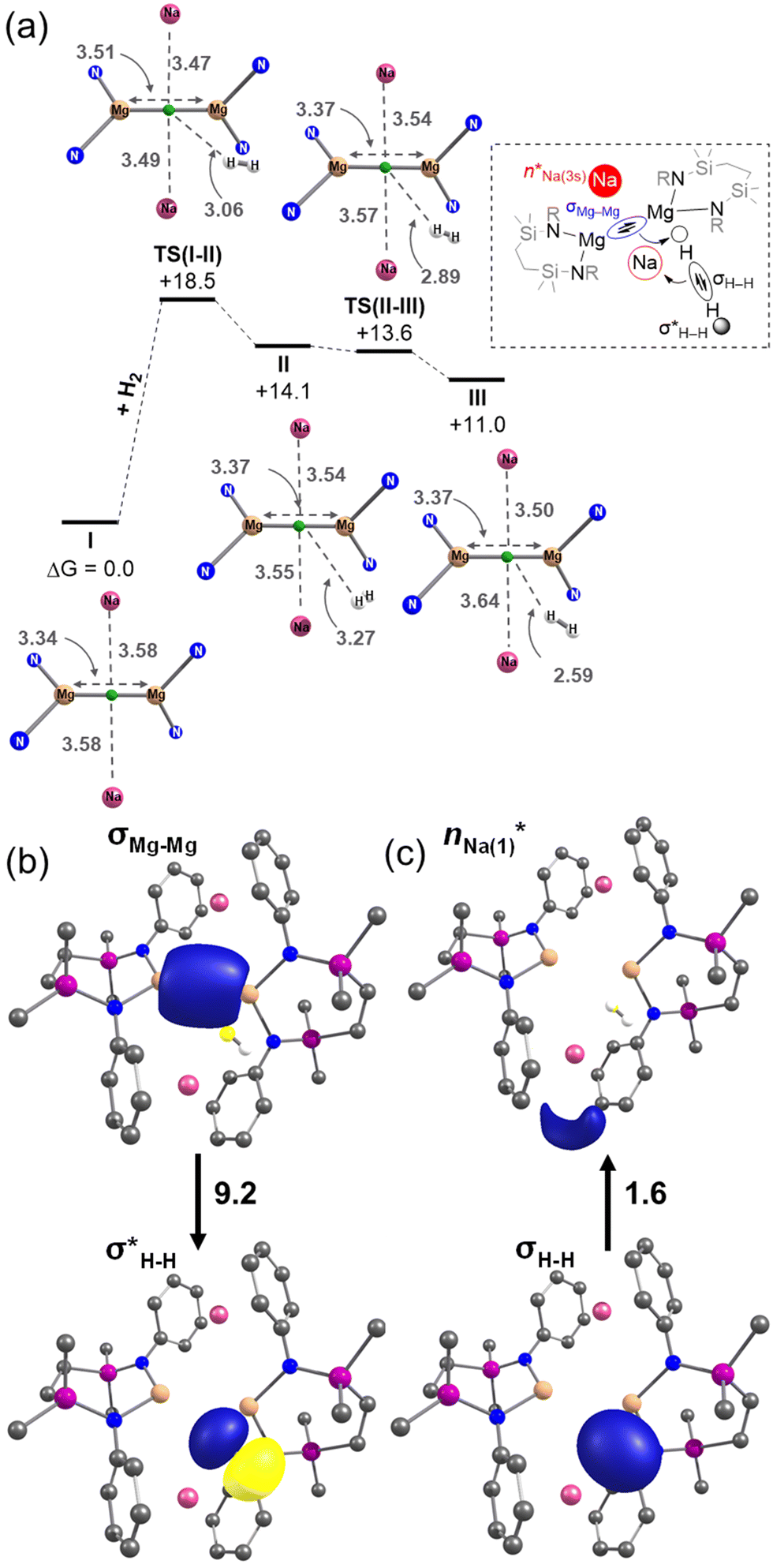 | ||
Fig. 5 (a) Computed free energy profile (BP86-D3BJ/BS2(benzene)//BP86/BS1), in kcal mol−1 for initial addition of H2 to 6/I. (SiNDipp ligand backbone and aromatic rings removed for clarity, and interatomic distances, and distances between each Na and the Mg–Mg midpoint (green) quoted in Å). (b)  interaction of III, identified by second-order perturbation energy analysis of the Fock matrix in NBO basis; (c) the corresponding interaction of III, identified by second-order perturbation energy analysis of the Fock matrix in NBO basis; (c) the corresponding 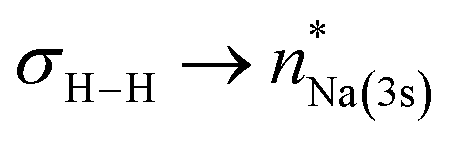 interaction of III. The donor–acceptor interaction energies, ΔE(2), are quoted in kcal mol−1. interaction of III. The donor–acceptor interaction energies, ΔE(2), are quoted in kcal mol−1. | ||
NBO-based donor–acceptor interaction analysis of III reveals two appreciable interactions between H2 and the tetrametallic Mg2Na2 unit; a 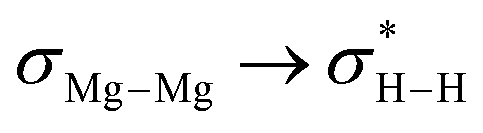 interaction (ΔE(2) = 9.2 kcal mol−1; Fig. 4b) supplemented by a subtle but still significant engagement via
interaction (ΔE(2) = 9.2 kcal mol−1; Fig. 4b) supplemented by a subtle but still significant engagement via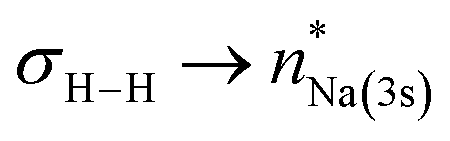 (ΔE(2) = 1.6 kcal mol−1; Fig. 5c).
(ΔE(2) = 1.6 kcal mol−1; Fig. 5c).
While we cannot yet discount alternative polarised metathesis or radical-based pathways,11,14 these deductions infer that the frontier orbital interactions invoked in the initial coordination of H2 to 6 bear some analogy to those of the generalised d- and p-block-derived systems depicted in Fig. 1. The semi-heterogeneous nature of the Mg(0) extrusion process and the structural complexity of 9, however, dictate that the onward process does not lend itself to further prudent mechanistic analysis.
These combined experimental and theoretical results imply that the H2 activation process invoked through its interaction with 6 may be rationalised as a consequence of the frontier orbitals arising from the low oxidation state assembly of the two dissimilar s-block elements (Fig. 3a). This perspective serves to further discriminate the chemistry of the low oxidation state heterobimetallic species, 6, from that so far deduced for the previously described Mg–Mg bonded Mg(I) derivatives (e.g.1–3 and 7).12,13 Similarly, the conceptual framework provided by the intermediate III signposts a potential ability to manipulate the frontier orbitals of related systems toward even more challenging small molecule transformations through the templated assembly of further low oxidation state arrays of dissimilar s-block element centres. We are continuing to explore these possibilities with a broader scope of complex types, metal identities and small molecule substrates.
HYL performed the synthesis and characterisation of the new compounds reported. MSH and CLM conceptualised the study and finalised the manuscript for submission. SEN and BLM performed the computational analysis and MFM finalised the X-ray diffraction analysis of 9 for publication.
We thank the EPSRC (EP/R020752/1) for support of this research. This research made use of the Balena and Anatra High Performance Computing (HPC) Services at the University of Bath. (University of Bath, Research Computing Group, DOI: https://doi.org/10.15125/b6cd-s854).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. J. Kubas, Acc. Chem. Res., 1988, 21, 120–128 CrossRef.
- G. J. Kubas, J. Organomet. Chem., 2009, 694, 2648–2653 CrossRef CAS.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042–6044 RSC.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- F. G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- T. A. Rokob, I. Bako, A. Stirling, A. Hamza and I. Papai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS PubMed.
- H. Bauer and S. Harder, in Early Main Group Metal Catalyzed Hydrogenation, in Early Main Group Metal Catalysis: Concepts and Reactions, ed., S. Harder, 2020, pp. 175–199 Search PubMed.
- (a) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed; (b) A. Datta, J. Phys. Chem. C, 2008, 112, 18727–18729 CrossRef CAS.
- For a recent review, see L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef.
- Here, we draw a demarcation between such divalent {Mg–Mg}-bonded Mg(I) compounds and radicular Mg(I) species. Notably, a recent report by Harder and co-workers has highlighted that ball milling of K/KI and [(BDI)MgI(OEt2)] under H2 provides a ca. 1.3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 mixture of compound 1 and [(BDI)MgH]2. While compound 1 remains unreactive, the latter species is interpreted to result from the generation and persistence of highly reactive ‘unquenched’ [(BDI)Mg˙], radicals under the solvent-free conditions and which are then reactive toward H2. See:
(a) D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, e202200138 CrossRef . The hydrogenation of Mg(I) dimers using 1,3-cyclohexadiene has also been described. See: ;
(b) R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
:1.0 mixture of compound 1 and [(BDI)MgH]2. While compound 1 remains unreactive, the latter species is interpreted to result from the generation and persistence of highly reactive ‘unquenched’ [(BDI)Mg˙], radicals under the solvent-free conditions and which are then reactive toward H2. See:
(a) D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, e202200138 CrossRef . The hydrogenation of Mg(I) dimers using 1,3-cyclohexadiene has also been described. See: ;
(b) R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed. - B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–719 CrossRef PubMed.
- H. Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- Y. Liu, S. Li, X.-J. Yang, P. Yang and B. Wu, J. Am. Chem. Soc., 2009, 131, 4210–4211 CrossRef CAS PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Angew. Chem., Int. Ed., 2023, 62, e202213670 CAS.
- (a) D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O’Hara, R. E. Mulvey and R. B. Rowlings, Chem. Commun., 2002, 376–377 RSC; (b) D. V. Graham, A. R. Kennedy, R. E. Mulvey and C. T. O’Hara, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, M366–M368 CrossRef PubMed; (c) D. J. Liptrot, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2014, 20, 9871–9874 CrossRef CAS PubMed ; for a review, see:; (d) M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed.
- Based on the electronic energy, TS(II–III) is 0.1 kcal higher than II with one imaginary frequency of −58.5 cm−1. Corrections have stabilised this TS structure relative to the zero species more than intermediate II.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compound 9, details of the computational analysis and atomic coordinates. CCDC 2217279. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00710c |
| This journal is © The Royal Society of Chemistry 2023 |