
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn organic cage controlling the dimension and stability of gold nanoparticles†‡
Erich Henrik
Peters
 a and
Marcel
Mayor
*abc
a and
Marcel
Mayor
*abc
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, Basel 4056, Switzerland. E-mail: marcel.mayor@unibas.ch
bInstitute for Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), P. O. Box 3640, Karlsruhe 76021, Germany
cLehn Institute of Functional Materials (LIFM), Sun Yat-Sen University (SYSU), Xingang Xi Rd. 135, Guangzhou 510275, P. R. China
First published on 24th March 2023
Abstract
A molecular cage encapsulating gold nanoparticles is presented. Six benzylic thioethers are pointing into its cavity, stabilizing the particles in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand-to-particle-ratio in excellent yields. They are bench-stable for several months and can withstand unprecedented thermal stress of up to 130 °C, documenting the advantages of the cage-type stabilization over open-chain analogues.
:1 ligand-to-particle-ratio in excellent yields. They are bench-stable for several months and can withstand unprecedented thermal stress of up to 130 °C, documenting the advantages of the cage-type stabilization over open-chain analogues.
Owing to their optical,1–4 physical4,5 and catalytic4,6–9 properties, gold nanoparticles (Au NPs) are of major importance in labeling applications4,10–16 and as functional building blocks in molecular devices and materials.5,17–25 Ever since the pioneering works of Brust et al.,26,27 great attention has been drawn to thiol-based organic ligands for the stabilization of Au NPs due to the thiolates’ affinity for gold.28,29
Based on those seminal works, Pankau et al. proposed the use of benzylic thioether-based structures,30,31 benefiting from the quasi-reversible character of the dispersion force-type bond between thioether and gold,29,32 allowing the ligand to adopt its most favorable conformation upon Au NP formation. This ligand design allowed the synthesis of more complex sulfur-based ligands such as linear,33–36 dendritic37–39 and branched40,41 oligomers that allow the synthesis of Au NPs passivated by a small, discrete number of ligands. While these oligomers were optimized to cover and thereby passivate a well-defined area of the particle surface, the particle itself acted as the template, shaping and arranging the multidentate oligomeric ligand by the attractive interaction between the ligand's thioether and the particle's gold surface.
As an alternative strategy, the synthesis of a cage-type structure with a well-defined cavity for the nanoparticle is considered in this work. In particular, the impact of a predefined organic cage on the particle size and stability is the focus of interest. The difference between both approaches is sketched in Fig. 1. Organic cages to grow metal nanoparticles have been reported for catalytic applications,42 and there is also a single example of a rigid cage as the template for Au NP growth.43
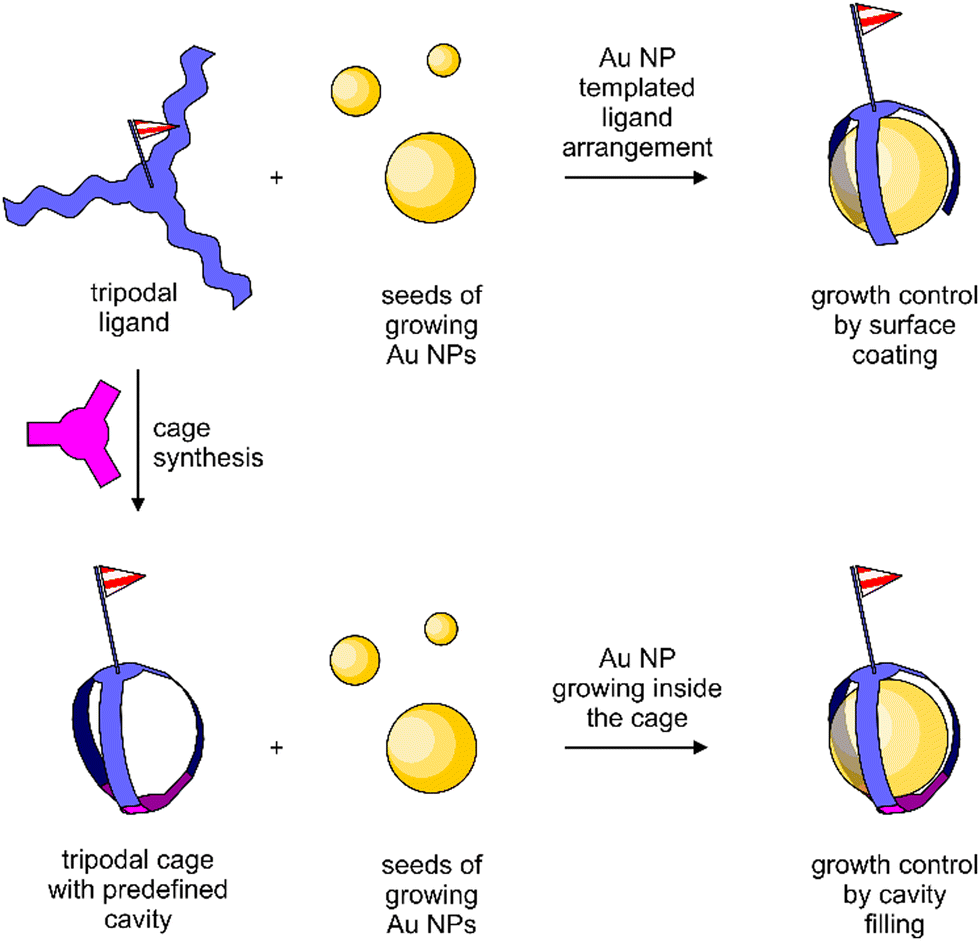 | ||
| Fig. 1 Conceptual difference between oligomeric ligands passivating the Au NP growth by surface coating (top) and Au NP growth inside an organic cage (bottom). | ||
The cage design 1 is a variation of our already reported threefold branched ligand structure for Au NPs (2)41 and is displayed in Scheme 1. In analogy to the threefold branched oligomer 2, the central tetraphenylmethane subunit 7 was decorated with three bifunctional tetraphenylmethane subunits 6 which were subsequently tied together to a cage by reacting with 1,3,5-tris(bromo methyl)benzene (10). The central tetraphenylmethane exposes an oligo(phenyleneethynylene) (OPE)-type molecular rod which turned out to be instrumental to define the outer side of the cavity. It is noteworthy that initial attempts without a sterically demanding molecular rod mounted at the central tetraphenylmethane had a pronounced tendency to close the cage with the remaining phenyl subunit of the central building block inside the cage, already occupying the cavity intended for the Au NP growth. Furthermore, the exposed triisopropylsilyl (TIPS)-protected terminal alkyne may act as a functional group, such that monofunctionalized Au NPs are expected.44
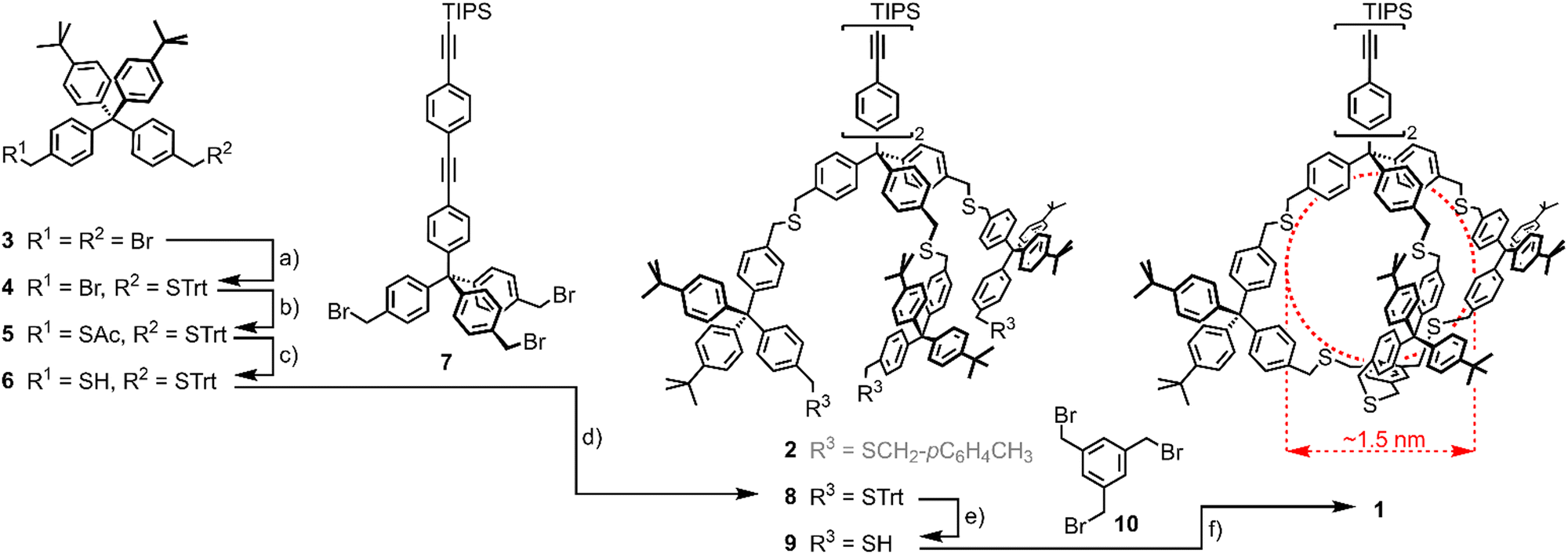 | ||
| Scheme 1 Synthesis of Cage-type ligand 1. (a) TrtSH, NaH, THF, rt, 2 h, 34%; (b) KSAc, DMF, rt, 30′, 93%; (c) (1) K2CO3, MeOH/THF 1:1, rt, 30′, (2) HCl (aq.), 96%; (d) THF, NaH, rt, 15 h, 84%; (e) SiEt3H, TFA, CH2Cl2, rt, 5′, 93%; (f) 10, NaH, THF, rt, 72 h, 52%. Trt = trityl, DMF = N,N-dimethylformamide, THF = tetrahydrofurane, TFA = trifluoroacetic acid. In red, cage 1's cavity's expected dimensions are displayed. Note that the threefold branched oligomer 2 has been previously synthesised41 and is thus displayed in grey. | ||
The synthesis of the cage target structure 1 is displayed in Scheme 1. The assembly of the central benzylic tribromide 7 bearing a lengthy OPE was already reported.41 The main cage building block 6 was obtained from the benzylic dibromide 333via the orthogonally protected disulfide 5. One bromide was substituted by trityl mercaptan (TrtSH) in THF, using sodium hydride (NaH) as a base to give precursor 4 in mediocre yields. The substitution of the second bromide by thioacetate provided the orthogonally protected precursor 5. Subsequent selective deprotection of the acetate-protected benzylic thiol under basic conditions gave the monoprotected cage building block 6. Treating the tribromide 7 with 4 equivalents (eq.) of thiol 6 in THF with NaH as the base provided 8 in good 84% isolated yield after gel permeation chromatography (GPC). Acidic deprotection with TFA and triethylsilane (SiEt3H) as a cation scavenger gave trithiol 9 in very good 93% isolated yield.
Threefold nucleophilic substitution of the bromines of 10 with the thiols of 9 resulted in the cage target structure 1. After a first SN2 reaction between 9 and 10, intramolecular ring closure was favored over intermolecular reactions under high-dilution conditions. A 1:1 mixture of 9 and 10 with about 10 eq. of NaH was stirred as 0.4 mM solution in THF for 72 hours at room temperature, giving cage 1 as a colorless solid after GPC in a very good 52% yield, considering the threefold macrocyclization.
To be able to benchmark the Au NP formation in cage 1 with the one passivated by the coating ligand 2, a similar Au NP synthesis protocol was used,41 which is a variation of the one reported by Brust et al.26 1 eq. of tetrachloroauric (III) acid (HAuCl4) for each sulfur atom of the stabilizing structure, i.e. 6 eq. with respect to cage 1, was dissolved in minimum amounts of water (0.3 ml). The transfer of the gold salt from the aqueous to the organic phase was enabled by addition of tetra-n-octylammonium bromide (TOAB, 2 eq. with respect to HAuCl4, i.e. 12 eq. with respect to 1) dissolved in minimum amounts of CH2Cl2 (0.3 ml). After completion of the phase transfer, indicated by complete decoloration of the aqueous phase, cage 1 as the stabilizing structure (1 eq.) was added as a solid. Nucleation and growth of the Au NPs was triggered by the quick addition of the reducing agent sodium borohydrate (NaBH4, 8 eq. with respect to HAuCl4, i.e. 48 eq. with respect to 1) dissolved in minimum amounts of water (0.3 ml). Upon addition of the reducing agent, an immediate color change of the organic phase from bright orange to auburn was observed. After rigorous stirring for 15 min, the organic phase was separated, dried in vacuo and the particles were suspended in ethanol. Separation of the Au NPs from excess TOAB, NaBH4 and cage molecules was achieved by three subsequent centrifugations in ethanol, followed by size-exclusion chromatography (SEC) to provide cage 1-stabilized Au NPs (referred to as Au-1) as black powder. The colloidal gold particles grown inside the cage were the only detectable form of gold and were collected in yields exceeding 90% with respect to the gold source. It is noteworthy that Au NPs lacking surface-passivating agents were inherently unstable in our hands and aggregated immediately, e.g. on the SEC column. Note that the determined masses were assuming a 1:1 ratio between cage 1 and Au NP, and this hypothesis that the cage structure is controlling the particle's growth by encapsulation was corroborated by the analyses discussed in the following. The observed small mass loss most likely occurs during the separation and purification steps.
The Au NPs of Au-1 were characterized by transmission electron microscopy (TEM, Fig. 2(a) and Fig. S2 in the ESI‡ displaying a larger area) from which the particle dimensions and their size distribution (Fig. 2(b)) was determined. The mass ratio between organic cage 1 and Au NP was determined by thermogravimetric analysis (TGA). UV-vis absorption spectroscopy (Fig. 2(c)) not only complemented the characterization of Au-1, but also allowed the investigation of the thermal stability of the hybrid architecture consisting of the particle inside the organic cage to be carried out.
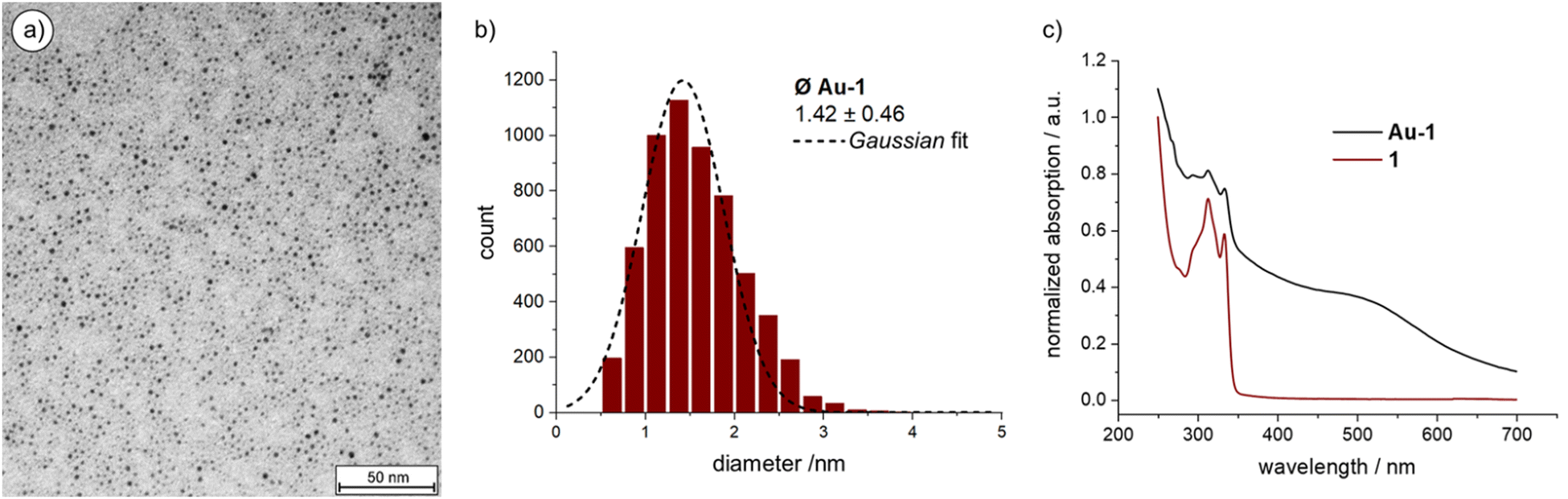 | ||
| Fig. 2 (a) TEM image of Au-1; (b) size-distribution of Au-1; (c) Normalized UV-vis absorption spectra of 1 and Au-1. Offset for clarity. | ||
Over 5000 Au NPs on the TEM micrograph gave an average NP diameter of 1.42 ± 0.46 nm, which is in good agreement with the estimated cavity size of cage 1 of about 1.5 nm obtained by simple MM2 simulation of the structure in Chem3D version 19 (sketched in red in Scheme 1; Fig. S4 in the ESI‡). The Au NP size distribution was analyzed using the threshold and particle analysis tools from ImageJ.45 The NPs of Au-1 are enlarged compared with the ones coated and stabilized by the threefold symmetric ligand 2 (1.21 ± 0.36 nm).41 This suggests a more compact arrangement of the three branches of 2 on the Au NP surface and that the closing of the cage with a mesitylene subunit widens their spatial arrangement in 1.
Thermogravimetric behavior of Au-1 was analyzed up to 900 °C with a temperature gradient of 10 °C min−1 (Table S1 and Fig. S3 in the ESI‡). The sample (2.053 mg) lost 18.1% (0.371 mg) which was attributed to the organic ligand 1. Thus, 0.16 μmol of 1 were burned from the Au-1 sample, while 1.682 mg of gold remained, corresponding to 8.54 μmol Au. Thus, on average 53.4 gold atoms are forming the Au NP in the cavity of 1 in the hybrid architecture Au-1. The analysis is in excellent agreement with the TEM-based particle size analysis, as the extensively investigated Au55 clusters are reported with diameters of 1.44 ± 0.40 nm.46–48 Combined TEM and TGA analyses not only corroborate the 1:1 ratio between organic cage 1 and Au NP in Au-1, but also support the hypothesis of Au NP growth inside the cage's cavity.
The comparison between the UV-vis absorption spectra of Au-1 and 1 (Fig. 2(c)) shows a broad and shallow bump between 450 and 600 nm, characteristic for the Au NPs’ plasmon band absorption. The broadness of the absorption points at particle diameters below 2 nm since this is the limit below which the rate of surface scattering exceeds bulk scattering, resulting in a drastic surface plasmon band broadening. The electron-donating nature of the sulfur–gold bond increases the surface electron density below this size, contributing in addition to the surface scattering.49 Interestingly, the absorption bands of the OPE subunit between 300 and 350 nm are well-defined for both, 1 and Au-1, documenting a well-dissolved OPE subunit in both cases and suggesting that the OPE rod of Au-1 is exposed to the solvent. At this stage, the accessibility of the OPE is less important but becomes instrumental in the currently ongoing studies, profiting from the exposed alkyne as a reactive handle allowing these monofunctionalized particles to be considered as “massive molecules”.
The remarkable stability of the caged Au NPs in Au-1 already became apparent in their separation by SEC. To evaluate their thermal stability, a sample was dispersed in o-xylene and gradually heated while investigated by UV-vis spectroscopy (Fig. 3). Of particular interest was the plasmon band bump between 450 and 600 nm. Starting at 60 °C, the temperature increment was 5 °C per 30 minutes. The series of recorded UV-vis absorption traces displays the first slight redshift of the bump at 135 °C. Exceeding this temperature threshold, the particles aggregated, resulting in a considerable redshift visible to the naked eye. Exposure of samples to temperatures above 135 °C for longer periods resulted in the precipitation of agglomerated Au NPs as a purple solid. Interestingly, the caged Au NPs in Au-1 have an increased decomposition temperature compared with the ones coated with 2, which are reported to decompose above 105 °C.41 We hypothesize that, in Au-1, the NP coating sulfides cannot easily detach from the particle surface due to their spatial pre-organization and fixation in the polycyclic cage structure, being reflected in the observed improved resistance against thermal stress. Furthermore, the superior thermal stability features also support the claim of the Au NP grown and caged inside the cavity in the Au-1 hybrid architecture.
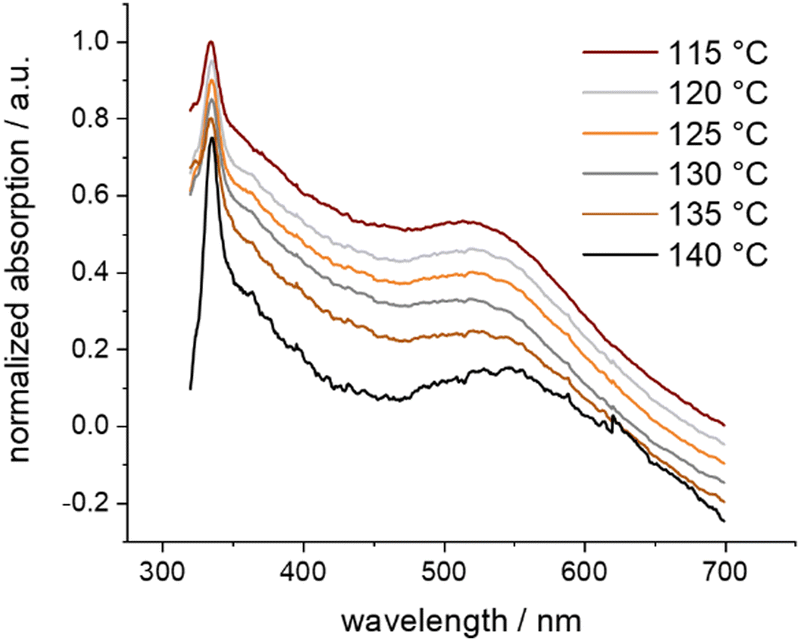 | ||
| Fig. 3 Normalized UV-vis absorption spectra of Au-1 in o-xylene at selected temperatures, showing thermal aggregation. Offset for clarity. | ||
In summary, the cage-type polycyclic ligand 1 is reported which acts as a growth and stabilization cavity for originating Au NPs. Cage 1 not only controls the dimension of the trapped Au NP in Au-1, but also protects it better against decomposition and aggregation than uncaged analogues. We are currently exploring the size limitation of the concept as well as the potential of these monofunctionalized Au NPs as labels and building blocks of devices. Furthermore, we intend to explore the catalytic activities of these structures and to investigate the entrapment of other metal clusters in the cage.
The authors thank the Swiss National Science Foundation (Grant no. 200020_207744) for financial support, Patrick Eckerst from FHNW Muttenz for providing the TGA measurements, and Dr Michael Pfeffer and Dr Heinz Nadig for measuring ESI-HRMS. M. M. acknowledges support from the 111 project (90002-18011002).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- P. Mulvaney, MRS Bull., 2001, 26, 1009–1014 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed.
- G. Schmid, M. Bäumle, M. Geerkens, I. Heim, C. Osemann and T. Sawitowski, Chem. Soc. Rev., 1999, 28, 179–185 RSC.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- L. Pasquato, P. Pengo and P. Scrimin, Supramol. Chem., 2005, 17, 163–171 CrossRef CAS.
- A. K. Ilunga and R. Meijboom, Catal. Lett., 2019, 149, 84–99 CrossRef CAS.
- B. I. Kharisov, H. V. R. Dias, O. V. Kharissova and A. Vázquez, J. Nanopart. Res., 2014, 16, 2665 CrossRef.
- D. Safer, J. Struct. Biol., 1999, 127, 101–105 CrossRef CAS PubMed.
- J. F. Hainfeld, Science, 1987, 236, 450–453 CrossRef CAS PubMed.
- J. F. Hainfeld and F. R. Furuya, J. Histochem. Cytochem., 1992, 40, 177–184 CrossRef CAS PubMed.
- D. Safer, L. Bolinger and J. S. Leigh, J. Inorg. Biochem., 1986, 26, 77–91 CrossRef CAS PubMed.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
- J. E. Reardon and P. A. Frey, Biochemistry, 1984, 23, 3849–3856 CrossRef CAS PubMed.
- H. Yang, J. E. Reardon and P. A. Frey, Biochemistry, 1984, 23, 3857–3862 CrossRef CAS PubMed.
- M. A. Mangold, M. Calame, M. Mayor and A. W. Holleitner, ACS Nano, 2012, 6, 4181–4189 CrossRef CAS PubMed.
- T. Dadosh, Y. Gordin, R. Krahne, I. Khivrich, D. Mahalu, V. Frydman, J. Sperling, A. Yacoby and I. Bar-Joseph, Nature, 2005, 436, 677–680 CrossRef CAS PubMed.
- J. Liao, S. Blok, S. J. van der Molen, S. Diefenbach, A. W. Holleitner, C. Schönenberger, A. Vladyka and M. Calame, Chem. Soc. Rev., 2015, 44, 999–1014 RSC.
- T. A. Gschneidtner, Y. A. D. Fernandez and K. Moth-Poulsen, J. Mater. Chem. C, 2013, 1, 7127–7133 RSC.
- Y.-S. Chen, M.-Y. Hong and G. S. Huang, Nat. Nanotechnol., 2012, 7, 197–203 CrossRef CAS PubMed.
- H. Gu, J. Chao, S.-J. Xiao and N. C. Seeman, Nature, 2010, 465, 202–205 CrossRef CAS PubMed.
- Md. A. Rahman, H.-B. Noh and Yoon-Bo Shim, Anal. Chem., 2008, 80, 8020–8027 CrossRef CAS PubMed.
- G. Schmid, Adv. Eng. Mater., 2001, 3, 737–743 CrossRef CAS.
- L. Ma, F. Li, T. Fang, J. Zhang and Q. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11024–11031 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- J. R. Reimers, M. J. Ford, S. M. Marcuccio, J. Ulstrup and N. S. Hush, Nat. Rev. Chem., 2017, 1, 0017 CrossRef CAS.
- W. M. Pankau, K. Verbist and G. von Kiedrowski, Chem. Commun., 2001, 519–520 RSC.
- W. M. Pankau, S. Mönninghoff and G. von Kiedrowski, Angew. Chem., Int. Ed., 2006, 45, 1889–1891 CrossRef CAS PubMed.
- M. S. Inkpen, Z.-F. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Nat. Chem., 2019, 11, 351 CrossRef CAS PubMed.
- M. Lehmann, E. H. Peters and M. Mayor, Chem. – Eur. J., 2016, 22, 2261–2265 CrossRef CAS PubMed.
- M. Lehmann, E. H. Peters and M. Mayor, Helv. Chim. Acta, 2017, 100, e1600395 CrossRef.
- T. Peterle, A. Leifert, J. Timper, A. Sologubenko, U. Simon and M. Mayor, Chem. Commun., 2008, 3438–3440 RSC.
- T. Peterle, P. Ringler and M. Mayor, Adv. Funct. Mater., 2009, 19, 3497–3506 CrossRef CAS.
- J. P. Hermes, F. Sander, T. Peterle, R. Urbani, T. Pfohl, D. Thompson and M. Mayor, Chem. – Eur. J., 2011, 17, 13473–13481 CrossRef CAS PubMed.
- F. Sander, U. Fluch, J. P. Hermes and M. Mayor, Small, 2014, 10, 349–359 CrossRef CAS PubMed.
- J. P. Hermes, F. Sander, U. Fluch, T. Peterle, D. Thompson, R. Urbani, T. Pfohl and M. Mayor, J. Am. Chem. Soc., 2012, 134, 14674–14677 CrossRef CAS PubMed.
- E. H. Peters, M. Lehmann and M. Mayor, Part. Part. Syst. Charact., 2018, 35, 1800015 CrossRef.
- E. H. Peters and M. Mayor, Eur. J. Inorg. Chem., 2020, 2325–2334 CrossRef CAS.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS PubMed.
- E. H. Peters and M. Mayor, Chimia, 2021, 75, 414–426 CrossRef PubMed.
- ImageJ, https://imagej.nih.gov/ij/download.html, (accessed December 27, 2019).
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- T. Mori and T. Hegmann, J. Nanopart. Res., 2016, 18, 295 CrossRef PubMed.
- D. H. Rapoport, W. Vogel, H. Cölfen and R. Schlögl, J. Phys. Chem. B, 1997, 101, 4175–4183 CrossRef CAS.
- M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706–3712 CrossRef CAS.
Footnotes |
| † This work is dedicated to the late Günter Schmid. |
| ‡ Electronic supplementary information (ESI) available: Experimental methods: Synthesis and characterization of compounds 1 and 3–9, Fig. S1: 1H NMR of Au-1, Fig. S2: TEM image of Au-1, Fig. S3: TGA trace of Au-1, and Fig. S4: Chem3D MM2 simulation rendering 1. See DOI: https://doi.org/10.1039/d3cc00277b |
| This journal is © The Royal Society of Chemistry 2023 |