
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface synthesis of metal–organic frameworks: the critical role of the reaction conditions†
Nerea
Ruiz del Árbol‡
a,
Carlos
Sánchez-Sánchez‡
 a,
José I.
Martínez
a,
Luis
Rodríguez
a,
David
Serrate
bc,
Alberto
Verdini
d,
Luca
Floreano
d,
Peter
Jacobson§
e,
Leonhard
Grill
e,
José A.
Martín-Gago
a and
María F.
López
*a
a,
José I.
Martínez
a,
Luis
Rodríguez
a,
David
Serrate
bc,
Alberto
Verdini
d,
Luca
Floreano
d,
Peter
Jacobson§
e,
Leonhard
Grill
e,
José A.
Martín-Gago
a and
María F.
López
*a
aInstituto de Ciencia de Materiales de Madrid (ICMM), CSIC, Sor Juana Inés de la Cruz 3, 28049 Madrid, Spain. E-mail: mflopez@icmm.csic.es
bInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-UNIZAR, 50009 Zaragoza, Spain
cLaboratorio de Microscopias Avanzadas (LMA), Universidad de Zaragoza, 50018 Zaragoza, Spain
dLaboratorio TASC, CNR-IOM, Basovizza SS-14, Km 163.5, I-34149 Trieste, Italy
eDepartment of Physical Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
First published on 13th February 2023
Abstract
Two different metal–organic frameworks with either a honeycomb or Kagome structure were grown on Cu(111) using para-aminophenol molecules and native surface adatoms. Although both frameworks are made up from the same chemical species, they are structurally different emphasizing the critical role being played by the reaction conditions during their growth. This work highlights the importance of the balance between thermodynamics and kinetics in the final structure of surface-supported metal–organic networks.
MOFs are porous polymeric materials that contain coordination bonds between organic ligands and metal ions or clusters.1,2 2D-MOFs, recently highlighted as a new class of low dimensional material, are typically synthesised either by exfoliation of bulk layered MOFs (top-down strategy)3,4 or by bottom-up strategies,5–8 such as two-phase interfacial or surfactant-assisted methods. However, all of these top-down and bottom-up methodologies typically lack accurate control of the thickness of the final flakes, making the achievement of atomically thin 2D layers challenging.9,10
In the last few years, On-surface Synthesis (OSS) has been used to achieve the bottom-up growth of truly 2D materials with atomic precision.11,12 OSS is based on the use of two main ingredients: tailored molecular building blocks and a surface that provides both a catalytic effect that lowers activation barriers and a 2D confinement that facilitates molecular encounter. The core of OSS is the formation of new covalent bonds upon on-surface molecular activation.13,14 However, it can also be used to activate the molecules and promote the metal–ligand coordination bond formation, thus emerging as an attractive strategy to explore the growth of 2D-MOF materials.15–19 In that case, another ingredient that is of particular importance is the availability of metallic adatoms on a surface. These adatoms typically originate at the step edges and diffuse over the surface,20 coordinating with the organic units without the need for an extra supply of external adatoms, although in some cases, external adatoms have been used to confer the final MOFs’ novel properties.21,22
In the MOFs’ growth process, the final structure is controlled by a competition between kinetic and thermodynamic mechanisms.23 Different experimental parameters such as the surface temperature, the annealing procedure, the molecular coverage or the density of surface adatoms are important agents that determine not only the reaction, but also its efficiency. For instance, it is clear that annealing a highly covered surface or using low temperatures during deposition hampers surface diffusion, and, therefore, the formed molecular structures could be stabilized into a metastable phase. Despite the huge importance that thermodynamic versus kinetic control can play on the final product of on-surface reactions, this field has been scarcely explored.24–26
In this work, we have selected para-aminophenol (p-AP), a simple aromatic molecule with two functional groups (alcohol and amine, see lower inset in Fig. 1a) previously used in OSS,27,28 as a precursor for growing two different 2D-MOFs on Cu(111) by rationally adjusting the synthesis protocol, evidencing the relevance of the reaction conditions on the final product. The two networks are a topological Kagome and a canonical honeycomb lattice, structures that are attracting increasing interest as they have been predicted to be potential organic topological insulators.29,30 To gather information about the two formation mechanisms, we combine in situ scanning tunnelling microscopy (STM), X-ray photoemission spectroscopy (XPS) and first-principles calculations. We have compared two protocols: (i) depositing the molecules onto a clean Cu(111) surface at room temperature (RT) and subsequent post-annealing to higher temperatures, and (ii) dosing the molecules directly on the hot surface. We show that, although the oxidation processes of the p-AP precursors on the surface exclusively depend on the final temperature (thermodynamics), the formed MOF structure depends on the particular experimental sequence (kinetics).
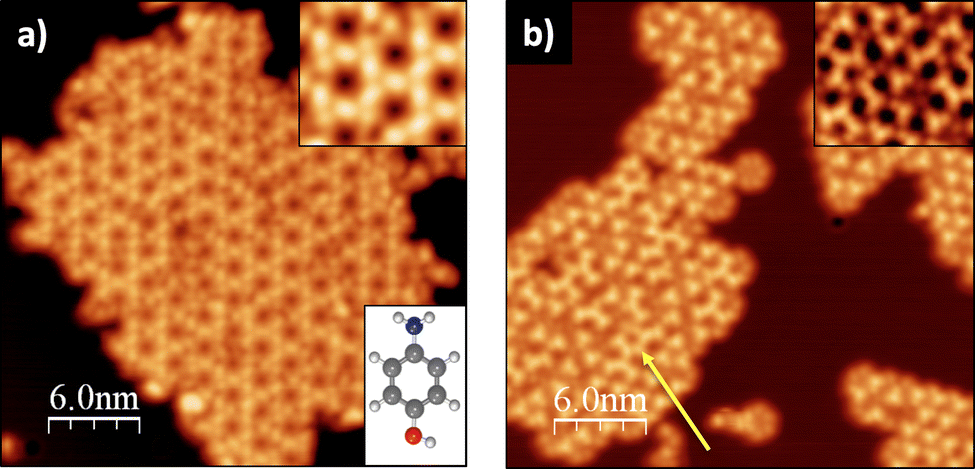 | ||
| Fig. 1 LT-STM image of p-AP on Cu(111) for: (a) molecular deposition at RT followed by postgrowth annealing at ∼500 K (Vbias = 1 V, It = 28 pA) and (b) molecular deposition on the substrate at ∼500 K (Vbias = 0.9 V, It = 0.13 nA). Top insets: enlarged details of the STM images. Bottom inset in (a): ball-and-stick model of the precursor molecule. | ||
Fig. 1 shows two STM images of the p-AP molecular networks formed on Cu(111) for two different experimental conditions: (a) molecular deposition with the substrate at RT and subsequent annealing to around 500 K; and (b) direct deposition on the hot (∼500 K) Cu(111) surface, each procedure resulting in a different molecular arrangement of the final networks. Fig. 1a exhibits a Kagome type lattice with a distance between pores of 2.11 ± 0.06 nm. On the other hand, Fig. 1b shows several two-dimensional networks based on trigonal arrangements that, in places, suggest the formation of a hexagonal lattice (see yellow arrow) with a distance between pores of 1.50 ± 0.05 nm.
To elucidate whether this different molecular arrangement depends on the experimental conditions or if it is induced by a chemical change in the molecules, XPS spectra of the two systems were recorded. Fig. 2 shows the O1s, N1s and C1s XPS core-level peaks just after molecular deposition at RT (top panels, red curves), after stepwise annealing from RT to 510 K (middle panels, green curves) and after molecular deposition on the hot Cu(111) surface (bottom panels, blue curves). For the RT case, the O1s spectrum presents two components. The main signal at a binding energy (BE) of 530.7 eV can be assigned to either a ketone or a phenoxy group, both indicating that the alcohol group is already dehydrogenated at RT.16 As we will see below, the theoretical calculations confirm the presence of phenoxy groups where the dehydrogenated alcohol is oxidized and stabilized by the surface. Thus, this component represents the bonding of the molecule to the Cu adatoms. Similar dehydrogenation reactions of alcohol groups in p-AP have been observed on the Cu(110) surface at RT.28 The small component located at 532.8 eV corresponds to the unreacted alcohol groups, indicating an incomplete chemical transformation into the phenoxy group.16 This component, however, vanishes at 330 K (see Fig. S3a, ESI†), yielding a molecular film with fully dehydrogenated alcohol groups. At higher temperatures, a new emission can be observed at 533.6 eV (see green and blue spectra of Fig. 2), which might be associated with O-containing organic residues, originated by the initial fragmentation of molecules at defects.31
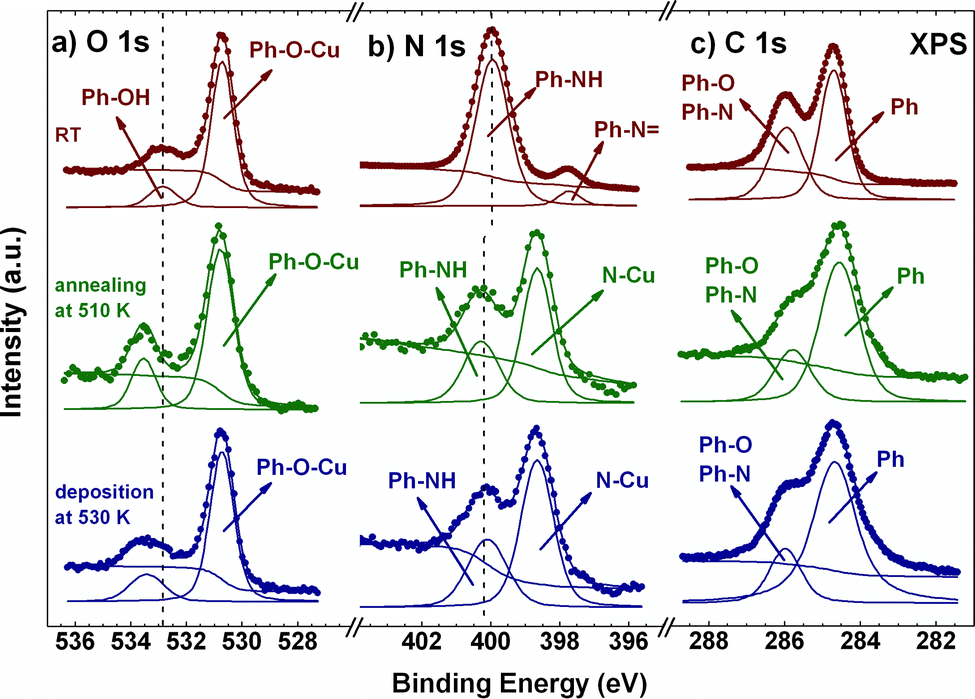 | ||
| Fig. 2 O1s (a), N1s (b) and C1s (c) XPS spectra of p-AP deposited on Cu(111) at RT (red), after subsequent annealing at 510 K (green) and for direct deposition at 530 K (blue). All spectra taken at RT. | ||
The N1s XPS spectrum at RT shows a main component at 399.9 eV, which is assigned to an NH group,32 indicating the partial dehydrogenation of the amine group upon molecular adsorption. From comparison with the case of free-base porphyrins on copper,32 the small emission detected at 397.5 eV might be associated with an iminic (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) type component, coming from molecules that undergo complete dehydrogenation of the amine group. This small component, however, disappears at 370 K and an intense component emerges at 398.7 eV, which grows at the expense of both pristine components and becomes most prominent at 510 K (see Fig. S3b and green and blue curves in Fig. 2, ESI†). This new peak is associated with a metalation reaction of the amine and the iminic components with a Cu adatom.32,33 On the other hand, the intensity of the signal at 399.9 eV decreases strongly at high temperature and undergoes a small shift, 0.2 eV towards high BE, which we attribute to the interaction with the substrate and the consequent charge transfer. We can envision three possible scenarios for such an NH component: (i) a small amount of molecular networks incorporating NH groups (compatible with their less favourable stabilization energy, as judged by first principle calculations, see below), (ii) molecules located at network edges, and (iii) isolated groups of molecules not forming a metal–organic framework (see regions marked in white in Fig. S1b and S2b, ESI†).
) type component, coming from molecules that undergo complete dehydrogenation of the amine group. This small component, however, disappears at 370 K and an intense component emerges at 398.7 eV, which grows at the expense of both pristine components and becomes most prominent at 510 K (see Fig. S3b and green and blue curves in Fig. 2, ESI†). This new peak is associated with a metalation reaction of the amine and the iminic components with a Cu adatom.32,33 On the other hand, the intensity of the signal at 399.9 eV decreases strongly at high temperature and undergoes a small shift, 0.2 eV towards high BE, which we attribute to the interaction with the substrate and the consequent charge transfer. We can envision three possible scenarios for such an NH component: (i) a small amount of molecular networks incorporating NH groups (compatible with their less favourable stabilization energy, as judged by first principle calculations, see below), (ii) molecules located at network edges, and (iii) isolated groups of molecules not forming a metal–organic framework (see regions marked in white in Fig. S1b and S2b, ESI†).
Finally, in the case of the C1s XPS core level spectra, two main emissions are observed for the sample deposited at RT. The signal located at 284.6 eV corresponds to aromatic C–C bonds, while the emission at 285.9 eV stands for the C–N and C–O components.34–36 These two components are also present for the two metal–organic networks obtained upon annealing (see blue and green spectra in the right panel of Fig. 2).
Therefore, our XPS results show that molecules deposited at RT undergo an incomplete chemical transformation (complete and partial dehydrogenation of the alcohol and the amine groups, respectively), becoming activated. In this case, the high initial p-AP coverage of the surface (close to a monolayer, see Fig. S1a, ESI†) hinders molecular movement and rotation, and thus kinetic reaction control plays the major role in the formation of the metal–organic network. In contrast, molecules deposited on the hot substrate are activated upon absorption, as indicated by XPS, while the molecular density increases gradually, favouring processes involving diffusion and rotation and allowing them to find the most favourable global reaction pathway in a thermodynamically controlled reaction.
To gain insights into the specific structural characteristics of the two different networks, a large battery of ab initio Density Functional Theory (DFT)-based calculations were performed. Fig. 3 summarizes the main results of these calculations, where the two ground-state networks with Kagome and honeycomb structures and lattice parameters of 2.07 and 1.42 nm are obtained, in excellent agreement with the experimental values of 2.11 ± 0.06 and 1.5 ± 0.05 nm, respectively. Both networks are formed by the p-AP precursors with their functional groups fully dehydrogenated, as evidenced by the main peaks of the XPS signals. It is worth noting that the computed stabilization energy for networks with partially dehydrogenated amine groups is less favourable by around 0.9 eV per molecule, complicating their formation. The geometry of the lattice shown in Fig. 3e, driven by the 4-fold coordination to the Cu adatoms, yields a Kagome arrangement. To circumvent steric interactions in the Kagome lattice, the ring of the AP molecule is tilted out of the surface plane by about 30°. Fig. 3f shows a honeycomb lattice with two Cu adatoms in the unit cell and a three-fold coordination of the p-AP building blocks to the Cu adatoms. It is important to mention that C–N bonds in both network structures are compatible with a Pauling bond order of approximately 1, which allows discarding double bonds –N in the N atoms. The results obtained from the theoretical STM simulations (Fig. 3c and d) present a good agreement with both experimental images (Fig. 3a and b), reinforcing the idea of the stability of the ground-state configurations achieved by the structural optimizations. Moreover, these simulations have permitted the dim nodal regions in the honeycomb structure of Fig. 3b to be assigned to Cu adatoms coordinated to terminal O atoms of the precursors (see Fig. 3d and f), whilst the centre of the triangular protrusions corresponds to Cu adatoms linked to terminal N atoms of the p-AP building blocks.
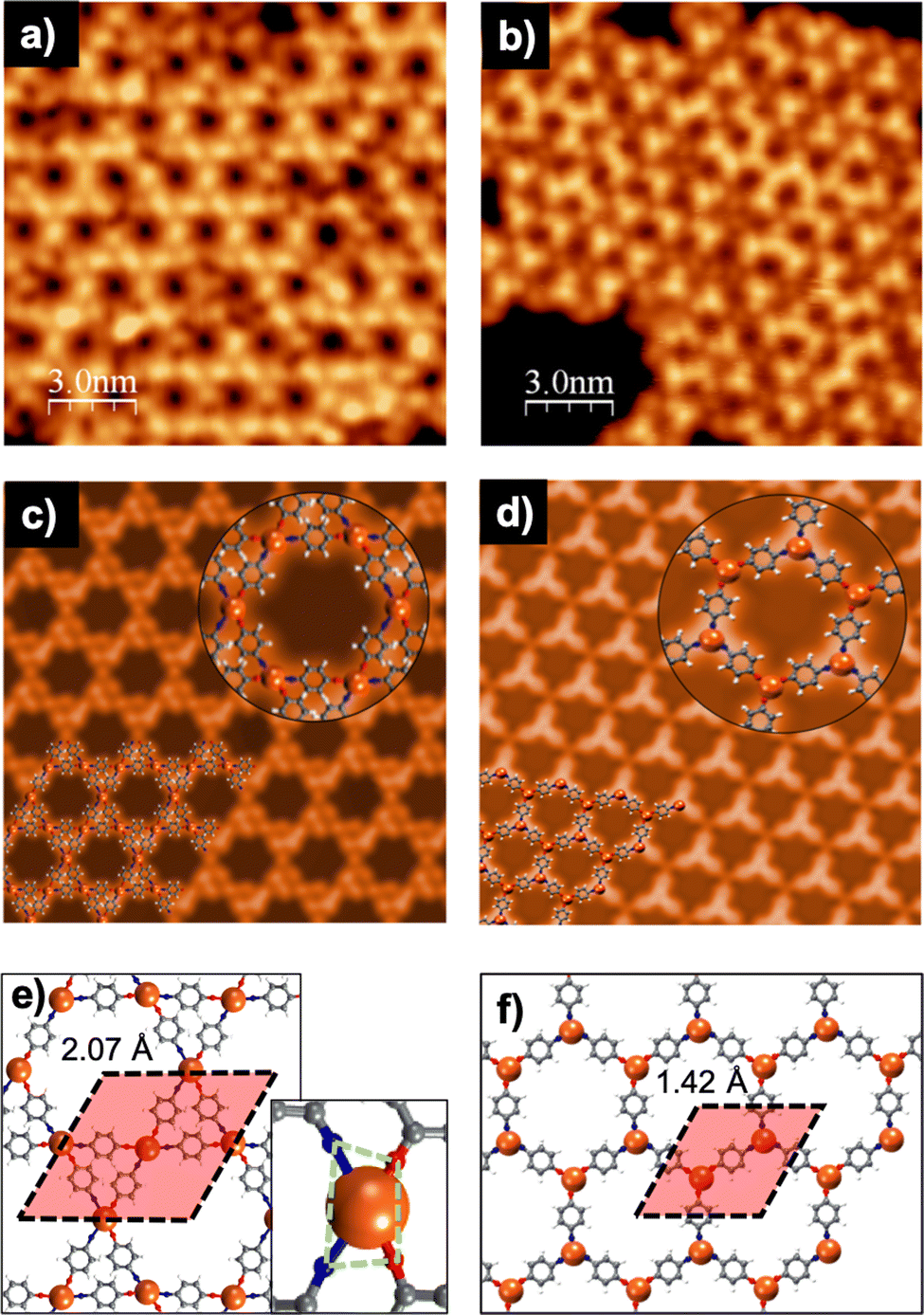 | ||
| Fig. 3 p-AP on Cu(111) for molecular deposition: (left panel) at RT and posterior annealing at ∼500 K, and (right panel) directly on the substrate at ∼500 K. (a and b) LT-STM images (Vbias = 1 V, It = 28 pA and Vbias = 1.1 V, It = 0.24 nA, respectively), (c and d) computed Keldish-Green STM images at constant-current regime (Vbias = 1 V, It = 0.1 nA) with the insets showing a zoomed-in view of the corresponding network structures, and (e and f) pictorial top view of the optimized on-surface network structures and unit cell used in the calculations with the inset in (e) showing a closer view of the Cu coordination. White, gray, blue, red, and tan spheres correspond to H, C, N, O and Cu atoms, respectively. | ||
We have also computed the stabilization energy per molecule in both networks (as the difference between the DFT total energies of the whole interfacial systems and the surfaces with the Cu adatoms and the precursors with no substrate), resulting in −2.04 and −2.32 eV per molecule for the networks in Fig. 3e and f, respectively. This difference may be explained in terms of their different Cu-coordination order, which has its origin in the different synthesis protocol. Additionally, in the Kagome network formed by post-annealing (Fig. 3e), each Cu adatom is bonded to N and O atoms at the same metal site, whereas in the honeycomb network formed directly at high temperature, the metal atom is either bonded to N or to O atoms. The presence of heteroatoms in the unit in 2D-MOFs, in particular N and O, is not unusual.37–39 Nevertheless, the values found in our work for the stabilization energies indicate greater stability for the case of the honeycomb lattice as compared to the Kagome framework. Usually, the 4-fold metal coordination in Kagome networks is square planar, which is highly stable. However, in our case, N and O form a distorted rectangle around the Cu atom (see green polygon in the inset of Fig. 3e). This configuration is probably caused by the hexagonal symmetry of the surface and could rationalize its lower stability. On the other hand, for the honeycomb network, the metal coordination is trigonal planar (see Fig. 3f), which is the most stable one for a 3-fold coordination. The obtained stabilization energies strongly support the assumption of thermodynamic control in the reaction process leading to the honeycomb network, while the Kagome one would be driven by kinetic control.
In summary, on-surface synthesis has been used as a strategy for bottom-up growth of different metal–organic frameworks on a Cu(111) surface. Starting from the same precursor, para-aminophenol molecules, we were able to tune the final network structure to Kagome or honeycomb lattice by controlling the reaction kinetically or thermodynamically, respectively. As evidenced by XPS, both metal–organic frameworks are composed of the same chemical species although they are structurally different, as determined by STM. DFT calculations rationalize both on-surface network structures. This work evidences the role of the reaction conditions on the growth mechanisms of the two metal–organic networks. The different precursor concentrations on the surface influence other factors such as the collision frequency, the degrees of rotation and the surface diffusion of the molecular activated species with a direct impact on the reaction process. This work suggests the possibility of stabilizing metastable 2D phases susceptible to the present tailored electronic structure.
This work was supported by the FotoArt-CM (S2018/NMT-4367) and FotoSurf-CM (Proyectos Sinérgicos I+D, Y2020/NMT-6469) projects funded by the “Comunidad de Madrid” and co-financed by European Structural Funds, and by grants PID2020-113142RB-C21, funded by MCIN/AEI/10.13039/501100011033, and PLEC2021-007906 and TED2021-129999B-C31, funded by MCIN/AEI/10.13039/501100011033 and the “European Union NextGenerationEU/PRTR”. CSS acknowledges grant RYC2018-024364-I funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future” and NRdA the Spanish MINECO for support from the FPI program (BES-2015-072642).
Conflicts of interest
There are no conflicts to declare.References
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., 2004, 116, 2388 CrossRef.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267 RSC.
- Y. Ding, Y.-P. Chen, X. Zhang, L. Chen, Z. Dong, H.-L. Jiang, H. Xu and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 9136 CrossRef CAS PubMed.
- Y. Z. Li, Z. H. Fu and G. Xu, Coord. Chem. Rev., 2019, 388, 79 CrossRef CAS.
- I. F. Chen, C. F. Lu and W. F. Su, Langmuir, 2018, 34, 15754 CrossRef CAS PubMed.
- Z.-Z. Ma, Q.-H. Li, Z. Wang, Z.-G. Gu and J. Zhang, Nat. Commun., 2022, 13, 6347 CrossRef CAS PubMed.
- L.-M. Chang, Q.-H. Li, P. Weidler, Z.-G. Gu, C. Wöll and J. Zhang, CCS Chem., 2022, 4, 3472 CrossRef CAS.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565 CrossRef CAS PubMed.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. J. Zhang, J. Am. Chem. Soc., 2016, 138, 6924 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- A. L. Pinardi, J. I. Martinez, A. Jančařík, I. G. Stará, I. Starý, M. F. López, J. Méndez and J. A. Martín-Gago, Chem. Commun., 2014, 50, 1555 RSC.
- Q. Fan, J. Dai, T. Wang, J. Kuttner, G. Hilt, J. M. Gottfried and J. Zhu, ACS Nano, 2016, 10, 3747 CrossRef CAS PubMed.
- L. Grill and S. Hecht, Nat. Chem., 2020, 12, 115 CrossRef CAS PubMed.
- A. Kumar, K. Banerjee, A. S. Foster and P. Liljeroth, Nano Lett., 2018, 18, 5596 CrossRef CAS PubMed.
- R. Zhang, J. Liu, Y. Gao, M. Hua, B. Xia, P. Knecht, A. C. Papageorgiou, J. Reichert, J. V. Barth, H. Xu, L. Huang and N. Lin, Angew. Chem., Int. Ed., 2020, 59, 2669 CrossRef CAS PubMed.
- S. O. Parreiras, D. Moreno, B. Cirera, M. A. Valbuena, J. I. Urgel, M. Paradinas, M. Panighel, F. Ajejas, M. A. Niño, J. M. Gallego, M. Valvidares, P. Gargiani, W. Kuch, J. I. Martínez, A. Mugarza, J. Camarero, R. Miranda, P. Perna and D. Ecija, Small, 2021, 17, 2170179 CrossRef CAS.
- J. D. Fuhr, L. I. Robino, L. M. Rodriguez, A. Verdini, L. Floreano, H. Ascolani and J. E. Gayone, J. Phys. Chem. C, 2020, 134, 416 CrossRef.
- A. Rochefort, L. Vernisse, A. C. Gómez-Herrero, C. Sánchez-Sánchez, J. A. Martín-Gago, F. Chérioux, S. Clair, J. Coraux and J. I. Martínez, J. Phys. Chem. C, 2021, 125, 17333 CrossRef CAS.
- Z. Zhanf and M. G. Lagally, Science, 1997, 276, 377 CrossRef PubMed.
- L. Alvarez, S. Pelaez, R. Caillard, P. A. Serena, J. A. Martin-Gago and J. Mendez, Nanotechnology, 2010, 21, 305703 CrossRef PubMed.
- D. Moreno, B. Cirera, S. O. Parreiras, J. I. Urgel, N. Gimenez-Agullo, K. Lauwaet, J. M. Gallego, J. R. Galan-Mascaros, J. I. Martinez, P. Ballester, R. Miranda and D. Ecija, Chem. Commun., 2021, 57, 1380 RSC.
- J. V. Barth, G. Constantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS PubMed.
- J. Eichhorn, D. Nieckarz, O. Ochs, D. Samanta, M. Schmittel, P. J. Szabelski and M. Lackinger, ACS Nano, 2014, 8, 7880 CrossRef CAS PubMed.
- M. Fritton, D. A. Duncan, P. S. Deimel, A. Rastgoo-Lahrood, F. Allegretti, J. V. Barth, W. M. Heckl, J. Björk and M. Lackinger, J. Am. Chem. Soc., 2019, 141, 4824 CrossRef CAS PubMed.
- C. K. Krug, Q. Fan, F. Fillsack, J. Glowatzki, N. Trebel, L. J. Heuplick, T. Koehler and J. M. Gottfried, Chem. Commun., 2018, 54, 9741 RSC.
- N. Ruíz del Árbol, C. Sánchez-Sánchez, G. Otero-Irurueta, J. I. Martínez, P. L. de Andrés, A. Gómez-Herrero, P. Merino, M. Piantek, D. Serrate, P. Lacovig, S. Lizzit, J. Alemán, G. Ellis, M. F. López and J. A. Martín-Gago, Angew. Chem., Int. Ed., 2020, 59, 23220 CrossRef PubMed.
- N. Ruiz del Árbol, I. Palacio, G. Otero-Irurueta, J. I. Martínez, P. L. de Andrés, O. Stetsovych, M. Moro-Lagares, P. Mutombo, M. Svec, P. Jelínek, A. Cossaro, L. Floreano, G. J. Ellis, M. F. López and J. A. Martín-Gago, Angew. Chem., Int. Ed., 2018, 57, 8582 CrossRef PubMed.
- Z. F. Wang, Z. Liu and F. Liu, Nat. Commun., 2013, 4, 1 CAS.
- L. Hernández-López, I. Piquero-Zulaica, C. A. Downing, M. Piantek, J. Fujii, D. Serrate, J. E. Ortega, F. Bartolomé and J. Lobo-Checa, Nanoscale, 2021, 13, 5216 RSC.
- M. N. Faraggi, C. Rogero, A. Arnau, M. Trelka, D. Écija, C. Isvoranu, J. Schnadt, C. Marti-Gastaldo, E. Coronado, J. M. Gallego, R. Otero and R. Miranda, J. Phys. Chem. C, 2011, 115, 21177 CrossRef CAS.
- R. González-Moreno, C. Sanchez-Sanchez, M. Trelka, R. Otero, A. Cossaro, A. Verdini, L. Floreano, M. Ruiz-Bermejo, A. García-Lekue, J. A. Martín-Gago and C. Rogero, J. Phys. Chem. C, 2011, 115, 6849 CrossRef.
- Q. Li, B. Yang, J. Björk, Q. G. Zhong, H. Ju, J. J. Zhang, N. Cao, Z. L. Shi, H. M. Zhang, D. Ebeling, A. Schirmeisen, J. F. Zhu and L. F. Chi, J. Am. Chem. Soc., 2018, 140, 6076 CrossRef CAS PubMed.
- G. Olivieri, A. Cossaro, E. Capria, L. Benevoli, M. Coreno, M. De Simone, K. C. Prince, G. Kladnik, D. Cvetko, B. Fraboni, A. Morgante, L. Floreano and A. Fraleoni-Morgera, J. Phys. Chem. C, 2015, 119, 121 CrossRef CAS.
- I. Palacio, A. L. Pinardi, J. I. Martínez, A. Preobrajenski, A. Cossaro, A. Jancarik, I. Stará, I. Starý, J. Méndez, J. A. Martín-Gago and M. F. López, Phys. Chem. Chem. Phys., 2017, 19, 22454 RSC.
- N. Martensson and A. Nilsson, J. Electron Spectrosc. Relat. Phenom., 1990, 52, 1 CrossRef.
- A. Goswami, D. Ghosh, V. V. Chernyshev, A. Dey, D. Pradhan and K. Biradha, ACS Appl. Mater. Interfaces, 2020, 12(30), 33679 CrossRef CAS PubMed.
- Z. H. Li, L. P. Xue, L. Wang, S. T. Zhang and B. T. Zhao, Inorg. Chem. Commun., 2013, 27, 119 CrossRef CAS.
- J. Wang, Y. Zheng, X. Nie, C. Xu, Z. Hao, L. Song, S. You, J. Xi, M. Pan, H. Lin, Y. Li, H. Zhang, Q. Li and L. Chi, J. Phys. Chem. Lett., 2021, 12, 8151 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00185g |
| ‡ Nerea Ruiz del Árbol and Carlos Sánchez-Sánchez contributed equally to this work as first authors. |
| § Current affiliation: School of Mathematics and Physics, The University of Queensland, Brisbane, Queensland 4072, Australia. |
| This journal is © The Royal Society of Chemistry 2023 |