
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereodefined synthesis of cyclic amidines by domino 1,7-H shift and 6π electrocyclisation†
Matthew L.
Martin
,
Claire
Wilson
 and
Alistair
Boyer
*
and
Alistair
Boyer
*
School of Chemistry, University of Glasgow, G12 8QQ, UK. E-mail: alistair@boyer-research.com
First published on 4th April 2023
Abstract
Unsaturated N2-sulfonyl amidines are transformed into valuable N-heterocyclic products when heated with BF2OTf. Mechanism studies suggest a domino 1,7-H shift, activating a C–H bond, followed by electrocylisation that results in stereodefined cyclic amidines.
The controlled transformation of simple molecules to complex products is a standing goal in the development of novel synthetic chemical methodology. Methods have been developed that activate the C–H bond1 adjacent to a nitrogen atom for functionalisation through intramolecular transfer and cyclisation (1, Scheme 1a).2,3 This approach to C–H activation is particularly useful because it allows access to complex valuable nitrogen-containing heterocycles4 (e.g.3) from simple building blocks (e.g.2). The majority of these reactions proceed through 1,5–H transfer, exploiting a favourable 6-membered transition state. In contrast, there are few examples of any reaction that exploit a 1,7-H transfer. The most notable 1,7-process is the biosynthesis of vitamin D 6 from dehydrosterols 4 (Scheme 1b).5 This synthesis comprises a light promoted 6π-conrotatory electrocyclisation followed by a pericyclic 1,7-H rearrangement. Although there have been extensive vitamin D synthesis studies and mechanistic studies of the 1,7-H shift, to the best of our knowledge, there are little or no applications of this reaction for chemical synthesis.6,7
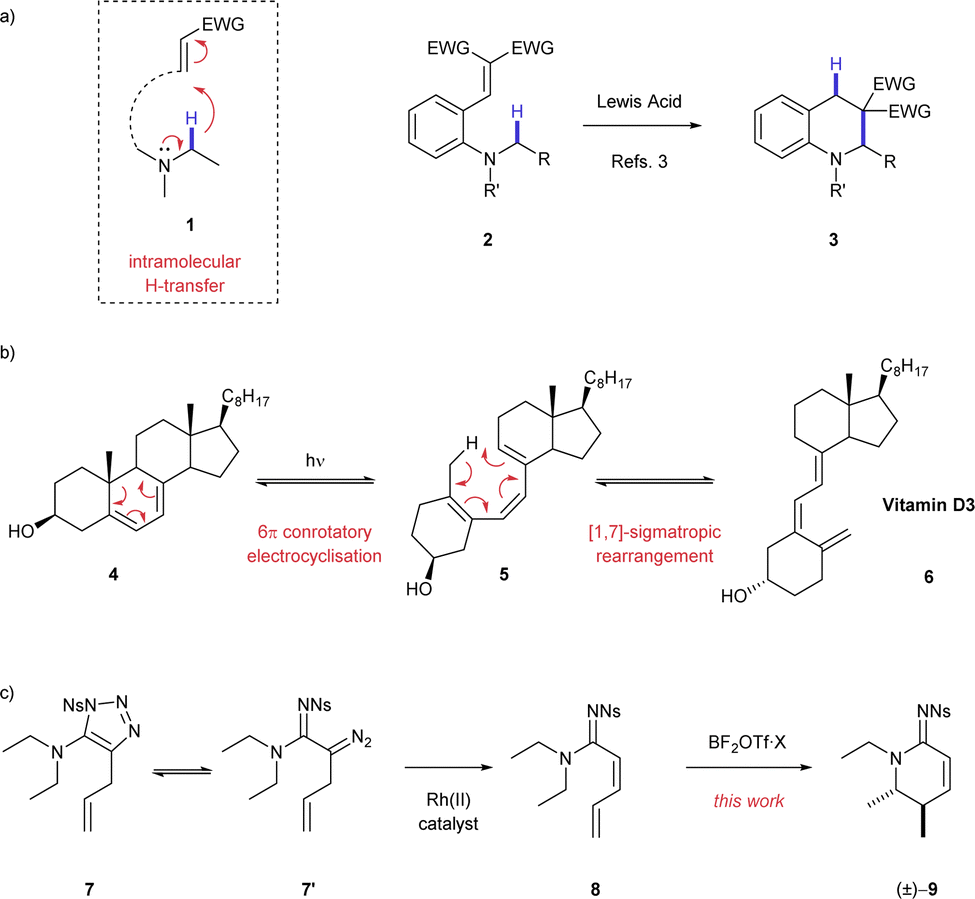 | ||
| Scheme 1 (a) General scheme for C–H activation by H-transfer adjacent to nitrogen atoms and overview of products accessed using this method; (b) Biosynthesis of vitamin D3. (c) Rhodium(II) catalysed 1,2–H shift to form unsaturated amidines 8 and their conversion to valuable nitrogen heterocycles 9 [this work]. Ns = 4-nitrobenzenesulfonyl. | ||
Here, we describe a reaction that proceeds through 1,7-H shift activation of a C–H bond adjacent to a nitrogen atom and cyclisation to deliver value-added products. As part of a study into using 1-sulfonyl-1,2,3-triazoles as carbene precursors through Dimroth equilibration,8 we developed a rhodium(II) catalysed 1,2–H shift reaction that created highly unsaturated amidine products (8, Scheme 1c).9 Using a boron Lewis acid, we were able to transform these unsaturated acyclic compounds 8 into valuable stereodefined cyclic amidine products 9.
The highly unsaturated amidine 8a was treated with a series of reagents, under a range of conditions to explore its reactivity (Fig. 1).10 In general, when treated with acids or Lewis acids the outcome was decomposition of the substrate or isomerisation of the Z-alkene into its E-geometry.
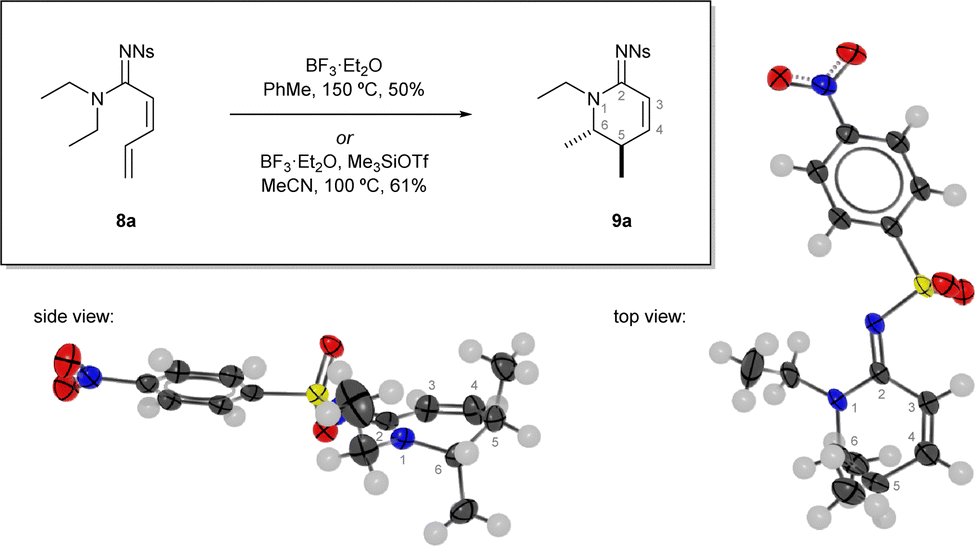 | ||
| Fig. 1 Discovery of the reaction and optimisation highlight. Reactions performed in sealed a vial, isolated yields reported. Single crystal x-ray diffraction structure of 9a from two views, atomic displacement ellipsoids at 50% probability, H atoms at 0.30 Å.11 | ||
However, when treated with boron trifluoride in toluene at elevated temperature (150 °C, sealed vial), a new product was formed 9a that was determined to be the unsaturated cyclic amidine 9a.10 Interestingly, a rearrangement had occurred in which one of the N-ethyl groups was incorporated into the newly formed ring. The high sp2 content of the hexacycle 9a resulted in the two methyl groups being axially disposed with respect to a relatively flat ring evidenced by a near-zero coupling constant between their respective methine protons in 1H NMR spectrum. The structure was also confirmed by a single crystal x-ray diffraction structure.11
The cyclic amidine product was formed in a reasonable yield but decomposition during the reaction was still a major outcome and significantly lower yields (< 30%) were achieved when the conditions were applied to substrates with anything other than N1,N1-diethyl substituents. The key piece of the optimisation puzzle came by switching to a different boron-based Lewis acid accessed by mixing Me3SiOTf and BF3·OEt2. The synergistic combination of Me3SiOTf and BF3·OEt2 in acetonitrile has been shown to be beneficial due to formation of BF2OTf·MeCN.12 The use of acetonitrile as solvent was also important because of limited substrate solubility in other solvents. Overall, these optimum conditions allowed the reaction to proceed at a reduced temperature (100 °C, sealed vial) and were able to deliver the cyclic amidine product 9a in 61% yield. Furthermore, these conditions were found to be more generally applicable.
The scope of the reaction was explored with respect to the N1-substituents and the sulfonyl group (Fig. 2). Under optimised reaction conditions (in situ generated BF2OTf, 100 °C, 18 h), the N1,N1 diethyl substrate with a N2-tosyl group gave a significantly reduced yield (18%) of its corresponding product 9aTs compared with the N2-nosyl group 9a (61%). This result demonstrated the importance of the electron-poor nitro-substituted sulfonyl group in this transformation. This fortuitously matches the requirements for a nosyl group in the synthesis of these substrates.9
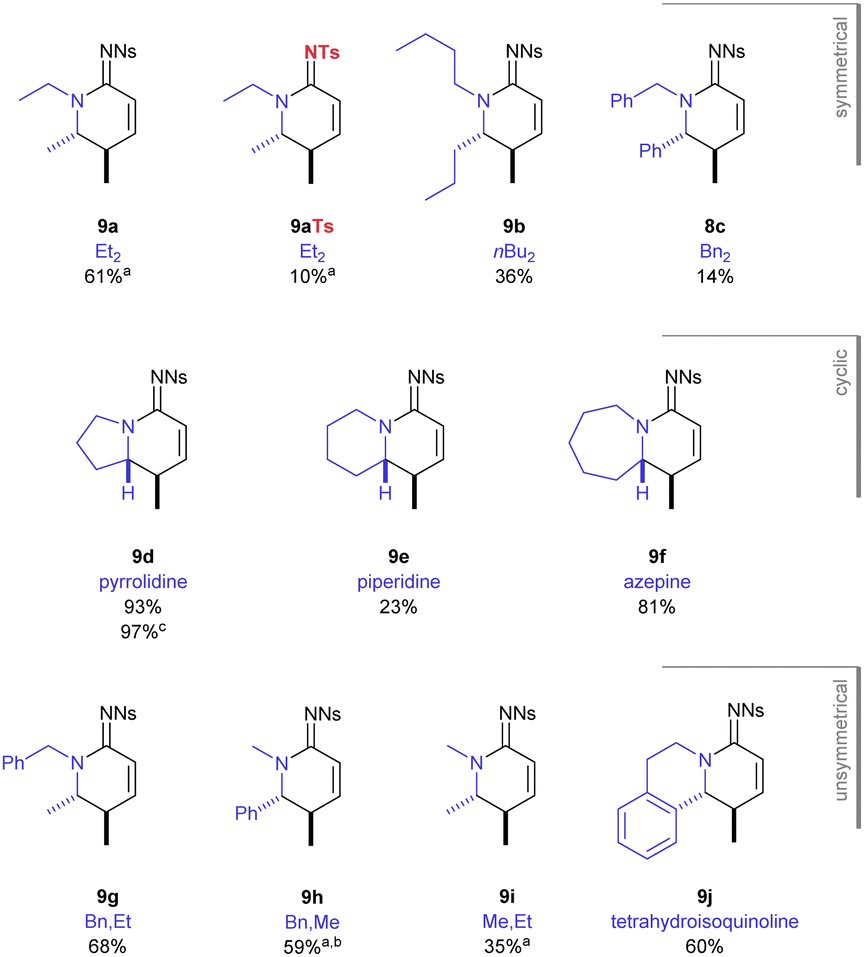 | ||
| Fig. 2 Cyclic amidine products. Labels refer to N1,N1-substitution in the substrate 5. Reagents and conditions: Diene substrate (1.0 equiv.), BF3·Et2O (3.0 equiv.), Me3SiOTf (6.0 equiv.), MeCN, 80 °C, 18 h; reactions were performed on 0.060 to 0.500 mmol scale. (a) Reaction performed at 100 °C; (b) reaction performed for 24 h; (c) reaction performed on 1.9 mmol scale. | ||
The temperature could be lowered to 80 °C for substrates that did not contain only N1-ethyl or N1-methyl groups and complete consumption of the starting material occurred in the same time frame (18 h). Despite proceeding at a lower temperature, the larger n-butyl and dibenzyl substrates gave a drop in yield, giving 38% and 14% of the amidine products 9b, 9c respectively. The best yield in this study was obtained for the pyrrolidine-containing precursor which gave the bicyclic product 9d (93%). Increasing the heterocyclic ring size by one atom to the piperidine gave a dramatic drop in yield (23%, 9e); but when adding a further atom, in form of an azepane ring, the excellent yield was restored (81%, 9f).
Using non-symmetric substituents gave an opportunity to study the behaviour of this process through intramolecular competition. With N1,N1-benzyl,ethyl substituents, the reaction occurred entirely at the ethyl C–H to give the N2-benzyl product 9g. In stark contrast, given a choice between benzyl and methyl the reaction occurred entirely at the benzylic C–H (9h). When a N1,N1-ethyl,methyl substrate was studied it was consistent that it was only the ethyl group that was functionalised in the reaction sequence (9i). Finally, the cyclic non-symmetric tetrahydroisoquinoline substrate gave reaction at the benzylic position entirely (9j). Importantly, in each of the cases studied, only the product described was observed during analysis of the crude reaction mixture and there was no trace of products resulting from alternative diastereoselectivity or chemoselectivity. When the N1,N1-substituents were very large (i.e. iPr or CH2CH2OSiPh2tBu) the reaction did not occur and decomposition of the highly unsaturated substrate occurred instead.13
The indolizidine product 9d, derived from pyrrolidine, is a particularly good example of the value of this transformation owing to its prevalence as a natural product core across several natural product families.14 The reaction worked just as well on a larger scale (97%, 1.9 mmol), which both demonstrates the versatility of the transformation and allowed investigation into how the unsaturated functional group could be further elaborated (Scheme 2).
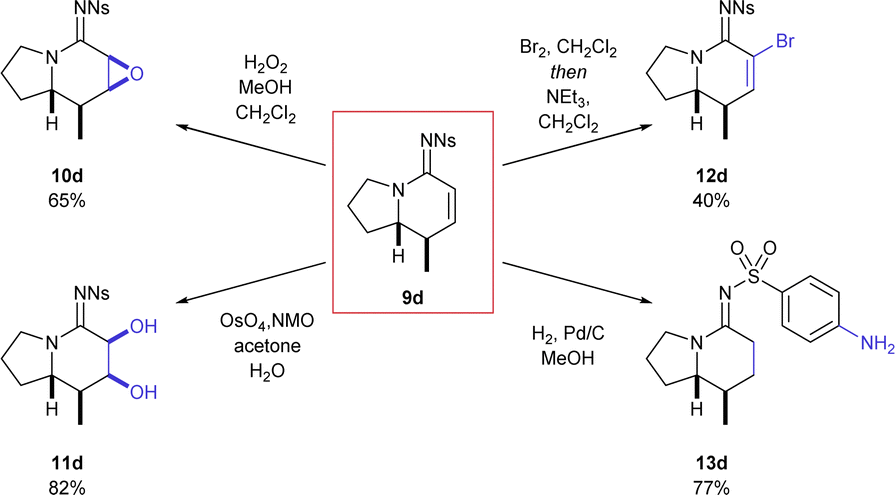 | ||
| Scheme 2 Selected reactivity of cyclic amidine 9d. | ||
Treatment of the bicyclic azaheterocycle 9d with hydrogen peroxide gave the epoxide 10d as only one detectable stereoisomer.15 Dihydroxylation also efficiently delivered a single diol product 11d in excellent yield. Dibromination with molecular bromine followed by elimination with base (NEt3) resulted in efficient bromine incorporation at the α-position (12d). Finally, hydrogenation with palladium on carbon efficiently resulted in reduction of the alkene as well as the nitro group (13d).
The behaviour of substrates at lower temperatures provided key insight into this mechanism (Scheme 3).
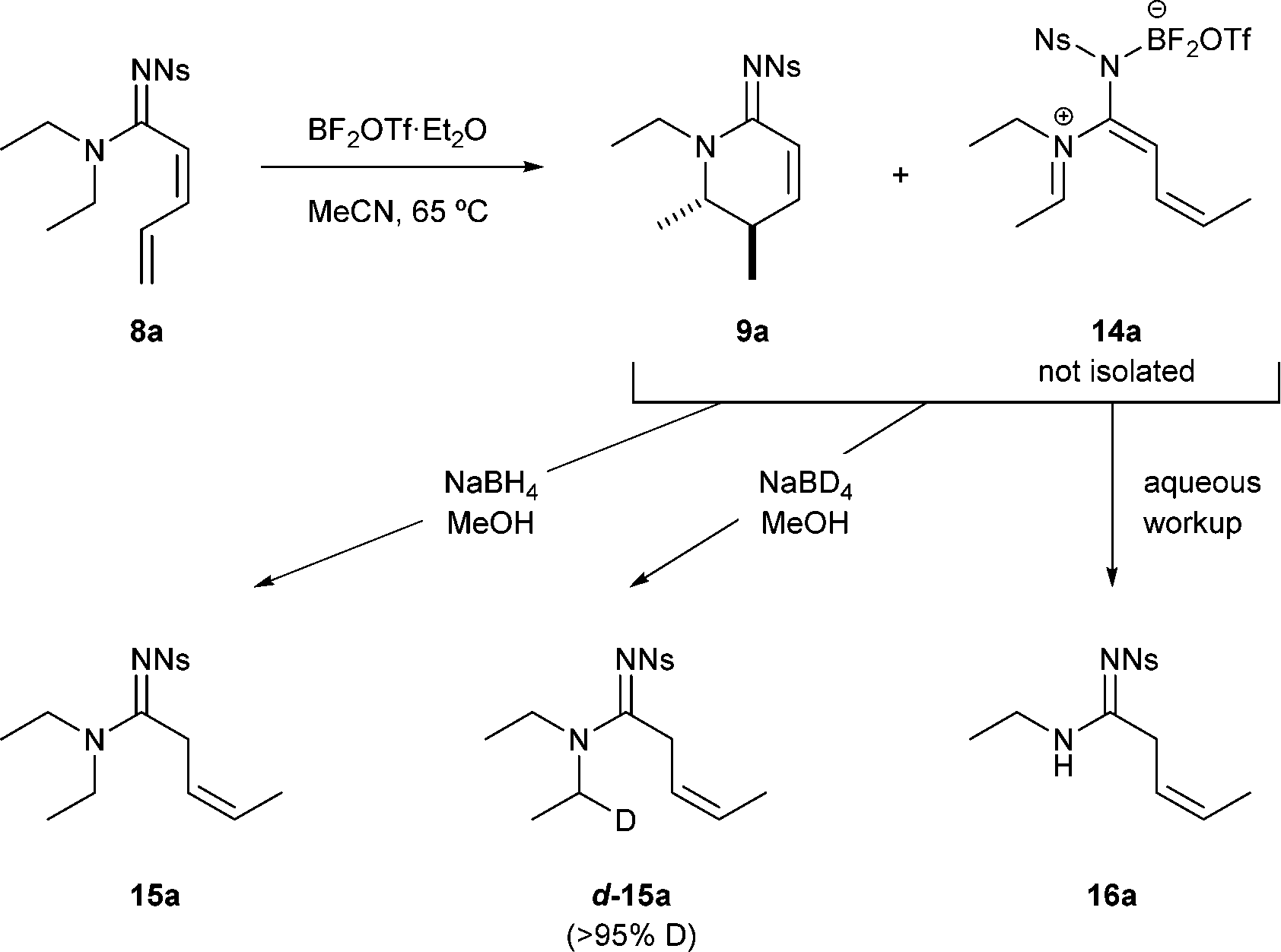 | ||
| Scheme 3 Experimental evidence of an azatriene intermediate 14a. | ||
1H NMR analysis of the reaction mixture following 18 h at 65 °C (instead of the optimum 100 °C) revealed a mixture of unreacted starting material 8a, cyclic amidine 9a and other intermediates that was believed to be related to an azatriene 14a. This compound 14a could neither be isolated nor observed directly but upon hydrolysis the secondary amide 16a, resulting from iminium hydrolysis, was observed. Further evidence for the existence of azatriene 14a came upon adding sodium borohydride to the crude mixture to give the reduced alkene product 15a. Simple conjugate reduction of the diene starting material was ruled out because only the Z-geometry (3JH–H = 10.6 Hz) of the alkene was observed; and using sodium borodeuteride resulted in deuterium incorporation in the N-substituent (d-15a).16
The mechanism for this transformation can be explained by a suggested sequence of two pericyclic reactions (Scheme 4).
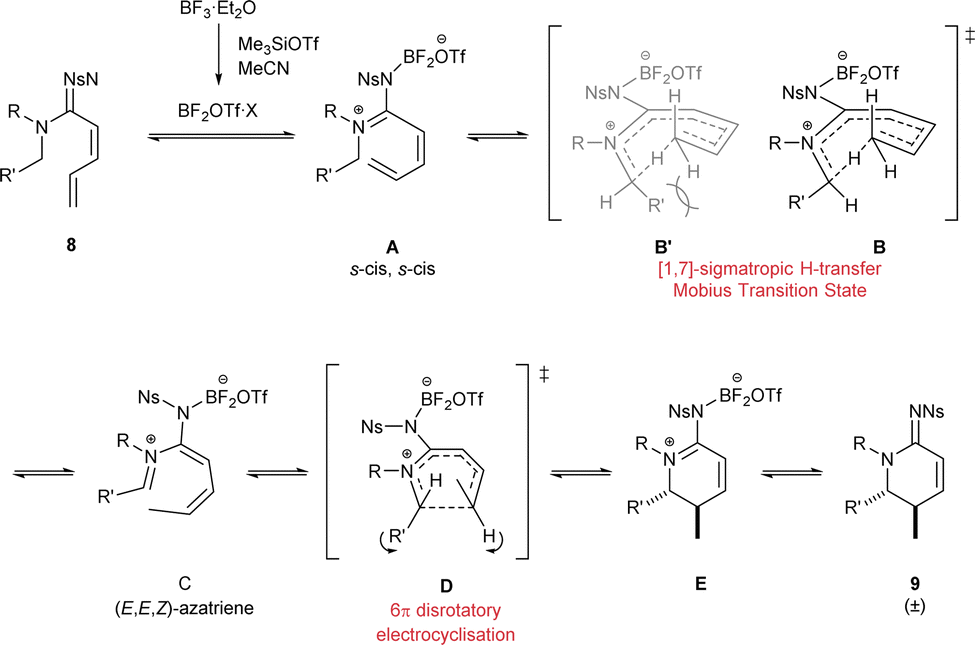 | ||
| Scheme 4 Proposed reaction mechanism. | ||
First, in situ generated BF2OTf Lewis acid would interact with the imine nitrogen atom (in 8) to populate the zwitterionic form A of the amidine resonance. Next, a 1,7-H shift would activate the position adjacent to the amidine N2-atom. The high unsaturation in the system A suggests a pericyclic process with twisted or Möbius-type17 transition state B to satisfy the Woodward-Hoffman rules.18 Both geometries of the iminium portion could be possible but in order to form Z-iminium geometry the N1-substituent would reach in towards the 8-membered ring (B′), presumably resulting in increased steric clash. The geometric requirements of the transition state are such only one geometry of the diene portion is attainable so the (E,E,Z)-azatriene C would be the favoured product of the first pericyclic process. Then, thermally allowed 6π-electrocyclisation D would translate of the (E,E,Z) geometry to a product that contains substituents on opposite sides of the closing ring E.18a,19 This would also be the expected thermodynamically favoured product due to minimisation of 1,2 strain. Finally, loss of the Lewis acid would release the cyclic amidine product 9. This mechanism explains both the product and stereocontrol observed in the reaction. Interestingly, the order of the two pericyclic processes are reversed in this mechanism compared with vitamin D biosynthesis.
However, alternative mechanistic pathways can not be ruled out. The requirement of a strong boron Lewis Acid suggests that the mechanism may have polar character. There has also been an example of a reaction where a bisborane Lewis acid played a critical role in a C–H transfer.20
In summary, this process represents the transformation of simple unsaturated building blocks into valuable stereodefined cyclic amidine products. A selection of valuable scaffolds was formed and the reaction was amenable to scale-up, which is a testament to the importance of BF2OTf as a Lewis acid. Characterisation of reaction intermediates suggested a mechanism including 1,7-H shift and electrocyclisation that represents a rare example of 1,7-H shift applied to synthetic methodology. Overall, this method represents a valuable approach to azaheterocycle synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (b) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092–10142 CrossRef CAS PubMed.
- (a) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010–5036 CrossRef CAS PubMed; (b) H. Liu, Y. Quan, L. Xie, X. Li and X. Xie, Front. Chem., 2022, 10, 840934 CrossRef CAS PubMed; (c) B. Peng and N. Maulide, Chem. – Eur. J., 2013, 19, 13274–13287 CrossRef CAS PubMed; (d) J. C. Duff, J. Chem. Soc., 1941, 547–550 RSC; (e) I. D. Jurberg, B. Peng, E. Wöstefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950–1953 CrossRef CAS PubMed; (f) D. Vasu, A. L. Fuentes de Arriba, J. A. Leitch, A. de Gombert and D. J. Dixon, Chem. Sci., 2019, 10, 3401–3407 RSC; (g) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2017, 10, 165–169 CrossRef PubMed.
- (a) W. H. N. Nijhuis, W. Verboom and D. N. Reinhoudt, Synthesis, 1987, 641–645 CrossRef CAS; (b) W. Verboom, M. R. J. Hamzink, D. N. Reinhoudt and R. Visser, Tetrahedron Lett., 1984, 25, 4309–4312 CrossRef CAS; (c) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180–12181 CrossRef CAS PubMed; (d) S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Org. Lett., 2008, 11, 129–132 CrossRef PubMed; (e) C. Zhang, S. Murarka and D. Seidel, J. Org. Chem., 2008, 74, 419–422 CrossRef PubMed; (f) J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, J. Am. Chem. Soc., 2009, 131, 3991–3997 CrossRef CAS PubMed; (g) K. Mori, Y. Ohshima, K. Ehara and T. Akiyama, Chem. Lett., 2009, 38, 524–525 CrossRef CAS; (h) G. Zhou and J. Zhang, Chem. Commun., 2010, 46, 6593–6595 RSC; (i) M. C. Haibach, I. Deb, C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 2100–2103 CrossRef CAS PubMed; (j) Y.-Y. Han, W.-Y. Han, X. Hou, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2012, 14, 4054–4057 CrossRef CAS PubMed; (k) Y. Wang, Y. Chi, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2012, 134, 2926–2929 CrossRef CAS PubMed; (l) K. Mori, K. Kurihara, S. Yabe, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2014, 136, 3744–3747 CrossRef CAS PubMed; (m) S. Yamazaki, T. Naito, T. Tatsumi and K. Kakiuchi, ChemistrySelect, 2018, 3, 4505–4511 CrossRef CAS; (n) E. R. Zaitseva, A. Y. Smirnov, V. I. Timashev, V. I. Malyshev, E. A. Zhigileva, A. A. Mikhaylov, M. G. Medvedev, N. S. Baleeva and M. S. Baranov, Eur. J. Org. Chem., 2022, e202200680 Search PubMed.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC; (c) R. H. F. Manske, R. G. A. Rodrigo, A. Brossi, G. A. Cordell and H.-J. Knolker, Book Series: The Alkaloids, Elsevier, vol. 1952–2021 Search PubMed; (d) S. Funayama and G. A. Cordell, Alkaloids: A Treasury of Poisons and Medicines, Elsevier, 2014 Search PubMed.
- (a) H. F. DeLuca, BoneKEy Rep., 2014, 3, 479 Search PubMed; (b) A. H. A. Windaus, Nachr. Ges. Wiss. Gottingen, Math., 1926, 1926, 174–184 Search PubMed; (c) M. Holick, J. MacLaughlin, M. Clark, S. Holick, J. Potts, R. Anderson, I. Blank, J. Parrish and P. Elias, Science, 1980, 210, 203–205 CrossRef CAS PubMed.
- (a) J. L. M. A. Schlatmann, J. Pot and E. Havinga, Recl. Trav. Chim. Pays-Bas, 1964, 83, 1173–1184 CrossRef CAS; (b) R. W. Murray and M. L. Kaplan, J. Am. Chem. Soc., 1966, 88, 3527–3533 CrossRef CAS; (c) V. Boekelheide and T. A. Hylton, J. Am. Chem. Soc., 1970, 92, 3669–3675 CrossRef CAS; (d) C. A. Hoeger and W. H. Okamura, J. Am. Chem. Soc., 1985, 107, 268–270 CrossRef CAS; (e) J. E. Baldwin and V. P. Reddy, J. Am. Chem. Soc., 1987, 109, 8051–8056 CrossRef CAS; (f) H. Y. Elnagar and W. H. Okamura, J. Org. Chem., 1988, 53, 3060–3066 CrossRef CAS; (g) M. E. Gurskii, I. D. Gridnev, Y. V. Il'ichev, A. V. Ignalenko and Y. N. Bubnov, Angew. Chem., Int. Ed. Engl., 1992, 31, 781–783 CrossRef; (h) G. A. Dushenko, I. E. Mikhailov, A. Zschunke, N. Hakam, C. Mügge and V. I. Minkin, Mendeleev Commun., 1997, 7, 50–51 CrossRef; (i) J. Clarke, P. W. Fowler, S. Gronert and J. R. Keeffe, J. Org. Chem., 2016, 81, 8777–8788 CrossRef CAS PubMed.
- (a) For an example of a 1,5-H shift electrocyclisation reaction, see: C. Zhu, X. Zhang, X. Lian and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 7817–7820 CrossRef CAS PubMed; (b) M. Alajarin, B. Bonillo, M.-M. Ortin, P. Sanchez-Andrada and A. Vidal, Eur. J. Org. Chem., 2011, 1896–1913 CrossRef CAS.
- (a) T. Miura and M. Murakami, Rhodium Catalysis in Organic Synthesis, 2019, pp. 449–470 Search PubMed; (b) V. Bakulev, W. Dehaen and T. Beryozkina, Chemistry of 1,2,3-triazoles, 2015, pp. 1–49 Search PubMed; (c) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (d) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 3004–3023 CrossRef CAS; (e) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed.
- M. L. Martin and A. Boyer, Eur. J. Org. Chem., 2021, 5857–5861 CrossRef CAS.
- Further details are included in the ESI†.
- There was a 0.8: 0.2 occupancy split between two confomers, the minor component is omitted from Fig. 1. Deposition number 2118194 contains the supplementary crystallographic data for 9a.
- (a) E. L. Myers, C. P. Butts and V. K. Aggarwal, Chem. Commun., 2006, 4434–4436 RSC; (b) K. C. Nicolaou, C. K. Hwang and M. E. Duggan, J. Am. Chem. Soc., 1989, 111, 6682–6690 CrossRef CAS; (c) C. Y. Hong and Y. Kishi, J. Am. Chem. Soc., 1991, 113, 9693–9694 CrossRef CAS.
- Unfortunately, examples of cyclic amidines having a group other than methyl at the 5-position were not accessible due to the requirements of the method that generated the substrates.
- (a) E. Gellert, J. Nat. Prod., 2004, 45, 50–73 CrossRef; (b) J. P. Michael, Nat. Prod. Rep., 2001, 18, 520–542 RSC; (c) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- (a) H. Zhang, Y.-K. Ni, G. Zhao and Y. Ding, Eur. J. Org. Chem., 2003, 1918–1922 CrossRef CAS; (b) J. Xie, T. Güveli, S. Hebbe and L. Dechoux, Tetrahedron Lett., 2004, 45, 4903–4906 CrossRef CAS.
- Treatment of the substrate 10a with sodium borohydride also did not result in any reaction.
- (a) C. W. Spangler, Chem. Rev., 1976, 76, 187–217 CrossRef CAS; (b) H. Jiao and P. von Ragué Schleyer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1763–1765 CrossRef; (c) B. A. Hess, L. J. Schaad and J. Pancir, J. Am. Chem. Soc., 2002, 107, 149–154 CrossRef.
- (a) R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS; (b) R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- S. Thompson, A. G. Coyne, P. C. Knipe and M. D. Smith, Chem. Soc. Rev., 2011, 40, 4217–4231 RSC.
- M. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2021, 60, 17185–17190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, data and copies of NMR spectra. CCDC 2118194. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01127e |
| This journal is © The Royal Society of Chemistry 2023 |